

Abstract
Introduction:
Pulmonary arterial hypertension (PAH) is a progressive disease characterized by remodeling of small pulmonary arteries leading to increased pulmonary arterial pressure. Existing treatments acts to normalize vascular tone via three signaling pathways: the prostacyclin, the endothelin-1, and the nitric oxide. Although over the past 20 years, there has been considerable progress in terms of treatments for PAH, the disease still remains incurable with a disappointing prognosis.
Areas covered:
This review summarizes the pathophysiology of PAH, the advantages and disadvantages of the inhalation route, and assess the relative advantages various inhaled therapies for PAH. The recent studies concerning the development of controlled-release drug delivery systems loaded with available anti-PAH drugs have also been summarized.
Expert opinion:
The main obstacles of current pharmacotherapies of PAH are their short half-life, stability, and formulations, resulting in reducing the efficacy and increasing systemic side effects and unknown pathogenesis of PAH. The pulmonary route has been proposed for delivering anti-PAH drugs to overcome the shortcomings. However, the application of approved inhaled anti-PAH drugs is limited. Inhalational delivery of controlled-release nanoformulations can overcome these restrictions. Extensive studies are required to develop safe and effective drug delivery systems for PAH patients.
Keywords: pulmonary arterial hypertension, inhaled route, targeted drug delivery
1. Introduction
Pulmonary arterial hypertension (PAH) is a complex and life-threatening vascular disorder that affects both pulmonary arteries and arterioles (PAs), which carry the blood from the right ventricle to the lungs[1]. The disease is defined hemodynamically by an elevation in mean pulmonary artery pressure (mPAP) ≥ 25 mmHg at rest and ≥ 30 mmHg at exercise with normal left ventricular filling pressures[2].
A pathogenic hallmark of this syndrome is an increased pulmonary vascular resistance (PVR) resulting from uncontrolled pulmonary vascular remodeling. High PVR leads to progressive elevations in PAP and sustained vasoconstriction, resulting in adaptive and maladaptive right ventricular hypertrophy (RVH) and ultimately, heart failure and death[3]. The underlying mechanisms of elevations in PVR in PAH include sustained vasoconstriction, increased vascular wall stiffness, uncontrolled pulmonary vascular remodeling, and in situ thrombosis[4]. PAH development is a multifactorial and heterogeneous process, and various genetic mutations in bone morphogenetic protein receptor II (BMPRII)[5] and activin receptor-like kinase 1 (ALK1)[6] are involved in the development of PAH.
Alterations in biomolecular pathways and various cell types within the pulmonary arteries walls, including pulmonary arterial endothelial cells (PAECs)[7], pulmonary arterial smooth muscle cells (PASMCs)[8], fibroblasts[9], inflammatory cells[10], and platelets[11], are implicated in the disease process. Three layers of PAs are the intimal layer comprised of PAECs, the medial layer comprised of PASMCs, and the adventitial layer consisting mainly of fibroblasts[12]. The dysfunction of all these cell types has been implicated in the development and progression of PAH (Figure 1). When damaged by shear stress, consistent constriction, hypoxia, inflammation, or toxins, PAECs decrease production of vasodilators such as prostacyclin and nitric oxide (NO) and increased production of the vasoconstrictors such as endothelin-1 (ET-1), which alters pulmonary vascular tone[13]. Disruption of the balance between endothelium-derived vasoconstrictors and vasodilators and inhibition of voltage-gated potassium channels in PASMCs, which leads to the opening of voltage-gated calcium channels and increasing the concentration of intracellular calcium, cause abnormal PASMCs contractility[14]. Additionally, impaired PAECs can either release factors that stimulate PASMCs proliferation, such as FGF-2, or fail to produce agents that usually suppress proliferation of PASMCs, such as apelin[9, 15]. Marked medial hypertrophy due to unrestrained PASMCs proliferation and contraction and apoptosis resistance and neointimal formation due to PAECs dysfunction cause vascular remodeling, obstructive lesions, which narrow the luminal space of the vessels, and impede blood flow[16]. Lung tissue of PAH patients showed intimal fibrosis, medial hypertrophy, and adventitial changes with increased extracellular matrix deposition of elastin, collagen, and fibronectin because of high abnormal activity of proteases such as matrix metalloproteases (MMPs)[17].
Figure 1.

Vascular abnormalities associated with PAH. This schema depicts the abnormalities throughout the pulmonary circulation: abnormal muscularization of distal and medial precapillary arteries, loss of precapillary arteries, thickening of large pulmonary arteries, and neointimal formation.
In one-third of PAH patients, thrombotic lesions are observed[18]; thus, hypercoagulability and in situ microthrombosis, due to enhanced platelet activity, contribute to the initiation and progression of the disease. Furthermore, the interaction of circulating platelets and inflammatory cells with the vascular wall play major roles in the remodeling process and disease process. A large population of PAH patients show elevated serum levels of inflammatory cytokines, including interleukin-1 and −6, and increased macrophage inflammatory protein. In the severe stage of the disease, clonal expansion of apoptosis-resistant PAECs, dysregulated PASMCs proliferation, and inflammatory cells produce aberrant channels in the obliterated lumen of the vessel called plexiform lesion, which is found in two-third of lung tissue specimens of PAH patients[19].
This fatal disease has multiple etiologies that affects over 100 million people worldwide[20]. Even though relatively rare, its impact on society is significant; in fact, PAH kills patients at very young age that incur a tremendous economic cost. The epidemiology of PAH is evolving, and the demographics of PAH has been changing. Younger patients showed more severe and hemodynamic impairment but better survival rates compared to older patients with more comorbidities[21]. The Fifth World Symposium of Pulmonary Hypertension in Nice, France, in 2013, classified PAH into 5 groups (Table 1). Group 1 contains PAH, which can be heritable, idiopathic, or associated with congenital heart disease, connective tissue disease, human immunodeficiency virus (HIV) infection, schistosomiasis, portal hypertension, and exposure to toxins/drugs. Group 2 is related to systolic, diastolic, or valvular left-heart disease and contributed to combined pre- and post-capillary pulmonary hypertension. Group 3 is associated with parenchymal lung diseases such as chronic obstructive pulmonary disease (COPD), obstructive sleep apnea, idiopathic pulmonary fibrosis, and interstitial lung disease. Group 4 is chronic thromboembolic pulmonary hypertension, caused by the unresolved accumulation of thromboembolic lesions in the pulmonary arteries. Finally, Group 5 is a miscellaneous category, including sarcoidosis and sickle cell-related pulmonary hypertension[22].
Table 1:
Updated clinical classification, causes, and treatments of pulmonary hypertension
| WHO PH Group | Causes | Treatment |
|---|---|---|
| Group1 Pulmonary Arterial Hypertension |
- Unknow (Idiopathic) - Genetic - Drug and toxin exposure - Diseases associated with PAH: - Connective tissue (scleroderma, lupus, etc.) - HIV - Portal Hypertension - Congenital Heart Disease |
Medications (pills, inhalers, and continuous infusions) developed specifically for the treatment of PAH to dilate and reduce the inappropriate growth of cells in the pulmonary arteries. |
| Group 2 Left Heart Disease |
This often a result of: - Coronary Artery Disease - High Blood Pressure - Damage to the Heart Muscle - Heart Valve Disease - Age |
Therapies are focused on treating the underlying heart disease. |
| Group 3 Lung Disease |
Pulmonary vessels are tightened in response to other lung diseases, such as: - COPD - Interstitial lung Disease - Any other lung disease causing low blood oxygen level |
Therapies are focused on treating the underlying lung disease. |
| Group 4 Chronic Thromboembolic Pulmonary Hypertension |
PH caused by old organized blood clots in the lungs that form a physical barrier to blood flow within the pulmonary arteries. |
Surgical removal of the clots, when possible, or oral medication if surgery is not possible or if PH remains after surgery. |
| Group 5 PH Resulting from Unclear Mechanisms |
Associated Diseases: - Sarcoidosis - Blood disease (sickle cell anemia, chronic hemolytic anemia, etc.) - History of spleen removal - Metabolic disease (Gaucher disease, thyroid disease) |
Therapies are focused on treating the underlying disease. |
PAH is associated with poor prognosis and it is diagnosed very late in the course of the disease[23]. The symptoms primarily result from decreased cardiac output and so tend to be nonspecific, including shortness of breath, weakness, syncope with exertion, and fluid retention, and bluish color in the lips and skin (cyanosis)[24]. However, the presence of different symptoms depends on the severity of the disease and the patient’s stage. Hence, the pulmonary hypertension functional classification divides the stages of the disease into various clinical classes that helps physicians identify the disease earlier. The most applicable and well-known functional assessment classifications are the World Health Organization Functional Class (WHO FC) and the New York Health Association Functional class (NYHA FC)[25] (Table 2). The class system is mainly focused on how affected the patients are by the disease. Class 1 refers to patients that do not experience any symptoms with usual physical activity. Class 2 includes patients with symptoms while performing regular activities and a slight limitation of movement. Class 3 refers to patients that experience symptoms with less than the ordinary activity and marked restriction of activity. Class 4 is the last and includes patients that suffer the symptoms of the disease at rest. The poorly understood disease processes, when left untreated, can significantly contribute to patient morbidity and mortality; thus, accurate diagnosis is imperative. Numerous invasive and noninvasive procedures are available to diagnose PAH, such as electrocardiography, pulmonary function testing such as 6-minute walk distance (6MWD), chest radiography, echocardiography, serologic testing such as brain natriuretic peptide (BNP), and right heart catheterization[26]. Therapy for PAH has improved over the last 20 years and thus ten FDA approved drugs are currently available for the treatment of PAH (Table 3). PAH therapies act on the following major pathways: the prostacyclin pathway (prostacyclin and its analogs), the NO pathway (inhaled NO, phosphodiesterase-5 inhibitors, and soluble guanylate cyclase modulators), or endothelin pathway (ET receptor antagonists, ERA)[27]. Despite the advancement in diagnostic procedure and understanding of the underlying pathogenic mechanisms of PAH, current therapies are limited to supportive treatments and targeting pulmonary vasoconstriction to alleviate the symptoms of the disease, not to resolve the pathogenesis of PAH and improve the mortality rate. Thus, PAH is seriously in need of a new treatment strategy that could reduce mortality and ameliorate functional deterioration associated with it.
Table 2:
Functional assessment of pulmonary arterial hypertension
| NYHA Class | WHO Class | Activity Limitations |
|---|---|---|
| 1 | I | None |
| 2 | II | Sight limitation of activity |
| 3 | III | Marked limitation of activity |
| 4 | IV | Symptoms with any activity or at rest; discomfort is increased by any physical activity |
NYHA: New York Heart Association; WHO: World Health Organization.
Table 3:
Currently Approved Medications for Treatment of Pulmonary Arterial
| Class | Drug | Route of Administration | Dose |
|---|---|---|---|
| Prostacyclin derivatives | Epoprostenol | IV infusion | 2 ng/kg/min Increased as tolerated |
| Iloprost | Inhaled | 2.5 – 5.0 µg 6–9 inhalations /day |
|
| Treprostinil | Oral | 0.25 mg bid or 0.125 mg tid Increase 0.125 mg bid every 3–4 days |
|
| Inhaled | 18–54 µg (3–9 inhalations) 4 times daily |
||
| Subcutaneous or IV infusion | 1.25 ng/kg/min; increase 1.25 ng/kg/min per week based on clinical response; after week 4 increase by 2.5 ng/kg/min per week based on clinical response | ||
| Endothelin receptor antagonists | Bosentan | Oral | 125 mg twice daily |
| Ambrisentan | Oral | 5 or 10 mg once daily | |
| Macitentan | Oral | 10 mg once daily | |
| Phosphodiesterase type-5 inhibitors | Sildenafil | Oral IV injection |
20 mg every 8 h |
| Tadalafil | Oral | 40 mg once daily | |
| Soluble cGMP stimulators | Riociguat | Oral | 0.5–1.0 mg every 8 h (increase 0.5 mg every 2 wk as tolerated to maximum dose 2.5 mg) |
| Prostacyclin receptor agonists | Selexipag | Oral | 200 mg twice daily Increase as tolerated to maximum dose of 16,000 mg twice daily |
In the present review, we discuss the advantages and disadvantages of the inhalation route and then evaluate various approved inhaled therapies according to their biochemical pathway, reviewing their evidence for efficacy, pharmacology, practical applications, indications, and limitations. Additionally, we attempted to bring the latest available therapy or investigational inhaled formulations of anti-PAH medications up to October 2019.
2. Therapeutic targets for PAH management
2.1. The prostacyclin pathway
Prostaglandin I2 (PGI2, prostacyclin) is derived from arachidonic acid through the action of prostacyclin synthase in the vascular endothelial layer[28] (Figure 3A). It induces PASMCs relaxation by increasing the concentration of intracellular cyclic adenosine 3′,5′-monophosphate (cAMP), and inhibiting the growth of the medial layer of PAs. Also, prostacyclin is a potent antiproliferative, antithrombotic, and antimitogenic agent [29]. The published report suggests that the loss of PGI2 synthase activity and its metabolite in pulmonary arteries isolated from patients with PAH[30]. Therefore, the exogenous administration of prostacyclin analogs has been applied to patients with PAH.
Figure 3.

(a) Schematic representation of the prostacyclin pathway, (b) Schematic representation of the guanosine monophosphate (GMP) and nitric oxide pathways., (c) Schematic representation of the endothelin system in vascular tissue. (d) Mechanisms through which current drugs elicit pulmonary vasodilatation to treat pulmonary hypertension.
2.1.1. Epoprostenol
Epoprostenol (Flolan®) was the first prostacyclin cleared by the FDA for the treatment of PAH in 1995. It is administered by continuous intravenous (IV) infusion via a central venous catheter connected to an infusion pump, which requires careful preparation and strict hygiene standards. The pivotal randomized clinical trial proved that epoprostenol improved exercise capacity, 6MWD, hemodynamics, and survival in PAH patients with NYHA FC III and IV[31]. In spite of its role in improving clinical function in some patients, epoprostenol infusion is far from the ideal treatment for PAH due to its short half-life, complication, patient compliance, and systemic adverse effect. Symptoms of systemic vasodilatation, such as flushing, headache, jaw pain, or diarrhea, are the main dose-dependent side-effects of this treatment[32]. Fortunately, the IV formulation of epoprostenol can be aerosolized and used therapeutically off-label. One study examined the short-term effects of inhaled epoprostenol on hemodynamics and gas exchange in a group of subjects with PAH following surgery, including cardiac procedures and lung transplantation[33]. After 4–6 hrs of inhaled epoprostenol, mPAP fell remarkably, cardiac output rose, and the oxygenation index increased. Furthermore, there was reduced effect on the mean systemic arterial pressure (mSAP), indicating pulmonary selectivity. Thus, based on this clinical trial and other studies, minimal toxicity and systemic side-effect, relatively low cost, lack of dedicated administration equipment, and higher efficacy favor this route of epoprostenol administration for short-term in-hospital applications and management of critically ill patients with PAH[34]. However, one of the limitations of nebulized epoprostenol is its very short half-life (3–5 min)[35]. As a result, it needs continuous nebulization, rendering it impracticable for long-term applications. Therefore, further investigation is required to synthesize a sustained-release formulation of the drug for long-term use.
2.1.2. Iloprost
Iloprost (Ventavis®) is a chemically stable derivative of prostacyclin with a biological half-life of approximately 30 minutes[36]. In the Aerosolized Iloprost Randomized (AIR) study, the use of inhaled iloprost in patients with PAH on NYHA FC III or IV enhanced the quality of life, dyspnea scores, hemodynamic characteristics and exercise capacity after 12 weeks[37]. In the other prospective, multicenter, single-arm trial, Chon et al. demonstrated that after 48 weeks of inhaled iloprost therapy mPAP, mSAP, and PVR were significantly decreased in patients with exertional dyspnea WHO functional Class III–IV and Eisenmenger physiology[38]. The inhaled route is associated with fewer adverse effects on systemic symptoms compared with IV drug delivery. However, unfortunately, a minority of patients could be stabilized with this monotherapy during a follow-up period of up to 5 years. Besides, iloprost has to be inhaled six to nine times a day, up to 15 min each, to maintain the drug levels within the therapeutic range; these multiple daily nebulizations may be responsible for frequent airway symptoms such as cough, flushing, jaw pain and headache[39]. Development of controlled- release formulation of iloprost can be a solution for the obstacle of multiple dosing.
2.1.3. Treprostinil
Treprostinil (Tyvaso®), a prostacyclin analog with higher stability, can be administered subcutaneously, intravenously, orally, and by inhalation. Sandifer et al. compared the efficacy of aerosolized and IV-administered treprostinil on a PAH-induced sheep. Aerosolized treprostinil was more effective than IV treprostinil as a pulmonary vasodilator even at a lower dose (250 ng/kg/min). Treprostinil decreased both mPAP and PVR to baseline levels; pulmonary delivery also resulted in a more localized delivery of treprostinil into the alveolar regions[40]. Compared with other prostacyclins analogs, treprostinil has the most prolonged half-life (3–4 h), which translates into a longer dosing interval when administered by inhalation. FDA approved inhaled treprostinil in 2011 based on a double-blind placebo-controlled clinical trial of inhaled treprostinil sodium in patients with severe PAH (TRIUMPH I) Trial, which randomized 235 subjects into inhaled treprostinil versus placebo groups[41]. This trial demonstrates that of the PAH patients who remain symptomatic after treatment with bosentan or sildenafil, inhaled treprostinil improves exercise capacity, reduces BNP level, and enhances the quality of life, and is safe and well-tolerated.
