

Abstract
Peroxisome proliferator-activated receptor γ (PPARγ) is a transcriptional coactivator that binds to a diverse range of transcription factors. PPARγ coactivator 1 (PGC-1) coactivators possess an extensive range of biological effects in different tissues, and play a key part in the regulation of the oxidative metabolism, consequently modulating the production of reactive oxygen species, autophagy, and mitochondrial biogenesis. Owing to these findings, a large body of studies, aiming to establish the role of PGC-1 in the neuromuscular system, has shown that PGC-1 could be a promising target for therapies targeting neuromuscular diseases. Among these, some evidence has shown that various signaling pathways linked to PGC-1α are deregulated in muscular dystrophy, leading to a reduced capacity for mitochondrial oxidative phosphorylation and increased reactive oxygen species (ROS) production. In the light of these results, any intervention aimed at activating PGC-1 could contribute towards ameliorating the progression of muscular dystrophies. PGC-1α is influenced by different patho-physiological/pharmacological stimuli. Natural products have been reported to display modulatory effects on PPARγ activation with fewer side effects in comparison to synthetic drugs. Taken together, this review summarizes the current knowledge on Duchenne muscular dystrophy, focusing on the potential effects of natural compounds, acting as regulators of PGC-1α.
KEY WORDS: Muscular dystrophy, Natural product, Peroxisome proliferator-activated receptor γ coactivator 1α, PPARγ activation, Reactive oxygen species, Mitochondrial oxidative phosphorylation
Abbreviations: AAV, adeno-associated virus; AMP, adenosine monophosphate; AMPK, 5′ adenosine monophosphate-activated protein kinase; ASO, antisense oligonucleotides; ATF2, activating transcription factor 2; ATP, adenosine triphosphate; BMD, Becker muscular dystrophy; cGMP, cyclic guanosine monophosphate; CnA, calcineurin a; COPD, chronic obstructive pulmonary disease; CREB, cyclic AMP response element-binding protein; DAGC, dystrophin-associated glycoprotein complex; DGC, dystrophin–glycoprotein complex; DMD, Duchenne muscular dystrophy; DRP1, dynamin-related protein 1; DS, Down syndrome; ECM, extracellular matrix; EGCG, epigallocatechin-3-gallate; ERRα, estrogen-related receptor alpha; FDA, U. S. Food and Drug Administration; FGF, fibroblast growth factor; FOXO1, forkhead box class-O1; GABP, GA-binding protein; GPX, glutathione peroxidase; GSK3b, glycogen synthase kinase 3b; HCT, hydrochlorothiazide; HDAC, histone deacetylase; HIF-1α, hypoxia-inducible factors; IL, interleukin; iPSCs, induced pluripotent stem cells; LDH, lactate dehydrogenase; MCP-1, monocyte chemoattractant protein-1; MD, muscular dystrophy; MEF2, myocyte enhancer factor 2; MSCs, mesenchymal stem cells; MyoD, myogenic differentiation; NADPH, nicotinamide adenine dinucleotide phosphate; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; NMJ, neuromuscular junctions; NO, nitric oxide; NOS, NO synthase; p38 MAPK, p38 mitogen-activated protein kinase; PDGF, platelet derived growth factor; PGC-1, peroxisome proliferator-activated receptor γ coactivator 1; PPARγ, peroxisome proliferator-activated receptor γ; ROS, reactive oxygen species; SIRT1, silent mating type information regulation 2 homolog 1; SOD, superoxide dismutase; SPP1, secreted phosphoprotein 1; TNF-α, tumor necrosis factor-α; UCP, uncoupling protein; VEGF, vascular endothelial growth factor
Graphical abstract
This review summarizes the current knowledge on Duchenne muscular dystrophy, focusing on the potential effects of natural compounds, acting as regulators of PGC-1α.
1. Introduction
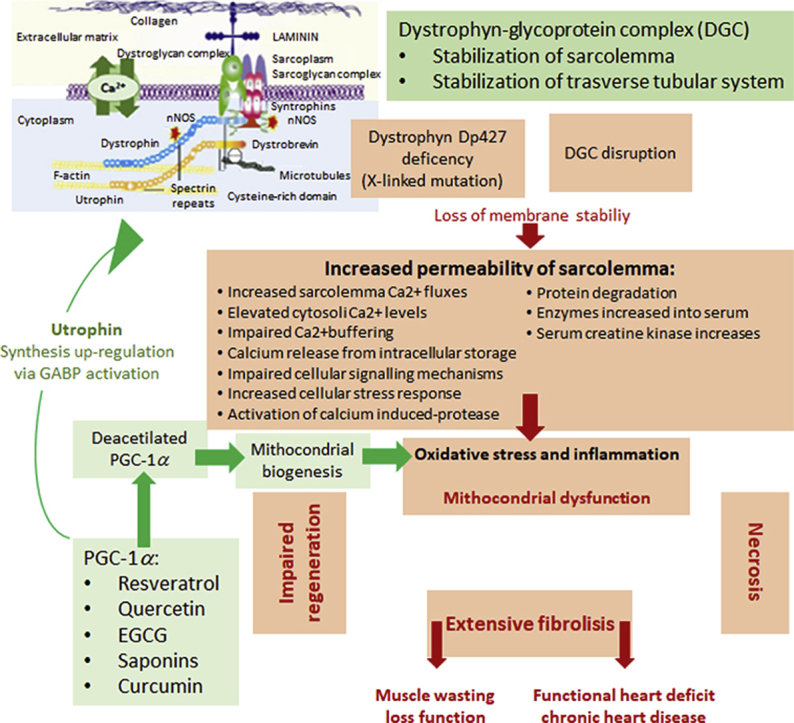
Peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1α (PGC-1α) is an important coactivator of several nuclear receptors regulating mitochondrial function in various tissues including brain, heart, skeletal muscle, and liver1. PGC-1α was initially discovered as one of the PPARγ-binding proteins present in brown adipose tissue as a response to cold treatment. PGC-1 coactivators were reported to possess a vast range of biological effects in different tissues. Indeed, PGC-1 transcriptional coactivators were revealed to have an essential role in physiological adaptation via many signaling cascades2. PGC-1α regulates nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathway by modulating inflammatory processes in muscle cells, and the targeting of PGC-1α in chronic diseases may thus reduce inflammation3. PGC-1α has been found to act as a reactive oxygen species (ROS) scavenging enzyme regulator that contributes to the survival of neurons4. More to the point, in earlier reports, PGC-1 coactivators were found to possess an important role in skeletal muscle biology by inducing mitochondrial biogenesis, muscle fiber-type switching4,5, and functional angiogenesis in skeletal muscle6 (Fig. 1). Indeed, PGC-1 was reported to enhance GA-binding protein (GABP) which is an important transcription factor controlling the genes involved in forming neuromuscular junctions (NMJ)7. Furthermore, GABP activation has been shown to induce utrophin promoter activity in muscle cells and in muscle tissues8.
Figure 1.
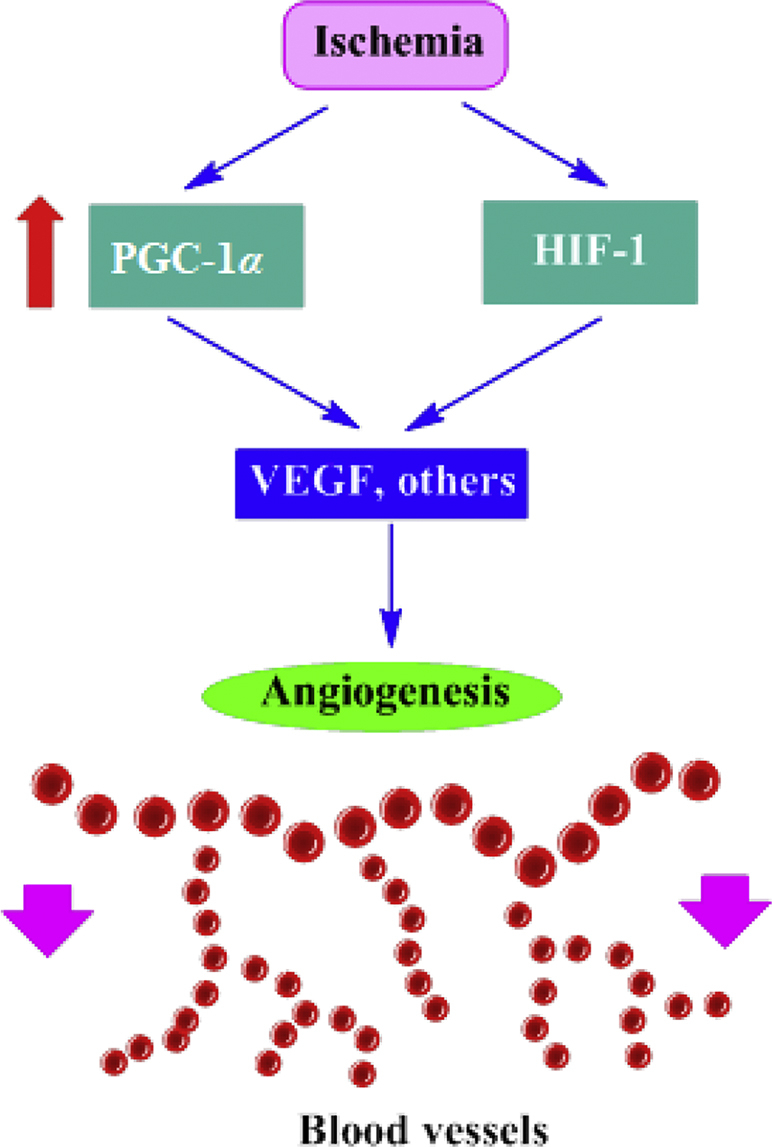
Speculative model of the role of PGC-1α in the regulation of angiogenesis during exercise and in response to ischemia.
A variety of studies have looked into the PPARγ-activating effects of natural products derived from medicinal plants. Resveratrol, honokiol, biochanin A, genistein, sargahydroquinoic acid, amorphastilbol, sargaquinoic acid, magnolol, amorfrutin 1, and amorfrutin B were demonstrated to display modulatory effects on PPARγ activation with fewer side-effects compared to synthetic drugs9. Therefore, in this review, we aimed to summarize the current knowledge on muscular dystrophy (MD), focusing on the potential effects of natural compounds which act as regulatory agents on PGC-1α.
