

Abstract
Infections caused by multidrug-resistant bacteria represent a significant and ever-increasing cause of morbidity and mortality. There is thus an urgent need to develop efficient and well-tolerated antibacterials targeting unique cellular processes. Numerous studies have led to the identification of new biological targets to fight bacterial resistance. Two-component signal transduction systems are widely employed by bacteria to translate external and cellular signals into a cellular response. They are ubiquitous in bacteria, absent in the animal kingdom and are integrated into various virulence pathways. Several chemical series, including isothiazolidones, imidazolium salts, benzoxazines, salicylanilides, thiophenes, thiazolidiones, benzimidazoles and other derivatives deduced by different approaches have been reported in the literature to have histidine kinase inhibitory activity. In this review, we report on the design and the synthesis of these histidine kinases inhibitors and their potential to serve as antibacterial agents.
Keywords: Histidine kinase, antibacterial agents, histidine kinase inhibitors, two-component signal transduction systems
I. INTRODUCTION
The discovery of antibiotics in the 1940s is considered the major therapeutic advance of the 20th century. This medical breakthrough changed the future of mankind by significantly reducing the mortality rate caused by bacterial infections. Consequently, the benefits of these “miracle drugs” for the treatment of infectious diseases have been taken for granted, and are essential for modern medicine as a whole.1 Misuse of antibiotics2 combined with a diminished lack of interest by the pharmaceutical industry has led to the emergence of bacterial strains that have become resistant to most families of antibiotics available on the market. In 2004, antibiotics represented fewer than 2% of drugs in clinical development by the 15 largest drug companies.3,4
Exposure to antibiotics leads to rapid selection of bacterial strains with the ability to resist clinical concentrations of antibiotics. Common resistance mechanisms employed by bacteria are: (1) antibiotic destruction or modification, (2) target modification and/or target over-expression, (3) bypass of the target through alternative mechanisms and (4) alteration in drug concentration by blocking uptake or by actively removing the drug from the site of action5 The resistance problem is so dire that the World Health Organization (WHO) has identified multidrug-resistant bacteria as one of the top three threats to human health.6 Hence there is an urgent need to find new antibacterial targets and antibacterial agents to combat multidrug-resistance bacteria.
To survive and adapt to environmental changes, prokaryotic and eukaryotic cells alike use a flurry of signal transduction systems ranging from relatively simple to very complex in terms of number and types of proteins involved.7,8 The essence of signal transduction mechanisms in general is to couple a molecular or physical stimulus with an appropriate cellular response. Bacteria have evolved a variety of mechanisms to detect and respond to environmental changes. The communication between those proteins that detect signals and those that execute the response is commonly mediated by a phosphorylation/dephosphorylation-type mechanism.9,10 Prokaryotes use phosphorylation dependent signal transduction to detect, respond to and adapt to numerous stresses including but not limited to temperature, pH, nutrient concentration and the presence of antibiotics.7,11
In bacteria the most common phosphorylation-dependent signal transduction pathway is the so-called two-component system (TCS) with an average bacterium harboring 20 or more individual systems to initiate various different responses.12 The TCS uses as the receptor protein the sensor histidine kinase (HK) (Figure 1), which modifies its autophosphorylation rate of a conserved histidine residue in response to a stimulus (Figure 1, 1 and 2). Subsequently, the phosphohistidine on the HK serves as a substrate for phosphoryl-group transfer to a conserved aspartate residue on the receiver domain of an associated response regulator (RR) protein (Figure 1, 3). Phosphorylation of the receiver domain on the RR induces conformational changes typically resulting in dimerization of the protein and as a consequence its activation. While some RR proteins have associated enzymatic or protein interaction domains, most RR proteins feature DNA-binding domains and dimerization enhances DNA-binding to specific target sequences in promoter regions. Thus, in the prototypical TCS, the presence of a signal leads to the appropriate changes in transcription of target genes.10,13 (Figure 1, 4) Given a crucial role in bacterial adaptation and survival combined with the absence of similar proteins from the animal kingdom, TCS proteins are considered a possible antimicrobial target. Studies of biological processes governed by TCS have in past decades highlighted the involvement of TCS in bacterial growth, sporulation, biofilm formation, virulence factor expression and even antibiotic resistance itself. 14,15, 16 Involvement in the totality of these processes thus further singles out the TCS as a promising antibacterial-drug discovery target. A significant body of literature now exists describing efforts to chemically target the HK protein of the bacterial TCS. In the first part of this mini-review, the TCS mechanism in general will be explained and specific systems that have served as targets in drug discovery efforts will be highlighted. In the second part the synthesis of HK inhibitors and their activities will be presented to explain the current state of efforts targeting the TCS.
Figure 1 :

Two-component systems structure and signaling pathway. 1: Detection of an external stimulus; 2: ATP phosphorylate histidine kinase residue present DHp domain; 3: Transfer of phosphoryl group to the aspartate residue of RR; 4: Phosphorylated response regulator activates a specific gene transcription
II. ARCHITECTURE AND STUCTURE OF TCS PROTEINS
TCSs in their simplest architecture employ two proteins: a membrane-spanning or cytosolic HK and a cytosolic protein called the RR that most commonly acts as a transcription factor. These two proteins are generally monogamously paired and often organized in operons.
HK are stable homodimeric proteins (Figure 1) that catalyze autophosphorylation either in cis or in trans under activating conditions.10,13 They are composed of a variable number of signal detection domains that can either be cytoplasmic or extra-cytoplasmic, flanked by transmembrane helices. The catalytic core of the HK is at the C-terminus.17 The catalytic core is composed of two domains: a domain directly linked to the N-terminus sensing domains called dimerization and histidine phosphotransferase (DHp) and an ATP binding domain, called catalytic and ATPase domain (CA).18 A stimulus, detected by the N-terminal domain, is translated into a conformational change in the DHp domain. This subsequently leads to a change in the rate of autophosphorylation.
The RR is the second component that participates in the signaling pathway and mediates the adaptive response (Figure 1). There are numerous architectures but most commonly the RR has a two-domain architecture with a receiver domain at the N-terminus and a DNA-binding domain at the C-terminus. The receiver domain catalyzes its own autophosphorylation on an aspartyl residue by utilizing the phosphohistidine from the HK as a substrate. Phosphorylation subsequently leads a conformational change and activation of the DNA-binding domain, often by mediating dimerization.19,20
HK proteins have become attractive therapeutic targets to fight bacterial resistance due to several intrinsic characteristics: (1) The catalytic domain of the HK has a high level of structural conservation and sequence homology at key residues in the ATP binding pocket. This structural homology is a key point for the design of novel HK inhibitors as they may have broad spectrum antibacterial activity.21,22 (2) HK proteins are absent from mammalian genomes. Other types of kinases such as serine/threonine kinases and tyrosine kinases do not share structural homology with HK proteins. It can thus be expected that inhibitors specific to HK will show little toxicity to mammalian cells. (3) TCS and HK are embedded in many important bacterial processes, some even essential for viability.23
III. BIOLOGICAL GOAL
TCSs are the most prevalent signal transduction system in the bacteria mediating a flurry of important adaptation and developmental decision. They control directly or indirectly the expression of numerous genes responsible for cell wall homeostasis, stress adaptation, motility regulation, adaptation to nutrient deprivation, sporulation, bacterial virulence, antibiotic resistance and others.24
Despite their importance for bacterial adaptation and fitness, only a few have proven to be essential for cell viability.25,26 The most prevalent is the YycG/YycF regulatory system (also known as WalK/WalR, VicK/VicR, MicA/MicB) present in Gram-positive bacteria of the order Firmicutes.25,26,27 Orthologs of this TCS have been shown to be essential for B. subtilis, S. aureus, E. faecalis, S. pneumoniae and S. mutans where it serves a crucial role in connecting cellular growth with cell wall turnover and homeostasis28 For its involvement in cell wall homeostasis the name WalK/WalR has gained traction in the literature in recent years and will be used hereafter.29 Given its prevalence in important pathogens along with its essentiality, this study has been targeted the most by TCS inhibitor studies.
While most TCS are not essential for viability in laboratory conditions, they certainly improve fitness since bacteria must be able to adapt to environmental changes. Pathogenic bacteria express specific responses to a host environment, and this response in many cases depends on TCSs. One important TCS pathway is the sporulation pathway of the Bacilli and Costridia leading to the development of dormant endospores30 Sporulation has mainly been studied in Bacillus subtilis where multiple HK, including KinA phosphorylate the RR Spo0F.31,32,33 Beyond spore formation, the pathway also contributes to biofilm formation which is often a complication in clinical infections. The elimination of biofilms has become a major goal in clinical treatment and inactivating this HK could be a possible solution.34,35
Aside from the frequently targeted or assayed TCS above, we will mention other TCS that have been targeted in isolated studies throughout, some of which were also implicated in the governance of virulence of bacteria (summarized in Table I).
Table I:
Two-component systems targeted in inhibition studies
| Two-component systems | Bacteria where is expressed |
Responsible for: | |
|---|---|---|---|
| Histidine kinase | Response regulator |
||
| PhoP | PhoQ | Salmonella typhimurium | Regulation in response to changes in Mg2+ levels and antimicrobial peptides.36 |
| PhoR | PhoB | Gram-negative bacteria | Regulation of phosphate in phosphate-limiting conditions.37 |
| WalK | WalR | Firmicutes | Essential system involved in cell wall homeostasis.29 |
| PhoR | PhoP | Firmicutes | Adaptation to phosphate-limiting conditions.38,39 |
| ResE | ResD | Firmicutes | Regulation of aerobic and anaerobic respiration.40 |
| EnvZ | OmpR | Escherichia coli and relatives | Regulation of osmotic pressure or environmental stress.41 |
| AlgR2 | AlgR1 | Pseudomonas aeruginosa | Production of the polysaccharide alginate that serves to evade the human immune system.42 |
| VanS | VanR | E. faecium (VRE) and S. aureus (VRSA) | Expression of genes responsible for vancomycin resistance.43 Direct detection of Vancomycin. |
| QseC | QseB | E. coli and relatives | Induces virulence factor expression in response to host signals including epinephrine 44,45 |
| KinA | Spo0F | Bacillus subtilis and relatives | Regulators of endospore formation.46 |
| CheA | CheY | Motile bacteria of all phyla | Mediation of motility. CheY interacts with the flagellum to coordinate directional swimming. 47 |
| NtrB or NRI | NtrC or NRII | Escherichia coli and relatives | Regulation of the expression of the Ntr regulon important nitrogen fixation.48 |
| HK853 | RR468 | Thermotoga maritima | Function unknown, but HK853 was the first HK structure to be solved by X-ray crystallography.49 |
| CckA | CckA | Caulobacter crescentus and relatives | Cell cycle regulator. This is a hybrid kinase that has both, HK and RR domains.50 |
IV. SYNTHESIS OF HISTIDINE KINASE INHIBITORS
Given the now well-appreciated importance of TCS proteins to bacterial survival and adaptation, numerous research groups have worked on the design of inhibitors that deactivate TCS by inhibition of the HK and rarely the RR protein. This mini-review focuses and summarize studies on the synthesis and biological evaluation of HK inhibitors (Figure 2) with the goal of identifying new antimicrobial agents. The assay utilized to quantify inhibitory activities in vitro typically measured inhibition of HK autophosphrylation autoradiographically utilizing γ-32P- ATP. Of note is that in the context of this assay, the HK serves as both, enzyme and substrate. This has important implications on the minimal IC50 values that can be observed in this assay. In order to detect radio-labeled protein, the assay typically requires a minimum of about 1 μM HK protein. Thus, the theoretical minimum for an IC50 for a competitive inhibitor is 0.5 μM. In other words, IC50 values reported for HK inhibitors cannot be compared to those for other types of kinases.
Figure 2:

General structures of HK inhibitors: Trityles (A), benzoxazines (B), salicylanilides (C), triazoles (D), 6-oxa isosteres (E), terpenes derivatives (F), benzimidazoles (G), benzoxazoles (H), thiazolidiones (I), thiophenes (J), adenines (K) and heterocycle (L).
A). Isothiazolidone and imidazolium
The first reported HK inhibitors were isothiazolone and imidazolium salt 51 discovered in 1993 by Roychoudhury et al., in a search for compounds that modulate the activity of the Pseudomonas aeruginosa AlgR2/AlgR1 TCS.52 AlgR2 is the HK that phosphorylates RR, AlgR1. The phosphorylation of AlgR1 leads to binding of the algD promoter and induces the transcription of algD, an alginate biosynthetic gene. The exoplysaccharide alignate is an important virulence factor of this bacterium.
Out of about 25 000 compounds screened for inhibition only 15 compounds appeared to be effective. Among these 15 compounds, four (1, 2, 3 and 4, Table II and III) exhibited significant inhibition of alginate production with IC50 values of 1-2 μM. Isothiazolones 1 and 2 showed inhibition of autophophorylation of AlgR2 and phosphorylation of AlgR1 in vitro. Inhibition of 95% of algD promoter activity was observed for 1 and 2 in vivo at concentrations 10 fold above the IC50. Meanwhile, compounds 3 and 4 showed strong inhibition of the autophosphorylation activity of several other HK proteins, including CheA (involved in motility), NRII (involved in nitrogen fixation) and KinA (involved in sporulation).52,53 Moreover, they demonstrated that compound 3 is the only inhibitor that inhibited the DNA-binding activity of AlgR1. These compounds, despite good inhibitory activities on HK did not effectively block bacterial growth, perhaps because they targeted non-essential pathways. The synthesis of the four compounds was not described by Roychoudhury.52
Table II:
Inhibitors classified by molecular target and drug discovery of each class of inhibitors
| TCS target | Inhibitors | Drug discovery or drug design |
|---|---|---|
| AlgR2/AlgR1 | Isothiazolones 1 and 2 | HTS (natural and synthetic compounds tested in vivo)52 |
| Imidazoliums 3 and 4 | HTS (natural and synthetic compounds tested in vivo)52 | |
| YycG/YycF or WalK/WalR | Imidazoliums 7a | SAR inspired on compounds 3 and 455 |
| Zerumbones 41, 43 and 47a |
41: Isolated from wild ginger Zingiber zerumbet71 43 and 47a: SAR inspired on compound 4158,74 |
|
| Thiazolidiones 63, 64, 68a-b and 74a |
63 and 64: SBVS from molecular lead-compound library using a 3D model of YycG HATPase_c domain. Docked into ATP-binding pocket to understand interaction with the protein.80 68a-b and 74a: SAR inspired on compounds 63 and 6483,84 |
|
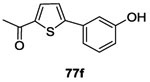
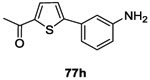
| Thiophenes 77a-h | 3D HK catalytic domain and 3D structure of a complex Gyrase B-inhibitor were superposed. Thiophenes 77a-h were designed from Gyrase B structural-modifications using 3D software. Inhibitors were docked into ATP-binding pocket to better understand interaction with the protein 87 | |
| Thienopyridine 92 | HTS of diverse small molecules. In silico screens targeting ATP-binding.93 | |
| Walkmycin 93 and 94 | Isolated from Streptomyces sp. strain MK632-100F11 and extracted with methanol96,97 | |
| Signermycin 95 | Isolated from Streptomyces sp. strain MK851-mF8 and extracted with methanol98 | |
| Waldiomycin 96 | Isolated from Streptomyces sp. strain MK844-mF10 and extracted with ethanol99 | |
| KinA/Spo0F | Trityles 11 and 14 | HTS from a chemical inventory56 |
| Benzoxazines 24a-c | HTS from a chemical inventory59 | |
| Salicynilides 27a-d |
27a-b: HTS from a chemical inventory65 27c-d: SAR inspired on compounds 27a-b65,66 |
|
| Salicynilides 32a-b | 32a-b: SAR inspired on compounds 27a-b | |
| Diaryltriazoles 36a-b | 36a-b: SAR inspired on compounds 27a-b67 | |
| 6-oxa isosteres 40a-c | SAR inspired on anacardic acids70 | |
| Indoles 48 and 49 | HTS from a chemical library75 | |
| Benzimidazoles 52a-f | SAR inspired on compounds 48 and 4975 | |
| Benzoxazoles 57a-b | SAR inspired on compounds 52a-f75 | |
| Qsec/QseB | Sulfonamide 62 | HTS using small organic molecules77 |
| HK853 | Adenines 78-81 and 84a |
78-81: HTS of diverse small molecules. In silico screens targeting ATP-binding.88 84a: SAR inspired on compounds 78-81. Docked on the receptor cavity to understand interaction with the protein.91,104 |
| Benzothiazoles 98-101 | HTS of diverse small molecules. In silico screens targeting ATP-binding.88 | |
| 5H-Pyrrolo[2,3-b]pyrazine 102 | HTS of diverse small molecules105 | |
| NRII/NRI | Diaryltriazoles 36a-b | |
| 6-oxa isosteres 40a-c | ||
| PhoR/PhoB | Thiophenes 77a-h | |
| Phenol 97 | In silico screenings of small molecules and in vitro FBS by DSF. 102 | |
| ResE/ResD | Thiophenes 77a-h | |
| PhoP/PhoQ | Diaryloxazole 85 | Purchased Hsp90 inhibitor and evaluated on CrkA HK92 |
| Diarylpyrazoles 86 and 91a-c |
86: Purchased Hsp90 inhibitor and evaluated on CrkA HK92 91a-c: SAR inspired on compound 8692 |
|
| CckA | Diarylpyrazoles 86 and 91a-c | |
| HpkA/DrrA | Thienopyridine 92 | |
| EnvZ/OmpR | Thienopyridine 92 | |
| VanS/VanR | Thienopyridine 92 | |
| VicK/VicR | Benzothiazoles 98-101 |
TABLE III:
Structure of HK inhibitors and their inhibitory activity on HK proteins.
| Structure | Synthesis | TCS | IC50 (μM) | Mechanism of inhibition | Antibacterial activity MIC (μg/ml) |
|
|---|---|---|---|---|---|---|
| A) Isothiazolones | ||||||
 |
NO | AlgR2/ AlgR1 | 1-252 | -Autophosphrylation of AlgR2 -Activity of algD promoter -Phosphorylation of AlgR1 |
Data not shown | |
 |
NO | AlgR2/ AlgR1 | 1-252 | - Autophosphrylation of AlgR2 -Phosphorylation of AlgR1 |
Data not shown | |
| A) Imidazoliums | ||||||
 |
NO | AlgR2/ AlgR1 | 1-252 |
-DNA-binding of AlgR1 -Activity of algD promoter -Autophosphorylation of CheA, NRII and KinA |
Data not shown | |
 |
NO | AlgR2/ AlgR1 | 1-252,53 | -Autophosphorylation of CheA, NRII and KinA | Not determined | |
 |
✓ | WalK/WalR | 6.655 | -Autophosphorylation of multiple HK : EnvZ, PhoQ, BvgS, EvgS and WalK | 3 0.39 0.78 1.56 3.12 0.39 0.39 |
(E. coli)54 (S. aureus)54 (MRSA)54 (S. pneumoniae)54 (PRSP)54 (E. faecalis)54 (VRE)54 |
| B) Trityles | ||||||
 |
✓ | KinA/Spo0F | 556,57 1.658 |
-ATP-competitive -Autophosphorylation of KinA |
1-2 2 1-2 2 1 |
(S. aureus)58
(MRSA)58 (PRSP)58(E. faecalis)58 (VRE)58 |
 |
✓ | KinA/Spo0F | 656,57 | Mechanism was not described | Not determined | |
| C) Benzoxazines | ||||||
 |
✓ | KinA/Spo0F | 959 | Mechanism was not described | 2 4 to 128 |
Gram-positive strains59 Gram-negative strains59 |
 |
✓ | KinA/Spo0F | 1159 | Mechanism was not described | Not determined | |
 |
✓ | KinA/Spo0F | 1359 | Mechanism was not described | 2 | (S. aureus)61 |
| D) Salicylanilides | ||||||
 |
✓ | KinA/Spo0F | 3.865 | -Autophosphorylation of KinA | 1 2 1 1 |
(S. aureus)65
(MRSA)65 (E. faecalis)65 (VRE)65 |
 |
✓ | KinA/Spo0F | 4565 | -Autophosphorylation of KinA | 0.5 0.25 0.5 0.5 |
(S. aureus)65
(MRSA)65 (E. faecalis)65 (VRE)65 |
 |
✓ | KinA/Spo0F | 4.865 | -Autophosphorylation of KinA | 1 1 2 2 |
(S. aureus)66 (MRSA)66 (E. faecalis)66 (E. faecium)66 |
 |
✓ | KinA/Spo0F | 2265 | -Autophosphorylation of KinA | 1 0.25 0.5 0.5 |
(S. aureus)66
(MRSA)66 (E. faecalis)66 (E. faecium)66 |
 |
✓ | KinA/Spo0F | 2.866 | -Autophosphorylation of KinA | 16 16 16 16 |
(S. aureus)66 (MRSA)66 (E. faecalis)66 (E. faecium)66 |
 |
✓ | KinA/Spo0F | 6.366 | -Autophosphorylation of KinA | 8 8 8 32 |
(S. aureus)66
(MRSA)66 (E. faecalis)66 (E. faecium)66 |
| E) Diaryl triazoles | ||||||
 |
✓ | KinA/Spo0F NRII/NRI |
867 2867 |
Mechanism was not described | >64 | Gram-positive strains67 |
 |
✓ | KinA/Spo0F NRII/NRI |
1667 6567 |
Mechanism was not described | 0.25-1 | Gram-positive strains67 |
| F) 6-oxa isosteres | ||||||
 |
✓ | KinA/Spo0F NRII/NRI |
2.270 5.070 |
-Autophosphorylation of KinA | >64 >32 4 4 |
(S. aureus)70
(MRSA)70 (E. faecalis)70 (E. faecium)70 |
 |
✓ | KinA/Spo0F NRII/NRI |
3.070 1.570 |
-Autophosphorylation of KinA | >128 128 8 8 |
(S. aureus)70
(MRSA)70 (E. faecalis)70 (E. faecium)70 |
 |
✓ | KinA/Spo0F NRII/NRI |
4.470 1770 |
-Autophosphorylation of KinA | 8 8 8 8 |
(S. aureus)70
(MRSA)70 (E. faecalis)70 (E. faecium)70 |
| G) Zerumbones | ||||||
 |
NO | WalK/WalR | -71 | Mechanism was not described | Not determined | |
 |
✓ | WalK/WalR (E) WalK/WalR (E/Z) |
63.573 75058 |
-Autophosphorylation of YycG | 62.5 | (B. subtilis)73 |
 |
✓ | WalK/WalR | 43.974 | -Autophosphorylation of WalK | 100 50 |
(MRSA)74 (VRE)74 |
| H) Indoles | ||||||
 |
NO | KinA/Spo0F | 3175 | Mechanism was not described |
1 0.5 1 0.5 |
(S. aureus)75 (MRSA)75 (E. faecalis)75 (E. faecium)75 |
 |
NO | KinA/Spo0F | 7.775 | Mechanism was not described | 4 4 4 4 |
(S. aureus)75 (MRSA)75 (E. faecalis)75 (E. faecium)75 |
| H) Benzimidazoles | ||||||
 |
✓ | KinA/Spo0F | 4275 | Mechanism was not described | 8 16 16 16 |
(S. aureus)75 (MRSA)75 (E. faecalis)75 (E. faecium)75 |
 |
✓ | KinA/Spo0F | 1775 | Mechanism was not described | 4 16 16 16 |
(S. aureus)75 (MRSA)75 (E. faecalis)75 (E. faecium)75 |
 |
✓ | KinA/Spo0F | 1275 | Mechanism was not described | 1 2 2 1 |
(S. aureus)75 (MRSA)75 (E. faecalis)75 (E. faecium)75 |
 |
✓ | KinA/Spo0F | 675 | Mechanism was not described | 1 1 2 2 |
(S. aureus)75 (MRSA)75 (E. faecalis)75 (E. faecium)75 |
 |
✓ | KinA/Spo0F | 1075 | Mechanism was not described | 1 1 1 1 |
(S. aureus)75 (MRSA)75 (E. faecalis)75 (E. faecium)75 |
 |
✓ | KinA/Spo0F | 1075 | Mechanism was not described | Not determined | |
| H) Benzoxazoles | ||||||
 |
✓ | KinA/Spo0F | 1475 | Mechanism was not described | 1 1 1 1 |
(S. aureus)75 (MRSA)75 (E. faecalis)75 (E. faecium)75 |
 |
✓ | KinA/Spo0F | 15.475 | Mechanism was not described | 2 2 2 1 |
(S. aureus)75 (MRSA)75 (E. faecalis)75 (E. faecium)75 |
| I) Sulfonamides | ||||||
 |
✓ | QseC/ QseB | -77 | Receptor antagonist interfering with signal binding of QseC | No antimicrobial activity but inhibition of virulence | |
| J) Thiazolidiones | ||||||
 |
NO | WalK/WalR | 2980 | -Autophosphorylation of WalK (Binds to HK) |
13 13 51 |
(S. epidermidis)80 (S. aureus)80 (S. pyogenes)80 |
 |
NO | WalK/WalR | 1480 | -Autophosphorylation of WalK (Binds to HK) |
4 4 1 |
(S. epidermidis)80 (S. aureus)80 (S. pyogenes)80 |
 |
✓ | WalK/WalR | 34.883 | -Autophosphorylation of WalK | 6.2 3.1-6.2 |
(S. epidermidis)83 (S. aureus)83 |
 |
✓ | WalK/WalR | 22.183 | -Autophosphorylation of WalK | 3.1 3.1-6.2 |
(S. epidermidis)83 (S. aureus)83 |
 |
✓ | WalK/WalR | 12.684 | -Autophosphorylation of WalK | 0.93 1.90 3.8 |
(S. epidermidis)84 (S. epidermidis)84 (S. aureus)84 |
| K) Thiophenes | ||||||
 |
✓ |
WalK/WalR PhoR/PhoP ResE/ResD |
196.987 122.687 124.387 |
-Autophosphorylation of WalK -ATP-competitve |
7 16 7 10 16 10 32 32 32 |
(B. subtilis)87 (S. aureus)87 (B. anthracis)87 (S. pyogenes)87 (S. agalactiae)87 (L. monocytogenes)87 (E. faecalis)87 (S. enterica)87 (E. coli)87 |
 |
✓ |
WalK/WalR PhoR/PhoP ResE/ResD |
52.887 13.187 89.387 |
-Autophosphorylation of WalK -ATP-competitve |
7 32 7 16 32 32 >64 32 32 |
(B. subtilis)87 (S. aureus)87 (B. anthracis)87 (S. pyogenes)87 (S. agalactiae)87 (L. monocytogenes)87 (E. faecalis)87 (S. enterica)87 (E. coli)87 |
 |
✓ |
WalK/WalR PhoR/PhoP ResE/ResD |
181.187 21.387 52.287 |
-Autophosphorylation of WalK -ATP-competitve |
No antibacterial activity | |
 |
✓ |
WalK/WalR PhoR/PhoP ResE/ResD |
80.187 11.387 243.987 |
-Autophosphorylation of WalK -ATP-competitve |
No antibacterial activity | |
 |
✓ |
WalK/WalR PhoR/PhoP ResE/ResD |
134.487 1.6387 102.387 |
-Autophosphorylation of WalK -ATP-competitve |
No antibacterial activity | |
 |
✓ |
WalK/WalR PhoR/PhoP ResE/ResD |
78.387 44.087 32.887 |
-Autophosphorylation of WalK -ATP-competitve |
No antibacterial activity | |
 |
✓ |
WalK/WalR PhoR/PhoP ResE/ResD |
121.387 33.387 40.387 |
-Autophosphorylation of WalK -ATP-competitve |
No antibacterial activity | |
 |
✓ |
WalK/WalR PhoR/PhoP ResE/ResD |
145.687 46.187 20.387 |
-Autophosphorylation of WalK -ATP-competitve |
No antibacterial activity | |
| L) Adenines | ||||||
 |
NO | HK853 | 49.688 | -Autophosphorylation of HK853 -ATP-competitive |
Not determined | |
 |
NO | HK853 | 95.388 | -Autophosphorylation of HK853 -ATP-competitive |
Not determined | |
 |
NO | HK853 | 14588 | -Autophosphorylation of HK853 -ATP-competitive |
Not determined | |
 |
NO | HK853 | 13188 | -Autophosphorylation of HK853 -ATP-competitive |
Not determined | |
 |
✓ | HK853 | 7.389,91 | -Autophosphorylation of HK853 -ATP-competitive |
Not determined | |
| M) Diaryl oxazoles | ||||||
 |
NO | CckA PhoP/PhoQ |
7.392 73 |
Mechanism was not described | 17-35 >140 >140 |
(E. coli)92 (C. crescentus)92 (B. subtilis)92 |
| M) Diaryl pyrazoles | ||||||
 |
NO | CckA PhoP/PhoQ |
2892 992 |
Mechanism was not described | 13-26 53-79 40 |
(E. coli)92 (C. crescentus)92 (B. subtilis)92 |
 |
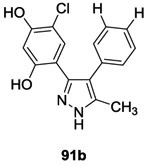
✓ | CckA PhoP/PhoQ |
12.392 6692 |
-Autophosphorylation of CckA and PhoQ -ATP-competitve |
12-25 74 50-74 |
(E. coli)92 (C. crescentus)92 (B. subtilis)92 |
 |
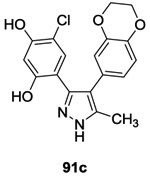
✓ | CckA PhoP/PhoQ |
17.592 4592 |
-Autophosphorylation of CckA and PhoQ -ATP-competitve |
11-23 68 45 |
(E. coli)92 (C. crescentus)92 (B. subtilis)92 |
 |
✓ | CckA PhoP/PhoQ |
11.292 3592 |
-Autophosphorylation of CckA and PhoQ -ATP-competitve |
27 81 40-54 |
(E. coli)92 (C. crescentus)92 (B. subtilis)92 |
| N) Miscellaneous | ||||||
| N) Thienopyridine | ||||||
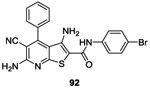 |
NO |
HpkA/DrrA WalK/WalR EnvZ/OmpR VanS/VanR |
5.593 13.293 26.893 103.893 |
-Autophosphorylation of HpkA, WalK, EnvZ and VanS -ATP-competitve |
>256 | (S. pneumoniae, E. faecium, E. faecalis, H. influenza,
E. coli)93 |
| N) WalKmycins | ||||||
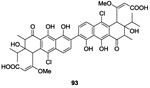 |
NO | WalK/WalR (Bs) WalK/WalR (Sa) |
1.696 5.796 |
-Autophosphorylation of WalK by binding cytoplasmic domain | 0.39 0.2 0.39 6.25 0.39 |
(B. subtilis)96 (S. aureus)96 (MRSA)96 (E. faecalis)96 (VRE)96 |
 |
NO |
VicK/VicR CiaH/CiaR LiaS/LiaR |
2.8797 4.8797 5.6397 |
-Autophosphorylation of VicK, CiaH, LiaS by binding cytoplasmic domain | 0.06-0.25 0.06-1 2-8 8-16 0.13-0.25 0.25-1 |
(S. aureus)97
(B. subtilis)97 (E. faecalis)97 (E. faecium)97 (S. pneumoniae)97 (S. pyogenes)97 |
| N) Signermycin | ||||||
 |
NO |
WalK/WalR (Bs) WalK/WalR (Sa) WalK/WalR (Ef) WalK/WalR (Sm) |
4398 3798 6198 6298 |
-Autophosphorylation of WalK -Binding the dimerization domain of WalK |
3.13 3.13 6.25 6.25 3.13 |
(B. subtilis)98
(S. aureus)98 (E. faecalis)98 (S. mutans)98 (MRSA)98 (VRE)98 |
| N) Waldiomycin | ||||||
 |
NO |
WalK/WalR (Bs) WalK/WalR (Sa) WalK/WalR (Ef) WalK/WalR (Sm) |
10.299,100 8.899,100 9.2100 25.8100 |
-Autophosphorylation of WalK -Targeting the dimerization domain of WalK |
8 8-16 16 128 |
(B. subtilis)99
(S. aureus)99 (MRSA)99 (E. faecalis)99 |
| N) Phenol | ||||||
 |
NO | PhoR/PhoB (Ec) PhoR/PhoP (Sa) |
16102,103 212102,103 |
-Autophosphorylation of PhoR -ATP-competitive |
8-16 1-16 8 16 |
(S. aureus)102,103 (S. epidermidis)102,103 (S. suis)102,103 (S. pneumoniae)102,103 |
| N) Benzothiazole | ||||||
 |
NO | HK853 VicK/VicR |
1.2188 7588 |
-Autophosphorylation of HK853 and VicK -ATP-competitive |
32-64 49-64 |
(E. coli)88(B. subtilis)88 |
 |
NO | HK853 VicK/VicR |
7.1588 61888 |
-Autophosphorylation of HK853 and VicK -ATP-competitive |
128 293 |
(E. coli)88 (B. subtilis)88 |
 |
NO | HK853 | 8.388,91,104 | Mechanism was not described Reduce production of virulence factors |
No antibacterial activity | |
 |
NO | HK853 | 1.5688,91,104 | Mechanism was not described Reduces production of virulence factors |
No antibacterial activity | |
| N) 5H-Pyrrolo[2,3-b]pyrazine | ||||||
 |
NO | HK853 | 23.7105 | Mechanism was not described | Not determined | |
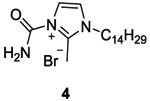
In 2000, Yamamoto et al. synthesized twenty analogs of imidazolium 3 in order to enhance antibacterial activity.54 Among the twenty imidazolium analogs, only one compound 7a (Table II and III), commonly known as NH125, presented promising result as HK inhibitor, effective against Escherichia coli HK proteins EnvZ (involved in regulation of osmotic pressure), PhoQ, BvgS and EvgS (IC50 values not shown). Moreover, 7a showed a WalK autophosphorylation inhibition with an IC50 value around 6.6 μM.55 Therefore, they investigated the effect of 7a on Gram-positive bacteria that have an essential WalK protein (S. aureus, Methicilline-resistant S. aureus (MRSA), S. pneumonia, Penicillin-resistant S. pneumoniae (PRSP), E. faecalis and Vancomycin-resistant Enterococci (VRE)). Compound 7a showed significant minimal inhibitory concentration (MIC) ranging 0.39 to 3 μg/mL.
The synthesis of these imidazolium salts 7 is shown in scheme 1. The first step was the alkylation of methylimidazole 5 leading to 6. Then a second alkylation using benzyl bromide in the presence of potassium iodide led to the desired imidazolium salt 7.54 Yields were not given for this synthetic pathway (Scheme 1).
Scheme 154: Reagents and conditions:

(i) RBr, CHCl3, 70°C; (ii) KI, benzyl bromide, CHCl3, 70°C.
B). Trityles
In 1995 and 1998, Demers and Barrett developed a new series of molecules with potential inhibitory activity of the KinA / Spo0F sporulation system.56,57,58 A total of 49 potential trityle inhibitors (Figure 2, A) of HK proteins were synthesized in order to improve the results obtained previously with the imidazolium series.
These 49 compounds were evaluated for their activity in inhibiting the autophosphorylation of KinA and the transphophorylation of Spo0F. Most of the potential inhibitors presented some inhibitory activity but only 2 compounds, 11 (named RWJ-49815) and its analog 14 presented promising results on KinA inhibition with IC50 values of 5 μM and 6 μM, respectively.56,57,58 Further studies were performed on compound 11 that showed ATP-competitive inhibition on the autophosphorylation of KinA with IC50 values of 1.6 μM.58 This molecule was also assessed as antibacterial agent on Gram-positive and Gram-negative strains. Trityle 11 inhibited growth of Gram-positive bacteria (S. aureus, MRSA, PRSP, E. faecalis and VRE) with MIC values of 1 to 2 μg/mL. E. coli only proved sensitive following a membrane-permeabilizing treatment (MIC not indicated), indicating that the compound is not able to penetrate the outer membrane of Gram-negative bacteria.
The synthesis of molecule 11 and 14 is described in scheme 2. The first step was a Mitsunobu reaction between the triphenylpropanol 8 and the appropriate phenol (9a or 9b) to give 10a and 10b in 31% and 58% yield, respectively. Compound 10a was deprotected in the presence of anisole and anhydrous HCl leading to guanidine 11. No yield is indicated for this step. Reduction of 10b using sodium borohydride gave compound 12 in 97% yield. Reaction of 12 with sulfuryl chloride in benzene led to chloride 13 in 83% yield. Finally, pyridinium chloride 14 was prepared from 13 using pyridine in 86% yield.56,57
Scheme 256,57: Reagents and conditions:

(i) PPh3, DEAD, THF anhydrous, −15°C; (ii) Anisole, HCl anhydrous, iPrOH, 50°C; (iii) NaBH4, EtOH; (iv) SO2Cl2, Benzene, reflux; (v) Pyridine, reflux.
C). Benzoxazines
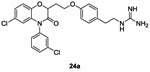
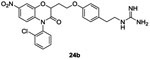
In 1997, Frechette et al. patented a series of benzoxazine derivatives (Figure 2, B) that inhibited the bacterial two-component signal transduction system.59,60 They synthesized 144 potentials inhibitors of HK proteins using the strategy described in scheme 3. These 144 benzoxazine derivatives were first evaluated for their phosphorylation activity inhibition (autophosphorylation of KinA and the transphosphorylation of Spo0F). In this system, most of the compounds presented moderate to good inhibitory activity on the autophosphorylation of KinA with three molecules 24a, 24b and 24c with IC50 values around 15 μM.59 Antibacterial activities were assessed on Gram-positive (E. faecalis, E. faecium, S. epidermidis and S. aureus) and Gram-negative (E. coli, K. pneumoniae and P. aeroginosa) strains and only the MIC of compound 24a was reported to be about 2μg/mL for Gram-positive strains and ranging from 4 to 128 μg/mL for Gram-negative strains.59 Further studies were carried out by Hilliard et al. where they reported MIC values of 24b (2 μg/mL)61 on a S. aureus strain. The synthesis of 24a-c is described in scheme 3.
Scheme 359,62: Reagents and conditions:

(i) K2CO3, DMF, rt ; (ii) H2, Pd/C, EtOH or NaBH4, NiCl2.6H2O, MeOH; (iii) Chloro t-butyldimethylsilane, imidazole, DMF, N2; (iv) NaH, R1Cl, DMF; (v) tetrabutylammonium fluoride (1N), THF, rt ; (vi) n-Bu3P, ADDP, Benzene, 0°C; (vii) HCl, iPrOH, rt.
Reaction of commercially available substituted nitrophenols 15 with α-bromo-γ-butyrolactone 16 in DMF under alkaline conditions led to compounds 17 in 60-70% yield. Then, a cyclization step occurred upon reduction of the nitro function by a catalytic hydrogenation using H2/Pd/C in EtOH or NaBH4, NiCl2.6H2O in MeOH and benzoxazine leading to heterocycles 18 obtained in 45-87% yield. Protection of the alcohols 18 with chloro t-butyldimethylsilylane in DMF led to compounds 19 in moderate to good yield (44-91%). The alkylation of the lactam of 19 was performed with NaH and the corresponding chloride (R1Cl) to give compounds 20, which was used without further purification in the next step. Deprotection of the alcohol of compounds 20 with tetrabutylammoniumfluoride (1M) in THF gave the respective deprotected alcohols 21 with 45% overall yield for these two deprotection steps. A coupling reaction of the alcohols 21, by Mitsunobu reaction, with Boc-protected 4-(2-aminoethyl)phenol 22 gave ethers 23 in 34-52% yield. Finally, protected guanidines 23 were deprotected under acidic conditions to give the desired compounds 24a, 24b and 24c in 28%, 79% and 72% yield, respectively.59,62 (Scheme 3)
D). Salicylanilides
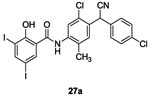
The synthesis of various salicylanilides (Figure 2, C) was first published in 1955 by Bindler et al. who demonstrated that these compounds have antibacterial activity when one of the two benzene rings was substituted by a halogen in para-position while the other one was substituted by an halogen in meta-position.63 Closantel 27a (Table II and III) is one of these compounds and is an antiparasitic agent belonging to the salicylanilides first used in veterinary medicine. Its synthesis was described by Janssen et al. in 1977.64
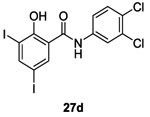
In 1998, Hlasta et al. demonstrated by screening studies that the veterinary agent Closantel 27a and one of its analogs 3,3’,4’,5-tetrachlorosalicylanilides 27b inhibited autophosphorylation of the sporulation kinase KinA with an IC50 of 3.8 μM and 45 μM, respectively.65
Consequently, 12 new analogues of 27a and 27b were synthesized in order to evaluate a structure-activity relationship. They found that inhibitors 27c and 27d with hydrophobic/electron-withdrawing substituents on either aromatic ring increased potency of inhibitory activity on the autophosphorylation of KinA (IC50 of 4.8 μM and 22 μM) showing competitive inhibitory activity compared with those obtained with 27a and 27b.65 Compounds 27a-d demonstrated good antibacterial inhibition of growth of Gram-positive strains (S. aureus, MRSA, E. faecalis, E. faecium and VRE) with MIC values around 1-2 μg/mL.65 However, they didn’t affect the growth of Gram-negative strains possibly because they cannot penetrate the outer membrane.
The authors used the method described by Janssen et al. to synthesize Closantel 27a and its analog 27b. These compounds were prepared by an acylation reaction of an amine 25 with the corresponding acid chloride 26 in dioxane to obtain 27a-d in 45-91% yield.64, 65 (Scheme 4)
Scheme 464,65: Reagents and conditions:

(i) Dioxane, 50°C.
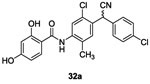
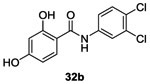
Salicylanilides were known to also interfere with the phosphorylation of mitochondria leading to potentially undesirable consequences for mammalian cells. In 1998, Macielag et al. tried to overcome such undesirable side effects. They developed two series of analogs of each compound. In total, 22 derivatives of 27a and 27b were synthesized by acylation as described above with slight modifications.66 Salicylanilides 31 were synthesized in two steps. First, compound 28 in the presence of oxalyl acid and catalytic DMF led to intermediate acyl chloride 29 which was used without any further purification and reacted with the appropriated aniline 30 in the presence of pyridine to give salicylanilides 31. No yield is indicated for this step. Finally, the desired deprotected salicylanilides 32a and 32b were obtained (in 10% and 20% yield, respectively) by demethylation of the methoxy groups with pyridine hydrochloride.66 (Scheme 5)
Scheme 566: Reagents and conditions:

(i) (COCl)2, 60 °C; DMF (cat.), THF; (ii) Pyridine, THF, 50 °C; (iii) Pyridine hydrochloride, 190 °C.
They showed that the presence of electro-donating or bulky substituents on the salicyloyl ring dramatically reduced or completely abolished activity on mitochondrial protein phosphorylation. For this new series, the substitution of the salicyloyl ring by hydroxy groups (leading to catechols 32a-b) allowed to improve the in vitro inhibition of KinA with IC50 values ranging from 2.8 to 6.3 μM.66 However, these molecule presented reduced antibacterial (MIC= 8-16 μg/mL) activity in comparison with 27a-d.
E). Diaryltriazole
A diaryltriazole series (Figure 2, D) was developed by Sui et al. in 1998 and was designed from the salicylanilide derivatives, in order to improve the biological activity and avoid the oxidative properties of salicylanilides series described previously that could be a danger for mammalian cells.67 Triazoles (Figure 2, D) were chosen because of their structural similarity to the amide bond, their relative higher aqueous solubility, and their hydrogen bond donating ability.68,69
Sui et al. synthesized a total of 17 triazole derivatives of compounds 27a and 27b to avoid the adverse effects observed in the salicylanilide series. Only two molecules 36a and 36b (Table II and III) exhibited competitive inhibition of HK proteins comparable to those obtained by salicylanilides 27a and 27b. Triazoles 36a and 36b inhibited KinA/Spo0F with IC50= 8 μM and 16 μM, respectively and NRII/NRI with IC50= 28 μM and 65 μM, respectively.67 Triazole 36b possessed good antibacterial activity on Gram-positive strains (S. epidermidis, S. aureus, MRSA, E. feacalis, E. faecium and VRE) however compound 36a didn’t present antibacterial properties maybe due to a complete dissociation in phenolate.67 None of the compounds had antimicrobial activity against Gram-negative bacteria possibly due to impermeability of the outer membrane to these compounds.
The diphenyltriazoles 36 were synthesized by the pathway shown in scheme 6. The intermediates 35 were prepared by O-acylation of salicylanilides 33 with benzoyl chlorides 34 in the presence of pyridine in refluxing xylene. No yield was indicated for this step. Then, the preparation of triazole 36a and 36b was performed by the opening of the benzoxazinone ring 35 followed by a cyclisation in 40-70% yield.67
Scheme 667: Reagents and conditions:

(i) Pyridine, Xylene; (ii) NH2NH2, EtOH
F). Anacardic acids analogs: 6-oxa isosteres
In 1999, Kanojia et al. decided to investigate anacardic acids for inhibitory activity against HK proteins (Figure 2, E). Anacardic acids are natural compounds found in plants and in the shell of cashew nuts. Previous studies had shown that anacardic acids presented antibacterial activity on several Gram-positive bacteria but the mechanism of action was unknown. Kanojia et al. hypothesized that its activity was due to inhibition of TCSs.70
In total, Kanojia et al. synthesized 17 analogs of anacardic acids, the 6-oxa isosteres family, new potential inhibitors of HK. Only three molecules 40a, 40b and 40c presented promising results as inhibitors of autophosphorylation of sporulation kinase KinA with an IC50= 2.2 μM, 3.0 μM and 4.4 μM, respectively. They also evaluated these compounds on the NRII/NRI system and compounds 40a, 40b and 40c presented inhibitory activity against this TCS, too, with IC50= 5.0 μM, 1.5 μM and 17 μM, respectively.70 Nevertheless, compounds 40a-c presented only weak antibacterial activities on Gram-positive strains (S. aureus, MRSA, E. feacalis and E. faecium). Compound 40a-b were inactive against S. aureus strains, but active against E. feacalis and E. faecium strains suggesting that inhibitors had different penetration or interaction with targets in these strains.70
For the synthesis, the protection of carboxylic acid 37 by an acid-catalyzed esterification reaction with MeOH gave 38. No yield is indicated for this step. A Mitsunobu reaction for the selective mono-O-alkylation of the γ-resorcylic ester 38 led to alkoxy-2-hydrobenzoate 39 in 15-55% yield. Finally, 39 was deprotected by saponification of ester group to obtain 40 in 45-95% yield.70 (Scheme 7)
Scheme 770: Reagents and conditions:

(i) MeOH, H2SO4; (ii) DEAD, PPh3, R-OH, THF; (iii) KOH/EtOH, heat, overnight.
G). Terpenes derivatives
In 1999, Yamamoto and Kitayama were interested in Zerumbone 41 (Table II and III), a natural compound found in wild ginger. It had been reported that Zerumbone exhibited a variety of physiological effects including anti-cancer activity, HIV inhibition, anti-inflammatory activity and plant growth regulation. Yamamoto and Kitayama hypothesized that Zerumbone might have useful biological activity. The group developed a simple method to isolate this natural molecule by steam distillation and recrystallization, which allowed to deduce a crystallographic structure of the molecule.71 Subsequently, they synthesized a large number of zerumbone derivatives, which were evaluated for antibacterial properties.
In 2001, Kitayama et al. developed 100 cyclic derivatives of zerumbone 41. Unfortunately, the synthetic pathway was not described in the article.72 Zerumbone derivatives were tested as inhibitors of the autophosphorylation of two HK proteins, EnvZ and PhoQ.72 Whereas cyclic derivatives of zerumbone 41 did not inhibit HK autophosphorylation, one open ring derivative, 43 (E/Z= 3/2 by 1H NMR), selectively inhibited these two kinases. IC50 were not reported in this paper for either system. Concurrently, Yamamoto et al. found that compound 43 (E/Z= 3/2) also had inhibitory activity on autophosphorylation of WalK with an IC50 of 750 μM.55,72,73 Compound 43 (E/Z= 3/2) had antibacterial activity against the Gram-positive B. subtilis with MIC= 62.5 μg/mL but no against the Gram-negative E. coli.73 The antibacterial effect could be related to HK inhibition since WalK is essential for B. subtilis growth.72
To synthesize zerumbone derivatives, a regiospecific bromination of one double bond of zerumbone 41 was performed to give 42 in 95% yield. Ring opening and debromination of 42 in the presence of an aqueous solution of KOH and α-cyclodextrin led to 43 with a 90% yield (C10 geometrical mixture, 10E/10Z= 3/2).72 (Scheme 8)
Scheme 872: Reagents and conditions:

(i) Br2, CCl4; (ii) aqueous KOH, α-cyclodextrine, CH3CN.
Further work on a new series of open ring derivatives was pursued. In 2004, Kitayama et al. isolated isomer (E)-4373 and evaluated the inhibitory activity of each isomer on WalK. They noticed that 43 with E configuration had greater inhibitory activity than E/Z mixture on WalK with an IC50= 63.5 μM.55,73,74 Nevertheless, compound (E)-43 and zerumbone 41 did not present good antibacterial properties possibly due to a lack of cytoplasmic membrane permeability for the molecules. Therefore, Kitayama et al. decided, in 2007, to develop a new series of derivatives from molecule (E)-43 with the goal to enhance antibacterial activity; 17 amino acid (AA) derivatives 47 (Figure 2, F) were synthesized following the synthetic pathway described in scheme 9.
Scheme 974: Reagents and conditions:

(i) SOCl2, CCl4; (ii) pyridine, CH3CN, rt, 24h; (iii) NaOH(aq), CH3CN, rt, 18h.
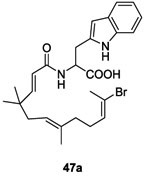
Compound (E)-43 reacted with SOCl2 in CCl4 to afford acyl chloride 44. The resulting intermediate 44 was used without any further purification and was added to a mixture of the appropriate amino acid (AA) methyl ester 45 and pyridine in CH3CN leading to 46 in 33-92% yield. The methyl ester derivatives 46 were saponified with an aqueous solution of NaOH in CH3CN to obtain the corresponding carboxylic acid derivatives 47 in 32-99% yield.74 (Scheme 9)
These derivatives were evaluated for the inhibition of the autophosphorylation of HK WalK. Of all 17 synthesized AA derivatives, the substituted tryptophan 47a compound was the only derivative that showed inhibition of WalK with an IC50 of 43.9 μM, which was slightly stronger than that of (E) 43.74 (Table II and III) Moreover, compound 47a had moderate antibacterial activity against Gram-positive strains resistant to common antibiotics (Methicilline-resistant S. aureus (MRSA) and Vancomycin-resistant Enterococci (VRE)) with MIC = 50-100 μg/mL.74
H). Benzimidazole and benzoxazole
Employing an in vitro screen Weidner-Wells et al. identified in 2001 a bisamidine indole 48 (Table II and III) as a lead inhibitor of KinA autophosphorylation activity with an IC50 of 31 μM and MIC value ranging 0.5 to 1 μg/mL on Gram-positive strains. (Table II and III) This class of molecules was reported to have antibacterial and antifungal properties and was structurally related to the bisbenzamidine class of DNA binders. In order to avoid the DNA-binding property due to the similarity with bisbenzamidine, Weidner-Wells et al. developed a series of monoamidine derivatives to study the structure-activity relationship as HK inhibitor.75 First, they designed a simplified scaffold of molecule 48 by the removal of one of the amidine groups to reduce the potential binding with DNA. This led to molecule 49 (Table II and III) with an enhanced inhibitory activity against KinA (IC50 of 7.7 μM) and a MIC of about 4 μg/mL against Gram-positive strains. The authors did not synthesize the indole series because of potential difficulties and low yield and developed benzimidazole derivatives 52 instead. (Figure 2, G)
Compounds 52 were synthesized in one step reaction by condensing the bisulfite adduct of the appropriately substituted benzaldehyde 51 with substituted benzene-1,2-diamine 50 in EtOH in 44% yield. Over 200 compounds were prepared this way in a parallel synthetic effort.75,76 (Scheme 10)
Scheme 1075,76: Reagents and conditions:

(i) 40% aq NaHSO3, ethanol, 80°C.
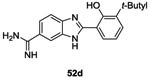
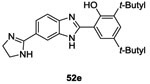
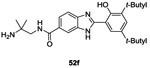
Several strategies were undertaken to evaluate a structure-activity relationship such as shortening the side chain, changing substituents on the phenyl ring as well as substitution at the benzimidazole ring. Only 6 molecules (52a-f, Table II and III) presented good results as HK inhibitors. First, analog 52a of molecule 49 was less potent with an IC50= 42 μM. However, introduction of bulky groups like diphenylamino, tert-butyle or a hydroxyl group increased the HK inhibitory activity to IC50= 17 μM (52b), 12 μM (52c) and 6 μM (52d), respectively on KinA. This reflected the importance of the presence of hydrophobic groups and hydrogen bond capacity to improve the inhibitory activities. With regard to the substitution of the benzimidazole ring, as demonstrated by compounds 52e (IC50= 10 μM) and 52f (IC50= 10 μM), the presence of atoms with the capacity to form hydrogen bonds was essential, especially the presence of a nitrogen atom.75
In a second step, the authors changed the benzimidazole core to a benzoxazole core (Figure 2, H) to assess the importance of the heterocycle; the synthesis is depicted in scheme 11.
Scheme 1175: Reagents and conditions:

(i) NH4OC(O)H, Pd/C, THF; (ii) ArC(O)Cl, dioxane, 100°C; (iii) PPTS, xylenes, reflux; (iv) HCl/EtOH, 0°C; NH3/EtOH, reflux; 1N HCl.
First, reduction of the nitro function of 53 by an in situ catalytic hydrogenation generated by ammonium formate with Pd/C in THF led to 54 in 94% yield. Then amino compound 54 in the presence of the appropriately substituted aryl acid chloride in dioxane gave compound 55 in 90-95% yield. Amide 55 was then cyclized to benzoxazole 56 with pyridium p-toluenesulfonate (PPTS) in refluxing xylene and 56 was obtained in 75-80% yield. Finally, amidines 57 were obtained by the aminolysis of 56 in acidic conditions in 65-75% yield.75 (Scheme 11)
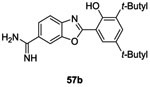
Benzimidazole 52c (12 μM) and benzoxazoles 57a (14 μM) and 57b (15.4 μM) displayed comparable inhibitory activity on KinA thus indicating that the NH group is not essential for activity. Comparable antibacterial activities were obtained for benzimidazoles 52 and benzoxazoles 57 against Gram-positive bacteria with MIC values ranging from 0.5 to 16 μg/mL.
I). Sulfonamides/Thiourea
In 2008, a high-throughput in vivo screen of 150 000 molecules was performed by Rasko et al. to find inhibitors of the quorum sensing TCS QseC/QseB.77,78 QseC induces virulence factor expression in response to host epinephrine and norepinephrine and the screen was set up to identify receptor antagonists. This is in contrast to all other studies described in this review, which targeted the catalytic activity of HK proteins. Among all molecules assessed, 75 molecules showed inhibitory activity with median inhibitory concentration values (IC50) at or below 10 μM. The derivatives have been synthesized using the general synthetic route shown in scheme 12 but here only the synthesis of N-phenyl-4(3-phenylthioureido)benzenesulfonamide (LED 209) 62 will be described. The IC50 of compound 62 was not reported in the article. They evaluated LED209 62 on S. typhimurium and expectedly 62 did not influence bacterial growth. However, the authors determined in animal models of infection that this receptor antagonist markedly inhibited virulence of several pathogens, consistent with a reduced expression of virulence factors. Importantly, compound 62 had no toxicity in mice and did not interfere with human β-adrenergic receptors.77
Scheme 1277,79: Reagents and conditions:

(i) CH3CO2Na, rt; (ii) (6N) HCl, reflux; (iii) NEt3, Ph-NCS, reflux.
N-acetylsulfanilyl chloride 58 reacted with aniline 59 in the presence sodium acetate to give compound 60 in 60% yield. Hydrolysis of acetamide 60 was performed in ethanol in strong acidic conditions to give compound 61 in 65% yield. Finally, 61 reacted with triethylamine and phenyl isothiocyanate in THF leading to 62 (LED 209) in 35%.77,79 (Scheme 12)
J). Thiazolidione
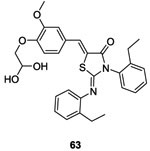
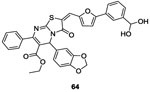
In 2006, Qin et al. identified potential WalK inhibitors by using a structured-based virtual screening employing the structure of the CA domain of the WalK HK from the opportunistic pathogen S. epidermidis. They identified seven inhibitors belonging to different classes of chemical structures: three derivatives of the thiazolidione family, two derivatives of benzamide and one furan derivative. Experimentally, they demonstrated that thiazolidiones 63 and 64 (Table II and III and figure 2, I) could inhibit the auto-phosphorylation of WalK with IC50 of 29 μM and 14 μM, respectively, likely due to competitive inhibition. Further, the compounds showed antibacterial activities against Gram-positive pathogens (S. epidermidis, S.aureus and S. pyogenes) and exerted anti-biofilm activity of S. epidermidis. They also evaluated hemolytic activity and cytotoxicity on mammalian cells and neither 63 nor 64 showed any toxicity. Docking simulations suggested structures of the interaction for 63 and 64 with the target protein. These inhibitors were considered promising lead-compounds for the development of new antibacterial reagents.80 A few years later, the same group synthesized structures related to thiazolidiones 63 and 64.
Two different strategies were developed to find the most effective WalK inhibitors among the thiazolidione derivatives. (Schemes 13 and 14)
Scheme 1381,82: Reagents and conditions:

(i) CS2, Lac.Sulfur, triethanolamine, water, reflux, 6 h; (ii) ClCH2COOH, NaOAc, absolute EtOH, reflux, 10 h, (iii) R2CHO, β-alanine, acetic acid, reflux.
Scheme 1482: Reagents and conditions:

(i) SnCl2 .2H2O, EtOH, reflux; (ii) bromoacetic acid, NaOAc, AcOH/Ac2O, reflux; (iii) β-alanine, acetic acid, reflux.
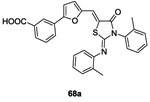
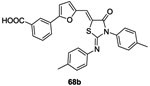
In 2010 and 2011, Pan et al. synthesized a series of analogs of molecule 63 in order to improve the inhibitory activity against WalK.81,82 Twenty-four derivatives were synthesized from the reaction pathway described in scheme 13 in order to evaluate a structure-activity relationship. In 2012, Huang et al. evaluated the potential inhibitory activity of these molecules. Only two molecules 68a, 68b showed similar or better results than 63 with IC50 values of 34.8 μM and 22.1 μM,83 respectively (IC50 of the compound 63 was 29 μM80 on YycG/YycF system). These molecules, 68a and 68b, inhibited bacterial growth of S. epidermidis and S. aureus strains with MIC values ranging 3.1-6.2 μg/mL. It appeared that the introduction of a furan ring on the double bond increased the rigidity of the structure and thus stabilized the thiazolidione in the bonding pocket.83
The synthesis of compounds 68a,b was described in references (81,82) although yields were only given in reference (82). A simple one-pot reaction consisting of the appropriately substituted aniline (R1) 65, carbon disulfide and triethanolamine catalyzed by Lac Sulfur, gave 1,3-diarylthiourea 66. Formation of a thiazolidione ring 67 from the di-substituted symmetrical thioureas 66 was accomplished in EtOH with chloracetic acid and NaOAc to lead to 67. No yields were given for this step. An aldol condensation of corresponding substituted arylaldehyde with an equivalent amount of molecule 67 by using β-alanine in acidic conditions gave molecule 68a and 68b in 90% and 93% yield, respectively.81,82(Scheme 13)
In 2014 Zhao et al. pursued the work on the thiazolidione family.80 Following the rational strategy used for compound 63, they derived molecule 64 (IC50= 14 μM on WalK autophosphorylation).80 They developed a series of analogs of molecule 64 in order to improve the inhibitory activity on WalK inspired by in silico results.
In order to improve the biological activity, several structural modifications and different substitutions on the aromatic ring were considered.82 Following this strategy, 18 new derivatives were synthesized. The authors introduced an electronegative atom such as chlorine or fluorine because these groups have previously shown interesting properties as antimycobacterial agents. The introduction of the chlorine atom in one of the phenyl rings (see 74a on Table II and III) improved inhibitory activity on WalK with an IC50= 12.6 μM, a very similar activity than that obtained with molecule 64.80,84 This molecule 74a also presented good antimicrobial activity against Gram-positive strains (MIC = 0.93-3.8 μg/mL), however it did not have antibacterial activity against Gram-negative bacteria possibly due to inability to penetrate the outer membrane or due to of lack of a WalK ortholog in Gram-negatives. The synthesis of compound 74 is described in scheme 14.
The synthesis of compound 72 consisted of a simple one-pot Biginelli reaction from the corresponding substituted aldehydes 69, thiourea 70 and ethyl benzoylacetate 71 and gave different dihydropyrimidine 72 scaffolds. No more details are indicated for this step. Cyclization with bromoacetic acid afforded molecules 73 in 72% yield. An acid-catalyzed aldol condensation of substituted aromatic aldehydes with one equivalent of 73 gave 74 in 92% yield.82 (Scheme 14)
K). Thiophene
In 2016, Boibessot et al. tried to deactivate TCSs by targeting the catalytic and adenosine triphosphate (ATP)-binding (CA) domain. The ATP-binding domain is characterized by a Bergerat fold, which is a domain shared with DNA-gyrases, Hsp90 and the mutator protein MutL which constitute the GHKL superfamilly.85 The CA domain is a hallmark of all HK proteins and features a high degree of homology across proteins from diverse bacteria.15 The authors argued that simultaneous inhibition of multiple TCSs is desirable to prevent bacterial resistance development and increase the potential antimicrobial spectrum of such inhibitors.86 To this end, they designed a series of thiophene derivatives (Figure 2, J) starting with structures of existing DNA-gyrase inhibitors and aided by in silico design and screening. They then evaluated the potential HK inhibitory activities of these thiophene compounds on multiples HK proteins.87
Twenty-four potential inhibitors of TCSs were evaluated in biological studies. Among these inhibitors, eight compounds 77a-h (Table II and III) showed an ATP-competitive inhibiton of B. subtilis WalK autophosphorylation with IC50 values of 196.9 μM, 52.8 μM, 181.1 μM, 80.11 μM, 134.4 μM, 78.3 μM, 121.3 μM and 145.6 μM, respectively.87 These eight compounds were next tested for inhibition of HK PhoR, involved in the detection of phosphate-limitation, with IC50 values of 122.6 μM, 13.11 μM, 21.3 μM, 11.3 μM, 1.6 μM, 44 μM, 33.3 μM and 46.1 μM, respectively and against HK ResE, involved in the adaptation to low oxygen environments with IC50 values of 124.3 μM, 89.3 μM, 52.2 μM, 243.9 μM, 102.3 μM, 32.8 μM, 40.3 μM and 20.3 μM, respectively. Of these eight HK inhibitors, two molecules 77a and 77b featured antibacterial activity against a wide variety of Gram-positive and Gram-negative human pathogens (MIC= 7-32 μg/mL). None of the compounds had hemolytic activity and lack of activities against ser/thr kinases and DNA-Gyrases proofed specificity for HK proteins. Perhaps most interesting, compound 77a at sublethal concentrations was able to restore antibiotic sensitivity for two diverse antimicrobial resistant clinical bacterial isolates. Thus, Boibessot et al. demonstrated that HK inhibitors might also serve as adjuvants to existing antibiotics.87
The synthesis of the eight compounds is described in scheme 15. Thiophenes 77a-h were synthesized in one simple step using the palladium-catalyzed Suzuki–Miyaura cross-coupling reaction. Commercially available boronic acid 76 derivatives reacted with the bromo-nitrothiophene 75 derivatives and desired compound 77 were obtained in moderate to good yields (31-77%).87
Scheme 1587: Reagents and conditions:

Pd(PPh3)4 or PdCl2(PPh3)2, Na2CO3 or K2CO3 or Cs2CO3, Toluene/EtOH or 1,4-dioxane/H2O or DMF/H2O or DMF, 70-90°C.
L). Adenine
In 2015, Wilke et al. rationally designed a selective adenine-based scaffold by in silico studies 88 (Figure 2, K).
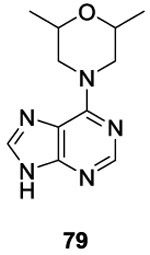
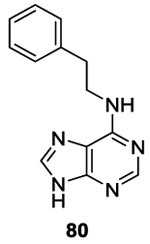
Similar to Boibessot et al. they argued that inactivation of several HK simultaneously is a desirable traint for HK inhibitors to improve the spectrum of antibiotic activity and make the emergence of resistance less likely. First, Wilke et al. tested for inhibition of HK853 (Thermotoga maritima) and identified 119 hits by high-throughput screenings. Among the 119 compounds, only nine were able to inhibit multiple HK proteins. They noticed that among the nine hits, four had an adenine core scaffold (Table II and III, 78, 79, 80 and 81).88,89 This purine series had been reported before as potential ATP-competitive kinase inhibitors 90 so this result was of no surprise. Based on this result, Goswami et al. studied in 2017 the structure-activity relationship of this adenine series. They evaluated substitutions on the pyrimidine core (R3 and R4), the addition of ribose (R2) and the substitution of imidazole (R1). Best results were obtained by the substitution on R3, with an IC50 of 7.3 μM for compound 84a.89, 91 Finally, they showed that purine 84a did not inhibit mammalian kinases (Ser/Thr, Tyr) or Hsp90, thus proving that their compounds had good selectivity for HK proteins.89 This class of molecules was not evaluated as antibacterial agents.
To synthesize this class of molecules, they used the following strategy (Scheme 16). Adenine 84 was synthesized by nucleophile substitution of the chloropurine 82 and substituted amine 83 to obtain compounds 84 in 55-85%.89,90
Scheme 1689,90: Reagents and conditions:

(i) ethanol, reflux, overnight.
M). Diaryl pyrazole
In a strategy reminiscent to that of Boibessot et al., Vo et al. attempted to identify in 2017 new HK inhibitors by using the shared structural and sequence homology in the Bergerat fold of HK and Hsp90. Their design was based on six commercially available Hsp90 inhibitors and evaluated on the CckA HK of Caulobacter crescentus. This kinase is essential for controlling cell-cycle progression as well as the transcription of more than 90 genes.92 They noticed that two commercial Hsp90 inhibitors (Table II and III, 85 and 86) had good inhibitory activity against CckA with IC50 of 7.3 μM and 28 μM, respectively.92 The two compounds had structural similarity with a central heterocycle and two aromatic rings (Figure 2, L).
Based on this general structure, they synthesized nine potential HK inhibitors. Three pyrazole compounds 91a, 91b and 91c had ATP-competitive inhibitory activity on CckA with IC50 of 12.3 μM, 17.5 μM and 11.2 μM, respectively and on PhoQ with IC50 of 66 μM, 45 μM and 35 μM, respectively. Moreover they predicted the binding mode of 91a by molecular docking studies to try to understand which molecular interactions were involved in ATP-binding. In addition, molecules 91a-c also had moderate antimicrobial activities of Gram-positive (B. subtilis) and Gram-negative (E. coli and C. crescentus) strains with MICs ranging 11-81 μg/mL, comparable or even better than MICs obtained for 85 and 86.
The preparation of compounds 91a-c is disclosed in scheme 17. Friedel-Crafts acylation of the respective resorcinols 87 and susbtitued phenyl acetic acid 88 using boron trifluoride diethyletherate as a Lewis acid catalyst, gave the corresponding dihydroxyphenyl ketones 89a-c. Ketones 89 in the presence of acetic anhydride and potassium carbonate were refluxed to form alkylated isoflavones 90a-c. Finally, compounds 90 were heated in the presence of hydrazine to give the desired diaryl pyrazole 91a-c in 24% (91a), 3% (91b) and 8% (91c) yield over three steps.92
Scheme 1792: Reagents and conditions:

(i) BF3.OEt2, 80°C, overnight; (ii) acetic anhydride, K2CO3, DMF, 160°C; (iii) hydrazine hydrate, ethanol, 60°C, 1 h.
N). Miscellaneous
Most TCSs inhibitors have been discovered by in vitro high-throughput screening or by structure based virtual screening experiments. Synthetic pathways are not described in the literature for some compounds and these are summarized in this section.
In 2005, Gilmour et al. described the structure of a pyridine derivative 92 (Table II and III), identified by high throughput screening. Compound 92 was purchased from AsInEx Company or ChemBridge Corporation and evaluated as an inhibitor of a large number of HK proteins. The authors showed that compound 92 exhibited ATP-competitive inhibitory activity against four HK proteins at concentrations between 5.5 and 103.8 μM. Specificity to HK was assured since the compound did not inhibit a panel of ten mammalian protein kinases (serine and threonine). The compound failed to significantly inhibit bacterial growth with a MIC of 256 μg/mL or worse against several gram-positive and gram-negative bacteria,93 suggesting that the compound might not permeate through the bacterial membrane.
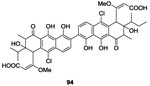
Filamentous actinobacteria of the genus Streptomyces were known for their propensity to produce bioactive secondary metabolites that have, e.g., antifungal, antiviral, antibacterial activities, etc.94 In 2010 and 2011 Utsumi’s research team focused on screening 1368 broths of Streptomyces cultures for TCS inhibitory activity. By this method,95 in 2010, Okada et al., isolated and evaluated two molecules subsequently referred to as walkmycins for their biological properties. Walkmycin 93 and 94 were obtained by fermentation. Walkmycin B 93 inhibited WalK autophosphorylation by binding to the WalK cytoplasmic regions of B. subtilis and S. aureus with concentration values in the micromolar range (IC50= 1.6 μM and 5.7 μM, respectively).96 Walkmycin B 93 also presented antibacterial growth inhibition with MIC values ranging 0.2-6.25 μg/mL only against Gram-positive strains (B. subtilis, S. aureus and E. faecalis). On the other hand, the inhibitory effect of walkmycin C 94, against the autophosphorylation activity of the cytoplasmic regions of various HK proteins, namely VicK, CiaH and LiaS was analyzed and 94 showed IC50 of 2.87 μM, 4.87 μM and 5.63 μM, respectively.97 Antibacterial evaluations on Gram-positive strains (B. subtilis, S. aureus, E. faecalis, E. faecium, S. pneumoniae and S. pyogenes) revealed that Walkmycin 94 inhibited bacterial growth with MIC values ranging 0.06 to 16 μg/mL.
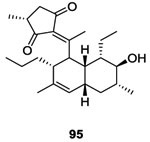
Continuing their search for new inhibitors of HK proteins, in 2012, Watanabe et al. developed a high-throughput genetic system to find potential inhibitors of WalK. For this they screened natural products that could target the DHp domain. Isolated from a culture of Streptomyces, signermycin B 95 inhibited the dimerization domain and subsequent autophosphorylation of WalK. The structure of the natural compound was confirmed by NMR, IR, Mass spectrometry and UV. Signermycin B 95, showed IC50s of 37-62 μM against different HK proteins from Gram-positive strains of S. aureus, E. faecalis, B. subtilis and S. mutans.98 The bacterial growth of these Gram-positive strains was inhibited by Signermycin B with MICs ranging from 3.13 to 6.25 μg/mL.
In 2013 and 2015, new studies by Igarashi and Fakhruzzaman carried out on metabolites synthesized by Actinomycetes, revealed new inhibitors of the HK WalK. Waldiomycin 96 (obtained by fermentation) is a hexacyclic molecule belonging to the family of angucyclines, compounds that have a tetracyclic ring. The authors showed that 96 had inhibitory activity against WalK HKs purified from B. subtilis, S. aureus, E. faecalis and S. mutans with IC50 values ranging from 8.8 μM to 25.8 μM.99,100 Waldiomycin 96 also inhibited the bacterial growth of Gram-positive strains with MIC values ranging from 8-16 μg/mL.. These results were confirmed in 2017 by Kato et al. that revealed Waldiomycin 96 also inhibited PhoR and ResE kinases by targeting the DHp region, demonstrating the desirable inhibition of multiple HK proteins.101 Unlike most other studies before, this studies also showed that Waldiomycin 96 inhibited the expression of genes under regulation by the WalK/WalR system in vivo, thus confirming that this compound can reach its cellular target.
In 2016 by employing in vitro fragment-based screening (FBS) with differential scanning fluorimetry (DSF), Velikova et al. identified molecules that inhibited autophosphorylation of different HK proteins in vitro. Those molecules also showed antibacterial activity against various laboratory strains of bacteria. In order to identify compounds with broad spectrum activity they screened compounds against two HK from diverse bacteria: a Gram-negative representative (E. coli PhoRE) and a Gram-positive representative (S. aureus PhoRS). Twenty-five components were evaluated and one potent compound, 97, was identified (Table II and III). Compound 97, which is a commercially available molecule with antibacterial effect against Gram-positive bacteria (MIC= 8-16 μg/mL) showed competitive inhibition of autophosphorylation activity of these kinases with IC50= 16 μM (PhoRE) and 212 μM (PhoRS).102,103
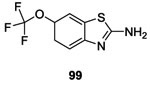
In 2015, Wilke et al. described a high-throughput fluorescence polarization displacement assay to identify potential competitive HK inhibitors from a library of 53,000 compounds. Among 9 positive hits two compounds, 98 and 99 possessed a benzothiazole core and presented good inhibitory activity on binding CA domain of multiples HK and antibacterial effects on Gram-positive and Gram-negative bacteria. The IC50 and MIC values of these compounds are summarized in Table II and III. 88 Following, in 2017, Goswami et al. generated a small library of compounds with a benzothiazole core by high-throughput screening to evaluate their biochemical potency and the structure-activity relationship. They showed that compounds 100 and 101 had IC50 values of 8.3 μM and 1.56 μM, respectively.88,91,104 However, 100 and 101 did not possess antimicrobial activity. Nonetheless, these molecules could be promising because not only did they not promote protein aggregation, they also significantly reduced the production of virulence factors of various pathogenic bacteria.
In 2018, Wilke et al. pursued their studies on ATP-binding domain inhibition. 105 They argued that some serine/threonine or tyrosine kinase inhibitors could be repurposed to specifically target HK CA domain that is unique to the latter (along with Gyrases, topoisomerases and heat shock proteins). Wilke et al. showed that mammalian kinase inhibitors could inspire the design of new HK inhibitors. To this end, they selected 222 published serine/threonine and tyrosine kinase inhibitors and identified nine compounds that presented good inhibitory activity on HK853 by high-throughput screening assays. They noticed that several among the nine compounds inhibited autophosphorylation by promoting promiscuous protein aggregation. One of the compounds 102 showed good inhibitory activity on HK853 without promoting protein aggregation. However, this molecule did not inhibit bacterial virulence factor production in vivo, had no antimicrobial activity and was expected to be a mammalian kinases inhibitor. They noticed that molecule 102 shares some structural similarities with the adenine series of HK inhibitors (see section L). Thus, this class of molecules could serve to inspire the design of future inhibitors with HK specificty. 105
V. CONCLUSION
The bacterial TCS has been recognized as a potential target for novel antibiotics some 25 years ago. The realization that multi-drug resistant bacteria are a significant threat to human health has led to the numerous studies included in this review to identify inhibitors of TCS proteins, primarily focusing on HK inhibition. These studies have identified a wide range of molecules with diverse chemical structures, capable of inhibiting HK activity. Yet, despite these extensive efforts, none of the identified inhibitors have made it into clinic, nor are any of these compounds even in clinical trials, to the best of our knowledge. To move forward and ultimately accomplish the goal of identifying new viable antimicrobial agents, obstacles have to be understood and addressed. We will use the conclusion section to speculate what these obstacles might be.
Since the pharmaceutical industry all but abended the pursuit of new antibiotics in the late 90ies, the studies described here were almost exclusively carried out by academic laboratories that often lack the resources and expertise to move compounds forward. This might be one reason, why no follow up manuscripts of many of the published compounds were described and gives hope that some of the already discovered compounds might have utility as new antibiotics.
Perhaps subsequent toxicity studies were pursued in some cases and identified compounds did prove to be only semi-specific inhibitors of enzyme activity with undesired off-target toxicity to mammalian systems. Negative results are commonly not published and the scientific public would thus nor be informed why no follow up studies were published. One study that did address negative properties of some HK inhibitors was published in 2000 by Stephenson et al.15 The authors found that several known HK inhibitors at the time, including Closantel and RWJ-49815 exerted their activity by partially denaturing HK proteins in vitro, leading to protein aggregation. Furthermore, some of these compounds had hemolytic activity on sheep red blood cells, suggesting general cytotoxicity. Subsequent studies were more conscious of this problem and often eliminated inhibitors that merely caused protein aggregation or had hemolytic activity. Yet toxicity studies on mammalian cell lines rarely accompanied any of the above studies.
Many of the described inhibitors were tested for and often exerted antimicrobial activity with MIC values in the range of antibiotics in clinical use (typically in the low μg/mL range). However, for most of these compounds it is unclear whether the ability to inhibit HK proteins as identified in in vitro assays is the causation for antimicrobial activities or whether killing of bacteria is independent of in vitro HK inhibitory activity. To this end, the recent discovery a phosphohistidine specific antibody106 should prove useful to establish that in vitro inhibition actually translates to in vivo inhibition of the same HK proteins. Such assays might single out certain molecules among the described that should receive increased attention and eliminate others from further pursuit. The antibody can also be utilized to avoid in vitro assays in future approaches and immediately start with screening of compounds that actually have confirmed in vivo effects.
Several of the more recent studies used to identify HK inhibitors utilized known structures of HK proteins and in silico screening in order to identify hit compounds that were further pursued experimentally. Yet to date, an experimental crystal structure of a HK in complex with any of the inhibitors has remained elusive. HK proteins had been notoriously difficult to crystalize, evident by a relatively small number of structures in the PDB, until recently. This might be one of the reasons why an inhibitor complexed HK structure is not yet available. Such a structure however could prove invaluable in further improving specificity and affinity of known HK inhibitors and should be pursued for the most promising compounds.
In summary, there are several advance in recent years that give hope to the notion that some of the described compounds or perhaps new ones might eventually make it from laboratory to bedside. Increased efforts and resources will be necessary to make this happen.
VI. Acknowlegments
Miyanou Rosales Hurtado, Pr. Patrick Meffre and Dr. Zohra Benfodda thank the French « ministère de l’éducation nationale, de l’enseignement supérieur et de la recherche » and the University of Nîmes for their financial support.
Dr. Szurmant was supported by grant GM106085 from the US National Institute of General Medical Sciences, National Institutes of Health.
Biography
Miyanou Rosales-Hurtado holds a master’s degree in Molecular and Macromolecular Chemistry at University of Bordeaux, in Talence, France. She is now a PhD student at the University of Nîmes (France) under the supervision of Prof. Patrick MEFFRE and Dr. Zohra BENFODDA. Now, she is interested on how overcome the bacterial resistance. That is the reason why, her doctoral research investigates the design, synthesis and evaluation of new potential agents to combat antibacterial resistance.