2.2. Nitric Oxide/cGMP Pathway
2.2.1. Gaseous NO and NO donors
Endothelial-derived nitric oxide (eNO) is a highly diffusible gaseous nitrogen radical synthesized due to conversion of L-arginine to citrulline under the regulation of a family of 3 enzymes, collectively called endothelial nitric oxide synthases (eNOS). Afterward, at the medial layer of pulmonary vasculatures, NO binds to and activates the soluble guanylate cyclase (sGC), through the stimulation of cyclic guanosine monophosphate (cGMP) production. cGMP acts as a second messenger regulating smooth muscle contractility by activating cGMP-dependent protein kinases leading to vascular relaxation and inhibition of PASMCs proliferation and platelet aggregation[42, 43]. In the pulmonary vasculature, phosphodiesterase (PDEs), especially PDE5, provide a counter-regulatory function by converting and inactivating cGMP to 5′-GMP via hydrolysis[44]. Perturbations in the NO-sGC-cGMP pathway represents an important theoretical mechanism leading to PAH (Figure 3B). This pathway is dysregulated at many steps, which leads to increase in PAP. This finding is consistent with the published report showing that patients with PAH have a reduced expression of eNOS and its activity and diminished NO bioavailability[45, 46].
Inhaled NO has been shown to reduce mPAP, improve oxygenation, decrease PVR, and intrapulmonary shunting with minimal systemic side effects in a variety of adult populations with PAH in the intensive care unit (ICU) [47, 48]. Although the effects of inhaled NO, documented in several clinical trials[49, 50], makes it a feasible option for the short-term management of PAH, FDA approved inhaled NO is used only in hospitalized pediatric patients with PAH. Unfortunately, inhaled NO has several disadvantages, including cost, short half-life due to rapid inactivation after binding to hemoglobin, the rebound of PAH symptoms with abrupt discontinuation, production of toxic metabolites such as nitrate, the development of methemoglobinemia, and the need for sophisticated equipment for administration[51].
NO donor compounds can be administered locally to increase NO availability and consequently circumvent the rebound phenomenon. NO donors are compounds formed by reacting NO with various nucleophiles such as NONOates and release NO spontaneously and induce vasodilation. Jacob et al. examined the effect of NO donors after aerosol treatment of NONOate in PAH-induced pigs and demonstrated that NONOate improved oxygenation and reduced PVR significantly without causing significant systemic toxicity[52]. Further investigations are required to develop a sustained-release formulation of NO and NO donors to apply these therapies for long-term treatment.
2.2.2. Phosphodiesterase Inhibitors
PDEs, predominately PDE1 and PDE5, inactivate cGMP, the intracellular second messenger of NO, leading to vasoconstriction in pulmonary arteries[44]. Since PDE5 is profusely expressed in PASMCs of PAH patients, inhibition of PDE5 is an attractive therapeutic option [53]. The observation that PDE5 inhibitors, such as sildenafil and tadalafil, have potent vasodilatory and antiproliferative effects on PASMCs underscores the role of PDE in regulating the NO pathway[54]. Oral sildenafil (Revatio®) received FDA approval after two multi-center, multinational, long-term clinical trials, SUPER-1 and −2, showing its effect on the improvements in the 6MWD, pulmonary hemodynamics, and survival rate in PAH patients of NYHA FC II and III[54, 55]. Researchers examined inhaled sildenafil in animal models. In one study using the PAH-induced lamb model, Ichinose et al. proved that inhaled sildenafil, independent of the tested dose, lowered the mPAP as much as inhaled NO and had no significant effect on mSAP, indicating selective pulmonary vasodilation. Also, inhaled sildenafil had an additive effect with inhaled NO[56]. The practice of treating PAH patients with oral sildenafil suffers from the shortcomings such as of large dose, short dosing intervals due to its short half-life, unwanted systemic vasodilation, and restricted use in pediatric patients[57]. Thus, to fill this unmet gap, our lab has developed an inhaled long-acting formulation of sildenafil as a viable alternative to oral tablets of sildenafil, which is explained in section 5.3.
2.2.3. Soluble Guanylate Cyclase stimulator/activator
sGC, a heterodimeric enzyme with a heme-containing prosthetic group, plays a critical role in NO-mediated signaling selective to the pulmonary circulation[58]. In patients with PAH, the function of sGC is diminished by decreased NO bioavailability, generation of reactive oxygen species (ROS), sGC oxidation via oxidation of the heme-iron due to oxidative stress, and the related loss of enzyme’s heme group[59]. Since the therapeutic effect of PDE5 inhibitors depends on baseline NO expression, which is decreased in PAH patients, treatments that act directly on sGC may have greater efficacy than other anti-PAH treatments on NO-pathway[60]. Pharmacological modulators of sGC are divided into two categories based on their mechanisms of action: sGC stimulators (riociguat) require heme-containing sGC to catalyze cGMP production, whereas sGC activators (cinaciguat) activate heme-free sGC[61]. Riociguat (Adempas®) is approved for PAH and yields functional and hemodynamic benefits similar to other therapies. Phase III, international, double-blind, randomized, placebo-controlled trial conducted over 12 weeks in 443 patients with symptomatic Group 1 PH, PATENT-1 and- 2, studies showed sustained improvements in exercise capacity and 6MWD, and functional ability for up to 1 year after using 2.5 mg TID riociguat[62, 63]. Unfortunately, sGC activators and stimulators lack specificity for the pulmonary circulation and cause dose-dependent hypotension. Researchers suggested inhaled delivery of the drug to overcome this obstacle, which we explain in section 5.4.
2.3. Endothelin pathway
ET-1 ubiquitously and predominantly produced by the PAECs and, to a lesser extent, by other cell types, including PASMCs and fibroblasts[64, 65]. Additionally, various promoters, including cytokines, hypoxia, shear stress, growth factors, and angiotensin II, stimulate ET-1 synthesis[66]. ET-1 is a potent pulmonary vasoconstrictor with a proliferative effect on vascular SMCs after interaction with two types of ET receptors: ET receptor A (ETA), mainly on PASMCs; and ET receptor B (ETB) mostly on the vascular PAECs and less on PASMCs[67] (Figure 3C). Activation of the ETA isoform induces vasoconstriction and proliferation of vascular SMCs[68]. Since the ETB receptors are involved in the clearance of ET-1, it may induce vasodilation via the release of NO and prostacyclin from the ECs[69]. All categories of PAH patients proved activation of the ET pathway in all types of human PAH. PAH patients showed higher plasma and pulmonary arterial ET-1 expression, which directly correlate with the severity of the disease, including the degree of vascular remodeling[70]. Currently, both nonselective and selective ET receptor antagonists (ERAs) are approved for treating PAH. Theoretically, selective ETA-ERAs should be more effective than nonselective ERA, given the role played by ETB in both vasodilation and ET-1 clearance. However, there is no clear evidence that receptor selectivity is relevant concerning the clinical effects of these drugs.
2.3.1. Bosentan
Bosentan (Tracleer®) is the first approved oral nonselective ERA used for PAH treatment since the beginning of the last decade. BREATHE and EARLY are clinical studies that evaluated the efficacy of bosentan at two different doses, 125 and 250 mg bid, in NYHA FC II, III, and IV patients with PAH[71, 72]. BREATHE-1, which included 213 patients with PAH, showed an enhanced 6MWD, PVR, NYHA FC, and hemodynamics with both 125 and 250 mg daily doses of bosentan[72].
2.3.2. Ambrisentan
Ambrisentan (Volibris®) is an oral selective ETA receptor antagonist with a lower rate of hepatic injury[73]. In two large, multicenter, clinical trials ARIES-1 and −2, ambrisentan showed improved 6MWD, NYHA FC, quality of life scores, and tolerability profile for a 2-year period[74, 75]. The drug has been approved for patients with NYHA FC II and III and IV in the USA.
2.3.3. Macitentan
Macitentan (Opsumit®) is a new FDA approved dual oral ERA that demonstrated sustained receptor binding and tissue penetration properties compared with other ERA[76]. The phase III SERAPHIN clinical trial proved the long-term efficacy of macitentan. Analysis of the results indicated that treatment with 3 and 10 mg macitentan reduced the risk of morbidity and mortality events, enhanced 6MWD, NYHA FC for up to 3.5 years[77].
Although all three currently approved ERAs demonstrated potent prolonged efficacy in PAH, the main limiting factors for the widespread application of ERA for treatment of PAH patients are life-threatening adverse effects associated with long-term therapy, including elevation of liver transaminase levels, decrease in hemoglobin levels[71]. The inhalation route of application has been suggested to minimize systemic side effects and to restrict the action of these substances to the lungs and maximize their deposition. Leuchte et al. demonstrated that aerosolization of selective ETA (BQ-123) and a dual selective ETA and ETB receptor blocker (Tezosentan) attenuated pulmonary vasoconstriction, improved arterial oxygenation, and decreased mPAP without significant systemic side-effect[78].
2.4. Other pathways
2.4.1. Vasoactive intestinal peptide (VIP) pathway
The VIP, a 28 amino acids neuropeptide, primarily functions as a neurotransmitter with an essential role in the water and electrolyte secretion in the gut[79]. Also, VIP acts as a potent systemic and pulmonary vasodilator and mild inhibitor of platelet activation[80] and PASMCs proliferation[81]. The biological effects are mediated by specific VIP receptors (VPAC-1 and −2) expressed on the cell surface membrane of subintima of pulmonary arteries and veins and macrophages surrounding capillaries[82]. Stimulation of VIP receptors results in the activation of the cGMP and cAMP systems; analogously, cGMP- and cAMP-dependent pathways have been shown to mediate the action of prostacyclin, NO, and PDE inhibitors in the PAH treatment. Since the lung tissue and serum of patients with PAH showed decreased concentrations of the VIP, investigators applied exogenous VIP as a novel target for PAH treatment[83]. In a small cohort study of 8 idiopathic PAH patients, inhaled VIP showed impressive favorable effects on exercise capacity and pulmonary arterial hemodynamics, as evidenced by decreasing the mPAP, increasing cardiac output, and improving 6MWD[83]. Another clinical trial conducted by Leuchte et al. showed a reduction in mPAP and PVR and improved oxygenation without affecting systemic hemodynamic parameters following nebulization of 100 µg of aviptadil, a synthetically inhaled form of VIP[84]. Unfortunately, the main limitation of frequent use of VIP is its short half-life, which causes the rapid degradation of the peptides by endogenous proteases. Thus, to overcome this problem, the slow release of VIP formulation was developed by Hajos et al. explained in section 5.7.
2.4.2. Rho kinases pathway
Rho kinases (ROCK), ubiquitously expressed serine/threonine kinases, are intracellular signaling molecules that regulate vasoconstriction and cell proliferation[85]. ROCK has two isoforms: ROCK1 mainly in the lung, liver, and ROCK2, mostly in the heart, brain[86]. RhoA activates ROCK after binding to extracellular G-protein coupled receptor and increases calcium sensitivity of the contractile apparatus via phosphorylation and suppression of myosin light chain (MYPT-1), leading to smooth muscle contraction. Additionally, ROCK involves in various essential cellular functions, including proliferation, motility, and migration[85, 87]. Because of its impact on smooth muscle cell contraction and proliferation, endothelial cell damage, and inflammation, ROCK plays a vital role in the pathogenesis of various cardiovascular diseases such as PAH. Many studies revealed that a significant elevation in ROCK expression and its activities in the lungs, platelets, PASMCs, and thickened intima and media of small PAs of patients with idiopathic PAH[88, 89]. Inhaled inhibitors of Rock (Y-27632 and fasudil) potently lowered mPAP in several PAH-induced rat models[90] (Figure 3D).
In one randomized animal trial, Nagaoka et al. compared the effectiveness and selectiveness of inhaled and oral Y-27632 in PAH-induced rats[90]. Despite oral Rock inhibitor caused a marked decrease in mPAP, it also decreased mSAP. In contrast, the inhaled drug reduced mPAP without lowering mSAP. A series of clinical trials confirmed fasudil’s acute vasodilatory effect[91, 92]. In summary, most of these studies demonstrated that inhalation of fasudil slightly decreased mPAP, significantly decreased PVR, and partially increased cardiac index without remarkable systemic adverse effects. Although the benefits of acute vasodilator response have been identified, it is challenging to evaluate fasudil’s impact on the prognosis PAH due to the drug’s short half-life. Therefore, the long-acting formulation of inhalational fasudil is required to develop. The development of a liposomal delivery system provides a new method for the clinical use of fasudil, although it will require further clinical evaluation.
2.4.3. Tyrosine kinases pathway
Tyrosine kinases such as Platelet-Derived Growth Factor (PDGF), Fibroblast Growth Factor 2 (FGF-2), Vascular Endothelial Growth Factor (VEGF) belong to a ubiquitous family of proteins involved in various cell process and repair mechanisms, including cellular proliferation, migration, differentiation, apoptosis, angiogenesis, and ECM production[93]. Lung-tissue isolates from PAH patients and PAH-induced lambs highlighted excessive production of these growth factors, receptor expression, and alterations in the intracellular mitogenic signals[94, 95]. These abnormalities may result from local production with autocrine or paracrine effects or excessive release of growth factors encrypted in the ECM of remodeled pulmonary arteries.
One of the boundaries of our current PAH armamentarium is that currently approved drugs act mainly on arterial vasodilation and do not primarily target the pulmonary arterial remodeling characteristic of the disease, which leads to a treatment fashion that is aimed at stabilization rather than regression of disease (Figure 3D). Tyrosine kinase inhibitors (TKI) give us the opportunities to attack vascular remodeling leading to the prevention of PAH progression. A randomized, double-blind, placebo-controlled 24-week trial (IMPRES), evaluated the efficacy of imatinib in patients with PAH. Although imatinib improved exercise capacity (6MWD), hemodynamics (PVR and RVH), and NYHA FC in patients, serious cardiotoxic and adverse events were typical, which caused drug discontinuations[96]. These collateral effects were likely the result of the pleiotropy of the TKI, with disrupted signaling in multiple organs giving rise to numerous off-target adverse effects. However, other approaches aimed at inhibiting tyrosine kinases may demonstrate to be promising, including the application of TKI via the inhaled route to affect more specifically and limit side-effects on off-target organs. Pitsiou et al. indicate that current TKI, imatinib, can be modified to be produced as aerosols that could be applied as an aerosol treatment for PAH[97].
For the initial therapy, drugs are classified according to WHO FC, reflecting the severity of PAH pathology. Initial treatment with approved anti-PAH drugs needs to be initiated in PAH patients who are nonresponsive to calcium channel blockers (CCBs). Nonresponders to CCBs who are in WHO FC II should be treated with an oral ERA, PDE5 inhibitors, or rociguate. Nonresponders to high dose CCBs, or responders who remain in WHO FC III, should be considered candidates for treatment with inhaled iloprost, inhaled treprostinil, oral ERAs, and oral/IV sildenafil. Continuous IV epoprostenol is recommended as first-line therapy for WHO FC IV PAH patients because of the survival benefit in this subset. In the case of inadequate clinical response, sequential combination therapy should be considered. Combination therapy can either include an ERA plus a PDE5 inhibitor, a prostanoid plus an ERA, or a prostanoid plus a PDE5 inhibitor. Upon regulatory approval, the sGC stimulator riociguat can be considered as a potential alternative to a PDE5 inhibitor in the different types of double combinations. In the case of inadequate clinical response with dual combination therapy, triple combination therapy should be attempted[98].
3. Advantages and disadvantages of inhaled drug delivery for PAH
Pulmonary delivery of drugs has become an attractive target and of tremendous biomedical interest in the health care systems as the lung can absorb pharmaceuticals for localized effects. The primary treatment goal for patients with PAH is enhancing vasodilation of PAs and pulmonary blood circulation. Although several drugs are available that improve 6MWD and hemodynamics, their tolerated doses are limited in critically ill PAH patients because of unwanted systemic side-effects such as systemic vasodilation resulting in hypotension[99]. The use of inhaled vasodilators may be an option in providing effective and selective pulmonary vasodilation without affecting systemic pressures. In this respect, growing attention has been paid to the potential of the inhalation route as a non-invasive administration for local delivery of anti-PAH therapeutic agents such as inhaled NO and prostacyclin analogs, which selectively reduce PAP without systemic hypotension[100].
Compared with other routes of drug administration, inhalational delivery offers several advantages in the PAH treatment (Table 4). First, it delivers medication directly to the lung, enabling higher pulmonary drug concentrations, which determines the efficacy of the drug. Therefore, drug inhalation is typically associated with elevated efficacy in reducing PAP and reduced dosing intervals and thus enhanced patient compliance[101]. Second, since the inhalation route puts the drug directly into the alveolar region of the lung, there should be lower systemic drug concentration, and less severe systemic adverse effect[102]. Thus, inhalation delivery of the drugs can minimize systemic hypotension, a common adverse effect of anti-PAH therapies in critically ill patients, because most of the drugs are both pulmonary and systemic vasodilators. Lower systemic drug levels conferred by inhalation leads to improved pulmonary hemodynamics and thus reduced risk of unwanted systemic side-effects. Third, inhaled vasodilators improve gas exchange. Systemic administration of vasodilator agents indiscriminately dilates the pulmonary arterial vasculature, leading to high blood flow to poorly ventilated areas, which impair gas exchange. On the other hand, pulmonary administration of vasodilators delivered the drugs to the ventilated areas, where their specific vasodilatory action can enhance blood flow to the ventilated regions, improving ventilation and/or perfusion [103]. Fourth, inhalation can provide a more rapid drug absorption and fast onset of action, within minutes, in the lungs compared to other routes of administration because of the excellent permeability, high blood flow, and large absorptive surface area of the lungs (approximately 70–140 m in adult humans having thin absorptive epithelial barrier)[104]. Further advantages of inhalation over other pharmaceutical administration are the avoidance of the first-pass metabolism due to the comparatively low enzymatic activity of the lung[105]. Sixth, by localization of therapeutic agents directly to the lung (target organ), inhalation allows administering a lower dose than what is required for systemic delivery (oral or injection), potentially lowering cost.