2. Duchene muscular dystrophy (DMD)
Degenerative disorders targeting muscles, which arise from genetic anomalies in the expression of genes in muscle tissues, are known as MDs10. MDs are a set of diseases indicated by the extensive wasting of skeletal muscles due to mutations in genes responsible for linkage formation between the cytoskeleton and the basal lamina11,12. Dystrophin protein (427 kDa) is a cytoplasmic protein which links with numerous other proteins in the formation of the dystrophin-associated glycoprotein complex (DAGC) responsible for hooking up the cytoskeleton of muscle fibers with the surrounding extracellular matrix (ECM) by means of the cell membrane11. DAGC connects muscle fiber cytoskeletal structures with the extracellular matrix and thus ensures the structural integrity of muscle fibers13,14. MDs are considered to be a group of heterogeneous disorders, of which the most severe and common form is DMD15, named in 1868 after Guillame Duchenne, a renowned neurologist who described 13 boys affected by this disorder16. Despite the passing of two centuries from its first description, no cure has been found for DMD to date and it remains a fatal condition. DMD is accepted as an X-linked recessive disorder caused by a dystrophin gene mutation which leads to a loss in expression of functional dystrophin, a cytoskeletal protein responsible for facilitating the functionality, stability, and strength of myofibres18. The key characteristic of DMD is irreversible and extreme skeletal muscle degeneration associated with physical incapacitation. Moreover, several patients also suffer from weakness of the intercostal muscles and diaphragm which leads to terminal respiratory failure before 25 years of age10. The consequences of dystrophin inexpression in cardiac purkinje fibers and cardiomyocytes give rise to progressive cardiomyopathy and arrhythmia, leading to heart failure in approximately 10% DMD patients10,19. DMD affects approximately 1:5000 newborn males and occurs in one in every 3500–5000 males born across the world20,21.
DMD's prevalence has been reported as 19.5 cases in 100,000 live male births in the UK. The incidence in the USA is 15.9 cases per 100,00022, 23, 24. A recent study reported that the methods for diagnosing DMD have remained the same since around 201018. DMD is sometimes considered to be an X-linked myopathy, leading to progressive wasting of respiratory and locomotor muscles. The resultant chronic ventilatory failure has been identified as a major cause of death25. Absence or deficiency of dystrophin may lead to progressive wasting of skeletal muscle and lethal heart failure.
Effective treatment and overall care for DMD is required to prevent the death of children during adolescence or early adulthood. Despite the exploration of several therapies, only corticosteroids showed effective benefits17. Novel therapeutic approaches based on prior studies in animal models are currently undergoing clinical experimentation with encouraging results. These include therapies aiming at restoring dystrophin expression and others aiming at compensating for the deleterious consequences of its absence in skeletal muscle. Description of novel therapeutic approaches is far beyond the scope of this review26.
Read-through, exon skipping, and vector-mediated genetic and cell therapies have been used to restore the expression of dystrophin, whereas pharmacological treatments include anti-inflammatory, anti-fibrotic, antioxidant agents, myostatin pathway inhibition, neuronal nitric oxide synthase pathway enhancement, and utrophin upregulation therapies. Read-through therapy applies to 10% of patients with nonsense mutations, resulting in a premature stop codon in dystrophin mRNA27.
Of the compounds that can read through premature nonsense codons, ataluren (PTC124) is currently available online in Europe28.
In patients with DMD, exon-skipping, applicable to 80% of mutations, can correct the altered reading frame by targeting the exon to be skipped with antisense oligonucleotides (ASOs). Skipping a gene would hypothetically be of therapeutic relevance for various mutation series29 as DMD is one of the largest genes known with 79 exons30,31. A functional, shorter, dystrophin is produced by this process, effectively converting a severe DMD phenotype to a milder Becker muscular dystrophy (BMD) phenotype. The major ASO developed to skip exon 51 was eteplirsen32.
Conditional approval was granted to eteplirsen by the U.S. Food and Drug Administration (FDA) in 2016. Exon skipping therapies have grown to target other exons with time. Dystrophin protein production is restored in DMD by means of vector-mediated gene therapy, delivering micro/mini domains of dystrophin through either viral or non-viral vectors21,33, 34–35. Clinical trials are currently underway for adeno-associated virus (AAV) vectors, finalized for micro-dystrophin gene transfer36.
Cell therapy involves the transplanting of either cells provided by healthy donors or genetically corrected autologous cells, both capable of producing functional dystrophin37. Myoblasts38, CD133+ stem cells39, and mesoangioblasts40 are undergoing clinical trials as potential therapeutic candidates, which are chosen from a wider range of viable cell types. Of the cell therapy options, investigations are currently ongoing on induced pluripotent stem cells (iPSCs) taken from DMD, umbilical cord mesenchymal stem cells, and bone marrow-derived autologous stem cells. Human iPS-derived myogenic cells have been transplanted into mdx mice (the most popular animal model for DMD carrying a point mutation in DMD gene), resulting in human-derived dystrophin-positive muscle fibers and an improvement in muscle strength41.
CRISPR/Cas9 technology has been used to induce frame shifting, exon knock-in, and exon skipping in patient-derived human iPS cells, raising the possibility of ex-vivo gene correction followed by autologous cell transplantation for DMD patients42, 43, 44. However, there are serious limitations on treating DMD patients with current cell therapy technology, including limits on cell availability, low survival, and migration rates for injected cells, the risk of tumor formation, and the immune response to donated cells, with no effective treatment available at present for the prevention of the progression and occurrence of this lethal disease condition13,22,45,46.
Pharmacological therapy represents an additional fundamental approach mainly utilized to limit complications, i.e., inflammation, atrophy, and fibrosis, generally associated with the dystrophic process, which can worsen the evolution of the disease and severely limit the effectiveness of other therapies such as gene or cell therapy. In particular, it has been shown that the evolution to fibrosis in DMD is mainly promoted by an excessive inflammatory response47. Several therapeutic strategies, including prednisone, deflazacort, and NF-κB inhibitors, have been used to address inflammation in combination with cell therapy and exon skipping, with promising results48,49.
A novel steroid analog, vamorolone, which lacks hormonal or immunosuppressive effects, is also under recent investigation for its safety and efficacy50.
Relevant results have been also obtained with nitric oxide (NO) donors and deacetylase inhibitors targeting events downstream of the genetic defect in both a mouse model of limb girdle muscular dystrophy and in adult dystrophic patients. In particular, the use of nonsteroidal drugs (hydrochlorothiazide, HCT) which act as NO donors has been shown to counteract the progression of the dystrophic process in a mouse model of limb girdle MD and in humans51,52.
Studies in animal models have also reported that insufficient dystrophin deregulates histone deacetylase (HDAC) activity, and this is followed by progression of the disease through activation of compensatory regeneration and/or fibroadipogenic degeneration, thus supporting histone deacetylase inhibitors as potentially effective pharmacological interventions to counteract disease progression in dystrophic patients. The beneficial effects of long lasting HDAC inhibitor exposure have been found in mdx mice α-sarcoglycan null mice in a model of limb girdle muscular dystrophy53.
The effects of HDAC inhibitor givinostat have recently been investigated in DMD patients and a phase 3, randomized, double-blind, placebo-controlled study is currently ongoing54.
Important emerging research highlights that deregulation of NO signaling to HDAC contributes to the pathogenesis of the disease. It has been found that the absence of dystrophin at the sarcolemma in mdx mice downregulates NO synthase (NOS), leading to the deficient S-nitrosylation of HDAC2, which controls the follistatin gene, and this event may ultimately promote muscle degeneration.
Direct inhibition of HDAC2 by HDAC inhibitors, or inactivation by either NO donors, may lead to the derepression of follistatin, thus promoting myogenesis and disease amelioration55.
These results indicate that HDAC, with HDAC2 in particular, is an important common pharmacological target for distinct pharmacological interventions (for histone deacetylase inhibitors, see the other review56).
3. The PGC-1 coactivators
Coactivators can regulate target gene expression by specifically affecting protein–protein interactions through known transcription factors possessing DNA-binding activity. Therefore, they act as transcriptional regulators which do not directly bind to target gene promoters. For example, myogenic differentiation (MyoD) or PPARγ can induce the differentiation of muscle or adipose cells57.
Transcription may be enhanced by association with RNA polymerase machinery, or by altering the chromatin structure in target gene promoters57.
A coactivator may sometimes interact with several transcription factors and vice versa57. Although these interactions are versatile, they depend on certain protein interacting interfaces and signaling cues that promote the activation of transcription factors58.
The expression of PGC-1 coactivators was first implicated in thermogenesis and regulation of energy metabolism49. However, studies have concluded that it acts in a broader context, and the overexpression of PGC-1 promotes mitochondrial biogenesis as well as key mitochondrial functions57,59. In fact, these coactivators are important in maintaining the homeostasis between glucose, lipids, and energy60. PGC-1 is also involved in other pathological conditions, such as obesity, cardiomyopathy, and neurodegeneration58.
PGC-1α was the first member of the PGC-1 family identified. It was found as a PPARγ-interacting protein in brown adipose tissue. PGC-1β is another member of this family and the closest homolog of PGC-1α. Other members have limited homology58.
Overall, studies on Pgc-1α transgenic mice have showed remarkable tissue effects due to its overexpression, thus stimulating subsequent analysis of the role of its physiological expression in fundamental mechanisms in skeletal muscle and fat61.
In particular, PGC-1α has been found to exert a role in brown adipose tissue, unlike transdifferentiation. Moreover, PGC-1 coactivators were found to be important in differentiation-induced mitochondrial biogenesis59.
PGC-1α has interactions with a wide range of transcription factors, including nuclear respiratory factors, nuclear hormone receptors, and muscle-specific transcription factors, reacting to environmental stimuli60.
Summermatter et al.62 reported that PGC-1α is responsible for the estrogen-related-α-dependent expression of lactate dehydrogenase B (LDH B) in skeletal muscle, and for the repression of lactate dehydrogenase A (LDH A). Therefore, PGC-1α coordinates lactate homeostasis, alters the composition of the LDH complex, and prevents the increase of lactase in blood during exercise.
ROS, such as superoxides, can damage DNA, lipids, and proteins, and are the originators of ischemia–reperfusion injury, aging, and neurodegenerative diseases, such as Alzheimer's disease, Parkinson's disease, and Huntington's disease. St-Pierre et al.63 reported that PGC-1α, which has the ability to increase the activity of mitochondrial electrons, is a regulator of ROS metabolism. Therefore, new therapies should be investigated for controlling these pathophysiological conditions and thus inducing PGC-1α in the brain. While this is not an easy task, PGC-1α is inducible in many tissues and responds to important metabolic pathways of calcium and cyclic adenosine monophosphate (AMP) signaling63. In fact, Zheng et al.64 identified PGC-1α as a promising factor in the early treatment of Parkinson's disease, as PGC-1α are underexpressed in these patients. The capacity of PGC-1α to control energy homeostasis indicates its possible suitability as a target for antidiabetic or antiobesity drugs60,65.
