

Keywords: endoplasmic reticulum, gastric epithelium, Helicobacter pylori, Na-K-ATPase, protein maturation
Abstract
Helicobacter pylori infection always induces gastritis, which may progress to ulcer disease or cancer. The mechanisms underlying mucosal injury by the bacteria are incompletely understood. Here, we identify a novel pathway for H. pylori-induced gastric injury, the impairment of maturation of the essential transport enzyme and cell adhesion molecule, Na-K-ATPase. Na-K-ATPase comprises α- and β-subunits that assemble in the endoplasmic reticulum (ER) before trafficking to the plasma membrane. Attachment of H. pylori to gastric epithelial cells increased Na-K-ATPase ubiquitylation, decreased its surface and total levels, and impaired ion balance. H. pylori did not alter degradation of plasmalemma-resident Na-K-ATPase subunits or their mRNA levels. Infection decreased association of α- and β-subunits with ER chaperone BiP and impaired assembly of α/β-heterodimers, as was revealed by quantitative mass spectrometry and immunoblotting of immunoprecipitated complexes. The total level of BiP was not altered, and the decrease in interaction with BiP was not observed for other BiP client proteins. The H. pylori-induced decrease in Na-K-ATPase was prevented by BiP overexpression, stopping protein synthesis, or inhibiting proteasomal, but not lysosomal, protein degradation. The results indicate that H. pylori impairs chaperone-assisted maturation of newly made Na-K-ATPase subunits in the ER independently of a generalized ER stress and induces their ubiquitylation and proteasomal degradation. The decrease in Na-K-ATPase levels is also seen in vivo in the stomachs of gerbils and chronically infected children. Further understanding of H. pylori-induced Na-K-ATPase degradation will provide insights for protection against advanced disease.
NEW & NOTEWORTHY This work provides evidence that Helicobacter pylori decreases levels of Na-K-ATPase, a vital transport enzyme, in gastric epithelia, both in acutely infected cultured cells and in chronically infected patients and animals. The bacteria interfere with BiP-assisted folding of newly-made Na-K-ATPase subunits in the endoplasmic reticulum, accelerating their ubiquitylation and proteasomal degradation and decreasing efficiency of the assembly of native enzyme. Decreased Na-K-ATPase expression contributes to H. pylori-induced gastric injury.
INTRODUCTION
Helicobacter pylori colonizes the normal acid-secreting stomach of ~50% of the world’s population, leading to gastritis, gastric and duodenal ulcers, gastric carcinoma, and mucosa-associated lymphoid tissue (MALT) lymphoma (9, 46, 50, 51, 61). The bacteria induce gastric inflammation in 100% of those infected (41). Initial infection often occurs early in life and typically persists lifelong without treatment (37). Chronic inflammation is known to be a trigger for further gastric injury, including decreased barrier function and gastric cancer (17). H. pylori infection is the greatest risk factor for gastric cancer development and has been classified by the World Health Organization as a class I, or definite, carcinogen, with a 75% attributable risk (46, 81a). Gastric cancer confers a significant worldwide health burden, as it is the fifth most common cancer and third most common cause of cancer death (51, 71). H. pylori is also the most common cause of gastric and duodenal ulcer disease (61). Gastric ulcers do not develop spontaneously in the normal stomach. Ulceration is a multifactorial process with acidity as a dominant factor (73), and H. pylori-induced gastric epithelial injury allows susceptibility of underlying serosa to acid-related damage. Successful treatment of H. pylori infection leads to ulcer healing rates of over 90% and is effective in preventing bleeding recurrence (24, 27, 38).
The Na-K-ATPase is an essential membrane transport enzyme expressed in the vast majority of animal cells. The Na-K-ATPase comprises a catalytic α-subunit and a structural N-glycosylated β-subunit that is essential for maturation, trafficking, and activity of the enzyme. There are four isoforms of the Na-K-ATPase α-subunit and three isoforms of the β-subunit, but the major isoforms found in epithelial cells are the α1- and β1-subunits (8, 19). The transporter uses ATP to move sodium ions out of cells and potassium ions in against their concentration gradients (60), generating ion gradients that are used to enable secondary active transport of various molecules (sugars, neurotransmitters, amino acids, waste products, etc.) and other ions (H+, Ca2+, Cl−) (33, 35, 42). The ion gradients are critical for the establishment of resting membrane potential and for rapid signaling by opening voltage-gated ion channels (15). Via establishment and maintenance of ion gradients, the Na-K-ATPase is important for multiple cell functions and processes, including cell growth, differentiation, migration, contraction, secretion, and volume regulation (10, 26, 39, 62).
Generation and maintenance of sodium and potassium gradients by the Na-K-ATPase are required for proper intercellular adhesion (52). In addition, the Na-K-ATPase has properties of a cell adhesion molecule independent of ion transport activity: The β-subunit connects to the β-subunit in neighboring cells through the specific trans-interactions between several amino acid residues and N-glycans in the extracellular domain (49, 59, 65, 66, 77, 78), while the α-subunit links to the cytoskeleton, and the trans-bridge between β-subunits stabilizes the junctional complex. The Na-K-ATPase also functions as a signaling and scaffolding molecule by interacting with extracellular cardiotonic steroids and their circulating endogenous analogs and with numerous cytoplasmic partners (42, 54, 82).
Ion-transporting, adhesive, and signaling functions of the Na-K-ATPase require proper maturation and delivery of newly synthesized enzymes to the plasma membrane. Both α- and β-subunits undergo cotranslational and posttranslational maturation, which is assisted by the endoplasmic reticulum (ER) chaperones (5, 6, 64, 68). This chaperone-assisted folding of the subunits is required for proper assembly of the α/β-heterodimers. Only intact α/β-complexes are exported from the ER, whereas unassembled subunits are retained in the ER and degraded (2, 67). The ER chaperone glucose-regulated protein (GRP)78, or BiP, is an abundant chaperone that facilitates folding of newly made transmembrane and secreted proteins, prevents early ER exit of orphan subunits of multisubunit proteins, and aids in the degradation of misfolded proteins (25, 29). Binding of the β1-subunit to BiP has specifically been shown to prevent exit of the subunit from the ER when it is not yet bound to the α-subunit, which ensures proper stoichiometry and plasma membrane delivery of the Na-K-ATPase (68). BiP also interacts with the unassembled α-subunit, facilitating subunit folding and assembly into heterodimers (5, 6).
Since the Na-K-ATPase plays a critical role in multiple cell functions, modulation of the enzyme by H. pylori could represent a potential source of gastric injury. An effect of H. pylori on the Na-K-ATPase has been suggested in the past, in two studies with data demonstrating that broth culture filtrate from cytotoxin-producing strains of H. pylori leads to a decrease in K+-dependent phosphatase activity (55, 58). However, these studies did not directly measure Na-K-ATPase abundance or activity, so the aim of the present study was to determine whether H. pylori indeed targets the Na-K-ATPase in gastric epithelial cells. The results demonstrate that the attachment of H. pylori to gastric cells impairs chaperone-assisted maturation of the Na-K-ATPase in the ER, leading to the defective assembly of α/β-heterodimers, accelerates ER-associated degradation of unassembled subunits, and decreases levels of mature Na-K-ATPase molecules in the plasma membrane. These findings represent a potential mechanism for epithelial injury by H. pylori.
MATERIALS AND METHODS
Bacterial strains and culture conditions.
H. pylori strain G27, which is cagA and vacA positive, was used for all experiments (3, 18). Before infection of cultured cells, bacteria were grown overnight on trypticase soy agar plates with 5% sheep blood (TSA plates; Fisher Scientific, Hampton, NH) in a mixed-gas incubator with 10% CO2 and 5% O2. For experiments where bacterial lysates were used, bacteria were harvested from plates, resuspended in 25 mM sodium phosphate buffer, pH 7.4, and passed through a French press three times at 10,000 psi. For experiments where conditioned medium was used, bacteria were grown in culture for 16 h or incubated with cells for the same time period, followed by removal and filtering of medium to remove intact bacteria. The filtered medium (“conditioned medium”) was then applied to cells.
Cell culture.
AGS (ATCC, Manassas, VA) and HGE-20 cells (13) were grown in 50:50 DMEM-F12 (DMEM, Cellgro Mediatech, Manassas, VA; F12, Life Technologies, Carlsbad, CA) with 10% FBS (Gemini Bio-Products, West Sacremento, CA) and 100 U/mL penicillin, and 0.1 mg/mL streptomycin (Sigma, St. Louis, MO). HGE-20 cells were provided by Dr. Daniel Mènard, who kindly granted permission to use the cells for this work. HGT-1 (12) cells were grown in DMEM (Cellgro Mediatech) containing 4.5 g/L glucose, 2 mM l-glutamine, 8 mg/L phenol red, 100 U/mL penicillin, 0.1 mg/mL streptomycin, and 10% FBS. The cell line used in each experiment was chosen based on the characteristics needed for the experiment. Most notably, HGE-20 cells form cell junctions and have polarized basolateral and apical plasma membrane domains (40), but are difficult to transfect, requiring use of the other cell lines for transfection experiments.
Infection of gastric cells with H. pylori.
Gastric epithelial cells were grown to confluence on 24-mm diameter, 0.4-µm pore size transwell inserts in six-well plates (Corning Costar, Tewksbury, MA). After reaching confluence, cells were washed with medium without antibiotics, and some wells were infected with H. pylori wildtype at a MOI of >100:1 to ensure adherence. Medium pH was adjusted with HCl to the desired pH when needed and then filter sterilized. For experiments with pH adjustments, 5 mM urea was added to the basolateral side of all wells to mimic physiological exposure of H. pylori to urea from the blood. After 16 h of cell incubation with H. pylori, nonadherent bacteria were washed off. Cells were biotinylated followed by cell lysis and isolation of biotinylated proteins as described in Cell lysis below or lysed immediately, and biotinylated proteins or total cell lysates were used as described for individual experiments.
Gerbil infection.
All animal work was approved by the Veterans Affairs Greater Los Angeles Healthcare System Institutional Animal Care and Use Committee. Male Mongolian gerbils were obtained from Charles River Laboratories (Wilmington, MA). After appropriate quarantine, they were infected with H. pylori by oral gavage. Bacteria were grown on TSA plates to a confluent lawn. Bacteria were scraped from plates and suspended in Brain Heart Infusion Medium (BD, Franklin Lakes, NJ) to a ratio of one plate per 0.5 mL medium. Target inoculate for each gerbil was a concentration of 2 × 109 colony-forming units (CFU)/mL (47); 0.5 mL of bacteria suspension was used for gavage, and medium alone was used as vehicle for uninfected control animals. Gavage was completed every other day for three inoculations. After 5 mo, gerbils were euthanized and stomachs fixed for Giemsa stain and immunofluorescence.