Table 4:
Advantages and disadvantages of the inhaled route for administration of PAH Medications
| Advantages | Disadvantages |
|---|---|
| Local delivery, potentially higher concentration of medication in the target organ, which enhance the efficacy | Irritant effects on airways |
| Avoidance of systemic adverse effects, including systemic hypotension | Limitation of medication dose due to airway symptoms |
| Delivery to ventilated areas, vasodilatation improves V˙/Q˙ and gas exchange | Delivery systems can be cumbersome and time consuming |
| Potentially lower total dose of medication with less frequent administrations and lower cost | May be very costly |
| Rapid drug absorption and fast onset of effect | |
| Avoidance of the first-pass metabolism due to the low enzymatic activity of the lung |
V˙/Q˙= ventilation/perfusion ratio
The inhaled route also has disadvantages. Intolerance of inhaled drug administration due to direct irritant effects of the drugs on the airways may result in airway symptoms such as cough. Also, patients cannot precisely control drug dosing due to variability in breathing patterns and the complexity in determining the exact concentration of medication that reaches the pulmonary arteries of the lung. Pulmonary delivery systems introduce the potential for personal error and inaccurate dose due to difficulty in equipment operation. There are different breathing maneuvers for various inhalers or nebulizers to prevent exhaled or ambient loss of drug[106]. These limitations, coupled with cost considerations, may limit the practical application of inhaled drugs in patients with PAH. The other disadvantage of this route includes a small fraction of aerosolized drugs reaches the target site. Once deposited in the lungs, a significant fraction of inhaled medications is either cleared from the lungs, absorbed into the systemic circulation, or degraded via drug metabolism. Drug particles deposited in the conducting airways are primarily removed through mucociliary clearance and, to a lesser extent, are absorbed through the airway epithelium into the blood or lymphatic system[101]. Insoluble particles mainly are entrapped in the mucous of the conducting airways and are moved toward the pharynx (and ultimately to the gastrointestinal tract) by the upward movement of mucus generated by the metachronous beating of cilia. Factors such as particle size, solubility, lipophilicity, and charge govern the ability of the drug to penetrate this mucous barrier. In addition to mucociliary clearance, soluble particles also can be removed by absorptive mechanisms in the conducting airways. Lipophilic molecules pass quickly through the airway epithelium via passive transport. Hydrophilic molecules cross via extracellular pathways, such as tight junctions, or by active transport via endocytosis and exocytosis[107]. From the submucosal region, particles are absorbed into either the systemic circulation, bronchial circulation or lymphatic system, which elicits side effects. Drugs deposited in the alveolar region may be phagocytosed and cleared by alveolar macrophages or absorbed into the pulmonary circulation. Abnormal airways, bronchoconstriction, alveolar flooding, interstitial fibrosis, and inflammation influence the deposition and distribution of aerosols and therefore serve as a barrier to effective aerosolized drugs therapy when the pathologically affected sites are the lung tissue. Chronic inflammation may result in airway remodeling and obstruction, which changes the dynamics of airflow, impair mucociliary clearance, thus reducing the pulmonary drug deposition[108]. A decrease in the cross-sectional area of the lung caused by obstruction increases air velocities and turbulence in regions where the airflow usually is laminar. Airway obstruction diverts inspired air to unobstructed airways and, thus, the minimal drug is deposited in obstructed areas, often the areas that need to be reached to achieve the optimal therapeutic effect of the drug[109]. These changes lead to a proximal shift in the airway deposition pattern of the aerosols.
Many disadvantages of inhalation applications can be avoided by administrating the drug parenterally. IV administration, either by direct injection or infusion, is one of the most reliable methods for delivering medication to the systemic circulation to treat pulmonary diseases because it bypasses many of the absorption barriers, mucous layer, and metabolic mechanisms. In fact, by definition, the bioavailability of drugs is 100% by IV injection because the drug is administered directly into the vascular space and blood central compartment. It is also one of the preferred routes of administration to achieve therapeutically effective drug concentrations fast and, consequently, rapid action[110]. IV drug delivery provides stable and predicted pharmacokinetic profile and permits a maximal degree of control over administration rate and the dose and concentration of the drug on the site of action, which is not merely possible in the inhaled drug delivery. IV infusions may be used to achieve a constant level of medicine in the bloodstream for short half-life drugs, especially anti-PAH therapeutics[111].
However, the IV route of drug administration is not always viable for the treatment of pulmonary diseases because of several limitations, including higher systemic adverse effect, patient discomfort, and cost. IV administered drugs are inevitably associated with the lack of pulmonary selectivity results in significant systemic side effects on gas exchange and systemic blood pressure, and drug tolerance leads to tachyphylaxis and progressive increases in dose. For PAH-specific IV administration, the most common adverse effects include systemic hypotension, headaches, flushing, skin hyperemia, and jaw pain[112]. Also, infusion-site pain was common in patients administered IV infusion of treatments. The risks of infection and thrombosis must be considered after IV administration; a notable complication of IV anti-PAH medications is recurrent infections of the IV catheter and increased risk of bloodstream infection with gram-negative organisms[113]. The other disadvantage is that the application of this route is limited to highly soluble drugs, which must be in aqueous solution or suspensions to avoid the possibility of embolism and the propensity to form precipitates. Furthermore, the preparation of IV medicines requires the use of an aseptic technique, often in a ward environment that is unsuited to such work.
Treatment of PAH by inhalation is an optimal approach to target drugs to the lungs and thus reduce the systemic side effects. However, some studies showed a transient drop in mSAP after inhalation of anti-PAH drugs[114]. Therefore, there is an unmet need to clarify the mechanism by which inhaled anti-PAH drugs improve pulmonary hemodynamics and reduce systemic hypotension. If we are to exploit the anatomical properties of the lung to treat PAH, we need to understand the factors that affect the absorption, deposition, metabolism, and local pharmacodynamics and systemic pharmacokinetic kinetics of drugs following inhalation delivery.
The first critical feature that affects the pulmonary drug efficacy is the nature of the aerosol deposition in the airways, following inhalation. Drug absorption occurs from both the conducting airways and the alveolar region. A significant amount of the aerosolized dose gets trapped in the alveolar compartment, but a fraction deposit on the mucosal surface of the conducting bronchi. A small portion of absorbed drug transfers to the gut via mucociliary clearance of conducting airways and degrade in the GI tract (Figure 2). A large part of deposited drugs in the epithelium of conductive airway and bronchioles penetrate the PAs and directly diffuse into terminal arterioles and capillaries, which are surrounded by alveolar surfaces. Aerosols that have been deposited at the alveolar region do not experience significant clearance, but diffuse to the blood vessels of the lung, and dilate pulmonary vasculature directly. Drugs can reside in this area for an extended period leading to prolonged effects in the PAs. Hemodynamic effects and arterial blood concentrations of inhaled prostanoids and NO confirmed the suggested deposition pathways, indicating that inhaled anti-PAH medicines directly act on the pulmonary arterial wall (from the adventitial side), not by drug recirculation from the bronchial arteries[115]. The additional fact that the lung takes the complete cardiac output from right ventricle via pulmonary arteries and returns a portion of the absorbed drug directly to the heart that subsequently moves to the systemic circulation, which causes systemic hypotension following inhalation of a high dose of anti-PAH medicines with extended half-life (Figure 2). However, aerosolization of the drug into the lung reduces the required dose, which is too small to produce a measurable systemic effect following inhalation of the majority anti-PAH treatments within the inhaled dose. Thus, the net outcome would be reduced exposure of the drug in the systemic circulation and side effects.
Figure 2.

Diagrammatic representation of drug distribution in the respiratory system following inhalation.
The second important feature that influences pulmonary drug delivery is the pharmacokinetics of the inhaled drug. Olschewski et al. showed the serum half-life of iloprost was much shorter than the half-life of the pulmonary vasodilatory effects exerted by this drug, suggesting that the inhaled iloprost caused prolonged local vasodilation not reflected by the serum levels[115]. Several mechanisms may explain this observation. First, perivascular lung tissue (pulmonary artery cells, alveolar lining layer, and interstitial space) may trap a percentage of iloprost following inhalation, and this fraction may possess a longer half-life than the systemic iloprost because it is not exposed to the catabolic enzyme in the lower lung. Second, since the interstitium of the lung alveolar and lining layer are small spaces, even a low quantity of drug result in high local concentrations inducing vasorelaxation of PAs. In summary, the ideal vasodilator therapeutic formulation would be cost-effective, safe, and selective to the pulmonary vasculature.
4. Novel systems for controlled delivery of anti-PAH drugs
Although multiple treatments are available to cure PAH, still it is a life-threatening disease with a high mortality rate. While current pharmacotherapies have improved the quality of life of the patients, PAH drugs suffer from limitations in the form of instability, poor organ specificity, short-term pharmacokinetics, and formulation limitations. Approved anti-PAH therapies have improved patient hemodynamics and quality of life, but are not without remarkable restrictions. Chief among these are drug stability, half-life, and formulation limitations, resulting in deleterious side effects. As an example, epoprostenol has instability at low pH values and a short half-life of 3–5 min[35]. As a result, the drug should be continuously maintained under refrigeration, and the medicine must be continually infused IV employing an implanted catheter and infusion pump, increasing risk of infections, sepsis, and thrombosis[116]. Because of limitations associated with the conventional treatment of anti-PAH treatments, growing attention has been given to the development of controlled, targeted drug delivery systems, including liposomes, micelles, and solid polymer particles.
4.1. Liposomes
Liposomes are widely used nanocarriers on the order of 100–500 nm in size, which composed of various natural and synthetic phospholipids with polar heads and hydrophobic tails, forming bilayer constructs with an aqueous core. Liposomal formulations with compositions similar to lung surfactants could serve as potential carriers for pulmonary delivery of therapeutics, owing to their low local irritation to the lung parenchyma[117], controllable surface charge[118], as well as capability of encapsulating hydrophilic, amphiphilic, or hydrophobic compounds. Hydrophobic drugs can be captured within the bi-phospholipid membrane, and water-soluble drugs can be encapsulated in aqueous compartment[119]. Functionalization of liposomes with polyethylene glycol (PEG) on the particle’s surface led to significant enhancement of circulation lifetimes via sterical stabilization of liposomes from phagocytosis of macrophages[120]. Liposomes could also be produced in both dry powder and liquid suspension forms based on the pulmonary delivery setup[121]. Liposomes have myriad of advantages for pulmonary drug delivery, including reduction of the nonspecific side-effects and toxicity of encapsulated drugs, thus improving their efficacy and therapeutic index, prolonged-release rates and extending the drug retention half-life following pulmonary administration, improving their intracellular diffusion and bioavailability, compatibility with biodegradable and non-toxic materials, enhanced solubility of the encapsulated drugs, and ease of chemical modifications minimizing the pulmonary clearance via protecting the chemotherapeutics from enzymatic degradation[119]. The therapeutic activity, transferred drug dosage, deposition/release rates, and cytotoxicity of the liposomes in the lung depending on the lipid composition including, phospholipid type and content, rigidity, cholesterol content, saturated–unsaturated lipid ratio, and other additives), size, surface properties, drug characteristics, drug-to-lipid ratio, process parameters, and delivery technique[122, 123]. Hence, alteration of the lipid composition and fabrication protocol could result in highly customized physicochemical properties that substantially affect the efficacy of the final nanocarrier in the target site.
A significant issue in nebulizing liposomes is the stability of vesicles during nebulization. In principle, shearing provided by a nebulizer to convert liposome dispersions into fine aerosol droplets results in vesicle fragmentation, with concomitant loss of the entrapped initially hydrophilic material. A range of strategies has been used to minimize the instability of liposomes and the failure of the entrapped hydrophilic drug. The inclusion of cholesterol or high-phase transition phospholipids in liposome formulations has improved the stability of vesicles during nebulization[124]. Unlike hydrophilic drugs, many studies have demonstrated that the issue of the physical stability of liposomes is less significant when the entrapped drug is hydrophobic[125].
Safety of liposome formulations for pulmonary drug for pulmonary delivery has attracted a marked interest since three liposomal drugs entrapping doxorubicin (Doxil®), daunorubicin (DaunoXome®), and amphotericin B (Ambisome®) are already licensed in several countries. Many others are in advanced clinical trials[126]. Few studies have revealed a hypersensitivity reaction to certain liposomes that develops immediately after the start of infusion due to activation of the complement system, leading to cardiovascular and pulmonary adverse responses[127–129]; in contrast, other studies have demonstrated the safety of liposomes for pulmonary administration. For example, Myers et al. have shown using animal models that inhalation of hydrogenated soy phosphatidylcholine (HSPC) liposomes caused no pathological effects on alveolar macrophages[130]. Many studies using human volunteers have established the safety of liposomes for inhalation[124, 131]. Arikace® is a novel anti-pseudomonal liposome formulation that has shown safety and suitability for inhalation by human cystic fibrosis subjects in Phase II clinical trials[124]. Many studies have also shown that drugs entrapped in liposomes are safe for pulmonary delivery since liposomes can control the mode of drug release, hence reducing the drug amount available to exert adverse effects[132, 133]. Lee et al. confirmed the safety of cerivastatin in nano-liposome formulations given via inhalation for PAH treatment. The nano-liposomes demonstrated significantly less cellular cytotoxicity as compared to free cerivastatin; furthermore, liposome delivery produced significantly lower levels of the lactone metabolite that is considered a safety risk at high statin doses. In summary, the inhaled liposomal formulation is safe to deliver the therapeutics locally to the lung[132].
4.2. Solid polymeric particles
Solid polymeric particles, normally comprised of the polyester, hydroxypropyl methacrylate, and polylactide-co-glycolide (PLGA), have long been employed in inhalable, controlled-release formulations of drugs. These particles are spherical in morphology, can be formulated into the drug-carrying devices at all scales ranging from the nano to micrometer dimensions, and can be used for delivery of water-soluble and -insoluble drugs, with agents dissolved or encapsulated within the polymer matrix[134, 135]. Polymeric particles offer potential advantages over liposomes in terms of physical and chemical stability, drug encapsulation capability, sustained drug release, and prolonged pharmacological activity of their cargos. In particular, low aggregation tendency, deep deposition in the lower lung regions, and high mechanical stability under the shear forces make polymeric particles suitable for pulmonary delivery of therapeutics. Different drugs such as doxorubicin, paclitaxel, cisplatin, and sildenafil have been encapsulated within the polymeric particles[136–138]. A variety of carrier parameters, including morphology, hydrophilicity, porosity, and molecular weight of the polymeric carriers, as well as the environmental factors such as pH, temperature, moisture content, solvent polarity, and physicomechanical stresses, show significant effects on the efficacy of pulmonary delivery by polymeric carriers. PLGA, a polymer approved by the FDA for various biomedical applications, remains the constituent polymer of choice for these particles due to several advantages: Chief among these is the controlling degradation of PLGA for sustained drug release at desirable doses, as well as the biodegradability and biocompatibility[139]. Drug release from PLGA particles occurs through initial diffusion followed by degradation of the polymer matrix. Thus, applying various ratios of hydrophobic to hydrophilic polymers helps to achieve highly controllable and sustained release profiles[140]. Many studies confirmed the safety of PLGA formulation applications[141, 142]. For example, Rashid et al. proved that the PLGA micro-particle containing sildenafil is not cytotoxic for the pulmonary arterial cells. Also, intratracheal administration of the formulation to the rat did not change the weight of the lung or the level of the lung enzymes and proteins, indicating the safety of the formulation for acute treatment[137].
4.3. Polymer micelles
Polymeric micellar structures have also been broadly utilized in pulmonary drug delivery due to their structural similarity to biological compounds such as lipoproteins and viruses. Polymeric micelles are supramolecular, core-shell nanoparticles (10–100 nm) that offer considerable advantages for targeted drug delivery. Polymer micelles are formed from the self-assembly of amphiphilic block copolymers in aqueous environments. The core-shell morphology of polymer micelles contains a hydrophobic core and a hydrophilic shell (the constituent polymer is typically PEG)[143]. The micellar structures generally exhibit high loading capacity and biocompatibility, which reflect their high potential for pulmonary administration[144]. Due to their hydrophilic shell, micelles could evade clearance by the alveolar macrophages, diffuse through the mucus layer, and penetrate the epithelial cells. various micellar formulations have been used to deliver several drugs, including calcitonin, sirolimus, and polymyxin B[145–147]. In terms of stability, targeting efficiency, and prolongation of drugs’ half-lives, these micelles are superior to their conventional counterparts, such as microparticles and liposomes[148, 149]. For example, intratracheally administered paclitaxel-loaded DSPE–PEG micelles showed a greater accumulation in rat lungs compared with microparticles, solid lipid nanoparticles, or lipid nanocapsules containing paclitaxel[148]. Micellar carriers demonstrated a favorable safety profile for the treatment of PAH. Gupta et al. evaluated the short-term safety of the formulations after intratracheal administration. Micelles did not cause any significant increase in lung weight, the formation of edema, or elevation of the levels of lung enzymes, suggesting the safety of the formulation for pulmonary local drug delivery[150]. In summary, their relatively small size, ability to solubilize hydrophobic drugs, stimuli-responsive and tailored drug release, and improved pharmacokinetics provide a useful bioengineering platform for pharmaceutical applications[151, 152].
4.4. Effect of carrier physical properties on targeting efficiency of inhaled particles
A desirable inhalation performance requires the optimization of various particle properties. The particle size, morphology, surface characteristics, and penetrability through the airway mucus are the most studied physicochemical parameters for the optimization of the particle-based pulmonary delivery systems performance.