The involvement of PGC-1 in controlling mitochondrial biogenesis and energy metabolism was hotly pursued after its discovery in the 1990s66. The three PGC-1 coactivators were reported to control the mitochondrial metabolism in different ways despite their structural similarities, and thus different modes of action. PGC-1α is induced by various stimuli; whereas PGC-1β is stimuli-unresponsive and maintains basal mitochondrial function; the action of the third PGC-1 member, PGC-1-related coactivator, is apparently limited to regulating gene expression and mitochondrial metabolism in highly proliferating cells. PGC-1 coactivators bind to various transcription factors or nuclear receptors which orchestrate target genes expression67.
The role of the transcriptional coactivator PGC-1 in oxidative stress, inflammation, and angiogenesis is discussed in the following sub-sections, along with its mechanisms of action.
3.1. The role PGC-1α in oxidative stress
PGC-1α and PGC-1β have emerged as major players that integrate signaling pathways and exert protective actions against oxidative stress. They are also recognized as key factors in many oxidative stress response programs.
In particular, PGC-1α can serve as a master regulator of the oxidative metabolism and mitochondrial biogenesis, thus acting as a key regulator of ROS primarily generated by mitochondria. It is widely reported that PGC-1α modulates ROS scavenging enzymes, including the mitochondrial proteins manganese superoxide dismutase 1 and 2 (SOD1 and SOD2) and the uncoupling proteins 1 and 2 (UCP1 and UCP2)66,68. Indeed, overexpression of PGC-1α results in an increased expression of SOD2 in SH-SY5Y neuroblastoma cells69, and depletion of PGC-1α by siRNAs prevents the upregulation of SOD1 and SOD2, as well as glutathione peroxidase 1 (GPX1), another ROS scavenging enzyme, together with UCP1 and UCP2, indicating that PGC-1α controls cytoprotective responses63,70.
Furthermore, PGC-1 (both α and β) gene expression increases alongside antioxidant defences, under induction of oxidative stress by H2O2 treatment of mouse embryonic fibroblasts; conversely, PGC-1 depletion prevents the increased expression of genes responsible for ROS detoxification in response to H2O263, suggesting a protective role of PGC-1 under effects of oxidative stress.
In many diseases, overproduction of ROS by dysfunctional mitochondria has been associated with decreased PGC-1α expression. For example, PGC-1α levels were found to be lower in the kidneys of diabetic rats; while by inducing PGC-1α overexpression, ROS generation was inhibited via the modulation of dynamin-related protein 1 (DRP1)-mediated mitochondrial dynamics, with a consequent improvement in glomerular mesangial cell function71.
Interestingly, in Down syndrome (DS), a neurodevelopmental disease associated with mitochondrial dysfunction and oxidative stress72, down-regulation of PGC-1α protein levels and activity has been found in fibroblasts as well as in neural cells taken from the hippocampus of DS mouse73,74. DS is a genetic disease characterized by cognitive impairment associated with early neurodegeneration and aging as well as muscular weakness75.
PGC-1α activation is regulated by three systems: PPARs, 5′ adenosine monophosphate-activated protein kinase (AMPK), and silent mating type information regulation 2 homolog 1 (SIRT1)76. AMPK phosphorylation promotes expression and activation of PGC-1α77; similarly, SIRT1 deacetylates and activates PGC-1α, thereby regulating mitochondrial biogenesis78.
NO has been found to activate PGC-1α through AMPK or in a P53-dependent manner, with the possible involvement of multiple pathways simultaneously79. NO has also been found to prompt the expression of SIRT1 deacetylase inducer, acting via cyclic guanosine monophosphate (cGMP)80,81. Thus, the alteration of AMPK/SIRT1 pathways, proven to occur in DS74,82, has been suggested to account for a decrease in PGC-1α activity, and therefore for mitochondrial dysfunction and ROS, which play central roles in the pathogenesis of DS72 and in that of neurodegenerative diseases, including Alzheimer's, Huntington's, and Parkinson's diseases75,83. Activation of PGC-1α by pharmacological agents has been proven to improve oxidative stress and clinical phenotype in oxidative stress-related diseases. For instance, nutraceuticals including resveratrol, administered via SIRT1 induction, and thiazolidinediones, which induce PPARγ activation, have been reported to counteract oxidative stress and resultant mitochondrial dysfunction in mouse models of neurodegenerative disease84,85. In addition, polyphenols, resveratrol, and epigallocatechin-3-gallate (EGCG), have been demonstrated to reverse mitochondrial dysfunction and reduce oxidative stress in DS cells by acting through the PGC-1α/SIRT1/AMPK axis74,86. Metformin, which activates PGC-1α via AMPK induction, has recently been shown to restore the mitochondrial network and to counteract mitochondrial dysfunction and ROS generation in heart fibroblasts with chromosome 21 trisomy87.
Notably, several signaling pathways related to PGC-1α activation/function are deregulated in MD, which in turn results in a reduced capacity for mitochondrial oxidative phosphorylation88 and the detoxification of ROS89. In the light of these results, an early pharmacological intervention aimed at activating PGC-1α and thus reducing oxidative stress and mitochondrial dysfunction could potentially limit muscle damage and degeneration.
3.2. PGC-1 and inflammation
Inflammation is a tissue process consisting of a series of molecular, cellular, and vascular phenomena with defensive purposes against physical, chemical, or biological aggression90. As a process, it is focused on a specific area, though exceptions may occur in the case of systemic inflammation. In addition, inflammation is an immediate and unspecific response which can facilitate the development of a specific immune response. The inflammatory process is characterized by a migration of immune cells to the inflammatory focus. As a consequence of the inflammation, vasodilatation occurs along with an increase in the permeability of the blood vessels near the affected area, facilitating the arrival of leukocytes to the place of inflammation and the influx of other mediator molecules89,91. The ultimate goal of inflammation is to eliminate or inhibit the agent causing the infection or cell damage, and to allow the organism to recover to normal conditions, restoring the functionality of the affected tissue or organ92. Inflammation is receiving increasing consideration due to its likely involvement in the development of various disorders, including MD93, 94, 95. Since PGC-1 contributes to cellular respiration and mitochondrial biogenesis, its adequate function is essential for skeletal muscle. In this way, repression of PGC-1 and its associated inflammatory responses are present in many skeletal muscle diseases96. In addition, PGC-1 also induces the expression and activation of several antioxidant enzymes, including catalase, Mn-superoxide dismutase (Mn-SOD), and GPX, contributing to the improvement of oxidative damage and inflammation59,97.
Studies performed in different organs such as the heart, liver or kidneys evidenced that LPS injection significantly represses expression of Pgc-1α mRNA98, 99, 100. In skeletal muscle, acute treatment with LPS or palmitate seems to exert a dual regulation, with increased PGC-1 expression in the short-term, followed by long-term repression101,102. Using myotubes (C2C12) and mouse primary myotubes as cell models, chronic treatment with tumor necrosis factor-α (TNF-α) has been found to significantly decrease expression of PGC-1 (both α and β isoforms)103. In accordance with these data, chronic exposure to cigarette smoke resulted in TNF-α-mediated down-regulation of PGC-1α in C2C12 cells and in the soleus muscle of mice104. In an interesting study, patients with advanced chronic obstructive pulmonary disease (COPD), with features of muscle wastage and elevated levels of TNF-α, showed lower expression of PGC-1α in vastus lateralis muscle biopsies with respect to healthy smoking control subjects105. Taken together, this indicates that chronic inflammation leads to a repression of PGC-1 coactivators.
Conversely, diverse studies have reported that PGC-1 could prevent inflammation. In this manner, muscle-specific PGC-1α deficient mice presented increased interleukin (IL)-6 in both circulating blood and in skeletal muscle106. A study investigated the effects of PGC-1α and β on inflammatory cytokines in C2C12 cells after stimulation with TNF-α, toll-like receptor agonists, and free fatty acids107. PGC-1s activation suppressed the generation of several proinflammatory cytokines by targeting the NF-κB signalling pathway and reducing the phosphorylation of P65. The same research group also found similar anti-inflammatory effects of PGC-1α/-1β in muscle-specific Pgc-1α/1β double transgenic mice. PGC-1s induced the expression of anti-inflammatory factors while suppressing pro-inflammatory IL-12, which is likely associated with a M2-type macrophage polarization in skeletal muscle108. Similarly, PGC-1β has been reported as being capable of suppressing palmitic acid-induced inflammation by inhibiting macrophage NF-κB P65109. These results also suggest that elevated PGC-1s expression contributes to the instauration of an anti-inflammatory environment.
3.3. The role of PGC-1α in angiogenesis
Angiogenesis, an intricate and highly regulated process, is defined as the generation of newly formed blood vessels from preexisting vessels in order to supply oxygen and nutrients to various tissues. The purpose of angiogenesis is to meet metabolic demands under physiological (pregnancy, exercise, and embryonic development), and pathological conditions (such as tumor progression and metastasis)110, 111, 112. Several growth factors, including vascular endothelial growth factor (VEGF), platelet derived growth factor (PDGF), fibroblast growth factor (FGF), and angiopoietin are the main triggers of this process. Regulation of nutrient supply so as to provide an appropriate volume of metabolic demand is a highly essential factor for tissues such as skeletal muscle, due to their high metabolic rates. Due to the regulatory effects of PGC-1 on various metabolic functions in numerous tissues, recent studies have focused on the involvement of this coactivator in angiogenesis, as demonstrated in various in vivo and experimental studies. For example, Chinsomboon et al.113 reported that PGC-1α is involved in exercise-induced angiogenesis.
When compared with Pgc-1α+/+ mice, which had robust angiogenesis during voluntary exercise, Pgc-1α–/– mice failed to increase their microvessel density. Exercise was shown to induce β-adrenergic signaling-dependent expression of PGC-1α. Overexpression of VEGF also contributes to PGC-1α induction, and this was found to be mediated by orphan nuclear receptor estrogen-related receptor alpha (ERRα). These findings illustrate that β-adrenergic activation of a PGC-1α/ERRα/VEGF pathway is involved in exercise-induced physiological angiogenesis in skeletal muscles114. In another study by Geng et al.114, it was found that PGC-1α plays an important part in exercise-induced angiogenesis, and mitochondrial biogenesis in mouse skeletal muscles. In a study on mesenchymal stem cells (MSCs), Lu et al.115 found that PGC-1α upregulation resulted in the overexpression of hypoxia-inducible factors (HIF-1α), increased BCL-2/BAX ratio, downregulation of caspase 3, as well as consequent enhanced survival rates and increased expression of proangiogenic factors. In ischemic hindlimbs of diabetic rats, PGC-1α overexpression was found to significantly increase the perfusion of blood and microvessel density115. Thom et al.116 showed truncated-PGC-1α induced VEGF expression and angiogenesis under hypoxic conditions. Conditioned media from cells expressing the truncated form of PGC-1α strongly prompts migration of endothelial cells and formation of capillary-like tubes. Transgenic expression of PGC-1α, specifically in skeletal muscle, induces angiogenesis in vivo116. VEGF and HIF-1 were demonstrated to be direct targets of a PGC-1α-mediated increase in angiogenesis in various studies110, 111, 112. Additionally, Rowe et al.6 demonstrated the effects of PGC-1α in muscle, significantly increasing angiogenesis in aged and in diabetic mice. PGC-1α induces the secretion of secreted phosphoprotein 1 (SPP1), and monocyte chemoattractant protein-1 (MCP-1) directed macrophage recruitment, which activates endothelial cells, smooth muscle cells, and pericytes. On the contrary, PGC-1α induction in Spp1−/− mice leads to blunted arteriolarization as well as immature capillarization6. The importance of PGC-1α in angiogenesis is significant to the point that deletion of this coactivator within skeletal muscle was reported to result in an impaired angiogenic program113,114,117, 118, 119, 120.