Pr. Patrick Meffre was born in Marseille, France, in 1964. After studies at the Ecole Nationale Supérieure de Chimie de Paris (ENSCP, Chimie ParisTech, a member of Université Paris-Sciences et Lettres, PSL, France) he receives his Ph.D. at the Pierre and Marie Curie University (Paris) in 1991. After post-doctoral training with Prof S. Hanessian (Montreal University, Québec, Canada) he was appointed Assistant Professor at the Ecole Nationale Supérieure de Chimie de Paris in 1993. Since 2004 he moved to university of Nîmes (France) where he was appointed as Professor. His current interest concerns the synthesis of unusual amino acids, chiral synthons derived from natural amino acids, chiral or non-chiral biologically active compounds, extractions of natural products from plants.
Dr. Hendrik Szurmant is an Associate Professor for Microbiology at Western University of Health Sciences in Pomona, California, USA and an Adjunct Professor at The Scripps Research Institute in La Jolla, California, USA. Dr. Szurmant has over 20 years of experience working on diverse topics related to bacterial signal transduction, with an emphasis on two-component systems. His primary expertise is in molecular microbiology, biochemistry and structural biology. Applying various techniques from these fields, he characterized the essential WalRK two-component system of the Gram-positive model organism Bacillus subtilis. The WalRK system has commonly been described as a promising target for anti-invectives and is a focus of the current review article. Beyond his work on individual signal transduction systems, Dr. Szurmant and colleagues have devised a now commonly utilized bioinformatic technique termed Direct-Coupling Analysis, which can deduce structures and function of proteins and their complexes from extensive sequence alignments.
Dr. Zohra Benfodda received her PhD in organic chemistry at Montpellier University in 2006 under the direction of Dr H. Blancou on the synthesis of fluorinated sulfonamides, sulfonimides and sulfonylureas and their biological properties. After post-doctoral training with Prof. Lluis Fajas (cancer research institute of Montpellier), she was an associate professor at Nîmes university where her research focuses on the syntheses of heterocycles derivatives that inhibit bacterial histidine kinase and on syntheses of unusual amino acids with biological properties.
VII References
- 1.Chellat MF, Raguž L, Riedl R. Targeting Antibiotic Resistance. Angew Chemie - Int Ed 2016;55:6600–6626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mitscher LA, Pillai SP, Gentry EJ, Shankel DM. Multiple drug resistance. Med Res Rev 1999;19:477–496. [DOI] [PubMed] [Google Scholar]
- 3.Stanton TB. A call for antibiotic alternatives research. Trends in Microbiology. 2013;21:111–113. [DOI] [PubMed] [Google Scholar]
- 4.Jabes D The antibiotic R&D pipeline: An update. Current Opinion in Microbiology. 2011;14:564–569. [DOI] [PubMed] [Google Scholar]
- 5.Lee VJ, Hecker SJ. Antibiotic resistance versus small molecules, the chemical evolution. Med Res Rev. 1999;19:521–542. [DOI] [PubMed] [Google Scholar]
- 6.Worthington RJ, Melander C. Combination approaches to combat multidrug-resistant bacteria. Trends in Biotechnology. 2013;31:177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomason P, Kay R. Eukaryotic signal transduction via histidine-aspartate phosphorelay. J Cell Sci. 2000;113:3141–3150. [DOI] [PubMed] [Google Scholar]
- 8.Ni N, Li M, Wang J, Wang B. Inhibitors and antagonists of bacterial quorum sensing. Med Res Rev. 2009;29:65–124. [DOI] [PubMed] [Google Scholar]
- 9.Cashin P, Goldsack L, Hall D, O’Toole R. Contrasting signal transduction mechanisms in bacterial and eukaryotic gene transcription. FEMS Microbiology Letters 2006;261:155–164. [DOI] [PubMed] [Google Scholar]
- 10.Casino P, Rubio V, Marina A. The mechanism of signal transduction by two-component systems. Current Opinion in Structural Biology. 2010;20:763–771. [DOI] [PubMed] [Google Scholar]
- 11.Laub MT, Goulian M. Specificity in Two-Component Signal Transduction Pathways. Annu Rev Genet. 2007;41:121–145. [DOI] [PubMed] [Google Scholar]
- 12.Gross R, Beier D. Two-Component Systems in Bacteria. 2012. xii–426 p. [Google Scholar]
- 13.Zschiedrich CP, Keidel V, Szurmant H. Molecular Mechanisms of Two-Component Signal Transduction. Journal of Molecular Biology. 2016;428:3752–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gotoh Y, Eguchi Y, Watanabe T, Okamoto S, Doi A, Utsumi R. Two-component signal transduction as potential drug targets in pathogenic bacteria. Current Opinion in Microbiology. 2010;13:232–239. [DOI] [PubMed] [Google Scholar]
- 15.Stephenson K, Yamaguchi Y, Hoch JA. The mechanism of action of inhibitors of bacterial two-component signal transduction systems. J Biol Chem. 2000;275:38900–38904. [DOI] [PubMed] [Google Scholar]
- 16.Stephenson K, Hoch JA. Virulence- and antibiotic resistance-associated two-component signal transduction systems of Gram-positive pathogenic bacteria as targets for antimicrobial therapy. Pharmacoloy Ther. 2002;93:293–305. [DOI] [PubMed] [Google Scholar]
- 17.Capra EJ, Laub MT. Evolution of Two-Component Signal Transduction Systems. Annu Rev Microbiol. 2012;66:325–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang S Protein Phosphorylation in Human Health. Intech; 2012. 439 p. [Google Scholar]
- 19.Stock AM, Robinson VL, Goudreau PN. Two-Component Signal Transduction. Annu Rev Biochem. 2000;69:183–215. [DOI] [PubMed] [Google Scholar]
- 20.West AH, Stock AM. Histidine kinases and response regulator proteins in two-component signaling systems. Trends in Biochemical Sciences. 2001;26:369–376. [DOI] [PubMed] [Google Scholar]
- 21.Bem AE, Velikova N, Pellicer MT, Van Baarlen P, Marina A, Wells JM. Bacterial histidine kinases as novel antibacterial drug targets. ACS Chem Biol. 2015;10:213–224. [DOI] [PubMed] [Google Scholar]
- 22.Gossage L, Eisen T. Targeting multiple kinase pathways: A change in paradigm. Clinical Cancer Research. 2010;16:1973–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takada H, Yoshikawa H. Essentiality and function of WalK/WalR two-component system: The past, present, and future of research. Bioscience, Biotechnology and Biochemistry. 2018;82:741–751. [DOI] [PubMed] [Google Scholar]
- 24.Velikova N, Bem AE, Van Baarlen P, Wells JM, Marina A. Walk, the path towards new antibacterials with low potential for resistance development. ACS Medicinal Chemistry Letters. 2013;4:891–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Szurmant H, Nelson K, Kim EJ, Perego M, Hoch JA. YycH regulates the activity of the essential YycFG two-component system in Bacillus subtilis. J Bacteriol. 2005;187:5419–5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dubrac S, Boneca IG, Poupel O, Msadek T. New insights into the WalK/WalR (YycG/YycF) essential signal transduction pathway reveal a major role in controlling cell wall metabolism and biofilm formation in Staphylococcus aureus. J Bacteriol. 2007;189:8257–8269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Szurmant H Two-component systems in Bacteria. Caister Academic Press; 2012. 127 p. [Google Scholar]
- 28.Bisicchia P, Noone D, Lioliou E, Howell A, Quigley S, Jensen T, Jarmer H, Devine KM. The essential YycFG two-component system controls cell wall metabolism in Bacillus subtilis. Mol Microbiol. 2007;65:180–200. [DOI] [PubMed] [Google Scholar]
- 29.Dubrac S, Bisicchia P, Devine KM, Msadek T. A matter of life and death: cell wall homeostasis and the WalKR (YycGF) essential signal transduction pathway. Mol Microbiol. 2008;70:1307–1322. [DOI] [PubMed] [Google Scholar]
- 30.Grossman AD. Genetic Networks Controlling the Initiation of Sporulation and the Development of Genetic Competence in Bacillus subtilis. Annu Rev Genet. 1995;29:477–508. [DOI] [PubMed] [Google Scholar]
- 31.Jiang M, Shao W, Perego M, Hoch JA. Multiple histidine kinases regulate entry into stationary phase and sporulation in Bacillus subtilis. Mol Microbiol. 2000;38:535–542. [DOI] [PubMed] [Google Scholar]
- 32.Eswaramoorthy P, Duan D, Dinh J, Dravis A, Devi SN, Fujita M. The threshold level of the sensor histidine kinase KinA governs entry into sporulation in Bacillus subtilis. J Bacteriol. 2010;192:3870–3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Winnen B, Anderson E, Cole JL, King GF, Rowland SL. Role of the PAS sensor domains in the Bacillus subtilis sporulation kinase KinA. J Bacteriol. 2013;195:2349–2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mielich-Süss B, Lopez D. Molecular mechanisms involved in Bacillus subtilis biofilm formation. Environmental Microbiology. 2015;17:555–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parsek MR, Singh PK. Bacterial Biofilms: An Emerging Link to Disease Pathogenesis. Annu Rev Microbiol. 2003;57:677–701. [DOI] [PubMed] [Google Scholar]
- 36.Cai X, Zhang J, Chen M, Wu Y, Wang X, Chen J, Zhang J, Shen X, Qu D, Jiang H. The effect of the potential phoq histidine kinase inhibitors on shigella flexneri virulence. PLoS One.2011;6:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Crépin S, Chekabab SM, Le Bihan G, Bertrand N, Dozois CM, Harel J. The Pho regulon and the pathogenesis of Escherichia coli. Vet Microbiol. 2011;153:82–88. [DOI] [PubMed] [Google Scholar]
- 38.Smirnov A, Esnault C, Prigent M, Holland IB, Virolle MJ. Phosphate homeostasis in conditions of phosphate proficiency and limitation in the wild type and the phoP mutant of Streptomyces lividans. PLoS One. 2015;10:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ghorbel S, Kormanec J, Artus A, Virolle MJ. Transcriptional studies and regulatory interactions between the phoR-phoP operon and the phoU, mtpA, and ppk genes of Streptomyces lividans TK24. J Bacteriol. 2006;188:677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Härtig E, Jahn D. Regulation of the Anaerobic Metabolism in Bacillus subtilis. Adv Microb Physiol. 2012;61:195–216. [DOI] [PubMed] [Google Scholar]
- 41.Foo YH, Spahn C, Zhang H, Heilemann M, Kenney LJ. Single cell super-resolution imaging of E. coli OmpR during environmental stress. Integr Biol (United Kingdom). 2015;7:1297–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Whitchurch CB, Erova TE, Emery JA, Sargent JL, Harris JM, Semmler ABT, Young MD, Mattick JS, Wozniak DJ. Phosphorylation of the Pseudomonas aeruginosa response regulator AlgR is essential for type IV fimbria-mediated twitching motility. J Bacteriol. 2002;184:4544–4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koteva K, Hong HJ, Wang XD, Nazi I, Hughes D, Naldrett MJ, Buttner MJ, Wright GD. A vancomycin photoprobe identifies the histidine kinase VanSsc as a vancomycin receptor. Nat Chem Biol. 2010;6:327–329. [DOI] [PubMed] [Google Scholar]
- 44.Sperandio V, Mellies JL, Nguyen W, Shin S, Kaper JB. Quorum sensing controls expression of the type III secretion gene transcription and protein secretion in enterohemorrhagic and enteropathogenic Escherichia coli. Proc Natl Acad Sci USA. 1999;96:15196–15201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Clarke MB, Hughes DT, Zhu C, Boedeker EC, Sperandio V. The QseC sensor kinase: A bacterial adrenergic receptor. Proc Natl Acad Sci USA. 2006;103:10420–10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shafikhani SH, Núñez E, Leighton T. Hpr (ScoC) and the phosphorelay couple cell cycle and sporulation in Bacillus subtilis. FEMS Microbiol Lett. 2004;231:99–110. [DOI] [PubMed] [Google Scholar]
- 47.Wang X, Vu A, Lee K, Dahlquist FW. CheA-receptor interaction sites in bacterial chemotaxis. J Mol Biol 2012;422:282–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Weiss V, Kramer G, Dunnebier T, Flotho A. Mechanism of Regulation of the Bifunctional Further Reading. J Mol Microbrol Biotechnol. 2002;4:229–233. [PubMed] [Google Scholar]
- 49.Liu Y, Rose J, Huang S, Hu Y, Wu Q, Wang D, Li C, Liu M, Zhou P, Jiang L. A pH-gated conformational switch regulates the phosphatase activity of bifunctional HisKA-family histidine kinases. Nat Commun. 2017;8:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Narayanan S, Kumar L, Radhakrishnan SK. Sensory domain of the cell cycle kinase CckA regulates the differential DNA binding of the master regulator CtrA in Caulobacter crescentus. Biochim Biophys Acta - Gene Regul Mech. 2018;1861:952–961. [DOI] [PubMed] [Google Scholar]
- 51.Watanabe T, Okada A, Gotoh Y, Utsumi R. Bacterial signal transduction: Network and drug targets. Springer Sciences; 2008. 229 p. [Google Scholar]
- 52.Roychoudhury S, Zielinski NA, Ninfa AJ, Allen NE, Jungheim LN, Nicas TI, Chakrabarty AM. Inhibitors of two-component signal transduction systems: inhibition of alginate gene activation in Pseudomonas aeruginosa. Proc Natl Acad Sci USA. 1993;90:965–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matsushita M Histidine kinases as targets for new antimicrobial agents. Bioorganic & Med Chem. 2002;10:855–867. [DOI] [PubMed] [Google Scholar]
