

Abstract
Background
High-grade gliomas are aggressive and immunosuppressive brain tumors. Molecular mechanisms that regulate the inhibitory immune tumor microenvironment (TME) and glioma progression remain poorly understood. Fyn tyrosine kinase is a downstream target of the oncogenic receptor tyrosine kinase pathway and is overexpressed in human gliomas. Fyn’s role in vivo in glioma growth remains unknown. We investigated whether Fyn regulates glioma initiation, growth and invasion.
Methods
We evaluated the role of Fyn using genetically engineered mouse glioma models (GEMMs). We also generated Fyn knockdown stem cells to induce gliomas in immune-competent and immune-deficient mice (nonobese diabetic severe combined immunodeficient gamma mice [NSG], CD8−/−, CD4−/−). We analyzed molecular mechanism by RNA sequencing and bioinformatics analysis. Flow cytometry was used to characterize immune cellular infiltrates in the Fyn knockdown glioma TME.
Results
We demonstrate that Fyn knockdown in diverse immune-competent GEMMs of glioma reduced tumor progression and significantly increased survival. Gene ontology (GO) analysis of differentially expressed genes in wild-type versus Fyn knockdown gliomas showed enrichment of GOs related to immune reactivity. However, in NSG and CD8−/− and CD4−/− immune-deficient mice, Fyn knockdown gliomas failed to show differences in survival. These data suggest that the expression of Fyn in glioma cells reduces antiglioma immune activation. Examination of glioma immune infiltrates by flow cytometry displayed reduction in the amount and activity of immune suppressive myeloid derived cells in the Fyn glioma TME.
Conclusions
Gliomas employ Fyn mediated mechanisms to enhance immune suppression and promote tumor progression. We propose that Fyn inhibition within glioma cells could improve the efficacy of antiglioma immunotherapies.
Keywords: antitumor immune responses, Fyn tyrosine kinase, glioma, myeloid-derived suppressor cells
Key Points.
1. Inhibition of Fyn tyrosine kinase in genetically engineered mouse glioma models delays tumor initiation and progression.
2. The oncogenic effects of Fyn in vivo are mediated by downregulation of antiglioma immunity.
Importance of the Study.
Fyn is an effector of receptor tyrosine kinase signaling in glioma. However, its role in vivo remains unknown. Our study demonstrates that Fyn tyrosine kinase is a novel regulator of the antiglioma immune response. We show that Fyn inactivation suppresses glioma growth, increases survival, and enhances antitumor immune reactivity. Our findings suggest that suppressing the expression of Fyn in glioma cells could provide a novel therapeutic target.
Glioblastoma multiforme (GBM), or high-grade glioma, is the most frequent and aggressive primary tumor of the central nervous system. It is characterized by extensive infiltrative growth and resistance to therapy.1 Mutated/activated driver genes such as the receptor tyrosine kinases (RTKs) (epidermal growth factor receptor [EGFR], platelet derived growth factor receptor [PDGFR], hepatocyte growth factor (HGF)/MET), tumor suppressor genes (tumor protein 53 [TP53], phosphatase and tensin homolog/neurofibromatosis type 1[ PTEN/NF1]), and downstream Ras/mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) or phosphatidylinositol-3 kinase (PI3K)/Akt pathways contribute to the malignity of glioma.2 Fyn, a non-RTK member of the Src family kinase (SFK), is a downstream proto-oncogene target of the RTK pathway.3–6 However, the specific mechanisms by which Fyn stimulates glioma growth and invasion remain unknown. Fyn is rarely mutated7 in human high-grade glioma, but is significantly overexpressed.
Several in vitro studies showed that Fyn knockdown is associated with decreased cell migration and proliferation of glioma cells.3,8,9 Nevertheless, in vivo human glioma xenograft models of Fyn knockdown in immune-suppressed animals failed to show differences in survival.8 Therefore, an immune-competent mouse model that enables study of gliomas with Fyn knockdown was established.
In this study we demonstrate that Fyn, a downstream target of RTK signaling, inhibits the antiglioma immune response. We demonstrated that Fyn tyrosine kinase promotes glioma initiation and growth utilizing both immune-competent and immune-deficient mouse glioma models. We observed that genetically engineered mouse glioma models (GEMMs) of gliomas and implantable gliomas, both with Fyn knockdown, displayed an extended survival compared with wildtype Fyn tumor bearing immune-competent mice. However, Fyn knockdown tumors implanted into immune-deficient mice did not extend survival compared with controls. Molecular analysis revealed a significant overrepresentation of immune-related gene ontologies (GOs) in short hairpin (sh)Fyn tumors. The overrepresentation of immune-related GOs suggests the possibility that Fyn is somehow suppressing immune function. Our data suggest that Fyn expression in glioma cells suppresses the immune responses by stimulating the expansion and activity of myeloid-derived suppressor cells (MDSCs) in the glioma tumor microenvironment (TME). Gliomas have been demonstrated to employ a variety of immunosuppressive mechanisms which promote tumor progression, thus reducing the effectiveness of immunotherapies.10,11
Our data uncover a new paradigm of how Fyn tyrosine kinase expressed within the tumor cells regulates antiglioma immune responses. We propose that tumor cell–specific inhibition of Fyn tyrosine kinase will increase the sensitivity of gliomas to immune attack, and represents a potential target for future treatments of glioma patients.
Materials and Methods
Glioma Cells
Mouse neurosphere cells were derived from GEMMs of gliomas. These cells were generated in our lab using the Sleeping Beauty (SB) transposon system.12,13 All cells were cultured as described in the Supplementary Material.
Generation of Stable Cell Lines with Fyn Knockdown
Neurospheres of shp53 and NRAS (NP) and shp53, NRAS and shATRX (NPA) cell lines were used to generate stable cell lines with Fyn knockdown. The pLenti pLKO-nontarget shRNA control vector (SHC002) and 2 different pLenti-mouse shRNA vectors for Fyn were selected from Sigma Aldrich MISSION shRNA vectors. The Fyn shRNA identification numbers are: TRCN0000023383 (shFyn #1) and TRCN0000361213 (shFyn #2). Cells were infected with the lentivirus as described previously by us.14 Immunoblotting was used to confirm Fyn knockdown. Fyn shFyn #2 cells were selected for in vivo experiments. Moreover, to validate the specificity of the shFyn and discard any potential off-target effects, we performed a rescue experiment, as described in detail in the Supplementary Material.
Intracranial Implantable Syngeneic Mouse Glioma Model
Studies were conducted according to the guidelines approved by the Institutional Animal Care and Use Committee (IACUC) at the University of Michigan (approved protocol, PRO00007666 for C57BL/6 immune-competent mice and PRO00007669 for immune-suppressive mice). Implanted were 3.0 × 104 neurospheres into the striatum of mouse brains to generate tumors. See the Supplementary Material.
Generation of Genetically Engineered Mouse Glioma Model for Fyn Knockdown
The animal model studies were conducted in C57BL/6 mice (Jackson Laboratory), according to IACUC approved protocol PRO00007617. A Fyn knockdown glioma murine model and the appropriate controls were created by the SB transposon system as described.12,13 The genotypes of SB-generated tumors were: (i) shp53 and NRAS, (ii) shp53, NRAS, and shFyn (NPF), (iii) shp53, NRAS, and shATRX, (iv) shp53, NRAS, shATRX, and shFyn (NPAF), (v) shp53, NRAS, and PDGFβ (NPD), (vi) shp53, NRAS, PDGFβ, and shFyn (NPDF). Design and cloning of shFyn vector are in the Supplementary Material.
Immunoblotting
Glioma cells (1.0 × 106 cells) were seeded in a 100 mm dish and grown at various time points as shown in the Supplementary Material.
Immunohistochemistry of Paraffin Embedded Brains
Immunohistochemistry assay was performed on paraffin embedded tissue as described previously.15
Immunofluorescence of Paraffin Embedded Brains
Brains that were fixed in 4% paraformaldehyde were processed, embedded in paraffin, and sectioned as described previously.15 Fyn antibody was conjugated with the Alexa Fluor 488 Tyramide SuperBoost Kit, goat anti-rabbit immunoglobulin G (# B40922) following the manufacturer’s instructions (Invitrogen/Thermo Fisher Scientific).
