

Abstract
Mitochondrial dysfunction and oxidative stress characterize major factors involved in the activation of complex processes corresponding to apoptosis-mediated neuronal senescence of dopaminergic neurons (DA) in Parkinson's disease (PD). Here, we evaluated the molecular mechanisms participating in the treatment of a 1-methyl-4-phenyl-1,2,3,6-tetrahydopyridine- (MPTP-) intoxicated PD mouse model in response to chlorogenic acid (CGA). The results indicate that CGA treatment significantly improved the motor coordination of the MPTP-intoxicated mice. CGA also alleviated the fall in activity of mitochondrial complexes I, IV, and V in accordance with ameliorating the level of superoxide dismutase and mitochondrial glutathione in the midbrain of MPTP-induced mice. CGA inhibited the activation of proapoptotic proteins including Bax and caspase-3, while elevating the expression of antiapoptotic protein like Bcl-2 consequently preventing the MPTP-mediated apoptotic cascade. The study also revealed the improved phosphorylation state of Akt, ERK1/2, and GSK3β which was downregulated as an effect of MPTP toxicity. Our findings signify that CGA may possess pharmacological properties and contribute to neuroprotection against MPTP induced toxicity in a PD mouse model associated with phosphorylation of GSK3β via activating Akt/ERK signalling in the mitochondrial intrinsic apoptotic pathway. Thus, CGA treatment may arise as a potential therapeutic candidate for mitochondrial-mediated apoptotic senescence of DA neurons in PD.
1. Introduction
Research has made it quite prominent that aging plays a prime factor in the onset and progression of the sporadic form of the neurodegenerative disorder Parkinson's disease (PD) characterized by the progressive depletion in the dopaminergic (DA) neurons along with the formation and accumulation of Lewy bodies primarily in the substantia nigra pars compacta (SNpc) region of the midbrain and striatum [1, 2]. While the exact mechanism of PD is yet to be elucidated, promising evidences indicate that oxidative stress and mitochondrial dysfunction play a crucial part in its pathogenesis [3–5]. Lewy body formation, comprising aggregates of abnormal or misfolded α-synuclein protein, is a hallmark feature of both familial and sporadic forms of PD. α-Synuclein undergoes aggregation predominantly in the cytoplasm of the neurons present in SN and leads to multifactorial etiopathogenesis of PD [6, 7]. Numerous pathogenic signals including increased oxidative stress, mitochondrial impairment, ubiquitin-proteasomal dysfunction, and activation of apoptotic cascade along with progressive loss of DA neurons cause the pathogenesis of PD [8, 9]. The clinical evidences from PD patients show the formation of free radicals consisting of reactive oxygen species (ROS), reactive nitrogen species (RNS), mitochondrial dysfunction, adenosine triphosphate (ATP) depletion, and the initiation of caspase-mediated apoptotic cascade in the SN [10].
At the present time, the chief treatment options for PD are dopamine agonists or L-DOPA which simply provide symptomatic relief and show less efficiency with prolonged use resulting in disabling fluctuations and dyskinesias [11]. Therefore, finding treatments with neuroprotective potential against PD in correspondence with effectiveness in relieving symptoms is the main therapeutic target for slowing disease progression. Emerging evidences prove the vital role of apoptosis in PD pathogenesis. Hence, the apoptotic pathway can be prohibited by providing neuroprotection through the use of phytochemicals [12, 13].
Various cell culture models and animal models of PD have been developed using environmental neurotoxins including rotenone, 6-hydroxydopamine, and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) [14]. The charged metabolite of MPTP is 1-methyl-4-phenylpyridinium (MPP+) acting as the main toxic agent against the DA neurons in the brain of MPTP-intoxicated in vivo models [15, 16]. MPP+ readily enters the nigrostriatal neurons via dopamine transporters and affects the SN neuronal loss contributing to striatal dopamine depletion further causing a parkinsonian syndrome. It also disturbs the mitochondrial membrane potential causing mitochondrial stress. It is reported to be an inhibitor of complex I of the electron transport chain (ETC) and contributes to considerable oxidative stress along with ATP depletion, consequently leading to neuronal loss [17]. MPP+ activates the proapoptotic proteins, making the mitochondrial membrane permeable for cytochrome c to be released into the cytosol. Associated signalling inactivates protein kinases (MAPKs) which might have a neuroprotective role.
Akt and ERK signalling pathways play crucial parts in neuroprotection via cell differentiation, proliferation, survival, and apoptosis [18, 19]. Numerous recent researches have highlighted the neuroprotective role of plant-based compounds and polyphenols in PD against neurotoxins and neuroinflammation, by promoting cell survival through their antioxidative, anti-inflammatory, and immunomodulatory effects [20–22]. Evidences prove the pivotal role of polyphenols in replenishing the neurons via increased activity of ETC, regulating the effects on redox potential, and restraining the apoptosis as a result of increased mitochondrial biogenesis [23–25]. CGA, a type of phenolic acid, is reported to demonstrate antiapoptotic, anti-inflammatory, antioxidative, neurotrophic, anticancerous, and neuroprotective properties. CGA is formed as an ester of the cinnamic acids including caffeic acid and quinic acid forming the second major component of coffee after caffeine [26–28]. In our previous study, we reported the antioxidative and anti-inflammatory property of CGA against the neurotoxic effect of MPTP in mice [26]. The present study was undertaken to assess the neuroprotective effect of CGA in an MPTP-induced PD mouse model via modulation of Akt, ERK1/2, and GSK3β signalling pathways. Under this context, the effect of CGA was evaluated against MPP+-mediated DA neuronal injury in MPTP-intoxicated mice showing its neuroprotective role via the activation of Akt and ERK1/2 signalling pathways by inhibiting the mitochondrial dysfunction. Our findings may be helpful in designing a CGA-based treatment strategy against PD.
2. Chemical and Reagents
2.1. Reagents and Antibodies
Mannitol, sodium dihydrogen phosphate, bovine serum albumin (BSA), oxidized cytochrome c, digitonin, disodium hydrogen phosphate, potassium chloride, ammonium chloride, and reduced nicotinamide adenine dinucleotide phosphate (NADPH) were purchased from Sisco Research Laboratories (SRL, Mumbai, India). MPTP, DABCO, 5,5-dithiobis 2-nitrobenzoic acid (DTNB), normal goat serum (NGS), and chlorogenic acid were obtained from Sigma-Aldrich (St. Louis, MO, United States). Sucrose and sodium carbonate were purchased from Merck (Darmstadt, Germany), and EGTA and sodium dodecyl sulphate (SDS) were obtained from HiMedia (Mumbai, India). Glycylglycine buffer, sodium fluoride, paraformaldehyde, Tris buffer, and trichloroacetic acid were bought from LobaChemie, India. Primary antibodies for Akt, p-Akt, caspase-3, ERK, p-ERK, and Bcl-2 were acquired from Abcam Life Science, Biogenuix Medsystems Pvt. Ltd. (New Delhi, India), and primary antibodies for GSK3β, p-GSK3β, TH, β-actin, and Bax were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, United States).
2.2. Experimental Animals
The experiment was conducted using male Swiss albino mice (25 ± 5 g), obtained from the Institute of Medical Sciences' animal facility, Banaras Hindu University, Varanasi (India). The mice were kept in clean polypropylene cages with constant light-dark cycles of 12 h prior to the start of experiment. Mice were provided with ad libitum water and standard diet pellet until the dosing was completed. The experimental protocol was established according to the Animal Ethics Committee's guidelines of Banaras Hindu University, Varanasi, India.
2.2.1. Animal Dosing
Experimental animals were randomized into four groups: control, MPTP, MPTP+CGA, and CGA (n = 10/group). The dosing was administered in accordance with our previous study with some modifications [26]. The mice in the control group received normal saline orally, once consequently for 24 days. In MPTP+CGA and CGA groups, the oral administration of 50 mg/kg of CGA was done, once daily for 24 days. Except for the control and drug-only groups, all the mice received MPTP (30 mg/kg) intraperitoneally, dissolved in normal saline for five consecutive days (from the 20th to 24th day of CGA dosing). After completion of dosing, a behavioral test was performed from the 25th to 28th day, and thereafter, mice were sacrificed to isolate the brains for the mitochondrial dysfunction assay, Western blotting, and immunohistochemical analysis.
2.2.2. Behavioral Test
To assess the effect of MPTP intoxication on motor function impairment in the parkinsonian mouse model, four behavioral tests were conducted, including the rotarod test, pole test, catalepsy test, and traction test.
2.2.3. Rotarod Test
The training for 3 consecutive days was given to the animals from all the groups at a speed of 5 rpm for the rotarod test. Thereafter, the time on the rotarod for each animal was noted down for maximum 5 min. The test was done four times, and finally, the average time for which the mice stayed on the rotarod was noted down [29]. After the completion of dosing, the rotarod test was again conducted and the time spent on the instrument was noted down for each mouse.
