

Abstract
Purpose of review:
Heterotopic ossification (HO) is associated with inflammation. The goal of this review is to examine recent findings on the roles of inflammation and the immune system in HO. We examine how inflammation triggers HO in fibrodysplasia ossificans progressiva, in traumatic HO, and in other clinical conditions of HO. We also discuss how inflammation may be a target for treating HO.
Recent findings:
Both genetic and acquired forms of HO show similarities in their inflammatory cell types and signaling pathways. These include macrophages, mast cells, and adaptive immune cells; along with hypoxia signaling pathways, mesenchymal stem cell differentiation signaling pathways, vascular signaling pathways, and inflammatory cytokines.
Summary:
Because there are common inflammatory mediators across various types of HO, these mediators may serve as common targets for blocking HO. Future research may focus on identifying new inflammatory targets and testing combinatorial therapies based on these results.
Keywords: heterotopic ossification, inflammation, immune activation, macrophages, cytokines
Introduction
Bone homeostasis is closely linked to the immune system. Osteoblasts and osteoclasts are the main cell types thought to maintain the balance of bone formation and resorption. Osteoblasts are thought to be derived from mesenchymal stem cells (MSCs) in the long bones, or MSC-like cells in the neural crest lineage in the craniofacial bones [1]. In contrast, osteoclasts are thought to originate from hematopoietic progenitors within the monocyte/macrophage lineage [2], and their formation is mediated through the receptor activator of NF-κB ligand (RANKL) binding to the RANK receptor [3, 4]. In addition, macrophages appear to promote the formation of osteoblasts because macrophage-deficient mice show a significant reduction in bone density and impaired ability of MSCs to differentiate into osteoblasts [5]. During the fracture repair process, osteal macrophages (sometimes referred to as “osteomacs”), which represent a subtype of macrophages residing in bone tissues, can promote anabolic bone formation in collaboration with pro-inflammatory macrophages [6]. Additionally, multiple diseases affecting the immune system or causing autoimmunity are associated with bone loss [7]. These observations suggest that innate immune cells such as macrophages may be crucial for the pathogenesis of bone disorders and may represent potential therapeutic targets for bone diseases.
Potential links between the immune system and abnormal bone formation, such as in heterotopic ossification (HO), have been discussed for more than twenty years. HO is a debilitating process of abnormal bone formation at a non-bone site [8, 9]. HO often occurs after trauma and in settings of severe inflammation such as in rheumatologic diseases or in burns [9–13].
Inflammatory cytokines secreted by immune cells are thought to be involved in injuries or pathologies prone to HO [14–19]. Pro-inflammatory cytokines including IL-3 are elevated in patients with combat wounds [17]. Interleukin 6 (IL-6), interleukin 10 (IL-10), and monocyte chemoattractant protein 1 (MCP-1) are also reported to be associated with non-genetic HO after high-energy penetrating war injuries [16]. It is also clinically recognized that anti-inflammatory treatment can decrease the incidence of HO after major hip procedures, although it cannot completely block bone formation [15, 18, 20].
These clinical observations indicate that immune and inflammatory processes are major contributors to HO pathogenesis. Here, we will examine the current data supporting the potential roles of the immune and inflammatory systems as drivers of both genetic and acquired HO, and explore how the similarities and differences may help identify therapeutic targets for these devastating conditions.
Fibrodysplasia ossificans progressiva (FOP) as a prototypical disease of genetic heterotopic ossification
Fibrodysplasia ossificans progressiva (FOP) is a rare inherited disease, occurring at an incidence of one per 1.36 million to 2 million people [21]. This disease is characterized by abnormal bone formation in skeletal muscle and in connective tissues which are normally not mineralized. The ossifications are cumulative, leading to immobility and severe pain through progressive extra-skeletal bone formations in skeletal muscles, tendons, and cartilage. There are currently no effective treatments.
FOP is caused by activating mutations in the Activin A type 1 receptor (ACVR1), which is a bone morphogenetic protein (BMP) type 1 receptor [22]. The majority of FOP patients carry the classical single point mutation (ACVR1 R206H), which results in constitutive activation of BMP signaling pathways [22]. Recently, Activin A was identified as a ligand for ACVR1. Normally, Activin A binding to the wild type ACVR1 receptor leads to signaling through the SMAD2/3 pathway. However, Activin A binding to the FOP ACVR1 receptor activates signaling through the SMAD1/5/9 pathway. In addition, Activin A is normally a competitive inhibitor of BMP signaling at the wild type ACVR1 [23–26].
A striking clinical feature of FOP is that patients can develop massive inflammatory lesions, even with relatively mild trauma [21]. These inflammatory lesions occur as “flare-ups” that are accompanied by clinical symptoms of inflammation and can result in significant bone formation. Anti-inflammatory agents, including nonsteroidal anti-inflammatory drugs (NSAIDs) and corticosteroids, can help reduce HO formation [21]. Although these anti-inflammatory medications are the standard therapy in both FOP and non-genetic HO, they show poor efficacy in blocking HO formation.
Inflammation in FOP
Potential roles for the immune system in FOP have been discussed for many years. Shafritz et al. reported that overexpression of bone morphogenetic protein 4 (BMP4) in lymphocytes is associated with the osteogenesis observed in FOP [27]. Recently, studies using mouse models have shown how immune cells contribute to bone formation in FOP. Kaplan et al. reported that cells of hematopoietic origin contributed to the early inflammatory and late marrow-repopulating phases of BMP4-induced HO but were not represented in the fibroproliferative, chondrogenic, or osteogenic stages of HO [28]. They later suggested that inflammation may trigger Tie-2 expressing precursor cells to form abnormal bone formation [29]. Chakkalakal et al. created a mouse model possessing an R206H mutation in ACVR1 and found that various kinds of immune cells, including macrophages and mast cells, were present in sites showing abnormal bone formation [30]. Wang et al. recently showed that systemic suppression of TGF-β attenuates HO in FOP mouse models, implicating that the TGF-β pathway is an inducer and promoter of HO [31]. As TGF-β is a cytokine secreted by M2 tissue-repair macrophages, these reports suggest that myeloid cell lineages may play a crucial role in driving the early phase of inflammation in FOP.
