

Abstract
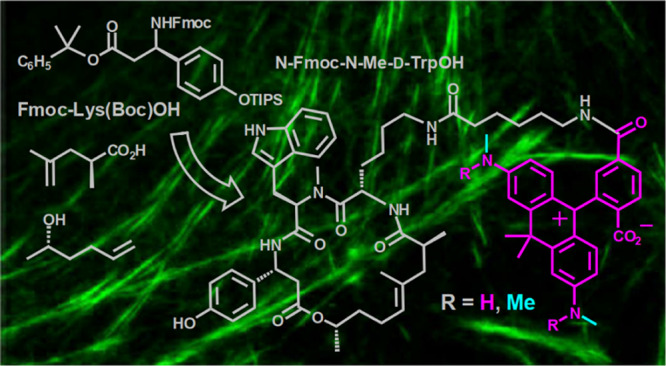
The nanometer thickness of filaments and the dynamic behavior of actin—a protein playing a crucial role in cellular function and motility—make it attractive for observation with super-resolution optical microscopy. We developed the solution-phase synthesis of des-bromo-des-methyl-jasplakinolide-lysine, used as the “recognition unit” (ligand) for F-actin in living cells. The first amino acid—Fmoc-O-TIPS-β-tyrosine—was prepared in 78% yield (two steps in one pot). The new solution-phase synthesis involves 2-phenylisopropyl protection of the carboxyl group and does not require excesses of commercially unavailable amino acids. The overall yield of the target intermediate obtained in nine steps is about 8%. The 2-phenylisopropyl group can be cleaved from carboxyl with 2–3% (v/v) of TFA in acetonitrile (0–10 °C), without affecting TIPS protection of the phenolic hydroxyl in β-tyrosine and N-Boc protection in lysine. Des-bromo-des-methyl-jasplakinolide-lysine was coupled with red-emitting fluorescent dyes 580CP and 610CP (via 6-aminohexanoate linker). Actin in living cells was labeled with 580CP and 610CP probes, and the optical resolution measured as full width at half-maximum of line profiles across actin fibers was found to be 300–400 nm and 100 nm under confocal and STED conditions, respectively. The solution-phase synthesis of des-bromo-des-methyl-jasplakinolide-lysine opens a way to better fluorescent probe perspective for actin imaging.
Introduction
Actin protein plays a crucial role in cellular function and motility.1 It can be present either as a monomer (G-actin; globular) or, upon polymerization, it may form filaments (F-actin): flexible fibers with a diameter of 4–7 nm and length of up to several micrometers. In living cells, both forms of actin are present in equilibrium; they are essential for the proper mobility and contraction of cells during cell division, cell motility, cytokinesis, vesicle and organelle movement, cell signaling, as well as the establishment and maintenance of cell junctions and cell shapes. The nanometer thickness and dynamic behavior of actin filaments make them an attractive object for observation with super-resolution optical microscopy.
The fluorescent probes for super-resolution and live imaging of actin2−5 incorporate the so-called des-bromo-des-methyl-jasplakinolide-lysine (Figure 1),6 as the ligand or “recognition unit” for F-actin in living cells. This macrocyclic depsipeptide has a reactive amino group, and its salts can be readily generated from N-tert-butoxycarbonyl derivative (7-H in Scheme 3) which represents the key intermediate and stable precursor of the conjugates with organic dyes. Compound 7-H is commercially unavailable, and the solid-phase synthesis of 7-H has been outlined only briefly.2,6 The aim of the present work was to develop the new and productive route to macrocyclic depsipeptide 7-H, compare the syntheses on a solid phase and in solution, prepare the conjugates of compound 7-H with fluorescent dyes, and apply them as fluorescent probes for the super-resolution microscopy of actin filaments in living cells. As cell-permeate fluorescent dyes, we have chosen carbopyronines 580CP and 610CP which demonstrated high imaging performance as conjugates with various ligands.5,7,8 The absorption and emission spectra of these dyes are given in Figure 1 and the photophysical properties in Table 1.
Figure 1.
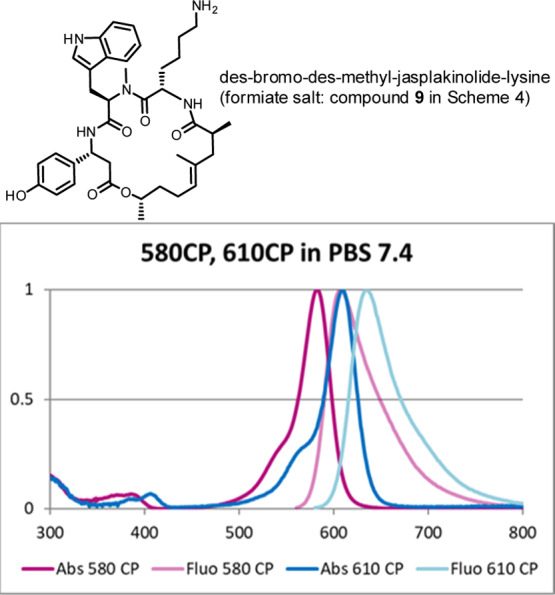
Des-bromo-des-methyl-jasplakinolide-lysine for conjugation with cell-permeate fluorescent dyes; absorption and emission spectra of 580CP and 610CP are shown (see also Table 1 and Scheme 4).5,7,8
Scheme 3. Final Assembly of Jasplakinolide Analog 7-H.

Table 1. Spectral Properties of Carbopyronines 580CP, 610CP, and SiR (Reference Dye) in Aqueous PBS Buffer (pH 7.4) at Room Temperature.
Results and Discussion
Both synthesis routes—on the solid phase and in solution—involve Fmoc-O-TIPS-β-tyrosine as the first amino acid (AA1; Schemes 1 and 2). This compound was initially obtained via a multistep procedure including the Michael addition of a chiral dibenzyl amine to p-coumaric acid ester, separation of the diastereomers, N-debenzylation, and manipulation with O-protecting groups.6 We found a shorter route to Fmoc-O-TIPS-β-tyrosine, which starts from commercially available Fmoc-β-tyrosine (Scheme 2). The two-step procedure includes silylation with triisopropylsilyl chloride on both oxygen centers9 followed by the hydrolytic cleavage of the more labile (triisopropylsilyl)ester group under mild basic conditions and affords the required amino acid AA1 (Scheme 2). The solid-phase synthesis (Scheme 1) provides triamide 1-H-TIPS as the key intermediate. Compound 1-H-TIPS was isolated with an overall yield of about 22% (52%, when calculated on the loading degree of the first amino acid AA1). However, these yields are based on the use of large excess of amino acids AA1, AA2, and AA3. The first two are not commercially available and have to be prepared separately. Therefore, the use of large excess of AA1 and AA2 is not cost- and time-efficient. Another important detail of the solid-phase synthesis is that the cleavage from the resin is performed in the presence of weakly acidic hexafluoroisopropanol. The latter (b. p. 58 °C) concentrates in the reaction mixture in the course of solvent evaporation (DCM) and causes (partial) removal of the triisopropyl silyl group. We added a higher boiling solvent (ethyl acetate) into the solution in order to prevent this undesirable effect and suppress the formation of deprotected phenol 1-H-H.
Scheme 1. Solid-Phase Synthesis Affords the Key Intermediate 1-H-TIPS.

Scheme 2. Solution-Phase Synthesis of Acyclic Amides 5-C(CH3)2C6H5–H and 5-H-H.