2.2.4. Pole Test
Bradykinesia in PD mouse models is usually evaluated by this test [30]. The test was done after the last MPTP injection was given. Mice were supported on the top of a pole (diameter 10 mm, height 52 cm, with a rough surface). The T-turn (time to turn) was recorded accordingly to the time taken by mice to step down the length of the pole.
2.2.5. Traction Test
Equilibrium and the muscle strength are measured by performing the traction test [30], and it was also conducted on the same day as the pole test. The forepaws of a mouse were located on a suspended horizontal bar, while its hind limb placements were scored from 1 to 3, with the lowest score indicating the severe motor and balance impairment. The following criteria were used to evaluate the score: the score was 3, if both hind limbs seized the bar, while it was 2 or 1, if one or no hind limb seized the bar, respectively.
2.2.6. Catalepsy Test
Catalepsy is a state of the behavioral test that characterizes the muscle stiffness in which the mice fail to change its posture which is imposed to them and is estimated by making the animals to stand on a wooden platform where their hind limbs were placed on a wood and forelimbs on the ground [31]. The intensity of catalepsy was measured when a mouse moved its hind limbs from a wooden platform (3 cm). The mice were adapted for 3 min, and if the latency exceeds more than 180 sec, the test was ended.
2.3. Mitochondrial Parameter Analysis
2.3.1. Isolation of Mitochondria
Differential centrifugation was done to isolate the mitochondrial pellet from the mouse brain [32]. The midbrain regions of the mice were homogenized in homogenizing buffer, prepared by mixing 5 mM HEPES, 225 mM mannitol, 1 mM ethylene glycol tetra acetic acid (EGTA), 75 mM sucrose, and 1 mg/ml BSA (pH 7.4). Centrifugation of the homogenates was done at 2,000 g for 30 min at 4°C. The pellet was disposed, and the supernatant obtained was again set to centrifugation for 10 min at 12,000 g. The resultant pellet constituting the mixture of mitochondria and synaptosomes was dissolved in the homogenizing buffer containing digitonin (0.02%). Crude mitochondrial fraction was finally obtained by the centrifugation of the suspended mixture for 10 min at 12,000 g. Homogenizing buffer without BSA and EGTA was used to wash the crude mitochondrial fraction twice, and finally, the resuspension was done in phosphate buffer (50 mM, pH 7.4). The Bradford assay was performed to estimate proteins in all the samples. The assays were done within 24 h of the mitochondrial isolation using 20 μg proteins.
2.3.2. Complex I-III Assay
The assay of complexes I-III was done by incorporating the method of Sood et al. in our study [33]. In this assay, the catalytic oxidation of NADH to NAD+ is done by subsequently reducing cytochrome c. The reaction mixture was prepared by the addition of glycylglycine buffer (0.2 M, pH 8.5), NADH (6 mM in 2 mM glycylglycine buffer), and cytochrome c (10.5 mM). 20 μg mitochondrial protein was added to the reaction mixture, and the absorbance change was recorded at 550 nm for 2 min. At 340 nm, the extinction coefficient for NADH is 6.22/mM/cm. The activity of the enzyme was expressed in terms of nmol NADH oxidized/min/mg protein.
2.3.3. Complex IV Assay
Complex IV activity was determined by measuring the oxidation of reduced cytochrome c by complex IV at 550 nm [33]. The reaction mixture consisted of mitochondrial protein (20 μg) along with 10 mM phosphate buffer (pH 7.4), reduced cytochrome c, and potassium ferricyanide. Cytochrome c (oxidized) solution (10 mg/ml) was used to prepare cytochrome c (reduced), by adding few crystals of sodium borohydride in it. Absorbance change was monitored for about 3 min. The activity of complex IV was calculated in nmol cytochrome c oxidized/min/mg of protein.
2.3.4. Complex V Assay
Griffiths and Houghton's method was used to perform the mitochondrial ATPase test [34]. In this assay, the amount of inorganic phosphorus liberated is measured that is obtained by the hydrolysis of ATP to ADP. The incubation of the reaction mixture (1.0 ml) containing the mitochondrial sample and ATPase buffer (2 mM MgCl2, 5 mM ATP, and 50 mM Tris HCl, pH 8.5) was done for 5 min at 30°C. After the addition of 10% TCA, the reaction mixture was spun at 3,000 g for 10 min. Phosphorus was assayed, and the results were expressed as nmol inorganic phosphate (Pi) liberated/min/mg protein.
2.3.5. Mitochondrial GSH
Mitochondrial GSH (reduced) content was estimated by using the mitochondrial protein sample (20 μg) suspended in phosphate buffer. The addition of 25% trichloroacetic acid (TCA) was done to the samples, followed by a brief centrifugation at 1,500 g. The supernatant was collected, and 5,5-dithiobis 2-nitrobenzoic acid (DTNB) was added that reacts with thiol groups to form 2-nitro-5mercapto benzoic acid. Absorbance was recorded at 412 nm. Further, commercially available GSH was used to prepare the standard curve, and the amount was expressed as mmol of GSH/g mitochondrial protein [35].
2.3.6. Manganese SOD
The activity of manganese superoxide dismutase (Mn-SOD) was estimated by adding hydrogen peroxide that selectively inhibits Cu/Zn-SOD, in order to distinguish the enzyme's activity from that of Cu/Zn-SOD. The reaction mixture consists of sodium carbonate (50 mM), EDTA (0.1 mM), Triton-X (0.6%), and NBT (90 mM) and hydroxylamine hydrochloride [35]. Hydroxylamine hydrochloride undergoes photooxidation to produce superoxide that further reduces nitroblue tetrazolium (NBT) in the reaction medium, and this reaction was inhibited by SOD. The reading was taken at 560 nm for 3 min, and the SOD activity was expressed as units. The one enzymatic unit of SOD was defined as the amount of enzyme required for 50% inhibition.
2.4. Western Blot (WB) Analysis
The SN of the mouse brain was homogenized using the lysis buffer (RIPA) and agitated for 2 h at 4°C. The homogenate was centrifuged (12,000 rpm, 30 min) to collect the supernatant, and the Bradford assay was performed to quantify the protein concentration. The total protein extract (50 μg) was loaded onto the polyacrylamide gels. After electrophoresis, proteins were transblotted to PVDF membranes and sequentially incubated overnight at 4°C with Bax (1 : 1000), Bcl-2 (1 : 800), caspase-3 (1 : 1000), p-Akt (1 : 1000), Akt (1 : 1000), p-ERK1/2 (1 : 1000), ERK1/2 (1 : 800), p-GSK3β (1 : 1000), GSK3β (1 : 1000), and β-actin (1 : 1000) and then with a horseradish peroxidase- (HRP-) conjugated secondary antibody for 2 h at room temperature. TBST washing (twice) separated each step. Blots were visualized using the DAB system (buffer+substrate+chromogen), and for each band, the relative density was calculated with respect to that of β-actin. Quantity One software (Windows, Bio-Rad) was used to indicate the expression of relative density.
2.5. Immunohistochemistry (IHC)
Mice from each group were sacrificed using pentobarbital perfused intracardially with chilled 0.9% saline solution and further using 4% paraformaldehyde solution (prepared in 0.1 M phosphate-buffered saline, pH 7.4). Again, paraformaldehyde (10%) was used to postfix the brains overnight, and further, 30% sucrose solution was used to immerse the brains. Using the standard protocol, immunohistochemical staining was performed for TH, p-Akt, p-Erk1/2, and p-GSK3β [36]. Coronal brain sections were cut to 20 μm thickness using a cryotome (Leica, Wetzlar, Germany). Tissue sections were washed with 0.01 M PBS (pH 7.4) 2 × 10 min, followed by 1 h blocking with 10% NGS in PBST and then 1% BSA-PBST. The sections were rinsed with PBS and incubated with primary antibodies for TH (1 : 1000), p-Akt1 (1 : 500), p-ERK1/2 (1 : 500), and p-GSK3β (1 : 500) at 4°C (16 h). The brain sections were then treated with FITC-conjugated (for anti-mice primary) and TRITC-conjugated (for anti-rabbit primary) secondary antibodies (diluted in 1% BSA-PBS) at room temperature (2 h). Tissue sections were washed thrice at each step with 1% BSA-PBS and PBS, respectively, and mounted with polyvinyl alcohol with DABCO. Images were captured with a fluorescent microscope (Nikon, Thermo Fisher Scientific). All images were then processed by ImageJ software (NIH, United States). Results expressed as a % area were reported.
2.6. Statistical Analysis
The analysis of data was done by one-way analysis of variance (ANOVA) using the Student–Newman–Keuls test and by Student's two-tailed t-test using GraphPad Prism software. The results are expressed as the mean ± SEM. p values < 0.05 were considered statistically significant.
