

Abstract
Skin injury is a highly inflammatory process that is carefully regulated to mitigate tissue damage and allow for proper barrier repair. Regulatory T cells (Tregs) are crucial coordinators of the immune response to injury in several organs. Here, we review the emerging role of Tregs in facilitating skin repair after injury. We focus on recently discovered interactions between lymphocytes and nonhematopoietic cells during wound healing and discuss how these interactions are regulated both by “classical” suppressive mechanisms of Tregs and by “nonclassical” reparative Treg functions.
One sentence summary:
This review summarizes the recent advances in our understanding of the roles of regulatory T cells in skin injury and tissue repair.
Introduction
The skin is the largest organ in the body and the first line of defense against the outside world. It undergoes constant trauma due to direct contact with the external environment, requiring dynamic and tightly regulated tissue repair processes to maintain barrier integrity. The cutaneous immune system is considerably diverse and plays a complex role in wound healing. While the role of macrophages and neutrophils in pathogen clearance and tissue repair has long been appreciated, only recently has the adaptive immune system been more thoroughly investigated, sometimes with seemingly paradoxical results. Lymphocytes are abundant in wounded skin but may be neither necessary nor sufficient for wound healing to occur: wounds heal more quickly in athymic nude mice (which lack T cells) and fetal skin (which is relatively lymphocyte-poor).1 This phenomenon has been generally attributed to the pro-inflammatory effects of lymphocytes, as excessive inflammation can hamper tissue repair.
Why then is it advantageous for lymphocytes to participate in skin injury? Unlike in relatively sterile experimental wounding models, cutaneous injury in natural environments carries a substantial risk of infection, rendering lymphocyte participation in the healing process essential. To counterbalance the potentially detrimental effects of inflammation, specialized subsets of lymphocytes have evolved to mitigate tissue damage by suppressing excessive inflammation, directing myeloid cell participation in healing, and facilitating tissue regeneration from stem cell populations. Regulation is therefore the key to productive participation of lymphocytes in tissue repair in order to fine-tune the balance of inflammation, regeneration, and remodeling necessary to restore the skin to homeostasis.
Regulatory T cells (Tregs) maintain the balance between immune homeostasis and inflammation systemically and are particularly abundant in skin.2 In addition to the classical role of Tregs in enforcing immune tolerance and suppressing excessive inflammation, pioneering studies over the last decade have uncovered novel tissue reparative functions of these cells in many organs, repositioning Tregs as crucial regulators of the immune response to injury. Herein, we review the process of cutaneous wound healing, summarize recent work on the tissue reparative roles of lymphocytes, and discuss the mechanisms by which Tregs facilitate wound repair in skin. Finally, we touch upon outstanding questions regarding the response of Tregs and the wider skin immune system to cutaneous injury.
I. Overview of Wound Healing in Skin
Structure and Function of Skin
Mammalian skin is comprised of two layers, the epidermis and dermis, situated above a layer of adipose tissue and fascia, known as the subcutis, hypodermis, and/or dermal white adipose tissue (dWAT). The skin forms a physical barrier between the host and the external environment, which protects against mechanical and chemical insults, pathogens, and regulates insensible water loss. The primary cellular constituent of the epidermis are keratinocytes, which undergo sequential differentiation and upward migration from basal layer stem cells, terminating in an anucleate cornified layer that interfaces with the external environment.3 The epidermal surface is studded with hair follicles that undergo homeostatic cycling through resting and growth phases. The latter is mediated by hair follicle stem cells (HFSC) residing within a region of the follicle known as the bulge.4
The dermis is a collagen-rich tissue that houses a plethora of stromal and immune cell populations. Dermal fibroblasts produce collagen, elastin and other components of the extracellular matrix (ECM) that are critical to maintain the structural integrity of skin in the steady-state and during skin repair. The subcutis is largely composed of mature adipocytes that have thermoregulatory and mechanoprotective functions. Both the dermis and subcutis are lined with a dense network of blood vessels, neurons, and lymphatics, which form migratory conduits for immune cells and play important roles in skin homeostasis, inflammation, and repair.
Cutaneous Wounds and Repair
Human skin is subject to a wide spectrum of traumatic injury, ranging from superficial abrasions of the epidermis to blunt/crush injury to deep penetrating wounds that damage both the skin and underlying tissue. In addition, a variety of chronic wounds are associated with vasculopathy and diabetes; however, little is known about the role of the immune system in many of these injuries. We therefore focus on the acute wound healing response, which is most commonly studied in animal models by excising circular skin biopsies to create full-thickness wounds. These wounds initiate a repair response that results in epidermal regeneration and dermal scarring, with a relative loss of adnexal structures (i.e., hair follicles, sebaceous glands, etc).5
The process of wound repair is classically divided into four phases: 1) hemostasis, 2) inflammation, 3) proliferation and 4) remodeling (see Figure 1).6 In the first phase, traumatic damage results in endothelial cell injury and activation, leading to platelet activation, initiation of the coagulation cascade, and formation of a fibrin clot over the wounded area. This sequence of events prevents excessive red blood cell loss from the circulation and provides a scaffold for wound repair. The second phase is dominated by neutrophils, which rapidly traffic into the wound bed and reach maximal numbers at 24 to 48 hours following injury.7 Molecules released by degenerating cells immediately following injury known as damage-associated molecular patterns (DAMPs) result in the secretion of neutrophilic and macrophage/monocyte chemoattractants.8 Neutrophilic inflammation is a double-edged sword: while necessary to eliminate offending microbes, it has the potential to inflict collateral damage to the tissue through production of reactive oxygen species, nitric oxide, and neutrophilic extracellular traps.9 During the inflammatory phase, tissue-resident macrophages and monocytes become activated DAMPs and alarmin cytokines such as the IL-1 family members IL-1α/β. The quantity and quality of this response can have lasting impacts on proliferative and remodeling phases that follow.10 Macrophages prevent infection of the wound environment by engulfing pathogens and apoptotic/necrotic cells. They influence tissue repair by modulating the functions of other immune cells and promoting angiogenesis and fibroblast activation.11 Historically, tissue macrophages have been split into classically (pro-inflammatory/M1) and alternatively activated (reparative/M2) subsets; however, it is increasingly appreciated that this binary classification is oversimplified, and that much greater heterogeneity and plasticity exists within the macrophage lineage in vivo. “Pro-inflammatory” macrophages can be activated by a variety of DAMPs and alarmins, which result in the expression of pro-inflammatory mediators including TNFα, IL-1β, IL-6, IL-8, and IL-12. “Reparative” macrophages are polarized by the cytokines IL-4, IL-13, and IL-10, resulting in the expression of TGFβ, IL-10, and various matrix remodeling mediators.12 Distinct macrophage subsets fluctuate depending on the different phases of the wound healing response, and perturbation of these subsets can have profound consequences on the quality of the repaired skin.13
Figure 1-. Progression of Acute Wound Healing in Skin.

(A) Immediately after wounding, a fibrin clot forms to plug the damaged tissue. (B) For several days after injury, local release of tissue damage signals leads to the recruitment of neutrophils and monocytes/macrophages from circulation to prevent infection and phagocytose dead cells and debris. Recent evidence suggests that nearby tissue-resident lymphocytes can also mobilize to wound margins during this time period. (C) As inflammation resolves, keratinocytes at the wound margins proliferate, migrate, and differentiate to re-epithelialize the injury site and restore barrier integrity. Granulation tissue forms in the wound bed and is vascularized by angiogenesis of nearby blood vessels. Myofibroblasts accumulate in granulation tissue and contract to draw the wound closed. The influx of lymphocytes into the wound reaches its peak, and macrophage polarization shifts from pro-inflammatory to pro-reparative states. (D) After the wound closes, cellularity of the wound bed decreases and a scar forms through a combination of collagen deposition, cross-linking, and remodeling. This process strengthens the wound site at the expense of regenerating normal dermis.
During the proliferative phase, keratinocytes, fibroblasts and endothelial cells increase in number and differentiate and migrate to cover the wound site. Keratinocytes re-epithelialize the wound to replace fibrin clot with regenerated epidermis. Lineage tracing experiments in mice have shown that several epithelial populations, including stem cells and committed progenitors, can contribute to patch the regenerating epidermis. Interfollicular keratinocytes are the first responders to injury and the primary contributors to re-epithelialization.14 HFSCs that reside in the isthmic/infundibulum region and bulge also undergo differentiation and migration in response to skin wounding at later time points.15 Beneath the regenerating epithelium, fibroblasts from the reticular dermis migrate into the wound bed and deposit collagen and other ECM components to plug the wound,16 initially forming a scaffold of granulation tissue. This phase is also marked by the accumulation of myofibroblasts, which express alpha smooth muscle actin (α-SMA) and contract to draw the wound together and decrease its surface area.17 Recent evidence suggests that in deep, full-thickness skin wounds, a subset of fibroblasts within the fascia of the subcutis migrates upward and produces ECM components that act as a provisional scar-like matrix, after which some of these cells differentiate into myofibroblasts.18
Tissue remodeling is the final phase of the wound healing response and is an attempt to restore the skin’s architecture to the original pre-wounded state, which can last for weeks to years. It is characterized by a diminution in myofibroblast numbers, regression of blood vessels, and a decreased recruitment of immune cells. There is also a notable shift in the production of type III to type I collagen by fibroblasts.19 The ECM undergoes extensive remodeling by production of growth factors and matrix metalloproteinases, in part by wound-associated macrophages.20 The end result is collagenous scar formation and increased overall strength of the repaired skin. This process comes at the cost of the tissue’s original composition and function, as key components of skin such as hair follicles, sweat glands, and adipocytes are infrequently regenerated.5
II. The Lymphocyte Response to Cutaneous Injury
Although lymphocytes are not required for any of the above wound healing processes to occur, they are capable of modulating each phase of tissue repair. Our understanding of lymphocyte-driven inflammation in tissues has evolved by studying infectious models that elicit strongly polarized type 1, type 2, or type 3 immune responses. The driving forces behind each subset of immune response are “signature” cytokines: IFN-γ for type 1 immunity; IL-4, IL-5, and IL-13 for type 2; and IL-17 and IL-22 for type 3 (Table 1). Lymphocytes coordinate polarized immune responses through differentiation into specialized subsets of helper T cells (Th1, Th2, Th17), innate lymphoid cells (ILC1, ILC2, ILC3), and unconventional T cells (γδ T cells, iNKT cells, MAIT cells, etc.) that produce their respective signature cytokines to combat specific types of pathogens.21 It is increasingly appreciated that these arms of the immune system are not only relevant to host defense and inflammation, but also to maintaining tissue homeostasis and facilitating the repair and regeneration of nonlymphoid organs.22 The type 1/2/3 paradigm of immunity is therefore a useful organizing framework for understanding the immune response to tissue injury, keeping in mind that wounding induces a pleiotropic immune response that is far less polarized than typical infections. It is also important to acknowledge that plasticity exists between type 1, type 2 and type 3 immune responses in some contexts, prompting the notion that these are semi-stable cell states more so than terminally differentiated cell fates.23
Table 1-. Summary of lymphocyte-driven immune responses following skin injury.