Human tissue.
Under an Institutional Review Board-approved protocol (UCLA IRB no. 15-001367), the pathology database was searched to retrospectively identify pediatric patients who had undergone upper gastrointestinal endoscopy and were positive for H. pylori infection, along with age-matched negative controls. Paraffin blocks were obtained and slides made with the help of the Translational Pathology Core Laboratory at UCLA. Fluorescent immunohistochemistry was completed as outlined below (Immunoprecipitation). Staining was quantified using the LSM 510 confocal software (Zeiss, Oberkochen, Germany). Three patients were used for each condition, and 10 regions were quantified from each slide for the final calculation.
Antibodies and inhibitors.
The antibodies used were the following: ErbB2 (1:1,000 Western blot; mouse, Ab-17, clone E2-4001+3B5, Thermo Fisher, Waltham, MA), β-actin (1:1,000; mouse, clone C4, Millipore, Billerica, MA and 1:1,000; rabbit, Cell Signaling Technologies, Beverly, MA), ubiquitin (1:500; mouse, clone P4D1, Santa Cruz Biotechnology, Dallas, TX), Na-K-ATPase β1-subunit (1:1,000; mouse, clone M17-P5-F11, Affinity Bioreagents, Golden, CO), cofilin (1:1,000; rabbit, Abcam, Cambridge, MA), Na-K-ATPase α-subunit (mouse, clone: C464.6, Millipore); Western blot (1:1,000), immunofluorescence (1:20). The inhibitors and concentrations used were the following: 20 µg/mL cycloheximide, 2.5 µM lactacystin, 50 nM bafilomycin, and 100 µM CA074-me (all purchased from Sigma).
Immunofluorescent staining of paraffin-embedded tissue.
Human or gerbil stomach tissue sections were deparaffinized in xylenes using four exchanges for 2 min each. Sections were hydrated gradually through graded alcohols: 100% twice for 5 min each, and 95% twice, 80% and 70% ethanol for 3 min each. For antigen retrieval, slides were heated in 1 mM EDTA in 10 mM Tris buffer, pH 9.0, at 90°C for 15 min. To quench endogenous peroxidase, the slides were incubated in 0.3% H2O2 in methanol for 15 min. Permeabilization and blocking nonspecific binding were performed by incubating the tissue with Dako Protein Block serum-free solution (Dako, Carpinteria, CA) containing 0.3% Triton for 30 min. Immunofluorescent staining was performed by a 1-h incubation with the primary antibody against the Na-K-ATPase α1-subunit (mouse, clone C464.6, Millipore) followed by a 1-h incubation with Alexa fluor 647- anti-mouse antibodies (Thermo Fisher Scientific).The actin filaments were visualized using Alexa fluor 488-phalloidin (Thermo Fisher Scientific), as described previously (77). Confocal microscopy images were acquired using a Zeiss LSM 510 laser scanning confocal microscope and analyzed using the ZEN 2009 software platform.
Intracellular sodium ion measurements.
HGE-20 cells were grown on MatTek dishes (MatTek, Ashland, MA) but otherwise using conditions described previously and infected with H. pylori compared with uninfected controls. Infected and uninfected cells were loaded with CoroNa Green-AM (Invitrogen/Molecular Probes, Eugene, OR) to a final concentration of 21 μM for 45 min and then washed with PBS. Each condition was repeated in triplicate. Ten fields were generated on the confocal microscope for each condition. Total fluorescence for each field was calculated to account for individual differences in loading of cells. Means and SD were calculated for each condition, and statistical significance was determined by t test.
Adenoviral expression constructs for NaK-α1 and NaK-β1.
For the adenoviral expression construct Ad-GFP-NaK-α1, a PCR fragment (3842 kb) from pCMV-GFP-NaK-α1, a gift from Dr. Laura Dada, coding for GFP fused in-frame to the amino terminus of the rat Na-K-ATPase α1-subunit, and a PCR fragment (1748 kb) coding for YFP fused in-frame to the amino terminus of the Na-K-ATPase β1-subunit (79), were cloned into HindIII/XbaI sites of pVQAd CMV K-NpA (ViraQuest, Inc., North Liberty, IA), respectively. The construct Ad-YFP-NaK-β1 was generated previously (69). Cells were infected with 20 plaque-forming units (pfu)/cell control adenovirus (Ad-null) or with Ad-GFP-NaK-α1 or Ad-WT-YFP-β1, as previously described (75).
Creation of stable cell line expressing GFP-NaK-α1.
Construction of the Na-K-ATPase α1-subunit tagged with GFP (GFP-α1) and the establishment of AGS cells stably expressing the rodent Na-K-ATPase α1-subunit isoform were completed as described previously (21, 22). Cells were transfected with the expression construct using the Lipofectamine 2000 Transfection Reagent (Thermo Fisher Scientific). Selection for cells expressing the highest level of rodent GFP-Na-K-ATPase α1-subunit was achieved by exposing them to a medium containing 3 µM ouabain. Since the endogenous Na-K-ATPase of AGS cells is completely inhibited by this concentration of ouabain, only successful recipients of the transfected rodent GFP-Na-K-ATPase α1-subunit would be able to survive. Resistant colonies were expanded and maintained in DMEM containing 3 µM ouabain.
Cell lysis.
In all experiments except for surface biotinylation, after incubation at the indicated conditions, cells were washed twice with ice-cold PBS containing 1 mM MgCl2 and 1 mM CaCl2 (PBS++) on ice and lysed by incubating with 50 mM Tris, pH 7.5, containing 150 mM NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate and Complete Protease Inhibitor Cocktail, 1 tablet/50 mL, (Roche Diagnostics, Indianapolis, IN) at 4°C for 30 min. For samples intended for use in experiments where ubiquitylation would be studied, 10 mM iodoacetamide (Sigma) was added to lysis buffer to inhibit deubiquitylating enzymes. Cells were scraped from the plates or transwell inserts, and insoluble material was removed by centrifugation (20,000 g, 15 min) at 4°C.
Surface biotinylation.
Cells grown on transwell inserts were washed twice with ice-cold PBS on ice, and then a membrane-impermeable biotinylation reagent (EZ-Link Sulfo-NHS-Biotin, Thermo Fisher) at 1 mg/mL in PBS was added to the basolateral chamber for 30 min. Biotinylation reaction was quenched by 2 × 15-min incubations with 50 mM ammonium chloride. Cells were washed and lysed with 1% Triton X-100 in PBS. Protein concentration was determined using BCA (Thermo Fisher). Biotinylated proteins were extracted with streptavidin-agarose (Sigma). Cell lysates were incubated overnight with beads, washed three times with 0.5% Triton X-100 in PBS, and then eluted from the beads by boiling in 4% SDS, 20% glycerol, and 1% β-mercaptoethanol in 0.1 M Tris, pH 6.8, for 5 min.
Immunoprecipitation.
Total cell lysates of HGE-20 cells expressing GFP-Na-K-α1 or YFP-Na-K-β1 incubated with or without H. pylori were incubated with 2 µL of polyclonal GFP antibody (TaKaRa, Mountain View, CA) as described previously (64, 68); 35 μL of protein A-agarose suspension (Roche Diagnostics) was used. Immunoprecipitates were washed three times with lysis buffer. Adherent proteins were eluted from the beads by incubation in 35 μL of SDS-PAGE sample buffer (4% SDS, 0.05% bromophenol blue, 20% glycerol, 1% β-mercaptoethanol in 0.1 M Tris, pH 6.8) for 5 min at 80°C. A similar procedure was used with ErbB2 antibody in untransfected cells for control experiments. Negative controls for immunoprecipitation (IP) included controls with no lysate to allow delineation of IgG bands from bands of interest and controls with beads, cell lysate and unrelated antibody or no antibody to ensure there was no nonspecific binding to the beads. Proteins separated by SDS-PAGE were analyzed by Western blotting.
Modifications of IP for nanoscale liquid chromatography-tandem mass spectrometry of immunoprecipitated proteins.
To analyze proteins coimmunoprecipitated with GFP-Na-K-α1 or YFP-Na-K-β1 by nanoscale liquid chromatography-tandem mass spectrometry (nLC-MS/MS), IP was performed as described above with the exception that each sample was run in triplicate and the eluted proteins from the three samples were combined in one volume of elution buffer as follows to concentrate the sample. The elution buffer (0.5% sodium deoxycholate, 12 mM sodium lauryl sarcosine, 50 mM triethylammonium bicarbonate) was added to the first tube and boiled for 5 min, and then the eluate was transferred into the second tube, boiled, and then transferred to the third tube and boiled for 5 min.
Modifications of IP for determination of ubiquitylation of GFP-Na,K-α1 or YFP-Na,K-β1.
For experiments designed to measure ubiquitylation, to allow for isolation of the Na-K-ATPase subunits alone without any interacting partners, cells were lysed in 2% SDS buffer (72). The lysate was boiled for 2 min in a water bath and immediately diluted with 1% Triton X-100 buffer [50 mM Tris, pH 7.4, 150 mM NaCl, 1% Triton X-100, 10 mM iodoacetamide (to inhibit deubiquitylating enzymes), 50 μM MG-132 (to inhibit proteasomal degradation), and protease inhibitors] to a final concentration of 0.1% SDS. Insoluble materials were removed by centrifugation at 15,000 g for 5 min, and clarified lysate was precleared with protein A before IP. IP of GFP-tagged proteins was performed as described above with exception that 10 mM iodoacetamide and 50 μM MG-132 were present in all washing buffers.
Western blotting.
Proteins in cell lysates, biotinylated fractions, or immunoprecipitated samples were size fractionated by SDS-PAGE and transferred to nitrocellulose membranes for Western blotting. Primary antibodies were used as described above. Secondary antibodies were horseradish peroxidase-linked goat anti-mouse or goat anti-rabbit (1:10,000; American Qualex, San Clemente, CA). Bands were detected using a SuperSignal West Pico Chemiluminescence Kit (Thermo). Immunoblots were quantified by densitometry using Image Studio Software (LI-COR, Lincoln, NE). Loading controls were used for all blots as indicated in figures and figure legends.
RNA extraction, cDNA construction, and qPCR.