4. Role of natural products as PGC-1α modulators in DMD
Natural products have been found to exert protective activity against MD. Various natural products have been tested, such as green tea polyphenolics, quercetin, resveratrol, curcumin, and saponins, using cell and animal models of MD.
A primary culture of skeletal muscle cells was experimentally tested for the protective activity of green tea polyphenol mixture and EGCG against oxidative stress121. Treatment of dystrophic cells with green tea polyphenol blend/EGCG improved the cell survival, whereas normal cells were protected by green tea polyphenol blend and not by EGCG.
Using an mdx mouse model, EGCG was tested for its protective activity against DMD. EGCG was injected subcutaneously (5 mg/kg) for 8 weeks122. Improvements in blood chemistry (the reduced activity of serum kinase), muscle histological changes, and the electrophysiology of treated mdx mice were recorded. The numbers of fluorescent lipofuscin granules in soleus and diaphragm muscle were significantly reduced to 50% of control. With the EGCG treatment, the relative area of normal muscle fibers increased up to two-fold above control. The level of utrophin in the diaphragm muscles of treated mice was significantly increased by 17%. In a similar experiment, an mdx mouse dystrophy model was tested with green tea (0.01% or 0.05%, by weight) administered as a diet supplement123. The treatment was found to significantly reduce necrosis in the fast-twitch muscle elongator digitorum longue, but with negligible impact on the slow-twitch soleus muscle. The treatment was also found to decrease oxidative stress in the mdx mouse model, thereby improving muscle health by retarding necrosis.
In a similar study, a dystrophic mdx mouse model was used to assess the pharmacological effects of green tea extract, including its major polyphenol compounds, EGCG, and pentoxifyllin124. This revealed that the tested mice exhibited retarded necrosis of the extensor digitorum longus muscle. Phasic and tetanic tension was also increased, rising to normal values. Moreover, the ratio of phasic to tetanic tension was found to be corrected after the treatment. In a fatigue assay, the treated mice were found to exhibit 30%–50% more resistant phenotypes. There is no evidence in the literature indicating the mechanism for this protective activity of EGCG against DMD. However, a recent in vitro study has shown that an EGCG pre-treatment could exert a protective effect against oxidative damage induced by 1-methyl-4-phenyl-pyridine in a rat adrenal pheochromocytoma (PC12) cell line via the SIRT1/PGC-1α signaling pathway. The authors showed that mRNA expression of Pgc-1α, Sod, and Gpx1 are upregulated following pre-treatment with EGCG. In addition, the protein levels of PGC-1α and SIRT1 were increased, suggesting that the SIRT1/PGC-1α pathway is one of the mechanisms by which EGCG exerts PC12 cellular protection against 1-methyl-4-phenyl-pyridine damage125.
As reported in section 3, SIRT1, which is found in the nucleus, acts as a deacetylase on PGC-1α, leading to the formation of SIRT1-deacetylated PGC-1α.
Saponins, such as digitonin and tomatine, were found to increase the activity of antisense oligonucleotides against DMD126. In the presence of saponins, a significant enhancement in antisense phosphorodiamidate morpholino oligomer which targeted delivery to dystrophin exon 23 was found in mdx mice. This improvement was up to 7 times that of phosphorodiamidate morpholino oligomer alone. Saponin was also found to have lower cytotoxicity. It was found that saponins increased the efficiency of antisense oligonucleotide delivery and treatment of MD. Furthermore, a particular kind of saponin, chikusetsu saponin V, was found to be able to attenuate oxidative stress induced by H2O2 in human neuroblastoma SH-SY5Y cells, by increasing the activation of SIRT1, PGC-1α, and Mn-SOD127. Chikusetsu saponin V is found in Panax japonicus rhizome. The anti-oxidative and anti-inflammatory effects of P. japonicus saponins have been under investigation for the past two decades. He et al.128 showed that these compounds exert a cardioprotective effect in a rat model of acute myocardial ischemia.
Resveratrol was found to be effective against DMD. It increases the expression of nucleocytoplasmic shuttling protein SIRT1126 which positively impacts the activity of SOD, decreasing ROS levels, and increasing expression of PGC-1α85,128. PGC-1α was found to regulate type specification of muscle fiber (switching from fast to slow fiber types), oxidative capacity, and mitochondrial biogenesis129. It also targets utrophin, a dystrophin homolog130, and acts as a therapeutic active molecule against DMD.
In an experiment using the mdx mouse model, 500 mg/kg/day resveratrol was administered to mice for 32 weeks, and the authors observed that resveratrol decreased muscle wasting compared to control. It also reduced oxidative damage and downregulated nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, α-smooth muscle actin, and myofibroblast cells131.
In another experiment, mdx mice were treated with increasing dosages of resveratrol (0–400 mg/kg/day) for 8 weeks. It was found that at a 100 mg/kg/day dosage, the resveratrol increased fatigue resistance in slow-twitch soleus muscle and increased force in fast-twitch extensor digitorum longus muscle. However, resveratrol did not induce an increased resistance to injury in either muscle129. A higher 400 mg/kg dosage resulted in a number of deaths, indicating the toxicity of this compound. But other experiments did not show death at similar or higher dosages130.
In another study, resveratrol (100 mg/kg) treatment for 8 weeks led to an improvement in rotarod performance, centrally nucleated fibers damage, and oxidative stress using 4–5 week-old male mdx mice131. Using a similar experiment, mdx mice receiving reveratrol at a dosage of 100 mg/kg were found to possess higher activities of SIRT1 and PGC-1α after 6 weeks of treatment132. When the time period was increased to 12 weeks, resveratrol was found to be effective in increasing mitochondrial biogenesis and upregulating the expression of slower myosin heavy chain isoforms. However, an increased dosage of 500 mg/kg was not found to be effective compared to the 100 mg/kg dosage.
Overall, the efficacy of resveratrol in vivo may be hampered by its low bioavailability, and caution should be considered when extrapolating its laboratory effects to in vivo studies. In fact, resveratrol exhibits lipophilic characteristics, which promote high absorption but also subsequent rapid elimination after the first hepatic passage, contributing to its low bioavailability. Despite this, resveratrol shows efficacy in vivo. This is likely due to a variety of mechanisms, such as the reconversion of metabolites (sulfates and glucuronides) to resveratrol upon arrival in target organs, the enterohepatic recirculation of resveratrol metabolites and their subsequent deconjugation and reabsorption in the small intestine, and the activity of its metabolites133, 134, 135.
Quercetin was found to activate SIRT1 and then PGC-1α, and stimulate mitochondrial biogenesis in skeletal muscles136. It was found to exert antioxidant activity as well as mitochondrial biogenesis, and thus be beneficial against dystrophic skeletal muscle. In a study, supplementation with quercetin (0.2%) in three-month-old mdx mice for 6 months led to beneficial effects on the diaphragm136. Under this treatment, inflammatory response and pro-fibrotic infiltration was attenuated along with an increased expression of oxidative genes and mitochondrial transcription factors.
Curcumin was also found to be effective against MD. In an experiment, mdx mice were given a diet containing curcumin (1%, w/v)137. NF-κB activity was unchanged in the muscle, but muscular contractile activity was improved against control. In another study, curcumin was administered (0.1–1 mg/kg, i.p.) for 10 days to mdx mice137,138. Curcumin was found to reduce muscle damage in mdx mice and reduce skeletal muscle necrosis and muscle fiber size. The dose of 1 mg/kg was found to produce improvements in muscle function. In vascular smooth muscle cells, curcumin was found to increase SOD synthesis and adenosine triphosphate (ATP) reduction, leading to the formation of AMPK, which increases NAD+ levels and induces SIRT1 activation139. As a matter of fact, the beneficial effects of curcumin in MD could be due, at least in part, to its effect on SIRT1. Aside from these promising results obtained in animal models, the poor absorption, rapid metabolism, and systemic elimination of curcumin lead to lowered levels of the compound in plasma and tissue, blunting the possibility of implementing curcumin supplementation for human chronic disorders, such as DMD. Indeed, considering the progressive genetic nature of the disease, a short-term treatment is unlikely to positively impact long term muscular degeneration.
The identification of the molecular signaling contributing to the crosslink between natural products and PGC-1α remains a highly interesting subject. Trains of existing evidence suggest that PGC-1α transcription is highly regulated through the binding of transcription factors at its promoter, and these include myocyte enhancer factor 2 (MEF2), forkhead box class-O1 (FOXO1), activating transcription factor 2 (ATF2), and cyclic AMP response element-binding protein (CREB). These factors all enhance transcription of PGC-1α, and are each in turn modulated by different signaling pathways. In particular, AKT activation (i.e., by insulin) leads to cytoplasmic sequestration and inhibition of FOXO1, which physiologically binds and stimulates the promoter in skeletal muscle; p38 mitogen-activated protein kinase (p38 MAPK) activation (i.e., by exercise) phosporylates and activates MEF2 and ATF2, thus stimulates transcription; whereas calcineurin A (CnA) induces CREB and MEF2-mediated PGC-1α transcription. Importantly, posttranslational modifications of PGC-1α have also been shown to modulate its activity and levels by phosphorylation, acetylation, methylation, ubiquitination, and O-linked N-acetylglucosylation, in particular, by increasing transcription, p38 MAPK phosphorylation, and enhancement of PGC-1α activity140. Conversely, glycogen synthase kinase 3b (GSK3b), also phosphorylates PGC-1α, enhancing its proteasomal degradation due to oxidative stress while in the nucleus, thus inhibiting its activity141.
Importantly, most natural products interact with molecules involved in key intracellular pathways, including direct or indirect interactions with AKT, GSK3b, and p38 MAPK. For example, several compounds present in natural products negatively target AKT, p38 MAPK, and GSK-3 signaling activities, leading to inhibition of inflammation responses (i.e., quercetin, curcumin, and berberin). This regulatory behavior has been proposed for potential treatment of a range of disorders, in particular those which involve inflammatory processes potentially contributing to abnormal proliferation and cancer growth142, whereas their role as intermediates in modulating PGC-1α transcription and phosphorylation is as yet to be elucidated.
