

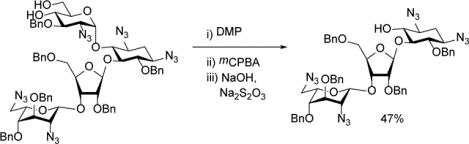
Abstract
To facilitate the synthesis of paromomycin and/or neomycin analogs we describe a cleavage of ring I from paromomycin that proceeds in the presence of azides and affords a glycosyl acceptor for the installation of a modified ring I. A paromomycin 4’,6’-diol is oxidized by the Dess-Martin periodinane followed by m-chloroperoxybenzoic acid. Base treatment then affords a protected pseudodisaccharide, which functions as a glycosyl acceptor. The method should also apply to the cleavage of pyranosyl 4,6-diols from oligosaccharides and glycoconjugates.
Graphical Abstract
Owing to the ever-increasing spread of multidrug-resistant infections there is much interest in the development of new and improved antibacterial agents.1 Next generation aminoglycoside antibiotics in particular are attracting attention because of their many attractive features2–4 including high efficacy and broad spectrum potency, rapid bactericidal potency, minimal protein binding and metabolic stability, lack of drug-related allergy, and absence of interaction with the hosts intestinal microbiome and with other pharmaceutical agents. Indeed, modification of ring I of the 2-deoxystreptamine aminoglycosides paromomycin and/or neomycin, which makes an essential contact with A1408 within the drug binding pocket on the small ribosomal subunit,5 has proven useful in increasing antibacterial activity while decreasing susceptibility to resistance and, in some cases, increasing selectivity for bacterial over eukaryotic ribosomes.6–13 Many modifications to ring I can be made by taking advantage of the extensive reported chemistry of these pseudotrisaccharides (Scheme 1),14, 15 but others are best addressed by a deglycosylation-reglycosylation strategy in which ring I is removed to give a pseudodisaccharide followed by reinstallation of a preformed modified ring I.10, 16 More drastic modifications involving the replacement of ring I by a heteroaromatic ring have to proceed via cleavage of ring I,17 or be prepared by synthesis from 2-deoxystreptamine or its derivatives (Scheme 1).18
Scheme 1.
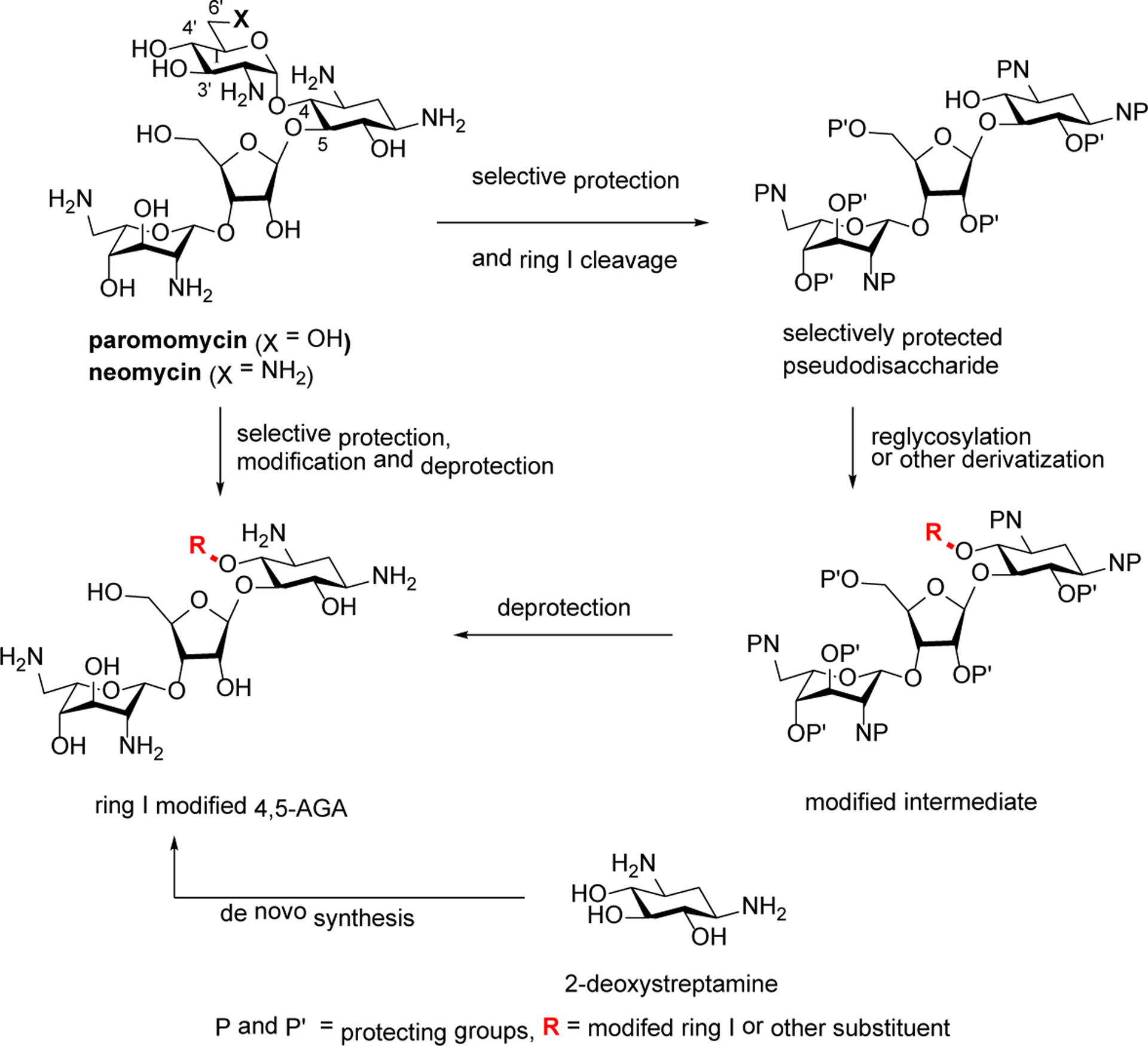
Strategies for the Synthesis of Ring I Modified Derivatives of Paromomycin and Neomycin.