RNA Isolation and RNA Sequencing
SB NP, NPF, NPA, NPAF, NPD, and NPDF tumors were studied by RNA sequencing (RNA-Seq) analysis. RNA was isolated using the RNeasy Plus Mini Kit (Qiagen) following the manufacturer’s instructions. RNA-Seq was performed at the University of Michigan DNA Sequencing Core. Detailed analysis is described in the Supplementary Material.
Flow Cytometry
For flow cytometry analysis of immune cells within the TME, we generated tumors by intracranial implantation of 3.0 × 104 NPA-nontreated (NT) and NPA-shFyn cells in C57Bl6 mice. Protocol was performed as described before16 and detailed in the Supplementary Material.
In Vitro MDSC Migration Assay
We generated mouse bone marrow–derived MDSCs as described by Marigo et al.17 We analyzed in vitro MDSC (monocytic [M]-MDSC and polymorphonuclear [PMN]-MDSC) migration using Transwell polycarbonate membrane inserts (Corning) of 6.5 mm diameter and 8 µm pore size. Detailed methodology is described in the Supplementary Material.
T-Cell Proliferation Assays
We measured MDSC immune suppressive activity using in vitro T-cell proliferation assay. MDSCs were purified from TME of shp53, NRAS and shATRX-non target (NPA-NT) and shp53, NRAS and shATRX-shFyn (NPA-shFyn) tumors from moribund mice. MDSCs were purified by flow sorting as Gr-1high (PMN-MDSC) and Gr 1low(M-MDSCs) as described.16 See detailed methodology in the Supplementary Material.
Statistical Analysis
All experiments were performed in at least 3 or more independent biological replicates, depending on the specific analysis. Data are presented as the mean ± SEM. All statistical tests used are indicated within the figure legends and in the Supplementary Material.
Results
Fyn Was Identified as a Potential Regulator of Glioma Progression
Fyn tyrosine kinase is activated by several RTKs, such as EGFR, PDGFR, and c-MET, commonly mutated genes in gliomas. Following Fyn activation, there are several downstream Ras-dependent and Ras-independent signaling pathways such as Ras/MEK/ERK, PIK3/Akt, focal adhesion kinase, paxillin, beta-catenin, signal transducer and activator of transcription 3 (STAT3), Src homology 2 domain-containing (SHC)-transforming protein member of the Src homology and Collagen family and vav guanine nucleotide exchange factor 2 (VAV2), leading to changes in proliferation, migration, invasion, and cell-cell adhesion (Fig. 1A).
Fig. 1.

Fyn is a potential regulator of aggressiveness in mouse gliomas. (A) Fyn tyrosine signals downstream from mutated membrane RTK and integrin driver genes. Red: mutation/amplification, green: mutation/deletion, orange: Fyn overexpression. (B) Kaplan–Meier survival curves of implantable mouse glioma models show difference in tumor malignancy. NPAI display an increased survival compared with NP and NPA glioma-bearing mice. NPAI (MS, 34 days; n = 4), NP (MS, 24 days; n = 4), NPA (MS, 23 days; n = 4). (C) Fyn levels correlate positively with glioma cell malignancy in western blot analysis comparing Fyn and Src levels in mouse (NP, NPA, NPAI) glioma cells; loading control = β-actin. (D) Network of DE genes in high malignancy NPA vs low malignancy NPAI mouse glioma neurospheres. Fyn is the largest yellow octagon. Clusters of nodes of identical color represent a module of highly interacting genes. The Fyn network is highlighted in yellow. (E) Right panel: the Fyn network; Fyn has a degree of 63. Red lines indicate edges connecting nodes to Fyn.
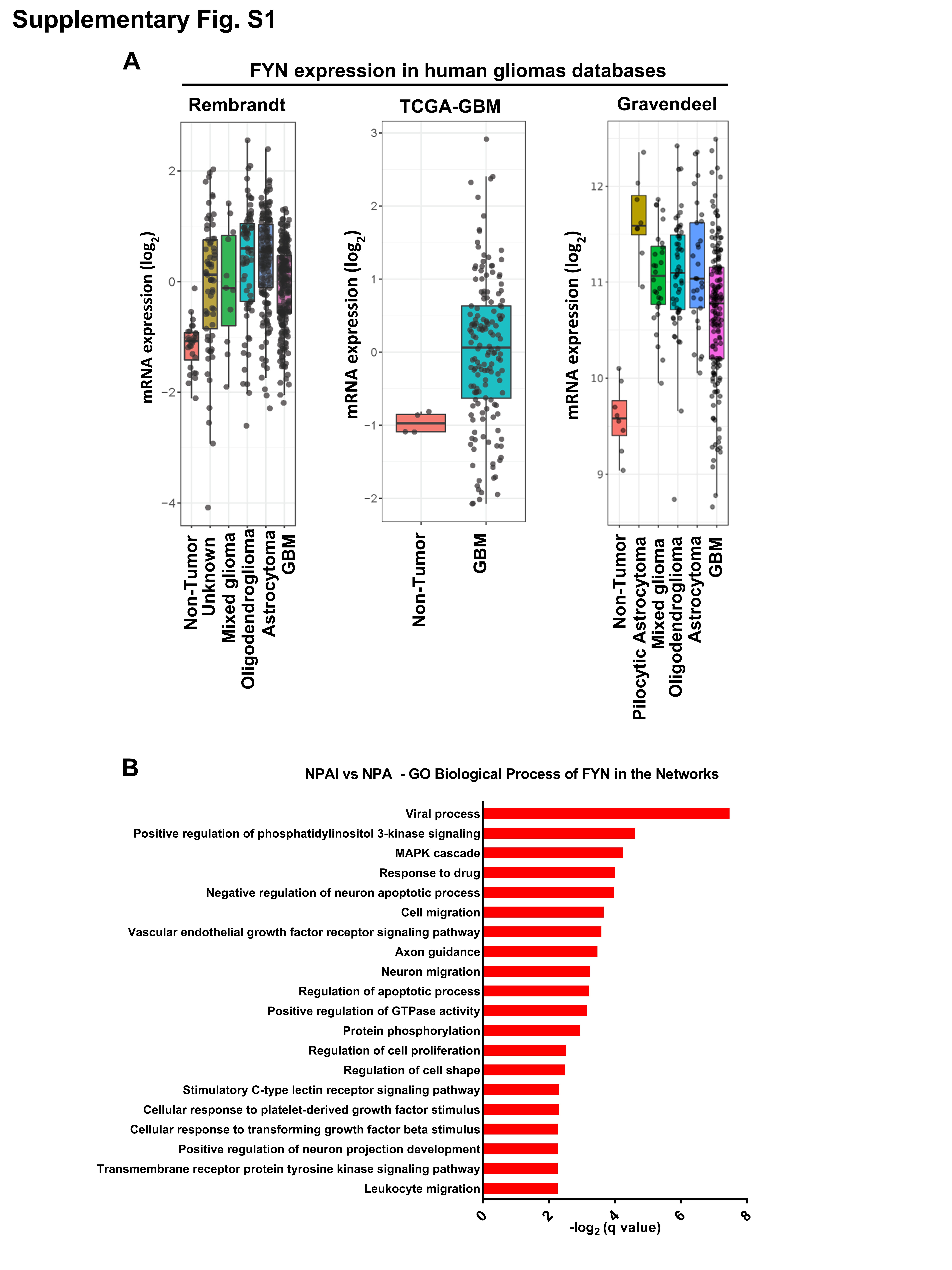
We analyzed RNA-Seq and Microarray human data from the Gliovis (http://gliovis.bioinfo.cnio.es) database. According to the Repository of Molecular Brain Neoplasia Data (REMBRANDT), The Cancer Genome Atlas (TCGA), and Gravendeel databases, Fyn mRNA expression levels were higher in different types of human gliomas compared with normal brain tissue (Supplementary Figure 1A). We observed that Fyn expression was positively correlated with mouse glioma cell aggressiveness. Figure. 1B shows that the survival of animals implanted with NP and NPA glioma cells is significantly shorter than that of animals implanted with NPAI cells. In accordance, western blot analysis in Figure. 1C demonstrates that the levels of Fyn, but not Src, are higher in NP and NPA cells compared with NPAI cells.