Planning the solution-phase synthesis of compound 1-H-TIPS (5-H-H in Schemes 2 and 3), we realized that its success is determined by the correct choice of carboxyl protection in AA1 (Scheme 2). We used the 2-phenylisopropyl protecting group10 because 2-chlorotrityl esters11 partially cleaved in the course of work-up and isolation procedures (chromatography), when the synthesis was carried out according to Scheme 2. The synthesis was carried out according to Scheme 2, using water-soluble carbodiimide (free base) in the presence of HOAt and 2,4,6-collidine in DCM.12 Under these conditions, no racemization was observed.12 9-Fluorenylmethylcarbamate groups were cleaved using diethylamine (the excess of which was removed by several evaporations with toluene); intermediate compounds with free amino groups were not isolated but used directly in the following amidation reactions. In this approach, the excess of N-protected amino acids is not required (which is an advantage over the solid-phase methodology). The final step—removal of 2-phenylpropyl protecting group—was effected using 2–3% solution of TFA in acetonitrile at 0... +5 °C. Under these conditions, N-tert-butoxycarbonyl protection of amines is stable.10,13
Moreover, TIPS protection of the phenolic hydroxyl group turned out to be stable as well. However, we detected and isolated compound 5-H-C(CH3)2C6H5 (17%), which was formed when 2-phenylpropyl residue was transferred to another nucleophilic center—nitrogen atom of tryptophan. The synthesis in solution is attractive not only because of the relatively high overall yield (35%; Scheme 2) but also because it is not necessary to apply (unrecoverable) excess of exotic and expensive amino and (S)-2,4-dimethylpent-4-enoic acids. The final steps of the assembly of macrocyclic depsipeptide 7-H are common for the solid- and solution-phase syntheses and are given in Scheme 3. The first reaction—formation of ester 6 from carboxylic acid 5-H-H (1-H-TIPS in Scheme 1) and (S)-5-hexen-2-ol in the presence of carbodiimide (EDC*HCl)—requires 4-(N,N-dimethylamino)pyridine (DMAP) as a catalyst. We found that the use of more than 10 mol % of DMAP is counterproductive, as it converts the intermediate O-acylurea (active acylating reagent) into the stable N-acylurea which is inert and does not react any further. The undesired O- to N-migration of the acetyl residue consumes carboxylic acid, and the required esterification does not take place. The metathesis of dialkene 6 was performed as reported.2,6 Along with macrocyclic alkene 7-TIPS, we isolated isomer 8-TIPS which, after removal of the TIPS group, gave compound 8-H (mixture of 2 epimers; the structure established by NMR spectroscopy). Formally, compound 8-TIPS is formed, if the methyl group at C-8 in 7-TIPS migrates to C-7 in 8-TIPS (see Scheme 3). This can be clearly seen from the appearance of two doublets of doublets at 3.97 and 3.70 ppm for H-8 in the 1H NMR spectrum of 8-H instead of a single multiplet (doublet of quartets of doublets, 4.78 ppm) in the spectrum of 7-H. However, we do not have a plausible explanation of this 1,2-shift. 5-Hexen-2-ol did not contain appreciable amounts of 2-methyl-5-hexen-1-ol. The overall yield of compound 7-H obtained in nine steps according to Schemes 2 and 3 is about 8%. Amine 9 (Scheme 4) was prepared from compound 7-H in the presence of formic acid. Deprotection with formic acid was found to be cleaner than the cleavage of the tert-butoxycarbonyl group with trifluoroacetic acid (TFA). Conjugates of fluorescent dyes 580CP and 610CP (their spectra are given in Figure 1) with ω-aminocaproic acid (linker) and amine 9 (actin ligands) were obtained in three steps, as outlined in Scheme 4 (via N-hydroxysuccinimidyl esters;3,4 for details, see the Supporting Information and ref (5)).
Scheme 4. Conjugation of Dyes 580CP and 610CP with Amine 9 via 6-Aminohexanoic Acid Linker: Synthesis of Actin Fluorescent Probes (See TOC Graph for Full Structures, Supporting Information and ref (5) for Experimental Details, Spectra, and Photophysical Properties).

We labeled actin in living human osteosarcoma cells (U-2 OS) (Figure 2A,B) and in kidney cells derived from the African green monkey (COS-7) (Figure 2C,D) using 580CP-jasplakinolide (Figure 2A,C) and 610CP-jasplakinolide (Figure 2B,D) probes, respectively. Both probes (for structures, see Scheme 4) performed well in confocal and STED (stimulated emission depletion) microscopy. The optical resolution (full width at half maximum of a line profile) in the STED mode improved: the apparent diameters of actin fiber bundles under confocal and STED conditions were 300–400 nm and ca. 100 nm, respectively (see Figure 2). Both dyes (for spectral properties, see Table 1) have some residual emission at 775 nm (wavelength of the STED laser; see Figure 1) but virtually no absorption at this wavelength. These valuable spectral features provide an efficient STED effect and, as a result, optical resolution improvement without undesirable re-excitation with the STED beam. Importantly, the conjugates of carbopyronine dye 580CP enable two-color STED microscopy in living cells with standard optical settings (e.g., in combination with SiR dye; see Table 1).5,7
Figure 2.

In vitro labeling of actin filaments in U-2 OS and COS-7 cells with 580CP-jasplakinolide and 610CP-jasplakinolide probes (see Scheme 4 and ref (5)). (A,B) Human osteosarcoma (U-2 OS) cells and (C,D) kidney cells derived from the African green monkey (COS-7) were incubated with (A,C) 580CP-jasplakinolide (5 μM for 30 min) or (B,D) 610CP-jasplakinolide (1 μM for 60 min) probes, respectively (followed by a washing step of additional 30 min). Live-cell STED images were acquired using a quad scanning STED microscope (Abberior Instruments, Göttingen, Germany) equipped with a UPlanSApo 100×/1, 40 Oil objective (Olympus, Tokyo, Japan). (1–4) Line profiles (with a line width of 60 nm (three pixels) for averaging) were taken at locations indicated by arrows in the enlarged sections (right). The averaged data were fitted with a Lorentzian function and plotted. Except for contrast stretching, no further image processing was applied. Scale bars: 5 μm (overviews), 500 nm (enlarged sections). Abbreviations: AU (arbitrary units), Conf (confocal), FWHM (full width at half maximum), STED (stimulated emission depletion).
Conclusions
In vitro labeling of actin filaments (Figure 2) with 580CP- and 610CP-jasplakinolide conjugates exhibits different patterns in different cell lines and at different concentrations: the best imaging results were achieved when 580CP probe was applied at 5 μM for 30 min and 610CP-jasplakinolide–at 1 μM for 60 min. Compared to 610CP-, 580CP-jasplakinolide enables an enhanced labeling of intricate actin structures. The overall performance can be affected by the specific dye residue coupled to the (same) jasplakinolide ligand, as the dye was shown to influence the core characteristics of the whole fluorescent probe, such as binding parameters (kinetics, affinity, equilibrium between F- and G-actin), cytotoxicity, and, most importantly, cell entry and/or retention.5 Other (less toxic, more specific, brighter) fluorescent probes for actin in cells and tissues may help further to understand the role of this protein in cell functions and motility.16 The proposed methodology enables the scalable synthesis of compounds 7-H, 9, their analogs (e.g., by varying the structure of unsaturated alcohol in Scheme 3), and their conjugates with fluorescent dyes, in order to reveal new important aspects of actin behavior in the living matter.
Experimental Section
General Remarks
The reactions (in solution phase synthesis) were performed with magnetic stirring under an argon atmosphere. Evaporations in vacuum were performed in a rotary evaporator with bath temperature not exceeding 45 °C. Automated flash column chromatography was carried out using cartridges with regular silica gel from Biotage (10, 25 or 50 g SiO2) on a Biotage Isolera One device. For analytical TLC, Merck Millipore ready-to-use plates with silica gel 60 (F254) were used. The spots were visualized by illumination with a UV lamp (λ = 254 and 365 nm), staining with phosphomolybdic acid or ninhydrin solutions. 1H and 13C{1H}NMR spectra were recorded at 25 °C on Agilent 400-MR (400 MHz 1H and 100.5 MHz 13C), Bruker AVANCE NEO 600 MHz (TBO probe) and Bruker AVANCE III HD 500 MHz (BBO Prodigy probe) instruments. Chemical shifts are given in parts per million (ppm) using the residual solvent peak(s) as references. Multiplicities of the signals are described as follows: s = singlet, d = doublet, t = triplet, q = quartet, quint = quintet, m = multiplet or overlap. J values are given in Hz. Mass-spectra with electro-spray ionization (ESI-MS) were recorded on a Varian 500-MS spectrometer (Agilent). ESI-HRMS were measured on a MICROTOF spectrometer (Bruker) equipped with an Apollo ion source and a direct injector with an LC-autosampler Agilent RR 1200. Analytical RP-HPLC was carried out with a Knauer Azura or Thermo Scientific (Ultimate 3000) systems equipped with diode array detectors. Solvent A: H2O + 0.1% v/v TFA; solvent B: MeCN + 0.1% v/v TFA. LC–MS analyses were performed with Thermo Fisher Scientific ISQ EM mass-spectrometer (coupled to Ultimate 3000 system) using a gradient of acetonitrile (20–100%, if not stated otherwise) in water (with addition of 0.1% v/v HCOOH to both solvents). Preparative HPLC separations were accomplished on an Interchim puriFlash 4250 device with a 250 × 21.2 mm column PF5C18AQ; flow rate 20 mL/min, gradient of acetonitrile in 50 mM aqueous of Et3N*H2CO3 buffer (pH = 7.5).