Several routes have been devised for the cleavage of ring I from paromomycin and/or neomycin analogs but all have limitations. The Farmitalia protocol involving diazotization of the 2’-amino group in ring I is effective but is preceded by a low yielding step involving selective protection of all other amino groups in the molecule.10, 16 Removal of ring I initiated by sodium metaperiodate cleavage of the 3’,4’-diol is effective but is typically conducted without protection of the various other hydroxyl groups, thereby necessitating subsequent selective protection steps before reinstallation of ring I.10, 19–21 Finally, reducing metal initiated cleavage of 6’-deoxy-6’-iodo derivatives of paromomycin require installation of the iodide, and are not compatible with azide protection of the multiple amino groups present.17, 22 We describe a protocol for the cleavage of ring I from paromomycin that takes advantage of the selective installation of a 4’,6’-O-benzylidene acetal in ring I and that functions in the presence of multiple azide groups.
Paromomycin was converted through three steps of azide installation, benzylidene acetal formation, and benzylation as described previously to give the fully protected derivative 1.23 Also as described previously,23 exposure of 1 to toluenesulfonic acid in methanol then gave the 4’,6’-diol 2 in excellent yield (Scheme 2). Stirring of 2 with the Dess Martin periodinane (DMP)24 in dichloromethane at room temperature was followed by addition of m-chloroperoxybenzoic acid and stirring for a further 24 h. Finally, the reaction mixture was worked-up by stirring with aqueous sodium hydroxide and sodium thiosulfate and purification by chromatography over silica gel. In this manner the selectively protected pseudodisaccharide 3 was isolated in 47% overall yield from 2 in the form of a white gum (Scheme 2).
Scheme 2.
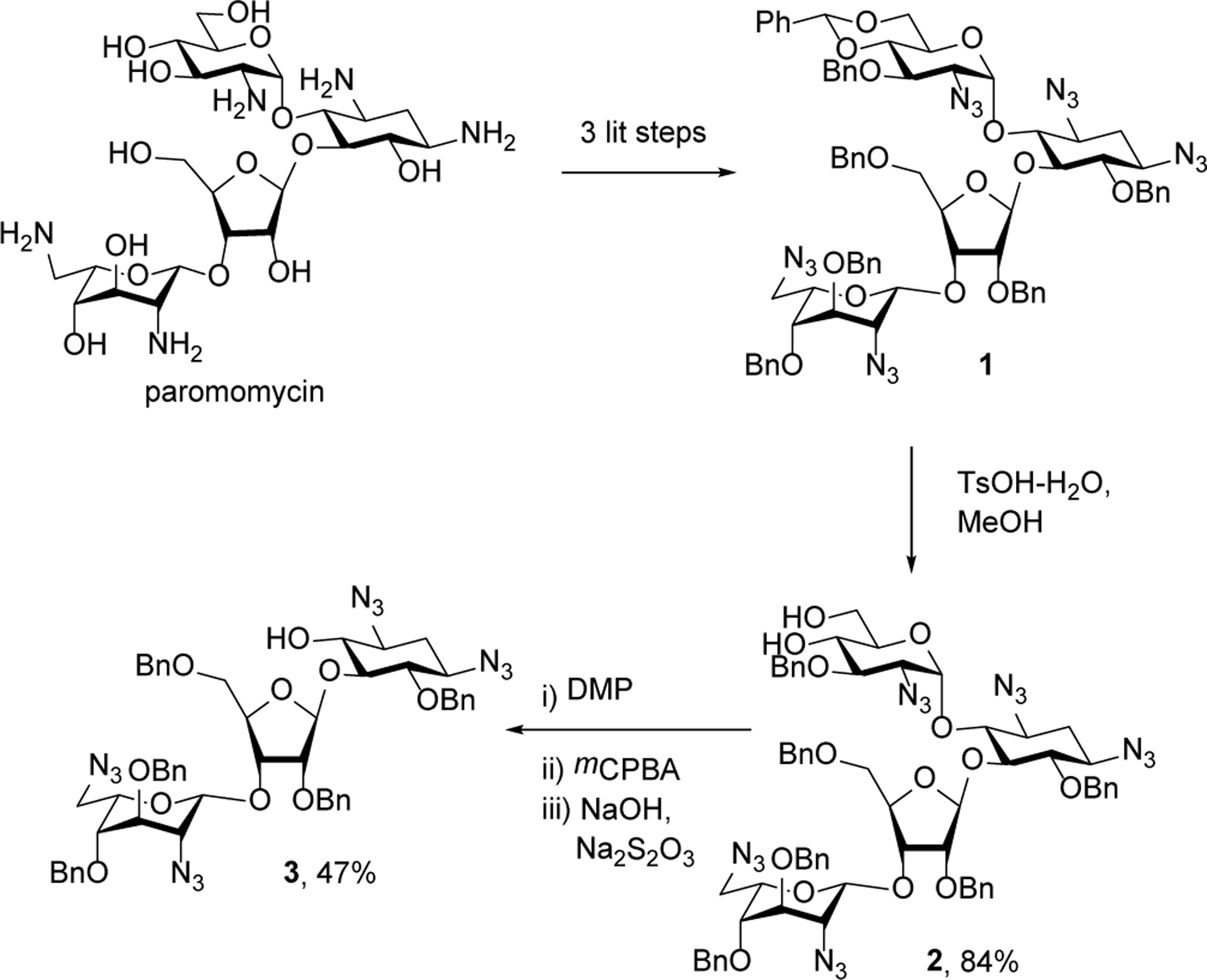
Synthesis of pseudodisaccharide 3 from paromomycin.
We envisage that this oxidative cleavage reaction takes place by oxidation of the 1,3-diol 2 to the corresponding α-formyl ketone, whose enolized form 4 is oxidized by the peracid to afford an α-hydroxy-α-formyl ketone 5. On treatment with base fragmentation of the hemiacetal ensues resulting overall in the cleavage of ring I and formation of 3 (Scheme 3).
Scheme 3.