RNA was isolated from HGE-20 cells following 16-h incubation with H. pylori or vehicle and using a combination of the TRIzol method (Thermo Fisher) and the RNeasy mini kit (Qiagen, Valencia, CA) as described previously (80). RNA concentration was measured on a nanodrop machine (Thermo Scientific) and RNA quality was confirmed on an Agilent Bioanalyzer using the RNA nano 600 kit (Agilent Technologies, Santa Clara, CA). All RNA used in this study had an RNA integrity number of >9. The qScript one-step SYBR Green qRT-PCR kit was used for cDNA construction and qPCR (Quanta Biosciences, Gaithersburg, MD). qPCR was completed on an iCycler CFX-96 machine (Bio-Rad, Hercules, CA) with annealing temperature of 60°. Primer design was aided by the Primer3 software available at bioinfo.ut.ee/primer3-0.4.0/ (74). Unique primers were designed for 100–300 base pair regions of the Na-K-ATPase α1- and β1-subunits and a housekeeping gene (GAPDH). All samples (vehicle and H. pylori) were run in triplicate, and PCRs were run in triplicate with each sample for a total of three biological replicates and three technical replicates for each condition. GAPDH was used as a housekeeping gene to normalize results. Results were analyzed using the comparative CT method (57). Primers for qPCR were as follows: Na-K-ATPase-α1-s, 5′-TCAGATGTGTCCAAGCAAGC-3′; Na-K-ATPase-α1-as, 5′-ACAGTCCCCAGTGGTAGTGG-3′; Na-K-ATPase-β1-s, 5′-TGGCTACAAAGAGGGCAAAC-3′; Na-K-ATPase-β1-as, 5′-TGCCCAGTCCAAAATACTCC-3′; GAPDH-s, 5′-ACCCAGAAGACTGTGGATGG-3′; GAPDH-as, 5′-TTCTAGACGGCAGGTCAGGT-3′.
Bottom-up proteomics using nLC-ESI-MS/MS.
Immunoprecipitated proteins were eluted by heating (95°C, 5 min) with lysis buffer [100 μL, 12 mM sodium lauryl sarcosine, 0.5% sodium deoxycholate, 50 mM triethylammonium bicarbonate (TEAB)]. An aliquot of the resulting solution (9 μL) was taken for measurement of total protein concentration (bicinchoninic acid assay; Micro BCA Protein Assay Kit (Thermo Fisher Scientific), using BSA as a standard). The remaining samples were diluted to equal concentrations with lysis buffer, and an aliquot of each (100 μL) was treated with tris(2-carboxyethyl) phosphine (10 μL, 55 mM in 50 mM TEAB, 30 min, 37°C) followed by treatment with chloroacetamide (10 μL, 120 mM in 50 mM TEAB, 30 min, 25°C in the dark). They were then diluted fivefold with aqueous 50 mM TEAB, and incubated overnight with Sequencing Grade Modified Trypsin (1 μg in 10 μL of 50 mM TEAB; Promega, Madison, WI), following which an equal volume of ethyl acetate-trifluoroacetic acid (TFA; 100:1, vol/vol) was added. After vigorous mixing (5 min) and centrifugation (13,000 g, 5 min), the supernatants were discarded and the lower phases were dried in a centrifugal vacuum concentrator. The samples were then desalted using a modified version of the Rappsilber protocol (53), in which the dried samples were reconstituted in acetonitrile-water-TFA (solvent A, 100 μL, 2:98:0.1, vol/vol/vol) and then loaded onto a small portion of a C18-silica disk (3M, Maplewood, MN) placed in a 200-μL pipette tip. Prior to sample loading, the C18 disk was prepared by sequential treatment with methanol (20 μL), acetonitrile-water-TFA (solvent B; 20 μL, 80:20:0.1, vol/vol/vol), and finally with solvent A (20 μL). After loading of the sample, the disk was washed with solvent A (20 μL, eluent discarded) and eluted with solvent B (40 μL). The collected eluent was dried in a centrifugal vacuum concentrator. The samples were then chemically modified using the in-solution stable isotopic dimethyl labeling protocol described by Boersema (11). The dimethyl-labeled peptides were dried and reconstituted in solvent A (50 μL), and an aliquot (2 μL) was taken for measurement of total peptide concentration (Pierce Quantitative Colorimetric Peptide, Thermo Fisher Scientific). The samples were then pooled according to protein content and desalted using the modified Rappsilber protocol described above (53). The eluants were then dried and reconstituted in water-acetonitrile-formic acid (FA; solvent E; 10 μL, 98:2:0.1, vol/vol/vol), and aliquots (5 μL) were injected onto a reverse-phase nanobore HPLC column (AcuTech Scientific, C18, 1.8 μm particle size, 360 μm × 20 cm, 150 μm ID), equilibrated in solvent E, and eluted (500 nL/min) with an increasing concentration of solvent F (acetonitrile-water-FA, 98:2:0.1, vol/vol/vol: min/%F; 0/0, 5/3, 18/7, 74/12, 144/24, 153/27, 162/40, 164/80, 174/80, 176/0, and 180/0) using an Eksigent NanoLC-2D system (Sciex, Framingham, MA). The effluent from the column was directed to a nanospray ionization source connected to a hybrid quadrupole-Orbitrap mass spectrometer (Q Exactive Plus, Thermo Fisher Scientific) acquiring mass spectra in a data-dependent mode alternating between a full scan (m/z 350–1,700, automated gain control (AGC) target 3 × 106, 50 ms maximum injection time, full width at half maximum (FWHM) resolution 70,000 at m/z 200), and up to 10 MS/MS scans (quadrupole isolation of charge states ≥2, isolation width 1.2 Th) with previously optimized fragmentation conditions (normalized collision energy of 32, dynamic exclusion of 30 s, AGC target 1 × 105, 100 ms maximum injection time, FWHM resolution 35,000 at m/z 200). The raw data were analyzed in Proteome Discoverer 2.2, which provided the heavy/light ratios, reflecting relative abundance of the identified proteins.
Overexpression of BiP.
pcDNA3.1(+)-GRP78/BiP was a gift from Richard C. Austin (Addgene plasmid no. 32701) (81). HGT-1 cells were plated on transwell inserts in six-well plates (Corning Costar, Tewksbury, MA). The following day, cells were transfected with pcDNA3.1(+)-GRP78/BiP plasmid DNA using the Lipofectamine 2000 transfection reagent (Thermo Fisher Scientific). Overexpression of BiP was confirmed by Western blot.
RESULTS
Direct attachment of H. pylori to gastric epithelial cells induces a decrease in surface and total cellular levels of Na-K-ATPase.
Infection of HGE-20 cells with H. pylori significantly decreased the levels of the α1- and β1-subunits in the basolateral membranes as measured by Western blot and densitometry. This effect was independent of pH (Fig. 1A). HGE-20 cells are highly polarized, with a definite barrier between the apical and basolateral surfaces (13, 40). Surface biotinylation was initially attempted on both the apical and basolateral sides, and, as expected, Na-K-ATPase subunits were not seen with apical labeling (not shown). Once this was confirmed, only basolateral biotinylation was used for all experiments going forward. Similarly to what was observed in the basolateral membranes of HGE-20 cells, H. pylori induced a decrease in total cellular levels of both α1- and β1-subunits of the Na-K-ATPase (Fig. 1B). Cellular levels of both α1- and β1-subunits were also decreased in two other gastric epithelial cell lines, HGT-1 and AGS (Fig. 1C). Since the β1-subunit of the Na-K-ATPase is a N-glycosylated protein, the size of N-glycans attached to this glycoprotein is different in the immature intracellular fraction of the β1-subunit and in the mature fraction of the subunit that reaches the plasma membrane, which allows a distinction between these two fractions by Western blot analysis [Fig. 1B, bands M (mature) and Im (immature)]. The immature but not the mature fraction was readily deglycosylated by Endo-H glycosidase, which is known to cleave high-mannose and hybrid, but not complex-type, N-glycans from glycoproteins (results not shown). We have demonstrated previously that the size of the lower band of the β1-subunit exactly corresponds to the size of the subunit produced in the presence of deoxymannojirimycin, which prevents the formation of hybrid and complex N-glycans but is significantly smaller than the size of the subunit produced in the presence of swainsonine, which allows the formation of hybrid N-glycans but prevents the formation of complex N-glycans (2, 67). Since the hybrid N-glycans can be formed only in the Golgi, these results indicate that the immature fraction of the β1-subunit mainly represents the ER-resident fraction of the subunit. H. pylori infection increased the amount of the ER fraction of the β1-subunit in HGE-20 cells (Fig. 1B) but not in HGT-1 or AGS cells (Fig. 1C). Also, no increase in the amount of the ER fraction of the β1-subunit was seen in AGS cells stably expressing GFP-α1 (Fig. 1D). However, incubation of these cells with H. pylori altered the distribution of GFP-α1 by increasing its fraction in the ER (Fig. 1E).
Fig. 1.

Helicobacter pylori (Hp) attachment to gastric epithelial cells decreases plasma membrane and total levels of Na-K-ATPase α1- and β1-subunits. HGE-20 cells grown on transwell inserts were infected with H. pylori. Apical pH was adjusted as indicated. A and B: cells were biotinylated on the basolateral side before cell lysis, basolateral surface proteins were extracted using streptavidin beads, and both surface proteins and total cell lysates were analyzed by Western blot. The amount of α1- and β1-subunits of Na-K-ATPase was decreased in the presence of H. pylori independently of pH, in the basolateral membranes (A) and in total cell lysates (B). CD29 was used as loading control for plasma membrane proteins, and cofilin was used as loading control for total cellular proteins. C: results from 2 other gastric cell lines (HGT-1 and AGS) demonstrate a similar decrease in total Na-K-ATPase levels. N-ethylmaleimide-sensitive fusion protein (NSF) was used as a loading control. M, mature fraction; Im, immature fraction. A stable cell line expressing green fluorescent protein (GFP)-α1 was infected with H. pylori. D: Western blot of total cell lysates demonstrated a decrease in the exogenous α1 in the presence of H. pylori. For A–D, representative blots and densitometry quantification results of 3 parallel experiments (means ± SD) are shown. Confocal microscopy demonstrates increased endoplasmic reticulum retention of GFP-α1. E: representative images and quantification of 12 microscopic fields per condition (means ± SD) are shown. ER, endoplasmic reticulum; PM, plasma membrane. *Significant difference from control (no Hp), P < 0.05, t test.
To determine whether live bacteria are required for the effect on the Na-K-ATPase, H. pylori were lysed using a French press, unbroken bacteria were filtered out, and serial dilutions of bacterial lysate were applied to AGS cells. Analysis of total AGS cell lysates performed after overnight incubation showed no effect of the bacterial lysate on the level of the Na-K-ATPase α-subunit (Fig. 2A). To determine whether factors secreted by H. pylori lead to the decrease in Na-K-ATPase, medium was removed from bacteria in culture (Fig. 2B) or from infected cells (Fig. 2C), filtered to remove intact bacteria, and applied to uninfected cells. Western blot showed no change in Na-K-ATPase in the presence of conditioned medium. Intact bacteria and vehicle were used as controls.