Th1, T helper 1 cell; ILC, innate lymphoid cell; AREG, amphiregulin; MAIT, mucosal-associated invariant T cell; AMP, antimicrobial peptide
| Immune Response | Effector Cytokines | Cytokine-producing celltypes in skin | Function during cutaneous repair | Mechanism of Action |
|---|---|---|---|---|
| Type 1 | IFN-γ | Th1, some γδ T cells | Increased inflammation, delayed/reduced repair | Classical macrophage polarization, increased production of inflammatory mediators |
| Type 2 | IL-4, IL-5, IL-13, AREG | ILC2, basophils, eosinophils, mast cells, Th2 | Reduced inflammation, re-epithelialization, wound contraction, scar formation | Alternative macrophage polarization, myofibroblast activation, increased collagen deposition/cross-linking |
| Type 3 | IL-17, IL-22 | Th17, ILC3, IL-17+ CD8 T cells, dermal γδ T cells, MAIT cells | Inflammation and tissue protection, re-epithelialization | Neutrophil recruitment, AMP production, keratinocyte proliferation and migration |
The type 2 immune module: orchestrator of full-thickness wound healing
Type 2 immunity is tightly integrated with organ homeostasis and directs reparative programs after tissue damage caused by metazoan parasites, venoms, toxins and mechanical wounding.24 Soon after injury, initial type 2 immune responses are coordinated by tissue-resident cells such as type 2 innate lymphoid cells (ILC2s) and mast cells at the site of damage. Additional effector cells associated with type 2 immune responses, including eosinophils, basophils, CD4+ Th2 cells, and GATA3+ Tregs are recruited from circulation in subsequent waves.25 This sequence of type 2 immune activation has largely been studied in helminth and allergic models of inflammation and is likely conserved to some degree in wound healing, though further studies are required to fully elucidate this. Healthy skin of naïve C57BL/6 mice contains a population of resident ILC2s that expand after wounding;26,27 however, Th2 cells are absent in this tissue at steady-state28 and their accumulation after cutaneous injury has not been quantified. After wounding, a large influx of type 2 immune cells enters the wound bed from circulation, including eosinophils, additional ILC2s and type 2-polarized Tregs.27,29,30 The bolstering of type 2 polarized lymphocytes and myeloid cells promotes both the regeneration of lost tissue and the deposition of fibrotic scar. The net result is to expediently restore the structural integrity of skin at the expense of full regeneration (Table 1).
Type 2 immune cells fulfill their tissue reparative functions via extensive cytokine-mediated crosstalk with most of the major parenchymal and stromal cell types in the tissue (Figure 2). Type 2 immune responses are initiated and propagated by the release of cytokines such as TSLP, IL-18, IL-25, and IL-33, which act as damage signals and are collectively referred to as “alarmins.” Epithelial cells, fibroblasts, endothelial cells, neurons, and lymphatics are all capable of producing alarmins to varying degrees in different tissues.31 These cytokines are present in basal amounts in healthy tissues, where they support tissue-resident cells like ILC2s.32 However, tissue damage induces a dramatic increase in alarmin release, which activates tissue-resident sentinels such as ILC2s, licenses the maturation of effector Th2 cells, and drives bone marrow production and tissue recruitment of myeloid cells such as basophils and eosinophils.33,34 Many parenchymal cell types in turn express receptors for the type 2 signature cytokines IL-4 and IL-13, which drive further release of inducing cytokines in an inflammatory feedback loop and effect profound changes in the cell state of nonhematopoietic cells. Following barrier disruption in skin, keratinocyte-derived TSLP and IL-33 induce Th2 and ILC2 responses.35,36 In return, amphiregulin, IL-4, and IL-13 are all capable of promoting keratinocyte proliferation,37,38 although the necessity of each these cytokines for re-epithelialization during physiologic wound healing is not clear. Stromal cells are likewise capable of activating and responding to type 2 immune cells during homeostasis and inflammation.32 In skin, dermal fibroblasts express the IL-13 receptor complex,39 and IL-13 potently drives myofibroblast activation and increased collagen deposition.25 Wound healing exemplifies the multifaceted effects of type 2 cytokines on skin: after excisional wounding, IL-33 drives both re-epithelialization and dermal collagen production to facilitate scarring, perhaps in part by activating ILC2 cells.27,40 Although type 2 cytokine signaling is clearly important for optimal wound healing, the relevant cellular sources of type 2 cytokines during wound healing have not been elucidated. Reparative crosstalk between type 2 immune cells and tissues has been best studied in the ILC2 literature, but neither ILC2s nor Th2 cells have been definitively shown to influence the progression of skin wound healing. Basophils, eosinophils, and mast cells can produce IL-4, are abundant in wounds, and affect tissue repair and fibrosis in other organs.41,42 The contribution of these celltypes to the global type 2 immune response after cutaneous injury merits further study.
Figure 2-. Participation of Type 2 and Type 3 Lymphocytes in Cutaneous Tissue Repair.

Left: The type 2 immune response to skin injury is triggered by release of alarmins such as TSLP, IL-18, and IL-33 from damaged keratinocytes, endothelial cells, and stromal cells. Locally resident type 2 lymphocytes such as ILC2s sense these tissue damage signals and produce IL-4, IL-13, and amphiregulin in response. These cytokines promote wound closure by activating wound-contracting myofibroblasts, both directly by signaling to nearby fibroblasts and indirectly by inducing alternative activation of macrophages (AAM). Type 2 cytokines are also capable of driving keratinocyte proliferation and therefore may promote re-epithelialization.
Right: Type 3 lymphocytes are abundant in skin and respond to both microbial ligands and tissue damage signals released by keratinocytes and myeloid cells, including IL-1β and IL-23. IL-17 and IL-22 produced by these cells act directly on keratinocytes, resulting in a two-pronged tissue protective response. First, antimicrobial immunity is bolstered by neutrophil recruitment and antimicrobial peptide production. Secondly, these cytokines reinforce the epidermal barrier by driving keratinocyte proliferation to cover injury sites at the expense of keratinocyte maturation.
Solid arrows, known interactions; Dotted arrows, likely interactions based on data from other tissues and contexts.
Alternatively activated macrophages (AAMs or “reparative”/M2 macrophages) are the foremost effectors of the type 2 immune response in full-thickness cutaneous wounding. These cells differentiate in response to IL-4 and IL-13, which induce the upregulation of effector molecules like RELM-α/FIZZ1, arginase-1, and Ym1/2.43 Ablation of AAMs by deleting IL-4Rα in macrophages causes a variety of defects in wound healing, including delayed wound closure, impaired formation of granulation tissue, and aberrant collagen deposition.44 Macrophages interact extensively with fibroblasts during the wound-healing process by driving the proliferation and differentiation of specific skin fibroblast subsets into myofibroblasts.45 AAM-derived RELM-α signals to fibroblasts to direct the proper assembly and cross-linking of collagen fibers in the scar.44 Deletion of the AAM effector molecule arginase-1 likewise impairs wound healing.46 Given the critical role of AAMs in wound healing, a principal function of type 2 lymphocytes is likely to serve as sources of IL-4 and IL-13 to drive AAM differentiation. The relative importance of Th2 cytokines in promoting wound healing by macrophage polarization versus direct signaling to nonhematopoietic cells remains to be elucidated.
The type 3 immune module: preserving the integrity of the epidermal barrier
Type 3 immunity evolved to combat extracellular microbial pathogens and maintain homeostasis with commensal microbiota. Type 3 immune responses in skin are thus focused at the interface between the host epidermis and the overlying microbiome. Skin is home to a rich array of specialized tissue-resident lymphocytes expressing the master transcriptional regulator RORγt and capable of producing the type 3 signature cytokines IL-17 and IL-22. These include dermal γδ T cells,47 ILC3s,48 mucosal-associated invariant T cells (MAIT cells),49 as well as Th17 and IL-17+ CD8+ T cells specific for microbial antigens.50,51 Imaging studies have demonstrated that these cell types predominantly localize within the epidermis or at the dermal-epidermal interface, with high densities near microbiota-rich hair follicles.48,52 Several different populations of type 3 lymphocytes have been shown to play a role in wound healing by accelerating re-epithelialization and wound closure,49,51,53,54 although the individual role of each subset may be partially compensated for by other cell types.