Fmoc-O-TIPS-β-tyrosine (AA1 in Schemes 1 and 2)2 was prepared from Fmoc-β-tyrosine (Fmoc-(R)-3-amino-3-(4-hydroxyphenyl)propionic acid, ABCR GmbH, Germany) and triisopropylsilyl chloride according to the modified procedure applied for the preparation of Boc-(S)-Tyr(TBS)-OH from Boc-(S)-Tyr-OH and tert-butyldimethylsilyl chloride.9 The modification was based on the addition of 5% aq. NaHCO3 to the reaction mixture of the second step (desilylation of the carboxyl group) so that the initial pH value was set to 11, in order to prevent the possible cleavage of the Fmoc group. Yield of AA1: 78% (1.7 g (2.5 mmol) of glassy foam from 1.8 g (3.2 mmol) of Fmoc-β-tyrosine); [α]D20 = +26.4 (c = 1.1, CHCl3); [α]D = +16.1 (c = 1.1, CHCl3)6 (1-methyl-1-phenyl)ethyl trichloroacetimidate (PhCMe2OC(=NH)Me, Scheme 2)10,13 was prepared from 2-phenylpropanol-2 and trichloroacetonitrile (both Merck) according to the general method.14 Fmoc-O-TIPS-β-tyrosine (1-methyl-1-phenyl)ethylester (2, Scheme 2) was synthesized according to the published method.13 Fmoc-N-Me-d-Trp-OH (AA2 in Schemes 1 and 2) was prepared as described in the Supporting Information to ref (2). Nα-Fmoc-Nε-Boc-l-lysine was from ABCR GmbH (Germany). N-ethyl-N′-[3-(dimethylamino)propyl]carbodiimide (EDC; free base) was from Sigma-Aldrich. HOAt (1-hydroxy-7-azabenzotriazole) was bought from Gen Script. (S)-2,4-Dimethylpent-4-enoic acid and (S)-hex-5-en-2-ol were purchased from Enamine (Ukraine) and Carbosynth (UK), respectively. Grubbs Catalyst M2a (C848) was from Sigma-Aldrich. All other starting materials and reagents were purchased from commercial suppliers (Acros, Alfa Aesar, Merck, Novabiochem) and used without purification. Anhydrous CH2Cl2 (DCM), N-methylpyrrolidone-2 (NMP), N,N-dimethylformamide (DMF), and hexafluoropropanol-2 (HFIP) were stored over molecular sieves (4 Å). The temperature “0 °C” corresponds to the cooling of the stirred reaction mixture with an ice bath.
Solid-Phase Peptide Synthesis
Loading
2-Chlorotrityl chloride resin (200–400 mesh, 1.2–1.4 mmol·g–1 Cl loading; 1% cross-linked polystyrene) was purchased from Sigma-Aldrich. Anhydrous DCM and DMF were used in the following protocol. Under an argon atmosphere, a fritted 20 mL syringe was charged with dried resin (1.29 mmol, 937 mg), 9.4 mL of DCM was added, and the resin swelled under shaking (300 rpm) for 3 h at 23 °C. The syringe was drained, and the coupling of the first amino acid (AA1; Scheme 1) performed by adding a solution of AA1 (0.83 equiv, 1.07 mmol, 602 mg) in DCM (6.4 mL) and N,N-diisopropylethylamine (DIPEA; 3.33 equiv, 4.28 mmol, 728 μL) followed by shaking for 20 h at 23 °C.
The syringe was drained and the resin washed with DCM (9.4 mL), DCM/MeOH/DIPEA (17:2:1 v/v; 3 × 9.4 mL), DCM (4 × 9.4 mL), DMF (3 × 9.4 mL), and DCM (3 × 9.4 mL). For better coupling efficiency, incubation with AA1 and washing steps were repeated with half of the amount of AA1.
Fmoc Cleavage
The solvent was removed and the resin dried for 12 h at 0.2 mbar. Two portions of the resin (5.5 mg each) were used to determine the loading degree of AA1 (0.44 mmol g–1) using a mixture of DBU/piperidine/NMP (2:2:96 v/v) for cleaving the Fmoc group and measuring the optical density (at 304 nm) against the blank sample.15 The main part of resin (1.25 g, 0.55 mmol) was washed with NMP (9.4 mL) and subjected to deprotection by refilling the syringe with a mixture of DBU/piperidine/NMP (2:2:96 v/v) and shaking (300 rpm) for 10 min at 23 °C. This operation was repeated (with 30 min exposure at 23 °C). Completion of the cleavage was controlled by TLC (hexane/EtOAc 75:25): the application of the second cleavage cocktail revealed no appreciable UV active spot(s) of the Fmoc derivatives.
Activation and Coupling
AA2 (AA3, A4) (1.65 mmol, 3 equiv) was added into an oven-dried round bottom flask filled with argon and dissolved in NMP (7.5 mL). Then, a solution of HOBt (4.0 equiv, 2.2 mmol, 299 mg) in NMP (750 μL) was added followed by DIC (4.0 equiv, 2.2 mmol, 343 μL), and the reaction solution was stirred for 5 min under argon. The syringe with the resin was filled with the reaction solution; NMP (1 mL) was used for rinsing the round-bottom flask. The suspension was shaken (300 rpm) for 20 h at 23 °C. The syringe was drained, the resin was washed with NMP (2 × 9.4 mL), CH2Cl2 (3 × 9.4 mL), and subjected to deprotection.
Cleavage from Resin and Purification
The resin was placed into a round-bottom flask (50 mL), suspended in abs. DCM/HFIP (4:1 v/v, 15 mL) and stirred (600 rpm) for 20 min at 23 °C. The suspension was filtered, and the resin was washed with DCM (4 × 10 mL). EtOAc (15 mL) was added to the filtrate to minimize cleavage of the TIPS group (in the course of solvent evaporation). The solvents were evaporated in vacuo followed by drying the residue (0.1 mbar) for 30 min to yield yellow foam. Column chromatography (DCM/MeOH 96:4–90:10) yielded amorphous solids of 1-H-TIPS (0.25 g, 0.28 mmol, 52% yield based on the loading degree of AA1) and the desilylated byproduct 1-H-H (42 mg, 0.06 mmol, 11%). HRMS (ESI) calcd for C39H53N5O8 [M + H]+, 720.3967; found, 720.3968.
Compound 1-H-TIPS (= 5-H-H)
Mixture of 2 amide rotamers in ratio ca. 10:1. 1H NMR (CD2Cl2, 500 MHz, major isomer): δ 9.58 (s, 1H, NH indole), 7.57 (d, 1H, J = 7.9), 7.34 (d, 1H, J = 8.0), 7.16 (d, 2H, J = 8.5), 7.13 (ddd, 1H, J = 8.2, 7.0, 1.2), 7.05 (ddd, 1H, J = 8.1, 7.1, 1.1), 6.99 (d, 1H, J = 2.3, NH), 6.84 (d, 2H, J = 8.6), 6.37 (d, 1H, J = 7.1, NH), 5.61 (m, 1H), 5.32 (m, 1H, masked by the solvent peak), 4.79 (m, 1H), 4.74 (t, 1H, J = 1.7, =CH2), 4.68 (dq, 1H, J = 2.0 and 1.0, =CH2), 4.56 (q, 1H, J = 6.7), 3.33 (dd, 1H, J = 16.1, 4.9 Hz), 3.25 (dd, 1H, J = 15.9, 11.7 Hz), 2.95 (m, 1H), 2.93 (s, 3H, NMe), 2.76 (dd, 1H, J = 15.0, 8.6), 2.47 (q, 1H, J = 7.0), 2.33 (dd, 1H, J = 14.3, 6.5 Hz), 2.01 (dd, 1H, J = 14.4, 7.9 Hz), 1.66 (dd, 3H, J = 1.5, 1.5, MeCH), 1.50 (s, 9H, tBu), 1.25 (m, 6H, 3× CHiPr + MeCH), 1.09 (m× 2, 21H, TIPS + Me), 0.87 (dd, 2H, J = 8.2 and 7.6, CH2); 13C NMR (CD2Cl2, 125 MHz, major isomer): δ 178.2, 173.4, 172.0, 169.5, 156.9, 155.7, 143.1, 136.8, 133.8 (all 9 signals—Cq), 127.5 (2× CH), 135.9, 128.9 (2×), 128.8, 128.5, 128.0, 127.5, 126.0, 125.1 (2×), 122.5, 121.8, 122.0, 120.1 (2× CH), 119.2, 118.7, 112.5 (=CH2), 110.8, 79.8 (Cq–O), 56.9 (CH), 54.0 (CH), 49.7 (CH), 50.2 (CH), 41.9 (CH2), 41.1 (CH2), 40.3 (CH2), 39.1 (CH), 30.9 (NMe), 28.5 (tBu), 22.9 (CH2), 22.3 (CH3), 18.0 (6× Me), 12.8 (3× CHSi). HRMS (ESI) calcd for C48H73N5O8Si [M + Na]+, 898.5121; found, 898.5118.