To further analyze the importance of Fyn in glioma malignancy, we investigated differential expression (DE) of genes in highly aggressive glioma NPA neurospheres (NRAS, shp53, shATRX, isocitrate dehydrogenase [IDH] wild-type) compared with NPAI neurospheres (NRAS, shp53, shATRX, IDH1R132H), of lower aggressiveness.13 The network of DE genes identified Fyn to be one of the most highly connected nodes (degree, 63; fourth node from the top), a hub in the network (Fig. 1C, D). Figure. 1D, E displays the network of Fyn, a set of nodes directly connected to Fyn. Higher magnification of the networks is shown in Supplementary Figure 1A, B. Analysis of the functional network GO terms discloses Cell Proliferation, Cell migration, MAPK cascade, Positive regulation of PI3K signaling, VEGF receptor signaling, Cellular response to PDGF as significant GO Biological Processes involving Fyn (Supplementary Figure 1B). The list of GO terms with their respective q and p values is shown in Supplementary Table 1.
Loss of Fyn Reduces Tumor Malignancy and Prolongs Survival of Mice Harboring GEMM Tumors
The SB Transposon System GBM model (GEMM) was used to understand the function of Fyn.12 We generated a Fyn-deficient genetically engineered mouse glioma model (Supplementary Figure 3A, B). We corroborated the efficacy of the shRNAs of Fyn by western blot analysis (Supplementary Figure 3C). The shFyn-(b) was selected for the GEMM glioma generation. We generated tumors harboring wildtype or Fyn knockdown in the presence of various genotype combinations (Supplementary Figure 3D).
Downregulation of Fyn increased median survival (MS) in comparison to wildtype Fyn control groups (Fig. 2A–C) in all genotype models. The NPF group displayed an increased MS of 131 days compared with 94 days in the NP control group (Fig. 2A). The experimental group with knockdown of Fyn in the context of loss of ATRX (alpha thalassemia/mental retardation syndrome X-linked) (NPAF) exhibited an increased survival (MS, 142 days) compared with the NPA control (MS, 80 days) (Fig. 2B). In the third experimental group, Fyn knockdown plus PDGFβ ligand upregulation (NPDF) also displayed an increased MS of 108 days compared with the NPD control group (MS, 69 days) (Fig. 2C). We corroborated the downregulation of Fyn protein in all experimental groups as shown in Figure. 2D–2F respectively. Histopathology analysis of tumors showed no evidence of significant differences in glioma malignant pathological markers (Fig. 2G–J). We further evaluated cellular proliferation of the tumor. Quantification demonstrated a significant decrease in the ratio of P-H3-S10 cells per total cells in Fyn knockdown groups (Fig. 2K, L). These data demonstrate that Fyn downregulation increases animal survival by decreasing tumor initiation, development, and proliferation.
Fig. 2.

Knocking down Fyn in GEMM prolongs animal survival. (A–C) Kaplan–Meier survival curves of SB mouse glioma models demonstrate that animals bearing Fyn knockdown tumors have increased median survival. (A) NP (MS, 94 days; n = 15) versus NPF (MS, 131 days; n = 29). (B) NPA (MS, 80 days; n = 16) versus NPAF (MS, 142 days; n = 28). (C) NPD (MS, 69 days; n = 15) versus NPDF (MS, 108 days; n = 23). Log-rank (Mantel–Cox) test; ***P < 0.001, ****P < 0.0001. (D–F, top) Fyn expression in tumors (green = tumor, blue = DAPI [4′,6′-diamidino-2-phenylindole] stained nuclei), quantified as fluorescence integrated density using ImageJ software (D–F, bottom); n = 5 per condition, scale bar = 50 μm. Ten random fields per tumor section per animal were imaged. Bars ± SEM are shown (***P < 0.001, *P < 0.05 using linear mixed effect models) (G–I). Histopathological analysis was performed in tumor sections stained with hematoxylin and eosin; shFyn tumors were compared with controls. Scale bars: 100 μm. P: pseudo-palisades, N: necrosis, H: hemorrhage, VP: vascular proliferation, MS, mesenchymal component, SC: small cells, G: giant cells. (J) Table representing histopathological semi-quantitative analysis: very low (+/−), low (+), medium (++), and high (+++). (K‒M) Cell proliferation analysis: Positive P-H3-S10 cells were counted by ImageJ software. Scale bars: 50 μm. P-H3-S10 positive cells per total cells in the visual field; n = 5. Ten fields of each section were selected at random. Error bars represent ± SEM; linear mixed effect models, ***P < 0.001, **P < 0.01.