In humans, we reported that FOP patient-derived blood samples showed significant elevations of cytokine levels, including interleukin 3 (IL-3), interleukin 7 (IL-7), interleukin 8 (IL-8), and granulocyte-macrophage colony-stimulating factor (GM-CSF) [19]. Purified monocytes from peripheral blood samples of FOP patients showed increased responsiveness to lipopolysaccharide (LPS) stimulation and prolonged activation of NF-kB compared with control samples, indicating that NF-kB activation underlies inflammation in FOP. Also, our study showed increased serum levels of TGF-β in FOP patients, compatible with a previous study in mice [31]. Although BMP receptors are robustly expressed on monocytes and tissue macrophages [32], how the ACVR1 mutation induces macrophage hyper-responsiveness to stimulation, and whether M1 and M2 macrophages have different functions during abnormal bone formation in FOP, are still unknown. Notably, a separate study based on the analysis of peripheral blood mononuclear cells from FOP patients showed that the expression level of DNAM1 is increased in FOP monocytes and that DNAM1 plays an important role in monocyte migration [33]. Since DNAM1 can be expressed by immune cells, these data suggest there are multiple pathways are likely involved in the inflammatory phase in FOP.
In addition to myeloid cells, other kinds of immune cells such as mast cells and lymphoid cells may contribute to the inflammation of FOP. Gannon et al. found that an intense perivascular lymphocytic infiltration could occur even in normal-appearing skeletal muscle in a 2-year-old child with FOP [34]. Kan et al. showed that RAG1 null mice, which have no mature B or T lymphocytes, developed HO after injury without delay, and that the loss of these specific lineages decreased the rate of spreading and overall amount of HO in heterozygous mice. These results indicated that the adaptive immune system was not necessary for the initial formation of HO but might be important for the spreading of HO [35, 36]. Ranganathan et al. also reported that HO growth and development after a burn injury are both attenuated in the absence of mature B- and T- lymphocytes [37]. Finally, early FOP lesions were shown to exhibit perivascular inflammatory infiltrates and cells expressing hypoxia inducible factor 1 alpha (HIF1α) [38]. These findings suggest that inflammation and hypoxia may be important drivers of FOP HO.
In 2001, Gannon et al. showed inflammatory mast cells were present at every stage of development of FOP lesions in humans [39]. In mice, Convente et al. showed that depletion of mast cells reduced HO volume to about 50% in conditional-on knock-in mouse Acvr1R206H. In addition, combined depletion of mast cells and macrophages reduced HO volume to about 25%, indicating that mast cells may also contribute to HO formation [40]. Our previous study supports this notion since blood from patients with FOP shows high levels of interleukin 9 (IL-9), a cytokine which can be secreted from mast cells [19]. Taken together, these human and mouse data indicate that macrophages, mast cells, and adaptive immune cells have different roles in different phases of inflammation in FOP.
Inflammation in traumatic heterotopic ossification
Inflammation is thought to be a key driver of non-genetic HO. Triggers of non-genetic HO include blast injuries, burns, spinal cord injuries, brain injuries, and some surgical procedures [41–43].
Blast injuries and burns cause high levels of systemic inflammation and predispose patients to HO [42]. Blast injuries may induce expression of substance P (SP-1), which is a neuroinflammatory peptide capable of promoting osteogenic differentiation of MSCs [44]. Blast injuries may also induce expression of various bone and osteoblast mineralization genes and inflammatory cytokine genes, including runt-related transcription factor 2 (Runx2), osteocalcin (Ocn), IL-6, interleukin 1β (IL-1β), C-C motif chemokine ligands 2 and 3 (Ccl2 and Ccl3), and C-X-C motif chemokine ligand 5 (Cxcl5) [44]. Large-surface-area burns that place patients at risk for HO have been associated with altered expression of genes involved in inflammation and vascularization, including HIF1α, von Willebrand factor (vWF), platelet endothelial cell adhesion molecule-1 (PECAM), cadherin 5 (CDH5), and vascular endothelial growth factor (VEGF) [41]. After burn injuries, Hif1α has been shown to colocalize with SRY-box transcription factor 9 (Sox9) in precartilage tissue, immature HO tissue, and mature HO tissue, suggesting Hif1α may be involved throughout development of HO [41]. Sung Hsieh et al. reported that a mouse model of trauma-induced HO with hind limb Achilles’ tenotomy and dorsal burn showed significantly increased levels of tumor necrosis factor alpha (TNF-α) and IL-1β, which are typically secreted from monocyte or M1 pro-inflammatory macrophages. In addition, elevated levels of monocyte chemoattractant protein-1 (MCP-1) and VEGF in saliva persisted for 1 week after injury in trauma-induced HO mice [45], suggesting that cytokines related to myeloid cell functions are important in HO.
Spinal cord injuries and traumatic brain injuries can result in development of HO, especially in peri-articular muscles [43]. In response to spinal cord injury, monocytes may infiltrate muscles and express oncostatin M [46]; macrophages may secrete oncostatin M to mediate inflammation [47, 48]; and muscle satellite and interstitial cells may express the oncostatin M receptor [46]. All these events may help trigger HO.
Some surgeries, such as total hip arthroplasty, are associated with increased HO risk from the tissue trauma that occurs during surgery and from associated inflammation [42, 49, 50]. In one study after total hip arthroplasty, bone marrow-derived cells positive for type 1 collagen and CD45 were identified as markers for a circulating osteogenic precursor cell population that may migrate to and seed sites of inflammation [51]. In contrast to the data in FOP, activation of adaptive immune cells may also be associated with suppression of HO formation in some cases. Hoff et al. analyzed the samples of patients who received preoperative radiation before total hip arthroplasty to prevent HO and found significantly increased CD8+ T cells and decreased naïve IgD+ B cells, indicating that an activation of cytotoxic T cells and B cells help prevent HO [52]. Although the human immune system might be different from mice, an increase in osteogenesis from allogenic MSCs was shown in T cell-deficient mice, indicating that T cells may be able to inhibit osteogenesis [53].