Fmoc-O-TIPS-β-tyrosine (1-Methyl-1-phenyl)ethylester (2)
The product 2 (1.73 g, 82%) was prepared as glass-like foam from Fmoc-O-TIPS-β-tyrosin2 (1.74 g; see above) and (1-methyl-1-phenyl)ethyl trichloroacetimidate10,13 according to the published procedure.131H NMR ([D6]DMSO + CD3CN, 400 MHz): δ 7.84 (d, 2H, J = 7.7), 7.75 (d, 2H, J = 8.8), 7.65 (d, 2H, J = 7.5), 7.38 (m, 2H), 7.25 (m, 9H), 6.84 (m, 2H), 4.95 (q, 1H, J = 8.1, CHN), 4.33 (m, 2H, CHCH2O), 4.20 (t, 1H, J = 6.8, CHCH2O), 2.73 (m, 2H, J = 15.1, 7.8; CH2CO), 1.59 (s, 6H), 1.26 (m, 3H, 3× CHMe2), 1.08 (d, 18H, J = 7.4, 3× CHMe2); 13C NMR ([D6]DMSO + CD3CN, 101 MHz): δ 169.7, 156.3, 155.7, 146.7, 144.8, 141.8 (2×), 135.9 (all 7 signals—Cq), 128.9 (2×), 128.8, 128.5, 128.0, 127.5, 126.0, 125.1 (2×), 121.0, 120.2 (2×; all 9 signals—CH), 82.3 (Cq–O), 66.3 (CH2O), 52.3 (CHN), 47.8 (CH), 42.9 (CH2), 29.1 (2× Me), 18.5 (6× Me), 13.3 (3× CHSi); HRMS (ESI) calcd for C42H51NO5Si [M + Na]+, 700.3429; found, 700.3411.
General Procedure (GP1) for Peptide Coupling in Solution12
Synthesis of Compounds 3,4 and 5-C(CH3)2-C6H5–H
Deprotection step. Compound 2 or Fmoc-protected peptides (3, 4; see Scheme 2 and text below) were dissolved in MeCN (3 mL per 1 mmol); diethylamine (2 mL) was added, and the reaction mixture was left at room temperature for 8–15 h. All volatile materials were removed in vacuum, and the residue was co-evaporated in vacuum with toluene (3 × 10 mL). After keeping in high vacuum (<0.1 mbar) for 2 h, the residue was dissolved in anhydrous DCM (ca. 3 mL per 1 mmol) and filtered from the polymeric materials (if there were any; obtained in the course of Fmoc removal). The filtrate was evaporated in vacuum, the residue was re-dissolved in DCM and used in the next (coupling) step. Coupling step. HOAt (1.1 equiv) and 2,4,6-collidine (TMP; 1.1 equiv) were added to a cooled (ice bath) solution of the corresponding Fmoc-protected amino acid (1.1 equiv) in anhydrous DCM (3.0 mL per 1.0 mmol). Then, EDC (free base, 1.1 equiv) was added dropwise with stirring at 0 °C. After 10–15 min, the solution of the appropriate N-deprotected peptide (1.0 equiv) and TMP (2.0 equiv) in DCM (ca. 2 mL per 1.0 mmol) was added at 0 °C. The ice bath was removed, the reaction mixture was stirred overnight at room temperature, diluted with ethyl acetate (EtOAc) (30 mL), washed with 1 M aq. KHSO4 (2 × 5 mL), water (2 × 5 mL), 5% aq. NaHCO3 (2 × 5 mL), and saturated brine (5 mL; all amounts for 1 mmol scale). After drying with anhydrous Na2SO4, the solvents were removed in vacuum, and the coupling product was isolated by flash chromatography on regular SiO2. Glass-like foamy solids are obtained after drying in high vacuum (<0.1 mbar).
Dipeptide 3
The product is isolated (0.69 g, GP1) by flash chromatography on regular SiO2 (25 g, Biotage cartridge with spherical SiO2) and elution with 10–90% EtOAc in the mixture of hexane/DCM (3:1) over 11 column volumes. 1H NMR (CD3CN, 400 MHz, major invertomer): δ 9.05 (m, 1H, NH indole), 7.81 (d, 1H, J = 7.9), 7.75 (d, 1H, J = 7.9), 7.58 (d, 2H, J = 8.0), 7.53–6.80 (m, 15H, Har + NH), 5.25 (m, 1H, CHN-Tyr), 4.95 (m, 1H, CHN-Trp), 4.36/4.17/3.93/3.78 (m× 4, Σ = 3H, CHCH2O in Fmoc), 3.34 (m, 1H, CHAHB-Trp), 3.03 (m, 1H, CHAHB-Trp), 2.81 (m, 2H, CH2-Tyr), 2.74 (s, 3H, NMe), 1.58 (br s, 6H, CMe2), 1.26 (m, 3H), 1.08 (d, 18H, J = 7.3); 13C NMR (CD3CN, 101 MHz, major invertomer): δ 169.3 (CO), 155.2 (NCOO), 145.8, 144.1 (br.), 141.0 (br.), 136.4 (4 signals—Cq), 128.1, 127.6, 127.1, 126.7, 124.3, 121.5, 119.9, 119.6, 118.8, 111.3 (all 10 signals—CH), 81.8 (Cq-O), 67.0 (CH2O), 59.5 (CHN), 49.2 (CHN), 47.0 (CH), 41.0 (CH2), 29.9 (NMe), 27.9 (Me2), 23.9 (CH2), 17.3 (6× Me), 12.5 (3× CHSi); HRMS (ESI) calcd for C54H63N3O6Si [M + Na]+, 878.4559; found, 878.4549.
Tripeptide 4
The product is isolated (0.72 g, GP1) by flash chromatography on regular SiO2 (25 g, Biotage cartridge with spherical SiO2) and elution with 10–90% EtOAc in the mixture of hexane/DCM (3:1) over 11 column volumes. 1H NMR (CDCl3, 400 MHz): δ 9.62 (br s, 1H, NH indole), 7.74 (d, 2H, J = 7.5), 7.58 (d, 1H, J = 7.7), 7.51 (m, 2H, J = 7.5 and 5.2), 7.36 (m, 3H), 7.30–7.02 (m, 11H, Har), 6.94 (s, 1H), 6.78 (m, 2H), 5.65 (dd, 1H, J = 12.2, 4.6; CHN-Tyr), 5.53 (d, 1H, J = 6.6, NH), 5.37 (q, 1H, J = 7.4, CHN-Lys), 4.64 (br s, 1H), 4.35 (dd, 2H, J = 11.0, 7.1), 4.23 (dd, 1H, J = 10.5, 6.9), 4.14 (m, 1H), 3.39 (dd, J = 16.2, 4.6, 1H, CHCHD-Trp), 3.24 (dd, 1H, J = 16.1, 12.3; CHCHD-Trp), 2.91 (m, 1H, CHAHB-Tyr), 2.81 (m, 1H, CHAHB-Tyr), 2.71 (s, 3H, NMe), 1.64/1.62 (2× s, 6H, CMe2), 1.52 (s, 9H, Boc), 1.39 (m, 1H), 1.23 (m, 5H), 1.07 (d, 18H, J = 7.3), 1.06 (m, 1H), 0.85 (m, 2H); 13C NMR (CDCl3, 101 MHz): δ 173.2, 169.3, 169.1, 156.5, 156.0, 155.2, 145.2, 143.8, 143.6, 141.3, 136.4, 133.1 (12 signals Cq), 128.2, 127.7, 127.5, 127.1, 126.8, 125.0, 124.4, 121.8, 121.7, 119.93, 119.85, 119.0, 118.4, 111.4 (14 signals—CH), 110.4 (Cq), 82.0, 79.8 (2× Cq-O), 67.0 (CH2O), 56.6, 51.1, 49.2, 47.1 (4 signals CH), 41.5 (CH2), 40.5 (CH2), 31.3 (CH2), 30.5 (NMe), 30.0 (CH2), 28.7 (Me2), 28.5 (tBu), 28.0, 23.0 (CH2), 21.3 (CH2), 17.9 (6× Me), 12.6 (3× CHSi); HRMS (ESI) calcd for C65H83N5O9Si [M + Na]+, 1128.5852; found, 1128.5845.