4.4.1. Size
The effect of size on the distribution pattern and deposition site of the inhaled particles has been investigated. In general, the particles with different sizes are deposited by inertial impaction, gravitational sedimentation, and Brownian diffusion mechanisms[153]. Considering the aerodynamic diameter of the drug carrier, the inertial impaction usually occurs during the passage of large-sized particles (>5 µm) through the oropharynx, trachea, and other large airways. However, sedimentation by gravitational forces is more likely for particles with a size range of about 1–5 µm and occurs in the smaller airways and respiratory bronchioles. Brownian motion by diffusion is the primary mode of particle distribution for small nanoscale drug carriers (≥500 nm)[154]. Besides, the size could significantly affect the activity of various lung clearance mechanisms that intensify during lung diseases. These mechanisms are considered as the major obstacle that the inhaled particles encounter, leading to rapid clearance from the lungs before reaching the target site. The inhaled particles are mostly cleared by mucociliary clearance or alveolar macrophages, depending on their deposition region. The particles larger than 6 µm that mostly accumulate at the upper airways are eliminated by mucociliary clearance in the epithelial tissue. When the foreign particles are trapped in mucus, cilia beat in a coordinated direction to remove the freight by either coughing or swallowing. Clearance by the alveolar macrophages is the second challenge that the particles encounter during their travel in the pulmonary system. Although the 1–5 µm particles are shown to escape the mucociliary clearance, they are prone to faster recognition and elimination by the resident alveolar macrophages[155]. In recent years, an increasing interest has been dedicated to the particles in the nano-sized range due to their deposition at the lining fluid and evading the lung clearance mechanisms[156]. Moreover, the nano-sized carriers are able to easily incorporation into the respirable ratio, leading to enhanced uniformity of drug distribution within the alveoli[157]. However, the application of nano-sized carriers in pulmonary drug delivery remains challenging due to their high aggregation tendency and low weight, which results in their rapid exhalation. In order to avoid both clearance mechanisms, while simultaneously obtaining high fractions of respirable delivery and sufficient chemotherapeutic dosages in the lower respiratory tract, it is suggested to encapsulate nano-sized carriers with particle sizes of less than 200 nm into the secondary carriers with the size ranging from 3 to 5 µm. Besides particle size, a polydispersibility index is significant in determining the uniformity of the particles in the formulation. The value higher than 0.2 shows the multiple sizes of particles in the formulation, while smaller polydispersibility index value shows more uniform size particles in the given formulation system.
4.4.2. Shape
In addition to particle size, the particle shape also influences the efficacy of inhaled particles through modulation of the alveolar macrophage clearance. Investigation on the particles with different morphological conformations, including oblate ellipsoids, elliptical disks, rectangular disks, spheres, and worm-like shape, indicated that the orientation and shape of these materials substantially affect their phagocytosis clearance hours[158, 159]. Orientation bias was observed in the macrophage function, where the elliptical disks where engulfed in less than 6 minutes due to the attachment of macrophages to their major axes. The spherical particles were also immediately cleared regardless of the macrophage attachment point. In contrast, macrophage attachment to the flat surfaces or minor axes of the rectangular disks, oblate ellipsoids, and elliptical disks could not clear these particles even in 2 hours[158]. Inhalation of the particles with a worm-like shape also led to significantly less phagocytosis clearance in contrast to spherical shape mainly because of their low curvature region[159].
4.4.3. Surface properties
The particles’ surface properties are known to influence their bioavailability significantly. In particular, the surface properties determine the hydrophilicity degree, surface charge (zeta potential), biocompatibility or cytotoxicity, and stealth characteristics. In general, the particle distribution in the extracellular matrix, as well as their penetration between and into the cells, significantly rely on their degree of hydrophilicity. Lipophilic particles could more readily enter the cells due to their higher diffusivity in the lipid membranes. The cell uptake is also controlled by the particles’ surface charge, where the positively charged particles could penetrate easily into the cells due to their higher binding activity. On top of that, when the zeta potential values are above ±30 Mv, the particles’ stability in the suspension is greatly improved[160] due to repulsion between particles and less aggregation helping in a uniform distribution of particles in the system thereby establishing better shelf life of the formulation.
The surface properties also substantially influence the clearance mechanism in the pulmonary system, where adhesion interactions between the particles and mucus usually occur via electrostatic, hydrophobic, and hydrogen bonding[161]. It is, therefore, necessary to manipulate the physicochemical properties of the carriers such as surface charge or hydrophobicity to provide stealth characteristics to minimize absorption, degradation, entrapment in the mucus, and thus increasing the penetration depth and prolonging the half-life of the inhaled drugs across the mucus layers. The stealth behavior is generally obtained by conjugation of hydrophilic polymers such as PEG, Pluronic F-127, or adding mucolytic agents such as polyacrylic acid (PAA) and papain on the particles’ surface to disrupt the mucoglycoprotein substructures and suppress the immune recognition, and phagocytosis[162, 163]. Wang et al. have demonstrated that coating nanoparticles with a high density of low molecular weight (MW) PEG can reduce the interactions between particle and mucus. The possible reasons are as follows: the MW of PEG was too small to support adhesion via polymer chains interpenetration, and the PEG density is sufficient to shield the hydrophobic core effectively[164].
5. Inhalable controlled-release treatments for PAH
Herein, we highlight controlled, targeted drug delivery systems aimed at delivering anti-PAH drugs on the site of action.
5.1. Polymeric micro and nano particulate drug delivery systems
Polymeric carriers have been extensively studied as carriers for inhalable, controlled-release formulations of drugs because this approach has tremendous potential for producing prolonged, localized delivery of anti-PAH agents. PLGA has chiefly been used to prepare micro- and nanoparticles for inhalable controlled-release formulations.
Prostaglandin E1 (PGE1) is a selective pulmonary vasodilator with a biological half-life of 3 minutes. To overcome the pharmacokinetic deficiency of this drug, our lab developed a PLGA formulation of prostaglandin E1 (PGE1), which remarkably increased the drug half-life following pulmonary administration[165, 166]. Long-acting formulation of PGE1 produced the same effects at a reduced dosing frequency compared to plain PGE1 and caused minimal systemic adverse effects. In the other study, Gupta et al. developed large porous PLGA microparticles modified with polyethyleneimine (PEI) as a carrier for pulmonary delivery of PGE1. Incorporation of PEI in the formulations resulted in an increased entrapment efficiency to 95.48±0.46%. Also, a remarkable extension of the circulation half-life up to 6.0–6.5 hours was observed when the formulations were administered via the lungs[167]. The same group evaluated the efficacy and viability of PLGA microspheres encapsulating an inclusion complex of prostaglandin E1 (PGE1) and 2-hydroxypropyl-β- cyclodextrin (HPβCD) for pulmonary delivery of PGE1Incorporation of HPβCD in the microparticles resulted in development of particles with internal pores, aerodynamic diameters of 1 to 5 μm and significant increase in the amount of in-vitro drug release compared to plain PLGA formulation. However, compared to plain PLGA microparticles, entrapment efficiency was decreased upon complexation with HPβCD. The pharmacokinetic profile showed prolonged availability of PGE1 in circulation following the pulmonary administration of the microparticulate formulations[166].
Similar to other PGI2 analogs, the oral beraprost has vasodilatory and antiplatelet activity, but similar to other prostanoids has a very short half-life (1 h). To overcome the pharmacokinetic limitations of the drug, Akagi et al. fabricated PLGA nanoparticles containing beraprost and evaluated the efficacy of the formulation in MCT- and Sugen/Hypoxia-induced PAH rats[168]. After a single intratracheal administration of nanoformulation of bepraprost, RVH, right ventricular systolic pressure, and the muscularization of small pulmonary arteries were significantly decreased compared to controls in both PAH models. Furthermore, the survival rate increased to 65% following the administration of beraprost-containing nanoparticles, compared to 27.8% in controls. This study effectively revealed the advantages offered by nanoparticle-based controlled drug delivery such as reduced frequency of administrations and lower doses to achieve similar efficacious responses.
Mohamed et al. developed a novel hydrogel like polymer composite nanoparticle to deliver NO[169]. The formulation released NO in a sustained fashion over time and showed pronounced concentration-dependent vasodilation effect in hypoxia-induced PAH mice compared to healthy mice.
Beck-Broichsitter et al. have fabricated PLGA microparticles of sildenafil using a novel spray drying technique[170]. The sustained release profile of these microparticles makes the formulation potentially beneficial for controlled pulmonary drug delivery in PAH. In another study, Rashid et al. developed a stable PLGA microparticle containing sildenafil (size< 1 µm) [137]. Intratracheal administration of the formulation loaded with sildenafil elicited more pulmonary specific and sustained vasodilation in SUGEN/hypoxia-induced PAH rats than other routes of administrations.
Peroxisome-proliferator-activated-receptor-gamma (PPAR-γ) is implicated, in some capacity, in the pathogenesis of pulmonary arterial hypertension (PAH). Rosiglitazone, an oral antidiabetic and PPAR-γ agonist has the potential to dilate pulmonary arteries and to attenuate arterial remodeling in PAH[171]. Rashid et al. prepared and optimized PLGA based particles of rosiglitazone to repurpose it as an inhaled formulation for the treatment of PAH. The optimized particles were porous and spherical and released 87.9%±6.7% of the drug in 24 hours. The elimination half-life of the drug formulated in PLGA particles was 2.5-fold higher than that of the plain drug administered via the pulmonary route at the same dose.
Additionally, the optimized formulation, given via the pulmonary route, produced selective pulmonary vasodilation in PAH animals[172]. To develop a new combination therapy, Rashid et al. prepared and characterized PLGA-based long-acting inhalable particles of sildenafil and rosiglitazone. Compared with the plain drugs, given via the pulmonary route as a single or dual combination, PLGA particles of the drugs caused a more pronounced reduction in mPAP without affecting mean systemic pressure, improved cardiac function, slowed down right heart remodeling, and reduced arterial muscularization[173].
sGC activators and stimulators lack specificity for the pulmonary circulation and cause dose-dependent hypotension. To overcome this obstacle, Evgenov et al. evaluated the efficacy of sGC stimulators on PAH-induced lambs following aerosolized delivery of the drug in microparticulate formulations[174]. Inhalation of the microparticles produced dose-dependent pulmonary vasodilation, enhanced oxygenation, and increased cGMP release without lowering mSAP, indicating selective local drug delivery of inhaled sGC stimulator.
The anti-proliferative application of TKI in a controlled-release formulation via the inhaled route may affect more specifically and limit side effects on off-target organs. Thus, Akagi et al. incorporated imatinib in PLGA nanoparticles and evaluated their efficacy in MCT-induced PAH rats[175]. Following a single intratracheal administration, imatinib-loaded nanoformulation reduced the muscularization of small pulmonary vessels by 50%, right ventricle systolic pressure by 40%, and RVH compared to vehicle controls.
Given that statin, specifically, pitavastatin has anti-proliferative effects, Chen et al. formulated a novel PLGA nanoparticle loaded with pitavastatin to examine its impact on the PAH[176]. A single intratracheal administration of the formulation resulted in the accumulation of nanoparticles into alveolar macrophages and small PAs for up to 14 days. Moreover, intratracheal treatment with pitavastatin-nanoparticle, but not with pitavastatin, reduced the inflammation, lowered pulmonary arterial remodeling, decreased RV pressure (Figure 4B), attenuated the development of PAH, and improved the survival rates (Figure 4A). Importantly, a Phase I clinical trial has evaluated the efficacy of PLGA nanoparticle-based delivery of pitavastatin (UMIN000014940). Future clinical trials may prove the safety and efficacy of a pitavastatin-PLGA nanoparticle for patients with PAH.
Figure 4.
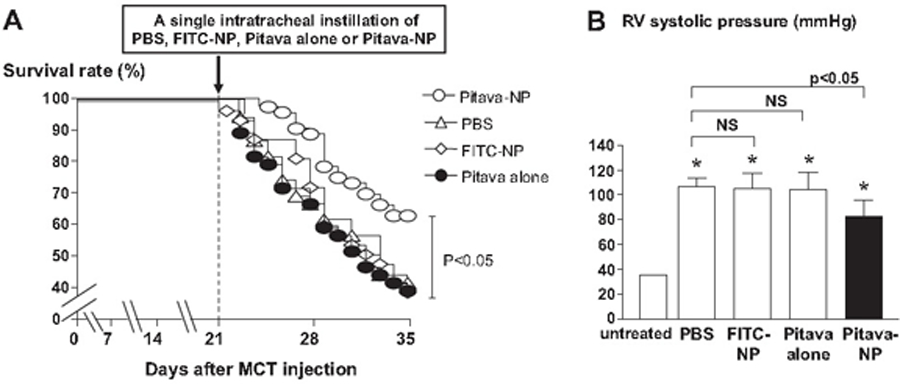
Effects of pitavastatin-NP on right ventricle (RV) systolic pressure and survival rate. (a) Survival curves analyzed by the Kaplan-Meier method in PBS, FITC-NP, pitavastatin only, and pitavastatin-NP groups. (b) RV systolic pressure (in mmHg) in the 4 experimental groups 2 weeks after treatment (at week 5 after MCT injection).
The above findings suggest that polymeric micro/nanocarriers may offer a viable alternative for delivery of anti-PAH medications directly to the lungs with sustained therapeutic benefits.
5.2. Liposomes
Liposomes have been studied as carriers for pulmonary delivery of various therapeutic agents for many years. Several studies have reported the use of liposomal drug-delivery systems for inhalational delivery of anti-PAH medications.
Since the clinically approved inhaled iloprost has a short half-life (7 min), it requires 12 inhalations per day, which primarily impact patient compliance. To increase iloprost bioavailability, Kleemann et al. developed an aerosolized liposomal formulation for sustained release of iloprost for PAH treatmnet[177]. Liposomes consisted of cholesterol and di-palmitoyl-phosphatidyl-choline to improve continuous delivery, and poly (ethylene glycol)-dipalmitoyl-phosphatidyl-ethanolamine to prevent clearance by pulmonary macrophages, which would decrease their bioavailability. Resulting liposomes contained 11 mg iloprost/ml, which would significantly reduce the number of inhalations required. In the other study, Jain et al. fabricated iloprost-containing liposomes with cationic lipids to enhance drug encapsulation efficiencies[178]. They evaluated the efficacy of their drug-formulation compared with free drug. Liposomal iloprost led to significant enhancement of vasodilation, with a lower concentration of liposomal iloprost required to bring about efficiencies similar to that of the free drug.
Fasudil has a very short half-life of 45 min, which limits the use of this potent vasodilator for long-term treatment of PAH[179]. In the hope of increasing drug half-life, Gupta et al. developed a liposomal formulation of fasudil for purposes of aerosolized delivery to lungs undergoing PAH[180]. Resulting liposomes were 180 nm in size, had loading efficiencies >50% with 80% in-vitro release. Intratracheal administration of the formulation increased the half-life by more than 10-fold, as well as the bioavailability of the drug, compared to a free drug administered via the IV route. Liposomal fasudil resulted in an increase in the duration of vasodilatory effects compared to controls in MCT-induced PAH rats.
The pulmonary vasculature plays a pivotal role in PAH, and therefore the diseases are likely to benefit from vascular-targeted drug delivery. Targeting lung-specific moieties provide an extraordinarily rapid and specific means to target pulmonary vasculature and potentially deliver therapeutic agents into the lung tissue. The roster of candidate molecules as endothelial targeting moieties includes peptidases such as aminopeptidase P (APP)[181], ECM component such as heparan sulfate[182], cell adhesion molecules and integrins[183, 184], localized in different domains of the endothelial plasmalemma and differentially distributed throughout the vasculature. For example, PECAM and VE-cadherin are localized in cell-cell borders[185–187], whereas VCAM-1 and ICAM-1 are found in micro-domains of the cellular apical surface[188–190]. Glycoprotein gp85 localizes to the luminal surface of the plasmalemma that belongs to a thin organelle-free part of the endothelial cell separating alveoli from blood[191]. Endowing carriers with a high affinity to a specific layer of pulmonary arteries enables an exceptional level of precision of control of drug delivery through binding to selected cell phenotypes, reducing unwanted effects, and adjusting the duration of therapeutic effects.
One of the strategy for targeted drug delivery for PAH is the CAR (CARSKNKDC) peptide which specifically homes to hypertensive pulmonary arteries of PAH-induced rats and accumulates in all layers of the remodeled arteries (endothelium, medial smooth muscle, and adventitia of pulmonary arteries) and vasculature and distributes in the extravascular space (interstitial space and alveolar lumen) and entire wall of hypertensive pulmonary arteries[192]. CAR is a cyclic peptide identified for homing to areas of angiogenesis during skin wound healing[193]. The CAR binding site is putatively heparan sulfate proteoglycans. Within two hours of IV or pulmonary administration, CAR binds many cell types, mainly pulmonary arterial (endothelial, smooth muscle cells, which overexpressed a specific pattern of heparan sulfate in the lungs of monocrotaline rats. CAR also can penetrate cells, which can facilitate particle movement from the alveolar ducts into the pulmonary arterioles, the site of action for anti-PAH drugs[194]. In an attempt to improve the site-specific accumulation of nanoparticles with prolonged effect, Keshavarz et al. and Nahar et al. subsequently developed fasudil liposomes with the homing cyclic peptide (CARSKNKDC), which binds to cell surface heparan sulfate which overexpressed in the diseased PAs[195, 196]. Liposomes were 200 nm in size, had loading efficiencies >90% with a sustained-release profile of fasudil. Peptide-coated liposomes resulted in a 32-fold increase in the half-life of the drug compared to an IV administered of free medications. The authors evaluated the effect of the various formulations on mPAP and mSAP in PAH rats. Fasudil in plain liposomes (Figure 5A) and CAR pretreatment followed by fasudil in plain liposomes (Figure 5B) administration reduced the mPAP to 28 % and 34 % of the initial mPAP. However, echoing the pharmacokinetics profile, the CAR-conjugated liposomes containing fasudil produced a much-pronounced reduction in mPAP (53%), and the vasodilation persisted over 6 hrs following the intratracheal administration (Figure 5C). Figure 5D showed that the lung targeting index (LTI) for fasudil-conjugated liposomes was about 3-fold higher than those for two other treatments, indicating a more considerable lung accumulation and reduced peripheral vasodilation. Also, the formulations were found to be safe for pulmonary administration, as demonstrated by acute cytotoxicity studies in the Calu-3 lung epithelial cell line and by studies of bronchoalveolar lavage (BAL) fluid. In the other study, Nahar et al. developed magnetic liposomes as a carrier for the pulmonary preferential accumulation of fasudil. Optimized safe and stable liposomes with a mean aerodynamic diameter of 164 nm and entrapment efficiency of 85% exhibited extended half-life compared with plain fasudil, indicating the feasibility of magnetic liposome for prolonged site-specific treatment of PAH[197].