5. Conclusions
MD comprises a heterogeneous collection of disorders which are characterized by progressive weakness, loss of muscle strength, and degeneration. Since most forms of MD are genetic diseases, no effective treatment currently exists for these pathologies. In this sense, the search for new therapeutic targets which may contribute to a reduction in the development and progression of MD is necessary, as long as we are not able to effectively treat the origin of these diseases. One of these targets is PGC-1 and its cofactors, particularly PGC-1α, because it is inducible under physiological variations and is useful for increasing oxidative metabolism, mitochondrial biogenesis, angiogenesis, reducing oxidative stress, and inflammation (Fig. 2). Several natural compounds have shown a protective activity against MD in various cellular and animal models, increasing cell survival and reducing necrosis and levels of oxidative stress. We can highlight the induction of PGC-1α expression among the mechanisms of action of certain natural compounds, such as resveratrol and quercetin. However, the mechanisms of action involved in the observed improvements have not been studied in depth. In addition, there are currently no clinical trials with which to compare the results obtained in cell and animal models, therefore, the therapeutic use of these compounds, while promising, is still in a very embryonic state.
Figure 2.

DMD pathogenesis and PGC-1α targets.
Acknowledgments
This work was supported by the crowd funding #Sport4Therapy to Giuseppe D'Antona (Italy). A. Sureda was supported by Instituto de Salud Carlos III, Grant Number: CIBEROBN CB12/03/30038.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
Contributor Information
Maria Daglia, Email: maria.daglia@unina.it.
Seyed Mohammad Nabavi, Email: maria.daglia@unina.it.
Author contributions
Maria Daglia, Seyed Mohammad Nabavi, and Giuseppe D'Antona were responsible for the conception and design of the review. Ipek Suntar, Antoni Sureda, Tarun Belwal, Ana Sanches Silva, Rosa Anna Vacca, Devesh Tewari, and Eduardo Sobarzo-Sánchez collected literatures. Ipek Suntar, Seyed Fazel Nabavi, Samira Shirooie, Ahmad Reza Dehpour, Suowen Xu, Bahman Yousefi, Maryam Majidinia, and Giuseppe D'Antona analyzed literatures and summarized results. Ipek Suntar, Antoni Sureda, Tarun Belwal, Ana Sanches Silva, Rosa Anna Vacca, Devesh Tewari, Eduardo Sobarzo-Sánchez, Seyed Fazel Nabavi, Samira Shirooie, Ahmad Reza Dehpour, Suowen Xu, Bahman Yousefi, Maryam Majidinia, Maria Daglia, Giuseppe D'Antona, and Seyed Mohammad Nabavi drafted the manuscript. Maria Daglia, Giuseppe D'Antona, and Seyed Mohammad Nabavi revised the manuscript.
Conflicts of interest
The authors declare that there is no conflict of interest.
References
- 1.Handschin C., Spiegelman B.M. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev. 2006;27:728–735. doi: 10.1210/er.2006-0037. [DOI] [PubMed] [Google Scholar]
- 2.Arany Z. PGC-1 coactivators and skeletal muscle adaptations in health and disease. Curr Opin Genet Dev. 2008;18:426–434. doi: 10.1016/j.gde.2008.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Eisele P.S., Salatino S., Sobek J., Hottiger M.O., Handschin C. The PGC-1 coactivators repress the transcriptional activity of NF-κB in skeletal muscle cells. J Biol Chem. 2012;288:2246–2260. doi: 10.1074/jbc.M112.375253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen S.D., Yang D.I., Lin T.K., Shaw F.Z., Liou C.W., Chuang Y.C. Roles of oxidative stress, apoptosis, PGC-1α and mitochondrial biogenesis in cerebral ischemia. Int J Mol Sci. 2011;12:7199–7215. doi: 10.3390/ijms12107199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lin J., Wu H., Tarr P.T., Zhang C.Y., Wu Z., Boss O. Transcriptional co-activator PGC-1α drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 6.Rowe G.C., Raghuram S., Jang C., Nagy J., Patten I.S., Goyal A. PGC-1α induces SPP1 to activate macrophages and orchestrate functional angiogenesis in skeletal muscle. Circ Res. 2014;115:504–517. doi: 10.1161/CIRCRESAHA.115.303829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kummer T.T., Misgeld T., Sanes J.R. Assembly of the postsynaptic membrane at the neuromuscular junction: paradigm lost. Curr Opin Neurobiol. 2006;16:74–82. doi: 10.1016/j.conb.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 8.Angus L.M., Chakkalakal J.V., Méjat A., Eibl J.K., Bélanger G., Megeney L.A. Calcineurin–NFAT signaling, together with GABP and peroxisome PGC-1α, drives utrophin gene expression at the neuromuscular junction. Am J Physiol Cell Physiol. 2005;289:C908–C917. doi: 10.1152/ajpcell.00196.2005. [DOI] [PubMed] [Google Scholar]
- 9.Wang L., Waltenberger B., Pferschy-Wenzig E.M., Blunder M., Liu X., Malainer C. Natural product agonists of peroxisome proliferator-activated receptor gamma (PPARγ): a review. Biochem Pharmacol. 2014;92:73–89. doi: 10.1016/j.bcp.2014.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gregorevic P., Chamberlain J.S. Gene therapy for muscular dystrophy-a review of promising progress. Expert Opin Biol Ther. 2003;3:803–814. doi: 10.1517/14712598.3.5.803. [DOI] [PubMed] [Google Scholar]
- 11.Cossu G., Sampaolesi M. New therapies for Duchenne muscular dystrophy: challenges, prospects and clinical trials. Trends Mol Med. 2007;13:520–526. doi: 10.1016/j.molmed.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 12.Meregalli M., Farini A., Torrente Y. Combining stem cells and exon skipping strategy to treat muscular dystrophy. Expert Opin Biol Ther. 2008;8:1051–1061. doi: 10.1517/14712598.8.8.1051. [DOI] [PubMed] [Google Scholar]
- 13.Ervasti J.M., Campbell K.P. A role for the dystrophin–glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. 1993;122:809–823. doi: 10.1083/jcb.122.4.809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petrof B.J., Shrager J.B., Stedman H.H., Kelly A.M., Sweeney H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci U S A. 1993;90:3710–3714. doi: 10.1073/pnas.90.8.3710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goyenvalle A., Seto J.T., Davies K.E., Chamberlain J. Therapeutic approaches to muscular dystrophy. Hum Mol Genet. 2011;20:R69–R78. doi: 10.1093/hmg/ddr105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Duchenne G. Recherches sur le paralysie musculaire pseudohypertrophique ou paralysie myosclerosique. I. Symptomatologie, marche, duree, terminaison. Arch Gen Med 6 ser. 1868;11:179. [Google Scholar]
- 17.Mendell J.R., Moxley R., Griggs R., Brooke M., Fenichel G., Miller J. Randomized, double-blind six-month trial of prednisone in Duchenne's muscular dystrophy. N Engl J Med. 1989;320:1592–1597. doi: 10.1056/NEJM198906153202405. [DOI] [PubMed] [Google Scholar]
- 18.Birnkrant D.J., Bushby K., Bann C.M., Apkon S.D., Blackwell A., Brumbaugh D. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and neuromuscular, rehabilitation, endocrine, and gastrointestinal and nutritional management. Lancet Neurol. 2018;17:251–267. doi: 10.1016/S1474-4422(18)30024-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bies R.D., Phelps S.F., Cortez M.D., Roberts R., Caskey C.T., Chamberlain J.S. Human and murine dystrophin mRNA transcripts are differentially expressed during skeletal muscle, heart, and brain development. Nucleic Acids Res. 1992;20:1725–1731. doi: 10.1093/nar/20.7.1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hoffman E., McNally E. Exon-skipping therapy: a roadblock, detour, or bump in the road? Sci Transl Med. 2014;6:230fs14. doi: 10.1126/scitranslmed.3008873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu X., Liu M., Wu L., Liang D. Gene therapy for hemophilia and Duchenne muscular dystrophy in China. Hum Gene Ther. 2018;29:146–150. doi: 10.1089/hum.2017.213. [DOI] [PubMed] [Google Scholar]
- 22.Mendell J.R., Shilling C., Leslie N.D., Flanigan K.M., al-Dahhak R., Gastier-Foster J. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol. 2012;71:304–313. doi: 10.1002/ana.23528. [DOI] [PubMed] [Google Scholar]
- 23.Moat S.J., Bradley D.M., Salmon R., Clarke A., Hartley L. Newborn bloodspot screening for Duchenne muscular dystrophy: 21 years experience in Wales (UK) Eur J Hum Genet. 2013;21:1049–1053. doi: 10.1038/ejhg.2012.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ryder S., Leadley R., Armstrong N., Westwood M., de Kock S., Butt T. The burden, epidemiology, costs and treatment for Duchenne muscular dystrophy: an evidence review. Orphanet J Rare Dis. 2017;12:79. doi: 10.1186/s13023-017-0631-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LoMauro A., Romei M., Gandossini S., Pascuzzo R., Vantini S., D'Angelo M.G. Evolution of respiratory function in Duchenne muscular dystrophy from childhood to adulthood. Eur Respir J. 2018;51:1701418. doi: 10.1183/13993003.01418-2017. [DOI] [PubMed] [Google Scholar]
- 26.Verhaart I.E.C., Aartsma-Rus A. Therapeutic developments for Duchenne muscular dystrophy. Nat Rev Neurol. 2019;15:373–386. doi: 10.1038/s41582-019-0203-3. [DOI] [PubMed] [Google Scholar]
- 27.Bladen C.L., Salgado D., Monges S., Foncuberta M.E., Kekou K., Kosma K. The TREAT–NMD DMD Global Database: analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum Mutat. 2015;36:395–402. doi: 10.1002/humu.22758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.European Medicines Agency Translarna. https://www.ema.europa.eu/en/medicines/human/EPAR/translarna
- 29.Aartsma-Rus A., Bremmer-Bout M., Janson A.A., den Dunnen J.T., van Ommen G.J., van Deutekom J.C. Targeted exon skipping as a potential gene correction therapy for Duchenne muscular dystrophy. Neuromuscul Disord. 2002;12:S71–S77. doi: 10.1016/s0960-8966(02)00086-x. [DOI] [PubMed] [Google Scholar]