3. Results
3.1. Behavioral Parameter Analysis
The results from our study suggested that the MPTP-intoxicated animal falls early from the rotarod in comparison to the control (p < 0.001, Figure 1(a)). On the other hand, when CGA was administered to the MPTP-treated mice, the time on the rotarod was significantly increased (p < 0.001). The pole test clued that MPTP-treated mice showed prolonged locomotor activity time (p < 0.01Figure 1(b)). The time duration was shortened significantly (p < 0.01) in pretreated mice with CGA, suggesting that bradykinesia induced by MPTP was alleviated by CGA treatment. Results of the traction test (Figure 1(c)) showed that PD mice have decreased strength and latency time as their hind limb grip score was lower (p < 0.01) than that of the control group. However, pretreatment with CGA increased the traction score (p < 0.05). This indicates that prophylactic treatment with CGA could mitigate the initial lesions induced by MPTP. Catalepsy was observed among the MPTP group of mice after MPTP injection. Latency in the catalepsy test among the MPTP group showed increased tendency with time (p < 0.001, Figure 1(d)), and the observed data was of considerable difference from that of the control group, while the mice treated with CGA showed little latency time (p < 0.001) than the MPTP-intoxicated group. Any significant changes did not appear in the neurobehavioral activity between the control and CGA-alone-treated animals. Thus, results have suggested that CGA improved the neurobehavioral lesion of MPTP-injected PD mice.
Figure 1.
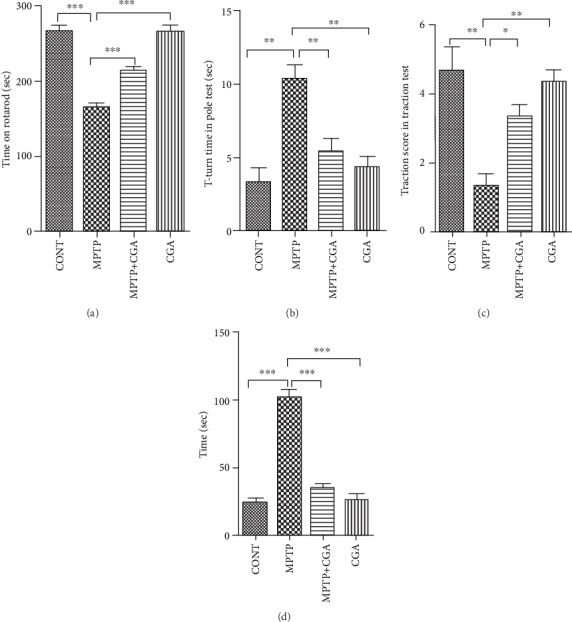
Mouse behavioral analysis. CGA mediated alteration in the neurobehavior of MPTP-injected mice. (a) Time on the rotarod test. (b) T-turn time in the pole test. (c) Traction score in the traction test. (d) Cataleptic test. Values are represented in the form of mean ± SEM (n = 10). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. Results were studied using the one-way ANOVA and further using the Newman-Keuls test.
3.2. CGA Modulates MPTP-Induced Mitochondrial ETS Impairment in Mice
Oxidative phosphorylation in mitochondria produces ATP as energy, and this process requires the organized action of five enzyme complexes located in the inner membrane. Catastrophic consequences can be observed by any defect in these energy-generating complexes, not only because of the ATP loss but also due to the downstream functional loss. Therefore, the consequences of MPTP intoxication on the energy metabolism in the mouse brain were studied. MPTP being a known complex I inhibitor of the electron transport chain (ETC) was analysed to study its effect on the activities of various complexes in the respiratory chain. As Figure 2 depicted, the effect of CGA treatment on MPTP induced alterations in the complex I-III (Figure 2(a)), IV (Figure 2(b)), and V activities (Figure 2(c)). There was significant attenuation in the activities of complexes I-III (p < 0.01), complex IV (p < 0.01), and complex V (p < 0.001) in MPTP-intoxicated PD mice as compared to control mice. CGA administration before MPTP intoxication to the animals helped improve the activities of complexes I-III (p < 0.01), complex IV (p < 0.01), and complex V (p < 0.001) in comparison with MPTP-intoxicated mice. No potential change was observed when the control group was treated with CGA.
Figure 2.
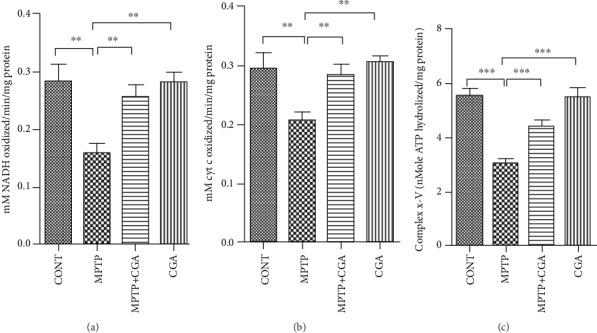
CGA mediated alterations on mitochondrial complexes I-III (a), IV (b), and V (c) of the electron transport chain in the midbrain of mice. Values are represented in the form of mean ± SEM (n = 5). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. Results were studied using the one-way ANOVA and further using the Newman-Keuls test.
3.3. Mitochondrial Glutathione and Superoxide Dismutase Level Analysis
Further, the increase in oxidative stress resulting in the reduced function of ETC leads to the utilization of mitochondrial GSH (mtGSH, reduced) and reduction in activity of Mn-SOD (Figures 3(a) and 3(b)). In the MPTP-treated group, a significant reduction (p < 0.001) in the levels of mtGSH was observed when compared to the control group. Consequently, a decline in the Mn-SOD activity (p < 0.001) was also analysed with MPTP intoxication (Figure 2(b)). On the other hand, CGA has reduced the ROS generation and ameliorated the function of ETS which has ultimately increased the level of mtGSH (p < 0.001) and Mn-SOD (p < 0.001) significantly.
Figure 3.
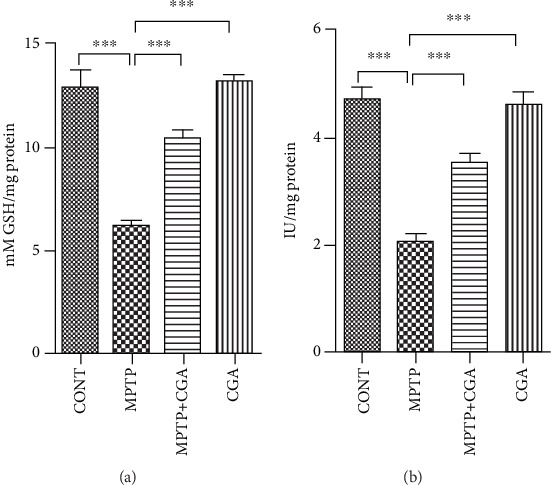
Alterations in mitochondrial antioxidant defence in respective treatment groups. (a) Mitochondrial reduced glutathione level. (b) Mitochondrial superoxide dismutase level. Values are represented in the form of mean ± SEM (n = 5). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. Results were studied using the one-way ANOVA and further using the Newman-Keuls test.
3.4. CGA Inhibited the MPTP-Induced Activation of Mitochondrial Apoptosis Signalling in SN of Mice
Various apoptosis-linked molecules were investigated to study the effect of CGA on MPTP-induced cell apoptosis rate (Figures 4(a)–4(c)). The MPTP-intoxicated group showed a considerable increment in the Bax/Bcl-2 ratio (p < 0.001) as compared to the control group whereas in CGA-treated PD mice, the Bax/Bcl-2 ratio (p < 0.001) was observed to be significantly reduced, which shows the antiapoptotic property of CGA in the parkinsonian mouse model (Figure 4(b)). The expression level of cleaved caspase-3 (p < 0.01) was observed to be elevated in the SN of PD mice whereas attenuated expression of cleaved caspase-3 (p < 0.05) was observed in the drug-treated group, which shows the antiapoptotic property of CGA in the parkinsonian mouse model (Figure 4(c)).
Figure 4.