Fig. 2.

Attachment of Helicobacter pylori (Hp) to gastric epithelial cells is required for decrease in Na-K-ATPase. A: serial dilutions of H. pylori cell lysate were added to AGS cells, followed by cell lysis and Western blot. No change in Na-K-ATPase was seen with addition of bacterial lysate. Conditioned medium taken from bacterial cultures (B) or from cells infected with H. pylori (C) was applied to AGS cells, followed by lysis and Western blot. Again, no difference was seen in the presence of conditioned medium. Representative blots and densitometry quantification results of 3 parallel experiments (means ± SD) are shown. Controls were vehicle (no Hp) and intact bacteria (Hp). NSF, N-ethylmaleimide-sensitive fusion protein loading control. *Significant difference from control (no Hp), P < 0.05, t test.
Therefore, H. pylori infection decreases the levels of Na-K-ATPase α1- and β1-subunits in total cell lysates and in the plasma membranes in gastric epithelial cells and alters the distribution of these subunits between the ER and the plasma membrane. The effect requires bacterial attachment to epithelial cells and is not driven by bacterial factors secreted into the medium.
Presence of H. pylori leads to increased intracellular sodium.
If H. pylori induces a decrease in membrane expression of the Na-K-ATPase, then, based on transport function, there should be a consequent increase in intracellular sodium, as there would be fewer pumps available to pump sodium out. To test this hypothesis, internal sodium was measured using the sodium indicator CoroNa Green-AM (Fig. 3). Gastric cells were incubated with or without H. pylori and then loaded with CoroNa Green-AM. There was a significant difference in internal fluorescence between conditions, with increased fluorescence, indicating increased internal sodium, seen in the presence of H. pylori.
Fig. 3.

Attachment of Helicobacter pylori (Hp) to gastric cells increases intracellular sodium concentration. HGE-20 cells were incubated for 16 h with or without H. pylori and then loaded with sodium dye coroNa Green-AM. Cells were viewed using confocal microscopy. In the presence of H. pylori, with fewer transporters on the membrane, intracellular sodium was increased compared with control. Experiments were done in triplicate, and fluorescence values represent the average of 10 full fields for each condition. Fluorescence is expressed as means ± SD. *Significant difference from control (no Hp), P < 0.95, t test.
H. pylori infection decreases Na-K-ATPase levels in vivo.
Immunofluorescence staining of stomachs isolated from Mongolian gerbils infected with H. pylori showed a decrease in Na-K-ATPase α1-subunit expression compared with uninfected controls (Fig. 4, A and C). Na-K-ATPase levels were also decreased in chronic H. pylori infection in humans. Immunofluorescence of gastric antrum biopsies obtained from children with chronic H. pylori infection demonstrated a decrease in Na-K-ATPase α1-subunit staining compared with biopsies from matched uninfected controls (Fig. 4, B and C). The results demonstrated that H. pylori decreases the Na-K-ATPase levels not only in cultured gastric epithelial cells but also in an in vivo model system and in the human stomach in the setting of well-established chronic infection.
Fig. 4.

Helicobacter pylori (Hp) induces decreased expression of Na-K-ATPase in chronic infection. A: gerbils were infected with H. pylori by oral gavage and euthanized after 5 mo. Stomachs were fixed and infection confirmed by Giemsa stain. Immunofluorescence was completed using antibodies against Na-K-ATPase-α1 with F-actin counterstain. B: antrum biopsy samples from pediatric patients with or without H. pylori infection (n = 3 per group) were acquired retrospectively following an IRB-approved pathology database search. Hematoxylin-eosin (H&E) staining was completed (right). Immunofluorescence was performed using antibodies against the α1-subunit of the Na-K-ATPase. Images (left) were obtained by confocal microscopy. C: quantification of 10 microscopic fields for each model is shown as means ± SD), confirming decreased expression of Na-K-ATPase in chronic infection. *Significant difference from control (no Hp), P < 0.05, t test.
H. pylori induces proteasomal degradation of newly made Na-K-ATPase subunits.
H. pylori could potentially decrease Na-K-ATPase levels by decreasing the rate of the synthesis of Na-K-ATPase subunits or by accelerating their degradation. H. pylori induced a decrease not only in endogenous subunits of the Na-K-ATPase, but also in the exogenous GFP-α1 subunits, which are under the control of an unregulated promoter (Fig. 1D), indicating that the decrease in the Na-K-ATPase is not due to the effect of H. pylori on transcription of the genes coding for Na-K-ATPase subunits. In support of such interpretation, qPCR demonstrated no difference in levels of mRNA of the Na-K-ATPase subunits in infected and uninfected cells. The fold change in both Na-K-α1- and -β1 subunits was not statistically significant in the presence of H. pylori compared with uninfected controls (0.95 ± 0.17 for α1 and 1.21 ± 0.31 for β1, means ± SD; n = 3 biological replicates and 3 technical replicates).
To determine whether H. pylori accelerates degradation of Na-K-ATPase, AGS cells were incubated with or without H. pylori in the presence of various inhibitors of protein degradation: bafilomycin, the inhibitor of lysosomal H-ATPase (83), CA-074, a specific inhibitor of lysosomal peptidase cathepsin B (44), and lactacystin, a specific inhibitor of proteasomal degradation (70). The presence of bafilomycin or CA-074 in the incubation medium had no effect on the H.pylori-induced decrease in the α1- and β1-subunits (Fig. 5A). However, the decrease was prevented in the presence of lactacystin. A similar protective effect of lactacystin was observed in AGS cells expressing the GFP-tagged α1-subunit for both exogenous GFP-α1 and endogenous β1-subunit (Fig. 5B). The results indicate that H. pylori accelerates proteasomal degradation of the Na-K-ATPase.
Fig. 5.

Helicobacter pylori (Hp) induces proteasomal degradation of newly-made Na-K-ATPase subunits. AGS cells (A) or AGS cells stably expressing exogenous green fluorescent protein (GFP)-α1 (B) were incubated with and without H. pylori and inhibitors of lysosomal degradation, bafilomycin (Baf) or CA074-me (CA), or inhibitor of proteosomal degradation lactacystin (Lact), followed by Western blot. V, vehicle. Lactacystin was protective against the effect of H. pylori on both endogenous (A) and exogenous (B) Na-K-ATPase, whereas the other inhibitors had no effect, suggesting a proteasomal degradation pathway. C: HGE-20 cells were biotinylated and then infected with H. pylori for 16 h, and Western blot of the extracted membrane fraction showed no change in rate of degradation of biotinylated Na-K-ATPase subunits in the presence of H. pylori, demonstrating mature transporters are unaffected. D: HGE-20 cells were incubated with and without H. pylori and protein synthesis inhibitor cycloheximide (CHX), followed by lysis and Western blot. CHX prevented the effect of H. pylori on Na-K-ATPase, indicating that H. pylori affects newly made protein. Representative blots and densitometry are shown. NSF (N-ethylmaleimide sensitive fusion protein), ErbB2, and cofilin, loading controls; n = 3 experiments, *Significant difference from control, P < 0.05, t test.
Both mature and immature forms of the Na-K-ATPase might be targets of H. pylori-induced proteasomal degradation. To determine whether H. pylori increases the rate of degradation of the mature Na-K-ATPase located in the plasma membrane of gastric epithelial cells, HGE-20 cells were biotinylated from the basolateral surface before cell incubation with or without H. pylori for 16 h. After incubation, cells were lysed, and basolateral proteins were isolated and analyzed by Western blot analysis. The membrane protein ErbB2 was used as a control. A comparison of the initial level of biotinylated subunits (a control experiment in which cells were lysed immediately after biotinylation) with their final levels demonstrated that a 16-h incubation of biotin-labeled cells resulted in an ~50% decrease in the amount of biotinylated α1- and β1-subunits (Fig. 5C, left, lanes 0 and 16). The comparison of cells incubated with or without H. pylori for 16 h demonstrated no difference in the amount of biotinylated α1- and β1-subunits (Fig. 5C, right), indicating no effect of H. pylori on mature Na-K-ATPase and suggesting that H. pylori causes degradation of newly synthesized Na-K-ATPase. To confirm this interpretation, HGE-20 cells were incubated with and without the protein synthesis inhibitor cycloheximide and with and without H. pylori. As expected, the ER form of the β1-subunit disappeared in both uninfected and infected cells after cell incubation with cycloheximide (Fig. 5D), because these newly made immature β1-subunits were either transported through the Golgi to the plasma membrane in a complex with α1-subunits or degraded. Importantly, the effect of H. pylori on the total level of Na-K-ATPase was abolished by inhibition of protein synthesis (Fig. 5D), consistent with H. pylori-induced degradation of newly synthesized Na-K-ATPase subunits.
Since proteasomal degradation of proteins is usually preceded by their modification by ubiquitin linkage, we determined whether H. pylori induces ubiquitylation of the Na-K-ATPase. HGE-20 cells expressing GFP-α1 subunit (by adenoviral infection as described in materials and methods) were incubated with or without H. pylori, and GFP-α1 subunit was immunoprecipitated with GFP antibody. Immunoprecipitates were boiled to disrupt the interactions with other proteins and analyzed by Western blot using anti-ubiquitin antibodies. In parallel, total cell lysates were analyzed by Western blot. The results demonstrated that H. pylori increased the amount of ubiquitylated forms of the Na-K-ATPase-α1 subunit 2.7-fold but did not alter the degree of ubiquitylation of total cellular proteins (Fig. 6A). A similar H. pylori-induced increase in in the amount of ubiquitylated forms of the Na-K-ATPase-α1 subunit, 2.6-fold, was found in AGS cells stably expressing GFP-α1 (Fig. 6B). Taken together, the results indicate that H. pylori decreases Na-K-ATPase levels by accelerating its degradation rather than by inhibiting its biosynthesis and strongly suggest that H. pylori induces ubiquitylation of newly made Na-K-ATPase subunits, followed by proteasomal degradation of these ubiquitylated forms.
Fig. 6.

Helicobacter pylori (Hp) induces ubiquitylation of Na-K-ATPase. A: green fluorescent protein (GFP)-α1 was expressed in HGE-20 cells using an adenovirus vector, followed by immunoprecipitation (IP) using GFP antibodies and Western blot with ubiquitin (Ub) antibodies. H. pylori increased the amount of ubiquitylated forms of the α1-subunit 2.7-fold, whereas total cellular protein ubiquitylation was unaffected. Actin was used as a loading control in total cell lysates. B: ubiquitylation of GFP-α1 stably expressed in AGS cells was also increased as shown. Representative blots and densitometry quantification results of 3 parallel experiments (means ± SD) are shown. *Significant difference from control (no Hp), P < 0.05, t test. Ub, ubiquitin; NC, nontransfected control.