Inflammation in other clinical conditions of heterotopic ossification
There are other examples of HO that appear to be triggered by inflammation including HO in rheumatic diseases [54–56] and HO in cardiovascular pathologies [57, 58].
Inflammation and HO can occur in rheumatic diseases such as scleroderma and dermatomyositis [54–56]. Scleroderma is an autoimmune disease mostly affecting the skin and musculoskeletal tissues [59]. In a case report, a patient with scleroderma exhibited extensive calcification and ossification of the right gluteal muscle and right thigh muscles [54]. This patient also exhibited arthritis in the right knee, which immobilized the right lower limb. One hypothesis for the disease mechanism in this patient is that limb immobility as a result of knee arthritis contributed to hypoxia of nearby muscles, leading to ossification. Also, dermatomyositis is an autoimmune disease characterized by chronic muscle inflammation and muscle weakness [60]. Multiple juvenile dermatomyositis patients have exhibited calcium deposits in their soft tissues, including the deep intermuscular fascial planes [55]. Juvenile dermatomyositis patients have also exhibited myofibers and macrophages that express HIF1α [56]. One particular dermatomyositis patient exhibited calcium deposits in soft tissues as a child and subsequent extensive HO in the left thigh muscle as an adult [55]. These observations from rheumatic diseases emphasize that hypoxia signaling pathways play a role in inflammation and HO.
Inflammation and HO have also been observed in cardiovascular pathologies such as atherosclerosis and aortic valve disease [57, 58]. Late-stage atherosclerotic plaques in human carotid arteries can show mineralized bone deposits [57] with associated smooth muscle-like cells, macrophages, mast cells, and multinucleated giant cells near the bone deposits. Some aortic valve disease lesions also showed bone deposits [58]. These bone deposits appeared in the leaflets of the aortic valves and contained organized calcification and bone marrow containing immune cells, including kappa light chain-secreting cells, lambda light chain-secreting cells, mast cells, neutrophils, and plasma cells that sometimes contained Russell bodies [58]. Vascular calcification has been observed in other conditions associated with increased cardiovascular risk, including diabetes, hypertension, hyperlipidemia, and kidney disease [57, 61–63].
The occurrence of inflammation and HO in cardiovascular-related diseases has prompted investigation into the cell types that may be involved in these cases. Cells important for vascular ossification are thought to originate from either the blood vessel walls, the surrounding tissue, or the circulating blood. In the blood vessel walls, calcifying vascular cells (CVCs) can differentiate into various mesenchymal cell lineages [64–67]. Endothelial cells may differentiate into mesenchymal stem-like cells and then into osteoblasts [68], and endothelial precursors may differentiate and form bone in response to inflammatory triggers [29]. Also, adventitial pericytes and myofibroblasts may migrate from surrounding tissue into the vessel wall [69], and particular subpopulations of CD14 monocytes circulating in the blood may lead to mesenchymal precursors that could play a role in HO [70]. Overall, these observations from cardiovascular diseases suggest that multiple cell types and cellular differentiation may be important contributors to HO.
Inflammation as a target for treating heterotopic ossification
Because inflammation is involved in FOP and other types of HO, new HO treatments are being tested for their effects on inflammation. Retinoid signaling is decreased during chondrogenesis, which is required for HO formation via the endochondral ossification pathway [71]. Therefore, one approach to treating HO involves using retinoid agonists to block chondrogenesis [72]. A potent retinoic acid receptor gamma (RARγ) agonist known as palovarotene has been shown to reduce HO in FOP mouse models [73, 74] and in a blast-related HO rat model [75]. In the blast-related HO mouse model, palovarotene dampened systemic and local inflammatory responses and specifically reduced levels of IL-6, TNFα, and IFNγ [75]. Clinical trials are underway for treatment of FOP patients using palovarotene (NCT03312634, NCT02190747, and NCT02279095). In addition, direct inhibition of Activin A by the neutralizing antibody REGN2477 is being tested in clinical trials as a potential target for blocking HO in FOP (NCT03188666).
Another method of treating blast-related HO may involve use of rapamycin. Rapamycin affects the mammalian target of rapamycin (mTOR) signaling pathway, and the mTOR signaling pathway is an effector of inflammation and hypoxia, both of which play an important role in traumatic injuries [76]. Rapamycin has been shown to prevent HO after blast-related injury in rats and has been shown to attenuate expression of inflammatory genes such as Cxcl5, C-X-C motif chemokine ligand 10 (Cxcl10), IL-6, and Ccl2 [77]. Rapamycin is also currently being tested in clinical trials as a therapy for FOP [78–80].
A method of treating HO after surgical trauma may involve use of celecoxib. Celecoxib is an NSAID and a cyclooxygenase-2 inhibitor, and global anti-inflammatory treatment of HO using celecoxib has been shown to reduce HO formation after surgical trauma in mice [81]. A clinical trial for celecoxib was performed and showed some positive results [82]. However, not all NSAIDs appear to reduce HO formation: for example, indomethacin did not prevent HO in a blast-related HO rat model [83]. Therefore, additional work on how NSAIDs can be used to treat HO may be helpful.
Recently, another strategy for blocking HO was identified for spinal cord injuries using ruxolitinib, a Janus kinase 1 and 2 (JAK1/2) tyrosine kinase inhibitor. Phosphorylation of Signal Transducer and Activator of Transcription 3 (Stat3), a part of the JAK-STAT signaling pathway, can be induced by the pro-inflammatory cytokine OSM, which is produced by myeloid cells after spinal cord injury [46]. In a spinal cord injury HO mouse model, ruxolitinib reduced phosphorylation of Stat3 and reduced development of HO [46].