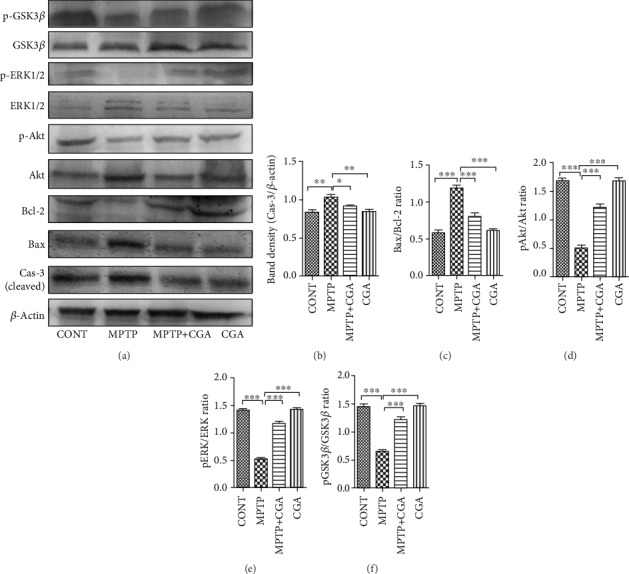
(a–f) Relative expression of Bax, Cas-3, Bcl-2, p-Akt, p-ERK1/2, and p-GSK3β in SN of mice was studied using the Western blot technique and densitometry analysis of proteins. CGA suppressed MPTP-induced apoptosis via Akt, GSK3β, and ERK1/2 signalling pathways in parkinsonian mice. It has normalized the deregulated expression of Bax, Bcl-2, and Cas-3 in MPTP-injected mice (a, b). β-Actin protein served as an internal control. Values are represented in the form of mean ± SEM (n = 5). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. Results were studied using the one-way ANOVA and further using the Newman-Keuls test. Abbreviations: CGA: chlorogenic acid; MPTP: 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine; SEM: standard error of the mean.
3.5. CGA Ameliorates the Dysregulation of Akt and ERK Activation and GSK3β Phosphorylation in the MPTP-Induced Parkinsonian Mouse Model
Expression of Akt, ERK1/2, and GSK3β was examined by WB (Figures 4(a) and 4(d)–4(f)) and immunohistochemical analysis techniques (Figures 5–7). Akt and ERK1/2 are the major downstream key components of the MAPK signalling pathway. Our WB data emphasised that MPTP treatment significantly attenuated the p-Akt/Akt ratio (p < 0.001) compared with the control group, whereas in CGA treatment, the p-Akt/Akt ratio (p < 0.01) was observed to be elevated in PD mice (Figure 4(d)). Similar results were also observed through the IHC technique showing reduced Akt phosphorylation in MPTP-treated mice (p < 0.001) whereas increased p-Akt was observed in the case of CGA-treated PD mice (Figure 5). Furthermore, WB results depicted downregulation in the p-ERK1/2/ERK1/2 ratio in MPTP-treated mice (p < 0.001). On the contrary, the p-ERK1/2/ERK1/2 ratio (p < 0.001) was upregulated in CGA-treated PD mice (Figure 4). Likewise, in IHC, the ERK1/2 phosphorylation was seen to be decreased on MPTP intoxication (p < 0.001) whereas CGA treatment has significantly increased the phosphorylation of ERK1/2 (p < 0.001) (Figure 7).
Figure 5.
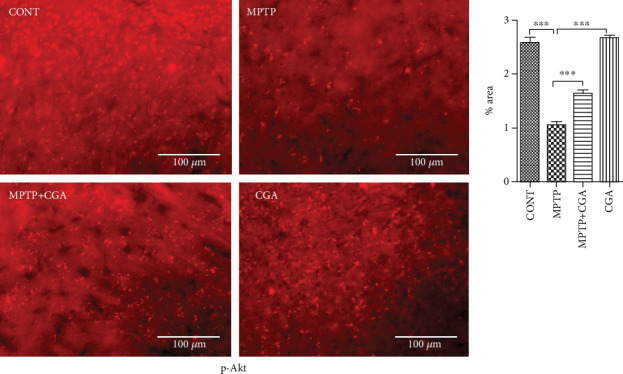
Immunohistochemical staining to analyse the expression of p-Akt in SN of different experimental groups. CGA administration enhanced the expression of p-Akt in SN of parkinsonian mice (20x magnification after staining). Values are expressed as mean ± SEM (n = 5). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. Results were studied using the one-way ANOVA and further using the Newman-Keuls test.
Figure 6.
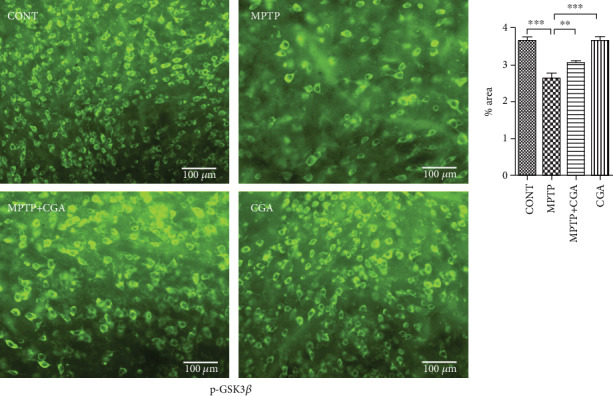
Immunohistochemical staining to analyse the expression of p-GSK3β in SN of different experimental groups (a, b). Profound expression of p-GSK3β in the CGA-administered group compared to the MPTP-intoxicated PD mice (20x). Values are represented as mean ± SEM (n = 5). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. Results were studied using the one-way ANOVA and further by the Newman-Keuls test.
Figure 7.
Immunohistochemical staining to analyse the expression of p-ERK1/2 in SN of different experimental groups. Upregulated expression of p-ERK1/2 due to CGA administration in parkinsonian mice (20x). Values are represented as mean ± SEM (n = 5). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. Results were studied using the one-way ANOVA and further by the Newman-Keuls test.
GSK3β has recently been linked to the mitochondrial intrinsic apoptosis. The WB result for GSK3β revealed a reduced p-GSK3β/GSK3β ratio (p < 0.001) in MPTP-intoxicated mice whereas in CGA-treated PD mice, the ratio of p-GSK3β/GSK3β was seen to be increased (p < 0.001, Figure 4(f)). To further confirm the results, IHC was performed presenting a substantial decrease in GSK3β phosphorylation after MPTP treatment in comparison with the control group (p < 0.01). On the other hand, CGA administration leads to the enhanced phosphorylation of GSK3β when compared to that of MPTP-intoxicated mice (p < 0.001, Figure 6).
3.6. CGA Ameliorates MPTP-Induced Degeneration of TH-Positive DA Neurons in SN of PD Mice
IHC of TH was performed to assess the loss of DA neurons in SN of MPTP-intoxicated mice (Figure 8). The data obtained in this study reveals the significant reduction of DA neurons in SN of an MPTP-intoxicated mouse as compared to control (p < 0.001). Alternatively, CGA pretreatment has done a significant increment in the expression of TH (p < 0.01) in SNpc of an MPTP-intoxicated mouse as compared with the MPTP-induced PD mouse model. Therefore, CGA administration was beneficial in protecting the DA neurons from MPTP-induced toxicity, as suggested by our findings.
Figure 8.
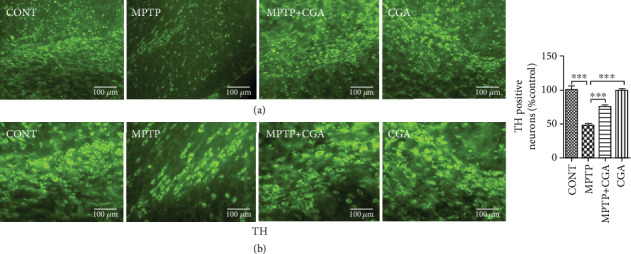
(a, b) Immunohistochemical staining to analyse the expression of TH-positive DA neurons in SN of different experimental groups. CGA protected the TH-positive DA neurons in MPTP-induced mice. (a) 10x and (b) 20x. TH immunoreactivity in SN of mice. Values are represented as mean ± SEM (n = 5). ∗p < 0.05, ∗∗p < 0.01, and ∗∗∗p < 0.001. Results were studied using the one-way ANOVA and further by the Newman-Keuls test.
4. Discussion
PD occurs due to multiple factors, and various pathways interact to induce the neurotoxic pathways that result in the loss of DA neurons in SN [6, 9]. The pathways that lead to neurodegeneration are influenced by each other, yet their contribution is independent of each other. All the involved processes work in an interdependent manner influencing one another and finally leading to the neuronal senescence [37, 38]. Hence, neuroprotection in the case of PD can only be achieved when combination of drugs or treatments targeting the collective pathways is used for PD therapy. In this context, it has been seen that CGA shows multiple biological effects such as antioxidant, anti-inflammatory, neuroprotective, and neurotrophic activities [27, 39–41]. In this study, CGA act as a potent neuroprotective agent by preventing mitochondrial dysfunction and suppressing the apoptotic death of DA neurons. Also, these neuroprotective effects of CGA were found to be mediated by Akt, ERK1/2, and GSK3β pathways. Different behavioral tests such as pole, catalepsy, rotarod, and traction tests have helped determine severe motor abnormalities in MPTP-intoxicated PD animal models [42–45] (Figure 1). Similar coordination deficits and motor impairment were also seen in our study too upon MPTP toxicity in mice, thereby validating our mouse model of PD in accordance with previous researches, whereas the neurobehavioral lesion was found to be alleviated by CGA as suggested by the behavioral tests which is in correspondence with earlier reports [30, 46–48].