H. pylori decreases the interaction of the α1- and β1-subunits with BiP.
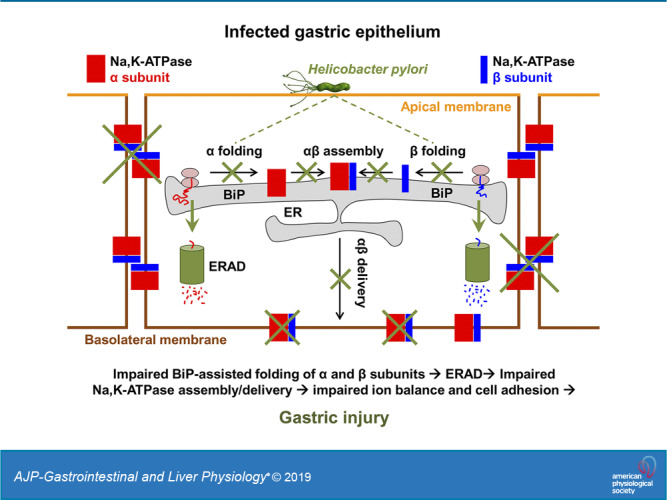
To understand how H. pylori affects the Na-K-ATPase in gastric cells, we determined whether H. pylori alters the interaction of the Na-K-ATPase subunits with other cellular proteins. To enable the isolation of subunit-interacting proteins by effective IP, GFP- and YFP-tagged α1- and β1-subunits were expressed in HGE-20 cells. The β1-subunit was fused to YFP and the α1-subunit to GFP, and adenovirus vectors were used to express both proteins. Expression of fluorescent tagged subunits at the membrane was confirmed by confocal fluorescence microscopy (Fig. 7A). Cells expressing either GFP-tagged α1-subunit or YFP-tagged β1-subunit were incubated with or without H. pylori, and then the subunit-interacting proteins were isolated by IP using GFP antibody. Protein complexes isolated from control cells and H. pylori-treated cells were fragmented using trypsin, and the peptides obtained were differentially labeled: “light” labels were added to peptides from control cells, and “heavy” labels were added to peptides from H. pylori-treated cells. Peptides from uninfected and infected cells were combined and analyzed by nLC-ESI-MS/MS (Fig. 7B). Analysis of MS/MS data by Proteome Discoverer 2.2 provided the ratio of heavy to light for each identified protein, which reflected the relative abundance of this protein in the infected cells compared with uninfected cells. These abundance ratios were normalized by the ratios obtained for the target immunoprecipitated protein (α1-subunit in GFP-α1 IP and β1-subunit in YFP-β1 IP). Therefore, the abundance ratio was set to 1 for the α1-subunit in GFP-α1 IP and for the β1-subunit in YFP-β1 IP (Fig. 7C). As expected, IP of the α1-subunit resulted in co-IP of the β-subunit. Two isoforms of the β-subunit, β1 and β3, were detected by MS/MS both in infected and in uninfected cells. According to the relative abundance ratios for β1- and β3-subunit isoforms, H. pylori infection decreased their co-IP with the α1-subunit by 40 and 50%, respectively (Fig. 7C). In addition, H. pylori infection significantly, by 60%, decreased co-IP of the ER chaperone BiP with the α1-subunit (Fig. 7C). Similarly, H. pylori infection decreased the co-IP of the α1-subunit and BiP with the β1-subunit, by 40 and 30%, respectively. The decreased interaction of the α1-subunit with BiP and with the β1-subunit was confirmed by IP of the α1-subunit followed by Western blot analysis using specific antibodies against the α1- and β1-subunits and BiP (Fig. 8A). Similarly, decreased interaction of the β1-subunit with BiP and with the α1-subunit was confirmed by IP of the β1-subunit followed by Western blot analysis using specific antibodies against the α1- and β1-subunits and BiP (Fig. 8B). Since both α1- and β1-subunits undergo BiP-assisted folding in the ER before the assembly of α1/β1-Na-K-ATPase heterodimers (5, 6, 64), the results suggest that H. pylori impairs chaperone-assisted maturation of individual subunits, which, in turn, attenuates the assembly of intact α1/β1-heterodimers.
Fig. 7.

Exposure of gastric cells to Helicobacter pylori decreases association of Na-K-ATPase subunits with endoplasmic reticulum (ER) chaperone BiP and with each other. A: to enable effective immunoprecipitation (IP) of Na-K-ATPase subunits, green fluorescent protein (GFP)-tagged α1-subunit and yellow fluorescent protein (YFP)-tagged β1-subunit were expressed in HGE-20 cells as confirmed by confocal microscopy. After infection with H. pylori, IP of the subunits was performed using GFP antibodies, and α1- or β1-coimmunoprecipitated proteins were subjected to quantitative mass spectrometry (MS) with dimethyl labeling. B: schema of the experiment is shown. In both α1- and β1-imunoprecipitates, MS results show decreased abundance of ER chaperone BiP and the respective partner Na-K-ATPase subunit(s) in the presence of H. pylori. C: results of 2 independent experiments for each α1- and β1-immunoprecipitate are shown.
Fig. 8.

Helicobacter pylori (Hp) decreases Na-K-ATPase by disrupting the Na-K-ATPase-BiP interaction. A and B: yellow fluorescent protein (YFP)-tagged β1-subunit or green fluorescent protein (GFP)-tagged α1-subunit was incorporated into HGE-20 cells and then immunoprecipitated with GFP antibody followed by Western blot, confirming the decrease in association of Na-K-ATPase subunits with BiP and with each other in the presence of H. pylori. C: there is no change in total cellular levels of BiP in the presence of H. pylori, confirming the response is specific to the interaction with the Na-K-ATPase subunits rather than decreased cellular levels of BiP. Cofilin was used as a loading control. D: ErbB2, another plasma membrane protein undergoing folding in the endoplasmic reticulum, does not change association with BiP in the presence of H. pylori. BiP was overexpressed in HGT-1 cells using pcDNA3.1(+)-GRP78/BiP. E: overexpression of BiP prevented the effect of H. pylori on Na-K-ATPase subunits. Representative blots and densitometry quantification results of 3 parallel experiments (means ± SD) are shown for all experiments. NSF (N-ethylmaleimide sensitive fusion protein), loading control. *Significant difference from control (no Hp), P < 0.05, t test. NC, nontransfected control.
H. pylori had no effect on total cellular level of BiP (Fig. 8C) and did not alter the interaction between another BiP client protein, ErbB2 (56), and BiP (Fig. 8D), demonstrating that the effect of H. pylori on the Na-K-ATPase is not a global ER stress response.
Overexpression of BiP protects against the effect of H. pylori on Na-K-ATPase levels.
The results presented above suggest that H. pylori decreases levels of Na-K-ATPase by impairing BiP-assisted folding of its newly made subunits. To test this hypothesis, we overexpressed BiP in gastric epithelial cells. HGT-1 cells were chosen for these experiments because the highly polarized HGE-20 cells are difficult to transfect and attempts to transfect AGS cells with BiP-coding plasmid did not lead to increased levels of BiP in these cells. Transient transfection of HGT-1 cells with the plasmid coding for BiP resulted in a twofold increase in the level of BiP in these cells and prevented a H. pylori-induced decrease in the levels of both α1- and β1-subunits of the Na-K-ATPase (Fig. 8E).
DISCUSSION
H. pylori causes gastric inflammation in 100% of those infected (41). Initial infection often occurs early in life and typically persists lifelong without treatment (37). It is not definitively known why only a subset of those infected will progress to more advanced disease. With the decline in H. pylori treatment efficacy to close to 70% (28), the incidence of advanced disease will increase over time. One major impediment to improving care of H. pylori-induced disease has been lack of a detailed understanding of how H. pylori actually causes gastric injury. More complete knowledge of how H. pylori is able to initiate the cascade of epithelial injury in the human stomach will lead to development of improved treatment and protective strategies. The work presented here identifies the biosynthetic trafficking of the Na-K-ATPase, a critical transport protein essential for numerous epithelial cell functions, and for the integrity of epithelial cell barrier, as a novel target of H. pylori in both acute and chronic infection models. The results indicate that direct attachment of H. pylori to gastric epithelial cells impairs the interaction of the Na-K-ATPase α- and β-subunits with the ER chaperone BiP, causing defective folding of individual subunits and accelerating their ER-associated proteasomal degradation, which, in turn, inhibits assembly of intact α1/β1-heterodimers and decreases total and plasma membrane levels of the Na-K-ATPase.
The outlined mechanism of H. pylori-induced decrease in Na-K-ATPase is supported by the following lines of evidence. No change in the amount of mRNA indicates that the decrease in the Na-K-ATPase is not due to transcriptional downregulation. This conclusion is supported by the H. pylori-induced decrease in the amount of the exogenous Na-K-ATPase that is under the control of an unregulated promoter (Fig. 1D). This focuses the mechanism to the translational or posttranslational level. The protective effect of the protein synthesis inhibitor cycloheximide and the lack of effect of H. pylori on the degradation rate of mature pumps (Fig. 5, C and D) demonstrate that H. pylori infection impacts newly synthesized Na-K-ATPase pumps rather than those already on the membrane. Quantitative mass spectrometry analysis of proteins interacting with either Na-K-ATPase subunit has revealed decreased association of both the α1- and β1-subunits with ER chaperone BiP in H. pylori-infected cells (Fig. 7C). The impaired interaction of the subunits with BiP was confirmed by IP followed by immunoblotting (Fig. 8, A and B). In addition, both mass spectrometry and Western blot of α1-immunoprecipitated and β1-immunoprecipitated complexes have demonstrated the impaired association between α1- and β1-subunits in H. pylori-infected cells (Fig. 8, A and B). BiP is a highly abundant protein that facilitates folding of nascent polypeptides, prevents unassembled subunits of multisubunit proteins from leaving the ER, and targets them for degradation when they accumulate out of proportion to need (25, 29). BiP has specifically been shown to assist folding of α1- and β1-subunits and to keep unassembled α1- and β1-subunits in the ER (5, 6, 68). By weakening the interaction of newly synthesized α1- and β1-subunits with BiP, H. pylori impairs the folding of individual subunits that is required for normal assembly of the α/β-heterodimer. This impedes the assembly of intact pumps. Since individual subunits cannot leave the ER unless they are assembled as α/β-heterodimers (2, 67), they are retained in the ER and eventually degraded (7, 67). A significant increase in ubiquitylated forms of the Na-K-ATPase α1-subunit and protective effect of lactacystin from H. pylori-induced decrease in Na-K-ATPase (Figs. 5, A and B, and 6) demonstrate that the ER-retained subunits are targeted by ubiquitylation for ER-associated proteasomal degradation. The ER-retained subunits appear to be degraded at different rates in different cell lines, resulting in different ratios between assembly-competent α- and β-subunits. In HGE-20 cells, apparently the number of properly folded α-subunits is a limiting factor for normal assembly, whereas in AGS cells, it is the number of β-subunits. Despite the decrease in total levels of the Na-K-ATPase in the setting of H. pylori infection, the ER fractions of the β-subunit were increased in HGE-20 cells as shown by immunoblotting (Fig. 1B), while the α-subunit was accumulated in the ER in AGS cells as shown by fluorescence microscopy (Fig. 1E).