Figure 5.
The effect of liposomal fasudil on the pulmonary hemodynamics in Sugen/hypoxia PAH rats after administration of (a) fasudil in no-CAR-liposomes, (b) CAR pretreatment followed by fasudil in no-CAR-liposomes and (c) fasudil in CAR-liposomes. (d) Targeting indices of fasudil after IT administration in various forms. The dose of fasudil was 3 mg/kg (rat body weight) or liposomes containing equivalent amount of fasudil and the dose of CAR was 0.5 mg/kg (data represent mean ± SD, n = 3; *p < 0.05, §p < 0.005, ?p < 0.001).
Since high ROS levels play a pivotal role in vascular remodeling in PAH, Gupta et al. incorporated superoxide dismutase (SOD), a ROS scavenger peptide, to enhance the efficacy of peptide-targeted fasudil liposomes[198]. In a Sugen/Hypoxia model of PAH, fractions of occluded blood vessels, arterial medial wall thickness, mPAP, and RVH declined in rats receiving targeted liposomes containing both fasudil and SOD compared to free drug controls.
Huang et al. developed a liposomal formulation of NO consisting of cholesterol, 1,2-dioleoyl-sn-glycerol-3-phosphocholine, and 1,2-dipalmitoyl-sn-glycerol-3-ethylphosphocholine [199]. These liposomes encapsulated 10 mL of NO/mg of lipids. In-vitro release profile showed a slower release pattern of NO from liposomes, resulting in a sustained release profile. Cultured rat vascular SMCs did not reveal any toxicity after incubation with liposomal NO, indicating the safety of the formulation. Additionally, a seven-fold increase in uptake of NO by SMCs was observed with the liposomal NO than free NO. Furthermore, formulation protected NO from microenvironmental scavengers such as hemoglobin. In-vivo efficacy was evaluated in a PAH rabbit model. After two weeks, diseased rabbits treated with liposomal NO demonstrated a significant decrease in intimal hyperplasia compared to vehicle controls, suggesting the feasibility of the delivery of bio-active gases in nanoparticles. Recently, Our lab investigated the feasibility of a combination therapy comprising fasudil and DETA NONOate (diethylenetriamine NONOate), a long-acting NO donor, both loaded in a liposomal formulation modified with a homing peptide, CAR, for the PAH treatment in MCT and SUGEN/hypoxia PAH models following intratracheal administration[200, 201]. Evaluation of tissue samples of the PAH lungs proved both drugs, formulated in CAR-modified liposomes, reduced the degree of collagen deposition, muscularization, and medial arterial wall thickness more than the combination of plain drugs. In-vivo efficacy results revealed that CAR-modified liposomes were the most selective formulation in reducing mPAP for a long period. In consistent with the in-vivo data, CAR-modified liposomes of fasudil and DETA NONOate increased the levels of the vasodilatory signaling molecule, cGMP, in the SMCs of PAH-afflicted PAs. This study highly suggested that fasudil and DETA NONOate, formulated in liposomes, has the potential to be applied as a combination therapy for the management of PAH.
Based on the above information, we anticipate that liposomal delivery systems of anti-PAH medications may eventually play a significant role in PAH treatment and in targeting the diseased cells of the pulmonary circulation.
5.3. Micelles
Micellar carriers promote specificity, improve efficacy, and enhance the safety of encapsulated therapeutic agents[202].
Adrenomedullin has been implicated as a key player in cardiovascular events and has vasodilatory and antiproliferative properties, resulting in a beneficial effect on PAH treatment[203]. Harada-Shiba et al. reported on the use of PEG-based cationic micelles (polyplex nanomicelles) for intratracheal, nonviral gene transfer to the lung of rats with monocrotaline-induced PAH. Polyplex nanomicelles were formulated with a therapeutic plasmid bearing the human adrenomedullin gene. Intratracheal administration of PEG-polyplex nanomicelles showed remarkable therapeutic efficacy with PAH animal models, as evidenced by significantly decreasing the right ventricular pressure three days after administration, which was confirmed by a notable increase of pulmonary human adrenomedullin mRNA levels[204].
Gupta et al. evaluated respirability and efficacy of CAR-conjugated peptide–micelle hybrid nanoparticles containing fasudil as carriers for inhalational therapy of PAH. The micelle −with the average size of 14 nm and entrapment efficiency of 58%− was made of polyethylene glycol–distearoyl-phosphoethanolamine (DSPE–PEG) conjugate. Compared with plain micelles, CAR-micelle increased the cellular uptake by approximately 1.7-fold and extended the drug half-life by fivefold, demonstrating that peptide–polymer hybrid micelles can serve as inhalational carriers for PAH therapy (Figure 6) [150].
Figure 6.
(a) Uptake of plain and CAR-conjugated micelles (green color) by TGF-?-activated rat PASM cells (actin stained red). (b) Flow cytometric analysis of cellular uptake of formulations by activated cells (data represent mean ± SD, n = 3). (c) Viability of rat PASM cells upon incubation with formulations or individual components of formulation (data represent mean ± SD, n = 12).
5.4. Other approaches
There are several new and exciting approaches that are being investigated for the development of inhalational sustained-release drug-delivery systems.
Nanoerythrosomes (NERs), an engineered derivative of erythrocytes, have long been used as drug delivery carriers. These cell-based carriers are biocompatible and biodegradable, and they demonstrate efficient drug loading, targeting specificity, and prolonged biological half-life. Gupta et al. developed NERs as inhalable carriers for the delivery of fasudil for the treatment of PAH. Spherical NERs with an average size of 154.1±1.31 nm and the drug loading efficiency of 48.76±2.18% were stable at 4 °C for three weeks. The drug entrapped in NERs inhibited the ROCK activity up to 50% and prolonged the half-life of fasudil 6–8 fold[205, 206].
To protects VIP against rapid enzymatic degradation and improve the drug’s half-life, Wernig et al. encapsulated VIP into the nanoparticle matrix of biodegradable protamine-oligonucleotide nanoparticles (proticles)[207]. Proticles demonstrated an appropriate nanoparticle size (177–251 nm), satisfying encapsulation efficiency (up to 80%), and sustained release profile. The pharmacological response to VIP revealed prolonged vasodilation following inhalation of VIP-containing formulation, indicating the suitability of proticles as a pulmonary VIP depot system.
6. Expert opinion
PAH is a progressive, lethal disorder characterized by thickening and consequently, high blood pressure in the PAs due to the imbalance of growth factors, inflammatory cytokines, and various neurochemical mediators secreted from diseased pulmonary arterial cells. Therapy for PAH has improved over the last 20 years that resulted in FDA approval of ten anti-PAH drugs. As the disease progresses, combination therapy is added according to the symptoms and functional class of the patients. Current treatments have been focused on reestablishing the right balance between vasoconstriction and vasodilatation, while some of them have anti-proliferative effects that tackles the abnormal proliferation of PAECs and PASMCs via augmenting the prostacyclin pathway, enhancing the NO pathway, or inhibiting the endothelin pathway. One of the significant limitations of our current PAH therapies is that currently approved drugs act mainly on arterial vasodilation and do not target the pulmonary arterial remodeling characteristic of the disease, which leads to a treatment approach that stabilize the condition rather than reversing the underlying pathology. This situation triggered consideration of other pathways responsible for PAH progression resulting in the advent of various investigational anti-PAH treatments such as Rock inhibitors, statins, and TKI. Despite these new approved and investigational drugs, the patient survival is still disappointing. Thus, the PAH patients is in dire need of a new treatment strategy that could reduce mortality and morbidity and ameliorate functional deterioration. The main shortcomings of current pharmacotherapies of PAH are drug half-life, stability, high dose, frequent administration, and formulation limitations, resulting in lowering the efficacy and deleterious side effects. Efforts have been made to develop novel approaches to deliver anti-PAH drugs to overcome the limitations of current anti-PAH therapies.
The pulmonary route has been proposed for delivering of anti-PAH drugs to avoid the systemic side effects. Using the inhaled route to deliver therapeutic aerosols and gases to patients with PAH offers potential advantages compared to the current injectable or oral treatments, including better-targeted therapy to the diseased region, which could be more effective, less toxic, and less expensive than other routes. Of the available inhaled therapies, inhaled NO was approved mainly for hospitalized pediatric patients with PAH. Two other inhaled medications, iloprost and treprostinil, are prostacyclin analogs that are FDA-approved for PAH and effective in increasing exercise capacity. However, they have not been used as widely as oral PAH therapies because of limited efficacy and the need for frequent administration.
These limitations can be overcome by applying novel nano/micro controlled-release technologies such as liposomes, micelles, and solid polymeric particles functionalized with a homing device to accurately deliver anti-PAH drugs to the site of action via inhalation administration. Nano/microparticles-based strategies for the treatment of PAH offer advantages of improving short-term pharmacokinetics associated with drugs and increased localization of therapy to diseased lung, in turn improving the efficacy and decreasing adverse effects of the treatment. Several studies have reported successful development of controlled-release drug delivery systems loaded with available anti-PAH drugs to treat PAH in human and rodent models. Biocompatibility, size, charge, and drug release rates of the particles must all be considered for the selection of drug carriers. Particle size dictates regional lung deposition of the particles after inhalation[208]. Large particles in size range of 1–5 µm deposit in bronchioles and smaller airways, particles in size range of 0.1–1 µm accumulate in alveolar regions and bypass alveolar macrophage clearance, and smaller particles (<0.1 µm) can undergo exhalation. Inhalational delivery of controlled-release nanoparticles containing targeting moiety represents an attractive strategy for mainly targeting pulmonary tissues and has several advantages, including selective short- and long-term vasodilation of the PAs, reduced dose, dosing frequency, enhanced patient compliance, higher bioavailability as well as avoiding the clearance mechanisms of the lung and the risk of being exhaled. As such, clinical trials can demonstrate whether long-term treatment with controlled-release, targeted, inhaled nano/micro formulations are both safe and efficacious in patients with PAH.
Article highlights.
Current PAH therapies have been focused on reestablishing the right balance between vasoconstriction and vasodilatation in pulmonary arteries, while some of them have anti-proliferative effects that tackle the abnormal proliferation of pulmonary arterial cells via augmenting the prostacyclin pathway, enhancing the NO pathway, or inhibiting the endothelin pathway.
While current pharmacotherapies have improved the quality of life of the patients, PAH drugs suffer from limitations in the form of instability, poor organ specificity, short-term pharmacokinetics, and formulation limitations, resulting in deleterious side effects and inadequate efficacy.
Inhalational delivery offers several advantages in the PAH treatment including, local drug delivery, higher concentration of therapeutics on the site of action, avoidance of systemic adverse effects, the lower total dose of medication with less frequent administrations, rapid drug absorption and fast onset of effect, and avoidance of the first-pass metabolism.
Polymeric carriers have been extensively studied as carriers for inhalable, controlled-release formulations of therapeutics because this approach has tremendous potential for producing prolonged, localized delivery of anti-PAH agents, including, sildenafil, prostaglandin E1, rosiglitazone, and pitavastatin.
Liposomes have been applied as carriers for inhaled, controlled, targeted drug delivery of various PAH treatments such as iloprost, NO, NO donors, and fasudil.
Inhalational delivery of controlled-release nanoparticles containing targeting moiety represents an attractive strategy for mainly targeting pulmonary tissues and has several advantages, including selective short- and long-term vasodilation of the pulmonary arteries, reduced dose and dosing frequency, enhanced patient compliance, higher bioavailability as well as avoiding the clearance mechanisms of the lung and the risk of being exhaled.
Acknowledgments
Funding
This work was supported in part by two NIH grants (R01HL114677 and R01HL144590) and the Cardiovascular Medical Research and Education Fund (CMREF) grant awarded to F Ahsan.
List of abbreviations
- PAH
pulmonary arterial hypertension
- Pas
pulmonary arteries
- mPAP
mean pulmonary artery pressure
- PVR
pulmonary vascular resistance
- RVH
right ventricular hypertrophy
- BMPRII
bone morphogenetic protein receptor II
- ALK
activin receptor-like kinase
- PAECs
pulmonary artery endothelial cells
- PASMCs
pulmonary artery smooth muscle cells
- NO
nitric oxide
- ET-1
endothelin-1
- MMPs
matrix metalloproteases
- HIV
human immunodeficiency virus
- COPD
chronic obstructive pulmonary disease
- WHO FC
World Health Organization Functional Class
- NYHA FC
New York Health Association Functional class
- 6MWD
6-minute walk distance
- BNP
brain natriuretic peptide
- ERA
ET receptor antagonists
- PGI2
Prostaglandin I2
- cAMP
cyclic adenosine 3′,5′-monophosphate
- IV
intravenous
- mSAP
mean systemic arterial pressure
- eNO
endothelial-derived nitric oxide
- eNOS
endothelial nitric oxide synthases
- sGC
soluble guanylate cyclase
- cGMP
cyclic guanosine monophosphate
- PDE
phosphodiesterase
- ROS
reactive oxygen species
- ETA
ET receptor A
- VIP
vasoactive intestinal peptide pathway
- ROCK
Rho kinases
- PDGF
platelet-derived growth factor
- FGF
fibroblast growth factor
- VEGF
vascular endothelial growth factor
- TKI
tyrosine kinase inhibitors
- PLGA
polylactide-co-glycolide
- PEG
polyethylene glycol
- PGE1
prostaglandin E1
- NERs
nanoerythrosomes
- SOD
superoxide dismutase
Footnotes
Declaration of interest
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as:
* of interest
** of considerable interest
- 1.Badesch DB, Champion HC, Sanchez MA, Hoeper MM, Loyd JE, Manes A, et al. Diagnosis and assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2009. June 30;54(1 Suppl):S55–66. [DOI] [PubMed] [Google Scholar]
- 2.McLaughlin VV, Archer SL, Badesch DB, Barst RJ, Farber HW, Lindner JR, et al. ACCF/AHA 2009 expert consensus document on pulmonary hypertension: a report of the American College of Cardiology Foundation Task Force on Expert Consensus Documents and the American Heart Association: developed in collaboration with the American College of Chest Physicians, American Thoracic Society, Inc., and the Pulmonary Hypertension Association. Circulation 2009. April 28;119(16):2250–94. [DOI] [PubMed] [Google Scholar]
- 3.Archer S, Rich S. Primary pulmonary hypertension: a vascular biology and translational research “Work in progress”. Circulation 2000. November 28;102(22):2781–91. [DOI] [PubMed] [Google Scholar]