Future research directions and conclusions
Although inflammation and trauma are linked to HO, the exact mechanisms linking these processes and the potential therapeutic targets embedded in these processes still remain largely unknown. So far, these mechanisms are known to involve macrophages, mast cells, and adaptive immune cells, and these cell types play different roles throughout the development of HO. There are commonalities between inflammation in FOP and inflammation in other types of HO, including the involvement of hypoxia signaling pathways, the triggering of MSC differentiation, and the triggering of vascular signaling pathways.
A number of major questions remain: Although some rheumatic diseases including systemic scleroderma are reported to be complicated by HO [54], how this differs from chronic inflammation and other rheumatic diseases associated with bone loss [7] is unclear. In addition, how innate immune cells ultimately trigger activation of a skeletal stem cell type to induce mineralization and ossification remains unknown. Finally, while pharmacologic immune modulation appears to be a useful strategy for mitigating HO, we still have large gaps in understanding exactly how some pharmacologic agents interact with the immune system to affect HO. Furthermore, finding additional specific targets that could be used to effectively block HO or developing combinatorial therapies for HO would be useful. Ongoing investigation into these mechanisms promises to reveal exciting insights that will help guide future directed therapies for preventing HO.
Figure 1:
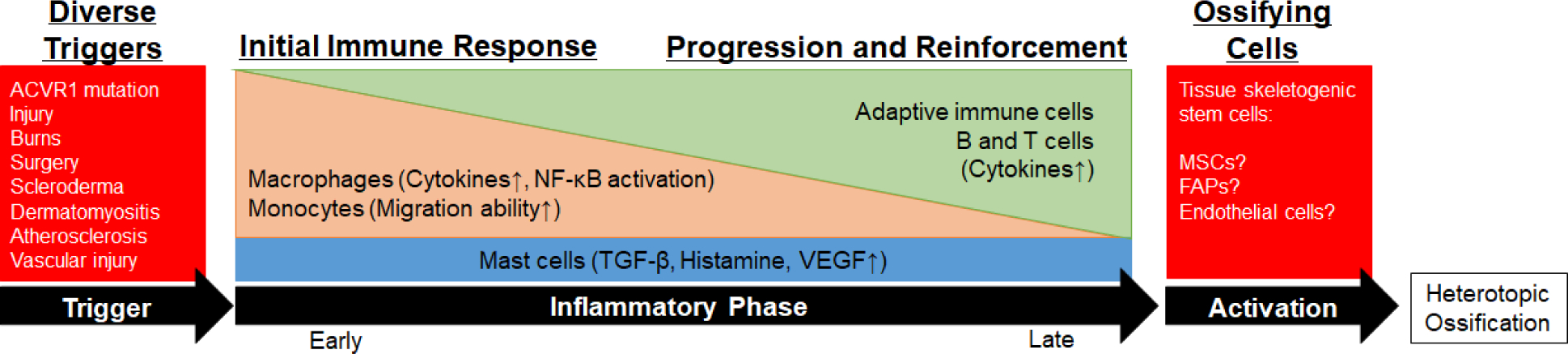
Inflammation and Immunity are common features in many forms of heterotopic ossification. Despite the diversity of initiating triggers, and differences in the specific immune responses that result, inflammation generally appears to serve as a common pathway for initiating the ossification process. The specific links between inflammation and the ossifying skeletogenic stem cells are largely unknown. MSCs, mesenchymal stem cells; FAPs, fibro-adipogenic progenitors.
Acknowledgements:
The authors thank Mary Nakamura for critical advice on this manuscript. ECH received support from 1R01AR066735 and 1R01AR073015. RDC received support from 5R21AR072778.
Footnotes
Conflict of Interest Statement:
ECH serves on the registry advisory board of the International Fibrodysplasia Ossificans Progressiva Association the International Clinical Council on FOP, and on the Fibrous Dysplasia Foundation Medical Advisory Board. ECH receives clinical trials research support from Clementia Pharmaceuticals Inc. to support clinical trials of palovarotene in FOP. These pose no conflicts for this study.
References
- 1.Long F Building strong bones: molecular regulation of the osteoblast lineage. Nature reviews Molecular cell biology 2011:13(1):27–38. doi: 10.1038/nrm3254 [DOI] [PubMed] [Google Scholar]
- 2.Udagawa N The mechanism of osteoclast differentiation from macrophages: possible roles of T lymphocytes in osteoclastogenesis. Journal of bone and mineral metabolism 2003:21(6):337–43. doi: 10.1007/s00774-003-0439-1 [DOI] [PubMed] [Google Scholar]
- 3.Nakagawa N, Kinosaki M, Yamaguchi K, Shima N, Yasuda H, Yano K, et al. RANK is the essential signaling receptor for osteoclast differentiation factor in osteoclastogenesis. Biochemical and biophysical research communications 1998:253(2):395–400. doi: 10.1006/bbrc.1998.9788 [DOI] [PubMed] [Google Scholar]
- 4.Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proceedings of the National Academy of Sciences of the United States of America 1998:95(7):3597–602. doi: 10.1073/pnas.95.7.3597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vi L, Baht GS, Whetstone H, Ng A, Wei Q, Poon R, et al. Macrophages promote osteoblastic differentiation in-vivo: implications in fracture repair and bone homeostasis. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2015:30(6):1090–102. doi: 10.1002/jbmr.2422 [DOI] [PubMed] [Google Scholar]
- 6.Raggatt LJ, Wullschleger ME, Alexander KA, Wu AC, Millard SM, Kaur S, et al. Fracture healing via periosteal callus formation requires macrophages for both initiation and progression of early endochondral ossification. The American journal of pathology 2014:184(12):3192–204. doi: 10.1016/j.ajpath.2014.08.017 [DOI] [PubMed] [Google Scholar]
- 7.Negishi-Koga T, Gober HJ, Sumiya E, Komatsu N, Okamoto K, Sawa S, et al. Immune complexes regulate bone metabolism through FcRgamma signalling. Nature communications 2015:6:6637. doi: 10.1038/ncomms7637 [DOI] [PubMed] [Google Scholar]
- 8.Kaplan FS, Chakkalakal SA, Shore EM. Fibrodysplasia ossificans progressiva: mechanisms and models of skeletal metamorphosis. Dis Model Mech 2012:5(6):756–62. doi: 10.1242/dmm.010280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCarthy EF, Sundaram M. Heterotopic ossification: a review. Skeletal radiology 2005:34(10):609–19. doi: 10.1007/s00256-005-0958-z [DOI] [PubMed] [Google Scholar]