RNA-Seq and Bioinformatics Analysis Reveal Increased Representation of Immune Ontologies in Fyn Knockdown Glioma Models
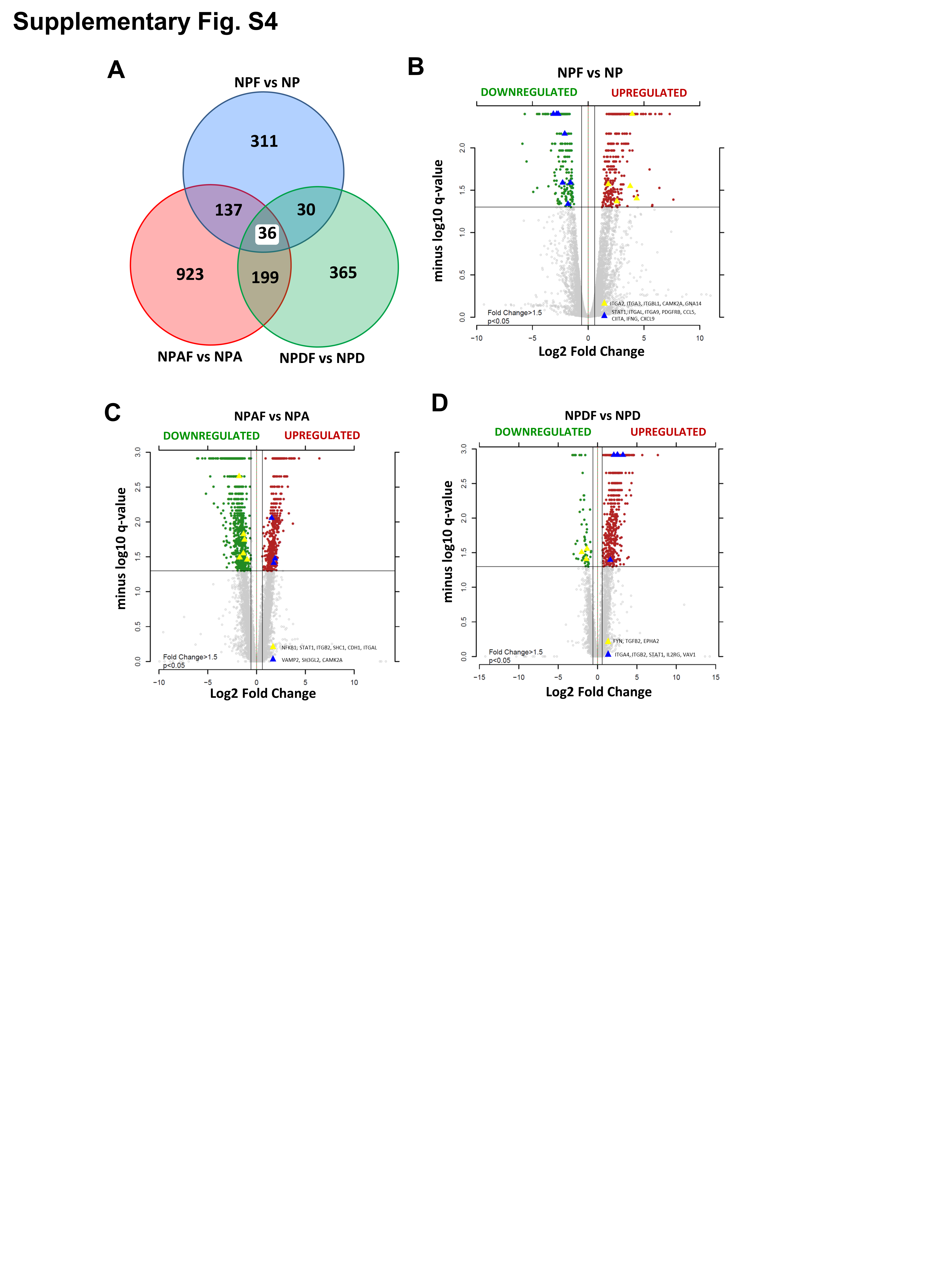
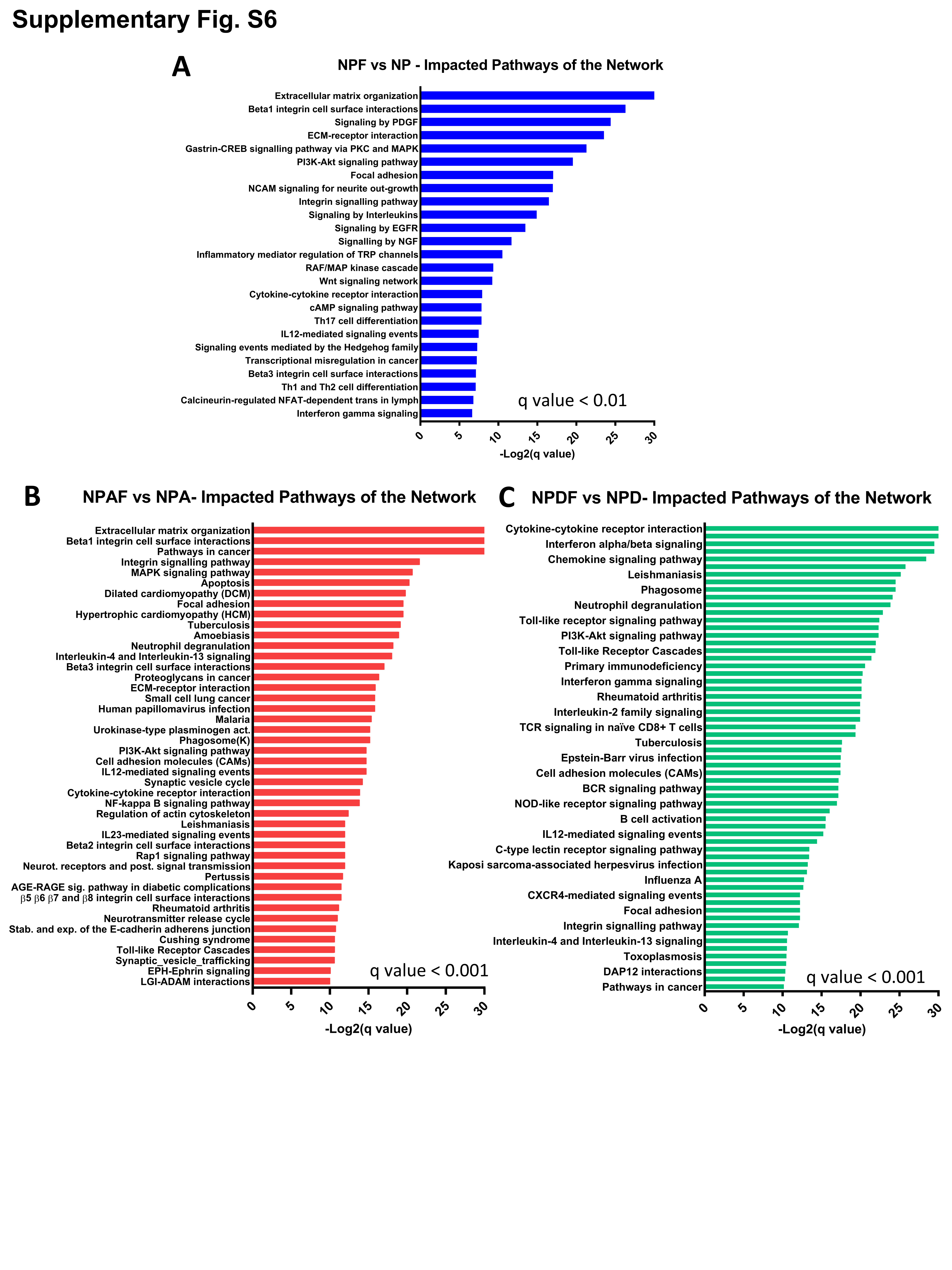
RNA-sequencing and bioinformatics analysis were used to discover changes in GOs that could help us understand the mechanism by which Fyn knockdown leads to the inhibition of tumor growth and progression. Genomic studies were performed in the following GEMM groups: NPF versus NP, NPAF versus NPA, and NPDF versus NPD. RNA-Seq analysis revealed a group of 515 DE genes in NPF versus NP (205 upregulated genes and 310 downregulated genes); 1295 DE genes in NPAF versus NPA (469 upregulated and 826 downregulated), and 630 DE genes in NPDF versus NPD (565 upregulated and 65 downregulated) (Supplementary Figure 4A–D). Using network analysis (Cytoscape) we analyzed the functional interaction of the DE genes resulting from Fyn knockdown (Supplementary Figure 4). The analysis of the network interactions revealed several genes which represent hubs and thus potential regulators of the network functions (Supplementary Figure 5 A–C). We found in NPF versus NP that STAT1, integrin subunit alpha (ITGA)2, ITGA3, ITGA9, G protein subunit alpha 14 (GNA14), calcium/calmodulin dependent protein kinase II alpha (CAMK2A) represent highly connected hubs of the network. In NPAF versus NPA, the most connected genes on the network were NFKB1, STAT1, Src homology 2 domain-containing transforming protein 1 (SHC1), ITGB2, fibronectin 1, vinculin, ITGB7, and CAMK2A. In NPDF versus NPD the hubs of the network are represented by Fyn, STAT1, spleen tyrosine kinase (SYK), Ras-related C3 botulinum toxin substrate 2 (RAC2), VAV1, phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit gamma [PIK3CG], and ITGB2. These genes play an essential role in gene regulation and biological processes. Moreover, the functional network analysis found the following pathways as commonly overrepresented in all 3 shFyn tumors independent of the genetic background of the tumors: extracellular matrix organization, focal adhesion, interleukin (IL)-12–mediated signaling events, integrin signaling pathway, pathways in cancer, PI3K/Akt signaling pathway, and cytokine-cytokine receptor interaction (Supplementary Figure 6A–C and Supplementary Table 2A–C ). As is shown in the figures, other pathways were specifically impacted for each genetic condition. Further, GO analysis performed by iPathwayGuide platform (Advaita Corporation), corrected for multiple comparisons using Elim pruning method, is compatible with the hypothesis that Fyn knockdown mediates an activation of immune response among all GEMM of glioma. The analysis shows that the set of top overrepresented GO terms, in shFyn glioma for each individual genetic background, are mostly related to immune functions (Fig. 3A–D and Supplementary Table 3A–C). Furthermore, to identify common gene ontologies shared between all GEMM glioma models for Fyn downregulation (NPF vs NP, NPAF vs NPA and NPDF vs NPD), DE genes of each genetic background were compared by meta-analysis. We encountered 58 common overrepresented GO terms shared by all 3 Fyn knockdown glioma models (Fig. 3A). Significantly overrepresented GOs in the meta-analysis were associated with immune biological functions, including “cellular response to interferon-gamma,” “cellular response to interferon-beta,” “antigen processing and presentation of exogenous peptide antigen via MHC class II,” “cellular response to interleukin-1,” “myeloid dendritic cell differentiation,” “positive regulation of T cell proliferation,” among others (Fig. 3E). These signaling pathways and GO terms represent potential mechanism by which Fyn knockdown decreases glioma malignancy in NPF, NPAF, and NPDF glioma models. Details of the selected GO terms are shown in Supplementary Table 4A–C.