- 30.Randeree L., Eslick G.D. Eteplirsen for paediatric patients with Duchenne muscular dystrophy: a pooled-analysis. J Clin Neurosci. 2017;49:1–6. doi: 10.1016/j.jocn.2017.10.082. [DOI] [PubMed] [Google Scholar]
- 31.Wilton S.D., Fletcher S. Exon skipping and Duchenne muscular dystrophy: hope, hype and how feasible? Neurol India. 2008;56:254–262. doi: 10.4103/0028-3886.43443. [DOI] [PubMed] [Google Scholar]
- 32.Khan N., Eliopoulos H., Han L., Kinane T.B., Lowes L.P., Mendell J.R. Eteplirsen treatment attenuates respiratory decline in ambulatory and non-ambulatory patients with Duchenne muscular dystrophy. J Neuromuscul Dis. 2019;6:213–225. doi: 10.3233/JND-180351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nelson C.E., Robinson-Hamm J.N., Gersbach C.A. Genome engineering: a new approach to gene therapy for neuromuscular disorders. Nat Rev Neurol. 2017;13:647–661. doi: 10.1038/nrneurol.2017.126. [DOI] [PubMed] [Google Scholar]
- 34.Ousterout D.G., Kabadi A.M., Thakore P.I., Perez-Pinera P., Brown M.T., Majoros W.H. Correction of dystrophin expression in cells from Duchenne muscular dystrophy patients through genomic excision of exon 51 by zinc finger nucleases. Mol Ther. 2015;23:523–532. doi: 10.1038/mt.2014.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ousterout D.G., Perez-Pinera P., Thakore P.I., Kabadi A.M., Brown M.T., Qin X. Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol Ther. 2013;21:1718–1726. doi: 10.1038/mt.2013.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davies K.E., Guiraud S. Micro-dystrophin genes bring hope of an effective therapy for Duchenne muscular dystrophy. Mol Ther. 2019;27:486–488. doi: 10.1016/j.ymthe.2019.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shimizu-Motohashi Y., Miyatake S., Komaki H., Takeda S., Aoki Y. Recent advances in innovative therapeutic approaches for Duchenne muscular dystrophy: from discovery to clinical trials. Am J Transl Res. 2016;8:2471–2489. [PMC free article] [PubMed] [Google Scholar]
- 38.Mendell J.R., Kissel J.T., Amato A.A., King W., Signore L., Prior T.W. Myoblast transfer in the treatment of Duchenne's muscular dystrophy. N Engl J Med. 1995;333:832–838. doi: 10.1056/NEJM199509283331303. [DOI] [PubMed] [Google Scholar]
- 39.Torrente Y., Belicchi M., Marchesi C., D'Antona G., Cogiamanian F., Pisati F. Autologous transplantation of muscle-derived CD133+ stem cells in Duchenne muscle patients. Cell Transplant. 2007;16:563–577. doi: 10.3727/000000007783465064. [DOI] [PubMed] [Google Scholar]
- 40.Cossu G., Previtali S.C., Napolitano S., Cicalese M.P., Tedesco F.S., Nicastro F. Intra-arterial transplantation of HLA-matched donor mesoangioblasts in Duchenne muscular dystrophy. EMBO Mol Med. 2015;7:1513–1528. doi: 10.15252/emmm.201505636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Darabi R., Arpke R.W., Irion S., Dimos J.T., Grskovic M., Kyba M. Human ES- and iPS-derived myogenic progenitors restore dystrophin and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell. 2012;10:610–619. doi: 10.1016/j.stem.2012.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Young C.S., Hicks M.R., Ermolova N.V., Nakano H., Jan M., Younesi S. A single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC-derived muscle cells. Cell Stem Cell. 2016;18:533–540. doi: 10.1016/j.stem.2016.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li H.L., Fujimoto N., Sasakawa N., Shirai S., Ohkame T., Sakuma T. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Rep. 2015;4:143–154. doi: 10.1016/j.stemcr.2014.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lim K.R.Q., Yoon C., Yokota T. Applications of CRISPR/Cas9 for the treatment of Duchenne muscular dystrophy. J Personalized Med. 2018;8:E38. doi: 10.3390/jpm8040038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Batchelor C.L., Winder S.J. Sparks, signals and shock absorbers: how dystrophin loss causes muscular dystrophy. Trends Cell Biol. 2006;16:198–205. doi: 10.1016/j.tcb.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 46.Ervasti J.M., Ohlendieck K., Kahl S.D., Gaver M.G., Campbell K.P. Deficiency of a glycoprotein component of the dystrophin complex in dystrophic muscle. Nature. 1990;345:315–319. doi: 10.1038/345315a0. [DOI] [PubMed] [Google Scholar]
- 47.Tidball J.G. Inflammatory processes in muscle injury and repair. Am J Physiol Regul Integr Comp Physiol. 2005;288:R345–R353. doi: 10.1152/ajpregu.00454.2004. [DOI] [PubMed] [Google Scholar]
- 48.Verhaart I.E., Heemskerk H., Karnaoukh T.G., Kolfschoten I.G., Vroon A., van Ommen G.J. Prednisolone treatment does not interfere with 2′-O-methyl phosphorothioate antisense-mediated exon skipping in Duchenne muscular dystrophy. Hum Gene Ther. 2012;23:262–273. doi: 10.1089/hum.2011.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cabrera D., Gutiérrez J., Cabello-Verrugio C., Morales M.G., Mezzano S., Fadic R. Andrographolide attenuates skeletal muscle dystrophy in mdx mice and increases efficiency of cell therapy by reducing fibrosis. Skelet Muscle. 2014;4:6. doi: 10.1186/2044-5040-4-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Heier C.R., Damsker J.M., Yu Q., Dillingham B.C., Huynh T., Van der Meulen J.H. VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects. EMBO Mol Med. 2013;5:1569–1585. doi: 10.1002/emmm.201302621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brunelli S., Sciorati C., D'Antona G., Innocenzi A., Covarello D., Galvez B.G. Nitric oxide release combined with nonsteroidal antiinflammatory activity prevents muscular dystrophy pathology and enhances stem cell therapy. Proc Natl Acad Sci U S A. 2007;104:264–269. doi: 10.1073/pnas.0608277104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.D'Angelo M.G., Gandossini S., Martinelli Boneschi F., Sciorati C., Bonato S., Brighina E. Nitric oxide donor and non steroidal anti inflammatory drugs as a therapy for muscular dystrophies: evidence from a safety study with pilot efficacy measures in adult dystrophic patients. Pharmacol Res. 2012;65:472–479. doi: 10.1016/j.phrs.2012.01.006. [DOI] [PubMed] [Google Scholar]
- 53.Minetti G.C., Colussi C., Adami R., Serra C., Mozzetta C., Parente V. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat Med. 2006;12:1147–1150. doi: 10.1038/nm1479. [DOI] [PubMed] [Google Scholar]
- 54.NIH U.S. National library of medicine. Clinical study to evaluate the efficacy and safety of givinostat in ambulant patients with Duchenne muscular dystrophy. Available from: https://clinicaltrials.gov/ct2/show/NCT02851797.
- 55.Colussi C., Mozzetta C., Gurtner A., Illi B., Rosati J., Straino S. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc Natl Acad Sci U S A. 2008;105 doi: 10.1073/pnas.0805514105. 19183–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Consalvi S., Saccone V., Giordani L., Minetti G., Mozzetta C., Puri P.L. Histone deacetylase inhibitors in the treatment of muscular dystrophies: epigenetic drugs for genetic diseases. Mol Med. 2011;17:457–465. doi: 10.2119/molmed.2011.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Knutti D., Kralli A. PGC-1, a versatile coactivator. Trends Endocrinol Metab. 2001;12:360–365. doi: 10.1016/s1043-2760(01)00457-x. [DOI] [PubMed] [Google Scholar]
- 58.Lin J., Handschin C., Spiegelman B.M. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metabol. 2005;1:361–370. doi: 10.1016/j.cmet.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 59.Uldry M., Yang W., St-Pierre J., Lin J., Seale P., Spiegelman B.M. Complementary action of the PGC-1 coactivators in mitochondrial biogenesis and brown fat differentiation. Cell Metabol. 2006;3:333–341. doi: 10.1016/j.cmet.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 60.Puigserver P., Spiegelman B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1 alpha): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- 61.Kang C., Li Ji L. Role of PGC-1α signaling in skeletal muscle health and disease. Ann N Y Acad Sci. 2012;1271:110–117. doi: 10.1111/j.1749-6632.2012.06738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Summermatter S., Santos G., Pérez-Schindler J., Handschin C. Skeletal muscle PGC-1α controls whole-body lactate homeostasis through estrogen-related receptor α-dependent activation of LDH B and repression of LDH A. Proc Natl Acad Sci U S A. 2013;110:8738–8743. doi: 10.1073/pnas.1212976110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.St-Pierre J., Drori S., Uldry M., Silvaggi J.M., Rhee J., Jäger S. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 64.Zheng B., Liao Z., Locascio J.J., Lesniak K.A., Roderick S.S., Watt M.L. PGC-1α, a potential therapeutic target for early intervention in Parkinson's disease. Sci Transl Med. 2010;2:52ra73. doi: 10.1126/scitranslmed.3001059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Soyal S., Krempler F., Oberkofler H., Patsch W. PGC-1α: a potent transcriptional cofactor involved in the pathogenesis of type 2 diabetes. Diabetologia. 2006;49:1477–1488. doi: 10.1007/s00125-006-0268-6. [DOI] [PubMed] [Google Scholar]
- 66.Austin S., St-Pierre J. PGC1α and mitochondrial metabolism-emerging concepts and relevance in ageing and neurodegenerative disorders. J Cell Sci. 2012;125:4963–4971. doi: 10.1242/jcs.113662. [DOI] [PubMed] [Google Scholar]
- 67.Villena J.A. New insights into PGC-1 coactivators: redefining their role in the regulation of mitochondrial function and beyond. FEBS J. 2015;282:647–672. doi: 10.1111/febs.13175. [DOI] [PubMed] [Google Scholar]
- 68.Marmolino D., Manto M., Acquaviva F., Vergara P., Ravella A., Monticelli A. PGC-1alpha down-regulation affects the antioxidant response in Friedreich's ataxia. PLoS One. 2010;5 doi: 10.1371/journal.pone.0010025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cowell R.M., Talati P., Blake K.R., Meador-Woodruff J.H., Russell J.W. Identification of novel targets for PGC-1α and histone deacetylase inhibitors in neuroblastoma cells. Biochem Biophys Res Commun. 2009;379:578–582. doi: 10.1016/j.bbrc.2008.12.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Valle I., Álvarez–Barrientos A., Arza E., Lamas S., Monsalve M. PGC-1α regulates the mitochondrial antioxidant defense system in vascular endothelial cells. Cardiovasc Res. 2005;66:562–573. doi: 10.1016/j.cardiores.2005.01.026. [DOI] [PubMed] [Google Scholar]