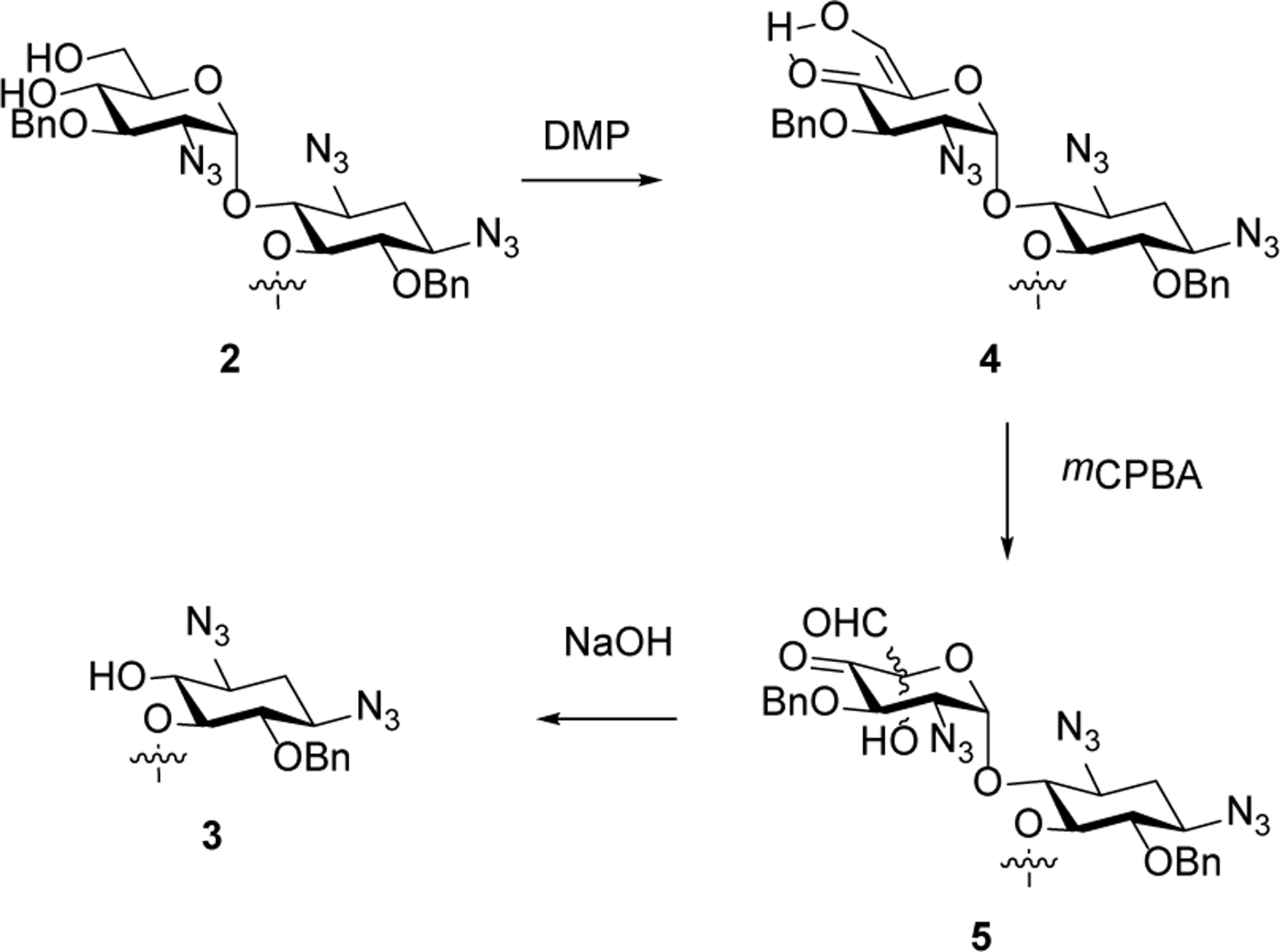
Proposed Mechanism for the Formation of 3 from diol 2.
To establish that 3 is suitable for the installation of a highly modified paromomycin ring I, we prepared a 3,4-dideoxy glycopyranosyl donor 11 as outlined in Scheme 4 from the readily available galactenitol derivative 6 and 1-octanol.25 Noteworthy in this preparation is the ability to carry the primary sulfonate ester through the hydrolysis of 10 to 11 in hot aqueous trifluoroacetic acid.
Scheme 4.
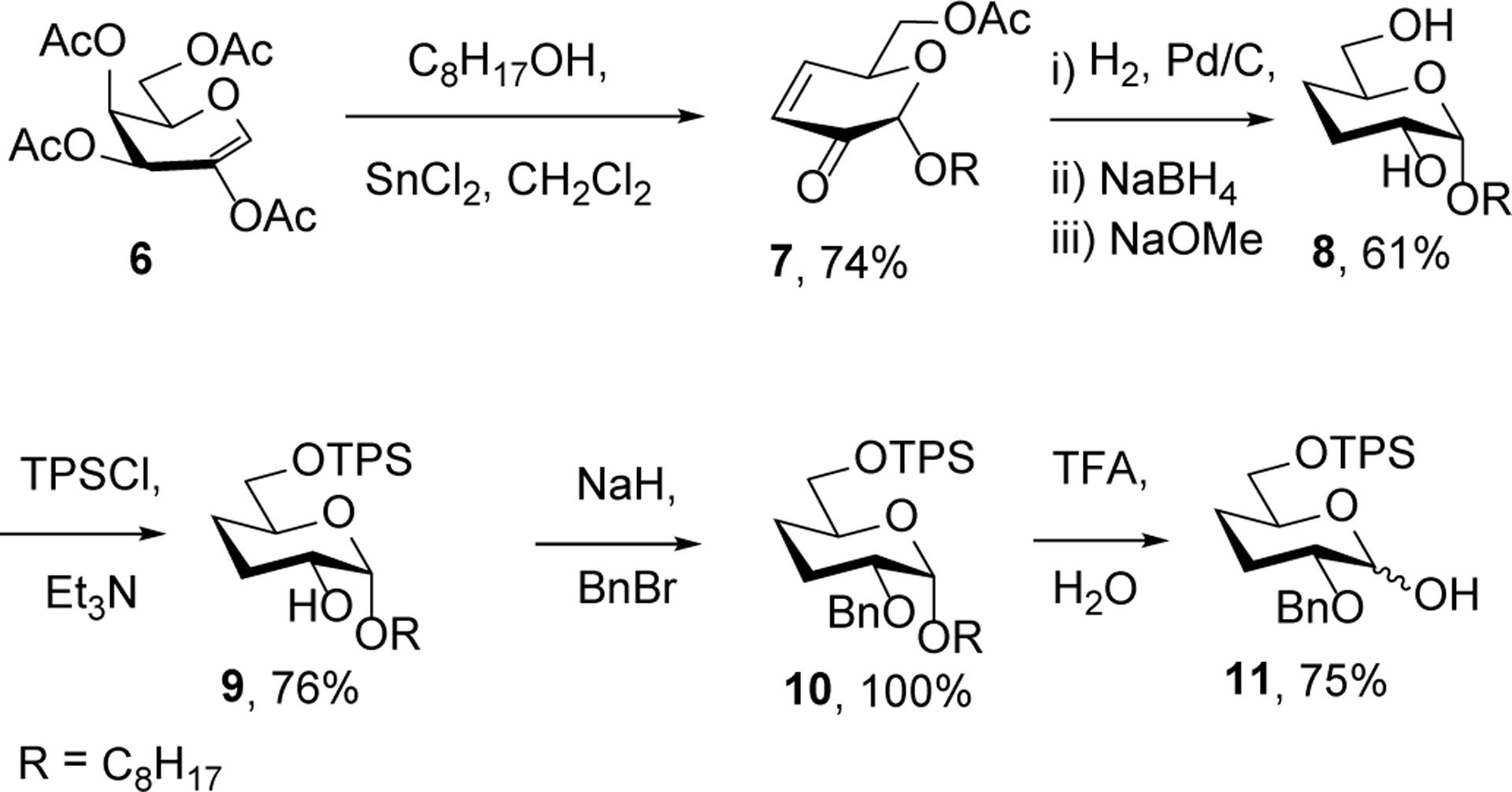
Glycosyl donor preparation.