Compound 5-C(CH3)2-C6H5–H
The product is isolated (0.60 g, GP1) by flash chromatography on regular SiO2 (25 g, Biotage cartridge with spherical SiO2) and elution with 10–90% (EtOAc + 3%v/v MeOH) in DCM over 11 column volumes. 1H NMR (CDCl3, 600 MHz): δ 9.54 (s, 1H, NH indole), 7.58 (d, 1H, J = 7.8, Trp 4), 7.32 (d, 1H, J = 8.1, Trp 7), 7.19–7.29 (m, 5H, C(CH3)2C6H5), 7.12–7.16 (m, 4H, Trp 6/βTyr 2/6/βTyr NH), 7.08 (ddd, 1H, J = 8.0, 7.0, 1.1; Trp 5), 6.94 (s, 1H, Trp 2), 6.81 (d, 2H, J = 8.6, βTyr 3/5), 6.19 (d, 1H, J = 6.0, Lys NH), 5.64 (dd, 1H, J = 12.3, 4.6; Trp α), 5.37 (dd, 1H, J = 7.8, 6.6; βTyr β), 4.75 (t, 1H, J = 1.8; 5), 4.68 (dd, 1H, J = 2.0, 1.0; 5), 4.64 (s, 1H, NH Lys side chain), 4.47 (q, 1H, J = 6.1, Lys α), 3.43 (dd, 1H, J = 16.2, 4.7; Trp β), 3.22 (dd, 1H, J = 16.2, 12.4; Trp β), 2.92 (dd, 1H, J = 15.3, 7.70; βTyr α), 2.91 (m, 1H, Lys ε), 2.87 (m, 1H, Lys ε), 2.77 (dd, 1H, J = 15.4, 6.30; βTyr α), 2.72 (s, 3H, Trp NMe), 2.37 (m, 1H, 2), 2.32 (dd, 1H, J = 14.1, 6.6; 3), 2.00 (dd, 1H, J = 14.0, 7.8; 3), 1.65–1.67 (m, 9H, C(CH3)2C6H5/7), 1.51 (s, 9H, Boc CH3), 1.37 (m, 1H, Lys β), 1.18–1.28 (m, 5H, TIPS CH/Lys δ), 1.04–1.11 (m, 1H, Lys β), 1.09 (d, 18H, J = 7.4, TIPS CH3), 1.07 (d, 3H, J = 6.8, 6), 0.70–0.80 (m, 2H, Lys γ); 13C NMR (CDCl3, 151 MHz): δ 176.3 (1), 173.4 (Lys CO), 169.5 (βTyr CO), 169.3 (Trp CO), 156.7 (Boc CO), 155.5 (βTyr 4), 145.7 (C(CH3)2C6H5 Cipso), 143.0 (4), 136.6 (Trp 7a), 133.3 (βTyr 1), 128.4 (C(CH3)2C6H5 Cmeta), 127.8 (βTyr 2/6), 127.5 (Trp 3a), 127.0 (C(CH3)2C6H5 Cpara), 124.6 (C(CH3)2C6H5 Cortho), 122.0 (Trp 2), 121.9 (Trp 6), 120.0 (βTyr 3/5), 119.2 (Trp 5), 118.6 (Trp 4), 112.6 (5), 111.5 (Trp 7), 110.7 (Trp 3), 82.1 (C(CH3)2C6H5), 79.9 (Boc Cq), 56.7 (Trp α), 49.8 (Lys α), 49.7 (βTyr β), 42.0 (3), 41.6 (βTyr α), 40.5 (Lys ε), 38.9 (2), 31.2 (Lys β), 30.7 (Trp NMe), 30.2 (Lys δ), 28.9 (C(CH3)2C6H5), 28.6 (Boc CH3), 28.3 (C(CH3)2C6H5), 23.2 (Trp β), 22.9, 22.4 (7), 21.6 (Lys γ), 18.1 (TIPS CH3), 17.4 (6), 12.8 (TIPS CH). HRMS (ESI) calcd for C57H83N5O8Si [M + Na]+, 1016.5903; found, 1016.5892.
Compound 5-H-H (= 1-H-TIPS)
Synthesis according to Scheme 2. Compound 5-C(CH3)2-C6H5–H (66 mg, 66 μmol) was dissolved in MeCN (0.90 mL), and TFA (0.1 mL of 20% v/v solution in MeCN) was added so that the final conc. of TFA became 2% v/v. The reaction mixture was kept in an ice bath. HPLC control: column US10C18HQ, Interchim, 10 μ, 250 × 4.6 mm, flow rate 1.2 mL/min, A/B = 50/50 (0–1 min), 50/50 → 0/100 (1–11 min). After 3 h at 0 °C, the starting material 5-C(CH3)2-C6H5–H with tR = 10.4 min was detected (23% HPLC area at 254 nm), compound 5-H-H with tR = 7.9 min (52% HPLC area), and compound 5-H-C(CH3)2C6H5 with tR = 9.4 min (15% HPLC area). Then, 0.1 mL of 20% v/v TFA in ACN was added, and the reaction mixture was warmed up to 10 °C. When HPLC displayed the presence of 7.5% (HPLC area at 254 nm) of ester 5-C(CH3)2-C6H5–H (19% of 5-H-C(CH3)2C6H5 and 72% 5-H-H), the reaction was quenched by the slow addition of Et3N (0.10 mL) at 0 °C, diluted with water (3.0 mL), and subjected to preparative HPLC; injection loop 5 mL, 30–100% acetonitrile with 50 mM aqueous of Et3N*H2CO3 buffer (pH = 7.5) in 30 min (for other conditions, see in General Remarks). Liophilization of appropriate fractions gave compound 5-H-H (42 mg, 71%) and 5-H-C(CH3)2C6H5 (11 mg, 17%) as colorless, voluminous, and very light powders. 5-H-H: 1H NMR (600 MHz, CDCl3): δ 9.47 (s, 1H, indole NH), 7.59 (d, 1H, J = 7.9, Trp 4), 7.48 (s, 1H, βTyr NH), 7.33 (d, 1H, J = 8.1, Trp 7), 7.12–7.19 (m, 3H, Trp 6/βTyr 2/6), 7.08 (t, 1H, J = 7.5, Trp 5), 6.96 (s, 1H, Trp 2), 6.79 (d, 2H, J = 8.6, βTyr 3/5), 6.61 (d, 1H, J = 7.2, Lys NH), 5.65 (dd, 1H, J = 11.2, 3.3, Trp α), 5.35 (q, 1H, J = 6.8, βTyr β), 4.73 (s, 1H, 5), 4.65–4.71 (m, 2H, 5/Lys ξ), 4.62 (q, 1H, J = 6.5, Lys α), 3.38 (dd, 1H, J = 16.0, 4.4, Trp β), 3.31 (dd, 1H, J = 16.0, 12.1, Trp β), 2.88–3.02 (m, 5H, Trp NMe/Lys ε), 2.72–2.80 (m, 2H, βTyr α), 2.41–2.48 (m, 1H, 2), 2.33 (dd, 1H, J = 14.3, 6.7, 3), 2.01 (dd, 1H, J = 14.3, 7.9, 3), 1.66 (s, 3H, 7), 1.50 (s, 9H, Boc CH3), 1.16–1.34 (m, 6H, TIPS CH/Lys β/δ), 1.02–1.14 (m, 22H, TIPS CH3/6/Lys β), 0.80–0.95 (m, 2H, Lys γ); 13C NMR (151 MHz, CDCl3): δ 177.3 (1), 173.8 (βTyr CO), 173.1 (Lys CO), 169.4 (Trp CO), 156.7 (Boc CO), 155.3 (βTyr 4), 142.9 (4), 136.5 (Trp 7a), 133.6 (βTyr 1), 127.5 (βTyr 2/6), 127.5 (Trp 3a), 122.1 (Trp 2), 121.9 (Trp 6), 119.9 (βTyr 3/5), 119.3 (Trp 5), 118.7 (Trp 4), 112.6 (5), 111.5 (Trp 7), 110.7 (Trp 3), 79.9 (Boc Cq), 56.7 (Trp α), 49.8 (Lys α), 49.6 (βTyr β), 41.8 (3), 41.3 (βTyr α), 40.3 (Lys ε), 39.0 (2), 31.1 (Lys β), 30.7 (Trp NMe), 30.1 (Lys δ), 28.6 (Boc Me), 23.0 (Trp β), 22.4 (7), 21.9 (Lys γ), 18.1 (TIPS Me), 17.5 (6), 12.8 (TIPS CH).
