

Abstract
Objective
Glucose-dependent insulinotropic polypeptide (GIP) conveys information from ingested nutrients to peripheral tissues, signaling energy availability. The GIP Receptor (GIPR) is also expressed in the bone marrow, notably in cells of the myeloid lineage. However, the importance of gain and loss of GIPR signaling for diverse hematopoietic responses remains unclear.
Methods
We assessed the expression of the Gipr in bone marrow (BM) lineages and examined functional roles for the GIPR in control of hematopoiesis. Bone marrow responses were studied in (i) mice fed regular or energy-rich diets, (ii) mice treated with hematopoietic stressors including acute 5-fluorouracil (5-FU), pamsaccharide (LPS), and Pam3CysSerLys4 (Pam3CSK4), with or without pharmacological administration of a GIPR agonist, and (iii) mice with global (Gipr−/−) or selective deletion of the GIPR (GiprTie2−/−) with and without bone marrow transplantation (BMT).
Results
Gipr is expressed within T cells, myeloid cells, and myeloid precursors; however, these cell populations were not different in peripheral blood, spleen, or BM of Gipr−/− and GiprTie2−/− mice. Nevertheless, gain and loss of function studies revealed that GIPR signaling controls the expression of BM Toll-like receptor (TLR) and Notch-related genes regulating hematopoiesis. Loss of the BM GIPR attenuates the extent of adipose tissue inflammation and dysregulates the hematopoietic response to BMT. GIPR agonism modified BM gene expression profiles following 5-FU and Pam3CSK4 whereas loss of the Gipr altered the hematopoietic responses to energy excess, two TLR ligands, and 5-FU. However, the magnitude of the cellular changes in hematopoiesis in response to gain or loss of GIPR signaling was relatively modest.
Conclusion
These studies identify a functional gut hormone-BM axis positioned for the transduction of signals linking nutrient availability to the control of TLR and Notch genes regulating hematopoiesis. Nevertheless, stimulation or loss of GIPR signaling has minimal impact on basal hematopoiesis or the physiological response to hematopoietic stress.
Keywords: Glucose-dependent insulinotropic polypeptide receptor, Bone marrow, Hematopoiesis, Myeloid cells, Inflammation
Graphical abstract
Highlights
-
•
Gipr is expressed in T cells and myeloid cells but is not essential for the development of myeloid lineages in mice.
-
•
GIPR signaling controls the expression of Bone Marrow (BM) TLR and Notch-related genes regulating hematopoiesis.
-
•
GIPR agonism modifies BM gene expression profiles of TLR and NOTCH ligand genes following 5-FU and PAM3CSK4 treatment.
-
•
Loss of the bone marrow GIPR dysregulates diet-induced adipose tissue inflammation and the hematopoietic response to BMT.
1. Introduction
Enteroendocrine cells (EECs) sense nutrients and play important roles as first responders to enable nutrient assimilation and maintenance of energy balance. Among the most extensively characterized EEC hormones are glucose-dependent insulinotropic polypeptide (GIP) and glucagon-like peptide 1 (GLP-1), secreted by gut K and L cells, respectively. Both GIP and GLP-1 communicate with islet cells to control hormone secretion and glucose metabolism within minutes of meal ingestion [1]. Notably, GLP-1 acts beyond the pancreas to reduce food intake, inhibit gastrointestinal motility, and attenuate inflammation, actions consistent with limiting excess energy intake [2]. The actions of GLP-1 extend to cardioprotection [3,4], supporting the clinical use of GLP-1R (GLP-1 Receptor) agonists for the treatment of type 2 diabetes (T2D) and, more recently, obesity [2].
The extrapancreatic actions of GIP encompass the central nervous system, the skeleton, the cardiovascular system, and adipose tissue [1]. GIP Receptor (GIPR) coagonists such as tirzepatide [5] are being studied in phase 3 clinical trials for the treatment of T2D, and GIPR antagonists are being explored for the treatment of metabolic disorders, including obesity [6,7]. Accordingly, delineating the consequences of enhanced or diminished GIPR signaling in peripheral tissues has immediate translational relevance. Within the brain, both gain and loss of GIPR signaling reduce food intake, actions mediated in part via the control of leptin sensitivity [8,9]. GIP also exerts anabolic actions within white adipose tissue (WAT) [10,11], and the whole body inactivation of the Gipr or GIPR antagonism promotes resistance to diet-induced obesity associated with reductions in adipose tissue mass [[12], [13], [14]].
GIPR is also expressed within multiple bone cell lineages [15,16] and in bone marrow-derived cells, predominantly within a subset of monocytes and macrophages [[17], [18], [19]]. Notably, Gipr−/− mice exhibited defective hematopoiesis, characterized by reduced myeloid progenitors in the bone marrow (BM) and decreased numbers of circulating monocytes and macrophages [20]. As both GIPR coagonists and antagonists are being explored clinically for the treatment of metabolic disorders [7], the impact of GIPR signaling on hematopoiesis in normal and pathophysiological contexts has translational relevance.