Subsequent coupling of 3 with 11 to give 12 was achieved smoothly in 95% yield as a single α-anomer under dehydrative conditions using diphenyl sulfoxide and triflic anhydride as reagents (Scheme 5).26 Although we have not made a study of the mechanism of this highly selective glycosylation reaction, it is reasonable in the light of current knowledge to suggest that it proceeds by displacement of an intermediate a β-O-glycosyl diphenylsulfonium ion.27–29
Scheme 5.
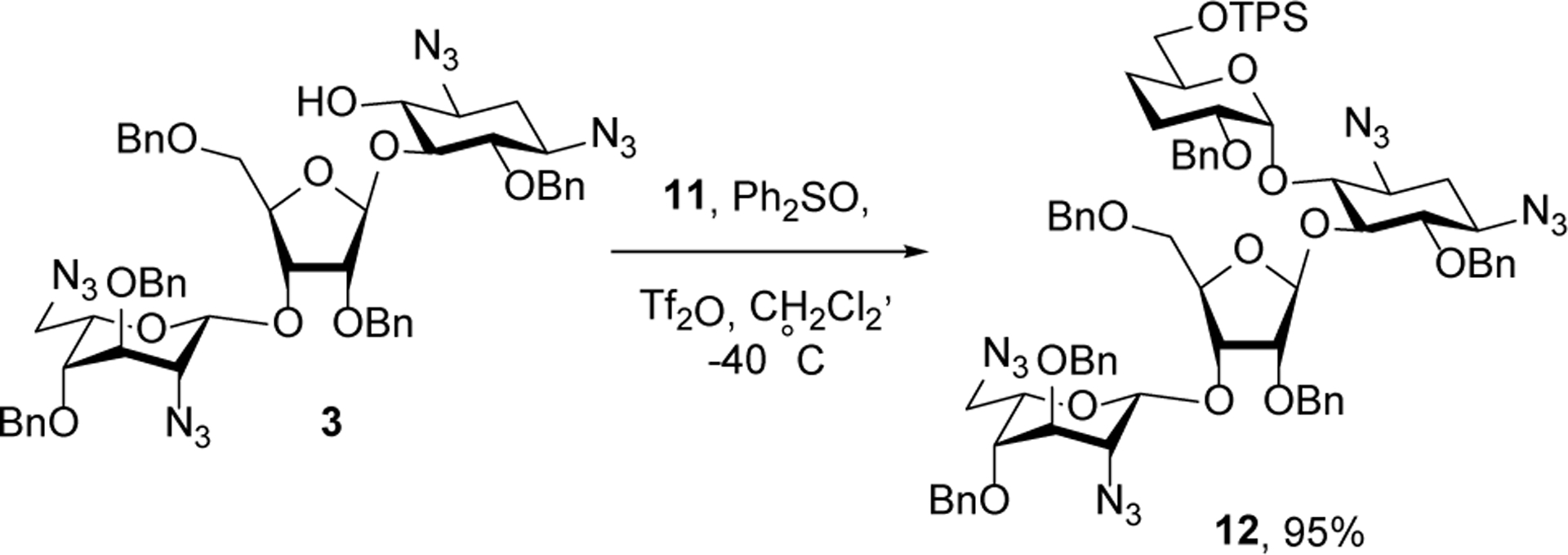
Glycosylation of Acceptor 3.
Conclusion
We have provided a straightforward method for the synthesis of the functioning glycosyl acceptor 3 from paromomycin in five simple steps. While the focus here is on controlled removal of a pyranosyl ring from an aminoglycoside antibiotic, the chemistry presented should be readily extended to the cleavage of a pyranoside ring capable of benzylidene acetal formation from the reducing terminus of any readily available oligosaccharide or glycoconjugate.
Experimental
GENERAL EXPERIMENTAL
High resolution mass spectra were collected on a Waters LCT Premier XE ESI-TOF mass spectrometer. Optical rotations were measured using an automated polarimeter. NMR spectra were collected at 400, 500, or 600 MHz as indicated and assigned by 1D and 2D techniques including COSY, HSQC, HMBC, and TOCSY.
1,3‐Diazido‐1,3‐dideamino‐5‐O‐[2’,5’‐dibenzyl‐3’‐O‐(2”,6”‐diazido‐2”,6”‐ dideamino‐3”,4”‐di‐O‐benzyl‐α‐L‐idopyranosyl)‐β‐D‐ribofuranosyl]‐6‐O‐benzyl‐2‐deoxystreptamine (3).