The key pathogenic events of PD mainly constitute oxidative stress and mitochondrial dysfunction, and these two further lead to the onset of cell apoptosis [49, 50]. The neurotoxin MPTP serves as an excellent animal model for PD as it mimics the symptoms of PD extensively. The neurotoxin after crossing the blood-brain barrier (BBB) is acted upon by monoamine oxidase B to get converted into MPP+ and enters into DA neurons specifically via the dopamine transporter. MPP+ is the toxic metabolite of MPTP, and its accumulation inside the neurons leads to the inhibition of complex I of mitochondria and induces the ROS generation and depletion of ATP, prompting to the loss of the DA neurons [51, 52]. Both in vivo and in vitro models have been studied for the effects of antioxidants on the reduced mitochondrial dysfunction along with the enhanced growth of certain bioactive compounds in the mitochondria [53, 54]. In this study, MPTP intoxication reduced the activity of complexes I-III, complex IV, and complex V of ETS (Figure 2). Thus, the pattern of electron transfer was disturbed by MPTP toxicity, which has led to the reduction in the effectiveness of the enzymes NADH dehydrogenase and cytochrome oxidase causing structural damage to the mitochondria leading to its dysfunction. Further, because of the reduced functioning of ETC, oxidative stress arises triggering the reduced activity of Mn-SOD and excessive consumption of mitochondrial GSH (mtGSH, reduced). A research done by Davey et al. in 1998 proposed that the reduced activity of complex I occurs because of the depletion in the level of GSH in PC12 cells, which also leads to the abolition of the threshold effect [55]. The mechanism involved in the threshold hindrance of complexes I-III by the GSH level is still unknown. The antioxidant GSH is known to protect the mitochondria from lipid peroxidation [56], and hence, its depletion might make complexes I-III prone to the attack of the free radicals. Also, in previous researches, it has been described that the depleted level of GSH causes the degeneration and enlargement of the brain mitochondria [57] and induces the reduced activity of complex IV in the purified brain mitochondrial preparations [58]. The decreased level of the reduced glutathione or the generation of reactive oxygen and nitrogen species by the activated glial cells might be the main factors for the decline in complex IV activity. Moreover, mitochondrial damage also takes place by the production of NO as a result of inflammation-mediated expression of iNOS induction, which causes the loss of the cytochrome oxidase pathway by producing RNS consequently inducing protein nitration in the ETC. Moreover, various in vivo studies have reported the pivotal role of GSH in protecting DA neurons against the adverse effects of MPTP on PD [59, 60]. Therefore, the findings suggest that the fall in the reduced GSH level increased the sensitization of mitochondria against the additional metabolic insult resulting in its dysfunction and cell death. However, in our study, CGA was found to be potent in maintaining the appropriate activity of the ETC complexes (Figure 3). Furthermore, as reflected by the enhanced levels of reduced mtGSH and Mn-SOD, CGA was also effective in reducing the burden on the mitochondrial antioxidative defence by inhibiting the generation of the ROS. Thus, the ability of CGA to improve the activities of the complexes of ETC might be due to increased availability of reduced glutathione which is known to be the primary defence of mitochondrial functions.
The mitochondrion, an intracellular powerhouse, plays various crucial roles including energy production, free reactive oxygen and nitrogen species production along with critically modulating the apoptosis, and cell survival which are essential factors of aging. During the pathological alterations, the leakage of cytochrome c and various proapoptotic molecules in the cytoplasm from the mitochondria, as a result of the mitochondrial membrane permeabilization, induces the cascade of events leading to cell death [61]. Mitochondrial dysfunction is generally estimated by measuring the membrane potential which is found impaired in the intrinsic apoptotic pathway [62]. The regulation of caspase activation is maintained as a result of the equilibrium between the expression levels of proapoptotic and prosurvival proteins in the Bcl-2 family [63], although Bcl-2 and Bax have contrasting functions while being from the same Bcl-2 family. While Bcl-2 is known for its antiapoptotic activity, Bax exhibits the proapoptotic function [64]. Numerous studies have suggested that the neurotoxin MPTP disturbs the balance of Bax/Bcl-2, which increases the activity of caspase-3 in the DA neurons [65, 66]. The release of apoptogenic proteins like cytochrome c into the cytosol from mitochondria mediates the activation of caspase-3 which occurs because of the loss of membrane integrity by the action of Bax, hence leading to cell death. Both in vivo and in vitro studies have suggested the increased expression of caspase-3 upon MPTP toxicity [67]. The activation of caspase-9 provokes the initiation of the apoptotic cascade on the release of cytochrome c into the cytosol after mitochondrial dysfunction. As a result, caspase-3 gets activated by changing the morphology of mitochondria leading to apoptosis [68]. However, CGA treatment has protected the DA neurons from MPTP-induced toxicity and inhibited the apoptosis (Figure 4).
Different cellular processes like growth, proliferation, survival, and apoptosis are regulated by the phosphorylation of some key proteins such as Akt, ERK1/2, and GSK3β [69, 70]. The activation of Akt is found to be linked with the survival pathways of various cell types including neuronal cells as reported in several in vitro studies. Moreover, the survival of different neurons is linked with different neurotrophic factors, whose activation is mediated by Akt [71–73]. Also, transfection with a constitutively activated form of Akt is observed to promote the survival of the neurons without any other support whereas the cultured neurons were found to be dead when the activation of the kinase was interfered [74]. Studies from different sources have suggested the role of GSK3β in the modulation of apoptosis, and the inhibition of GSK3β has protected the DA neurons from MPTP toxicity as suggested by different cell and animal models of PD [45, 75, 76]. The localization of GSK3β has been mainly observed in the cytosol, but its expression in lower amounts has also been seen in the nucleus and mitochondria [77, 78]. Furthermore, it is also linked with the regulation of the mitochondrial cell death pathway by eliciting numerous stressful conditions within the neurons [78]. Researchers have also suggested that inhibition of GSK3β rescues the DA neurons from MPTP toxicity, indicating its association with the pathogenesis of PD [45, 76]. The neurotoxin MPTP has been found to decrease the phosphorylation of Akt and GSK3β as suggested by previous studies, leading to the death of the DA neurons [70, 79]. MPTP-induced cell injury is mediated by GSK3β, which is the downstream target of Akt [75, 80]. Mitochondrial dysfunction is seen to be facilitated by GSK3β, and its inhibition prevents the loss of neurons mediated by the suppression of proapoptotic proteins [76, 81]. In accordance with previous studies, MPTP has decreased the phosphorylation of Akt (Figures 4 and 5) and GSK3β (Figures 4 and 6), and CGA on the other hand has protected Akt and GSK3β from dephosphorylation in the MPTP-intoxicated mouse model. It has been observed that Akt controls the phosphorylation of GSK3β and thereby inhibits its activity [82]. MAPK signalling pathways also play a pivotal role in many cellular events, such as proliferation, differentiation, and apoptosis. The three major MAPK subfamilies have been chiefly characterized including ERK, JNK, and p38 [83]. Among them, ERK is known to enhance the DA neuronal survival rate, as in SH-SY5Y cells, the phosphorylation of ERK was seen to be suppressed after 4 h of MPP+ toxicity [84, 85]. Our study also shows the dephosphorylation of ERK upon MPTP intoxication, while the neuroprotective agent CGA was potent in abolishing the neurotoxic effect in the PD mouse model (Figures 4 and 7).
TH immunoreactivity in this study was performed to examine the CGA-mediated alterations on the functions of DA neurons in the SN of the MPTP-intoxicated parkinsonian mouse model [86] (Figure 8). Our immunohistochemical study has shown the reduction expression of TH-positive DA neurons in SN of an MPTP-intoxicated mouse, i.e., the characteristic feature of PD, as suggested by numerous reports [44, 60, 86]. However, CGA has rescued this debilitating effect and hence conferred about protecting the DA neurons against MPTP-induced neurodegeneration in the mouse model of PD.
Hence, our results have therefore suggested that the neuroprotective effect of CGA is mediated by the GSK3β phosphorylation-associated Akt/ERK pathway and is potent in inhibiting the mitochondrial intrinsic apoptosis due to MPTP toxicity in the mouse brain.
5. Conclusion
This study demonstrates the neuroprotective effect of CGA against MPTP-induced apoptotic cell death in mice. The Bax/Bcl-2 ratio, mitochondrial dysfunction, and expression of caspase-3 were seen to be significantly increased on MPTP intoxication. Furthermore, the phosphorylation of Akt, ERK1/2, and GSK3β was also reduced by the neurotoxic action of MPTP. However, CGA was potent in protecting the neurons against MPTP-induced cytotoxicity through the phosphorylation of GSK3β via activation of Akt and ERK1/2 signalling pathways in the mouse model of PD. Thus, this study helps to understand the neuroprotective mechanism of CGA in PD which can be further explored for clinical interventions.