H. pylori decreases the interaction of BiP with the Na-K-ATPase subunits but not with all BiP clients. Particularly, the association of another plasma membrane protein, ErbB2, with BiP is unchanged in the presence of H. pylori (Fig. 8D). Furthermore, total cellular BiP levels are unaffected by H. pylori (Fig. 8C), demonstrating that the decrease in interaction between Na-K-ATPase subunits and BiP is not mediated by a generalized ER stress response. The effect of H. pylori on the Na-K-ATPase can be partially overcome by overexpression of BiP in gastric cells (Fig. 8E). Therefore, H. pylori decreases the amount of newly formed Na-K-ATPase heterodimers in the ER by impairing BiP-assisted folding of α- and β-subunits and accelerating ER-associated ubiquitylation and proteasomal degradation of unassembled subunits. This mechanism is distinct from that described for degradation of mature pumps, which involves ubiquitylation, proteasome-dependent endocytosis and trafficking, and lysosomal degradation (14, 16, 20, 36, 63, 76).
According to the results shown here, bacterial attachment is required for the decrease in Na-K-ATPase. This contradicts previous reports on the decrease in Na-K-ATPase activity in cultured human cells by H. pylori-secreted proteins (55, 58). Those reports, however, were not substantiated by sufficient experimental evidence: they lacked both the comparison of the effects of secreted proteins with those of intact bacteria and the direct measurement of Na-K-ATPase abundance or activity. The first study reported a decrease in K+-dependent ouabain-sensitive phosphatase (55), while the second reported a decrease in K+-dependent ATPase activity (58) in human cells exposed to broth culture filtrates from cytotoxin-positive H. pylori strains. The K+-dependent ATPase activity was measured in the absence of ouabain (1, 58) and, therefore, included the activities of all cellular ATPases rather than only the Na-K-ATPase. The concentration of ouabain used in the phosphatase activity assay, 1 mM (45, 55), was several orders of magnitude higher than the Ki for ouabain for human Na-K-ATPase (19, 43), which would inhibit other cellular phosphatases. Therefore, the reported decrease in phosphatase activity (55) or ATPase activity (58) by secreted cytotoxin could be attributed to inhibiting other phosphatases or ATPases. This puts into question the specificity of the effect of broth culture filtrates on the Na-K-ATPase and may explain the contradiction of those published data to our results.
The requirement for bacterial attachment is different from known effects of H. pylori on epithelial cells that are mediated through proteins secreted into media. For example, secretion of the serine protease HtrA into the medium leads to cleavage of E-cadherin between cells (30, 31). The decrease in Na-K-ATPase is not related to the effect of H. pylori on the mature surface-exposed transporter, ruling out a direct effect of bacteria on Na-K-ATPase and suggesting a mechanism involved with intracellular signaling triggered by bacterial factors. Adherence to the gastric epithelial layer via bacterial adhesins is a critical step in successful colonization and pathogenesis (34, 48), which enables injection of bacterial factors and activation of various signaling cascades (32). The specific signaling pathways leading to a decrease in Na-K-ATPase still need to be identified. Based on the data presented here, these pathways target folding and maturation of the newly synthesized subunits of the Na-K-ATPase in the ER.
The cell culture model used for this work is by nature a model of acute infection. Cells are exposed to bacteria long enough to induce attachment and allow changes in protein trafficking, but this raises the important question of what occurs in a more chronic infection model. Two in vivo models were used to confirm that the effect of H. pylori on the Na-K-ATPase is applicable in the fully intact, more chronically infected organism. A significant decrease in membrane expression of the Na-K-ATPase α-subunit was seen both in infected gerbils and in antrum biopsies from pediatric patients with chronic H. pylori infection, indicating that the effect of H. pylori on the Na-K-ATPase is sustained over time in vivo. A more chronic yet sublethal decrease in the Na-K-ATPase could potentially impair ion homeostasis and nutrient uptake in gastric epithelial cells, compromise the integrity of the cell junctions, and cause gastric injury. The effect of H. pylori on the Na-K-ATPase is independent of pH, suggesting that bacteria can continuously induce injury in the human stomach.
With decreased membrane expression of the Na-K-ATPase over time, the expected physiological consequence should be a decrease in the inward sodium gradient, with increased accumulation of sodium inside the cells and subsequent cell swelling. This would have to be a graded and mild increase in sodium and volume to ensure continued cell viability, although perhaps with some compromise. Shortly after the discovery of H. pylori, ultrastructural studies of the infected epithelium from human biopsy samples demonstrated cellular edema distinct from noted vacuoles. This was seen in conjunction with the presence of the bacteria clustered in proximity to the junctional complexes between swollen cells (23). A decrease in levels of the Na-K-ATPase at the membrane would impair ion homeostasis and nutrient uptake, increase cell swelling, and compromise the integrity of the cell junctions, causing gastric injury. The data presented here suggest that, in both acute and chronic infection models, one mechanism of epithelial injury by H. pylori involves targeting and degradation of the Na-K-ATPase, a critical transport protein. In the setting of decreased Na-K-ATPase expression, ion and nutrient homeostasis are disrupted, interfering with optimal cellular function. This provides critical insight into the multifactorial process by which the bacteria are able to injure the host, which will be important for optimization of treatment of the infection and associated sequelae.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants K08 DK-100661 (E. A. Marcus), R03 DK-110579 (E. A. Marcus), a UCLA Children’s Discovery and Innovation Institute Seed Grant, and Today’s and Tomorrow’s Children Fund (E. A. Marcus), R01 HL-113350 (O. Vagin), and UCSD/UCLA NIDDK Diabetes Research Center P30 DK-063491 (J. P. Whitelegge).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
E.A.M., J.P.W., and O.V. conceived and designed research; E.A.M., E.T., J.L.J., Y.W., B.V.N., A.N.H., S.K., J.C., W.C., and O.V. performed experiments; E.A.M., E.T., J.L.J., Y.W., B.V.N., A.N.H., S.K., J.C., W.C., J.P.W., and O.V. analyzed data; E.A.M., E.T., Y.W., and O.V. interpreted results of experiments; E.A.M., E.T., and O.V. prepared figures; E.A.M. and O.V. drafted manuscript; E.A.M., E.T., and O.V. edited and revised manuscript; E.A.M., E.T., J.L.J., Y.W., B.V.N., A.N.H., S.K., J.C., W.C., J.P.W., and O.V. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. G. Sachs, who initiated and supported our work on H. pylori. His memory will inspire our research moving forward. We thank Dr. L. Dada for providing cDNA coding for rat Na-K-ATPase α1-subunit.