DMP (1.58 g, 3.73 mmol) was added to a stirred solution of 223 (2.0 g, 1.55 mmol) in dry DCM (25 mL). The reaction mixture was stirred for 1.5 h at room temperature before mCPBA (402 mg, 2.3 mmol) was added, after which stirring was continued for 24 h. 3 N NaOH (5 mL) and aqueous Na2S2O3 solution (5 mL) were added to the reaction mixture and stirred for additional 1 h. After completion, the reaction mixture was diluted with EtOAc and the organic layer was washed with aqueous NaHCO3 followed by brine, dried with Na2SO4, and concentrated. The crude product was purified via silica gel chromatography eluting with 10% to 20% EtOAc in hexanes to give 3 (739 mg, 47%) as a white gum;[α]D25= +47.1 (c 5.2, DCM); 1H NMR (400 MHz, CDCl3) δ 7.50 – 7.14 (m, 23H, ArH), 7.13 – 7.02 (m, 2H, ArH), 5.28 (s, 1H, H-1’), 5.06 (s, 1H, OH), 4.95 (s, 1H, H-1”), 4.88 (d, J = 11.1 Hz, 1H, OCH2Ph), 4.84 – 4.75 (m, 2H, H-3’, OCH2Ph), 4.70 – 4.54 (m, 3H, OCH2Ph), 4.51 – 4.27 (m, 5H, H-4’, OCH2Ph), 4.23 (d, J = 11.7 Hz, 1H, OCH2Ph), 4.02 – 3.89 (m, 2H, H-2 ‘, H-5”), 3.86 – 3.77 (m, 2H, H-5’, H-3”), 3.75 – 3.60 (m, 2H, H-5’, H-6”), 3.49 (s, 1H, H-2”), 3.46 – 3.29 (m, 3H, H-1, H-3, H-5), 3.29 – 3.09 (m, 4H, H-4, H-6, H-4”, H-6”), 2.06 (dt, J = 13.4, 4.3 Hz, 1H, H-2), 1.22 (q, J = 13.0 Hz, 1H, H-2); 13C{1H} NMR (101 MHz, CDCl3) δ 137.9 (ArC), 137.5 (ArC), 137.2 (ArC), 128.7 (ArC), 128.6 (ArC), 128.5 (ArC), 128.4 (ArC), 128.3 (ArC), 127.9 (ArC), 127.8 (ArC), 127.7 (ArC), 127.3 (ArC), 127.2 (ArC), 106.4 (C-1’), 98.2 (C-1”), 85.0 (C-5), 83.4 (C-4), 80.64 (C-2’), 80.57 (C-4’), 75.3 (OCH2Ph), 74.9 (C-6), 74.34 (C-5”), 74.27 (C-3’), 73.7 (OCH2Ph), 73.1 (C-3”), 72.6 (OCH2Ph), 72.1 (OCH2Ph), 72.0 (OCH2Ph), 71.6 (C-4”), 67.5 (C-5’), 60.6 (C-3), 59.5 (C-1), 57.8 (C-2”), 51.0 (C-6”), 32.3 (C-2). ESI-HRMS: m/z calcd. for C52H56N12NaO10 [M+Na]+ 1031.4140; found, 1031.4126.
Octyl 6-O-acetyl-3,4-dideoxy-α-D-glycero-hex-3-enopyranosid-2-ulose (7).
A solution of 2,3,4,6-tetra-O-acetyl-1,5-anhydro-D-lyxo-hex-1-enitol25 6 (500 mg, 1.5 mmol) and octanol (0.45 mL, 3.0 mmol) in dry DCM (30 mL) was cooled to −78 °C and SnCl4 (0.2 mL, 1.8 mmol) was added. The reaction mixture was stirred at 0 °C for 2 h before dilution with DCM. The resulting organic solution was washed twice with saturated aqueous NaHCO3 solution, and then with brine. The organic extract was dried with sodium sulfate, filtered, and concentrated. The crude product was purified by gradient chromatography over silica gel (eluent: 5% to 8% EtOAc/hexanes) to yield 7 (335 mg, 74%) as a colorless oil; [α]D25= −3.6 (c 1.1, DCM); 1H NMR (400 MHz, CDCl3) δ 6.94 (dd, J = 10.6, 1.6 Hz, 1H), 6.17 (dd, J = 10.6, 2.5 Hz, 1H), 4.86 (s, 1H), 4.74 (tt, J = 4.6, 2.1 Hz, 1H), 4.35 (dd, J = 11.6, 5.5 Hz, 1H), 4.25 (dd, J = 11.6, 4.6 Hz, 1H), 3.80 (dt, J = 9.6, 6.8 Hz, 1H), 3.62 (dt, J = 9.6, 6.6 Hz, 1H), 2.10 (s, 3H), 1.68 – 1.52 (m, 2H), 1.38 – 1.18 (m, 10H), 0.93 – 0.81 (m, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 188.5, 170.6, 147.0, 126.3, 97.7, 69.9, 66.9, 64.6, 31.8, , 29.4, 29.3, 29.2, 25.9, 22.6, 20.7, 14.1. ESI-HRMS: m/z calcd. for C16H26NaO5 [M+Na]+ 321.1678; found, 321.1665.
Octyl 3,4-dideoxy-α-D-erythro-hexopyranoside (8).
Compound 7 (2.4 g, 7.47 mmol) dissolved in EtOAc (100 mL) and 10% Pd/C (200 mg) was subjected to catalytic hydrogenation at 45 psi for 3 h. The reaction mixture was filtered through Celite®. The crude compound (1.97 g, 83%) was pure enough to be used in the next step. A solution of this crude product (500 mg, 1.66 mmol) in dry MeOH (50 mL) was cooled to −30 °C and NaBH4 (70 mg, 1.8 mmol) was added. After 2 h, NaOMe (108 mg, 2 mmol) was added, and the reaction was continued for 1 h at room temperature. The solution was neutralized by the addition of glacial acetic acid and concentrated. The crude product was purified by gradient chromatography over silica gel (eluent: 40% to 50% EtOAc/hexanes) to yield 8 (335 mg, 74%, 61% overall from 7) as a white solid; [α]D25= +70.5 (c 1.0, DCM); 1H NMR (400 MHz, CDCl3) δ 4.77 (d, J = 3.6 Hz, 1H), 3.81 – 3.67 (m, 2H), 3.64 – 3.53 (m, 2H), 3.54 – 3.37 (m, 2H), 2.03 (dd, J = 7.4, 5.0 Hz, 1H), 1.94 (d, J = 11.2 Hz, 1H), 1.88 (dq, J = 12.2, 4.0 Hz, 1H), 1.72 (ddd, J = 13.3, 12.0, 4.3 Hz), 1.68 – 1.53 (m, 3H), 1.46 (tdd, J = 13.4, 11.6, 4.0 Hz, 1H), 1.40 – 1.18 (m, 10H), 0.94 – 0.83 (m, 3H); 13C NMR{1H} (101 MHz, CDCl3) δ 98.1, 68.8, 68.1, 67.8, 65.4, 31.8, 29.6, 29.4, 29.2, 27.1, 26.2, 26.0, 22.6, 14.1. ESI-HRMS: m/z calcd. for C14H28NaO4 [M+Na]+ 283.1885; found, 283.1882.
Octyl 3,4-dideoxy-6-O-trisyl-α-D-erythro-hexopyranoside (9).