Compound 5-H-C(CH3)2C6H5
1H NMR (CDCl3, 600 MHz): δ 7.54 (d, 1H, J = 7.9, Trp 4) 7.20–7.30 (m, 4H, Trp 2/C(CH3)2C6H5 Hmeta/Hpara), 7.07–7.16 (m, 5H, βTyr NH/βTyr 2/6/C(CH3)2C6H5 Hortho), 6.97 (ddd, 1H, J = 7.9, 7.0, 0.9; Trp 5), 6.84 (ddd, 1H, J = 8.3, 6.9, 1.2; Trp 6), 6.77 (d, 2H, J = 8.6, βTyr 3/5), 6.71 (d, 1H, J = 8.9, Lys NH), 6.56 (d, 1H, J = 8.5, Trp 7) 5.56 (m, 1H, Trp α), 5.37 (m, 1H, βTyr β), 4.76 (s, 1H, 5), 4.71 (s, 1H, 5), 4.62–4.68 (m, 1H, Lys α), 4.49–4.53 (m, 1H, NH Lys side chain), 3.47 (dd, 1H, J = 15.6, 5.9; Trp β), 3.24 (m, 1H, Trp β), 2.95–3.02 (m, 5H, Lys ε/Trp NMe), 2.83 (dd, 1H, J = 15.3, 4.86; βTyr α), 2.78 (dd, 1H, J = 15.3, 8.3; βTyr α), 2.50–2.59 (m, 1H, 2), 2.40 (dd, 1H, J = 14.4, 6.4; 3), 2.05 (dd, 1H, J = 14.3, 8.2; 3), 1.88 (s, 3H, C(CH3)2C6H5), 1.87 (s, 3H, C(CH3)2C6H5), 1.69 (s, 3H, 7), 1.43 (s, 9H, Boc CH3), 1.13–1.41 (m, 6H, Lys β/γ/δ), 1.21 (m, 3H, TIPS CH), 1.12 (d, 3H, J = 6.9; 6), 1.07 (d, 18H, J = 7.4, TIPS CH3); 13C NMR (151 MHz, CDCl3): δ 178.2 (1), 173.3 (Lys CO), 172.6 (βTyr CO), 169.2 (Trp CO), 156.5 (Boc CO), 155.4 (βTyr 4), 146.9 (C(CH3)2C6H5 Cipso), 142.8 (4), 135.5 (Trp 7a), 133.4 (βTyr 1), 129.6 (Trp 3a), 128.8 (C(CH3)2C6H5 Cmeta), 127.4 (βTyr 2/6), 127.1 (C(CH3)2C6H5 Cpara), 125.2 (C(CH3)2C6H5 Cortho), 123.8 (Trp 2), 121.0 (Trp 6), 112.0 (βTyr 3/5), 118.9 (Trp 5), 118.8 (Trp 4), 113.8 (Trp 7), 112.7 (5), 109.5 (Trp 3), 79.5 (Boc Cq), 60.4 (C(CH3)2C6H5), 57.3 (Trp α), 49.9 (Lys α), 49.6 (βTyr β), 41.8 (3), 40.9 (βTyr α), 39.6 (Lys ε), 39.0 (2), 31.2 (Lys β), 31.1 (Trp NMe), 30.4 (C(CH3)2C6H5), 30.3 (C(CH3)2C6H5), 29.7 (Lys δ), 28.6 (Boc CH3), 23.1 (Trp β), 22.5 (Lys γ), 22.4 (7), 18.1 (TIPS CH3), 17.5 (6), 12.8 (TIPS CH). HRMS (ESI) calcd for C57H83N5O8Si [M + Na]+, 1016.5903; found, 1016.5896.
Compound 6
Compound 5-H-H (160 mg, 0.18 mmol) was dissolved in anhydrous DCM (4.0 mL) under argon; then, (S)-hex-5-en-2-ol (73 mg, 0.73 mmol) was added followed by DMAP (2.2 mg, 0.018 mmol; as a stock solution in DCM) and EDC (free base, 63 μL, 56 mg, 0.36 mmol). EDC was added dropwise at 0... +5 °C. The reaction mixture was allowed to warm-up to room temp. and stirred overnight. Then, it was diluted with EtOAc (to ca. 15–20 mL), washed with cold. 1 M aq. KHSO4, water, 3% aq. NaHCO3, and brine. After drying over anhydrous Na2SO4, the solvents were evaporated in vacuo, and the residue was subjected to flash chromatography on regular SiO2 (Biotage cartridge with 25 g spherical silica gel). Elution with hexane-ethyl acetate (70:30–40:60) afforded ester 6 (74 mg, 43%) as a glass-like foam. The mixture of two amide rotamers in the ratio ca. 10:1. 1H NMR (CD2Cl2, 500 MHz, major isomer): δ 9.67 (s, 1H, NH indole), 7.58 (ddd, 1H, J = 7.9, 1.3 and 0.9), 7.35 (dt, 1H, J = 8.1 and 0.9), 7.28 (d, 1H, J = 8.4, NH), 7.16 (d, 2H, J = 8.5), 7.12 (ddd, 1H, J = 8.2, 7.0, 1.2), 7.05 (ddd, 1H, J = 8.0, 7.0, 1.1), 6.99 (m, 1H, NH), 6.82 (d, 2H, J = 8.6), 6.15 (d, 1H, J = 5.7, NH), 5.76 (ddt, 1H, J = 16.9, 10.2 and 6.6, CH=), 5.65 (dd, J = 12.2 and 4.1, 1H, CH), 5.35 (m, 1H, masked by the solvent peak, CH), 4.99 (m, 2H, CH2=), 4.79 (m, 1H), 4.83 (ddd, 1H, J = 7.5, 6.3, 5.1), 4.75 br s/4.67 dd (2H, J = 2.1 and 1.1, =CH2), 4.47 (dt, 1H, J = 7.2 and 5.7, CH), 3.46 (dd, 1H, J = 16.1, 4.3 Hz, CHH), 3.18 (dd, 1H, J = 16.0, 13.3 Hz, CHH), 2.95 (s, 3H, NMe), 2.91 (m, 1H, CHH), 2.75 (dd, 1H, J = 15.6, 6.2, CHH), 2.40 (dt, 1H, J = 7.7 and 6.5 Hz, CH), 2.30 (dd, 1H, J = 14.2, 6.3 Hz, CHH), 1.99 (m, 3H, CH2), 1.66 (d, 3H, J = 1.5, MeCH), 1.60 (m, 2H, CH2), 1.51 (s, 9H, tBu), 1.39 (m, 2H, CH2), 1.25 (m, 6H, 3× CHiPr + MeCH), 1.09 (m, 18H, TIPS), 1.09 (m, 3H, Me), 0.87 (m, 2H, CH2); 13C NMR (CD2Cl2, 125 MHz, major isomer): δ 176.5, 173.3, 170.7, 169.3, 156.9, 155.6, 143.4 (all—Cq), 138.3 (CH), 136.8, 134.0 (both Cq), 127.6 (Cq), 127.8 (2× CH), 122.4 (Cq), 121.8 (CH), 120.0 (2× CH), 118.7 (CH), 114.8 (=CH2), 112.4 (=CH2), 111.6 (CH), 79.8 (Cq-O), 70.9 (CH), 57.0 (CH), 50.1 (CH), 49.7 (CH), 42.0 (CH2), 41.2 (CH2), 40.5 (CH2), 38.8 (CH), 35.2 (CH2), 35.2 (CH2), 31.4 (CH2), 31.0 (NMe), 30.2 (CH2), 28.5 (tBu), 23.3 (CH2), 22.3 (CH3), 21.8 (CH2), 18.0 (6× Me), 17.3 (Me), 12.9 (3× CHSi). HRMS (ESI) calcd for C54H83N5O8Si [M + Na]+, 980.5903; found, 980.5883.