Fig. 3.

Enrichment in immune-related pathways as potential mechanisms involved in Fyn-mediated tumor growth control. Overrepresented GO biological term in the DE genes dataset was carried out using iPathwayGuide algorithms. Overrepresentation approach to compute the statistical significance was used. P-value is computed using the hypergeometric distribution and corrected for multiple comparisons using elim pruning methods. Minimum DE genes/term of 6. (A) Venn diagram of GO Biological Process. Meta-analysis of different genetic glioma models shows individual GO terms and 58 common biological processes shared between all groups. For GO analysis, genes with 0.05 P-value and a log fold change of at least 0.585 absolute value were considered significant. (B, C, D) Graph representing the top 20 most significant enriched GO terms identified for each individual genetic glioma model. (B) Enriched GO terms of NPF vs NP. (C) Enriched GO terms of NPAF vs NPA. (D) Enriched GO terms of NPDF vs NPD. (E) Bar graph showing the significant overrepresented 58 common GO terms Biological Process identified by meta-analysis.
The Role of Fyn in Glioma Growth, Malignancy, and Immune Response Interaction
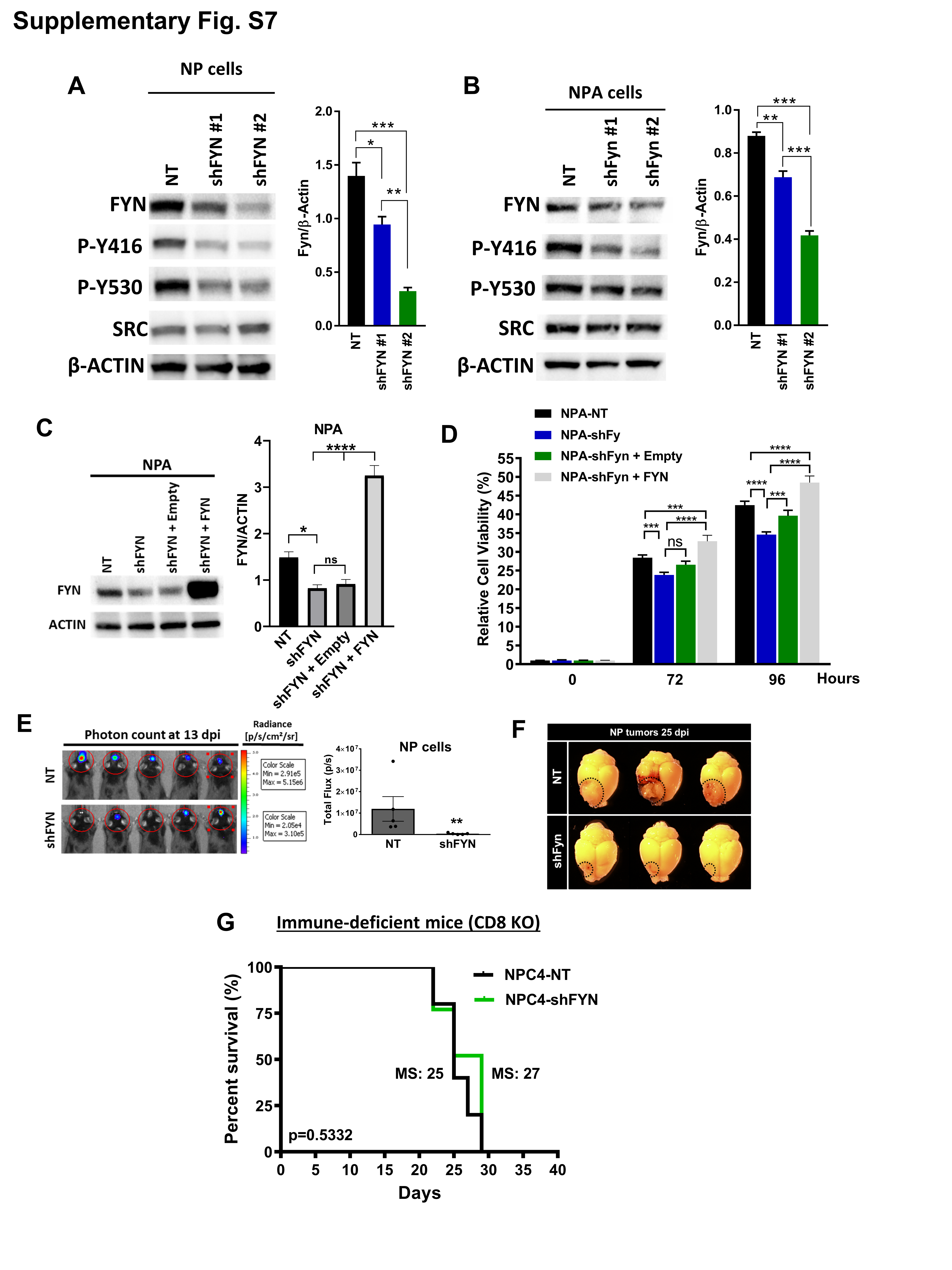
To test the hypothesis that Fyn depletion stimulates antitumor immune responses in vivo, we implanted Fyn knockdown cells in immune-competent and immune-deficient mice. Preceding the in vivo experiments, we identified by western blot that Fyn was successfully downregulated. Both shRNAs were specific for Fyn and do not have effect on Src tyrosine kinase. Decreased expression of SFK phosphorylation sites P-Y416 and P-Y530 was observed (Supplementary Figure 7A, B).
To test if potential off-target effects due to shFyn influence our results, we performed an in vitro rescue experiment. NPA-shFyn cells transduced with a plv-cherry-Fyn vector expressing a silent-mutated form of Fyn gene, designed to be resistant to the shRNA of Fyn, were used to rescue the shFyn induced knockdown. Expression of this construct counteracted the inhibition of shFyn (Supplementary Figure 7C). We then tested the biological effects of Fyn expression on cell viability. We observed a decreased proliferation in NPA-shFyn cells compared with NPA-NT. Fyn overexpression within NPA-shFyn cells reversed the effects of Fyn knockdown (Supplementary Figure 7D). The in vivo study showed that inactivation of Fyn in glioma cells strongly suppress tumor growth and increase survival in immune-competent mice (C57BL/6) (Fig. 4A, B), but the effects in immune-compromised (NSG) mice was negligible (Fig. 4C, D). Fyn knockdown group displayed a MS of 34 days compared with the NP- NT control group (MS, 27 days) (Fig. 4A ). Also, NPA-shFyn had a significantly higher median survival (MS, 30 days) than the NPA-NT control group (MS, 20) (Fig. 4B). Moreover, in vivo bioluminescence analysis of the tumors at 13 days post-implantation (dpi) showed that NP-Fyn knockdown tumors exhibited lower signal. Tumor size evaluation at necropsy showed a correlation with in vivo tumor bioluminescence analysis (Supplementary Figure 7E, F). These results validate the role of Fyn in immune-competent mice. However, tumor induction in NSG immune-deficient mice with NP-shFyn tumors did not exhibit a significant difference in survival compared with the control NP-wildtype Fyn tumors (MS, 24 vs 26 dpi) (Fig. 4C). In mice bearing NPA-shFyn tumors, a minor yet statistically significant increase in MS (22 vs 24 dpi) was observed (Fig. 4D). Moreover, implantation of NP-shFyn and NPA-shFyn tumors in cluster of differentiation (CD)8 knockout mice showed no difference in survival compared with the control tumor-bearing mice (Fig. 4E and Supplementary Figure 7G). Implantation of NPA-shFyn tumors in CD4 knockout mice showed no difference in survival (Fig. 4F). Collectively, these results demonstrate that enhanced survival in Fyn knockdown tumors is mediated by the immune system, including CD8 and CD4 T cells.
Fig. 4.