REFERENCES
- 1.Abdel-Latif AA, Smith JP, Hedrick N. Adenosinetriphosphatase and nucleotide metabolism in synaptosomes of rat brain. J Neurochem 17: 391–401, 1970. doi: 10.1111/j.1471-4159.1970.tb02226.x. [DOI] [PubMed] [Google Scholar]
- 2.Ackermann U, Geering K. Mutual dependence of Na-K-ATPase alpha- and beta-subunits for correct posttranslational processing and intracellular transport. FEBS Lett 269: 105–108, 1990. doi: 10.1016/0014-5793(90)81130-G. [DOI] [PubMed] [Google Scholar]
- 3.Baltrus DA, Amieva MR, Covacci A, Lowe TM, Merrell DS, Ottemann KM, Stein M, Salama NR, Guillemin K. The complete genome sequence of Helicobacter pylori strain G27. J Bacteriol 191: 447–448, 2009. doi: 10.1128/JB.01416-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beggah A, Mathews P, Beguin P, Geering K. Degradation and endoplasmic reticulum retention of unassembled alpha- and beta-subunits of Na,K-ATPase correlate with interaction of BiP. J Biol Chem 271: 20895–20902, 1996. doi: 10.1074/jbc.271.34.20895. [DOI] [PubMed] [Google Scholar]
- 6.Beggah AT, Geering K. Alpha and beta subunits of Na-K-ATPase interact with BiP and calnexin. Ann N Y Acad Sci 834: 537–539, 1997. doi: 10.1111/j.1749-6632.1997.tb52311.x. [DOI] [PubMed] [Google Scholar]
- 7.Béguin P, Hasler U, Staub O, Geering K. Endoplasmic reticulum quality control of oligomeric membrane proteins: topogenic determinants involved in the degradation of the unassembled Na-K-ATPase alpha subunit and in its stabilization by beta subunit assembly. Mol Biol Cell 11: 1657–1672, 2000. doi: 10.1091/mbc.11.5.1657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blanco G, Mercer RW. Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in function. Am J Physiol Renal Physiol 275: F633–F650, 1998. doi: 10.1152/ajprenal.1998.275.5.F633. [DOI] [PubMed] [Google Scholar]
- 9.Blaser MJ. Hypotheses on the pathogenesis and natural history of Helicobacter pylori-induced inflammation. Gastroenterology 102: 720–727, 1992. doi: 10.1016/0016-5085(92)90126-J. [DOI] [PubMed] [Google Scholar]
- 10.Blaustein MP. Endogenous ouabain: role in the pathogenesis of hypertension. Kidney Int 49: 1748–1753, 1996. doi: 10.1038/ki.1996.260. [DOI] [PubMed] [Google Scholar]
- 11.Boersema PJ, Raijmakers R, Lemeer S, Mohammed S, Heck AJ. Multiplex peptide stable isotope dimethyl labeling for quantitative proteomics. Nat Protoc 4: 484–494, 2009. doi: 10.1038/nprot.2009.21. [DOI] [PubMed] [Google Scholar]
- 12.Carmosino M, Procino G, Casavola V, Svelto M, Valenti G. The cultured human gastric cells HGT-1 express the principal transporters involved in acid secretion. Pflügers Arch 440: 871–880, 2000. doi: 10.1007/s004240000363. [DOI] [PubMed] [Google Scholar]
- 13.Chailler P, Ménard D. Establishment of human gastric epithelial (HGE) cell lines exhibiting barrier function, progenitor, and prezymogenic characteristics. J Cell Physiol 202: 263–274, 2005. doi: 10.1002/jcp.20124. [DOI] [PubMed] [Google Scholar]
- 14.Cherniavsky-Lev M, Golani O, Karlish SJ, Garty H. Ouabain-induced internalization and lysosomal degradation of the Na+/K+-ATPase. J Biol Chem 289: 1049–1059, 2014. doi: 10.1074/jbc.M113.517003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clausen MV, Hilbers F, Poulsen H. The structure and function of the Na-K-ATPase isoforms in health and disease. Front Physiol 8: 371, 2017. doi: 10.3389/fphys.2017.00371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Coppi MV, Guidotti G. Ubiquitination of Na-K-ATPase alpha1 and alpha2 subunits. FEBS Lett 405: 281–284, 1997. doi: 10.1016/S0014-5793(97)00182-8. [DOI] [PubMed] [Google Scholar]
- 17.Correa P, Haenszel W, Cuello C, Tannenbaum S, Archer M. A model for gastric cancer epidemiology. Lancet 306: 58–60, 1975. doi: 10.1016/S0140-6736(75)90498-5. [DOI] [PubMed] [Google Scholar]
- 18.Covacci A, Censini S, Bμgnoli M, Petracca R, Burroni D, Macchia G, Massone A, Papini E, Xiang Z, Figura N. Molecular characterization of the 128-kDa immunodominant antigen of Helicobacter pylori associated with cytotoxicity and duodenal ulcer. Proc Natl Acad Sci USA 90: 5791–5795, 1993. doi: 10.1073/pnas.90.12.5791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Crambert G, Hasler U, Beggah AT, Yu C, Modyanov NN, Horisberger JD, Lelièvre L, Geering K. Transport and pharmacological properties of nine different human Na, K-ATPase isozymes. J Biol Chem 275: 1976–1986, 2000. doi: 10.1074/jbc.275.3.1976. [DOI] [PubMed] [Google Scholar]
- 20.Dada LA, Welch LC, Zhou G, Ben-Saadon R, Ciechanover A, Sznajder JI. Phosphorylation and ubiquitination are necessary for Na,K-ATPase endocytosis during hypoxia. Cell Signal 19: 1893–1898, 2007. doi: 10.1016/j.cellsig.2007.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doné SC, Leibiger IB, Efendiev R, Katz AI, Leibiger B, Berggren PO, Pedemonte CH, Bertorello AM. Tyrosine 537 within the Na+,K+-ATPase alpha-subunit is essential for AP-2 binding and clathrin-dependent endocytosis. J Biol Chem 277: 17108–17111, 2002. doi: 10.1074/jbc.M201326200. [DOI] [PubMed] [Google Scholar]
- 22.Efendiev R, Bertorello AM, Pressley TA, Rousselot M, Féraille E, Pedemonte CH. Simultaneous phosphorylation of Ser11 and Ser18 in the alpha-subunit promotes the recruitment of Na(+),K(+)-ATPase molecules to the plasma membrane. Biochemistry 39: 9884–9892, 2000. doi: 10.1021/bi0007831. [DOI] [PubMed] [Google Scholar]
- 23.Fiocca R, Villani L, Turpini F, Turpini R, Solcia E. High incidence of Campylobacter-like organisms in endoscopic biopsies from patients with gastritis, with or without peptic ulcer. Digestion 38: 234–244, 1987. doi: 10.1159/000199597. [DOI] [PubMed] [Google Scholar]
- 24.Ford AC, Delaney BC, Forman D, Moayyedi P. Eradication therapy for peptic ulcer disease in Helicobacter pylori positive patients. Cochrane Database Syst Rev 2: CD003840, 2005. doi: 10.1002/14651858.CD003840.pub2. [DOI] [PubMed] [Google Scholar]
- 25.Forsayeth JR, Gu Y, Hall ZW. BiP forms stable complexes with unassembled subunits of the acetylcholine receptor in transfected COS cells and in C2 muscle cells. J Cell Biol 117: 841–847, 1992. doi: 10.1083/jcb.117.4.841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geering K. Functional roles of Na,K-ATPase subunits. Curr Opin Nephrol Hypertens 17: 526–532, 2008. doi: 10.1097/MNH.0b013e3283036cbf. [DOI] [PubMed] [Google Scholar]
- 27.Gisbert JP, Khorrami S, Carballo F, Calvet X, Gene E, Dominguez-Munoz JE. H. pylori eradication therapy vs. antisecretory non-eradication therapy (with or without long-term maintenance antisecretory therapy) for the prevention of recurrent bleeding from peptic ulcer. Cochrane Database Syst Rev 2: CD004062, 2003. doi: 10.1002/14651858.CD004062.pub2. [DOI] [PubMed] [Google Scholar]
- 28.Graham DY, Fischbach L. Helicobacter pylori treatment in the era of increasing antibiotic resistance. Gut 59: 1143–1153, 2010. doi: 10.1136/gut.2009.192757. [DOI] [PubMed] [Google Scholar]
- 29.Hebert DN, Molinari M. In and out of the ER: protein folding, quality control, degradation, and related human diseases. Physiol Rev 87: 1377–1408, 2007. doi: 10.1152/physrev.00050.2006. [DOI] [PubMed] [Google Scholar]
- 30.Hoy B, Geppert T, Boehm M, Reisen F, Plattner P, Gadermaier G, Sewald N, Ferreira F, Briza P, Schneider G, Backert S, Wessler S. Distinct roles of secreted HtrA proteases from gram-negative pathogens in cleaving the junctional protein and tumor suppressor E-cadherin. J Biol Chem 287: 10115–10120, 2012. doi: 10.1074/jbc.C111.333419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoy B, Löwer M, Weydig C, Carra G, Tegtmeyer N, Geppert T, Schröder P, Sewald N, Backert S, Schneider G, Wessler S. Helicobacter pylori HtrA is a new secreted virulence factor that cleaves E-cadherin to disrupt intercellular adhesion. EMBO Rep 11: 798–804, 2010. doi: 10.1038/embor.2010.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ishijima N, Suzuki M, Ashida H, Ichikawa Y, Kanegae Y, Saito I, Borén T, Haas R, Sasakawa C, Mimuro H. BabA-mediated adherence is a potentiator of the Helicobacter pylori type IV secretion system activity. J Biol Chem 286: 25256–25264, 2011. doi: 10.1074/jbc.M111.233601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jorgensen PL, Hakansson KO, Karlish SJ. Structure and mechanism of Na,K-ATPase: functional sites and their interactions. Annu Rev Physiol 65: 817–849, 2003. doi: 10.1146/annurev.physiol.65.092101.142558. [DOI] [PubMed] [Google Scholar]
- 34.Kao CY, Sheu BS, Wu JJ. Helicobacter pylori infection: an overview of bacterial virulence factors and pathogenesis. Biomed J 39: 14–23, 2016. doi: 10.1016/j.bj.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kaplan JH. Biochemistry of Na,K-ATPase. Annu Rev Biochem 71: 511–535, 2002. doi: 10.1146/annurev.biochem.71.102201.141218. [DOI] [PubMed] [Google Scholar]
- 36.Lecuona E, Sun H, Vohwinkel C, Ciechanover A, Sznajder JI. Ubiquitination participates in the lysosomal degradation of Na,K-ATPase in steady-state conditions. Am J Respir Cell Mol Biol 41: 671–679, 2009. doi: 10.1165/rcmb.2008-0365OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lehours P, Yilmaz O. Epidemiology of Helicobacter pylori infection. Helicobacter 12, Suppl 1: 1–3, 2007. doi: 10.1111/j.1523-5378.2007.00541.x. [DOI] [PubMed] [Google Scholar]
- 38.Leodolter A, Kulig M, Brasch H, Meyer-Sabellek W, Willich SN, Malfertheiner P. A meta-analysis comparing eradication, healing and relapse rates in patients with Helicobacter pylori-associated gastric or duodenal ulcer. Aliment Pharmacol Ther 15: 1949–1958, 2001. doi: 10.1046/j.1365-2036.2001.01109.x. [DOI] [PubMed] [Google Scholar]
- 39.Li Z, Langhans SA. Transcriptional regulators of Na,K-ATPase subunits. Front Cell Dev Biol 3: 66, 2015. doi: 10.3389/fcell.2015.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Marcus EA, Vagin O, Tokhtaeva E, Sachs G, Scott DR. Helicobacter pylori impedes acid-induced tightening of gastric epithelial junctions. Am J Physiol Gastrointest Liver Physiol 305: G731–G739, 2013. doi: 10.1152/ajpgi.00209.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Marshall BJ, Warren JR. Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 323: 1311–1315, 1984. doi: 10.1016/S0140-6736(84)91816-6. [DOI] [PubMed] [Google Scholar]
- 42.Matchkov VV, Krivoi II. Specialized functional diversity and interactions of the Na,K-ATPase. Front Physiol 7: 179, 2016. doi: 10.3389/fphys.2016.00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Müller-Ehmsen J, Juvvadi P, Thompson CB, Tumyan L, Croyle M, Lingrel JB, Schwinger RH, McDonoμgh AA, Farley RA. Ouabain and substrate affinities of human Na+-K+-ATPase α1β1, α2β1, and α3β1 when expressed separately in yeast cells. Am J Physiol Cell Physiol 281: C1355–C1364, 2001. doi: 10.1152/ajpcell.2001.281.4.C1355. [DOI] [PubMed] [Google Scholar]