Compounds 7-TIPS and 8-TIPS
Compound 6 (62 mg, 65 μmol) was dissolved in dry DCM (60 mL) in a screw-cap tube. The tube was closed with septum, and a gentle argon stream was blown through solution for 10 min with stirring and gradual heating up to 40 °C. Then, the solution of the Grubbs’ catalyst (2nd generation, 5.0 mg, 9% mol) in degassed anhydrous toluene (0.6 mL) was added under an argon atmosphere, the septum was replaced with a screw-cap, and the reaction mixture was stirred at 40 °C for 90 min. HPLC control: puriFlash column C18AQ (Interchim), 5 μ, 250 × 4.6 mm, flow rate 1.2 mL/min, A/B = 30/70 → 0/100 (20 min), then 100% B. Compound 6: tR = 24.7 min (3% HPLC area), compound 7-TIPS: tR = 23.8 min (∼87% HPLC area), and compound 8-TIPS: tR = 23.5 min (∼10% HPLC area); the toluene peak at 12.5 min was not integrated. The reaction mixture was evaporated in vacuo, and the residue subjected to flash chromatography on regular SiO2 (Biotage cartridge with 25 g spherical silica gel). Elution with hexane-ethyl acetate (80:20–30:70) afforded a mixture of compounds 7-TIPS and 8-TIPS (51 mg, 85%) as a glass-like foam (compound 6 has higher Rf value). Compound 7-TIPS (compound 11 from ref (2)). 1H NMR (CDCl3, 400 MHz): δ 9.89 (s, 1H, NH indole), 7.60 (dd, 1H, J = 7.5 and 1.3), 7.34 (d, 1H, J = 9.0), 7.17–7.04 (m, 5H), 6.93 (br s, 1H, NH), 6.81 (d, 2H, J = 8.6), 6.58 (d, 1H, J = 6.4, NH), 5.67 (dd, 1H, J = 12.1 and 4.7, CH=), 5.22 (dt, 1H, J = 7.8 and 4.1), 4.98 (t, J = 6.9, 1H), 4.86–4.65 (m, 3H), 3.36 (dd, 1H, J = 16.3 and 12.1), 3.26 (dd, 1H, J = 16.9 and 4.2), 3.03 (br s, 1H), 2.92 (d, 1H, J = 14.5 Hz), 2.88 (s, 3H, NMe), 2.76 (dd, 1H, J = 15.5 and 4.1), 2.55 (dd, 1H, J = 15.5, 4.1), 2.43 (m, 1H), 1.86–1.74 (m, 3H), 1.51 (s, 9H, tBu), 1.44 (d, J = 1.3, 3H, CH3), 1.32–1.15 (m, 6H, 3× CHiPr + CHCH3), 1.11 (d, 3H, J = 6, CH3CH), 1.08 (m, 20H, TIPS + CH2), 1.00 (m, 1H, CHH), 0.66 (m, 1H, CHH); 13C NMR (CDCl3, 101 MHz): δ 174.5, 173.9, 170.4, 169.3, 156.6, 155.4, 136.5, 133.9, 132.9, 127.2 (all—Cq), 127.1 (2× CH), 124.4 (CH), 121.7 (CH), 121.5 (CH), 119.9 (2× CH), 119.0 (CH), 118.3 (CH), 111.4 (CH), 79.8 (Cq-O), 69.4 (CHO), 56.0 (CH), 49.7 (CH), 49.0 (CH), 43.2 (CH2), 40.8 (CH2), 39.7 (CH), 35.5 (CH2), 30.8 (CH2), 30.2 (NMe), 30.1 (CH2), 28.5 (tBu), 23.1 (CH2), 22.3 (CH2), 21.0 (CH2), 20.4 (CH3), 20.3 (CH3), 17.9 (6× Me), 16.2 (Me), 12.6 (3× CHSi). HRMS (ESI) calcd for C52H79N5O8Si [M + Na]+, 952.5590; found, 952.5580.
Compounds 7-H and 8-H
The mixture of compounds 7-TIPS and 8-TIPS from the previous step (12.9 mg, 13.9 μmol) was dissolved in anhydrous THF (0.5 mL) under argon, and 1 M TBAF in THF (20 μL, 20 μmol) was added. The course of the reaction was monitored by TLC (hexane–EtOAc, 35/65; 7-H/8-H have lower Rf than 7-TIPS/8-TIPS). When the reaction was complete (more TBAF may require), the solvent was evaporated in vacuo, the residue was dissolved in aq. ACN (4.0 mL) and subjected to preparative HPLC; injection loop 5 mL, 30–80% acetonitrile with 50 mM aqueous of Et3N*H2CO3 buffer (pH = 8) in 30 min (for other conditions, see in General Remarks). Liophilization of appropriate fractions gave compound 8-H (“peak 1”, 1.8 mg, 17%) and 7-H (6.6 mg, 62%) as colorless solids. Compound 7-H (compound 12 from ref (2)). 1H NMR (CDCl3, 500 MHz): δ 9.88 (s, 1H, indole NH), 7.59 (d, 1H, J = 7.8, Trp 4), 7.42 (d, 1H, J = 8.1, βTyr NH), 7.34 (d, 1H, J = 8.2, Trp 7) 7.05–7.16 (m, 4H, Trp 5/6/βTyr 2/6), 6.93 (d, 1H, J = 2.2, Trp 2), 6.72–6.76 (m, 3H, βTyr 3/5/Lys NH), 5.68 (dd, 2H, J = 11.7, 5.1, Trp α), 5.18 (td, 1H, J = 8.0, 4.1, βTyr β), 4.97 (t, 1H, J = 7.1, 5), 4.83–4.89 (m, 1H, Lys α), 4.78 (dqd, 1H, J = 9.7, 6.3, 3.7, 8), 4.69–4.75 (m, 1H, NH Lys side chain), 3.33 (dd, 1H, J = 16.2, 12.1, Trp β), 3.28 (dd, 1H, J = 16.2, 5.3, Trp β), 2.97–3.08 (m, 1H, Lys ε), 2.89 (s, 3H, Trp NMe), 2.77–2.85 (m, 1H, Lys ε), 2.73 (dd, 1H, J = 15.5, 4.1, βTyr α), 2.55 (dd, 1H, J = 15.5, 8.0, βTyr α), 2.44–2.49 (m, 1H, 2), 2.36–2.43 (m, 1H, 3), 1.86 (d, 1H, J = 13.8, 3), 1.73–1.82 (m, 2H, 6), 1.46–1.56 (m, 11H, Boc CH3, 7, Lys β), 1.40 (s, 3H, 11), 1.28–1.36 (m, 1H, 7), 1.15–1.27 (m, 2H, Lys δ), 1.12 (d, 3H, J = 6.6, 10), 1.08 (d, 3H, J = 6.3, 9), 0.89–1.03 (m, 2H, Lys β/γ), 0.56–0.67 (m, 1H, Lys γ); 13C NMR (126 MHz, CDCl3): δ 175.3 (1), 173.7 (Lys CO), 170.6 (βTyr CO), 169.4 (Trp CO), 156.6 (Boc CO), 155.2 (βTyr 4), 136.5 (Trp 7a), 133.5 (4), 132.5 (βTyr 1), 127.3 (βTyr 2/6), 127.1 (Trp 3a), 124.9 (5), 121.7 (Trp 6), 121.6 (Trp 2), 119.0 (Trp 5), 118.3 (Trp 4), 115.5 (βTyr 3/5), 111.4 (Trp 7), 109.9 (Trp 3), 79.9 (Boc Cq), 69.6 (8), 56.2 (Trp α), 49.8 (Lys α), 49.2 (βTyr β), 43.1 (3), 40.7 (Lys ε), 39.6 (2), 39.6 (βTyr α), 35.4 (7), 30.8 (Lys β), 30.2 (Trp NMe), 30.0 (Lys δ), 28.5 (Boc CH3), 23.1 (6), 22.4 (Trp β), 20.9 (Lys γ), 20.4 (9), 20.2 (10), 15.9 (11). HRMS (ESI) calcd for C43H59N5O8 [M + Na]+, 796.4256; found, 796.4257.