Tumor growth delay and increased survival in Fyn downregulated gliomas are immune mediated. (A, B) Kaplan–Meier survival curves for glioma in C57BL/6 immune-competent mice. (A) NP-Fyn knockdown gliomas displayed significant increases in MS: 27 vs 34 days; *P = 0.011. (B) Mice bearing gliomas with NPA-Fyn knockdown displayed significant increase in MS: 20 vs 30 days; **P = 0.0031 compared with the control; n = 5. Fyn expression was detected by immunofluorescence. Tumor: green; nuclei: blue; scale bars: 50 μm. (C, D) Kaplan–Meier survival curves in NSG immune-compromised mice. (C) NP-Fyn knockdown gliomas displayed no significant increases in MS (24 vs 26 days; P = 0.06) n = 9/10. (D) NPA-Fyn knockdown displayed a minor increase in MS (22 vs 24 days; *P = 0.044); n = 5. (E, F) Kaplan–Meier survival curve for NPA-NT vs NPA-shFyn glioma in (E) CD8 and (F) CD4 knockout immune-deficient mice. No significant difference was observed in survival. For each implantable model, n = 5 was used. Statistics were assessed using the log-rank Mantel–Cox test.
As was observed above, the survival benefit of shFyn is larger in GEMM than in implantable tumors. The reason for the different survival benefit is that in the GEMMs, tumors originate de novo as a result of the genetic modification of neural stem cell progenitors in one-day-old pups. In the implantable glioma model, tumors are induced by intracranial implantation of 30 000 cells. Median survival is 80, 94 days in GEMM, and 27, 20 days in implantable tumors. Percentage-wise, shFyn increases survival by 139%, 177% in GEMM (NP, NPA), and by 125%, 150% in NP, NPA implantable tumors (Fig. 2A, B; Fig. 4A, B). We believe that the survival benefit of shFyn is reduced in implantable gliomas due to their increased growth rate.
Fyn Downregulation in Glioma Reduces MDSC Amount and Activation Markers and Inhibitory Potency within the TME
To determine whether downregulation of Fyn in gliomas has an effect on the immune response, we examined the immune cellular infiltrates in the TME. First, we analyzed the role of T cells in the TME of shFyn tumors. No significant difference in the frequency of CD4+ T cells (CD45+, CD3+, CD4+) or CD8+ T cells (CD45+, CD3+, CD8+) were observed in NPA-NT versus NPA-shFyn tumors (Fig. 5A–D). However, we found a reduction in CD8 T cells that express programmed cell death 1 (PD1), a marker of T-cell exhaustion (Fig. 5E, F). In gliomas, increases in MDSCs are an important mechanism of antitumor immune evasion.16 Therefore, we evaluated the expansion of MDSC-mediated immunosuppression in the glioma TME. MDSCs were identified as M-MDSC (CD45+, CD11b+, Ly6Chi, Ly6G−) or PMN-MDSC (CD45+, CD11b+, Ly6Clo, Ly6G+). Interestingly, we observed a 1.79-fold decrease of M-MDSCs (Fig. 5G, 5I) and a 3.04-fold decrease of PMN-MDSCs (Fig. 5H, 5I) in the TME of NPA-shFyn gliomas compared with NPA-NT controls. Further, we analyzed the immunosuppressive function of MDSC isolated from the TME of GEMM tumors. We first characterized MDSC by expression of T-cell immunosuppressive molecules (ie, as arginase 1 [ARG1] and CD80). We observed a significant decrease in the proportion of CD80+ and ARG+ M-MDSCs, and ARG+ PMN-MDSCs in shFyn tumors (Fig. 5J, K, L, M). To test whether decreased numbers of MDSCs in shFyn TME is due to reduced MDSC migration, we performed an in vitro bone marrow–derived MDSC migration assay (Fig. 6A). This experiment showed that shFyn conditioned media reduced M-MDSC migration. Migration of PMN-MDSCs was not decreased in the shFyn group (Fig. 6B). Finally, we analyzed the functional MDSC-mediated T-cell immune suppressive activity. We observed that PMN-MDSCs (GR1hi CD11b+) or M-MDSCs (Gr1low CD11b+) from the TME of NPA-NT and NPA-shFyn decreased T-cell proliferation stimulated by SIINFEKL peptide. However, MDSCs isolated from the shFyn tumors were significantly less inhibitory. These data suggest that the microenvironment of shFyn glioma tumors reduce the capacity of MDSCs to inhibit T-cell activation (Fig. 6C, 6D, 6F).
Fig. 5.

Downregulation of Fyn in glioma modulates immune responses through reduction in MDSC expansion. Immune cells within the TME of NPA-NT or NPA-shFyn tumors were analyzed by flow cytometry. (A, B) Percentage of CD4 T cells (CD4+, CD3+) within the CD45+ cell population. Representative flow plots for each group are displayed. (C, D) Percentage of CD8 T cells (CD8+, CD3+) within the CD45+ cell population. Representative flow plots for each group are displayed. (E, F) Percentage of PD1 + T cells within the CD8 + CD3 + and CD45+ cell population. Representative histogram flow plot for each group are displayed. (G) Percentage of M-MDSCs: CD11b+, Ly6Chi, Ly6G−) within the CD45+ cell population. (H) Percentage of PMN-MDSCs: CD11b+, Ly6Clo, Ly6G+ within the CD45+ cell population. (I) Representative flow plots of M-MDSC and PMN-MDSC cell analysis. (J–M) Percentage of CD80+ and ARG+ cells within (J, K) M-MDSC (CD45+, CD11b+, Ly6Chi, Ly6G−) cell population, and (L–M) PMN-MDSC (CD45+, CD11b+, Ly6Clo, Ly6G+) cell population. (K–M) Representative histogram of CD80+ and ARG+ cell analysis. Red-shaded: NPA-NT, gray-shaded: NPA-shFyn, solid black-dashed: FMO control. Each graph indicates individual values and mean ± SEM (n = 10). Data were analyzed using ANOVA; ns = nonsignificant; *P < 0.05.
Fig. 6.

Fyn knockdown in glioma decreases MDSC migration potential and immune-suppressive activity within the TME. (A) Diagram of experimental design of MDSC Transwell migration assay. MDSCs derived from bone marrow and induced with IL-6 and granulocyte-macrophage colony-stimulating factor were seeded on the top of the Transwell and incubated for 15 hours in NT and shFyn conditioned media. The migrated cells were analyzed using CellTiter-Glo. (B) MDSC migration assay results. Data are expressed as percentage of migrating cells relative to the control (plain media). Error bars represent ± SEM. Experiment was performed 3 times with 3 replicates per treatment. Statistical significance was determined using one-way ANOVA, followed by Duncan multiple comparisons. ns: nonsignificant, *P < 0.05, ***P < 0.001, ****P < 0.0001. (C) Diagram representing the experimental design to analyze the immunosuppressive potential of MDSCs. Gr-1high (PMN-MDSC) and Gr-1low (M-MDSC) were purified from the TME of moribund NPA-NT and NPA-shFyn tumor-bearing mice. They were cultured with carboxyfluorescein succinimidyl ester (CFSE)–labeled splenocytes from Rag2/OT-1 transgenic mice. Cells were stimulated with SIINFEKL peptide and proliferation was analyzed 4 days after by flow cytometry. (D) Representative flow plots for CFSE stains from splenocytes alone SIINFEKL stimulated and nonstimulated, and the effect of SIINFEKL-induced T-cell proliferation in the presence of MDSCs from the TME. Numbers in parentheses indicate the ratio of MDSCs to splenocytes. (E) Graph shows T-cell proliferation relative to SIINFEKEL + control. Experiment was repeated 3 times. Tumors from 5 mice per group were pooled together in each experiment to obtain sufficient number MDSCs. Mean ± SEM are indicated. Data were analyzed using one-way ANOVA, followed by Duncan multiple comparisons. ns: nonsignificant, *P < 0.05, **P < 0.005.