- 54.Yamamoto K, Kitayama T, Ishida N, Watanabe T, Tanabe H, Takatani M, Okamoto T, Utsumi R. Identification and Characterization of a Potent Antibacterial Agent, NH125 against Drug-resistant Bacteria. Biosci Biotechnol Biochem. 2000;64:919–923. [DOI] [PubMed] [Google Scholar]
- 55.Yamamoto K, Kitayama T, Minagawa S, Watanabe T, Sawada S, Okamoto T, Utsumi R. Antibacterial Agents that Inhibit Histidine Protein Kinase YycG of Bacillus subtilis. Biosci Biotechnol Biochem. 2001;65:2306–2310. [DOI] [PubMed] [Google Scholar]
- 56.Demers JP, Johnson S, Weidner-Wells MA, Kanojia RM, Fraga-Spano SA, Klaubert DH. Triphenylalkyl antimicrobial agents. US5643950A1 1995.
- 57.Demers JP. Triphenylalkyl antimicrobial agents. WO9748676A1.
- 58.Barrett JF, Goldschmidt RM, Lawrence LE, Foleno B, Chen R, Demers JP, Johnson S, Kanojia R, Fernandez J, Bernstein J, Licata L, Donetz A, Huang S, Hlasta DJ, Macielag MJ, Ohemeng K, Frechette R, Frosco MB, Klaubert DH, Whiteley JM, Wang L, Hoch JA. Antibacterial agents that inhibit two-component signal transduction systems. Proc Natl Acad Sci USA. 1998;95:5317–5322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Frechette R, Weidner-Wells MA. Benzoxazine antimicrobial agents. WO97/17333. 1997. [Google Scholar]
- 60.Macielag MJ, Goldschmidt R. Inhibitors of bacterial two-component signalling systems Inhibitors of bacterial two-component signalling systems. Exp Opin Invest Drugs. 2000;9:2351–2369. [DOI] [PubMed] [Google Scholar]
- 61.Hilliard JJ, Goldschmidt RM, Licata L, Baum EZ, Bush K. Multiple mechanisms of action for inhibitors of histidine protein kinases from bacterial two-component systems. Antimicrob Agents Chemother. 1999;43:1693–1699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Frechette RF, Beach & MJ. The Preparation of 2-Hydroxyethyl-2,3- dihydro-2H-1,4- benzoxazin-3(4H)-one Derivatives. Synth Commun. 1998;28:3471–3478. [Google Scholar]
- 63.Bindler J, Model E. Poly halo-salicylanilides. US2703332A 1955.
- 64.Janssen MAC, Sipido VK. Antiparasitic salicylanilide derivatives. US4005218USA 1977.
- 65.Hlasta DJ, Demers JP, Foleno BD, Fraga-Spano SA, Guan J, Hilliard JJ, Macielag MJ, Ohemeng KA, Sheppard CM, Sui Z, Webb GC, Weidner-Wells MA, Werblood H, Barrett JF. Novel inhibitors of bacterial two-component systems with gram positive antibacterial activity: Pharmacophore identification based on the screening hit closantel. Bioorganic Med Chem Lett. 1998;8:1923–1928. [DOI] [PubMed] [Google Scholar]
- 66.Macielag MJ, Demers JP, Fraga-Spano SA, Hlasta DJ, Johnson SG, Kanojia RM, Russell RK, Sui Z, Weidner-Wells MA, Werblood HM, Foleno BD, Goldschmidt RM, Loeloff MJ, Webb GC, Barrett JF. Substituted Salicylanilides as Inhibitors of Two-Component Regulatory Systems in Bacteria. J Med Chem. 1998;41:2939–2945. [DOI] [PubMed] [Google Scholar]
- 67.Sui Z, Guan J, Hlasta DJ, Macielag MJ, Foleno BD, Goldschmidt RM, Loeloff MJ, Webb GC, Barrett JF. Sar studies of diaryltriazoles against bacterial two-component regulatory systems and their antibacterial activities. Bioorg Med Chem Lett. 1998;8:1929–1934. [DOI] [PubMed] [Google Scholar]
- 68.Alvarez R, Velázquez S, San-Félix A, Aquaro S, De Clercq E, Perno CF, Karlsson A, Balzarini J, Camarasa MJ. 1,2,3-Triazole¯[2′,5′-Bis-O-(tert-butyldimethylsilyl)-β-D-ribofuranosyl]-3′-spiro-5″-(4″-amino-1″,2″-oxathiole 2″,2″-dioxide) (TSAO) Analogs: Synthesis and Anti-HIV-1 Activity. J Med Chem. 1994;37:4185–4194. [DOI] [PubMed] [Google Scholar]
- 69.Kharb R, Sharma PC, Yar MS. Pharmacological significance of triazole scaffold. J Enzyme Inhib Med Chem. 2011;26:1–21. [DOI] [PubMed] [Google Scholar]
- 70.Kanojia RM, Murray W, Bernstein J, Fernandez J, Foleno BD, Krause H, Lawrence L, Webb G, Barrett JF. 6-Oxa isosteres of anacardic acids as potent inhibitors of bacterial histidine protein kinase (HPK)-mediated two-component regulatory systems. Bioorganic Med Chem Lett. 1999;9:2947–2952. [DOI] [PubMed] [Google Scholar]
- 71.Kitayama T, Okamoto T, Hill RK, Kawai Y, Takahashi S, Yonemori S, Yamamoto Y, Ohe K, Uemura S, Sawada S. Chemistry of Zerumbone 1. Simplified isolation, conjugate addition reactions, and a unique ring contracting transannular reaction of its dibromide. J Org Chem. 1999;64:2667–2672. [DOI] [PubMed] [Google Scholar]
- 72.Kitayama T, Yamamoto K, Utsumi R, Takatani M, Kawai Y, Hill RK, Sawada S, Okamoto T. Chemistry of Zerumbone 2. Regulation of Ring Bond Cleavage and Unique Antibacterial Activities of Zerumbone Derivatives. Biosci Biotechnol Biochem. 2001;65:2193–2199. [DOI] [PubMed] [Google Scholar]
- 73.Kitayama T, Iwabuchi R, Minagawa S, Shiomi F, Cappiello J, Sawada S, Utsumi R, Okamoto T. Unprecedented olefin-dependent histidine-kinase inhibitory of zerumbone ring-opening material. Bioorganic Med Chem Lett. 2004;14:5943–5946. [DOI] [PubMed] [Google Scholar]
- 74.Kitayama T, Iwabuchi R, Minagawa S, Sawada S, Okumura R, Hoshino K, Cappiello J, Utsumi R. Synthesis of a novel inhibitor against MRSA and VRE: Preparation from zerumbone ring opening material showing histidine-kinase inhibition. Bioorganic Med Chem Lett. 2007;17:1098–1101. [DOI] [PubMed] [Google Scholar]
- 75.Weidner-Wells MA, Ohemeng KA, Nguyen VN, Fraga-Spano S, Macielag MJ, Werblood HM, Foleno BD, Webb GC, Barrett JF, Hlasta DJ. Amidino Benzimidazole Inhibitors of Bacterial Two-Component Systems. Bioorg Med Chem Lett. 2001;11:1545–1548. [DOI] [PubMed] [Google Scholar]
- 76.Ohemeng KA, Nguyen VN. 2-substituted phenyl-benzimidazole antibacterial agents. US5942532, 1999.
- 77.Rasko DA, Moreira CG, De RL, Reading NC, Ritchie JM, Waldor MK, Williams N, Taussig R, Wei S, Roth M, Hughes DT, Huntley JF, Fina MW, Falck JR, Sperandio V. Targeting QseC signaling and virulence for antibiotic development. Science. 2008;321:1078–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Worthington RJ, Blackledge MS, Melander C. Small-molecule inhibition of bacterial two-component systems to combat antibiotic resistance and virulence. Futur Med Chem. 2013;5:1265–1284. [DOI] [PubMed] [Google Scholar]
- 79.Sperandio V, Falck JR, Stewart D. Methods of inhibiting bacterial virulence and compounds relating thereto. WO2009088549A2 2007.
- 80.Qin Z, Zhang J, Xu B, Chen L, Wu Y, Yang X, Shen X, Molin S, Danchin A, Jiang H, Qu D. Structure-based discovery of inhibitors of the YycG histidine kinase: New chemical leads to combat Staphylococcus epidermidis infections. BMC Microbiol. 2006;6:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pan B, Huang RZ, Han SQ, Qu D, Zhu ML, Wei P, Ying HJ. Design, synthesis, and antibiofilm activity of 2-arylimino-3-aryl-thiazolidine-4-ones. Bioorganic Med Chem Lett. 2010;20:2461–2464. [DOI] [PubMed] [Google Scholar]
- 82.Pan B, Huang R, Zheng L, Chen C, Han S, Qu D, Zhu M, Wei P. Thiazolidione derivatives as novel antibiofilm agents: Design, synthesis, biological evaluation, and structure-activity relationships. Eur J Med Chem. 2011;46:819–824. [DOI] [PubMed] [Google Scholar]
- 83.Huang RZ, Zheng LK, Liu HY, Pan B, Hu J, Zhu T, Wang W, Jiang D Bin, Wu Y, Wu YC, Han SQ, Qu D. Thiazolidione derivatives targeting the histidine kinase YycG are effective against both planktonic and biofilm-associated Staphylococcus epidermidis. Acta Pharmacol Sin. 2012;33:418–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhao D, Chen C, Liu H, Zheng L, Tong Y, Qu D, Han S. Biological evaluation of halogenated thiazolo[3,2-a]pyrimidin-3-one carboxylic acid derivatives targeting the YycG histidine kinase. Eur J Med Chem. 2014;87:500–507. [DOI] [PubMed] [Google Scholar]
- 85.Dutta R, Inouye M. GHKL, an emergent ATPase/kinase superfamily. Trends Biochem Sci. 2000;25:24–28. [DOI] [PubMed] [Google Scholar]
- 86.Škedelj V, Tomašić T, Mašič LP, Zega A. ATP-binding site of bacterial enzymes as a target for antibacterial drug design. J Med Chem. 2011;54:915–929. [DOI] [PubMed] [Google Scholar]
- 87.Boibessot T, Zschiedrich CP, Lebeau A, Bénimèlis D, Dunyach-Rémy C, Lavigne JP, Szurmant H, Benfodda Z, Meffre P. The Rational Design, Synthesis, and Antimicrobial Properties of Thiophene Derivatives That Inhibit Bacterial Histidine Kinases. J Med Chem. 2016;59:8830–8847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wilke KE, Francis S, Carlson EE. Inactivation of multiple bacterial histidine kinases by targeting the ATP-binding domain. ACS Chem Biol. 2015;10:328–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Goswami M, Wilke KE, Carlson EE. Rational Design of Selective Adenine-Based Scaffolds for Inactivation of Bacterial Histidine Kinases. J Med Chem. 2017;60:8170–8182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Laufer SA, Domeyer DM, Scior TRF, Albrecht W, Hauser DRJ. Synthesis and biological testing of purine derivatives as potential ATP-competitive kinase inhibitors. J Med Chem. 2005;48:710–722. [DOI] [PubMed] [Google Scholar]
- 91.Carlson EE. Antibacterial agents including histidine kinase inhibitors. WO2018217884A1 2018.
- 92.Vo CD, Shebert HL, Zikovich S, Dryer RA, Huang TP, Moran LJ, Cho J, Wassarman DR, Falahee BE, Young PD, Gu GH, Heinl JF, Hammond JW, Jackvony TN, Frederick TE, Blair JA. Repurposing Hsp90 inhibitors as antibiotics targeting histidine kinases. Bioorg Med Chem Lett. 2017;27:5235–5244. [DOI] [PubMed] [Google Scholar]
- 93.Gilmour R, Foster JE, Sheng Q, McClain JR, Riley A, Sun PM, Ng WL, Yan D, Nicas TI, Henry K, Winkler ME. New class of competitive inhibitor of bacterial histidine kinases. J Bacteriol. 2005;187:8196–8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.de Lima Procópio RE, da Silva IR, Martins MK, de Azevedo JL, de Araújo JM. Antibiotics produced by Streptomyces. Brazilian Journal of Infectious Diseases. 2012;16:466–471. [DOI] [PubMed] [Google Scholar]
- 95.Okada A, Gotoh Y, Watanabe T, Furuta E, Yamamoto K, Utsumi R. Targeting two-component signal transduction: a novel drug discovery system. Methods Enzym. 2007;422:386–395. [DOI] [PubMed] [Google Scholar]
- 96.Okada A, Igarashi M, Okajima T, Kinoshita N, Umekita M, Sawa R, Inoue K, Watanabe T, Doi A, Martin A, Quinn J, Nishimura Y, Utsumi R. Walkmycin B targets WalK (YycG), a histidine kinase essential for bacterial cell growth. J Antibiot. 2010;63:89–94. [DOI] [PubMed] [Google Scholar]
- 97.Eguchi Y, Kubo N, Matsunaga H, Igarashi M, Utsumi R. Development of an Antivirulence Drug against Streptococcus mutans: Repression of Biofilm Formation, Acid Tolerance, and Competence by a Histidine Kinase Inhibitor, Walkmycin C. Antimicrob Agents Chemother. 2011;55:1475–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Watanabe T, Igarashi M, Okajima T, Ishii E, Kino H, Hatano M, Sawa R, Umekita M, Kimura T, Okamoto S, Eguchi Y, Akamatsu Y, Utsumi R. Isolation and characterization of signermycin B, an antibiotic that targets the dimerization domain of histidine kinase WalK. Antimicrob Agents Chemother. 2012;56:3657–3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Igarashi M, Watanabe T, Hashida T, Umekita M, Hatano M, Yanagida Y, Kino H, Kimura T, Kinoshita N, Inoue K, Sawa R, Nishimura Y, Utsumi R, Nomoto A. Waldiomycin, a novel WalK-histidine kinase inhibitor from Streptomyces sp. MK844-mF10. J Antibiot. 2013;66:459–464. [DOI] [PubMed] [Google Scholar]
- 100.Fakhruzzaman M, Inukai Y, Yanagida Y, Kino H, Igarashi M, Eguchi Y, Utsumi R. Study on in vivo effects of bacterial histidine kinase inhibitor, Waldiomycin, in Bacillus subtilis and Staphylococcus aureus. J Gen Appl Microbiol. 2015;61:177–84. [DOI] [PubMed] [Google Scholar]
- 101.Kato A, Ueda S, Oshima T, Inukai Y, Okajima T, Igarashi M, Eguchi Y, Utsumi R. Characterization of H-box region mutants of WalK inert to the action of waldiomycin in Bacillus subtilis. J Gen Appl Microbiol. 2017;63:212–221. [DOI] [PubMed] [Google Scholar]
- 102.Velikova N, Fulle S, Manso AS, Mechkarska M, Finn P, Conlon JM, Oggioni MR, Wells JM, Marina A. Putative histidine kinase inhibitors with antibacterial effect against multi-drug resistant clinical isolates identified by in vitro and in silico screens. Sci Rep. 2016;6:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Moreno Marina A, Velikova N, Finn P, Fulle S, Wells JM. Histidine kinase inhibitors with antibacterial activity. WO2016071552 2016.
- 104.Goswami M, Espinasse A, Carlson EE. Disarming the virulence arsenal of: Pseudomonas aeruginosa by blocking two-component system signaling. Chem Sci. 2018;9:7332–7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wilke KE, Fihn CA, Carlson EE. Screening serine/threonine and tyrosine kinase inhibitors for histidine kinase inhibition. Bioorganic Med Chem. 2018;26:5322–5326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fuhs SR, Meisenhelder J, Aslanian A, Ma L, Zagorska A, Stankova M, Binnie A, Al-Obeidi F, Mauger J, Lemke G, Yates JR, Hunter T. Monoclonal 1- and 3-Phosphohistidine Antibodies: New Tools to Study Histidine Phosphorylation. Cell. 2015;162:198–210. [DOI] [PMC free article] [PubMed] [Google Scholar]