As with type 2 immunity, signaling between parenchymal cells and immune cells maintains and/or drives activation of type 3 immune responses during the steady-state and after tissue injury (Figure 2). However, the best-characterized mechanisms in skin are largely confined to epidermal-immune cell crosstalk. Epidermal damage exposes the underlying dermis to microbes, resulting in the release of inflammatory cytokines and the activation of type 3 immune cells. Recognition of microbial molecules by toll-like receptors (TLRs) on keratinocytes and myeloid cells induces secretion of cytokines such as IL-1β and IL-23, which activate local tissue-resident type 3 lymphocytes and prime Th17 cells in skin-draining lymph nodes.50. After full-thickness wounding or epidermal abrasion by tape-stripping, IL-17 from lymphocytes drives the production of neutrophil chemoattractants by keratinocytes to promote neutrophil influx during the inflammatory phase of wounding.55,56 IL-22 acts on keratinocytes to increase proliferation and inhibit differentiation after epidermal damage.57,58 Both cytokines are further capable of inducing the production of numerous inflammatory cytokines and antimicrobial peptides.59,60
Although type 3 cytokines promote rapid reconstitution of the epidermal barrier, they hinder the formation of healthy epidermis and instead drive acanthosis (keratinocyte hyperplasia) and parakeratosis (abnormal retention of nucleated keratinocytes in the cornified layer).61 The effect of uncontrolled type 3 immunity on the epidermis is exemplified by topical application of the TLR7 ligand imiquimod, a model of human psoriasis that triggers IL-17 production primarily by dermal γδ T cells.47 This results in a pronounced neutrophilic infiltrate and epidermal acanthosis resembling that of the immature, reparative epidermis observed in wounded skin. Because the psoriatic plaques have highly acanthotic epidermis and can be triggered by physical trauma, some have characterized this disease as an “exaggerated wound healing response.”62 Taken together, the net effect of type 3 immune activation after skin injury is thus to bolster antimicrobial defense and to rapidly repair the epidermal barrier by strongly promoting epidermal proliferation at the expense of maturation (Table 1). It should be noted that although both type 2 and type 3 immune responses affect the epidermis after cutaneous injury, their cross-regulation is poorly understood.
Participation of Other Arms of Adaptive Immunity in Wound Repair
Type 1 immunity contributes most prominently to the inflammatory stage of tissue repair by promoting antimicrobial defense at the expense of tissue regeneration (Table 1). Production of the type 1 signature cytokine IFN-γ increases for the first several days after full-thickness wounding,63 although the relevant cellular sources of IFN-γ have not been documented. IFN-γ activates macrophages toward an inflammatory phenotype analogous to classically activated (“M1”) macrophages described in vitro.64 IFN-γ- activated macrophages produce elevated levels of inflammatory cytokines including TNF-α, IL-1β, and IL-6, which in combination with IFN-γ inhibit angiogenesis and re-epithelialization (possibly in part by sensitizing keratinocytes to apoptosis65). Signaling of IFN-γ to fibroblasts antagonizes pro-fibrotic TGF-β signaling and thus inhibits collagen deposition.63,66 Wounds of IFN-γ knockout mice therefore exhibit enhanced wound closure, increased granulation tissue, and reduced monocyte infiltration.63
Dendritic epidermal T cells (DETCs) are another lymphocyte population found in the epidermis of mice (but not humans) that play a well-established role in cutaneous tissue repair.54 These cells express a semi-invariant TCR that recognizes an unidentified ligand released from damaged keratinocytes, prompting them to produce reparative factors such as keratinocyte growth factor (KGF/FGF7) and insulin-like growth factor 1 (IGF-1) when activated.54 The hard-wired ability of DETCs to sense damage and directly enact tissue repair are instructive for understanding innate-like reparative functions of conventional lymphocytes in skin, including Tregs.
III. Tregs in Healthy Skin
Specialized subsets of Tregs are present in many peripheral (i.e., non-lymphoid) organs of the body.67 These cells are critical both for mitigating harmful inflammation68,69 and for promoting tissue homeostasis and organ function.67 Both murine and human skin harbor an extraordinarily high baseline frequency of Tregs, perhaps reflecting an increased need for immune regulation and reparative potential in this organ. In neonatal mice, Tregs comprise up to 90% of the skin CD4+ T cell compartment,70 and adult frequencies range between 20 and 60% depending on the phase of the hair follicle cycle.71 Frequencies in human skin are ~20%, with higher proportions in hair-bearing regions such as the scalp.72 The majority of skin Tregs exhibit surface markers suggestive of antigen experience and prolonged tissue residency.73 Skin grafting studies likewise confirm that skin Tregs recirculate far less than conventional CD4+ T cells.72 Attracted by hair follicle-secreted chemokines, the majority of Tregs localize to the epidermis and dermis near hair follicles,71,72 bringing them into close proximity with numerous antigen presenting cells, effector T cells and innate lymphoid cells that also localize in this area.48,74 The abundance, localization, and prolonged residency of Tregs in skin poise these cells to maintain homeostasis and rapidly respond to inflammation and injury.
Skin Treg suppressive and homeostatic functions
Constant Treg-mediated immune tolerance to tissue autoantigens is critical for preventing systemic autoimmunity.75 The autoimmune manifestations of Treg deficiency in skin are particularly severe: dermatitis driven by massive infiltration of T cells is one of the most salient features of genetic Treg deficiency.76 Tissue-resident Tregs express higher levels of suppressive molecules than lymphoid Tregs and are believed to be necessary for maintaining immune regulation within tissues. However, the precise mechanisms by which Tregs suppress autoimmunity in specific organs independent of their lymph node-resident counterparts are not fully understood. In skin, secretion of the anti-inflammatory cytokine IL-10 is likely to play a role, since lymph node Tregs express little IL-10, and Treg-specific deletion of IL-10 predisposes to allergic dermatitis.68 However, the skin phenotype of mice lacking IL-10 in Tregs is considerably milder than that seen in the gut or after systemic Treg cell ablation,68,75 suggesting that these cells may utilize multiple suppressive mechanisms that can be relatively tissue specific. The contribution of other mechanisms (e.g. CTLA-4 expression, IL-2 competition) that skin-resident Tregs may utilize remain to be elucidated, largely due to the lack of tools for deleting pathways specifically in skin Tregs without affecting Tregs in lymphoid organs or other tissues.
In addition to preventing spontaneous autoimmunity, Treg suppressive function is also critical for mitigating the extent of tissue inflammation after an inciting stimulus. Tregs accumulate alongside effector T cells at sites of infection and contribute to the resolution of inflammation, although this can lead to prolonged pathogen persistence.77,78 In some cases, the antigen specificity of Tregs during infection has been traced to specific microbial peptides, as in the case of Mycobacterium tuberculosis infections.79 Impairment of Treg differentiation or suppressive function during infections leads to increased production of effector T cell cytokines and more severe tissue pathology: for example, Tregs that lack the TNF superfamily members CD27 and OX40 develop more severe skin disease after cutaneous infection with Candida albicans.80,81 Infectious models thus highlight the ability of Tregs to limit collateral tissue damage in situations when some degree of inflammation is desirable. Tregs reprise this role during tissue repair after sterile or non-infectious injury as well, although the antigens that drive Treg accumulation in these contexts have not been identified (Box 1).
Box 1-. Antigen Specificity of Tregs During Tissue Repair.
The importance of antigen recognition for Treg function during tissue repair is currently unknown. Tissue injury is a highly inflammatory process that results in the release of “danger” signals alongside copious self-antigen from necrotic cells. Do Tregs play a role in preventing autoreactive cells from exacerbating an already bad situation? Th1 and Th2 cells that are presumptively autoreactive accumulate in wounded skin of Treg-depleted mice,30,81 but this phenomenon occurs in non-wounded skin after Treg depletion as well, making it difficult to tell whether these cells respond specifically to injury-associated antigens.
Unfortunately, the identities of the antigens recognized by skin Tregs at steady state and after injury are almost entirely unknown. Other tissue resident memory T cells (TRM) are specific for microbial antigens from pathogens or commensal microbes, and in some cases these cells have been shown to participate in tissue repair after being activated by tissue damage signals.51,52 Tregs specific for commensal microbes are generated in the skin and colon,70,83 so at least some Tregs may similarly react to non-injury associated antigens and trigger their reparative functions via innate stimuli. However, a significant number of Tregs recognize tissue-restricted autoantigens (TRAs) that are continuously expressed in tissues.84 It is tempting to speculate that recognition of TRAs by skin Tregs may be especially important during cutaneous injury to suppress an immune response against self-antigens. In support of this hypothesis, reparative Tregs that accumulate in wounded muscle have highly biased oligoclonal TCR repertoires, and mice bearing a muscle Treg TCR transgene exhibit increased accumulation of Tregs at injury sites, suggesting that they may be responding to a muscle-derived self-antigen released after injury.85,86
As with other tissue-resident T cell populations, Tregs possess non-classical functions that enable them to participate in organ homeostasis.67 In contrast to Treg suppressive functions, which are primarily adaptive (i.e. triggered by TCR recognition of cognate antigen), many non-classical Treg functions are innate (precipitated by nonspecific tissue signals independent of TCR stimulation), although this division is not absolute. A prominent homeostatic function of skin Tregs is to promote hair regeneration after depilation and during the natural hair follicle cycle.71 This occurs in part by direct paracrine signaling of the skin Treg-expressed Notch ligand Jagged-1 to hair follicle stem cells (HFSCs), facilitating their proliferation and differentiation without requiring classical Treg suppressive capabilities. In many cases, Tregs share tissue adaptations with effector lymphocyte populations to enable joint participation in tissue homeostasis. For instance, in visceral adipose tissue, Tregs, ILC2s, and AAMs are all necessary for maintaining metabolic homeostasis and preventing the development of glucose intolerance, a role that is enabled in part by expression of the transcription factor PPAR-γ in both Tregs and AAMs.87 Several lines of evidence suggest that reparative functions of skin Tregs are shared with other resident lymphocytes and primarily function innately, while suppressive Treg functions are predominantly (though not always) adaptive and are unique to Tregs during tissue repair. This will be discussed below.