- 10.Kocic M, Lazovic M, Mitkovic M, Djokic B. Clinical significance of the heterotopic ossification after total hip arthroplasty. Orthopedics 2010:33(1):16. doi: 10.3928/01477447-20091124-13 [DOI] [PubMed] [Google Scholar]
- 11.Leung C, Casey AT, Goffin J, Kehr P, Liebig K, Lind B, et al. Clinical significance of heterotopic ossification in cervical disc replacement: a prospective multicenter clinical trial. Neurosurgery 2005:57(4):759–63; discussion −63. doi: 10.1093/neurosurgery/57.4.759 [DOI] [PubMed] [Google Scholar]
- 12.Hurvitz EA, Mandac BR, Davidoff G, Johnson JH, Nelson VS. Risk factors for heterotopic ossification in children and adolescents with severe traumatic brain injury. Arch Phys Med Rehabil 1992:73(5):459–62. [PubMed] [Google Scholar]
- 13.Potter BK, Burns TC, Lacap AP, Granville RR, Gajewski DA. Heterotopic ossification following traumatic and combat-related amputations. Prevalence, risk factors, and preliminary results of excision. J Bone Joint Surg Am 2007:89(3):476–86. doi: 10.2106/JBJS.F.00412 [DOI] [PubMed] [Google Scholar]
- 14.Schett G Effects of inflammatory and anti-inflammatory cytokines on the bone. European journal of clinical investigation 2011:41(12):1361–6. doi: 10.1111/j.1365-2362.2011.02545.x [DOI] [PubMed] [Google Scholar]
- 15.Beckmann JT, Wylie JD, Potter MQ, Maak TG, Greene TH, Aoki SK. Effect of Naproxen Prophylaxis on Heterotopic Ossification Following Hip Arthroscopy: A Double-Blind Randomized Placebo-Controlled Trial. The Journal of bone and joint surgery American volume 2015:97(24):2032–7. doi: 10.2106/JBJS.N.01156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Evans KN, Forsberg JA, Potter BK, Hawksworth JS, Brown TS, Andersen R, et al. Inflammatory cytokine and chemokine expression is associated with heterotopic ossification in high-energy penetrating war injuries. Journal of orthopaedic trauma 2012:26(11):e204–13. doi: 10.1097/BOT.0b013e31825d60a5 [DOI] [PubMed] [Google Scholar]
- 17.Forsberg JA, Potter BK, Polfer EM, Safford SD, Elster EA. Do inflammatory markers portend heterotopic ossification and wound failure in combat wounds? Clinical orthopaedics and related research 2014:472(9):2845–54. doi: 10.1007/s11999-014-3694-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pakos EE, Ioannidis JP. Radiotherapy vs. nonsteroidal anti-inflammatory drugs for the prevention of heterotopic ossification after major hip procedures: a meta-analysis of randomized trials. International journal of radiation oncology, biology, physics 2004:60(3):888–95. doi: 10.1016/j.ijrobp.2003.11.015 [DOI] [PubMed] [Google Scholar]
- 19.•.Barruet E, Morales BM, Cain CJ, Ton AN, Wentworth KL, Chan TV, et al. NF-kappaB/MAPK activation underlies ACVR1-mediated inflammation in human heterotopic ossification. JCI insight 2018:3(22). doi: 10.1172/jci.insight.122958 [DOI] [PMC free article] [PubMed] [Google Scholar]; Analysis of FOP patient-derived sera and monocytes strongly indicates the contribution of myeloid cells in ACVR1-mediated inflammation.
- 20.The International Clinical Consortium on FOP The Medical Management of Fibrodysplasia Ossificans Progressiva: Current Treatment Considerations. Clin Proc Intl Clin Consort FOP 2011:4:1–100. [Google Scholar]
- 21.Baujat G, Choquet R, Bouee S, Jeanbat V, Courouve L, Ruel A, et al. Prevalence of fibrodysplasia ossificans progressiva (FOP) in France: an estimate based on a record linkage of two national databases. Orphanet journal of rare diseases 2017:12(1):123. doi: 10.1186/s13023-017-0674-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nature genetics 2006:38(5):525–7. doi: 10.1038/ng1783 [DOI] [PubMed] [Google Scholar]
- 23.Aykul S, Martinez-Hackert E. Transforming Growth Factor-beta Family Ligands Can Function as Antagonists by Competing for Type II Receptor Binding. The Journal of biological chemistry 2016:291(20):10792–804. doi: 10.1074/jbc.M115.713487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.•.Hino K, Ikeya M, Horigome K, Matsumoto Y, Ebise H, Nishio M, et al. Neofunction of ACVR1 in fibrodysplasia ossificans progressiva. Proceedings of the National Academy of Sciences of the United States of America 2015:112(50):15438–43. doi: 10.1073/pnas.1510540112 [DOI] [PMC free article] [PubMed] [Google Scholar]; This report reveals a novel mechaism of BMP signaling through mutated ACVR1 in FOP.
- 25.Olsen OE, Wader KF, Hella H, Mylin AK, Turesson I, Nesthus I, et al. Activin A inhibits BMP-signaling by binding ACVR2A and ACVR2B. Cell communication and signaling : CCS 2015:13:27. doi: 10.1186/s12964-015-0104-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.•.Hatsell SJ, Idone V, Wolken DM, Huang L, Kim HJ, Wang L, et al. ACVR1R206H receptor mutation causes fibrodysplasia ossificans progressiva by imparting responsiveness to activin A. Science translational medicine 2015:7(303):303ra137. doi: 10.1126/scitranslmed.aac4358 [DOI] [PMC free article] [PubMed] [Google Scholar]; Activin A, a cytokine produced by many cell types including immune cells, appears to be a key driver of heterotopic ossification in FOP.