Besides, we analyzed the expansion and activation status of macrophages. We did not observe a significant increase in the frequency of macrophages (CD45+, CD11c−, F4/80+) (Supplementary Figure 8A, B). The expression of major histocompatibility complex 2 (MHCII) on macrophages in the TME was increased by 1.4-fold in NPA-shFyn gliomas versus NPA-NT controls (Supplementary Figure 8C, D). It is likely that the lower proportion and suppressive activity of MDSCs in shFyn tumors lead to increases of M1 macrophages (MHCIIhi) and therefore reduced polarization to M2 macrophages. Overall, our data show that downregulating expression of Fyn in glioma tumors decreases expansion of MDSCs in the TME due to reduced migration potential, decreased expression of CD80 and ARG1, and lower functional immune-suppressive activity.
Discussion
In this study we demonstrate that glioma cell–specific genetic inhibition of Fyn tyrosine kinase increases antiglioma immune responses, thus significantly delaying tumor progression.
Fyn is an effector of the RTK (EGFR, MET, PDGFR) pathway in glioma and other cancers. Downstream of RTK, Fyn signals through Ras-dependent (via Ras/MEK/ERK) and Ras-independent pathways (via PIK3/Akt, β-catenin, FAK, paxillin, STAT3, VAV1, and/or SHC) (Fig. 1A). To activate several downstream molecular pathways, and physiological processes such as cellular proliferation, migration, and cell adhesion.4,18 RTK are commonly mutated drivers in high grade gliomas; yet their detailed downstream signaling pathways remain incompletely understood.3,4,18–20
Molecular analyses of high-grade glioma from the REMBRANDT, TCGA, and Gravendeel databases,7 and data from Lu et al,3 indicate increased expression of Fyn. These human data correlate with our results in mouse models of gliomas, in which increased levels of Fyn expression correlate with higher glioma aggressiveness. Further, our molecular gene interaction network analysis highlights Fyn as a central network hub, suggesting it might function as a potential regulator of glioma malignancy.
Previous studies have shown that Fyn is expressed by both tumor cells and immune cells.21 In T cells, Fyn regulates effector functions and amplifies T-cell antigen receptor signaling.4,19,20,22 However, it has been difficult to establish the role of Fyn within glioma in vivo. The function of Fyn can be studied genetically (ie, knockdown) or pharmacologically (ie, saracatinib or dasatinib inhibitors). Genetic inhibition is highly specific. Pharmacological inhibition, however, is nonspecific. Saracatinib or dasatinib will inhibit all SFK members and will inhibit SFK in all cell types (ie, Fyn in the immune cells).
For instance, in vitro studies using SFK inhibitors show that Fyn promotes cell proliferation and migration in gliomas.3,9,23,24 In vivo, effects of saracatinib treatment have been mixed, yet we are unable to determine any specific role of Fyn in glioma growth.3,8 Importantly, dasatinib did not extend survival of glioma patients in a phase II clinical trial.25 In vivo, however, there was no effect on animal survival.
As saracatinib and dasatinib are nonspecific inhibitors of individual SFK members,26 the genetic inhibition of Fyn remains the best option to study its functions in glioma biology. Indeed, genetic downregulation of Fyn expression inhibited glioma cell migration and proliferation in vitro,3,8,9,27 yet failed to affect glioma progression in in vivo immune-suppressive mice.8
To address this paradox, we developed a genetic approach to inhibit Fyn expression in tumor cells using a GEMM model of glioma in immune-competent animals. Our results suggest that shFyn delays tumor initiation and progression in vivo by inhibiting the antiglioma immune response.
We demonstrated the role that Fyn plays as central hub in antiglioma immune response and tumor progression by inducing tumors in immune-deficient animals. The effect of Fyn in delaying tumor growth was abolished in NSG, CD8−/− and CD4−/− immune-deficient animals. Both CD8 and CD4 T cells are necessary for increased tumor rejection of shFyn gliomas. These studies suggest that Fyn plays a crucial role in conveying immune inhibitory messages from within the glioma cells to the immune cells, thereby engineering the local immune response to favor tumor growth. Herein, we uncovered a new cell non-autonomous mechanism by which Fyn knockdown within glioma cells modulates the antiglioma immune responses.
Analysis of the molecular changes induced by shRNA-Fyn strongly suggests that the downregulation of Fyn in tumor cells activates the antitumor immune response. Meta-analysis of GO for NPF versus NP, NPAF versus NPA, and NPDF versus NPD revealed a significantly common immune-related biological process among all genetic glioma models of Fyn knockdown such as “cellular response to interferon-gamma,” “cellular response to interferon-beta,” “antigen processing and presentation of exogenous peptide antigen via MHC class II,” “cellular response to interleukin-1,” “myeloid dendritic cell differentiation,” “positive regulation of T cell proliferation,” and others. Moreover, further functional network analysis found that Fyn knockdown tumors display central hub regulators and overrepresented pathways as STAT1, a prominent regulator of the immune system.28 This module regulates immune functions, interferon-γ cellular signaling, Janus kinase–STAT pathways, cell differentiation of T helper cells (Th)1, Th2, and Th17, T-cell activation, and natural killer cell–mediated cytotoxicity.29,30 Although they exhibit the same final phenotype and common biological processes or signaling pathways, the specific mechanisms by which Fyn downregulation regulates the antiglioma immune response would be different for each GEMM of glioma.
Finally, our glioma TME analysis suggests that, Fyn downregulation in glioma tumors decreases MDSC expansion and their immune-suppressive activity. We and others have previously reported that glioma infiltrating MDSCs play a key role in inhibiting antitumor T-cell immune responses, thus promoting tumor progression.16 The inhibitory immune microenvironment in glioma is thought to contribute to the ineffectiveness of immunotherapies.31 Novel therapeutics approaches that reverse the inhibitory microenvironment are essential to counteract these effects, and we propose that the inhibition of Fyn within glioma cells could represent such a strategy. ShFyn gliomas determined a reduced MDSC migration potential, a decreased immunosuppressive cell phenotype (fewer CD80 and ARG1 cells), and lower functional immune-suppressive activity. T-cell depletion of L-arginine through arginase causes interference with the CD3-zeta chain and proliferative arrest of antigen-activated T cells. Inhibitory CD80 receptors of the B7.1 family were implicated in MDSC-mediated immune suppression.16,32 Glioma microenvironment of Fyn knockdown glioma diminished myeloid cell–mediated immunosuppression, leading to increased T-cell proliferation and cytotoxicity and decreased T-cell exhaustion and M1 to M2 macrophage polarization amplifying antiglioma immunity.
We propose that inhibition of Fyn within glioma cells could be an immune-mediated therapeutic target to restrict MDSC immune-suppressive expansion. Since Fyn is expressed by both immune cells and tumor cells, therapeutic approaches will need to target Fyn specifically in tumor cells. We propose that the combination of tumor cell–specific Fyn inhibition with other immune-stimulatory treatments—such as immune checkpoint blockade (PD1 and PD1 ligand inhibitors) and Ad-hCMV-TK and Ad-hCMV-Flt3L gene therapy11,33,34—is a promising avenue that is worth being explored in future experiments.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Funding
This work was supported by National Institutes of Health, National Institute of Neurological Disorders and Stroke (NIH/NINDS) grants: R37-NS094804, R01-NS105556, R21-NS107894 to M.G.C.; National Institute of Neurological Disorders and Stroke (NIH/NINDS) grants: R01-NS076991, R01-NS096756, R01-NS082311 to P.R.L.; National Institute of Biomedical Imaging and Bioengineering (NIH/NIBI): R01-EB022563; National Cancer Institute (NIH/NCI) U01CA224160; the Department of Neurosurgery, Rogel Cancer Center at The University of Michigan, ChadTough Foundation, and Leah’s Happy Hearts Foundation to M.G.C. and P.R.L. RNA Biomedicine grant: F046166 to M.G.C. Health and Human Services, National Institutes of Health, UL1 TR002240 for the Michigan Institute for Clinical and Health Research (MICHR), Postdoctoral Translational Scholars Program (PTSP), Project F049768 to A.C.
Conflict of interest statement
All authors of this paper declare no potential conflicts of interest.