IV. Tregs In Cutaneous Tissue Repair
Skin-resident Tregs are polarized toward a type 2 tissue-reparative phenotype
Activated Tregs are highly heterogeneous and are able to differentiate into subsets that mirror the helper T cell subsets Th1, Th2, and Th17. “Type 1” Tregs express the Th1 lineage master transcription factor T-bet and are required for suppressing Th1 immune responses; “type 2” Tregs express IRF4 and GATA-3 to suppress Th2 immunity; and “type 3” Tregs express RORγt, c-MAF, and STAT-3 to suppress Th17 cell responses.69,88 By expressing T helper lineage transcription factors, Tregs co-opt portions of T helper transcriptional modules, likely enabling them to adopt localization and tissue-specific adaptations similar to helper T cell and ILC subsets. Treg polarization is thus necessary both for suppressing inflammation caused by specific T helper subsets and for participating in organ homeostasis alongside helper T cells and ILCs.
Among all reported organs, skin bears the highest proportion of Tregs expressing the type 2 lineage defining transcription factor GATA-3. Different publications report that 50–80% of skin Tregs are GATA-3+ at baseline.52,82,89 GATA-3 and other co-expressed type 2-associated transcription factors are necessary both for Treg restraint of type 2 inflammation in skin and for participation in tissue repair. Mice with a Treg-specific deletion of GATA-3 gradually develop a lymphoproliferative disease characterized by Th2-driven inflammation in skin and gut, although disease severity varies somewhat by mouse facility.52,90,91 Notably, patients that lack functional Tregs due to genetic mutations in FoxP3 also develop severe atopic dermatitis, underscoring the role of Tregs in suppressing type 2 skin inflammation.76 GATA-3 promotes Treg suppressive function in part by binding to the CNS2 regulatory region of FoxP3 and stabilizing FoxP3 expression, which upregulates Treg signature genes such as ICOS without inducing Th2 cytokine expression.90,91 GATA-3 also represses Treg polarization toward other T helper-like fates by repressing non-type 2 lineage transcription factors like T-bet and RORγt.92 Accordingly, loss of GATA-3 in Tregs results in Treg instability and IL-17 production.90,93 GATA-3 likely also promotes the expression of Th2 chemokine receptors that facilitate localization of GATA-3+ Tregs near Th2 cells at sites of inflammation, as has been observed in T-bet+ type 1 Tregs.94
In addition to suppressing type 2 skin inflammation, GATA-3+ Tregs acquire innate tissue reparative functions that are shared by Th2 and ILC2 cells. These cells, which also require GATA-3 for their transcriptional programs, are able to sense tissue damage in a variety of organs by expressing receptors for the alarmins TSLP, IL-18, IL-25, and IL-33 released from nonhematopoietic cells upon stress or damage.31 In skin,89 wounded muscle,85 visceral adipose tissue,95 and several other organs, GATA-3+ Tregs express the IL-33 receptor ST2, which enables them to enact reparative functions in response to IL-33 released by stressed and dying cells. This method of Treg activation is independent of TCR stimulation.96 In addition to activating innate Treg functions, IL-33 is also a Treg trophic factor that is necessary for maximal Treg accumulation in organs like visceral fat.95 TSLP is similarly released from keratinocytes during inflammation and has been shown to promote skin Treg activation.97 IL-18 may be another relevant alarmin in skin, as skin ILC2s and CD8 T cells were recently shown to be activated by this cytokine.26,52 Treg tissue repair can be innately triggered by sensing of IL-18 in vitro,96 although Treg expression of the IL-18 receptor complex in skin has not yet been reported. The “type 2” Tregs that predominate in skin are thus rewired to utilize tissue reparative elements of the type 2 immune module.
Provenance and accumulation of Tregs in wounded skin
Wounding is a highly inflammatory process that results in recruitment of many circulating immune cells into injured tissue (as outlined for skin above). Even in tissues that normally do not contain resident Treg populations such as the brain and muscle, injury causes a marked influx of Tregs necessary for optimal organ repair.85,98 Tregs accumulate in skin after either epidermal abrasion or full-thickness wounding, reaching their maximal levels ~1 week after injury.30,55 The relative contributions of Treg recruitment from circulation versus local proliferation of the resident pool is currently unknown. In other settings, inflammation drives lymphocyte recruitment from circulation, local proliferation of tissue-resident cells, and egress of tissue-derived cells into lymph.99 Treg accumulation in wounded skin is likely a combination of all these factors, though it is unclear whether there are functional differences between Tregs derived from these different sources. Tregs recruited into injured muscle share many of the same reparative transcriptional signatures as non-recirculating tissue-resident Tregs in visceral adipose tissue, suggesting that regardless of ontogeny, Tregs in wounded tissues can ultimately achieve similar reparative capacities.85,100 However, there may be temporal or spatial differences between tissue-resident and tissue-infiltrating Tregs. Skin-resident Tregs are already present in the tissue and poised to respond quickly to injury, whereas Tregs derived from circulation likely require time to enable their activation, differentiation, and migration to the injury site. Thus, Tregs that are recruited to skin after injury may play more of a reinforcing role.
Skin repair by direct Treg-parenchymal cell interactions
Secretion of amphiregulin (AREG), a ligand for the epidermal growth factor receptor (EGFR), is a major mechanism of tissue repair shared by many type 2 immune cells including GATA3+ Tregs.38 Treg expression of AREG is driven innately in response to IL-18 and IL-33,96 and binding of AREG to EGFR induces proliferation and differentiation of target cells to regenerate tissue lost after injury. For instance, in wounded muscle, AREG expressed by reparative Tregs drives the regeneration of myofibers from satellite cell progenitors.85 Although AREG is expressed by many celltypes including Th2 cells, ILC2s, macrophages, and basophils (Figure 3), Treg expression of AREG plays a nonredundant role in some contexts, as mice with a Treg-specific ablation of AREG exhibit more severe lung pathology and higher mortality rates after influenza infection.96 Although Treg expression of AREG has not been functionally dissected in skin injury models, several lines of evidence argue for a role. Skin-resident Tregs express AREG89 and AREG drives proliferation of keratinocytes,38 consistent with its effect on other epithelial tissues. In addition to its effects on the epithelium, AREG has been shown to restore vascular integrity by augmenting pericyte activation of TGF-β, suggesting that AREG mediated signaling through EGFR may promote reparative programs in stromal compartments after skin wounding.101 Tregs in wounded skin express the AREG receptor EGFR, and Treg-specific ablation of EGFR results in decreased Treg numbers and slower wound closure, hinting at an autocrine function of AREG in promoting Treg accumulation during injury (Figure 3).30
Figure 3-. Reparative and Suppressive Effector Mechanisms of Type 2-Polarized Tregs.
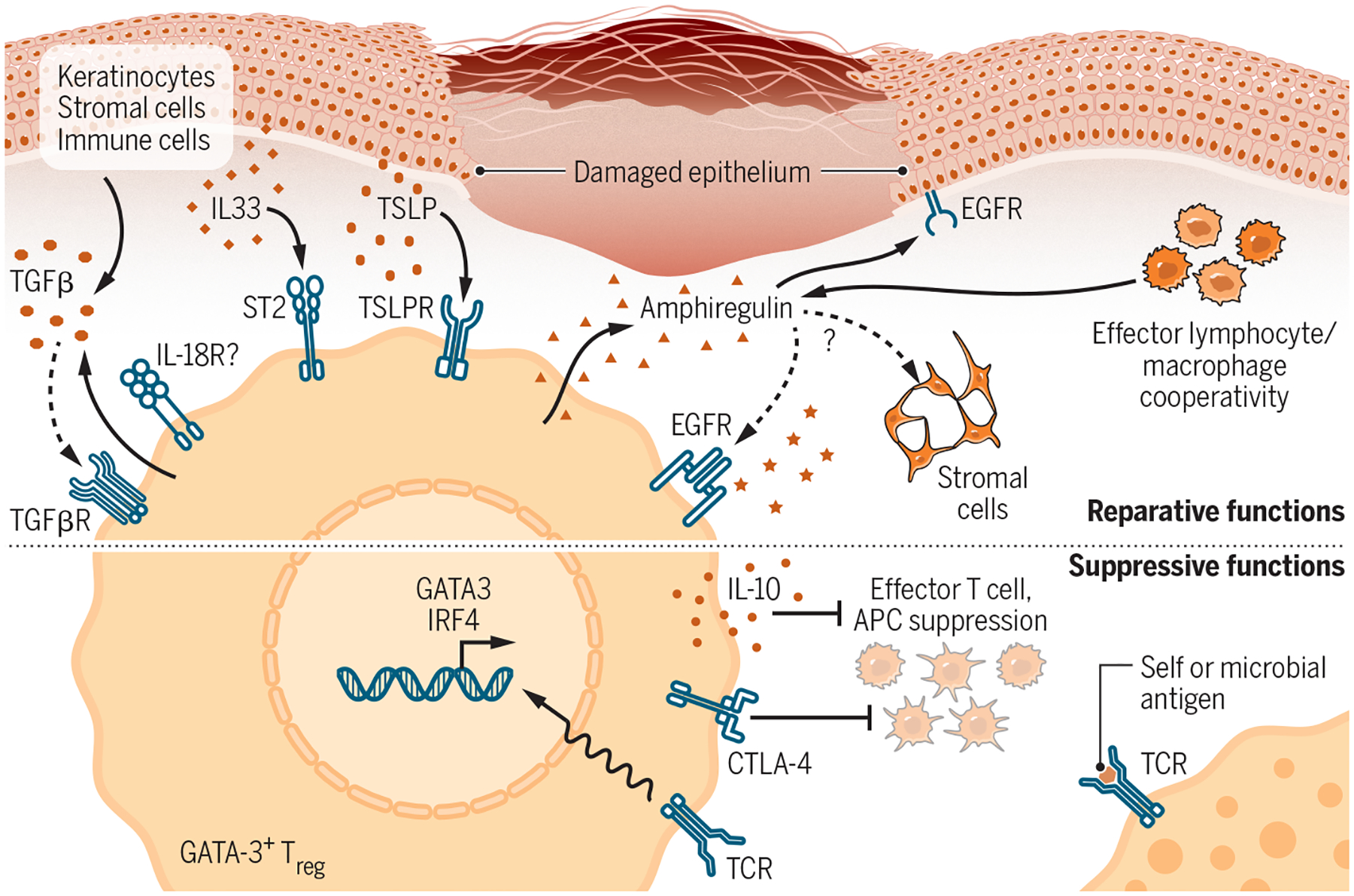
Skin bears a high proportion of type 2-polarized Tregs programmed by Th2-associated transcription factors such as GATA-3 and IRF4 that both confer tissue-reparative functions. Top: GATA-3+ Tregs in skin express receptors for alarmins such as TSLP, IL-33, and possibly IL-18 that are released upon tissue damage, enabling them to sense local injury. IL-18 and IL-33 can both stimulate Treg production of the reparative cytokine amphiregulin independent of TCR stimulation. Amphiregulin (AREG) drives keratinocyte proliferation and may promote regeneration of stromal populations in wounded skin. Skin Tregs additionally express both TGF-β and TGFβR, although the Treg-specific function of each is not entirely clear. Many of these reparative functions are shared by effector lymphocyte and macrophage populations. Bottom: In addition to conferring direct tissue-reparative function to Tregs, GATA-3 and IRF4 also bolster adaptive Treg functions in part by stabilizing FoxP3 expression. These “classical” Treg functions are linked to TCR stimulation and include production of the regulatory cytokine IL-10 and suppression of costimulation on antigen presenting cells by Treg-expressed CTLA-4. Solid arrows, known interactions; Dotted arrows, likely interactions based on data from other tissues and contexts.