Here, we assessed the importance of the murine GIPR for hematopoiesis and the control of BM and adipose tissue inflammation under basal conditions and after exposure to an energy-rich diet. Due to the importance of Toll-like receptor (TLR) and Notch signaling for the control of hematopoiesis [[21], [22], [23], [24], [25], [26]], we also assessed whether GIPR signaling regulates these pathways, using both gain and loss of function strategies. Furthermore, we studied whether the manipulation of GIPR signaling modifies the response to hematopoietic stressors including acute 5-Fluorouracil (5-FU), Lipopolysaccharide (LPS), Pam3CysSerLys4 (Pam3CSK4), and the success of bone marrow transplantation (BMT). Our results show that the Gipr is essential for the expression of BM genes regulating hematopoiesis and adipose tissue inflammation, and the loss of the BM GIPR alters the hematopoietic response to BMT. Nevertheless, gain or loss of GIPR signaling does not have a major impact on the bone marrow response to hematopoietic stress in mice.
2. Materials and methods
2.1. Animals
Mice were maintained on a 12 h light/dark cycle at room temperature, with free access to food and water, except when indicated. Mice were fed either a standard rodent chow diet (RCD) (18% kcal from fat, 2018 Harlan Teklad, Mississauga, ON, Canada) or a high-fat diet (HFD) (45% kcal from fat, D12451i, Research Diets, New Brunswick, NJ, USA). The generation and characterization of Gipr−/− as well as GiprFlox/Flox mice were previously described [10,27]. B6.Cg-Tg(Tek-cre)1Ywa/J (Tie2-cre) mice were obtained from Jackson Laboratories. As described for the B6.Cg-Tg(Tek-cre)1Ywa/J strain [28], germline deletion was prevented by restricting Cre expression to male breeders. To generate GiprTie2−/− mice, Tie2-cre hemizygous mice were bred with floxed Gipr mice (GiprFlox/Flox). WT B6.SJL-Ptprca Pepcb/BoyJ CD45.1+ mice for bone marrow transplant experiments were obtained from Jackson Laboratories. Animals were treated with 24 nmol/kg [DAla2]-GIP (Chi Scientific, Maynard, MA, USA) twice a day (9 am and 5 pm) for a total of 8 days when combined with 5-FU and a total of 6 days when combined with LPS and Pam3CSK4. Alternatively, 150 mg/kg of 5-FU (provided by Mount Sinai Hospital Pharmacy) was given once weekly (for a total of 2–3 doses as indicated). LPS 35 μg (Sigma–Aldrich, Cat# L3024, Oakville, ON, Canada) was administered for a total of three doses, administered 48 h apart. Pam3CSK4 (InvivoGen, Cat# tlr-pms, San Diego, CA, USA) 100 μg per injection was administered for a total of three doses, administered 48 h apart. Phosphate-buffered saline was administered as a vehicle control. Experiments were carried out in groups of male or female mice on a C57BL/6J background. As no differences were found between the control groups (Wild type, GiprFlox/Flox, and Tie2-cre), only data from Tie2-cre mice are shown as a control (unless otherwise stated).
2.2. Body composition using magnetic resonance imaging (MRI)
Body composition (fat and lean mass) was measured prior to and every 4 weeks after placing mice on an HFD, using an Echo MRI nuclear magnetic resonance system (Echo Medical Systems, Houston, TX, USA).
2.3. Blood and tissue collection
For terminal studies, mice were sacrificed by CO2 inhalation, blood was obtained by cardiac puncture, and tissues were dissected and immediately frozen in liquid nitrogen. All blood samples (50–100 μL) for measuring insulin, GLP-1, GIP, and triglycerides at indicated time points during metabolic tests were collected from tail vein into lithium-coated Microvette tubes (Sarstedt, Numbrecht, Germany) and mixed with a 10% volume of TED (5000 kIU/mL Trasylol (Bayer), 32 mM EDTA, and 0.01 mM Diprotin A (Sigma)). Samples were kept on ice and plasma was collected by centrifugation and stored at −80 °C. When blood was collected to perform a complete blood count analysis, ∼200 μL was collected from the tail vein into EDTA-coated Microvette tubes (Sarstedt, Numbrecht, Germany) and kept at room temperature (RT) prior to analysis.