Authorship statement
Conception and design: A. Comba and P. R. Lowenstein. Development of methodology: A. Comba, P. J. Dunn, A. E. Argento, P. Kadiyala, P. R. Lowenstein. Acquisition of data, analysis, and interpretation: A. Comba, P. J. Dunn, A. E. Argento, P. Kadiyala, M. Ventosa, P. Patel, D.B. Zamler, F. J. Nunez, L. Zhao, M. G. Castro, P. R. Lowenstein. Manuscript writing: A. Comba, P. J. Dunn, M. G. Castro, P. R. Lowenstein. Review and/or revision of the manuscript: A. Comba, P. J. Dunn, A. E. Argento, P. Kadiyala, M. Ventosa, P. Patel, D. B. Zamler, F. J. Nunez, L. Zhao, M. G. Castro, P. R. Lowenstein. Administrative, technical, or material support (ie, reporting or organizing data, constructing databases): A. Comba, P. J. Dunn, P. R. Lowenstein. Study supervision: M. G. Castro and P. R. Lowenstein.
References
- 1. Reifenberger G, Wirsching HG, Knobbe-Thomsen CB, Weller M. Advances in the molecular genetics of gliomas—implications for classification and therapy. Nat Rev Clin Oncol. 2017;14(7):434–452. [DOI] [PubMed] [Google Scholar]
- 2. Frattini V, Trifonov V, Chan JM, et al. . The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet. 2013;45(10):1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Lu KV, Zhu S, Cvrljevic A, et al. . Fyn and SRC are effectors of oncogenic epidermal growth factor receptor signaling in glioblastoma patients. Cancer Res. 2009;69(17):6889–6898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yadav V, Denning MF. Fyn is induced by Ras/PI3K/Akt signaling and is required for enhanced invasion/migration. Mol Carcinog. 2011;50(5):346–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jensen AR, David SY, Liao C, et al. . Fyn is downstream of the HGF/MET signaling axis and affects cellular shape and tropism in PC3 cells. Clin Cancer Res. 2011;17(10):3112–3122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zhang S, Fan G, Hao Y, Hammell M, Wilkinson JE, Tonks NK. Suppression of protein tyrosine phosphatase N23 predisposes to breast tumorigenesis via activation of FYN kinase. Genes Dev. 2017;31(19):1939–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bowman RL, Wang Q, Carro A, Verhaak RG, Squatrito M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol. 2017;19(1):139–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lewis-Tuffin LJ, Feathers R, Hari P, et al. . Src family kinases differentially influence glioma growth and motility. Mol Oncol. 2015;9(9):1783–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Han X, Zhang W, Yang X, et al. . The role of Src family kinases in growth and migration of glioma stem cells. Int J Oncol. 2014;45(1):302–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Preusser M, Lim M, Hafler DA, Reardon DA, Sampson JH. Prospects of immune checkpoint modulators in the treatment of glioblastoma. Nat Rev Neurol. 2015;11(9):504–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ratnam NM, Gilbert MR, Giles AJ. Immunotherapy in CNS cancers: the role of immune cell trafficking. Neuro Oncol. 2019;21(1):37–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Koschmann C, Calinescu AA, Nunez FJ, et al. . ATRX loss promotes tumor growth and impairs nonhomologous end joining DNA repair in glioma. Sci Transl Med. 2016; 8(328):328ra328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Núñez FJ, Mendez FM, Kadiyala P, et al. . IDH1-R132H acts as a tumor suppressor in glioma via epigenetic up-regulation of the DNA damage response. Sci Transl Med. 2019;11(479):eaaq1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yadav VN, Zamler D, Baker GJ, et al. . CXCR4 increases in-vivo glioma perivascular invasion, and reduces radiation induced apoptosis: a genetic knockdown study. Oncotarget. 2016;7(50):83701–83719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Calinescu AA, Yadav VN, Carballo E, et al. . Survival and proliferation of neural progenitor-derived glioblastomas under hypoxic stress is controlled by a CXCL12/CXCR4 autocrine-positive feedback mechanism. Clin Cancer Res. 2017;23(5):1250–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kamran N, Kadiyala P, Saxena M, et al. . Immunosuppressive myeloid cells’ blockade in the glioma microenvironment enhances the efficacy of immune-stimulatory gene therapy. Mol Ther. 2017;25(1):232–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Marigo I, Bosio E, Solito S, et al. . Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity. 2010;32(6):790–802. [DOI] [PubMed] [Google Scholar]
- 18. Saito YD, Jensen AR, Salgia R, Posadas EM. Fyn = a novel molecular target in cancer. Cancer. 2010;116(7):1629–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Elias D, Ditzel HJ. Fyn is an important molecule in cancer pathogenesis and drug resistance. Pharmacol Res. 2015;100:250–254. [DOI] [PubMed] [Google Scholar]
- 20. Zheng J, Li H, Xu D, Zhu H. Upregulation of tyrosine kinase FYN in human thyroid carcinoma: role in modulating tumor cell proliferation, invasion, and migration. Cancer Biother Radiopharm. 2017;32(9):320–326. [DOI] [PubMed] [Google Scholar]
- 21. Palacios EH, Weiss A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene. 2004;23(48):7990–8000. [DOI] [PubMed] [Google Scholar]
- 22. Yamauchi J, Miyamoto Y, Torii T, et al. . Phosphorylation of cytohesin-1 by Fyn is required for initiation of myelination and the extent of myelination during development. Sci Signaling. 2012; 5(243):ra69. [DOI] [PubMed] [Google Scholar]
- 23. Yamaguchi H, Takanashi M, Yoshida N, et al. . Saracatinib impairs the peritoneal dissemination of diffuse-type gastric carcinoma cells resistant to Met and fibroblast growth factor receptor inhibitors. Cancer Sci. 2014;105(5):528–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Liu KJ, He JH, Su XD, et al. . Saracatinib (AZD0530) is a potent modulator of ABCB1-mediated multidrug resistance in vitro and in vivo. Int J Cancer. 2013;132(1):224–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lassman AB, Pugh SL, Gilbert MR, et al. . Phase 2 trial of dasatinib in target-selected patients with recurrent glioblastoma (RTOG 0627). Neuro Oncol. 2015;17(7):992–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jelić D, Mildner B, Kostrun S, et al. . Homology modeling of human Fyn kinase structure: discovery of rosmarinic acid as a new Fyn kinase inhibitor and in silico study of its possible binding modes. J Med Chem. 2007;50(6):1090–1100. [DOI] [PubMed] [Google Scholar]
- 27. Zhang S, Qi Q, Chan CB, et al. . Fyn-phosphorylated PIKE-A binds and inhibits AMPK signaling, blocking its tumor suppressive activity. Cell Death Differ. 2016;23(1):52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Schindler C, Shuai K, Prezioso VR, Darnell JE Jr. Interferon-dependent tyrosine phosphorylation of a latent cytoplasmic transcription factor. Science. 1992;257(5071):809–813. [DOI] [PubMed] [Google Scholar]
- 29. Majoros A, Platanitis E, Kernbauer-Hölzl E, Rosebrock F, Müller M, Decker T. Canonical and non-canonical aspects of JAK-STAT signaling: lessons from interferons for cytokine responses. Front Immunol. 2017;8:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Avalle L, Pensa S, Regis G, Novelli F, Poli V. STAT1 and STAT3 in tumorigenesis: a matter of balance. JAKSTAT. 2012;1(2):65–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kamran N, Chandran M, Lowenstein PR, Castro MG. Immature myeloid cells in the tumor microenvironment: implications for immunotherapy. Clin Immunol. 2018;189:34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Youn JI, Nagaraj S, Collazo M, Gabrilovich DI. Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunology. 2008;181(8):5791–5802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ivashkiv LB. IFNγ: signalling, epigenetics and roles in immunity, metabolism, disease and cancer immunotherapy. Nat Rev Immunol. 2018;18(9):545–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kamran N, Alghamri MS, Nunez FJ, et al. . Current state and future prospects of immunotherapy for glioma. Immunotherapy. 2018;10(4):317–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.