Compound 8 (1.3 g, 5.0 mmol) and triethylamine (2.8 mL, 20 mmol) were dissolved in dry DCM (15 mL) and the resulting solution was stirred and ice cooled before trisyl chloride (1.9 g, 6.3 mmol) was added portionwise. The reaction mixture was stirred for 48 h at rt under argon, then diluted with ethyl acetate and the organic layer was washed with aqueous NaHCO3 followed by brine, dried with Na2SO4, and concentrated in vacuo. The crude product was purified by silica gel chromatography eluting with 5% to 15% EtOAc in hexanes to give 9 (2.0 g, 76%) as a white solid; [α]D25= +34.7 (c 1.1, DCM); 1H NMR (400 MHz, CDCl3) δ 7.18 (s, 2H), 4.71 (d, J = 3.6 Hz, 1H), 4.14 (hept, J = 6.8 Hz, 2H), 4.01 – 3.89 (m, 3H), 3.70 (dt, J = 9.7, 6.7 Hz, 1H), 3.55 (tt, J = 11.2, 4.6 Hz, 1H), 3.39 (dt, J = 9.7, 6.5 Hz, 1H), 2.91 (hept, J = 6.9 Hz, 1H), 1.93 – 1.79 (m, 2H, H-3), 1.76 – 1.65 (m, 2H), 1.57 (q, J = 6.8 Hz, 2H), 1.48 – 1.37 (m, 1H), 1.35 – 1.20 (m, 28H), 0.93 – 0.83 (m, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 153.7, 150.8, 123.7, 97.9, 70.8, 68.0, 67.7, 66.1, 34.2, 31.8, 29.6, 29.5, 29.4, 29.2, 27.0, 26.6, 26.2, 24.7, 24.7, 23.5, 22.6, 14.1. ESI-HRMS: m/z calcd. for C29H50NaO6S [M+Na]+ 549.3229; found, 549.3247.
Octyl 2-O-benzyl-3,4-dideoxy-6-O-trisyl-α-D-erythro-hexopyranoside (10).
Compound 9 (1.3 g, 2.5 mmol) was dissolved in dry THF (15 mL) and the resulting solution was stirred and ice cooled before benzyl bromide (1.5 mL, 12.3 mmol) and NaH (60% dispersion in oil) (200 mg, 5 mmol) was added portionwise. The reaction mixture was stirred for 3 h at 0 °C under argon and 3 h at rt, then diluted with ethyl acetate and the organic layer was washed with aqueous NaHCO3 followed by brine, dried with Na2SO4, and concentrated in vacuo. The crude product was purified via silica gel chromatography eluting with 3% to 15% EtOAc in hexanes to give 10 (1.5 g, quant.) as a white solid; [α]D25= +35.3 (c 0.5, DCM); 1H NMR (400 MHz, CDCl3) δ 7.37 – 7.26 (m, 5H), 7.17 (s, 2H), 4.77 (d, J = 3.2 Hz, 1H), 4.56 (q, J = 12.3 Hz, 2H), 4.13 (hept, J = 6.8 Hz, 2H), 4.04 – 3.89 (m, 3H), 3.64 (dt, J = 9.8, 7.0 Hz, 1H), 3.46 – 3.35 (m, 2H), 2.90 (hept, J = 6.7 Hz, 1H), 1.98 – 1.79 (m, 2H), 1.76 – 1.67 (m, 1H), 1.66 – 1.53 (m, 2H), 1.47 – 1.17 (m, 29H), 0.92 – 0.85 (m, 3H); 13C NMR{1H} (101 MHz, CDCl3) δ 153.6, 150.8, 128.3, 127.6, 123.7, 96.5, 74.7, 70.8, 70.6, 67.8, 65.9, 34.2, 31.8, 29.6, 29.5, 29.3, 26.8, 26.2, 24.7, 24.7, 23.5, 23.4, 22.7, 14.1. ESI-HRMS: m/z calcd. for C36H56NaO6S [M+Na]+ 639.3695; found, 639.3712.
2-O-Benzyl-3,4-dideoxy-6-O-trisyl-α,β-D-erythro-hexopyranose (11).
Compound 10 (263 mg, 0.43 mmol) was suspended in 50% aqueous trifluoroacetic acid (8 mL) and stirred at 60 °C for 6 h. The reaction mixture was co-evaporated with toluene three times. The crude product was purified via silica gel chromatography eluting with 5% to 15% EtOAc in hexanes to give 11 (160 mg, 75%) as a white solid (α:β = 1:0.5); (Major isomer)1H NMR (400 MHz, CDCl3) δ 7.40 – 7.24 (m, 5H), 5.17 (d, J = 2.4 Hz, 1H), 4.62 – 4.52 (m, 2H), 4.21 – 4.09 (m, 4H, H-5), 4.06 – 3.97 (m, 2H), 3.45 (ddd, J = 11.3, 5.1, 3.2 Hz, 1H), 2.91 (hept., J = 6.9 Hz, 1H), 2.85 (d, J = 7.3 Hz, 1H), 1.96 – 1.79 (m, 2H), 1.54 – 1.33 (m, 2H), 1.26 (d, J = 6.9 Hz, 18H); 13C{1H} NMR (101 MHz, CDCl3) δ 153.7, 150.8, 128.5, 127.9, 127.8, 127.7, 127.6, 123.8, 123.7, 90.9, 74.3, 70.7, 70.6, 65.9, 34.2, 29.6, 26.1, 24.8, 24.7, 22.6. ESI-HRMS: m/z calcd. for C28H40NaO6S [M+Na]+ 527.2443; found, 527.2433.
1,3,2”’,6”’-Tetraazido-6,2”,5”,3”’,4”’-penta-O-benzyl-1,3,2’,2”’,6”’-pentadeamino-2’-benzyloxy-3’,4’-dideoxy-6’-O-trisyl paromomycin (12).