- 4.Farber HW, Loscalzo J. Pulmonary arterial hypertension. N Engl J Med 2004. October 14;351(16):1655–65. [DOI] [PubMed] [Google Scholar]
- 5.Tian W, Jiang X, Sung YK, Shuffle E, Wu TH, Kao PN, et al. Phenotypically Silent Bone Morphogenetic Protein Receptor 2 Mutations Predispose Rats to Inflammation-Induced Pulmonary Arterial Hypertension by Enhancing the Risk for Neointimal Transformation. Circulation 2019. October 22;140(17):1409–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brenner L, Chung WK. Clinical and molecular genetic features of hereditary pulmonary arterial hypertension. Compr Physiol 2011. October;1(4):1721–8. [DOI] [PubMed] [Google Scholar]
- 7.Rabinovitch M, Bothwell T, Hayakawa BN, Williams WG, Trusler GA, Rowe RD, et al. Pulmonary artery endothelial abnormalities in patients with congenital heart defects and pulmonary hypertension. A correlation of light with scanning electron microscopy and transmission electron microscopy. Lab Invest 1986. December;55(6):632–53. [PubMed] [Google Scholar]
- 8.Dewachter L, Adnot S, Fadel E, Humbert M, Maitre B, Barlier-Mur AM, et al. Angiopoietin/Tie2 pathway influences smooth muscle hyperplasia in idiopathic pulmonary hypertension. Am J Respir Crit Care Med 2006. November 1;174(9):1025–33. [DOI] [PubMed] [Google Scholar]
- 9.Thompson K, Rabinovitch M. Exogenous leukocyte and endogenous elastases can mediate mitogenic activity in pulmonary artery smooth muscle cells by release of extracellular-matrix bound basic fibroblast growth factor. J Cell Physiol 1996. March;166(3):495–505. [DOI] [PubMed] [Google Scholar]
- 10.Price LC, Wort SJ, Perros F, Dorfmuller P, Huertas A, Montani D, et al. Inflammation in pulmonary arterial hypertension. Chest 2012. January;141(1):210–21. [DOI] [PubMed] [Google Scholar]
- 11.Singer ST, Kuypers FA, Styles L, Vichinsky EP, Foote D, Rosenfeld H. Pulmonary hypertension in thalassemia: association with platelet activation and hypercoagulable state. Am J Hematol 2006. September;81(9):670–5. [DOI] [PubMed] [Google Scholar]
- 12.Li N, Zhang S, Hou J, Jang IK, Yu B. Assessment of pulmonary artery morphology by optical coherence tomography. Heart Lung Circ 2012. December;21(12):778–81. [DOI] [PubMed] [Google Scholar]
- 13.Humbert M, Morrell NW, Archer SL, Stenmark KR, MacLean MR, Lang IM, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 2004. June 16;43(12 Suppl S):13S–24S. [DOI] [PubMed] [Google Scholar]
- 14.Yuan XJ, Wang J, Juhaszova M, Gaine SP, Rubin LJ. Attenuated K+ channel gene transcription in primary pulmonary hypertension. Lancet 1998. March 7;351(9104):726–7. [DOI] [PubMed] [Google Scholar]
- 15.Alastalo TP, Li M, Perez Vde J, Pham D, Sawada H, Wang JK, et al. Disruption of PPARgamma/beta-catenin-mediated regulation of apelin impairs BMP-induced mouse and human pulmonary arterial EC survival. J Clin Invest 2011. September;121(9):3735–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lai YC, Potoka KC, Champion HC, Mora AL, Gladwin MT. Pulmonary arterial hypertension: the clinical syndrome. Circ Res 2014. June 20;115(1):115–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jia D, He Y, Zhu Q, Liu H, Zuo C, Chen G, et al. RAGE-mediated extracellular matrix proteins accumulation exacerbates HySu-induced pulmonary hypertension. Cardiovasc Res 2017. May 1;113(6):586–97. [DOI] [PubMed] [Google Scholar]
- 18.Roldan T, Landzberg MJ, Deicicchi DJ, Atay JK, Waxman AB. Anticoagulation in patients with pulmonary arterial hypertension: An update on current knowledge. J Heart Lung Transplant 2016. February;35(2):151–64. [DOI] [PubMed] [Google Scholar]
- 19.Oshima K, Crockett ES, Joshi SR, McLendon JM, Matsumoto Y, McMurtry IF, et al. Aneurysm-type Plexiform Lesions Form in Supernumerary Arteries in Pulmonary Arterial Hypertension: Potential Therapeutic Implications. Am J Physiol Lung Cell Mol Physiol 2019. October 2. [DOI] [PMC free article] [PubMed]
- 20.Peacock AJ, Murphy NF, McMurray JJ, Caballero L, Stewart S. An epidemiological study of pulmonary arterial hypertension. Eur Respir J 2007. July;30(1):104–9. [DOI] [PubMed] [Google Scholar]
- 21.Ling Y, Johnson MK, Kiely DG, Condliffe R, Elliot CA, Gibbs JS, et al. Changing demographics, epidemiology, and survival of incident pulmonary arterial hypertension: results from the pulmonary hypertension registry of the United Kingdom and Ireland. Am J Respir Crit Care Med 2012. October 15;186(8):790–6. [DOI] [PubMed] [Google Scholar]
- 22.Simonneau G, Gatzoulis MA, Adatia I, Celermajer D, Denton C, Ghofrani A, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2013. December 24;62(25 Suppl):D34–41. [DOI] [PubMed] [Google Scholar]
- 23.Kylhammar D, Kjellstrom B, Hjalmarsson C, Jansson K, Nisell M, Soderberg S, et al. A comprehensive risk stratification at early follow-up determines prognosis in pulmonary arterial hypertension. Eur Heart J 2018. December 14;39(47):4175–81. [DOI] [PubMed] [Google Scholar]
- 24.Matura LA, Shou H, Fritz JS, Smith KA, Vaidya A, Pinder D, et al. Physical Activity and Symptoms in Pulmonary Arterial Hypertension. Chest 2016. July;150(1):46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schannwell CM, Steiner S, Strauer BE. Diagnostics in pulmonary hypertension. J Physiol Pharmacol 2007. November;58 Suppl 5(Pt 2):591–602. [PubMed] [Google Scholar]
- 26.Maron BA, Galie N. Diagnosis, Treatment, and Clinical Management of Pulmonary Arterial Hypertension in the Contemporary Era: A Review. JAMA Cardiol 2016. December 1;1(9):1056–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taichman DB, Ornelas J, Chung L, Klinger JR, Lewis S, Mandel J, et al. Pharmacologic therapy for pulmonary arterial hypertension in adults: CHEST guideline and expert panel report. Chest 2014. August;146(2):449–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moncada S, Gryglewski R, Bunting S, Vane JR. An enzyme isolated from arteries transforms prostaglandin endoperoxides to an unstable substance that inhibits platelet aggregation. Nature 1976. October 21;263(5579):663–5. [DOI] [PubMed] [Google Scholar]
- 29.Humbert M, Ghofrani HA. The molecular targets of approved treatments for pulmonary arterial hypertension. Thorax 2016. January;71(1):73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 1999. June;159(6):1925–32. [DOI] [PubMed] [Google Scholar]
- 31.Galie N, Hoeper MM, Humbert M, Torbicki A, Vachiery JL, Barbera JA, et al. Guidelines for the diagnosis and treatment of pulmonary hypertension: the Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS), endorsed by the International Society of Heart and Lung Transplantation (ISHLT). Eur Heart J 2009. October;30(20):2493–537. [DOI] [PubMed] [Google Scholar]
- 32.Sitbon O, Humbert M, Nunes H, Parent F, Garcia G, Herve P, et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol 2002. August 21;40(4):780–8. [DOI] [PubMed] [Google Scholar]
- 33.De Wet CJ, Affleck DG, Jacobsohn E, Avidan MS, Tymkew H, Hill LL, et al. Inhaled prostacyclin is safe, effective, and affordable in patients with pulmonary hypertension, right heart dysfunction, and refractory hypoxemia after cardiothoracic surgery. J Thorac Cardiovasc Surg 2004. April;127(4):1058–67. [DOI] [PubMed] [Google Scholar]
- 34.Buckley MS, Feldman JP. Inhaled epoprostenol for the treatment of pulmonary arterial hypertension in critically ill adults. Pharmacotherapy 2010. July;30(7):728–40. [DOI] [PubMed] [Google Scholar]
- 35.Gomberg-Maitland M, Olschewski H. Prostacyclin therapies for the treatment of pulmonary arterial hypertension. Eur Respir J 2008. April;31(4):891–901. [DOI] [PubMed] [Google Scholar]
- 36.Hoeper MM, Schwarze M, Ehlerding S, Adler-Schuermeyer A, Spiekerkoetter E, Niedermeyer J, et al. Long-term treatment of primary pulmonary hypertension with aerosolized iloprost, a prostacyclin analogue. N Engl J Med 2000. June 22;342(25):1866–70. [DOI] [PubMed] [Google Scholar]
- 37.Olschewski H, Simonneau G, Galie N, Higenbottam T, Naeije R, Rubin LJ, et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med 2002. August 1;347(5):322–9. [DOI] [PubMed] [Google Scholar]
- 38.Chon MK, Cho KI, Cha KS, Seo JS, Kim DS. Effects of long-term iloprost treatment on right ventricular function in patients with Eisenmenger syndrome. J Cardiol 2017. May;69(5):741–46. [DOI] [PubMed] [Google Scholar]
- 39.Opitz CF, Wensel R, Winkler J, Halank M, Bruch L, Kleber FX, et al. Clinical efficacy and survival with first-line inhaled iloprost therapy in patients with idiopathic pulmonary arterial hypertension. Eur Heart J 2005. September;26(18):1895–902. [DOI] [PubMed] [Google Scholar]
- 40.Sandifer BL, Brigham KL, Lawrence EC, Mottola D, Cuppels C, Parker RE. Potent effects of aerosol compared with intravenous treprostinil on the pulmonary circulation. J Appl Physiol (1985) 2005. December;99(6):2363–8. [DOI] [PubMed] [Google Scholar]
- 41.McLaughlin VV, Benza RL, Rubin LJ, Channick RN, Voswinckel R, Tapson VF, et al. Addition of inhaled treprostinil to oral therapy for pulmonary arterial hypertension: a randomized controlled clinical trial. J Am Coll Cardiol 2010. May 4;55(18):1915–22. [DOI] [PubMed] [Google Scholar]
- 42.Keil A, Blom IE, Goldschmeding R, Rupprecht HD. Nitric oxide down-regulates connective tissue growth factor in rat mesangial cells. Kidney Int 2002. August;62(2):401–11. [DOI] [PubMed] [Google Scholar]
- 43.Emerson M, Momi S, Paul W, Alberti PF, Page C, Gresele P. Endogenous nitric oxide acts as a natural antithrombotic agent in vivo by inhibiting platelet aggregation in the pulmonary vasculature. Thromb Haemost 1999. June;81(6):961–6. [PubMed] [Google Scholar]
- 44.Rybalkin SD, Yan C, Bornfeldt KE, Beavo JA. Cyclic GMP phosphodiesterases and regulation of smooth muscle function. Circ Res 2003. August 22;93(4):280–91. [DOI] [PubMed] [Google Scholar]
- 45.Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med 1995. July 27;333(4):214–21. [DOI] [PubMed] [Google Scholar]
- 46.Demoncheaux EA, Higenbottam TW, Kiely DG, Wong JM, Wharton S, Varcoe R, et al. Decreased whole body endogenous nitric oxide production in patients with primary pulmonary hypertension. J Vasc Res 2005. Mar-Apr;42(2):133–6. [DOI] [PubMed] [Google Scholar]
- 47.George I, Xydas S, Topkara VK, Ferdinando C, Barnwell EC, Gableman L, et al. Clinical indication for use and outcomes after inhaled nitric oxide therapy. Ann Thorac Surg 2006. December;82(6):2161–9. [DOI] [PubMed] [Google Scholar]
- 48.Walmrath D, Schneider T, Schermuly R, Olschewski H, Grimminger F, Seeger W. Direct comparison of inhaled nitric oxide and aerosolized prostacyclin in acute respiratory distress syndrome. Am J Respir Crit Care Med 1996. March;153(3):991–6. [DOI] [PubMed] [Google Scholar]
- 49.Clark RH, Kueser TJ, Walker MW, Southgate WM, Huckaby JL, Perez JA, et al. Low-dose nitric oxide therapy for persistent pulmonary hypertension of the newborn. Clinical Inhaled Nitric Oxide Research Group. N Engl J Med 2000. February 17;342(7):469–74. [DOI] [PubMed] [Google Scholar]
- 50.Roberts JD Jr., Fineman JR, Morin FC 3rd, Shaul, Rimar S, Schreiber MD, et al. Inhaled nitric oxide and persistent pulmonary hypertension of the newborn. The Inhaled Nitric Oxide Study Group. N Engl J Med 1997. February 27;336(9):605–10. [DOI] [PubMed] [Google Scholar]
- 51.Wetzel RC. Aerosolized prostacyclin. In search of the ideal pulmonary vasodilator. Anesthesiology 1995. June;82(6):1315–7. [DOI] [PubMed] [Google Scholar]
- 52.Jacobs BR, Brilli RJ, Ballard ET, Passerini DJ, Smith DJ. Aerosolized soluble nitric oxide donor improves oxygenation and pulmonary hypertension in acute lung injury. Am J Respir Crit Care Med 1998. November;158(5 Pt 1):1536–42. [DOI] [PubMed] [Google Scholar]
- 53.Murray F, MacLean MR, Pyne NJ. Increased expression of the cGMP-inhibited cAMP-specific (PDE3) and cGMP binding cGMP-specific (PDE5) phosphodiesterases in models of pulmonary hypertension. Br J Pharmacol 2002. December;137(8):1187–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Galie N, Ghofrani HA, Torbicki A, Barst RJ, Rubin LJ, Badesch D, et al. Sildenafil citrate therapy for pulmonary arterial hypertension. N Engl J Med 2005. November 17;353(20):2148–57. [DOI] [PubMed] [Google Scholar]
- 55.Rubin LJ, Badesch DB, Fleming TR, Galie N, Simonneau G, Ghofrani HA, et al. Long-term treatment with sildenafil citrate in pulmonary arterial hypertension: the SUPER-2 study. Chest 2011. November;140(5):1274–83. [DOI] [PubMed] [Google Scholar]
- 56.Ichinose F, Erana-Garcia J, Hromi J, Raveh Y, Jones R, Krim L, et al. Nebulized sildenafil is a selective pulmonary vasodilator in lambs with acute pulmonary hypertension. Crit Care Med 2001. May;29(5):1000–5. [DOI] [PubMed] [Google Scholar]
- 57.Beghetti M, Wacker Bou Puigdefabregas J, Merali S. Sildenafil for the treatment of pulmonary hypertension in children. Expert Rev Cardiovasc Ther 2014. October;12(10):1157–84. [DOI] [PubMed] [Google Scholar]
- 58.Vermeersch P, Buys E, Pokreisz P, Marsboom G, Ichinose F, Sips P, et al. Soluble guanylate cyclase-alpha1 deficiency selectively inhibits the pulmonary vasodilator response to nitric oxide and increases the pulmonary vascular remodeling response to chronic hypoxia. Circulation 2007. August 21;116(8):936–43. [DOI] [PubMed] [Google Scholar]
- 59.Schermuly RT, Stasch JP, Pullamsetti SS, Middendorff R, Muller D, Schluter KD, et al. Expression and function of soluble guanylate cyclase in pulmonary arterial hypertension. Eur Respir J 2008. October;32(4):881–91. [DOI] [PubMed] [Google Scholar]
- 60.O’Callaghan DS, Savale L, Yaici A, Natali D, Jais X, Parent F, et al. Endothelin receptor antagonists for the treatment of pulmonary arterial hypertension. Expert Opin Pharmacother 2011. July;12(10):1585–96. [DOI] [PubMed] [Google Scholar]
- 61.Dasgupta A, Bowman L, D’Arsigny CL, Archer SL. Soluble guanylate cyclase: a new therapeutic target for pulmonary arterial hypertension and chronic thromboembolic pulmonary hypertension. Clin Pharmacol Ther 2015. January;97(1):88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ghofrani HA, Galie N, Grimminger F, Grunig E, Humbert M, Jing ZC, et al. Riociguat for the treatment of pulmonary arterial hypertension. N Engl J Med 2013. July 25;369(4):330–40. [DOI] [PubMed] [Google Scholar]
- 63.Rubin LJ, Galie N, Grimminger F, Grunig E, Humbert M, Jing ZC, et al. Riociguat for the treatment of pulmonary arterial hypertension: a long-term extension study (PATENT-2). Eur Respir J 2015. May;45(5):1303–13. [DOI] [PubMed] [Google Scholar]
- 64.Markewitz BA, Farrukh IS, Chen Y, Li Y, Michael JR. Regulation of endothelin-1 synthesis in human pulmonary arterial smooth muscle cells. Effects of transforming growth factor-beta and hypoxia. Cardiovasc Res 2001. January;49(1):200–6. [DOI] [PubMed] [Google Scholar]
- 65.Shi-Wen X, Chen Y, Denton CP, Eastwood M, Renzoni EA, Bou-Gharios G, et al. Endothelin-1 promotes myofibroblast induction through the ETA receptor via a rac/phosphoinositide 3-kinase/Akt-dependent pathway and is essential for the enhanced contractile phenotype of fibrotic fibroblasts. Mol Biol Cell 2004. June;15(6):2707–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dupuis J, Hoeper MM. Endothelin receptor antagonists in pulmonary arterial hypertension. Eur Respir J 2008. February;31(2):407–15. [DOI] [PubMed] [Google Scholar]
- 67.Raja SG. Endothelin receptor antagonists for pulmonary arterial hypertension: an overview. Cardiovasc Ther 2010. October;28(5):e65–71. [DOI] [PubMed] [Google Scholar]
- 68.Pollock DM. Endothelin, angiotensin, and oxidative stress in hypertension. Hypertension 2005. April;45(4):477–80. [DOI] [PubMed] [Google Scholar]
- 69.Hirata Y, Emori T, Eguchi S, Kanno K, Imai T, Ohta K, et al. Endothelin receptor subtype B mediates synthesis of nitric oxide by cultured bovine endothelial cells. J Clin Invest 1993. April;91(4):1367–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Michel RP, Langleben D, Dupuis J. The endothelin system in pulmonary hypertension. Can J Physiol Pharmacol 2003. June;81(6):542–54. [DOI] [PubMed] [Google Scholar]