Acknowledgments
SSS, SNR, HB, RS, WZ, ASR, and HD are sincerely thankful to ICMR, UGC, DBT, CSIR, and BHU, India, for their respective fellowship. The authors are also thankful to the head of the Department of Biochemistry, IOS, BHU, for providing the basic departmental facility and ISLS, BHU, for their central instrument facility.
Data Availability
The data used to support the findings of this study are available from the corresponding author upon request.
Ethical Approval
This work has been approved by the institutional animal ethical committee, BHU, Varanasi, India. All efforts were made to minimize animal suffering.
Conflicts of Interest
All authors have no conflict of interest to report.
Authors' Contributions
SSS conceived and designed the study, wrote the manuscript, and analysed and interpreted the data. SNR, HB, WZ, ASR, HD, and RS help in animal dosing, behavioral study, and manuscript preparation. SPS supervised the study. All authors read and approved the final manuscript.
References
- 1.Sawada H., Hishida R., Hirata Y., et al. Activated microglia affect the nigro-striatal dopamine neurons differently in neonatal and aged mice treated with 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine. Journal of Neuroscience Research. 2007;85(8):1752–1761. doi: 10.1002/jnr.21241. [DOI] [PubMed] [Google Scholar]
- 2.Poewe W., Seppi K., Tanner C. M., et al. Parkinson disease. Nature Reviews Disease Primers. 2017;3(1, article 17013) doi: 10.1038/nrdp.2017.13. [DOI] [PubMed] [Google Scholar]
- 3.Mattson M. P. Apoptosis in neurodegenerative disorders. Nature Reviews Molecular Cell Biology. 2000;1(2):120–130. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- 4.Youn J. K., Kim D. W., Kim S. T., et al. PEP-1-HO-1 prevents MPTP-induced degeneration of dopaminergic neurons in a Parkinson’s disease mouse model. BMB Reports. 2014;47(10):569–574. doi: 10.5483/BMBRep.2014.47.10.286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Subramaniam S. R., Chesselet M.-F. Mitochondrial dysfunction and oxidative stress in Parkinson's disease. Progress in Neurobiology. 2013;106-107:17–32. doi: 10.1016/j.pneurobio.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ferrer I. Early involvement of the cerebral cortex in Parkinson's disease: convergence of multiple metabolic defects. Progress in Neurobiology. 2009;88(2):89–103. doi: 10.1016/j.pneurobio.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 7.Ma Q.-L., Chan P., Yoshii M., Uéda K. α-Synuclein aggregation and neurodegenerative diseases. Journal of Alzheimer's Disease. 2003;5(2):139–148. doi: 10.3233/JAD-2003-5208. [DOI] [PubMed] [Google Scholar]
- 8.Gong P., Deng F., Zhang W., et al. Tectorigenin attenuates the MPP+-induced SH-SY5Y cell damage, indicating a potential beneficial role in Parkinson's disease by oxidative stress inhibition. Experimental and Therapeutic Medicine. 2017;14(5):4431–4437. doi: 10.3892/etm.2017.5049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaur R., Mehan S., Singh S. Understanding multifactorial architecture of Parkinson’s disease: pathophysiology to management. Neurological Sciences. 2019;40(1):13–23. doi: 10.1007/s10072-018-3585-x. [DOI] [PubMed] [Google Scholar]
- 10.Moon H. E., Paek S. H. Mitochondrial dysfunction in Parkinson's disease. Experimental Neurobiology. 2015;24(2):103–116. doi: 10.5607/en.2015.24.2.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thanvi B., Lo N., Robinson T. Levodopa-induced dyskinesia in Parkinson’s disease: clinical features, pathogenesis, prevention and treatment. Postgraduate Medical Journal. 2007;83(980):384–388. doi: 10.1136/pgmj.2006.054759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tatton N. A., Maclean-Fraser A., Tatton W. G., Perl D. P., Warren C. O. A fluorescent double-labeling method to detect and confirm apoptotic nuclei in Parkinson's disease. Annals of Neurology. 1998;44(Supplement 1):S142–S148. doi: 10.1002/ana.410440721. [DOI] [PubMed] [Google Scholar]
- 13.Mochizuki H., Goto K., Mori H., Mizuno Y. Histochemical detection of apoptosis in Parkinson's disease. Journal of the Neurological Sciences. 1996;137(2):120–123. doi: 10.1016/0022-510X(95)00336-Z. [DOI] [PubMed] [Google Scholar]
- 14.Bové J., Prou D., Perier C., Przedborski S. Toxin-induced models of Parkinson's disease. NeuroRx. 2005;2(3):484–494. doi: 10.1602/neurorx.2.3.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Birla H., Rai S. N., Singh S. S., et al. Tinospora cordifolia suppresses neuroinflammation in parkinsonian mouse model. Neuromolecular Medicine. 2019;21(1):42–53. doi: 10.1007/s12017-018-08521-7. [DOI] [PubMed] [Google Scholar]
- 16.Chiba K., Trevor A. J., Castagnoli N., Jr. Active uptake of MPP+, a metabolite of MPTP, by brain synaptosomes. Biochemical and Biophysical Research Communications. 1985;128(3):1228–1232. doi: 10.1016/0006-291X(85)91071-X. [DOI] [PubMed] [Google Scholar]
- 17.Jantas D., Greda A., Golda S., et al. Neuroprotective effects of metabotropic glutamate receptor group II and III activators against MPP (+)-induced cell death in human neuroblastoma SH-SY5Y cells: the impact of cell differentiation state. Neuropharmacology. 2014;83:36–53. doi: 10.1016/j.neuropharm.2014.03.019. [DOI] [PubMed] [Google Scholar]
- 18.Nakaso K., Tajima N., Horikoshi Y., et al. The estrogen receptor β-PI3K/Akt pathway mediates the cytoprotective effects of tocotrienol in a cellular Parkinson's disease model. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2014;1842(9):1303–1312. doi: 10.1016/j.bbadis.2014.04.008. [DOI] [PubMed] [Google Scholar]
- 19.Teng L., Kou C., Lu C., et al. Involvement of the ERK pathway in the protective effects of glycyrrhizic acid against the MPP+-induced apoptosis of dopaminergic neuronal cells. International Journal of Molecular Medicine. 2014;34(3):742–748. doi: 10.3892/ijmm.2014.1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vauzour D., Vafeiadou K., Rodriguez-Mateos A., Rendeiro C., Spencer J. P. E. The neuroprotective potential of flavonoids: a multiplicity of effects. Genes & Nutrition. 2008;3(3-4):115–126. doi: 10.1007/s12263-008-0091-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams R. J., Spencer J. P. E. Flavonoids, cognition, and dementia: actions, mechanisms, and potential therapeutic utility for Alzheimer disease. Free Radical Biology & Medicine. 2012;52(1):35–45. doi: 10.1016/j.freeradbiomed.2011.09.010. [DOI] [PubMed] [Google Scholar]
- 22.Singh S. S., Rai S. N., Birla H., Zahra W., Rathore A. S., Singh S. P. NF-κB-mediated neuroinflammation in Parkinson’s disease and potential therapeutic effect of polyphenols. Neurotoxicity Research. 2020;37(3):491–507. doi: 10.1007/s12640-019-00147-2. [DOI] [PubMed] [Google Scholar]
- 23.Gibellini L., Bianchini E., de Biasi S., Nasi M., Cossarizza A., Pinti M. Natural compounds modulating mitochondrial functions. Evidence-based Complementary and Alternative Medicine. 2015;2015:13. doi: 10.1155/2015/527209.527209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Solanki I., Parihar P., Mansuri M. L., Parihar M. S. Flavonoid-based therapies in the early management of neurodegenerative diseases. Advances in Nutrition. 2015;6(1):64–72. doi: 10.3945/an.114.007500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Oliveira M. R., Nabavi S. F., Manayi A., Daglia M., Hajheydari Z., Nabavi S. M. Resveratrol and the mitochondria: from triggering the intrinsic apoptotic pathway to inducing mitochondrial biogenesis, a mechanistic view. Biochimica et Biophysica Acta (BBA)-General Subjects. 2016;1860(4):727–745. doi: 10.1016/j.bbagen.2016.01.017. [DOI] [PubMed] [Google Scholar]
- 26.Singh S. S., Rai S. N., Birla H., et al. Effect of chlorogenic acid supplementation in MPTP-intoxicated mouse. Frontiers in Pharmacology. 2018;9 doi: 10.3389/fphar.2018.00757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ito H., Sun X. L., Watanabe M., Okamoto M., Hatano T. Chlorogenic acid and its Metabolitem-Coumaric acid evoke neurite outgrowth in hippocampal neuronal cells. Bioscience, Biotechnology, and Biochemistry. 2014;72(3):885–888. doi: 10.1271/bbb.70670. [DOI] [PubMed] [Google Scholar]