- 27.Shafritz AB, Shore EM, Gannon FH, Zasloff MA, Taub R, Muenke M, et al. Overexpression of an osteogenic morphogen in fibrodysplasia ossificans progressiva. The New England journal of medicine 1996:335(8):555–61. doi: 10.1056/NEJM199608223350804 [DOI] [PubMed] [Google Scholar]
- 28.Kaplan FS, Glaser DL, Shore EM, Pignolo RJ, Xu M, Zhang Y, et al. Hematopoietic stem-cell contribution to ectopic skeletogenesis. The Journal of bone and joint surgery American volume 2007:89(2):347–57. doi: 10.2106/JBJS.F.00472 [DOI] [PubMed] [Google Scholar]
- 29.Lounev VY, Ramachandran R, Wosczyna MN, Yamamoto M, Maidment AD, Shore EM, et al. Identification of progenitor cells that contribute to heterotopic skeletogenesis. The Journal of bone and joint surgery American volume 2009:91(3):652–63. doi: 10.2106/JBJS.H.01177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chakkalakal SA, Zhang D, Culbert AL, Convente MR, Caron RJ, Wright AC, et al. An Acvr1 R206H knock-in mouse has fibrodysplasia ossificans progressiva. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2012:27(8):1746–56. doi: 10.1002/jbmr.1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.•.Wang X, Li F, Xie L, Crane J, Zhen G, Mishina Y, et al. Inhibition of overactive TGF-beta attenuates progression of heterotopic ossification in mice. Nature communications 2018:9(1):551. doi: 10.1038/s41467-018-02988-5 [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper suggests the importance of TGF-beta signaling in FOP, indicating TGF-beta is a potential therapeutic target.
- 32.Champagne CM, Takebe J, Offenbacher S, Cooper LF. Macrophage cell lines produce osteoinductive signals that include bone morphogenetic protein-2. Bone 2002:30(1):26–31. [DOI] [PubMed] [Google Scholar]
- 33.Del Zotto G, Antonini F, Azzari I, Ortolani C, Tripodi G, Giacopelli F, et al. Peripheral Blood Mononuclear Cell Immunophenotyping in Fibrodysplasia Ossificans Progressiva Patients: Evidence for Monocyte DNAM1 Up-regulation. Cytometry Part B, Clinical cytometry 2018:94(4):613–22. doi: 10.1002/cyto.b.21594 [DOI] [PubMed] [Google Scholar]
- 34.Gannon FH, Valentine BA, Shore EM, Zasloff MA, Kaplan FS. Acute lymphocytic infiltration in an extremely early lesion of fibrodysplasia ossificans progressiva. Clinical orthopaedics and related research 1998(346):19–25. [PubMed] [Google Scholar]
- 35.Kan L, Hu M, Gomes WA, Kessler JA. Transgenic mice overexpressing BMP4 develop a fibrodysplasia ossificans progressiva (FOP)-like phenotype. The American journal of pathology 2004:165(4):1107–15. doi: 10.1016/S0002-9440(10)63372-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kan L, Liu Y, McGuire TL, Berger DM, Awatramani RB, Dymecki SM, et al. Dysregulation of local stem/progenitor cells as a common cellular mechanism for heterotopic ossification. Stem cells 2009:27(1):150–6. doi: 10.1634/stemcells.2008-0576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ranganathan K, Agarwal S, Cholok D, Loder S, Li J, Sung Hsieh HH, et al. The role of the adaptive immune system in burn-induced heterotopic ossification and mesenchymal cell osteogenic differentiation. The Journal of surgical research 2016:206(1):53–61. doi: 10.1016/j.jss.2016.04.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang H, Lindborg C, Lounev V, Kim JH, McCarrick-Walmsley R, Xu M, et al. Cellular Hypoxia Promotes Heterotopic Ossification by Amplifying BMP Signaling. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2016:31(9):1652–65. doi: 10.1002/jbmr.2848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gannon FH, Glaser D, Caron R, Thompson LD, Shore EM, Kaplan FS. Mast cell involvement in fibrodysplasia ossificans progressiva. Human pathology 2001:32(8):842–8. doi: 10.1053/hupa.2001.26464 [DOI] [PubMed] [Google Scholar]
- 40.Convente MR, Chakkalakal SA, Yang E, Caron RJ, Zhang D, Kambayashi T, et al. Depletion of Mast Cells and Macrophages Impairs Heterotopic Ossification in an Acvr1(R206H) Mouse Model of Fibrodysplasia Ossificans Progressiva. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2018:33(2):269–82. doi: 10.1002/jbmr.3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Agarwal S, Loder S, Brownley C, Cholok D, Mangiavini L, Li J, et al. Inhibition of Hif1alpha prevents both trauma-induced and genetic heterotopic ossification. Proceedings of the National Academy of Sciences of the United States of America 2016:113(3):E338–47. doi: 10.1073/pnas.1515397113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meyers C, Lisiecki J, Miller S, Levin A, Fayad L, Ding C, et al. Heterotopic Ossification: A Comprehensive Review. JBMR plus 2019:3(4):e10172. doi: 10.1002/jbm4.10172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sullivan MP, Torres SJ, Mehta S, Ahn J. Heterotopic ossification after central nervous system trauma: A current review. Bone & joint research 2013:2(3):51–7. doi: 10.1302/2046-3758.23.2000152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qureshi AT, Crump EK, Pavey GJ, Hope DN, Forsberg JA, Davis TA. Early Characterization of Blast-related Heterotopic Ossification in a Rat Model. Clinical orthopaedics and related research 2015:473(9):2831–9. doi: 10.1007/s11999-015-4240-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sung Hsieh HH, Chung MT, Allen RM, Ranganathan