- 71.Dingemanse J, van Giersbergen PL. Clinical pharmacology of bosentan, a dual endothelin receptor antagonist. Clin Pharmacokinet 2004;43(15):1089–115. [DOI] [PubMed] [Google Scholar]
- 72.Rubin LJ, Badesch DB, Barst RJ, Galie N, Black CM, Keogh A, et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002. March 21;346(12):896–903. [DOI] [PubMed] [Google Scholar]
- 73.McGoon MD, Frost AE, Oudiz RJ, Badesch DB, Galie N, Olschewski H, et al. Ambrisentan therapy in patients with pulmonary arterial hypertension who discontinued bosentan or sitaxsentan due to liver function test abnormalities. Chest 2009. January;135(1):122–29. [DOI] [PubMed] [Google Scholar]
- 74.Galie N, Olschewski H, Oudiz RJ, Torres F, Frost A, Ghofrani HA, et al. Ambrisentan for the treatment of pulmonary arterial hypertension: results of the ambrisentan in pulmonary arterial hypertension, randomized, double-blind, placebo-controlled, multicenter, efficacy (ARIES) study 1 and 2. Circulation 2008. June 10;117(23):3010–9. [DOI] [PubMed] [Google Scholar]
- 75.Oudiz RJ, Galie N, Olschewski H, Torres F, Frost A, Ghofrani HA, et al. Long-term ambrisentan therapy for the treatment of pulmonary arterial hypertension. J Am Coll Cardiol 2009. November 17;54(21):1971–81. [DOI] [PubMed] [Google Scholar]
- 76.Gatfield J, Mueller Grandjean C, Sasse T, Clozel M, Nayler O. Slow receptor dissociation kinetics differentiate macitentan from other endothelin receptor antagonists in pulmonary arterial smooth muscle cells. PLoS One 2012;7(10):e47662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jansa P, Pulido T. Macitentan in Pulmonary Arterial Hypertension: A Focus on Combination Therapy in the SERAPHIN Trial. Am J Cardiovasc Drugs 2018. February;18(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Leuchte HH, Meis T, El-Nounou M, Michalek J, Behr J. Inhalation of endothelin receptor blockers in pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 2008. April;294(4):L772–7. [DOI] [PubMed] [Google Scholar]
- 79.Nellgard P, Bojo L, Cassuto J. Importance of vasoactive intestinal peptide and somatostatin for fluid losses in small-bowel obstruction. Scand J Gastroenterol 1995. May;30(5):464–9. [DOI] [PubMed] [Google Scholar]
- 80.Cox CP, Linden J, Said SI. VIP elevates platelet cyclic AMP (cAMP) levels and inhibits in vitro platelet activation induced by platelet-activating factor (PAF). Peptides 1984. Mar-Apr;5(2):325–8. [DOI] [PubMed] [Google Scholar]
- 81.Maruno K, Absood A, Said SI. VIP inhibits basal and histamine-stimulated proliferation of human airway smooth muscle cells. Am J Physiol 1995. June;268(6 Pt 1):L1047–51. [DOI] [PubMed] [Google Scholar]
- 82.Busto R, Prieto JC, Bodega G, Zapatero J, Carrero I. Immunohistochemical localization and distribution of VIP/PACAP receptors in human lung. Peptides 2000. February;21(2):265–9. [DOI] [PubMed] [Google Scholar]
- 83.Petkov V, Mosgoeller W, Ziesche R, Raderer M, Stiebellehner L, Vonbank K, et al. Vasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertension. J Clin Invest 2003. May;111(9):1339–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Leuchte HH, Baezner C, Baumgartner RA, Bevec D, Bacher G, Neurohr C, et al. Inhalation of vasoactive intestinal peptide in pulmonary hypertension. Eur Respir J 2008. November;32(5):1289–94. [DOI] [PubMed] [Google Scholar]
- 85.Loirand G, Guerin P, Pacaud P. Rho kinases in cardiovascular physiology and pathophysiology. Circ Res 2006. February 17;98(3):322–34. [DOI] [PubMed] [Google Scholar]
- 86.Julian L, Olson MF. Rho-associated coiled-coil containing kinases (ROCK): structure, regulation, and functions. Small GTPases 2014;5:e29846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, et al. Calcium sensitization of smooth muscle mediated by a Rho-associated protein kinase in hypertension. Nature 1997. October 30;389(6654):990–4. [DOI] [PubMed] [Google Scholar]
- 88.Do e Z, Fukumoto Y, Takaki A, Tawara S, Ohashi J, Nakano M, et al. Evidence for Rho-kinase activation in patients with pulmonary arterial hypertension. Circ J 2009. September;73(9):1731–9. [DOI] [PubMed] [Google Scholar]
- 89.Guilluy C, Eddahibi S, Agard C, Guignabert C, Izikki M, Tu L, et al. RhoA and Rho kinase activation in human pulmonary hypertension: role of 5-HT signaling. Am J Respir Crit Care Med 2009. June 15;179(12):1151–8. [DOI] [PubMed] [Google Scholar]
- 90.Nagaoka T, Fagan KA, Gebb SA, Morris KG, Suzuki T, Shimokawa H, et al. Inhaled Rho kinase inhibitors are potent and selective vasodilators in rat pulmonary hypertension. Am J Respir Crit Care Med 2005. March 1;171(5):494–9. [DOI] [PubMed] [Google Scholar]
- 91.Jiang X, Wang YF, Zhao QH, Jiang R, Wu Y, Peng FH, et al. Acute hemodynamic response of infused fasudil in patients with pulmonary arterial hypertension: a randomized, controlled, crossover study. Int J Cardiol 2014. November 15;177(1):61–5. [DOI] [PubMed] [Google Scholar]
- 92.Xiao JW, Zhu XY, Wang QG, Zhang DZ, Cui CS, Zhang P, et al. Acute effects of Rho-kinase inhibitor fasudil on pulmonary arterial hypertension in patients with congenital heart defects. Circ J 2015;79(6):1342–8. [DOI] [PubMed] [Google Scholar]
- 93.Godinas L, Guignabert C, Seferian A, Perros F, Bergot E, Sibille Y, et al. Tyrosine kinase inhibitors in pulmonary arterial hypertension: a double-edge sword? Semin Respir Crit Care Med 2013. October;34(5):714–24. [DOI] [PubMed] [Google Scholar]
- 94.Balasubramaniam V, Le Cras TD, Ivy DD, Grover TR, Kinsella JP, Abman SH. Role of platelet-derived growth factor in vascular remodeling during pulmonary hypertension in the ovine fetus. Am J Physiol Lung Cell Mol Physiol 2003. May;284(5):L826–33. [DOI] [PubMed] [Google Scholar]
- 95.Humbert M, Monti G, Fartoukh M, Magnan A, Brenot F, Rain B, et al. Platelet-derived growth factor expression in primary pulmonary hypertension: comparison of HIV seropositive and HIV seronegative patients. Eur Respir J 1998. March;11(3):554–9. [PubMed] [Google Scholar]
- 96.Hoeper MM, Barst RJ, Bourge RC, Feldman J, Frost AE, Galie N, et al. Imatinib mesylate as add-on therapy for pulmonary arterial hypertension: results of the randomized IMPRES study. Circulation 2013. March 12;127(10):1128–38. [DOI] [PubMed] [Google Scholar]
- 97.Pitsiou G, Zarogoulidis P, Petridis D, Kioumis I, Lampaki S, Organtzis J, et al. Inhaled tyrosine kinase inhibitors for pulmonary hypertension: a possible future treatment. Drug Des Devel Ther 2014;8:1753–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.McLaughlin VV, Shah SJ, Souza R, Humbert M. Management of pulmonary arterial hypertension. J Am Coll Cardiol 2015. May 12;65(18):1976–97. [DOI] [PubMed] [Google Scholar]
- 99.Zamanian RT, Haddad F, Doyle RL, Weinacker AB. Management strategies for patients with pulmonary hypertension in the intensive care unit. Crit Care Med 2007. September;35(9):2037–50. [DOI] [PubMed] [Google Scholar]
- 100.Preston IR, Sagliani KD, Roberts KE, Shah AM, Desouza SA, Howard W, et al. Comparison of acute hemodynamic effects of inhaled nitric oxide and inhaled epoprostenol in patients with pulmonary hypertension. Pulm Circ 2013. January;3(1):68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Labiris NR, Dolovich MB. Pulmonary drug delivery. Part I: physiological factors affecting therapeutic effectiveness of aerosolized medications. Br J Clin Pharmacol 2003. December;56(6):588–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tayab ZR, Hochhaus G. Pharmacokinetic/pharmacodynamic evaluation of inhalation drugs: application to targeted pulmonary delivery systems. Expert Opin Drug Deliv 2005. May;2(3):519–32. [DOI] [PubMed] [Google Scholar]
- 103.Olschewski H. Inhaled iloprost for the treatment of pulmonary hypertension. Eur Respir Rev 2009. March;18(111):29–34. [DOI] [PubMed] [Google Scholar]
- 104.Groneberg DA, Witt C, Wagner U, Chung KF, Fischer A. Fundamentals of pulmonary drug delivery. Respir Med 2003. April;97(4):382–7. [DOI] [PubMed] [Google Scholar]
- 105.Patil JS, Sarasija S. Pulmonary drug delivery strategies: A concise, systematic review. Lung India 2012. January;29(1):44–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rau JL. Determinants of patient adherence to an aerosol regimen. Respir Care 2005. October;50(10):1346–56; discussion 57–9. [PubMed] [Google Scholar]
- 107.Summers QA. Inhaled drugs and the lung. Clin Exp Allergy 1991. May;21(3):259–68. [DOI] [PubMed] [Google Scholar]
- 108.Fernandez Tena A, Casan Clara P. Deposition of inhaled particles in the lungs. Arch Bronconeumol 2012. July;48(7):240–6. [DOI] [PubMed] [Google Scholar]
- 109.Lippmann M, Yeates DB, Albert RE. Deposition, retention, and clearance of inhaled particles. Br J Ind Med 1980. November;37(4):337–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Preston IR, Feldman J, White J, Franco V, Ishizawar D, Burger C, et al. Safety and efficacy of transition from inhaled treprostinil to parenteral treprostinil in selected patients with pulmonary arterial hypertension. Pulm Circ 2014. September;4(3):456–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.LeVarge BL. Prostanoid therapies in the management of pulmonary arterial hypertension. Ther Clin Risk Manag 2015;11:535–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Barst RJ, Rubin LJ, McGoon MD, Caldwell EJ, Long WA, Levy PS. Survival in primary pulmonary hypertension with long-term continuous intravenous prostacyclin. Ann Intern Med 1994. September 15;121(6):409–15. [DOI] [PubMed] [Google Scholar]
- 113.Kallen AJ, Lederman E, Balaji A, Trevino I, Petersen EE, Shoulson R, et al. Bloodstream infections in patients given treatment with intravenous prostanoids. Infect Control Hosp Epidemiol 2008. April;29(4):342–9. [DOI] [PubMed] [Google Scholar]
- 114.Voswinckel R, Enke B, Reichenberger F, Kohstall M, Kreckel A, Krick S, et al. Favorable effects of inhaled treprostinil in severe pulmonary hypertension: results from randomized controlled pilot studies. J Am Coll Cardiol 2006. October 17;48(8):1672–81. [DOI] [PubMed] [Google Scholar]
- 115.Olschewski H, Rohde B, Behr J, Ewert R, Gessler T, Ghofrani HA, et al. Pharmacodynamics and pharmacokinetics of inhaled iloprost, aerosolized by three different devices, in severe pulmonary hypertension. Chest 2003. October;124(4):1294–304. [DOI] [PubMed] [Google Scholar]
- 116.McLaughlin VV, Palevsky HI. Parenteral and inhaled prostanoid therapy in the treatment of pulmonary arterial hypertension. Clin Chest Med 2013. December;34(4):825–40. [DOI] [PubMed] [Google Scholar]
- 117.Gandhi M, Pandya T, Gandhi R, Patel S, Mashru R, Misra A, et al. Inhalable liposomal dry powder of gemcitabine-HCl: Formulation, in vitro characterization and in vivo studies. Int J Pharm 2015. December 30;496(2):886–95. [DOI] [PubMed] [Google Scholar]
- 118.Joshi S, Cooke JR, Chan DK, Ellis JA, Hossain SS, Singh-Moon RP, et al. Liposome size and charge optimization for intraarterial delivery to gliomas. Drug Deliv Transl Res 2016. June;6(3):225–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Pattni BS, Chupin VV, Torchilin VP. New Developments in Liposomal Drug Delivery. Chem Rev 2015. October 14;115(19):10938–66. [DOI] [PubMed] [Google Scholar]
- 120.Hamilton A, Biganzoli L, Coleman R, Mauriac L, Hennebert P, Awada A, et al. EORTC 10968: a phase I clinical and pharmacokinetic study of polyethylene glycol liposomal doxorubicin (Caelyx, Doxil) at a 6-week interval in patients with metastatic breast cancer. European Organization for Research and Treatment of Cancer. Ann Oncol 2002. June;13(6):910–8. [DOI] [PubMed] [Google Scholar]
- 121.Chattopadhyay S. Aerosol generation using nanometer liposome suspensions for pulmonary drug delivery applications. J Liposome Res 2013. December;23(4):255–67. [DOI] [PubMed] [Google Scholar]
- 122.Kannan V, Balabathula P, Divi MK, Thoma LA, Wood GC. Optimization of drug loading to improve physical stability of paclitaxel-loaded long-circulating liposomes. J Liposome Res 2015;25(4):308–15. [DOI] [PubMed] [Google Scholar]
- 123.Kulkarni SB, Betageri GV, Singh M. Factors affecting microencapsulation of drugs in liposomes. J Microencapsul 1995. May-Jun;12(3):229–46. [DOI] [PubMed] [Google Scholar]
- 124.Clancy JP, Dupont L, Konstan MW, Billings J, Fustik S, Goss CH, et al. Phase II studies of nebulised Arikace in CF patients with Pseudomonas aeruginosa infection. Thorax 2013. September;68(9):818–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Verschraegen CF, Gilbert BE, Loyer E, Huaringa A, Walsh G, Newman RA, et al. Clinical evaluation of the delivery and safety of aerosolized liposomal 9-nitro-20(s)-camptothecin in patients with advanced pulmonary malignancies. Clin Cancer Res 2004. April 1;10(7):2319–26. [DOI] [PubMed] [Google Scholar]
- 126.Allen TM. Liposomes. Opportunities in drug delivery. Drugs 1997;54 Suppl 4:8–14. [DOI] [PubMed] [Google Scholar]
- 127.Alberts DS, Garcia DJ. Safety aspects of pegylated liposomal doxorubicin in patients with cancer. Drugs 1997;54 Suppl 4:30–5. [DOI] [PubMed] [Google Scholar]
- 128.Laing RB, Milne LJ, Leen CL, Malcolm GP, Steers AJ. Anaphylactic reactions to liposomal amphotericin. Lancet 1994. September 3;344(8923):682. [DOI] [PubMed] [Google Scholar]
- 129.Szebeni J. The interaction of liposomes with the complement system. Crit Rev Ther Drug Carrier Syst 1998;15(1):57–88. [PubMed] [Google Scholar]
- 130.Myers MA, Thomas DA, Straub L, Soucy DW, Niven RW, Kaltenbach M, et al. Pulmonary effects of chronic exposure to liposome aerosols in mice. Exp Lung Res 1993. Jan-Mar;19(1):1–19. [DOI] [PubMed] [Google Scholar]
- 131.Canonico AE, Plitman JD, Conary JT, Meyrick BO, Brigham KL. No lung toxicity after repeated aerosol or intravenous delivery of plasmid-cationic liposome complexes. J Appl Physiol (1985) 1994. July;77(1):415–9. [DOI] [PubMed] [Google Scholar]
- 132.Lee Y, Pai SB, Bellamkonda RV, Thompson DH, Singh J. Cerivastatin Nanoliposome as a Potential Disease Modifying Approach for the Treatment of Pulmonary Arterial Hypertension. J Pharmacol Exp Ther 2018. July;366(1):66–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Taylor KM, Taylor G, Kellaway IW, Stevens J. The influence of liposomal encapsulation on sodium cromoglycate pharmacokinetics in man. Pharm Res 1989. July;6(7):633–6. [DOI] [PubMed] [Google Scholar]
- 134.Danhier F, Ansorena E, Silva JM, Coco R, Le Breton A, Preat V. PLGA-based nanoparticles: an overview of biomedical applications. J Control Release 2012. July 20;161(2):505–22. [DOI] [PubMed] [Google Scholar]
- 135.Khoshraftar A, Noorani B, Yazdian F, Rashedi H, Ghaemi RV, Alihemmati Z, et al. Fabrication and Evaluation of Nanofibrous Polyhydroxybutyrate valerate Scaffolds Containing Hydroxyapatite Particles for Bone Tissue Engineering. Int J Polym Mater Po 2018. November;67(17):987–95. [Google Scholar]
- 136.Feng T, Tian H, Xu C, Lin L, Xie Z, Lam MH, et al. Synergistic co-delivery of doxorubicin and paclitaxel by porous PLGA microspheres for pulmonary inhalation treatment. Eur J Pharm Biopharm 2014. November;88(3):1086–93. [DOI] [PubMed] [Google Scholar]
- 137.Rashid J, Patel B, Nozik-Grayck E, McMurtry IF, Stenmark KR, Ahsan F. Inhaled sildenafil as an alternative to oral sildenafil in the treatment of pulmonary arterial hypertension (PAH). J Control Release 2017. March 28;250:96–106.**Brief explanation: The authors proved, for the first, that inhaled PLGA particles of sildenafil can be administered, as a substitute for the oral form of sildenafil, at a reduced dose and longer dosing interval, resulting in decreasing peripheral vasodilation (unwanted side effects) and enhancing the anti-PAH efficacy.