Compound 8-H
1H NMR (CDCl3, 500 MHz): δ 9.89 (s, 1H, indole NH), 7.60 (d, 1H, J = 7.8, Trp 4), 7.35 (d, 1H, J = 8.0, Trp 7), 7.06–7.20 (m, 5H, Trp 5/6/βTyr NH/βTyr 2/6), 6.94 (s, 1H, Trp 2), 6.80 (d, 2H, J = 8.5, βTyr 3/5), 6.66 (d, 1H, J = 6.78, Lys NH), 5.57 (dd, 1H, J = 12.2, 4.5, Trp α), 5.35 (td, 1H, J = 9.1, 3.7), 5.08 (dd, 1H, J = 9.0, 5.2, 5), 4.70–4.79 (m, 2H, Lys α/NH Lys side chain), 3.99 (dd, 1H, J = 10.6, 3.6, 8), 3.72 (dd, 1H, J = 10.8, 4.2, 8), 3.42 (dd, 1H, J = 16.0, 12.5, Trp β), 3.20 (dd, 1H, J = 16.2, 4.5, Trp β), 2.98–3.09 (m, 1H, Lys ε), 2.94 (s, 3H, Trp NMe), 2.74–2.86 (m, 1H, Lys ε), 2.78 (dd, 1H, J = 16.5, 3.6, 2.61, βTyr α), 2.61 (dd, 1H, J = 16.5, 9.8, βTyr α), 2.39–2.51 (m, 2H, 2/3), 1.87–1.92 (m, 2H, 3/6), 1.71–1.83 (m, 1H, 6), 1.63–1.71 (m, 1H, 7), 1.47–1.57 (m, 13H, 11/Lys β/Boc CH3), 1.15–1.27 (m, 2H, Lys δ), 1.13 (d, 3H, J = 6.4, 10), 0.91–0.98 (m, 2H, Lys β/γ), 0.85 (d, 3H, J = 6.8, 9), 0.55–0.66 (m, 1H, Lys γ); 13C NMR (126 MHz, CDCl3): δ 175.4 (1), 173.7 (Lys CO), 171.0 (βTyr CO), 169.7 (Trp CO), 156.8 (Boc CO), 155.4 (βTyr 4), 136.7 (Trp 7a), 134.1 (4), 132.9 (βTyr 1), 127.6 (βTyr 2/6), 127.2 (Trp 3a), 124.5 (5), 122.0 (Trp 6), 121.8 (Trp 2), 119.3 (Trp 5), 118.5 (Trp 4), 115.7 (βTyr 3/5), 111.7 (Trp 7), 110.0 (Trp 3), 80.1 (Boc Cq), 67.2 (8), 56.4 (Trp α), 50.0 (Lys α), 48.9 (βTyr β), 43.6 (3), 40.8 (Lys ε), 40.2 (2), 40.1 (βTyr α), 33.2 (7), 31.3 (6), 30.9 (Lys β), 30.6 (Trp NMe), 30.2 (Lys δ), 28.7 (Boc CH3), 22.7 (Trp β), 21.1 (Lys γ), 20.2 (10), 17.6 (9), 16.0 (11). HRMS (ESI) calcd for C43H59N5O8 [M + Na]+, 796.4256; found, 796.4256.
Acknowledgments
The authors are grateful to J. Bienert (MPI BPC) for performing HPLC analyses and recoding NMR spectra. The authors thank Dr. H. Frauendorf and his team (Institut für Organische und Biomolekulare Chemie, Georg-August-Universität Göttingen) for recording high-resolution mass spectra.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c00653.
Experimental details of cell culture, in vitro labeling, and STED microscopy; LC–MS analysis of the deprotection reaction of 5-C(CH3)2C6H5–H; HPLC trace and ESI-MS for amine 9 (Scheme 4); HPLC data for 580CP- and 610CP-jasplakinolide conjugates; full 1H- and 13C-NMR assignments for isomers 5-C(CH3)2C6H5–H and 5-H-C(CH3)2C6H5, as well as 7-H and 8-H; and copies of NMR spectra (PDF)
The authors declare no competing financial interest.
Footnotes
STED: stimulated emission depletion.
Supplementary Material
References
- Davidson A. J.; Wood W. Unravelling the Actin Cytoskeleton: A New Competitive Edge?. Trends Cell Biol. 2016, 26, 569–576. 10.1016/j.tcb.2016.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milroy L.-G.; Rizzo S.; Calderon A.; Ellinger B.; Erdmann S.; Mondry J.; Verveer P.; Bastiaens P.; Waldmann H.; Dehmelt L.; Arndt H.-D. Selective Chemical Imaging of Static Actin in Live Cells. J. Am. Chem. Soc. 2012, 134, 8480–8486. 10.1021/ja211708z. [DOI] [PubMed] [Google Scholar]
- Lukinavičius G.; Reymond L.; D’Este E.; Masharina A.; Gottfert F.; Ta H.; Güther A.; Fournier M.; Rizzo S.; Waldmann H.; Blaukopf C.; Sommer C.; Gerlich D. W.; Arndt H.-D.; Hell S. W.; Johnsson K. Fluorogenic probes for live-cell imaging of the cytoskeleton. Nat. Methods 2014, 11, 731–733. 10.1038/nmeth.2972. [DOI] [PubMed] [Google Scholar]
- Grimm F.; Nizamov S.; Belov V. N. Green-emitting rhodamine dyes for vital labeling of cell organelles: chemistry, optical spectra and microscopy. ChemBioChem 2019, 20, 2248–2254. 10.1002/cbic.201900177. [DOI] [PubMed] [Google Scholar]
- Gerasimaitė R.; Seikowski J.; Schimpfhauser J.; Kostiuk G.; Gilat T.; D́Este E.; Schnorrenberg S.; Lukinavičius G. Efflux pump insensitive rhodamine-jasplakinolide conjugates for G- and F-actin imaging in living cells. Org. Biomol. Chem. 2020, 18, 2929–2937. 10.1039/d0ob00369g. [DOI] [PubMed] [Google Scholar]
- Tannert R.; Milroy L.-G.; Ellinger B.; Hu T.-S.; Arndt H.-D.; Waldmann H. Synthesis and Structure–Activity Correlation of Natural-Product Inspired Cyclodepsipeptides Stabilizing F-Actin. J. Am. Chem. Soc. 2010, 132, 3063–3077. 10.1021/ja9095126. [DOI] [PubMed] [Google Scholar]
- Butkevich A. N.; Mitronova G. Y.; Sidenstein S. C.; Klocke J. L.; Kamin D.; Meineke D. N. H.; D’Este E.; Kraemer P.-T.; Danzl J. G.; Belov V. N.; Hell S. W. Fluorescent rhodamines and fluorogenic carbopyronines for super resolution STED microscopy in living cells. Angew. Chem., Int. Ed. 2016, 55, 3290–3294. 10.1002/anie.201511018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butkevich A. N.; Belov V. N.; Kolmakov K.; Sokolov V. V.; Shojaei H.; Sidenstein S. C.; Kamin D.; Matthias J.; Vlijm R.; Engelhardt J.; Hell S. W. Hydroxylated fluorescent dyes for live cell labeling: synthesis, spectra and super resolution STED microscopy. Chem.—Eur. J. 2017, 23, 12114–12119. 10.1002/chem.201701216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokokawa F.; Inaizumi A.; Shioiri T. Synthetic Studies of the Cyclic Depsipeptides bearing 3-Amino-6-hydroxy-2-piperidone (Ahp) unit. Total Synthesis of the proposed Structure of Micropeptin T-20. Tetrahedron 2005, 61, 1459–1480. 10.1016/j.tet.2004.12.009. [DOI] [Google Scholar]
- Yue C.; Thierry J.; Potier P. 2-Phenyl Isopropyl Esters as Carboxyl Terminus Protecting Groups in the Fast Synthesis of Peptide Fragments. Tetrahedron Lett. 1993, 34, 323–326. 10.1016/s0040-4039(00)60578-6. [DOI] [Google Scholar]
- Athanassopoulos P.; Barlos K.; Gatos D.; Hantzi O.; Tzavara C. Application of 2-Chlorotrityl Chloride in Convergent Peptide Synthesis. Tetrahedron Lett. 1995, 36, 5645–5648. 10.1016/00404-0399(50)1036h-. [DOI] [Google Scholar]
- Zlatopolskiy B. D.; de Meijere A. First Total Synthesis of Hormaomycin, a naturally Occurring Depsipeptide with Interesting Biological Activities. Chem.—Eur. J. 2004, 10, 4718–4727. 10.1002/chem.200400249. [DOI] [PubMed] [Google Scholar]
- Virta P.; Karskela M.; Lönnberg H. Orthogonally Protected Cyclo-β-tetrapeptides as Solid-Supported Scaffolds for the Synthesis of Glycoclusters. J. Org. Chem. 2006, 71, 1989–1999. 10.1021/jo052348o. [DOI] [PubMed] [Google Scholar]
- Wessel H.-P.; Iversen T.; Bundle D. R. Acid-catalysed Benzylation and Allylation by Alkyl Trichloroacetimidates. J. Chem. Soc., Perkin Trans. 1 1985, 2247–2250. 10.1039/p19850002247. [DOI] [Google Scholar]
- Gude M.; Ryf J.; White P. D. An accurate method for quantification of Fmoc-derivatized solid phase supports. Lett. Pept. Sci. 2002, 9, 203–206. 10.1007/bf02538384. [DOI] [Google Scholar]
- Borowiak M.; Küllmer F.; Gegenfurtner F.; Peil S.; Nasufovic V.; Zahler S.; Thorn-Seshold O.; Trauner D.; Arndt H.-D. Optical Manipulation of F-Actin with Photoswitchable Small Molecules. J. Am. Chem. Soc. 2020, 142, 9240–9249. 10.1021/jacs.9b12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.