K, Habbouche J, Cholok D, et al. Evaluation of Salivary Cytokines for Diagnosis of both Trauma-Induced and Genetic Heterotopic Ossification. Frontiers in endocrinology 2017:8:74. doi: 10.3389/fendo.2017.00074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Alexander KA, Tseng HW, Fleming W, Jose B, Salga M, Kulina I, et al. Inhibition of JAK1/2 Tyrosine Kinases Reduces Neurogenic Heterotopic Ossification After Spinal Cord Injury. Frontiers in immunology 2019:10:377. doi: 10.3389/fimmu.2019.00377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Genet F, Kulina I, Vaquette C, Torossian F, Millard S, Pettit AR, et al. Neurological heterotopic ossification following spinal cord injury is triggered by macrophage-mediated inflammation in muscle. The Journal of pathology 2015:236(2):229–40. doi: 10.1002/path.4519 [DOI] [PubMed] [Google Scholar]
- 48.Torossian F, Guerton B, Anginot A, Alexander KA, Desterke C, Soave S, et al. Macrophage-derived oncostatin M contributes to human and mouse neurogenic heterotopic ossifications. JCI insight 2017:2(21). doi: 10.1172/jci.insight.96034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Louie CE, Hong J, Bauer DF. Heterotopic ossification following suboccipital craniectomy decompression surgery for Chiari malformation type I: case report. Journal of neurosurgery Pediatrics 2019:1–4. doi: 10.3171/2019.1.PEDS18680 [DOI] [PubMed] [Google Scholar]
- 50.Newman EA, Holst DC, Bracey DN, Russell GB, Lang JE. Incidence of heterotopic ossification in direct anterior vs posterior approach to total hip arthroplasty: a retrospective radiographic review. International orthopaedics 2016:40(9):1967–73. doi: 10.1007/s00264-015-3048-4 [DOI] [PubMed] [Google Scholar]
- 51.Egan KP, Duque G, Keenan MA, Pignolo RJ. Circulating osteogentic precursor cells in non-hereditary heterotopic ossification. Bone 2018:109:61–4. doi: 10.1016/j.bone.2017.12.028 [DOI] [PubMed] [Google Scholar]
- 52.Hoff P, Rakow A, Gaber T, Hahne M, Senturk U, Strehl C, et al. Preoperative irradiation for the prevention of heterotopic ossification induces local inflammation in humans. Bone 2013:55(1):93–101. doi: 10.1016/j.bone.2013.03.020 [DOI] [PubMed] [Google Scholar]
- 53.Dighe AS, Yang S, Madhu V, Balian G, Cui Q. Interferon gamma and T cells inhibit osteogenesis induced by allogeneic mesenchymal stromal cells. Journal of orthopaedic research : official publication of the Orthopaedic Research Society 2013:31(2):227–34. doi: 10.1002/jor.22212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Botzoris VG, Argyropoulou MI, Voulgari PV, Zikou AK, Drosos AA. Heterotopic ossification in systemic sclerosis. Scandinavian journal of rheumatology 2009:38(4):317–9. doi: 10.1080/03009740902776919 [DOI] [PubMed] [Google Scholar]
- 55.Eckardt JJ, Ivins JC, Perry HO, Unni KK. Osteosarcoma arising in heterotopic ossification of dermatomyositis: case report and review of the literature. Cancer 1981:48(5):1256–61. doi: [DOI] [PubMed] [Google Scholar]
- 56.Preusse C, Allenbach Y, Hoffmann O, Goebel HH, Pehl D, Radke J, et al. Differential roles of hypoxia and innate immunity in juvenile and adult dermatomyositis. Acta neuropathologica communications 2016:4(1):45. doi: 10.1186/s40478-016-0308-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jeziorska M, McCollum C, Wooley DE. Observations on bone formation and remodelling in advanced atherosclerotic lesions of human carotid arteries. Virchows Archiv : an international journal of pathology 1998:433(6):559–65. [DOI] [PubMed] [Google Scholar]
- 58.Steiner I, Kasparova P, Kohout A, Dominik J. Bone formation in cardiac valves: a histopathological study of 128 cases. Virchows Archiv : an international journal of pathology 2007:450(6):653–7. doi: 10.1007/s00428-007-0430-7 [DOI] [PubMed] [Google Scholar]
- 59.Masi AT, Rodnan GP, Medsger TA, Altman RD, D’Angelo WA, Fries JF, et al. Preliminary criteria for the classification of systemic sclerosis (scleroderma). Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Arthritis and rheumatism 1980:23(5):581–90. [DOI] [PubMed] [Google Scholar]
- 60.Dalakas MC, Hohlfeld R. Polymyositis and dermatomyositis. Lancet 2003:362(9388):971–82. doi: 10.1016/S0140-6736(03)14368-1 [DOI] [PubMed] [Google Scholar]
- 61.Hunt JL, Fairman R, Mitchell ME, Carpenter JP, Golden M, Khalapyan T, et al. Bone formation in carotid plaques: a clinicopathological study. Stroke 2002:33(5):1214–9. [DOI] [PubMed] [Google Scholar]
- 62.Mohler ER 3rd, Gannon F, Reynolds C, Zimmerman R, Keane MG, Kaplan FS. Bone formation and inflammation in cardiac valves. Circulation 2001:103(11):1522–8. doi: 10.1161/01.cir.103.11.1522 [DOI] [PubMed] [Google Scholar]
- 63.Soor GS, Vukin I, Leong SW, Oreopoulos G, Butany J. Peripheral vascular disease: who gets it and why? A histomorphological analysis of 261 arterial segments from 58 cases. Pathology 2008:40(4):385–91. doi: 10.1080/00313020802036764 [DOI] [PubMed] [Google Scholar]
- 64.Campbell GR, Campbell JH. Vascular smooth muscle and arterial calcification. Zeitschrift fur Kardiologie 2000:89 Suppl 2:54–62. [DOI] [PubMed] [Google Scholar]
- 65.Cheng SL, Shao JS, Charlton-Kachigian N, Loewy AP, Towler DA. MSX2 promotes osteogenesis and suppresses adipogenic differentiation of multipotent mesenchymal progenitors. The Journal of biological chemistry 2003:278(46):45969–77. doi: 10.1074/jbc.M306972200 [DOI] [PubMed] [Google Scholar]