- 138.Singh DJ, Lohade AA, Parmar JJ, Hegde DD, Soni P, Samad A, et al. Development of Chitosan-based Dry Powder Inhalation System of Cisplatin for Lung Cancer. Indian J Pharm Sci 2012. November;74(6):521–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Makadia HK, Siegel SJ. Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers (Basel) 2011. September 1;3(3):1377–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Dinarvand R, Sepehri N, Manoochehri S, Rouhani H, Atyabi F. Polylactide-co-glycolide nanoparticles for controlled delivery of anticancer agents. Int J Nanomedicine 2011;6:877–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bivas-Benita M, Romeijn S, Junginger HE, Borchard G. PLGA-PEI nanoparticles for gene delivery to pulmonary epithelium. Eur J Pharm Biopharm 2004. July;58(1):1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Patel B, Gupta V, Ahsan F. PEG-PLGA based large porous particles for pulmonary delivery of a highly soluble drug, low molecular weight heparin. J Control Release 2012. September 10;162(2):310–20. [DOI] [PubMed] [Google Scholar]
- 143.Blanco E, Kessinger CW, Sumer BD, Gao J. Multifunctional micellar nanomedicine for cancer therapy. Exp Biol Med (Maywood) 2009. February;234(2):123–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Andrade F, das Neves J, Gener P, Schwartz S Jr., Ferreira D, Oliva M, et al. Biological assessment of self-assembled polymeric micelles for pulmonary administration of insulin. Nanomedicine 2015. October;11(7):1621–31. [DOI] [PubMed] [Google Scholar]
- 145.Baginski L, Gobbo OL, Tewes F, Salomon JJ, Healy AM, Bakowsky U, et al. In vitro and in vivo characterisation of PEG-lipid-based micellar complexes of salmon calcitonin for pulmonary delivery. Pharm Res 2012. June;29(6):1425–34. [DOI] [PubMed] [Google Scholar]
- 146.Brandenburg KS, Rubinstein I, Sadikot RT, Onyuksel H. Polymyxin B self-associated with phospholipid nanomicelles. Pharm Dev Technol 2012. Nov-Dec;17(6):654–60. [DOI] [PubMed] [Google Scholar]
- 147.Haeri A, Sadeghian S, Rabbani S, Anvari MS, Lavasanifar A, Amini M, et al. Sirolimus-loaded stealth colloidal systems attenuate neointimal hyperplasia after balloon injury: a comparison of phospholipid micelles and liposomes. Int J Pharm 2013. October 15;455(1–2):320–30. [DOI] [PubMed] [Google Scholar]
- 148.Gill KK, Nazzal S, Kaddoumi A. Paclitaxel loaded PEG(5000)-DSPE micelles as pulmonary delivery platform: formulation characterization, tissue distribution, plasma pharmacokinetics, and toxicological evaluation. Eur J Pharm Biopharm 2011. October;79(2):276–84. [DOI] [PubMed] [Google Scholar]
- 149.Sahib MN, Darwis Y, Peh KK, Abdulameer SA, Tan YT. Rehydrated sterically stabilized phospholipid nanomicelles of budesonide for nebulization: physicochemical characterization and in vitro, in vivo evaluations. Int J Nanomedicine 2011;6:2351–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Gupta N, Ibrahim HM, Ahsan F. Peptide-micelle hybrids containing fasudil for targeted delivery to the pulmonary arteries and arterioles to treat pulmonary arterial hypertension. J Pharm Sci 2014. November;103(11):3743–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Bae Y, Nishiyama N, Fukushima S, Koyama H, Yasuhiro M, Kataoka K. Preparation and biological characterization of polymeric micelle drug carriers with intracellular pH-triggered drug release property: tumor permeability, controlled subcellular drug distribution, and enhanced in vivo antitumor efficacy. Bioconjug Chem 2005. Jan-Feb;16(1):122–30. [DOI] [PubMed] [Google Scholar]
- 152.Jhaveri AM, Torchilin VP. Multifunctional polymeric micelles for delivery of drugs and siRNA. Front Pharmacol 2014;5:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Smola M, Vandamme T, Sokolowski A. Nanocarriers as pulmonary drug delivery systems to treat and to diagnose respiratory and non respiratory diseases. Int J Nanomedicine 2008;3(1):1–19. [PMC free article] [PubMed] [Google Scholar]
- 154.Patton JS, Brain JD, Davies LA, Fiegel J, Gumbleton M, Kim KJ, et al. The particle has landed--characterizing the fate of inhaled pharmaceuticals. J Aerosol Med Pulm Drug Deliv 2010. December;23 Suppl 2:S71–87. [DOI] [PubMed] [Google Scholar]
- 155.Usmani OS, Biddiscombe MF, Barnes PJ. Regional lung deposition and bronchodilator response as a function of beta2-agonist particle size. Am J Respir Crit Care Med 2005. December 15;172(12):1497–504. [DOI] [PubMed] [Google Scholar]
- 156.Kumar A, Bicer EM, Morgan AB, Pfeffer PE, Monopoli M, Dawson KA, et al. Enrichment of immunoregulatory proteins in the biomolecular corona of nanoparticles within human respiratory tract lining fluid. Nanomedicine 2016. May;12(4):1033–43. [DOI] [PubMed] [Google Scholar]
- 157.Yang W, Peters JI, Williams RO 3rd. Inhaled nanoparticles--a current review. Int J Pharm 2008. May 22;356(1–2):239–47. [DOI] [PubMed] [Google Scholar]
- 158.Champion JA, Mitragotri S. Role of target geometry in phagocytosis. Proc Natl Acad Sci U S A 2006. March 28;103(13):4930–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Champion JA, Mitragotri S. Shape induced inhibition of phagocytosis of polymer particles. Pharm Res 2009. January;26(1):244–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jacobs C, Muller RH. Production and characterization of a budesonide nanosuspension for pulmonary administration. Pharm Res 2002. February;19(2):189–94. [DOI] [PubMed] [Google Scholar]
- 161.Ensign LM, Schneider C, Suk JS, Cone R, Hanes J. Mucus penetrating nanoparticles: biophysical tool and method of drug and gene delivery. Adv Mater 2012. July 24;24(28):3887–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Muller C, Perera G, Konig V, Bernkop-Schnurch A. Development and in vivo evaluation of papain-functionalized nanoparticles. Eur J Pharm Biopharm 2014. May;87(1):125–31. [DOI] [PubMed] [Google Scholar]
- 163.Yang M, Lai SK, Wang YY, Zhong W, Happe C, Zhang M, et al. Biodegradable nanoparticles composed entirely of safe materials that rapidly penetrate human mucus. Angew Chem Int Ed Engl 2011. March 7;50(11):2597–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Wang YY, Lai SK, Suk JS, Pace A, Cone R, Hanes J. Addressing the PEG mucoadhesivity paradox to engineer nanoparticles that “slip” through the human mucus barrier. Angew Chem Int Ed Engl 2008;47(50):9726–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Gupta V, Gupta N, Shaik IH, Mehvar R, Nozik-Grayck E, McMurtry IF, et al. Inhaled PLGA particles of prostaglandin E(1) ameliorate symptoms and progression of pulmonary hypertension at a reduced dosing frequency. Mol Pharm 2013. May 6;10(5):1655–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Gupta V, Davis M, Hope-Weeks LJ, Ahsan F. PLGA microparticles encapsulating prostaglandin E1-hydroxypropyl-beta-cyclodextrin (PGE1-HPbetaCD) complex for the treatment of pulmonary arterial hypertension (PAH). Pharm Res 2011. July;28(7):1733–49. [DOI] [PubMed] [Google Scholar]
- 167.Gupta V, Ahsan F. Influence of PEI as a core modifying agent on PLGA microspheres of PGE(1), a pulmonary selective vasodilator. Int J Pharm 2011. July 15;413(1–2):51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Akagi S, Nakamura K, Matsubara H, Kondo M, Miura D, Matoba T, et al. Intratracheal Administration of Prostacyclin Analogue-incorporated Nanoparticles Ameliorates the Development of Monocrotaline and Sugen-Hypoxia-induced Pulmonary Arterial Hypertension. J Cardiovasc Pharmacol 2016. April;67(4):290–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Mohamed NA, Ahmetaj-Shala B, Duluc L, Mackenzie LS, Kirkby NS, Reed DM, et al. A New NO-Releasing Nanoformulation for the Treatment of Pulmonary Arterial Hypertension. J Cardiovasc Transl Res 2016. April;9(2):162–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Beck-Broichsitter M, Bohr A, Aragao-Santiago L, Klingl A, Kissel T. Formulation and process considerations for the design of sildenafil-loaded polymeric microparticles by vibrational spray-drying. Pharm Dev Technol 2017. September;22(6):691–98. [DOI] [PubMed] [Google Scholar]
- 171.Sutliff RL, Kang BY, Hart CM. PPARgamma as a potential therapeutic target in pulmonary hypertension. Ther Adv Respir Dis 2010. June;4(3):143–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rashid J, Alobaida A, Al-Hilal TA, Hammouda S, McMurtry IF, Nozik-Grayck E, et al. Repurposing rosiglitazone, a PPAR-gamma agonist and oral antidiabetic, as an inhaled formulation, for the treatment of PAH. J Control Release 2018. June 28;280:113–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Rashid J, Nozik-Grayck E, McMurtry IF, Stenmark KR, Ahsan F. Inhaled combination of sildenafil and rosiglitazone improves pulmonary hemodynamics, cardiac function, and arterial remodeling. Am J Physiol Lung Cell Mol Physiol 2019. January 1;316(1):L119–L30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Evgenov OV, Kohane DS, Bloch KD, Stasch JP, Volpato GP, Bellas E, et al. Inhaled agonists of soluble guanylate cyclase induce selective pulmonary vasodilation. Am J Respir Crit Care Med 2007. December 1;176(11):1138–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Akagi S, Nakamura K, Miura D, Saito Y, Matsubara H, Ogawa A, et al. Delivery of imatinib-incorporated nanoparticles into lungs suppresses the development of monocrotaline-induced pulmonary arterial hypertension. Int Heart J 2015. May 13;56(3):354–9. [DOI] [PubMed] [Google Scholar]
- 176.Chen L, Nakano K, Kimura S, Matoba T, Iwata E, Miyagawa M, et al. Nanoparticle-mediated delivery of pitavastatin into lungs ameliorates the development and induces regression of monocrotaline-induced pulmonary artery hypertension. Hypertension 2011. February;57(2):343–50.**Brief explanation: This study suggests that a single intratracheal administration of the PLGA nano-formulation containing pitavastatin reduced the inflammation, lowered pulmonary arterial remodeling, decreased RV pressure, attenuated the development of PAH, and improved the survival rates. Importantly, a Phase I clinical trial has evaluated the efficacy of PLGA nanoparticle-based delivery of pitavastatin.
- 177.Kleemann E, Schmehl T, Gessler T, Bakowsky U, Kissel T, Seeger W. Iloprost-containing liposomes for aerosol application in pulmonary arterial hypertension: formulation aspects and stability. Pharm Res 2007. February;24(2):277–87. [DOI] [PubMed] [Google Scholar]
- 178.Jain PP, Leber R, Nagaraj C, Leitinger G, Lehofer B, Olschewski H, et al. Liposomal nanoparticles encapsulating iloprost exhibit enhanced vasodilation in pulmonary arteries. Int J Nanomedicine 2014;9:3249–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Shibuya M, Hirai S, Seto M, Satoh S, Ohtomo E, Fasudil Ischemic Stroke Study G. Effects of fasudil in acute ischemic stroke: results of a prospective placebo-controlled double-blind trial. J Neurol Sci 2005. November 15;238(1–2):31–9. [DOI] [PubMed] [Google Scholar]
- 180.Gupta V, Gupta N, Shaik IH, Mehvar R, McMurtry IF, Oka M, et al. Liposomal fasudil, a rho-kinase inhibitor, for prolonged pulmonary preferential vasodilation in pulmonary arterial hypertension. J Control Release 2013. April 28;167(2):189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Chrastina A, Valadon P, Massey KA, Schnitzer JE. Lung vascular targeting using antibody to aminopeptidase P: CT-SPECT imaging, biodistribution and pharmacokinetic analysis. J Vasc Res 2010;47(6):531–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Sarrazin S, Lamanna WC, Esko JD. Heparan sulfate proteoglycans. Cold Spring Harb Perspect Biol 2011. July 1;3(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Goldenberg NM, Kuebler WM. Endothelial cell regulation of pulmonary vascular tone, inflammation, and coagulation. Compr Physiol 2015. April;5(2):531–59. [DOI] [PubMed] [Google Scholar]
- 184.Howard MD, Hood ED, Zern B, Shuvaev VV, Grosser T, Muzykantov VR. Nanocarriers for vascular delivery of anti-inflammatory agents. Annu Rev Pharmacol Toxicol 2014;54:205–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Giannotta M, Trani M, Dejana E. VE-cadherin and endothelial adherens junctions: active guardians of vascular integrity. Dev Cell 2013. September 16;26(5):441–54. [DOI] [PubMed] [Google Scholar]
- 186.Hood E, Simone E, Wattamwar P, Dziubla T, Muzykantov V. Nanocarriers for vascular delivery of antioxidants. Nanomedicine (Lond) 2011. September;6(7):1257–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Howard MD, Greineder CF, Hood ED, Muzykantov VR. Endothelial targeting of liposomes encapsulating SOD/catalase mimetic EUK-134 alleviates acute pulmonary inflammation. J Control Release 2014. March 10;177:34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Almenar-Queralt A, Duperray A, Miles LA, Felez J, Altieri DC. Apical topography and modulation of ICAM-1 expression on activated endothelium. Am J Pathol 1995. November;147(5):1278–88. [PMC free article] [PubMed] [Google Scholar]
- 189.Brenner JS, Bhamidipati K, Glassman PM, Ramakrishnan N, Jiang D, Paris AJ, et al. Mechanisms that determine nanocarrier targeting to healthy versus inflamed lung regions. Nanomedicine 2017. May;13(4):1495–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Shuvaev VV, Brenner JS, Muzykantov VR. Targeted endothelial nanomedicine for common acute pathological conditions. J Control Release 2015. December 10;219:576–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Murciano JC, Harshaw DW, Ghitescu L, Danilov SM, Muzykantov VR. Vascular immunotargeting to endothelial surface in a specific macrodomain in alveolar capillaries. Am J Respir Crit Care Med 2001. October 1;164(7):1295–302. [DOI] [PubMed] [Google Scholar]
- 192.Urakami T, Jarvinen TA, Toba M, Sawada J, Ambalavanan N, Mann D, et al. Peptide-directed highly selective targeting of pulmonary arterial hypertension. Am J Pathol 2011. June;178(6):2489–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Jarvinen TA, Ruoslahti E. Targeted Antiscarring Therapy for Tissue Injuries. Adv Wound Care (New Rochelle) 2013. March;2(2):50–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Toba M, Alzoubi A, O’Neill K, Abe K, Urakami T, Komatsu M, et al. A novel vascular homing peptide strategy to selectively enhance pulmonary drug efficacy in pulmonary arterial hypertension. Am J Pathol 2014. February;184(2):369–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Keshavarz A, Alobaida A, McMurtry IF, Nozik-Grayck E, Stenmark KR, Ahsan F. CAR, a Homing Peptide, Prolongs Pulmonary Preferential Vasodilation by Increasing Pulmonary Retention and Reducing Systemic Absorption of Liposomal Fasudil. Mol Pharm 2019. August 5;16(8):3414–29. **Brief explanation: This study proved that CAR, a homing peptide, aids in concentrating the anti-PAHdrug in the lungs, increasing the cellular uptake, extending the half-life of fasudil, and eliciting pulmonary specific vasodilation when the peptide remains conjugated on the liposomal surface.
- 196.Nahar K, Absar S, Gupta N, Kotamraju VR, McMurtry IF, Oka M, et al. Peptide-coated liposomal fasudil enhances site specific vasodilation in pulmonary arterial hypertension. Mol Pharm 2014. December 1;11(12):4374–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Nahar K, Absar S, Patel B, Ahsan F. Starch-coated magnetic liposomes as an inhalable carrier for accumulation of fasudil in the pulmonary vasculature. Int J Pharm 2014. April 10;464(1–2):185–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Gupta N, Rashid J, Nozik-Grayck E, McMurtry IF, Stenmark KR, Ahsan F. Cocktail of Superoxide Dismutase and Fasudil Encapsulated in Targeted Liposomes Slows PAH Progression at a Reduced Dosing Frequency. Mol Pharm 2017. March 6;14(3):830–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Huang SL, Kee PH, Kim H, Moody MR, Chrzanowski SM, Macdonald RC, et al. Nitric oxide-loaded echogenic liposomes for nitric oxide delivery and inhibition of intimal hyperplasia. J Am Coll Cardiol 2009. August 11;54(7):652–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Rashid J, Nahar K, Raut S, Keshavarz A, Ahsan F. Fasudil and DETA NONOate, Loaded in a Peptide-Modified Liposomal Carrier, Slow PAH Progression upon Pulmonary Delivery. Mol Pharm 2018. May 7;15(5):1755–65. *Brief explanation: The authors demonstrated that fasudil and NO donor, formulated in liposomes, delivered therapeutics to the site of action in the lung, increased the levels of the vasodilatory signaling molecule, prolonged the vasodilatory effect of anti-PAH drugs, indicating better management strategy for PAH combination therapy.
- 201.Nahar K, Rashid J, Absar S, Al-Saikhan FI, Ahsan F. Liposomal Aerosols of Nitric Oxide (NO) Donor as a Long-Acting Substitute for the Ultra-Short-Acting Inhaled NO in the Treatment of PAH. Pharm Res 2016. July;33(7):1696–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Trivedi R, Kompella UB. Nanomicellar formulations for sustained drug delivery: strategies and underlying principles. Nanomedicine (Lond) 2010. April;5(3):485–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Nagaya N, Kangawa K. Adrenomedullin in the treatment of pulmonary hypertension. Peptides 2004. November;25(11):2013–8. [DOI] [PubMed] [Google Scholar]
- 204.Harada-Shiba M, Takamisawa I, Miyata K, Ishii T, Nishiyama N, Itaka K, et al. Intratracheal gene transfer of adrenomedullin using polyplex nanomicelles attenuates monocrotaline-induced pulmonary hypertension in rats. Mol Ther 2009. July;17(7):1180–6. *Brief explanation: The authors developed a novel nonviral adrenomedullin gene delivery using the intratracheal administration of nanomicelles, which showed remarkable therapeutic efficacy with PAH animal models without compromising biocompatibility.
- 205.Gupta N, Patel B, Ahsan F. Nano-engineered erythrocyte ghosts as inhalational carriers for delivery of fasudil: preparation and characterization. Pharm Res 2014. June;31(6):1553–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Gupta N, Patel B, Nahar K, Ahsan F. Cell permeable peptide conjugated nanoerythrosomes of fasudil prolong pulmonary arterial vasodilation in PAH rats. Eur J Pharm Biopharm 2014. November;88(3):1046–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Wernig K, Griesbacher M, Andreae F, Hajos F, Wagner J, Mosgoeller W, et al. Depot formulation of vasoactive intestinal peptide by protamine-based biodegradable nanoparticles. J Control Release 2008. September 10;130(2):192–8. [DOI] [PubMed] [Google Scholar]
- 208.Paranjpe M, Muller-Goymann CC. Nanoparticle-mediated pulmonary drug delivery: a review. Int J Mol Sci 2014. April 8;15(4):5852–73. [DOI] [PMC free article] [PubMed] [Google Scholar]