- 28.Kwon S.-H., Lee H. K., Kim J. A., et al. Neuroprotective effects of chlorogenic acid on scopolamine-induced amnesia via anti-acetylcholinesterase and anti-oxidative activities in mice. European Journal of Pharmacology. 2010;649(1-3):210–217. doi: 10.1016/j.ejphar.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 29.Manna S., Bhattacharyya D., Mandal T. K., Dey S. Neuropharmacological effects of deltamethrin in rats. Journal of Veterinary Science. 2006;7(2):133–136. doi: 10.4142/jvs.2006.7.2.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hu M., Li F., Wang W. Vitexin protects dopaminergic neurons in MPTP-induced Parkinson’s disease through PI3K/Akt signaling pathway. Drug Design, Development and Therapy. 2018;Volume 12:565–573. doi: 10.2147/DDDT.S156920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Weihmuller F. B., Hadjiconstantinou M., Bruno J. P. Acute stress or neuroleptics elicit sensorimotor deficits in MPTP-treated mice. Neuroscience Letters. 1988;85(1):137–142. doi: 10.1016/0304-3940(88)90443-0. [DOI] [PubMed] [Google Scholar]
- 32.Berman S. B., Hastings T. G. Dopamine oxidation alters mitochondrial respiration and induces permeability transition in brain mitochondria: implications for Parkinson’s disease. Journal of Neurochemistry. 1999;73(3):1127–1137. doi: 10.1046/j.1471-4159.1999.0731127.x. [DOI] [PubMed] [Google Scholar]
- 33.Sood P. K., Nahar U., Nehru B. Curcumin attenuates aluminum-induced oxidative stress and mitochondrial dysfunction in rat brain. Neurotoxicity Research. 2011;20(4):351–361. doi: 10.1007/s12640-011-9249-8. [DOI] [PubMed] [Google Scholar]
- 34.GRIFFITHS D. E., HOUGHTON R. L. Studies on energy-linked reactions: modified mitochondrial ATPase of oligomycin-resistant mutants of Saccharomyces cerevisiae. European Journal of Biochemistry. 1974;46(1):157–167. doi: 10.1111/j.1432-1033.1974.tb03608.x. [DOI] [PubMed] [Google Scholar]
- 35.Khanna P., Nehru B. Antioxidant enzymatic system in neuronal and glial cells enriched fractions of rat brain after aluminum exposure. Cellular and Molecular Neurobiology. 2007;27(7):959–969. doi: 10.1007/s10571-007-9233-2. [DOI] [PubMed] [Google Scholar]
- 36.Gorbatyuk O. S., Li S., Sullivan L. F., et al. The phosphorylation state of Ser-129 in human α-synuclein determines neurodegeneration in a rat model of Parkinson disease. Proceedings of the National Academy of Sciences. 2008;105(2):763–768. doi: 10.1073/pnas.0711053105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Riess O., Krüger R. Diagnosis and Treatment of Parkinson’s Disease—State of the Art. Springer; 1999. Parkinson’s disease—a multifactorial neurodegenerative disorder; pp. 113–125. [DOI] [PubMed] [Google Scholar]
- 38.Michel P. P., Hirsch E. C., Hunot S. Understanding dopaminergic cell death pathways in Parkinson disease. Neuron. 2016;90(4):675–691. doi: 10.1016/j.neuron.2016.03.038. [DOI] [PubMed] [Google Scholar]
- 39.Huang S. M., Chuang H. C., Wu C. H., Yen G. C. Cytoprotective effects of phenolic acids on methylglyoxal-induced apoptosis in neuro-2A cells. Molecular Nutrition & Food Research. 2008;52(8):940–949. doi: 10.1002/mnfr.200700360. [DOI] [PubMed] [Google Scholar]
- 40.Hwang S. J., Jun S. H., Park Y., et al. Green synthesis of gold nanoparticles using chlorogenic acid and their enhanced performance for inflammation. Nanomedicine: Nanotechnology, Biology and Medicine. 2015;11(7):1677–1688. doi: 10.1016/j.nano.2015.05.002. [DOI] [PubMed] [Google Scholar]
- 41.Hwang S. J., Kim Y. W., Park Y., Lee H. J., Kim K. W. Anti-inflammatory effects of chlorogenic acid in lipopolysaccharide-stimulated RAW 264.7 cells. Inflammation Research. 2014;63(1):81–90. doi: 10.1007/s00011-013-0674-4. [DOI] [PubMed] [Google Scholar]
- 42.Sedelis M., Schwarting R. K. W., Huston J. P. Behavioral phenotyping of the MPTP mouse model of Parkinson's disease. Behavioural Brain Research. 2001;125(1-2):109–125. doi: 10.1016/S0166-4328(01)00309-6. [DOI] [PubMed] [Google Scholar]
- 43.Chesselet M.-F., Richter F. Modelling of Parkinson's disease in mice. The Lancet Neurology. 2011;10(12):1108–1118. doi: 10.1016/S1474-4422(11)70227-7. [DOI] [PubMed] [Google Scholar]
- 44.Rai S. N., Birla H., Singh S. S., et al. Mucuna pruriens protects against MPTP intoxicated neuroinflammation in Parkinson’s disease through NF-κB/pAKT signaling pathways. Frontiers in Aging Neuroscience. 2017;9:p. 421. doi: 10.3389/fnagi.2017.00421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang W., Yang Y., Ying C., et al. Inhibition of glycogen synthase kinase-3β protects dopaminergic neurons from MPTP toxicity. Neuropharmacology. 2007;52(8):1678–1684. doi: 10.1016/j.neuropharm.2007.03.017. [DOI] [PubMed] [Google Scholar]
- 46.Xia X.-J., Lian Y. G., Zhao H. Y., Xu Q. L. Curcumin protects from oxidative stress and inhibits α-synuclein aggregation in MPTP induced parkinsonian mice. International Journal of Clinical and Experimental Medicine. 2016;9(2):2654–2665. [Google Scholar]
- 47.Nataraj J., Manivasagam T., Thenmozhi A. J., Essa M. M. Lutein protects dopaminergic neurons against MPTP-induced apoptotic death and motor dysfunction by ameliorating mitochondrial disruption and oxidative stress. Nutritional Neuroscience. 2015;19(6):237–246. doi: 10.1179/1476830515y.0000000010. [DOI] [PubMed] [Google Scholar]
- 48.Rai S. N., Zahra W., Singh S. S., et al. Anti-inflammatory activity of ursolic acid in MPTP-induced parkinsonian mouse model. Neurotoxicity Research. 2019;36(3):452–462. doi: 10.1007/s12640-019-00038-6. [DOI] [PubMed] [Google Scholar]
- 49.Dawson T. M., Dawson V. L. Molecular pathways of neurodegeneration in Parkinson's disease. Science. 2003;302(5646):819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- 50.JIANG J., JIANG J., ZUO Y., GU Z. Rapamycin protects the mitochondria against oxidative stress and apoptosis in a rat model of Parkinson's disease. International Journal of Molecular Medicine. 2013;31(4):825–832. doi: 10.3892/ijmm.2013.1280. [DOI] [PubMed] [Google Scholar]
- 51.Smeyne R. J., Jackson-Lewis V. The MPTP model of Parkinson's disease. Molecular Brain Research. 2005;134(1):57–66. doi: 10.1016/j.molbrainres.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 52.Seo B. B., Nakamaru-Ogiso E., Flotte T. R., Matsuno-Yagi A., Yagi T. In vivo complementation of complex I by the yeast Ndi1 enzyme possible application for treatment of Parkinson disease. Journal of Biological Chemistry. 2006;281(20):14250–14255. doi: 10.1074/jbc.M600922200. [DOI] [PubMed] [Google Scholar]
- 53.Naoi M., Wu Y., Shamoto-Nagai M., Maruyama W. Mitochondria in neuroprotection by phytochemicals: bioactive polyphenols modulate mitochondrial apoptosis system, function and structure. International Journal of Molecular Sciences. 2019;20(10):p. 2451. doi: 10.3390/ijms20102451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jin H., Kanthasamy A., Ghosh A., Anantharam V., Kalyanaraman B., Kanthasamy A. G. Mitochondria-targeted antioxidants for treatment of Parkinson's disease: preclinical and clinical outcomes. Biochimica et Biophysica Acta (BBA)-Molecular Basis of Disease. 2014;1842(8):1282–1294. doi: 10.1016/j.bbadis.2013.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Davey G. P., Peuchen S., Clark J. B. Energy thresholds in brain mitochondria potential involvement in neurodegeneration. Journal of Biological Chemistry. 1998;273(21):12753–12757. doi: 10.1074/jbc.273.21.12753. [DOI] [PubMed] [Google Scholar]
- 56.Meister A. Glutathione deficiency produced by inhibition of its synthesis, and its reversal; applications in research and therapy. Pharmacology & Therapeutics. 1991;51(2):155–194. doi: 10.1016/0163-7258(91)90076-X. [DOI] [PubMed] [Google Scholar]