- 44.Murata M, Miyashita S, Yokoo C, Tamai M, Hanada K, Hatayama K, Towatari T, Nikawa T, Katunuma N. Novel epoxysuccinyl peptides. Selective inhibitors of cathepsin B, in vitro. FEBS Lett 280: 307–310, 1991. doi: 10.1016/0014-5793(91)80318-W. [DOI] [PubMed] [Google Scholar]
- 45.Murer H, Ammann E, Biber J, Hopfer U. The surface membrane of the small intestinal epithelial cell. I. Localization of adenyl cyclase. Biochim Biophys Acta 433: 509–519, 1976. doi: 10.1016/0005-2736(76)90277-7. [DOI] [PubMed] [Google Scholar]
- 46.Nomura A, Stemmermann GN, Chyou PH, Kato I, Perez-Perez GI, Blaser MJ. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N Engl J Med 325: 1132–1136, 1991. doi: 10.1056/NEJM199110173251604. [DOI] [PubMed] [Google Scholar]
- 47.Noto JM, Romero-Gallo J, Piazuelo MB, Peek RM. The Mongolian gerbil: a robust model of Helicobacter pylori-induced gastric inflammation and cancer. Methods Mol Biol 1422: 263–280, 2016. doi: 10.1007/978-1-4939-3603-8_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ohno T, Sμgimoto M, Nagashima A, Ogiwara H, Vilaichone RK, Mahachai V, Graham DY, Yamaoka Y. Relationship between Helicobacter pylori hopQ genotype and clinical outcome in Asian and Western populations. J Gastroenterol Hepatol 24: 462–468, 2009. doi: 10.1111/j.1440-1746.2008.05762.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Padilla-Benavides T, Roldán ML, Larre I, Flores-Benitez D, Villegas-Sepúlveda N, Contreras RG, Cereijido M, Shoshani L. The polarized distribution of Na+,K+-ATPase: role of the interaction between beta subunits. Mol Biol Cell 21: 2217–2225, 2010. doi: 10.1091/mbc.e10-01-0081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parsonnet J. Gastric adenocarcinoma and Helicobacter pylori infection. West J Med 161: 60, 1994. [PMC free article] [PubMed] [Google Scholar]
- 51.Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentreich N, Sibley RK. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med 325: 1127–1131, 1991. doi: 10.1056/NEJM199110173251603. [DOI] [PubMed] [Google Scholar]
- 52.Rajasekaran SA, Palmer LG, Moon SY, Peralta Soler A, Apodaca GL, Harper JF, Zheng Y, Rajasekaran AK. Na,K-ATPase activity is required for formation of tight junctions, desmosomes, and induction of polarity in epithelial cells. Mol Biol Cell 12: 3717–3732, 2001. doi: 10.1091/mbc.12.12.3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rappsilber J, Mann M, Ishihama Y. Protocol for micro-purification, enrichment, pre-fractionation and storage of peptides for proteomics using StageTips. Nat Protoc 2: 1896–1906, 2007. doi: 10.1038/nprot.2007.261. [DOI] [PubMed] [Google Scholar]
- 54.Reinhard L, Tidow H, Clausen MJ, Nissen P. Na+,K+-ATPase as a docking station: protein-protein complexes of the Na+,K+-ATPase. Cell Mol Life Sci 70: 205–222, 2013. doi: 10.1007/s00018-012-1039-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ricci V, Sommi P, Cova E, Fiocca R, Romano M, Ivey KJ, Solcia E, Ventura U. Na+,K+-ATPase of gastric cells. A target of Helicobacter pylori cytotoxic activity. FEBS Lett 334: 158–160, 1993. doi: 10.1016/0014-5793(93)81703-3. [DOI] [PubMed] [Google Scholar]
- 56.Rouillard AD, Gundersen GW, Fernandez NF, Wang Z, Monteiro CD, McDermott MG, Ma'ayan A. The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database (Oxford) 2016: baw100, 2016. doi: 10.1093/database/baw100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc 3: 1101–1108, 2008. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 58.Shanjana A, Archana A. Cytotoxic isolates of Helicobacter pylori from peptic ulcer diseases decrease K+-dependent ATPase activity in HeLa cells. BMC Gastroenterol 3: 31, 2003. doi: 10.1186/1471-230X-3-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shoshani L, Contreras RG, Roldán ML, Moreno J, Lázaro A, Balda MS, Matter K, Cereijido M. The polarized expression of Na+,K+-ATPase in epithelia depends on the association between beta-subunits located in neighboring cells. Mol Biol Cell 16: 1071–1081, 2005. doi: 10.1091/mbc.e04-03-0267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Skou JC. The influence of some cations on an adenosine triphosphatase from peripheral nerves. Biochim Biophys Acta 23: 394–401, 1957. doi: 10.1016/0006-3002(57)90343-8. [DOI] [PubMed] [Google Scholar]
- 61.Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med 347: 1175–1186, 2002. doi: 10.1056/NEJMra020542. [DOI] [PubMed] [Google Scholar]
- 62.Sweadner KJ. Isozymes of the Na+-K+-ATPase. Biochim Biophys Acta 988: 185–220, 1989. doi: 10.1016/0304-4157(89)90019-1. [DOI] [PubMed] [Google Scholar]
- 63.Thévenod F, Friedmann JM. Cadmium-mediated oxidative stress in kidney proximal tubule cells induces degradation of Na+-K+-ATPase throμgh proteasomal and endo-/lysosomal proteolytic pathways. FASEB J 13: 1751–1761, 1999. doi: 10.1096/fasebj.13.13.1751. [DOI] [PubMed] [Google Scholar]
- 64.Tokhtaeva E, Munson K, Sachs G, Vagin O. N-glycan-dependent quality control of the Na,K-ATPase beta(2) subunit. Biochemistry 49: 3116–3128, 2010. doi: 10.1021/bi100115a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tokhtaeva E, Sachs G, Souda P, Bassilian S, Whitelegge JP, Shoshani L, Vagin O. Epithelial junctions depend on intercellular trans-interactions between the Na,K-ATPase β1 subunits. J Biol Chem 286: 25801–25812, 2011. doi: 10.1074/jbc.M111.252247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tokhtaeva E, Sachs G, Sun H, Dada LA, Sznajder JI, Vagin O. Identification of the amino acid region involved in the intercellular interaction between the β1 subunits of Na+-K+-ATPase. J Cell Sci 125: 1605–1616, 2012. doi: 10.1242/jcs.100149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tokhtaeva E, Sachs G, Vagin O. Assembly with the Na,K-ATPase alpha(1) subunit is required for export of beta(1) and beta(2) subunits from the endoplasmic reticulum. Biochemistry 48: 11421–11431, 2009. doi: 10.1021/bi901438z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tokhtaeva E, Sachs G, Vagin O. Diverse pathways for maturation of the Na,K-ATPase β1 and β2 subunits in the endoplasmic reticulum of Madin-Darby canine kidney cells. J Biol Chem 285: 39289–39302, 2010. doi: 10.1074/jbc.M110.172858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tokhtaeva E, Sun H, Deiss-Yehiely N, Wen Y, Soni PN, Gabrielli NM, Marcus EA, Ridge KM, Sachs G, Vazquez-Levin M, Sznajder JI, Vagin O, Dada LA. The O-glycosylated ectodomain of FXYD5 impairs adhesion by disrupting cell-cell trans-dimerization of Na,K-ATPase β1 subunits. J Cell Sci 129: 2394–2406, 2016. doi: 10.1242/jcs.186148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tomoda H, Omura S. Lactacystin, a proteasome inhibitor: discovery and its application in cell biology. Yakμgaku Zasshi 120: 935–949, 2000. doi: 10.1248/yakushi1947.120.10_935. [DOI] [PubMed] [Google Scholar]
- 71.Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin 65: 87–108, 2015. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 72.Tsai YC, Maditz R, Kuo CL, Fishman PS, Shoemaker CB, Oyler GA, Weissman AM. Targeting botulinum neurotoxin persistence by the ubiquitin-proteasome system. Proc Natl Acad Sci USA 107: 16554–16559, 2010. doi: 10.1073/pnas.1008302107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tytgat GN. Etiopathogenetic principles and peptic ulcer disease classification. Dig Dis 29: 454–458, 2011. doi: 10.1159/000331520. [DOI] [PubMed] [Google Scholar]
- 74.Untergasser A, Cutcutache I, Koressaar T, Ye J, Faircloth BC, Remm M, Rozen SG. Primer3–new capabilities and interfaces. Nucleic Acids Res 40: e115, 2012. doi: 10.1093/nar/gks596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vadász I, Dada LA, Briva A, Trejo HE, Welch LC, Chen J, Tóth PT, Lecuona E, Witters LA, Schumacker PT, Chandel NS, Seeger W, Sznajder JI. AMP-activated protein kinase regulates CO2-induced alveolar epithelial dysfunction in rats and human cells by promoting Na,K-ATPase endocytosis. J Clin Invest 118: 752–762, 2008. doi: 10.1172/JCI29723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vadász I, Weiss CH, Sznajder JI. Ubiquitination and proteolysis in acute lung injury. Chest 141: 763–771, 2012. doi: 10.1378/chest.11-1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vagin O, Tokhtaeva E, Sachs G. The role of the beta1 subunit of the Na,K-ATPase and its glycosylation in cell-cell adhesion. J Biol Chem 281: 39573–39587, 2006. doi: 10.1074/jbc.M606507200. [DOI] [PubMed] [Google Scholar]
- 78.Vagin O, Tokhtaeva E, Yakubov I, Shevchenko E, Sachs G. Inverse correlation between the extent of N-glycan branching and intercellular adhesion in epithelia. Contribution of the Na,K-ATPase beta1 subunit. J Biol Chem 283: 2192–2202, 2008. doi: 10.1074/jbc.M704713200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Vagin O, Turdikulova S, Sachs G. Recombinant addition of N-glycosylation sites to the basolateral Na,K-ATPase beta1 subunit results in its clustering in caveolae and apical sorting in HGT-1 cells. J Biol Chem 280: 43159–43167, 2005. doi: 10.1074/jbc.M508262200. [DOI] [PubMed] [Google Scholar]
- 80.Wen Y, Marcus EA, Matrubutham U, Gleeson MA, Scott DR, Sachs G. Acid-adaptive genes of Helicobacter pylori. Infect Immun 71: 5921–5939, 2003. doi: 10.1128/IAI.71.10.5921-5939.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Werstuck GH, Lentz SR, Dayal S, Hossain GS, Sood SK, Shi YY, Zhou J, Maeda N, Krisans SK, Malinow MR, Austin RC. Homocysteine-induced endoplasmic reticulum stress causes dysregulation of the cholesterol and triglyceride biosynthetic pathways. J Clin Invest 107: 1263–1273, 2001. doi: 10.1172/JCI11596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81a.World Health Organization Infection with Helicobacter pylori. In: IARC Monographs on the Evaluation of the Carcinogenic Risk to Humans. Schistosomes, Liver Flukes, and Helicobacter Pylori. 1994, vol. 61, p. 177–240. [Google Scholar]
- 82.Xie Z, Askari A. Na+-K+-ATPase as a signal transducer. Eur J Biochem 269: 2434–2439, 2002. doi: 10.1046/j.1432-1033.2002.02910.x. [DOI] [PubMed] [Google Scholar]
- 83.Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. Bafilomycin A1, a specific inhibitor of vacuolar-type H+-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J Biol Chem 266: 17707–17712, 1991. [PubMed] [Google Scholar]