- 66.Liu T, Lin J, Ju T, Chu L, Zhang L. Vascular smooth muscle cell differentiation to an osteogenic phenotype involves matrix metalloproteinase-2 modulation by homocysteine. Molecular and cellular biochemistry 2015:406(1–2):139–49. doi: 10.1007/s11010-015-2432-0 [DOI] [PubMed] [Google Scholar]
- 67.Tintut Y, Alfonso Z, Saini T, Radcliff K, Watson K, Bostrom K, et al. Multilineage potential of cells from the artery wall. Circulation 2003:108(20):2505–10. doi: 10.1161/01.CIR.0000096485.64373.C5 [DOI] [PubMed] [Google Scholar]
- 68.Medici D, Shore EM, Lounev VY, Kaplan FS, Kalluri R, Olsen BR. Conversion of vascular endothelial cells into multipotent stem-like cells. Nature medicine 2010:16(12):1400–6. doi: 10.1038/nm.2252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Doherty MJ, Ashton BA, Walsh S, Beresford JN, Grant ME, Canfield AE. Vascular pericytes express osteogenic potential in vitro and in vivo. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 1998:13(5):828–38. doi: 10.1359/jbmr.1998.13.5.828 [DOI] [PubMed] [Google Scholar]
- 70.Kuwana M, Okazaki Y, Kodama H, Izumi K, Yasuoka H, Ogawa Y, et al. Human circulating CD14+ monocytes as a source of progenitors that exhibit mesenchymal cell differentiation. Journal of leukocyte biology 2003:74(5):833–45. doi: 10.1189/jlb.0403170 [DOI] [PubMed] [Google Scholar]
- 71.Weston AD, Rosen V, Chandraratna RA, Underhill TM. Regulation of skeletal progenitor differentiation by the BMP and retinoid signaling pathways. The Journal of cell biology 2000:148(4):679–90. doi: 10.1083/jcb.148.4.679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Pacifici M, Cossu G, Molinaro M, Tato F. Vitamin A inhibits chondrogenesis but not myogenesis. Experimental cell research 1980:129(2):469–74. doi: 10.1016/0014-4827(80)90517-0 [DOI] [PubMed] [Google Scholar]
- 73.•.Chakkalakal SA, Uchibe K, Convente MR, Zhang D, Economides AN, Kaplan FS, et al. Palovarotene Inhibits Heterotopic Ossification and Maintains Limb Mobility and Growth in Mice With the Human ACVR1(R206H) Fibrodysplasia Ossificans Progressiva (FOP) Mutation. Journal of bone and mineral research : the official journal of the American Society for Bone and Mineral Research 2016:31(9):1666–75. doi: 10.1002/jbmr.2820 [DOI] [PMC free article] [PubMed] [Google Scholar]; Palovalone is a potential therapeutic agent which is currently being tested in clinical trials in FOP patients. It may function by both blocking chondrocyte mineralization as well as immune cell function.
- 74.Lees-Shepard JB, Nicholas SE, Stoessel SJ, Devarakonda PM, Schneider MJ, Yamamoto M, et al. Palovarotene reduces heterotopic ossification in juvenile FOP mice but exhibits pronounced skeletal toxicity. eLife 2018:7. doi: 10.7554/eLife.40814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wheatley BM, Cilwa KE, Dey D, Qureshi AT, Seavey JG, Tomasino AM, et al. Palovarotene inhibits connective tissue progenitor cell proliferation in a rat model of combat-related heterotopic ossification. Journal of orthopaedic research : official publication of the Orthopaedic Research Society 2018:36(4):1135–44. doi: 10.1002/jor.23747 [DOI] [PubMed] [Google Scholar]
- 76.Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012:149(2):274–93. doi: 10.1016/j.cell.2012.03.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Qureshi AT, Dey D, Sanders EM, Seavey JG, Tomasino AM, Moss K, et al. Inhibition of Mammalian Target of Rapamycin Signaling with Rapamycin Prevents Trauma-Induced Heterotopic Ossification. The American journal of pathology 2017:187(11):2536–45. doi: 10.1016/j.ajpath.2017.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hino K, Zhao C, Horigome K, Nishio M, Okanishi Y, Nagata S, et al. An mTOR Signaling Modulator Suppressed Heterotopic Ossification of Fibrodysplasia Ossificans Progressiva. Stem cell reports 2018:11(5):1106–19. doi: 10.1016/j.stemcr.2018.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.IFOPA. Rapamycin Clinical Trial for FOP 2019. Available from: https://www.ifopa.org/rapamycin.
- 80.Kaplan FS, Zeitlin L, Dunn SP, Benor S, Hagin D, Al Mukaddam M, et al. Acute and chronic rapamycin use in patients with Fibrodysplasia Ossificans Progressiva: A report of two cases. Bone 2018:109:281–4. doi: 10.1016/j.bone.2017.12.011 [DOI] [PubMed] [Google Scholar]
- 81.Peterson JR, De La Rosa S, Eboda O, Cilwa KE, Agarwal S, Buchman SR, et al. Treatment of heterotopic ossification through remote ATP hydrolysis. Science translational medicine 2014:6(255):255ra132. doi: 10.1126/scitranslmed.3008810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lavernia CJ, Contreras JS, Villa JM, Rossi MD. Celecoxib and heterotopic bone formation after total hip arthroplasty. The Journal of arthroplasty 2014:29(2):390–2. doi: 10.1016/j.arth.2013.06.039 [DOI] [PubMed] [Google Scholar]
- 83.Robertson AD, Chiaramonti AM, Nguyen TP, Jaffe DE, Holmes RE, Hanna EL, et al. Failure of Indomethacin and Radiation to Prevent Blast-induced Heterotopic Ossification in a Sprague-Dawley Rat Model. Clinical orthopaedics and related research 2019:477(3):644–54. doi: 10.1097/CORR.0000000000000594 [DOI] [PMC free article] [PubMed] [Google Scholar]