Donor 11 (90 mg, 0.18 mmol), diphenyl sulfoxide (58 mg, 0.23 mmol) and TTBP (88 mg, 0.35 mmol) were charged to a round bottom flask, co-evaporated with toluene three times and dried in vacuo overnight. The flask was purged with argon and the mixture was dissolved in dry DCM (1 mL) and stirred with activated 4 Å M.S. for 1 h. The reaction mixture was cooled to −78 °C and treated with Tf2O (39 μL, 0.23 mmol) then stirred for 1.5 h at −40 °C. A solution of compound 3 (90 mg, 0.08 mmol) in dry DCM (1 mL) was stirred with activated 4 Å M.S. for 3 h before added to the reaction mixture. The reaction mixture was stirred for 3 h at −40 °C before the reaction was quenched with triethylamine (0.2 mL), diluted with EtOAc and washed with aqueous NaHCO3 and brine then concentrated. The crude product was purified using silica gel column chromatography (eluent: 5% - 15% EtOAc/hexanes) give 12 (128 mg, 95%) as a white foam; [α]D25= +28.6 (c 0.8, DCM); 1H NMR (600 MHz, CDCl3) δ 7.44 – 7.13 (m, 32H, ArH), 5.87 (d, J = 3.4 Hz, 1H, H-1’), 5.54 (d, J = 3.2 Hz, 1H, H-1”), 4.82 (d, J = 11.2 Hz, 1H, OCH2Ph), 4.76 (d, J = 2.0 Hz, 1H, H-1”’), 4.72 – 4.55 (m, 4H, CH2Ph), 4.52 – 4.48 (m, 2H, OCH2Ph), 4.48 – 4.44 (m, 2H, OCH2Ph), 4.42 (d, J = 11.9 Hz, 1H, CH2Ph), 4.38 – 4.34 (m, 2H, OCH2Ph), 4.34 – 4.24 (m, 5H, H-5’, H-3”, H-4”, OCH2Ph), 4.20 – 4.07 (m, 2H, o-CH(CH3)2), 4.04 – 4.01 (m, 1H, H-2”), 4.00 (t, J = 5.0 Hz, 1H, H-6’), 3.95 (dd, J = 10.0, 4.7 Hz, 1H, H-6’), 3.78 – 3.69 (m, 4H, H-4, H-5, H-3”’, H-5”’), 3.66 (d, J = 9.6 Hz, 1H, H-5”’), 3.62 – 3.53 (m, 2H, H-5”, H-6”’), 3.48 (ddt, J = 12.6, 9.2, 4.6 Hz, 1H, H-3), 3.39 – 3.32 (m, 3H, H-1, H-2’, H-2”’), 3.22 (t, J = 8.8 Hz, 1H, H-6), 3.14 (s, 1H, H-4”’), 2.98 (dd, J = 12.8, 4.5 Hz, 1H, H-6”’), 2.90 (hept, J = 7.0 Hz, 1H, p-CH(CH3)2), 2.20 (dt, J = 13.2, 4.8 Hz, 1H, H-2), 1.89 (qd, J = 12.7, 12.0, 4.1 Hz, 1H, H-3’), 1.78 – 1.70 (m, 2H, H-3’, H-4’), 1.43 – 1.33 (m, 2H, H-2, H-4’), 1.26 (dd, J = 6.9, 3.6 Hz, 18H, 3 × CH(CH3)2); 13C{1H} NMR (151 MHz, CDCl3) δ 153.6 (ArC), 150.8 (ArC), 138.5 (ArC), 137.9 (ArC), 137.8 (ArC), 137.0 (ArC), 136.9 (ArC), 128.7 (ArC), 128.5 (ArC), 128.4 (ArC), 128.3 (ArC), 128.2 (ArC), 128.0 (ArC), 127.9 (ArC), 127.7 (ArC), 127.5 (ArC), 127.4 (ArC), 127.3 (ArC), 123.7 (ArC), 107.2 (C-1”), 98.2 (C-1”’), 95.3 (C-1’), 83.0 (C-6), 81.9 (C-4”), 81.5 (C-5), 81.2 (C-2”), 76.0 (C-4), 75.6 (C-3”), 74.8 (C-2’), 74.7 (OCH2Ph), 74.0 (C-5”), 73.1 (OCH2Ph), 73.0 (C-3”), 72.7 (OCH2Ph), 72.5 (OCH2Ph), 71.9 (OCH2Ph), 71.6 (C-4”’), 71.2 (OCH2Ph), 70.9 (C-6’), 70.8 (C-5”), 66.0 (C-5’), 60.4 (C-1), 59.6 (C-3), 57.6 (C-2”’), 51.0 (C-6”’), 34.2 (CH(CH3)2), 32.1 (C-2), 29.7 (CH(CH3)2), 29.6 (C-4’), 26.7 (CH(CH3)2), 24.8 (CH(CH3)2), 22.7 (C-3’). ESI-HRMS: m/z calcd. for C80H94N12NaO15S [M+Na]+ 1517.6580; found, 1517.6512.
Supplementary Material
Acknowledgments.
We thank the NIH (AI123352) for support of this work.
Footnotes
Supporting Information. The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.
Copies of the 1H and 13C NMR spectra of all new compounds (PDF)
References
- 1.CDC. Antibiotic Resistance Threats in the United States, 2019; US Dept of Health and Human Services, CDC: Atlanta, GA, 2019. [Google Scholar]
- 2.Böttger EC; Crich D, Aminoglycosides: Time for Resurrection of a Neglected Class of Antibacterials? ACS Infect. Dis 2020, 6, 168–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Serio AW; Keepers TR; Andrews L; Krause KM, Aminoglycoside Revival: Review of a Historically Important Class of Antimicrobials Undergoing Rejuvenation. EcoSalPlus 2018, doi: 10.1128/ecosalplus.ESP-0002-201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takahashi Y; Igarashi M, Destination of Aminoglycoside Antibiotics in the ‘Post-Antibiotic Era’. J. Antibiotics 2018, 71, 4–14. [DOI] [PubMed] [Google Scholar]
- 5.Carter AP; Clemons WM; Brodersen DE; Morgan-Warren RJ; Wimberly BT; Ramakrishnan V, Functional Insights from the Structure of the 30S Ribosomal Subunit and its Interactions with Antibiotics. Nature 2000, 407, 340–348. [DOI] [PubMed] [Google Scholar]
- 6.Duscha S; Boukari H; Shcherbakov D; Salian S; Silva S; Kendall A; Kato T; Akbergenov R; Perez-Fernandez D; Bernet B; Vaddi S; Thommes P; Schacht J; Crich D; Vasella A; Böttger EC, Identification and Evaluation of Improved 4’-O-(Alkyl) 4,5-Disubstituted 2-Deoxystreptamines as Next Generation Aminoglycoside Antibiotics. mBio 2014, 5, 10.1128/mBio.01827-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Matsushita T; Chen W; Juskeviciene R; Teo Y; Shcherbakov D; Vasella A; Böttger EC; Crich D, Influence of 4′-O-Glycoside Constitution and Configuration on Ribosomal Selectivity of Paromomycin. J. Am. Chem. Soc 2015, 137, 7706–7717. [DOI] [PubMed] [Google Scholar]