- 57.Jain A., Martensson J., Stole E., Auld P. A., Meister A. Glutathione deficiency leads to mitochondrial damage in brain. Proceedings of the National Academy of Sciences. 1991;88(5):1913–1917. doi: 10.1073/pnas.88.5.1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heales R., Davies S. E. C., Bates T. E., Clark J. B. Deoletion of brain glutathione is accompanied by impaired micochondrial function and decreased N-acetyl aspartate concentration. Neurochemical Research. 1995;20(1):31–38. doi: 10.1007/BF00995149. [DOI] [PubMed] [Google Scholar]
- 59.Li X. H., Dai C. F., Chen L., Zhou W. T., Han H. L., Dong Z. F. 7, 8-Dihydroxyflavone ameliorates motor deficits via suppressing α-synuclein expression and oxidative stress in the MPTP-induced mouse model of Parkinson's disease. CNS Neuroscience & Therapeutics. 2016;22(7):617–624. doi: 10.1111/cns.12555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mani S., Sekar S., Barathidasan R., et al. Naringenin decreases α-synuclein expression and neuroinflammation in MPTP-induced Parkinson’s disease model in mice. Neurotoxicity Research. 2018;33(3):656–670. doi: 10.1007/s12640-018-9869-3. [DOI] [PubMed] [Google Scholar]
- 61.Liu X., Kim C. N., Yang J., Jemmerson R., Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86(1):147–157. doi: 10.1016/S0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 62.Joshi D. C., Bakowska J. C. Determination of mitochondrial membrane potential and reactive oxygen species in live rat cortical neurons. Journal of Visualized Experiments. 2011;(51, article e2704) doi: 10.3791/2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cory S., Adams J. M. The Bcl2 family: regulators of the cellular life-or-death switch. Nature Reviews Cancer. 2002;2(9):647–656. doi: 10.1038/nrc883. [DOI] [PubMed] [Google Scholar]
- 64.Singh L., Pushker N., Saini N., et al. Expression of pro-apoptotic Bax and anti-apoptotic Bcl-2 proteins in human retinoblastoma. Clinical & Experimental Ophthalmology. 2015;43(3):259–267. doi: 10.1111/ceo.12397. [DOI] [PubMed] [Google Scholar]
- 65.Li Y., Liu W. Z., Li L., Hölscher C. Neuroprotective effects of a GIP analogue in the MPTP Parkinson's disease mouse model. Neuropharmacology. 2016;101:255–263. doi: 10.1016/j.neuropharm.2015.10.002. [DOI] [PubMed] [Google Scholar]
- 66.Wang X., Zhao L., Han T., Chen S., Wang J. Protective effects of 2, 3, 5, 4′-tetrahydroxystilbene-2-O-beta-d-glucoside, an active component of Polygonum multiflorum Thunb, on experimental colitis in mice. European Journal of Pharmacology. 2008;578(2-3):339–348. doi: 10.1016/j.ejphar.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 67.Hartmann A., Hunot S., Michel P. P., et al. Caspase-3: a vulnerability factor and final effector in apoptotic death of dopaminergic neurons in Parkinson's disease. Proceedings of the National Academy of Sciences. 2000;97(6):2875–2880. doi: 10.1073/pnas.040556597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Friedlander R. M. Apoptosis and caspases in neurodegenerative diseases. New England Journal of Medicine. 2003;348(14):1365–1375. doi: 10.1056/NEJMra022366. [DOI] [PubMed] [Google Scholar]
- 69.Chen G., Bower K. A., Ma C., Fang S., Thiele C. J., Luo J. Glycogen synthase kinase 3β (GSK3β) mediates 6‐hydroxydopamine‐induced neuronal death. The FASEB Journal. 2004;18(10):1162–1164. doi: 10.1096/fj.04-1551fje. [DOI] [PubMed] [Google Scholar]
- 70.Zhao Q., Ye J., Wei N., Fong C., Dong X. Protection against MPP+-induced neurotoxicity in SH-SY5Y cells by tormentic acid via the activation of PI3-K/Akt/GSK3β pathway. Neurochemistry International. 2016;97:117–123. doi: 10.1016/j.neuint.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 71.Dudek H., Datta S. R., Franke T. F., et al. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275(5300):661–665. doi: 10.1126/science.275.5300.661. [DOI] [PubMed] [Google Scholar]
- 72.Duronio V. The life of a cell: apoptosis regulation by the PI3K/PKB pathway. Biochemical Journal. 2008;415(3):333–344. doi: 10.1042/BJ20081056. [DOI] [PubMed] [Google Scholar]
- 73.Orike N., Middleton G., Borthwick E., Buchman V., Cowen T., Davies A. M. Role of PI 3-kinase, Akt and Bcl-2–related proteins in sustaining the survival of neurotrophic factor–independent adult sympathetic neurons. The Journal of Cell Biology. 2001;154(5):995–1006. doi: 10.1083/jcb.200101068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ries V., Cheng H. C., Baohan A., et al. Regulation of the postnatal development of dopamine neurons of the substantia nigra in vivo by Akt/protein kinase B. Journal of Neurochemistry. 2009;110(1):23–33. doi: 10.1111/j.1471-4159.2009.06101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Petit-Paitel A., Brau F., Cazareth J., Chabry J. Involvment of cytosolic and mitochondrial GSK-3β in mitochondrial dysfunction and neuronal cell death of MPTP/MPP+-treated neurons. PLoS One. 2009;4(5, article e5491) doi: 10.1371/journal.pone.0005491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.King T. D., Bijur G. N., Jope R. S. Caspase-3 activation induced by inhibition of mitochondrial complex I is facilitated by glycogen synthase kinase-3β and attenuated by lithium. Brain Research. 2001;919(1):106–114. doi: 10.1016/S0006-8993(01)03005-0. [DOI] [PubMed] [Google Scholar]
- 77.Xavier I. J., Mercier P. A., McLoughlin C. M., Ali A., Woodgett J. R., Ovsenek N. Glycogen synthase kinase 3β negatively regulates both DNA-binding and transcriptional activities of heat shock factor 1. Journal of Biological Chemistry. 2000;275(37):29147–29152. doi: 10.1074/jbc.M002169200. [DOI] [PubMed] [Google Scholar]
- 78.Li D. W., Liu Z. Q., Wei-Chen, Min-Yao, Li G. R. Association of glycogen synthase kinase-3β with Parkinson's disease (review) Molecular Medicine Reports. 2014;9(6):2043–2050. doi: 10.3892/mmr.2014.2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Doo A.-R., Kim S. N., Park J. Y., et al. Neuroprotective effects of an herbal medicine, Yi-Gan San on MPP+/MPTP-induced cytotoxicity in vitro and in vivo. Journal of Ethnopharmacology. 2010;131(2):433–442. doi: 10.1016/j.jep.2010.07.008. [DOI] [PubMed] [Google Scholar]
- 80.Cross D. A. E., Alessi D. R., Cohen P., Andjelkovich M., Hemmings B. A. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378(6559):785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 81.Park S.-S., Zhao H., Mueller R. A., Xu Z. Bradykinin prevents reperfusion injury by targeting mitochondrial permeability transition pore through glycogen synthase kinase 3β. Journal of Molecular and Cellular Cardiology. 2006;40(5):708–716. doi: 10.1016/j.yjmcc.2006.01.024. [DOI] [PubMed] [Google Scholar]
- 82.Medina M., Garrido J. J., Wandosell F. G. Modulation of GSK-3 as a therapeutic strategy on tau pathologies. Frontiers in Molecular Neuroscience. 2011;4:p. 24. doi: 10.3389/fnmol.2011.00024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Lewis T. S., Shapiro P. S., Ahn N. G. Advances in Cancer Research. Elsevier; 1998. Signal transduction through MAP kinase cascades; pp. 49–139. [DOI] [PubMed] [Google Scholar]
- 84.Zhu J., Horbinski C., Guo F., Watkins S., Uchiyama Y., Chu C. T. Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. The American Journal of Pathology. 2007;170(1):75–86. doi: 10.2353/ajpath.2007.060524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Weng Z., Signore A. P., Gao Y., et al. Leptin protects against 6-hydroxydopamine-induced dopaminergic cell death via mitogen-activated protein kinase signaling. Journal of Biological Chemistry. 2007;282(47):34479–34491. doi: 10.1074/jbc.M705426200. [DOI] [PubMed] [Google Scholar]
- 86.Lee J.-M., Hwang D. S., Kim H. G., Lee C. H., Oh M. S. Dangguijakyak-san protects dopamine neurons against 1-methyl-4-phenyl-1, 2, 3, 6-tetrahydropyridine-induced neurotoxicity under postmenopausal conditions. Journal of Ethnopharmacology. 2012;139(3):883–888. doi: 10.1016/j.jep.2011.12.015. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available from the corresponding author upon request.