- 71.Guo K., Lu J., Huang Y., Wu M., Zhang L., Yu H. Protective role of PGC-1α in diabetic nephropathy is associated with the inhibition of ROS through mitochondrial dynamic remodeling. PLoS One. 2015;10 doi: 10.1371/journal.pone.0125176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Vacca R.A., Bawari S., Valenti D., Tewari D., Nabavi S.F., Shirooie S. Down syndrome: neurobiological alterations and therapeutic targets. Neurosci Biobehav Rev. 2019;98:234–255. doi: 10.1016/j.neubiorev.2019.01.001. [DOI] [PubMed] [Google Scholar]
- 73.Izzo A., Manco R., Bonfiglio F., Calì G., De Cristofaro T., Patergnani S. NRIP1/RIP140 siRNA-mediated attenuation counteracts mitochondrial dysfunction in Down syndrome. Hum Mol Genet. 2014;23:4406–4419. doi: 10.1093/hmg/ddu157. [DOI] [PubMed] [Google Scholar]
- 74.Valenti D., de Bari L., de Rasmo D., Signorile A., Henrion-Caude A., Contestabile A. The polyphenols resveratrol and epigallocatechin-3-gallate restore the severe impairment of mitochondria in hippocampal progenitor cells from a Down syndrome mouse model. Biochim Biophys Acta. 2016;1862:1093–1104. doi: 10.1016/j.bbadis.2016.03.003. [DOI] [PubMed] [Google Scholar]
- 75.Valenti D., Braidy N., De Rasmo D., Signorile A., Rossi L., Atanasov A. Mitochondria as pharmacological targets in Down syndrome. Free Radic Biol Med. 2018;114:69–83. doi: 10.1016/j.freeradbiomed.2017.08.014. [DOI] [PubMed] [Google Scholar]
- 76.Cantó C., Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol. 2009;20:98–105. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jäger S., Handschin C., St-Pierre J., Spiegelman B.M. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1α. Proc Natl Acad Sci U S A. 2007;104:12017–12022. doi: 10.1073/pnas.0705070104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rodgers J.T., Lerin C., Haas W., Gygi S.P., Spiegelman B.M., Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature. 2005;434:113–118. doi: 10.1038/nature03354. [DOI] [PubMed] [Google Scholar]
- 79.Komen J.C., Thorburn D.R. Turn up the power—pharmacological activation of mitochondrial biogenesis in mouse models. Br J Pharmacol. 2014;171:1818–1836. doi: 10.1111/bph.12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nisoli E., Tonello C., Cardile A., Cozzi V., Bracale R., Tedesco L. Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science. 2005;310:314–317. doi: 10.1126/science.1117728. [DOI] [PubMed] [Google Scholar]
- 81.Valerio A., Nisoli E. Nitric oxide, interorganelle communication, and energy flow: a novel route to slow aging. Front Cell Dev Biol. 2015;3:6. doi: 10.3389/fcell.2015.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Valenti D., Rossi L., Marzulli D., Bellomo F., De Rasmo D., Signorile A. Inhibition of Drp1-mediated mitochondrial fission improves mitochondrial dynamics and bioenergetics stimulating neurogenesis in hippocampal progenitor cells from a Down syndrome mouse model. Biochim Biophys Acta (BBA) – Mol Basis Dis. 2017;1863:3117–3127. doi: 10.1016/j.bbadis.2017.09.014. [DOI] [PubMed] [Google Scholar]
- 83.Petrozzi L., Ricci G., Giglioli N.J., Siciliano G., Mancuso M. Mitochondria and neurodegeneration. Biosci Rep. 2007;27:87–104. doi: 10.1007/s10540-007-9038-z. [DOI] [PubMed] [Google Scholar]
- 84.Dong W., Gao D., Zhang X. Mitochondria biogenesis induced by resveratrol against brain ischemic stroke. Med Hypotheses. 2007;69:700–701. doi: 10.1016/j.mehy.2007.01.017. [DOI] [PubMed] [Google Scholar]
- 85.Lagouge M., Argmann C., Gerhart-Hines Z., Meziane H., Lerin C., Daussin F. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α. Cell. 2006;127:1109–1122. doi: 10.1016/j.cell.2006.11.013. [DOI] [PubMed] [Google Scholar]
- 86.Vacca R.A., Valenti D., Caccamese S., Daglia M., Braidy N., Nabavi S.M. Plant polyphenols as natural drugs for the management of Down syndrome and related disorders. Neurosci Biobehav Rev. 2016;71:865–877. doi: 10.1016/j.neubiorev.2016.10.023. [DOI] [PubMed] [Google Scholar]
- 87.Izzo A., Nitti M., Mollo N., Paladino S., Procaccini C., Faicchia D. Metformin restores the mitochondrial network and reverses mitochondrial dysfunction in Down syndrome cells. Hum Mol Genet. 2017;26:1056–1069. doi: 10.1093/hmg/ddx016. [DOI] [PubMed] [Google Scholar]
- 88.Timmons J.A., Larsson O., Jansson E., Fischer H., Gustafsson T., Greenhaff P.L. Human muscle gene expression responses to endurance training provide a novel perspective on Duchenne muscular dystrophy. FASEB J. 2005;19:750–760. doi: 10.1096/fj.04-1980com. [DOI] [PubMed] [Google Scholar]
- 89.Disatnik M.H., Dhawan J., Yu Y., Beal M.F., Whirl M.M., Franco A.A. Evidence of oxidative stress in mdx mouse muscle: studies of the pre-necrotic state. J Neurol Sci. 1998;161:77–84. doi: 10.1016/s0022-510x(98)00258-5. [DOI] [PubMed] [Google Scholar]
- 90.Kumar V., Abbas A.K., Fausto N., Aster J.C. Elsevier Health Sciences; Amsterdam: 2014. Robbins and Cotran pathologic basis of disease, professional edition.https://www.elsevier.com/books/robbins-and-cotran-pathologic-basis-of-disease-professional-edition/kumar/978-1-4377-0792-2 8th edition. e-book. Available from: [Google Scholar]
- 91.Geering B., Stoeckle C., Conus S., Simon H.U. Living and dying for inflammation: neutrophils, eosinophils, basophils. Trends Immunol. 2013;34:398–409. doi: 10.1016/j.it.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 92.Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428–435. doi: 10.1038/nature07201. [DOI] [PubMed] [Google Scholar]
- 93.Tidball J.G., Wehling-Henricks M. Damage and inflammation in muscular dystrophy: potential implications and relationships with autoimmune myositis. Curr Opin Rheumatol. 2005;17:707–713. doi: 10.1097/01.bor.0000179948.65895.1a. [DOI] [PubMed] [Google Scholar]
- 94.De Paepe B., De Bleecker J.L. Cytokines and chemokines as regulators of skeletal muscle inflammation: presenting the case of Duchenne muscular dystrophy. Mediat Inflamm. 2013;2013:540370. doi: 10.1155/2013/540370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Woodman K.G., Coles C.A., Lamandé S.R., White J.D. Nutraceuticals and their potential to treat Duchenne muscular dystrophy: separating the credible from the conjecture. Nutrients. 2016;8:E713. doi: 10.3390/nu8110713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Eisele P.S., Handschin C. Functional crosstalk of PGC-1 coactivators and inflammation in skeletal muscle pathophysiology. Semin Immunopathol. 2014;36:27–53. doi: 10.1007/s00281-013-0406-4. [DOI] [PubMed] [Google Scholar]
- 97.Lu Z., Xu X., Hu X., Fassett J., Zhu G., Tao Y. PGC-1α regulates expression of myocardial mitochondrial antioxidants and myocardial oxidative stress after chronic systolic overload. Antioxidants Redox Signal. 2010;13:1011–1022. doi: 10.1089/ars.2009.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tran M., Tam D., Bardia A., Bhasin M., Rowe G.C., Kher A. PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J Clin Investig. 2011;121:4003–4014. doi: 10.1172/JCI58662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Feingold K., Kim M.S., Shigenaga J., Moser A., Grunfeld C. Altered expression of nuclear hormone receptors and coactivators in mouse heart during the acute-phase response. Am J Physiol Endocrinol Metab. 2004;286:E201–E207. doi: 10.1152/ajpendo.00205.2003. [DOI] [PubMed] [Google Scholar]
- 100.Kim M.S., Shigenaga J.K., Moser A.H., Feingold K.R., Grunfeld C. Suppression of estrogen-related receptor alpha and medium-chain acyl-coenzyme A dehydrogenase in the acute-phase response. J Lipid Res. 2005;46:2282–2288. doi: 10.1194/jlr.M500217-JLR200. [DOI] [PubMed] [Google Scholar]
- 101.Coll T., Jové M., Rodríguez-Calvo R., Eyre E., Palomer X., Sαnchez R.M. Palmitate-mediated downregulation of peroxisome proliferator-activated receptor-gamma coactivator 1alpha in skeletal muscle cells involves MEK1/2 and nuclear factor-kappaB activation. Diabetes. 2006;55:2779–2787. doi: 10.2337/db05-1494. [DOI] [PubMed] [Google Scholar]
- 102.Yu X.X., Barger J.L., Boyer B.B., Brand M.D., Pan G., Adams S.H. Impact of endotoxin on UCP homolog mRNA abundance, thermoregulation, and mitochondrial proton leak kinetics. Am J Physiol Endocrinol Metab. 2000;279:E433–E446. doi: 10.1152/ajpendo.2000.279.2.E433. [DOI] [PubMed] [Google Scholar]
- 103.Remels A.H., Gosker H.R., Bakker J., Guttridge D.C., Schols A.M., Langen R.C. Regulation of skeletal muscle oxidative phenotype by classical NF-κB signalling. Biochim Biophys Acta. 2013;1832:1313–1325. doi: 10.1016/j.bbadis.2013.03.018. [DOI] [PubMed] [Google Scholar]
- 104.Tang K., Wagner P.D., Breen E.C. TNF-α-mediated reduction in PGC-1α may impair skeletal muscle function after cigarette smoke exposure. J Cell Physiol. 2010;222:320–327. doi: 10.1002/jcp.21955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Remels A., Gosker H., Schrauwen P., Hommelberg P., Sliwinski P., Polkey M. TNF-α impairs regulation of muscle oxidative phenotype: implications for cachexia?. FASEB J. 2010;24:5052–5062. doi: 10.1096/fj.09-150714. [DOI] [PubMed] [Google Scholar]