In addition to amphiregulin, Tregs have been reported to express a variety of other growth factors that act directly on nonhematopoietic cells to promote repair after injury. Tregs express KGF to promote alveolar regeneration by type II alveolar epithelial cells after lung injury,102 and colonic Tregs perform a similar function by secreting FGF2 to increase proliferation of the intestinal epithelium during colitis.103 In zebrafish, Tregs facilitate regeneration of a variety of organs after injury, with one predominant growth factor driving repair in each organ (NTF in spinal cord, NRG1 in heart, and IGF-1 in retina).104 Notably, KGF and IGF-1 are among the factors secreted by DETCs to promote epidermal regeneration after skin wounding.54 Thus, it is quite possible that skin Tregs also utilize these mechanisms to promote repair after skin injury.
Regulation of Skin Repair by Treg Suppressive Functions
Although inflammation early in the wound healing process is necessary to prevent infection, excessive inflammation is counterproductive to wound healing. Therefore, the classical function of Tregs—suppressing inflammation—is critical in regulating the repair process. Early during wound healing, Tregs are necessary for the transition from the inflammatory to the reparative stages of healing. Treg depletion immediately after wounding delays wound closure and increases the amount of granulation tissue and eschar formed in the wound bed.30 This effect is due to the accumulation of IFN-γ+ T effector cells in Treg-depleted skin, which increases the accumulation of pro-inflammatory Ly6C+ monocytes. Treg suppression of type 1 immunity facilitates the transition from the inflammatory phase to the reparative phase of wound healing, during which alternatively activated macrophages predominate and drive tissue remodeling. Some reports further suggest that Tregs can direct AAM polarization by secreting IL-4 and IL-13.105 Treg-derived IL-13 has also been proposed to increase macrophage efferocytosis,106 a key feature of AAMs that enables clearance of debris.107 However, in vivo secretion of T helper cytokines by Tregs is controversial and may only occur in pathologic situations that destabilize Treg cell state.108 Further studies using in vivo cytokine reporters and Treg-specific cytokine knockouts are therefore needed to validate Treg cytokine production as a mechanism of tissue repair.
Skin Tregs were recently shown to suppress myofibroblast activation at steady state and during experimental models of cutaneous fibrosis,82 suggesting that these cells may prevent excessive scarring by restraining type 2 immunity during later stages of wound healing. GATA-3 expression by Tregs is necessary to prevent excessive myofibroblast activation and collagen deposition, and Treg-specific deletion of GATA-3 leads to the accumulation of Th2 cells in skin that robustly produce the pro-fibrotic cytokines IL-4 and IL-13.82 Since fibrosis results from dysregulation of normal wound healing programs, GATA-3+ Treg-mediated suppression of Th2 cells is likely to regulate physiologic wound healing as well as pathologic fibrosis.
Treg suppression of type 3 immunity is important for re-epithelialization during the initial stages of tissue repair. In a tape-stripping model of epidermal abrasion, Treg depletion causes delayed recovery of barrier function.55 In the absence of Tregs, Th17 cells accumulate in skin and cause increased expression of the neutrophil chemoattractant CXCL5.55 Barrier damage normally prompts Lgr5+ stem cells within the hair follicle bulge to proliferate and migrate into the interfollicular epidermis in order to regenerate the epithelium.109 However, the exuberant CXCL5-IL-17 inflammatory response to tape stripping in Treg depleted mice impedes bulge HFSC differentiation and migration into the interfollicular epidermis, possibly accounting for the defect in barrier function recovery after Treg depletion.55 These results demonstrate an indirect role for Tregs in promoting normal epidermal regeneration from stem cells by attenuating excessive type 3 immunity.
In addition to Treg suppression of subset-specific helper T cell responses, two broadly anti-inflammatory and highly pleiotropic cytokines secreted by Tregs—IL-10 and TGF-β—have been extensively implicated in wound healing.110 IL-10 is a potent suppressor of both inflammation and fibrosis, and global loss of IL-10 results in both faster wound closure and increased scar formation due to elevated myofibroblast accumulation.111 Treg expression of IL-10 is significantly higher in skin than in lymphoid organs,89 and Treg-derived IL-10 is crucial to preventing excessive inflammation in other contexts.68 It is therefore plausible that Treg-derived IL-10 contributes to the suppression of excessive fibrosis after cutaneous wounding.
TGF-β is perhaps the most potent pro-fibrotic cytokine and is a master regulator of numerous reparative processes including physiologic scar formation and pathologic fibrosis.110 Treg involvement in TGF-β mediated reparative processes can be conceptually divided into three aspects: TGF-β signaling to Tregs via TGF-β receptors, Treg secretion of TGF-β and Treg activation of latent TGF-β. TGF-β is required for generation of both thymic and peripherally-generated Tregs and helps maintain Tregs (along with numerous other immune cells) in tissues.112 In line with these data, skin Tregs express higher levels of TGF-β receptors than lymph node Tregs (Rosenblum lab, unpublished observation). Thus, TGF-β may play a role in maintaining skin Tregs and/or promoting their suppressive activity during wound healing. Tregs themselves produce TGF-β in many organs, but it is important to note that this alone does not constitute evidence of a pro-fibrotic role for Tregs. Many cells secrete TGF-β and Treg contribution to the tissue pool of TGF-β is likely to be small (Figure 3). It is further possible that TGF-β secreted by Tregs primarily acts to maintain Tregs in cis. The difficulty in elucidating the function of Treg production of TGF-β is exemplified by studies of this cytokine’s role in Treg suppressive function, which conflict as to whether Treg-derived TGF-β is required to suppress inflammation in vivo.112 An additional complexity is that TGF-β can be presented on the cell surface or deposited on extracellular matrix in a latent form and must be activated by proteases or integrins expressed in trans.113 The function of TGF-β from any given celltype is thus dependent on the mode of presentation, the timing of cytokine activation, and the recipient cell identity. Tregs additionally express the TGF-β activating integrin αvβ8, and Treg expression of β8 integrin is necessary for optimal suppression of inflammation in the intestine.114 It is therefore possible that Tregs activate latent TGF-β following tissue injury. Thus, although Tregs clearly sense, secrete, and activate TGF-β in a variety of inflammatory contexts, the in vivo significance of all three functions during tissue repair requires further investigation.
Future Directions
Despite our growing understanding of the role of Tregs in wound healing, several key questions remain. Treg suppressive function clearly plays a role in promoting tissue repair, but the relative contributions of lymph node versus skin-resident Tregs to suppressing excessive inflammation is unknown. This question remains unsolved both for wound healing and for a variety of other inflammatory models due to the lack of tools to specifically manipulate Tregs in a tissue-specific manner. It is also unknown whether specific Treg subsets in skin play different and specialized roles in tissue repair. Different populations of Tregs co-reside in tissues and in some cases have highly disparate functions.115,116 Do subsets of skin resident Tregs differentially localize to distinct microanatomic niches within this tissue, and do they have different roles in the wound healing process? Do Tregs derived from circulation in wounded skin form spatially and/or functionally distinct subsets? The anatomical diversity of human skin adds yet another layer of complexity to these questions. Tregs are found at different sites in human skin, but is their phenotype (and possibly function) different between these sites? The tissue architecture,117 wound healing-capacity,118 and resident microbiota119 of skin likewise varies by body location, and these factors may have important implications for the participation of the immune system in wound healing, as well as the choice of location and model organism for animal wound healing experiments.