- 8.Mandhapati AR; Yang G; Kato T; Shcherbakov D; Hobbie SN; Vasella A; Böttger EC; Crich D, Structure-Based Design and Synthesis of Apramycin-Paromomycin Analogues. Importance of the Configuration at the 6′-Position and Differences Between the 6′-Amino and Hydroxy Series. J. Am. Chem. Soc 2017, 139, 14611–14619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matsushita T; Sati GC; Kondasinghe N; Pirrone MG; Kato T; Waduge P; Kumar HS; Cortes Sanchon A; Dobosz-Bartoszek M; Shcherbakov D; Juhas M; Hobbie SN; Schrepfer T; Chow CS; Polikanov YS; Schacht J; Vasella A; Böttger EC; Crich D, Design, Multigram Synthesis, and in Vitro and in Vivo Evaluation of Propylamycin: A Semisynthetic 4,5-Deoxystreptamine Class Aminoglycoside for the Treatment of Drug-Resistant Enterobacteriaceae and Other Gram-Negative Pathogens. J. Am. Chem. Soc 2019, 141, 5051–5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sati GC; Sarpe VA; Furukawa T; Mondal S; Mantovani M; Hobbie SN; Vasella A; Böttger EC; Crich D, Modification at the 2’-Position of the 4,5-Series of 2-Deoxystreptamine Aminoglycoside Antibiotics to Resist Aminoglycoside Modifying Enzymes and Increase Ribosomal Target Selectivity. ACS Infect. Dis 2019, 5, 1718–1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Battistini C; Franceschi G; Zarini F; Cassinelli G; Arcamone F; Sanfilippo A, 3’,4’-Dideoxyparamomycin and Their Analogues. J. Antiobiotics 1982, 25, 98–101. [DOI] [PubMed] [Google Scholar]
- 12.Hanessian S; Saavedra OM; Vilchis-Reyes MA; Maianti JP; Kanazawa H; Dozzo P; Matias RD; Serio A; Kondo J, Synthesis, Broad Spectrum Antibacterial Activity, and X-ray Co-crystal Structure of the Decoding Bacterial Ribosomal A-site with 4’-Deoxy-4’-Fluoro Neomycin Analogs. Chem. Sci 2014, 5, 4621–4632. [Google Scholar]
- 13.Hanessian S; Giguere A; Grzyb J; Maianti JP; Saavedra; Aggen JB; Linsell MS; Goldblum AA; Hildebrandt DJ; Kane TR; Dozzo P; Gliedt MJ; Matias RD; Feeney LA; Armstrong ES, Toward Overcoming Staphylococcus aureus Aminoglycoside Resistance Mechanisms with a Functionally Designed Neomycin Analogue. ACS Med. Chem. Lett 2011, 2, 924–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berkov-Zrihen Y; Fridman M, Synthesis of Aminoglycosides. In Modern Synthetic Methods in Carbohydrate Chemistry; From Monosaccharides to Complex Glycoconjugates, Werz DB; Vidal S, Eds. Wiley: Weinheim, 2014; pp 161–190. [Google Scholar]
- 15.Chandrika NT; Garneau-Tsodikova S, Comprehensive Review of Chemical Strategies for the Preparation of New Aminoglycosides and their Biological Activities. Chem. Soc. Rev 2018, 47, 1189–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cassinelli G; Julita P; Arcamone F, Semisynthetic Aminoglycoside Antibiotics II. Synthesis of Analogues of Paromomycin Modified in the Glucosamine Moiety. J. Antibiotics 1978, 31, 382–384. [DOI] [PubMed] [Google Scholar]
- 17.Hanessian S; Adhikari S; Szychowski J; Pachamuthu K; Wang X; Migawa MT; Griffey RH; Swayze EE, Probing the Ribosomal RNA A-site with Functionally Diverse Analogues of Paromomycin—Synthesis of Ring I Mimetics. Tetrahedron 2007, 63, 827–846. [Google Scholar]
- 18.Ding Y; Hofstadler SA; Swayze EE; Risen L; Griffey RH, Design and Synthesis of Paromomycin-Related Heterocycle-Substituted Aminoglycoside Mimetics Based on a Mass Spectrometry RNABinding Assay. Angew. Chem. Int. Ed 2003, 42, 3409–3412. [DOI] [PubMed] [Google Scholar]
- 19.Hanessian S; Takamoto T; Masse R, Oxidative Degradations Leading to Novel Biochemical Probes and Synthetic Intermediates. J. Antibiotics 1975, 28, 835–836. [DOI] [PubMed] [Google Scholar]
- 20.Hanessian S; Takamoto T; Masse R; Patil G, Aminoglycoside Antibiotics: Chemical Conversion of Neomycin B, Paromomycin, and Lividomycin B into Bioactive Pseudosaccharides Can. J. Chem 1978, 56, 1482–1491. [Google Scholar]
- 21.Zárate SG; Bastida A; Santana AG; Revuelta J, Synthesis of Ring II/III Fragment of Kanamycin: A New Minimum Structural Motif for Aminoglycoside Recognition. Antibiotics 2019, 8, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pathak R; Böttger EC; Vasella A, Design and Synthesis of Aminoglycoside Antibiotics to Selectively Target 16S Ribosomal RNA Position 1408. Helv. Chim. Acta 2005, 88, 2967–2984. [Google Scholar]
- 23.Pathak R; Perez-Fernandez D; Nandurdikar R; Kalapala SK; Böttger EC; Vasella A, Synthesis and Evaluation of Paromomycin Derivatives Modified at C(4’). Helv. Chim. Acta 2008, 91, 1533–1552. [Google Scholar]
- 24.Dess PB; Martin JC, Readily Accessible 12-I-5 Oxidant for the Conversion of Primary and Secondary Alcohols to Aldehydes and Ketones. J. Org. Chem 1983, 48, 4155–4156. [Google Scholar]
- 25.Manzano VE; Repetto E; Uhrig ML; Barath M; Varela O, Synthesis of 2,3,4,6-Tetra-O-acetyl-1,5-anhydro-D-lyxo-hex-1-enitol and Its Conversion into a Hex-3-enopyranosid-2-ulose Analogue of Levoglucosenone. Carbohydr. Chem.: Proven Synth. Methods 2012, 1, 295–301. [Google Scholar]
- 26.Garcia BA; Poole JL; Gin DY, Direct Glycosylations with 1-Hydroxy Glycosyl Donors using Trifluoromethanesulfonic Anhydride and Diphenyl Sulfoxide. J. Am. Chem. Soc 1997, 119, 7597–7598. [Google Scholar]
- 27.Garcia BA; Gin DY, Dehydrative Glycosylation with Activated Diphenyl Sulfonium Reagents. Scope, Mode of C(1)-Hemiacetal Activation, and Detection of Reactive Glycosyl Intermediates. J. Am. Chem. Soc 2000, 122, 4269–4279. [Google Scholar]
- 28.Fascione MA; Brabham R; Turnbull WB, Mechanistic Investigations into the Application of Sulfoxides in Carbohydrate Synthesis. Chem. Eur. J 2016, 22, 3916–3928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adero PO; Amarasekara H; Wen P; Bohé L; Crich D, The Experimental Evidence in Support of Glycosylation Mechanisms at the SN1-SN2 Interface. Chem. Rev 2018, 118, 8242–8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.