- 106.Handschin C., Choi C.S., Chin S., Kim S., Kawamori D., Kurpad A.J. Abnormal glucose homeostasis in skeletal muscle-specific PGC-1α knockout mice reveals skeletal muscle-pancreatic β cell crosstalk. J Clin Investig. 2007;117:3463–3474. doi: 10.1172/JCI31785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Eisele P.S., Salatino S., Sobek J., Hottiger M.O., Handschin C. The peroxisome proliferator-activated receptor γ coactivator 1α/β (PGC-1) coactivators repress the transcriptional activity of NF-κB in skeletal muscle cells. J Biol Chem. 2013;288:2246–2260. doi: 10.1074/jbc.M112.375253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Eisele P.S., Furrer R., Beer M., Handschin C. The PGC-1 coactivators promote an anti-inflammatory environment in skeletal muscle in vivo. Biochem Biophys Res Commun. 2015;464:692–697. doi: 10.1016/j.bbrc.2015.06.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chen H., Liu Y., Li D., Song J., Xia M. PGC-1β suppresses saturated fatty acid-induced macrophage inflammation by inhibiting TAK1 activation. IUBMB Life. 2016;68:145–155. doi: 10.1002/iub.1470. [DOI] [PubMed] [Google Scholar]
- 110.Hu X.Q., Zhang L. Angiogenesis during pregnancy: all routes lead to MAPKs. J Physiol. 2017;595:4571–4572. doi: 10.1113/JP274489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ronca R., Benkheil M., Mitola S., Struyf S., Liekens S. Tumor angiogenesis revisited: regulators and clinical implications. Med Res Rev. 2017;37:1231–1274. doi: 10.1002/med.21452. [DOI] [PubMed] [Google Scholar]
- 112.Kissane R.W.P. University of Leeds; 2017. Animal models of exercise therapy: mechanisms of activity-induced angiogenesis [dissertation] [Google Scholar]
- 113.Chinsomboon J., Ruas J., Gupta R.K., Thom R., Shoag J., Rowe G.C. The transcriptional coactivator PGC-1α mediates exercise-induced angiogenesis in skeletal muscle. Proc Natl Acad Sci U S A. 2009;106:21401–21406. doi: 10.1073/pnas.0909131106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Geng T., Li P., Okutsu M., Yin X., Kwek J., Zhang M. PGC-1α plays a functional role in exercise-induced mitochondrial biogenesis and angiogenesis but not fiber-type transformation in mouse skeletal muscle. Am J Physiol Cell Physiol. 2010;298:C572–C579. doi: 10.1152/ajpcell.00481.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Lu D., Zhang L., Wang H., Zhang Y., Liu J., Xu J. Peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) enhances engraftment and angiogenesis of mesenchymal stem cells in diabetic hindlimb ischemia. Diabetes. 2012;61:1153–1159. doi: 10.2337/db11-1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Thom R., Rowe G.C., Jang C., Safdar A., Arany Z. Hypoxic induction of vascular endothelial growth factor (VEGF) and angiogenesis in muscle by truncated peroxisome proliferator-activated receptor γ coactivator (PGC)-1α. J Biol Chem. 2014;289:8810–8817. doi: 10.1074/jbc.M114.554394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Arany Z., Foo S.Y., Ma Y., Ruas J.L., Bommi-Reddy A., Girnun G. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1α. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- 118.Leick L., Hellsten Y., Fentz J., Lyngby S.S., Wojtaszewski J.F., Hidalgo J. PGC-1α mediates exercise-induced skeletal muscle VEGF expression in mice. Am J Physiol Endocrinol Metab. 2009;297:E92–E103. doi: 10.1152/ajpendo.00076.2009. [DOI] [PubMed] [Google Scholar]
- 119.Rowe G.C., Jang C., Patten I.S., Arany Z. PGC-1β regulates angiogenesis in skeletal muscle. Am J Physiol Endocrinol Metab. 2011;301:E155–E163. doi: 10.1152/ajpendo.00681.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Fan W., He N., Lin C.S., Wei Z., Hah N., Waizenegger W. ERRγ promotes angiogenesis, mitochondrial biogenesis, and oxidative remodeling in PGC1α/β-deficient muscle. Cell Rep. 2018;22:2521–2529. doi: 10.1016/j.celrep.2018.02.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dorchies O.M., Wagner S., Buetler T.M., Ruegg U.T. Protection of dystrophic muscle cells with polyphenols from green tea correlates with improved glutathione balance and increased expression of 67LR, a receptor for (–)-epigallocatechin gallate. Biofactors. 2009;35:279–294. doi: 10.1002/biof.34. [DOI] [PubMed] [Google Scholar]
- 122.Nakae Y., Hirasaka K., Goto J., Nikawa T., Shono M., Yoshida M. Subcutaneous injection, from birth, of epigallocatechin-3-gallate, a component of green tea, limits the onset of muscular dystrophy in mdx mice: a quantitative histological, immunohistochemical and electrophysiological study. Histochem Cell Biol. 2008;129:489–501. doi: 10.1007/s00418-008-0390-2. [DOI] [PubMed] [Google Scholar]
- 123.Buetler T.M., Renard M., Offord E.A., Schneider H., Ruegg U.T. Green tea extract decreases muscle necrosis in mdx mice and protects against reactive oxygen species. Am J Clin Nutr. 2002;75:749–753. doi: 10.1093/ajcn/75.4.749. [DOI] [PubMed] [Google Scholar]
- 124.Dorchies O.M., Wagner S., Vuadens O., Waldhauser K., Buetler T.M., Kucera P. Green tea extract and its major polyphenol (–)-epigallocatechin gallate improve muscle function in a mouse model for Duchenne muscular dystrophy. Am J Physiol Cell Physiol. 2006;290:C616–C625. doi: 10.1152/ajpcell.00425.2005. [DOI] [PubMed] [Google Scholar]
- 125.Ye Q., Ye L., Xu X., Huang B., Zhang X., Zhu Y. Epigallocatechin-3-gallate suppresses 1-methyl-4-phenyl-pyridine-induced oxidative stress in PC12 cells via the SIRT1/PGC-1α signaling pathway. BMC Complement Altern Med. 2012;12:82. doi: 10.1186/1472-6882-12-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang M., Wu B., Shah S.N., Lu P., Lu Q. Saponins as natural adjuvant for antisense morpholino oligonucleotides delivery in vitro and in mdx mice. Mol Ther Nucleic Acids. 2018;11:192–202. doi: 10.1016/j.omtn.2018.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wan J., Deng L., Zhang C., Yuan Q., Liu J., Dun Y. Chikusetsu saponin V attenuates H2O2-induced oxidative stress in human neuroblastoma SH-SY5Y cells through Sirt1/PGC-1α/Mn-SOD signaling pathways. Can J Physiol Pharmacol. 2016;94:919–928. doi: 10.1139/cjpp-2015-0262. [DOI] [PubMed] [Google Scholar]
- 128.He H., Xu J., Xu Y., Zhang C., Wang H., He Y. Cardioprotective effects of saponins from Panax japonicus on acute myocardial ischemia against oxidative stress-triggered damage and cardiac cell death in rats. J Etnopharmacol. 2012;140:73–82. doi: 10.1016/j.jep.2011.12.024. [DOI] [PubMed] [Google Scholar]
- 129.Hori Y.S., Kuno A., Hosoda R., Horio Y. Regulation of FOXOs and p53 by SIRT1 modulators under oxidative stress. PLoS One. 2013;8 doi: 10.1371/journal.pone.0073875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Selsby J.T., Morine K.J., Pendrak K., Barton E.R., Sweeney H.L. Rescue of dystrophic skeletal muscle by PGC-1α involves a fast to slow fiber type shift in the mdx mouse. PLoS One. 2012;7 doi: 10.1371/journal.pone.0030063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hori Y.S., Kuno A., Hosoda R., Tanno M., Miura T., Shimamoto K. Resveratrol ameliorates muscular pathology in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. J Pharmacol Exp Ther. 2011;338:784–794. doi: 10.1124/jpet.111.183210. [DOI] [PubMed] [Google Scholar]
- 132.Gordon B.S., Delgado-Diaz D.C., Carson J., Fayad R., Wilson L.B., Kostek M.C. Resveratrol improves muscle function but not oxidative capacity in young mdx mice. Can J Physiol Pharmacol. 2014;92:243–251. doi: 10.1139/cjpp-2013-0350. [DOI] [PubMed] [Google Scholar]
- 133.Gambini J., Inglés M., Olaso G., Lopez-Grueso R., Bonet-Costa V., Gimeno-Mallench L. Properties of resveratrol: in vitro and in vivo studies about metabolism, bioavailability, and biological effects in animal models and humans. Oxid Med Cell Longev. 2015;2015:837042. doi: 10.1155/2015/837042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ljubicic V., Burt M., Lunde J.A., Jasmin B.J. Resveratrol induces expression of the slow, oxidative phenotype in mdx mouse muscle together with enhanced activity of the SIRT1–PGC-1α axis. Am J Physiol Cell Physiol. 2014;307:C66–C82. doi: 10.1152/ajpcell.00357.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kuno A., Hori Y.S., Hosoda R., Tanno M., Miura T., Shimamoto K. Resveratrol improves cardiomyopathy in dystrophin-deficient mice through SIRT1-mediated modulation of p300. J Biol Chem. 2013;288:5963–5972. doi: 10.1074/jbc.M112.392050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Hollinger K., Shanely R.A., Quindry J.C., Selsby J.T. Long-term quercetin dietary enrichment decreases muscle injury in mdx mice. Clin Nutr. 2015;34:515–522. doi: 10.1016/j.clnu.2014.06.008. [DOI] [PubMed] [Google Scholar]
- 137.Durham W.J., Arbogast S., Gerken E., Li Y.P., Reid M.B. Progressive nuclear factor-κB activation resistant to inhibition by contraction and curcumin in mdx mice. Muscle Nerve. 2006;34:298–303. doi: 10.1002/mus.20579. [DOI] [PubMed] [Google Scholar]
- 138.Pan Y., Chen C., Shen Y., Zhu C.H., Wang G., Wang X.C. Curcumin alleviates dystrophic muscle pathology in mdx mice. Mol Cells. 2008;25:531–537. [PubMed] [Google Scholar]
- 139.Grabowska W., Suszek M., Wnuk M., Lewinska A., Wasiak E., Sikora E. Curcumin elevates sirtuin level but does not postpone in vitro senescence of human cells building the vasculature. Oncotarget. 2016;7:19201–19213. doi: 10.18632/oncotarget.8450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Knutti D., Kressler D., Kralli A. Regulation of the transcriptional coactivator PGC-1 via MAPK-sensitive interaction with a repressor. Proc Natl Acad Sci U S A. 2001;98:9713–9718. doi: 10.1073/pnas.171184698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Anderson R.M., Barger J.L., Edwards M.G., Braun K.H., O'Connor C.E., Prolla T.A. Dynamic regulation of PGC-1alpha localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell. 2008;7:101–111. doi: 10.1111/j.1474-9726.2007.00357.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.McCubrey J.A., Lertpiriyapong K., Steelman L.S., Abrams S.L., Cocco L., Ratti S. Regulation of GSK-3 activity by curcumin, berberine and resveratrol: potential effects on multiple diseases. Adv Biol Regul. 2017;65:77–88. doi: 10.1016/j.jbior.2017.05.005. [DOI] [PubMed] [Google Scholar]