Regeneration of tissue after wounding is ultimately carried out by skin parenchymal and stromal cells, yet the effect of Tregs (and the immune system more broadly) on the ontogeny, heterogeneity, and function of nonhematopoietic cells during wounding is poorly understood. The epidermal and dermal compartments of skin contain considerable cellular heterogeneity, and not all cells have equal regenerative capacity. After wounding, newly regenerated keratinocytes and myofibroblasts are derived from specific populations of stem and progenitor cells that can interact with Tregs and other lymphocytes in certain contexts. For instance, Tregs promote hair regeneration from HFSCs71 that contribute to epidermal barrier repair after wounding.109 In the lung, IL-13 from ILC2 and Th2 cells can expand perivascular Gli-1+ stromal cells120 that give rise to myofibroblasts during fibrosis.121 Recent work has only scratched the surface of immune-stem cell interactions, particularly in tissue injury contexts.122 Fully defining the precursor populations that contribute to tissue repair and dissecting the effect of the immune system on these cells will be critical to understanding how inflammation can be modulated to maximize regeneration.
Finally, our understanding of the immune response to cutaneous injury can be further leveraged to understand human disease, and vice versa. The clinical manifestations of several human skin diseases display aspects of injury and barrier repair processes gone awry. In both psoriasis, a Th17-driven skin disease, and atopic dermatitis (AD), a Th2-predominant disease, cutaneous lesions are characterized by abnormal alterations in the epidermal barrier that can be viewed as dysfunctional tissue repair responses.37 In AD, barrier damage itself is a mediator of disease pathogenesis. Itching causes epidermal damage that drives a Th2 immune response, but rather than contributing to barrier repair, uncontrolled Th2 immunity perpetuates disease by inducing further itching and driving the formation of immature epidermis that is hyperplastic but fragile.37. Quantitative and qualitative Treg deficiencies that have been observed in both diseases may therefore contribute not only to immune pathology but also to aberrant tissue repair responses in these conditions. Research into the immunologic mechanisms behind abnormal tissue repair in AD and psoriasis has shed light on the interactions between the immune system and skin during normal wound healing. This approach may be similarly productive in less well-understood diseases that involve dysfunctional tissue repair. For instance, scleroderma is a fibrotic disease with cutaneous manifestations akin to uncontrolled scarring in the setting of immunologic dysfunction and autoantibody production.123 Th2 cells are increased and Tregs are reduced in scleroderma lesions, suggesting that Treg deficiencies may underlie fibroblast activation in this disease.124 In agreement with this possibility, Tregs restrain Th2-mediated fibroblast activation in murine models of cutaneous fibrosis,82 and low dose IL-2 therapy (which clinically boosts Treg numbers and activation) may alleviate established skin fibrosis in patients with chronic GVHD.125 The links between excessive T cell-driven inflammation and aberrant tissue repair in human disease, along with the pro-reparative functions of Tregs after injury, suggests that Treg augmentation may have utility as a novel treatment modality.
Figure 4-. Regulation of Wound Healing by Classical Treg Suppressive Functions.
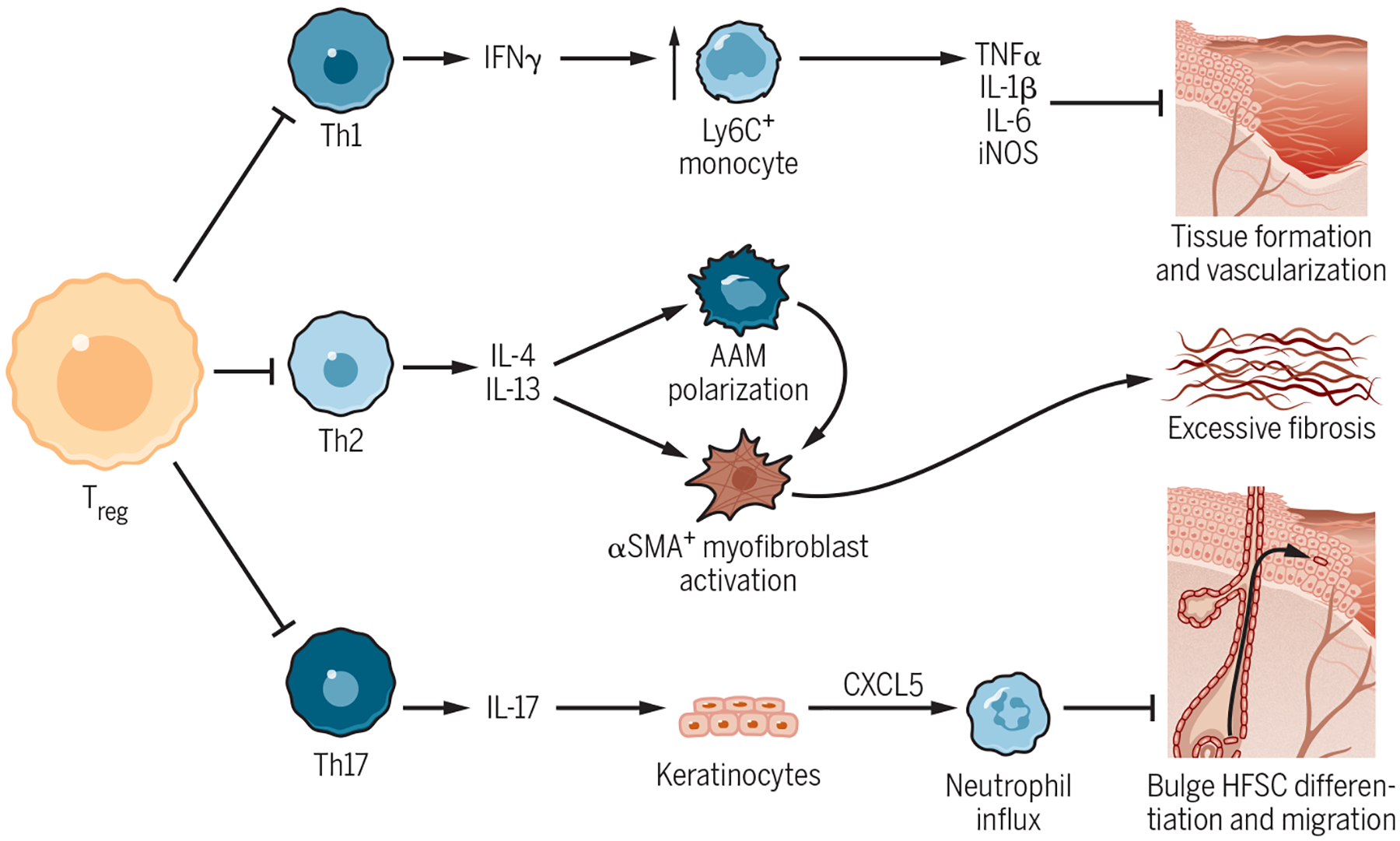
(A) Early after injury, Tregs suppress excessive production of IFN-γ by Th1 cells and thus restrain the accumulation of pro-inflammatory Ly6C+ monocytes/macrophages. These cells impede the transition to the proliferative phase of healing by secreting highly inflammatory cytokines such as TNF-α, IL-1β, and IL-6. (B) Impaired Treg control of type 2 immunity leads to Th2 accumulation in wounded skin and excessive production of IL-4 and IL-13. These cytokines drive collagen deposition by activating pro-fibrotic α-SMA+ myofibroblasts and alternatively activated macrophages. Treg depletion consequently worsens cutaneous fibrosis. (C) After epidermal barrier damage, Tregs promote re-epithelialization by hair follicle stem cells (HFSCs) by suppressing excessive type 3 immunity. Loss of Tregs results in uncontrolled Th17 and neutrophil accumulation, which inhibit the differentiation and migration of HFSCs from the hair follicle bulge into the interfollicular epidermis.
Acknowledgements:
We are grateful to Drs. Ari Molofsky, Dean Sheppard, and Joshua Moreau for thoughtful conversations and valuable suggestions for this manuscript. This work was supported by funding from NIH F30AI147364 and R01AR071944.
Works Cited
- 1.Eming SA, Hammerschmidt M, Krieg T & Roers A Interrelation of immunity and tissue repair or regeneration. Semin. Cell Dev. Biol 20, 517–527 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Ali N & Rosenblum MD Regulatory T cells in skin. Immunology (2017). doi: 10.1111/imm.12791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Blanpain C & Fuchs E Epidermal homeostasis: a balancing act of stem cells in the skin. Nat Rev Mol Cell Bio 10, nrm2636 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cotsarelis G, Sun TT & Lavker RM Label-retaining cells reside in the bulge area of pilosebaceous unit: Implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell (1990). doi: 10.1016/0092-8674(90)90696-C [DOI] [PubMed] [Google Scholar]
- 5.Wang X et al. Principles and mechanisms of regeneration in the mouse model for wound-induced hair follicle neogenesis. Regeneration 2, 169–181 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sun BK, Siprashvili Z & Khavari PA Advances in skin grafting and treatment of cutaneous wounds. Science (80-.) 346, 941–945 (2014). [DOI] [PubMed] [Google Scholar]
- 7.Kim MH et al. Dynamics of neutrophil infiltration during cutaneous wound healing and infection using fluorescence imaging. J. Invest. Dermatol 128, 1812–1820 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kaczmarek A, Vandenabeele P & Krysko DV Necroptosis: The Release of Damage-Associated Molecular Patterns and Its Physiological Relevance. Immunity 38, 209–223 (2013). [DOI] [PubMed] [Google Scholar]
- 9.Nathan C Neutrophils and immunity: Challenges and opportunities. Nature Reviews Immunology (2006). doi: 10.1038/nri1785 [DOI] [PubMed] [Google Scholar]
- 10.Dinarello CA Immunological and inflammatory functions of the interleukin-1 family. 27, 519–550 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Wynn TA & Vannella KM Macrophages in Tissue Repair, Regeneration, and Fibrosis. Immunity 44, 450–462 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mills CD, Kincaid K, Alt JM, Heilman MJ & Hill AM M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J. Immunol 164, 6166–6173 (2000).10843666 [Google Scholar]
- 13.Lucas T et al. Differential Roles of Macrophages in Diverse Phases of Skin Repair. J. Immunol 184, 3964–3977 (2010). [DOI] [PubMed] [Google Scholar]
- 14.Mascré G et al. Distinct contribution of stem and progenitor cells to epidermal maintenance. Nature 489, 257–262 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Page ME, Lombard P, Ng F, Göttgens B & Jensen KB The epidermis comprises autonomous compartments maintained by distinct stem cell populations. Cell Stem Cell (2013). doi: 10.1016/j.stem.2013.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Driskell RR et al. Distinct fibroblast lineages determine dermal architecture in skin development and repair. Nature (2013). doi: 10.1038/nature12783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Darby IA & Hewitson TD Fibroblast Differentiation in Wound Healing and Fibrosis. International Review of Cytology (2007). doi: 10.1016/S0074-7696(07)57004-X [DOI] [PubMed] [Google Scholar]
- 18.Correa-Gallegos D et al. Patch repair of deep wounds by mobilized fascia. Nature 24, 23–31 (2019). [DOI] [PubMed] [Google Scholar]
- 19.Lovvorn HN et al. Relative distribution and crosslinking of collagen distinguish fetal from adult sheep wound repair. in Journal of Pediatric Surgery (1999). doi: 10.1016/S0022-3468(99)90261-0 [DOI] [PubMed] [Google Scholar]
- 20.Caley MP, Martins VLC & O’Toole EA Metalloproteinases and Wound Healing. Adv. Wound Care (2015). doi: 10.1089/wound.2014.0581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eberl G Immunity by equilibrium. Nat. Rev. Immunol 16, 524–532 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Fan X & Rudensky AY Hallmarks of Tissue-Resident Lymphocytes. Cell (2016). doi: 10.1016/j.cell.2016.02.048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.O’Shea J & Paul WE Mechanisms underlying lineage commitment and plasticity of helper CD4 + T cells. Science (80-.) 327, 1098–1102 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allen JE & Wynn TA Evolution of Th2 Immunity: A Rapid Repair Response to Tissue Destructive Pathogens. PLoS Pathog. 7, 5–8 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gieseck RL, Wilson MS & Wynn TA Type 2 immunity in tissue repair and fibrosis. Nat. Publ. Gr 18, 62–76 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Ricardo-Gonzalez R et al. Tissue signals imprint {ILC2} identity with anticipatory function. Nat. Immunol 19, 1093–1099 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rak GD et al. IL-33-Dependent Group 2 Innate Lymphoid Cells Promote Cutaneous Wound Healing. J. Invest. Dermatol 136, 487–496 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nussbaum JC et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature (2013). doi: 10.1038/nature12526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Todd R et al. The eosinophil as a cellular source of transforming growth factor alpha in healing cutaneous wounds. Am. J. Pathol 138, 1307–1313 (1991). [PMC free article] [PubMed] [Google Scholar]
- 30.Nosbaum A et al. Cutting Edge: Regulatory T Cells Facilitate Cutaneous Wound Healing. J. Immunol 196, 2010–2014 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lloyd CM & Snelgrove RJ Type 2 immunity: Expanding our view. Sci. Immunol 3, 1–12 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Dahlgren MW & Molofsky AB Adventitial Cuffs: Regional Hubs for Tissue Immunity. Trends Immunol. 40, 877–887 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hammad H & Lambrecht BN Barrier Epithelial Cells and the Control of Type 2 Immunity. Immunity 43, 29–40 (2015). [DOI] [PubMed] [Google Scholar]
- 34.Van Dyken SJ et al. A tissue checkpoint regulates type 2 immunity. Nat. Immunol 17, 1381–1387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim BS et al. TSLP Elicits IL-33 – Independent Innate Lymphoid Cell Responses to Promote Skin Inflammation. 5, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Salimi M et al. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J. Exp. Med 210, 2939–2950 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dainichi T et al. The epithelial immune microenvironment (EIME) in atopic dermatitis and psoriasis. Nat. Immunol 19, 1286–1298 (2018). [DOI] [PubMed] [Google Scholar]
- 38.Zaiss DMW, Gause WC, Osborne LC & Artis D Emerging functions of amphiregulin in orchestrating immunity, inflammation, and tissue repair. Immunity 42, 216–226 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akaiwa M et al. Localization of human interleukin 13 receptor in non-haematopoietic cells. Cytokine 13, 75–84 (2001). [DOI] [PubMed] [Google Scholar]
- 40.Yin H et al. IL-33 accelerates cutaneous wound healing involved in upregulation of alternatively activated macrophages. Mol. Immunol (2013). doi: 10.1016/j.molimm.2013.05.225 [DOI] [PubMed] [Google Scholar]
- 41.Blériot C et al. Liver-Resident Macrophage Necroptosis Orchestrates Type 1 Microbicidal Inflammation and Type-2-Mediated Tissue Repair during Bacterial Infection. Immunity 42, 145–158 (2015). [DOI] [PubMed] [Google Scholar]
- 42.Morimoto Y et al. Amphiregulin-Producing Pathogenic Memory T Helper 2 Cells Instruct Eosinophils to Secrete Osteopontin and Facilitate Airway Fibrosis. Immunity 49, 134–150.e6 (2018). [DOI] [PubMed] [Google Scholar]
- 43.Van Dyken SJ & Locksley RM Interleukin-4- and Interleukin-13-Mediated Alternatively Activated Macrophages: Roles in Homeostasis and Disease. Annu. Rev. Immunol 31, 317–343 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Knipper JA et al. Interleukin-4 Receptor α Signaling in Myeloid Cells Controls Collagen Fibril Assembly in Skin Repair. Immunity 43, 803–816 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shook BA et al. Myofibroblast proliferation and heterogeneity are supported by macrophages during skin repair. Science (80-.) 362, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Campbell L, Saville CR, Murray PJ, Cruickshank SM & Hardman MJ Local arginase 1 activity is required for cutaneous wound healing. J. Invest. Dermatol 133, 2461–2470 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cai Y et al. Pivotal Role of Dermal IL-17-Producing γδ T Cells in Skin Inflammation. Immunity 35, 596–610 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kobayashi T et al. Homeostatic Control of Sebaceous Glands by Innate Article Homeostatic Control of Sebaceous Glands by Innate Lymphoid Cells Regulates Commensal Bacteria Equilibrium. Cell 1–16 (2019). doi: 10.1016/j.cell.2018.12.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Constantinides MG et al. MAIT cells are imprinted by the microbiota in early life and promote tissue repair. Science (80-.) 366, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Naik S, Bouladoux N, Wilhelm C & Molloy MJ Compartmentalized control of skin immunity by resident commensals. … (2012). [DOI] [PMC free article] [PubMed]
- 51.Linehan JL et al. Non-classical Immunity Controls Microbiota Impact on Skin Immunity and Tissue Repair. Cell 172, 784–796.e18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Harrison OJ et al. Commensal-specific T cell plasticity promotes rapid tissue adaptation to injury. Science (80-.) 363, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li Z et al. Epidermal Notch1 recruits RORγ+ group 3 innate lymphoid cells to orchestrate normal skin repair. Nat. Commun 7, 11394 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nielsen MM, Witherden DA & Havran WL γδ T cells in homeostasis and host defence of epithelial barrier tissues. Nat. Rev. Immunol 17, 733–745 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mathur AN et al. Treg-Cell Control of a CXCL5-IL-17 Inflammatory Axis Promotes Hair-Follicle-Stem-Cell Differentiation During Skin-Barrier Repair. Immunity 50, 655–667.e4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McGeachy MJ, Cua DJ & Gaffen SL The IL-17 Family of Cytokines in Health and Disease. Immunity 50, 892–906 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Boniface K et al. IL-22 Inhibits Epidermal Differentiation and Induces Proinflammatory Gene Expression and Migration of Human Keratinocytes. J. Immunol 174, 3695–3702 (2005). [DOI] [PubMed] [Google Scholar]
- 58.Zheng Y et al. Interleukin-22, a TH17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature 445, 648–651 (2007). [DOI] [PubMed] [Google Scholar]
- 59.Sonnenberg GF, Fouser LA & Artis D Border patrol: Regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol 12, 383–390 (2011). [DOI] [PubMed] [Google Scholar]
- 60.Lai Y et al. The Antimicrobial Protein REG3A Regulates Keratinocyte Proliferation and Differentiation after Skin Injury. Immunity 37, 74–84 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nograles KE et al. Th17 cytokines interleukin (IL)-17 and IL-22 modulate distinct inflammatory and keratinocyte-response pathways. Br. J. Dermatol 159, 1092–1102 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nickoloff BJ et al. Lessons learned from psoriatic plaques concerning mechanisms of tissue repair, remodeling, and inflammation. J. Investig. Dermatology Symp. Proc 11, 16–29 (2006). [DOI] [PubMed] [Google Scholar]
- 63.Ishida Y, Kondo T, Takayasu T, Iwakura Y & Mukaida N The Essential Involvement of Cross-Talk between IFN-γ and TGF-β in the Skin Wound-Healing Process. J. Immunol 172, 1848–1855 (2004). [DOI] [PubMed] [Google Scholar]
- 64.Novak ML & Koh TJ Macrophage phenotypes during tissue repair. J. Leukoc. Biol 93, 875–881 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arnold R, Seifert M, Asadullah K & Volk HD Crosstalk between keratinocytes and T lymphocytes via Fas/Fas ligand interaction: modulation by cytokines. J. Immunol 162, 7140–7 (1999). [PubMed] [Google Scholar]
- 66.Ghosh AK, Yuan W, Mori Y, Chen SJ & Varga J Antagonistic regulation of type I collagen gene expression by interferon-γ and transforming growth factor-β: Integration at the level of p300/CBP transcriptional coactivators. J. Biol. Chem 276, 11041–11048 (2001). [DOI] [PubMed] [Google Scholar]
- 67.Panduro M, Benoist C & Mathis D Tissue Tregs. Annu. Rev. Immunol 34, 609–633 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rubtsov YP et al. Regulatory T Cell-Derived Interleukin-10 Limits Inflammation at Environmental Interfaces. Immunity 28, 546–558 (2008). [DOI] [PubMed] [Google Scholar]
- 69.Josefowicz SZ, Lu L-F & Rudensky AY Regulatory T Cells: Mechanisms of Differentiation and Function. Annu. Rev. Immunol (2012). doi: 10.1146/annurev.immunol.25.022106.141623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scharschmidt TC et al. A Wave of Regulatory T Cells into Neonatal Skin Mediates Tolerance to Commensal Microbes. Immunity 43, 1011–1021 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ali N et al. Regulatory T Cells in Skin Facilitate Epithelial Stem Cell Differentiation. Cell 169, 1119–1129.e11 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rodriguez RS, Pauli M, Neuhaus I & Al E Memory regulatory T cells reside in human skin. J. Clin. Invest 124, 1027–36 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rosenblum MD, Way SS & Abbas AK Regulatory T cell memory. Nature Reviews Immunology (2016). doi: 10.1038/nri.2015.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Collins N et al. Skin CD4+memory T cells exhibit combined cluster-mediated retention and equilibration with the circulation. Nat. Commun (2016). doi: 10.1038/ncomms11514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kim JM, Rasmussen JP & Rudensky AY Regulatory T cells prevent catastrophic autoimmunity throughout the lifespan of mice. Nat. Immunol 8, 191–197 (2007). [DOI] [PubMed] [Google Scholar]
- 76.Halabi-Tawil M et al. Cutaneous manifestations of immune dysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX) syndrome. Br. J. Dermatol (2009). doi: 10.1111/j.1365-2133.2008.08835.x [DOI] [PubMed] [Google Scholar]
- 77.Belkaid Y Role of Foxp3-positive regulatory T cells during infection. Eur. J. Immunol 38, 918–921 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Laidlaw BJ et al. Production of IL-10 by CD4 + regulatory T cells during the resolution of infection promotes the maturation of memory CD8 + T cells. Nat. Immunol 16, 871–879 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shafiani S et al. Pathogen-Specific Treg Cells Expand Early during Mycobacterium tuberculosis Infection but Are Later Eliminated in Response to Interleukin-12. Immunity 38, 1261–1270 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Remedios KA et al. The TNFRSF members CD27 and OX40 coordinately limit T H 17 differentiation in regulatory T cells. Sci. Immunol 3, 2042 (2018). [DOI] [PubMed] [Google Scholar]
- 81.Hall AOH et al. The Cytokines Interleukin 27 and Interferon-γ Promote Distinct Treg Cell Populations Required to Limit Infection-Induced Pathology. Immunity 37, 511–523 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kalekar LA et al. Regulatory T cells in skin are uniquely poised to suppress profibrotic immune responses. Sci. Immunol 4, 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Russler-Germain E, Rengarajan S & Hsieh C-S Antigen-Specific Regulatory T Cell Responses to Intestinal Microbiota. Mucosal Immunol. 10, 1375–86 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Metzger TC & Anderson MS Control of central and peripheral tolerance by Aire. Immunol. Rev 241, 89–103 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Burzyn D et al. A special population of regulatory T cells potentiates muscle repair. Cell 155, 1282–1295 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Cho J, Kuswanto W, Benoist C & Mathis D T cell receptor specificity drives accumulation of a reparative population of regulatory T cells within acutely injured skeletal muscle. 1–7 (2019). doi: 10.1073/pnas.1914848116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Brestoff JR & Artis D Immune regulation of metabolic homeostasis in health and disease. Cell 161, 146–160 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Levine AG et al. Stability and function of regulatory T cells expressing the transcription factor T-bet. Nature 546, 421–425 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Delacher M et al. Genome-wide DNA-methylation landscape defines specialization of regulatory T cells in tissues. Nat. Immunol 18, 1160–1172 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang Y, Su MA & Wan YY An Essential Role of the Transcription Factor GATA-3 for the Function of Regulatory T Cells. Immunity 35, 337–348 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rudra D et al. Transcription factor Foxp3 and its protein partners form a complex regulatory network. Nat. Immunol 13, 1010–1019 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wohlfert EA, Grainger JR & Belkaid Y GATA3 controls Foxp3+ regulatory T cell fate during inflammation in mice. J. Clin . … 121, 4503–4515 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yu F, Sharma S, Edwards J, Feigenbaum L & Zhu J Dynamic expression of transcription factors T-bet and GATA-3 by regulatory T cells maintains immunotolerance. Nat. Immunol 16, 197–206 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Koch MA et al. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat. Immunol 10, 595–602 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Li C et al. TCR Transgenic Mice Reveal Stepwise, Multi-site Acquisition of the Distinctive Fat-Treg Phenotype. Cell 174, 285–299.e12 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Arpaia N et al. A Distinct Function of Regulatory T Cells in Tissue Protection. Cell 162, 1078–1089 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kashiwagi M et al. Direct control of regulatory T cells by keratinocytes. Nat. Immunol 18, 334–343 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ito M et al. Brain regulatory T cells suppress astrogliosis and potentiate neurological recovery. Nature 565, 246–250 (2019). [DOI] [PubMed] [Google Scholar]
- 99.Masopust D & Schenkel JM The integration of T cell migration, differentiation and function. Nat. Rev. Immunol 13, 309–320 (2013). [DOI] [PubMed] [Google Scholar]
- 100.Cipolletta D et al. PPAR-γ is a major driver of the accumulation and phenotype of adipose tissue T reg cells. Nature 486, 549–553 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Minutti CM et al. A Macrophage-Pericyte Axis Directs Tissue Restoration via Amphiregulin-Induced Transforming Growth Factor Beta Activation. Immunity 50, 645–654.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Dial CF, Tune MK, Doerschuk CM & Mock JR FoxP3 Regulatory T Cell Expression of Keratinocyte Growth Factor Enhances Lung Epithelial Proliferation. Am. J. Respir. Cell Mol. Biol 57, 162–173 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Song X et al. Growth Factor FGF2 Cooperates with Interleukin-17 to Repair Intestinal Epithelial Damage. Immunity 43, 488–501 (2015). [DOI] [PubMed] [Google Scholar]
- 104.Hui SP et al. Zebrafish Regulatory T Cells Mediate Organ-Specific Regenerative Programs. Dev. Cell 43, 659–672.e5 (2017). [DOI] [PubMed] [Google Scholar]
- 105.Tiemessen MM et al. CD4+CD25+Foxp3+ regulatory T cells induce alternative activation of human monocytes/macrophages. Proc. Natl. Acad. Sci. U. S. A 104, 19446–19451 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Proto JD et al. Regulatory T Cells Promote Macrophage Efferocytosis during Inflammation Resolution. Immunity 49, 666–677.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Bosurgi L et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science (80-.) 356, 1072–1076 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Zhou X, Bailey-Bucktrout S, Jeker L & Bluestone J Plasticity of CD4+ FoxP3+ T cells- NIH Public Access. Curr Opin Immunol 34, 97–115 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ito M et al. Stem cells in the hair follicle bulge contribute to wound repair but not to homeostasis of the epidermis. Nat. Med (2005). doi: 10.1038/nm1328 [DOI] [PubMed] [Google Scholar]
- 110.Wynn T Cellular and molecular mechanisms of fibrosis. J. Pathol 214, 199–210 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Eming SA et al. Accelerated wound closure in mice deficient for interleukin-10. Am. J. Pathol 170, 188–202 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Travis MA & Sheppard D TGF-β Activation and Function in Immunity. Annu. Rev. Immunol 32, 51–82 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Robertson IB et al. Latent TGF-β-binding proteins. Matrix Biol. 47, 44–53 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Worthington JJ et al. Integrin αvβ8-Mediated TGF-β Activation by Effector Regulatory T Cells Is Essential for Suppression of T-Cell-Mediated Inflammation. Immunity 42, 903–915 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ohnmacht C et al. The microbiota regulates type 2 immunity through {RORγt+} T cells. Science (80-.) 349, 989–993 (2015). [DOI] [PubMed] [Google Scholar]
- 116.DiSpirito JR et al. Molecular diversification of regulatory T cells in nonlymphoid tissues. Sci. Immunol 3, 1–16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rittié L Cellular mechanisms of skin repair in humans and other mammals. J. Cell Commun. Signal 10, 103–120 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ito M & Cotsarelis G Is the Hair Follicle Necessary for Normal Wound Healing? J. Invest. Dermatol 128, 1059–61 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Grice EA et al. Topographical and Temporal Diversity of the Human Skin Microbiome. 1190–1193 (2009). [DOI] [PMC free article] [PubMed]
- 120.Dahlgren MW et al. Adventitial Stromal Cells Define Group 2 Innate Lymphoid Cell Tissue Niches Article Adventitial Stromal Cells Define Group 2 Innate Lymphoid Cell Tissue Niches. Immunity 1–16 (2019). doi: 10.1016/j.immuni.2019.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kramann R et al. Perivascular Gli1+ progenitors are key contributors to injury-induced organ fibrosis. Cell Stem Cell 16, 51–66 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Naik S, Larsen SB, Cowley CJ & Fuchs E Two to Tango: Dialog between Immunity and Stem Cells in Health and Disease. Cell 175, 908–920 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Gabrielli A, Avvedimento E & Krieg T Scleroderma. N. Engl. J. Med 360, 1989–2003 (2009). [DOI] [PubMed] [Google Scholar]
- 124.Frantz C, Auffray C, Avouac J & Allanore Y Regulatory T cells in systemic sclerosis. Front. Immunol 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Whangbo JS et al. Dose-escalated interleukin-2 therapy for refractory chronic graft-versus-host disease in adults and children. Blood Adv. 3, 2550–2561 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]