2.4. Glucose, insulin, and lipid tolerance tests
All metabolic tests were performed after a 4–5 h fast (∼9 am–1 pm). For oral and intraperitoneal glucose tolerance tests (OGTT and IPGTT, respectively), d-Glucose (2 g/kg; Sigma, Oakville, ON, Canada) was administered by oral gavage (OGTT) or IP injection (IPGTT). During insulin tolerance tests (ITTs), animals received a single IP injection of 0.75 U/kg BW of insulin (Humalog, VL7510, Eli Lily, Scarborough, ON, Canada). Blood glucose was measured in tail vein samples using a handheld glucose meter (Contour, Bayer, Mississauga, ON, Canada) at baseline (time 0) and 15, 30, 45, 60, 90, and 120 min after glucose or insulin administration. For oral lipid tolerance tests (OLTTs), animals received a 200 μL oral gavage of olive oil (Sigma) at time 0, and blood samples were collected from the tail vein prior to and 1, 2, and 3 h after olive oil gavage.
2.5. Hormone and enzymatic assays
Plasma insulin (Ultrasensitive Mouse Insulin ELISA, Cat# 80-INSMSU-E01 Alpco Diagnostics, Salem, NH, USA), total GLP-1 (Meso Scale Diagnostics, Cat# K150JVC-2 Rockville, MD, USA), and total GIP (Crystal Chem, Cat# 81517, Elk Grove Village, IL, USA) levels were assessed in plasma samples collected at baseline (time 0), 5, 15, or 30 min after glucose or insulin administration during metabolic tests, as indicated. Triglycerides (TGs) were assayed using the Trig-GB kit (Cat# 11877771216, Roche, Mississauga, ON, Canada), at baseline (time 0), 1, 2, and 3 h after oral lipid administration
2.6. Cell preparation for flow cytometry analysis and sorting
Samples for cell isolation from peripheral blood, spleen, or bone marrow were obtained from 8-week-old females. Immediately following sacrifice by CO2 inhalation, ∼700–800 μL of blood was obtained by cardiac puncture and added to 13 mL of red blood cell lysis solution (RBC solution) (BioLegend, Cat# 420301, San Diego, CA, USA) for 14 min at RT with shaking, and cells were pelleted by centrifugation at 1800 rpm, for 5 min at 4 °C. To isolate spleen cells, the entire spleen was placed in a 70 μM cell strainer (Falcon, Cat# 352350, NY, USA) and gently ground through the strainer using the plunger of a 1 mL syringe (BD Biosciences, Cat# 309659, NJ, USA). Spleen cells were rinsed through the cell strainer with three rounds of 3 mL of Iscove's Modified Dulbecco's Medium (IMDM) (STEMCELL Technologies, Cat# 36150, Vancouver, BC, Canada). To isolate bone marrow cells, two femurs were crushed in 3 mL of IMDM medium using a mortar and pestle and filtered through a 70 μM cell strainer. This process was repeated two more times. Spleen or bone cells were pelleted and resuspended in 1–5 mL of FACS buffer (PBS containing 2 mM EDTA, 25 mM HEPES, and 2% FCS) and then lysed with RBC solution for 5 min at RT. The lysis was stopped with 5 mL of FACS buffer and cells were pelleted by centrifugation (1800 rpm 5 min at 4 °C). Cells from spleen and bone marrow were resuspended with FACS buffer and filtered using a 5 mL polystyrene round-bottom tube with a cell strainer cap (Falcon, Cat# 352235, NY, USA) to obtain a single-cell solution. Fc receptors were blocked using TruStain fcX™ (CD16/32 antibody) (BioLegend) for 10 min. Cells were then incubated for 30 min with two panels of fluorophore-conjugated antibodies (Tables S1 and S2), to identify immune cell populations, specifically (i) a lymphocyte-myeloid panel (APC-CD45, FITC-CD45R/B220, FITC-Ly6G/Ly6C (Gr-1), FITC-CD18, PE-CD45R/B220, PE-CD8a, and PE-CD4) and (ii) a monocyte-neutrophil panel. To isolate hematopoietic stem and progenitor cells (HSPCs) from bone marrow, 2–4 x 107 cells were resuspended in 1 mL of PBS containing 2 mM EDTA and 0.5% BSA and were incubated for 10 min at 4 °C according to the manufacturer's instructions for the mouse direct lineage cell depletion kit (Miltenyi Biotec, Cat# 130-110-470, Auburn, CA, USA). Lineage-negative cells were separated using LS columns (Miltenyi Biotec, Cat# 130-042-401, Auburn, CA, USA) and a QuadroMACS™ separation unit (Miltenyi Biotec, Cat# 130-090-976, Auburn, CA, USA). Cells were then incubated for 30 min with the HSPC panel of fluorophore-conjugated antibodies (Biotin-Lineage cocktail (ter119, CD11b, Gr-1, CD3e, and B220), Alexa Fluor 488-Sca-1, APC/Fire750-cKit (CD117), PE-CD34, BV510-CD16/32, BV421-CD135, PE/Cy7-CD150 (SLAM), and APC-CD48), followed by a 30 min incubation with Streptavidin-PE-Cy5. Target populations of live cells (DAPI+) were isolated using a MoFlo Astrios EQ Cell Sorter equipped with 355 nm, 405 nm, 488 nm, 560 nm, and 642 nm lasers (Beckman Coulter, Miami, FL, USA) or analyzed using a Gallios flow cytometer equipped with 405 nm, 488 nm, 561 nm, and 638 nm lasers (Beckman Coulter). B cells are defined as CD45+, CD45R/B220+, Gr-1 (Ly-6G/Ly-6C)-, CD18-, CD8a−, and CD4-. T cells are CD45+, CD45R/B220-, Gr-1 (Ly-6G/Ly-6C)-, CD18-, CD8a+, and CD4+; myeloid (M) cells are gated as CD45+, CD45R/B220-, Gr-1 (Ly-6G/Ly-6C)+, CD18+, CD8a−, and CD4-. In bone marrow samples, LK is Lin− cKit+ and LKS is Lin− cKit+ Sca-1+. Short-term and long-term hematopoietic stem cells (ST-HSC and LT-HSC, respectively) are Lin− cKit+ Sca-1+ CD135-, CD48-, and CD150DIM (ST) and CD150HI (LT), while multipotent progenitor (MPP) cells are Lin− cKit+ Sca-1+ CD135- and CD48+. Common lymphoid progenitor (CLP) cells are defined as Lin− cKit+ Sca-1+ CD135+ CD150-. Common myeloid progenitor (CMP) cells are Lin− cKit+ Sca-1- CD34+ CD16/32-, granulocyte-monocyte progenitor (GMP) cells are Lin− cKit+ Sca-1- CD34+ CD16/32+, and megakaryocyte-erythroid progenitor (MEP) cells are Lin− cKit+ Sca-1- CD34- CD16/32- (see Tables S1 and S2 for details).
2.7. Colony-forming unit (CFU) assay
MethoCult CFU assays were performed with MethoCult™ GF M3434 (STEMCELL Technologies, Cat# 03434, Vancouver, BC, Canada) according to the manufacturer's protocol. Briefly, MethoCult was thawed at 4 °C overnight and mixed. To isolate bone marrow cells, two femurs from 8-week-old female mice were crushed and washed three times with 3 mL of IMDM medium containing 10% Fungizone®-Amphotericin B (250 μg/mL) (Gibco, Cat# 15290–018) and filtered through a 70 μM cell strainer, all under sterile conditions. Cells were lysed with RBC solution for 5 min at RT and resuspended in 3 mL of IMDM medium to a concentration of 2 × 105 cells/mL. A one-tenth volume of the prepared cells was mixed with 3 mL of MethoCult by vortexing. Cells were plated in duplicate in 35 mm grid dishes (Sarstedt, Cat# 83.3900.002, Newton, NC, USA) using a 3 cc syringe (STEMCELL Technologies, Cat# 28240, Vancouver, BC, Canada) and a 16-gauge blunt-end needle (STEMCELL Technologies, Cat# 28110, Vancouver, BC, Canada). Cells were incubated in a humidified chamber at 37 °C and 5% CO2 for 7 (for replating purposes) or 10 days, and then colonies were identified and counted under a bright field microscope. For replating assays, after 7 days of the initial plating, marrow cells were collected by pipetting and washed with 9 mL of IMDM medium. Cells were counted and plated in equal proportions as described above. Total colony numbers were compared between WT and Gipr−/− or Tie2-cre and GiprTie2−/− mice.
2.8. Bone marrow transplantation
Bone marrow chimeras were generated by irradiating 8-week-old WT B6.SJL-Ptprca Pepcb/BoyJ CD45.1+ recipient males, obtained from Jackson Labs (1,100 cGy, split into two equal doses separated 4 h apart) followed by tail vein injection of 5 × 106 congenic bone marrow cells from WT C57BL6/J or Gipr−/− donor males, as described [29,30]. The efficiency of reconstitution was assessed by flow cytometry analysis (Gallios, Beckman Coulter) of tail vein blood every 4 weeks until sacrifice at 16 weeks after BMT. For flow cytometry, CD45.1-PE-Cy7, CD45.2-APC, and CD45.2-FITC antibodies were added to the lymphocyte-myeloid and monocyte-neutrophil panels described above (see Tables S1 and S2 for details).
2.9. RNA isolation and gene expression analysis
Tissue samples and cell pellets were homogenized in Tri Reagent (Molecular Research Center, Cincinnati, OH, USA) using a TissueLyser II system (Qiagen, Germantown, MD, USA), for the extraction of total RNA. First-strand cDNA was synthesized from DNase I-treated total RNA using the SuperScript III and random hexamers (Thermo Fisher Scientific, Markham, ON, Canada). Reverse transcription reactions were carried out for 10 min at 25 °C, 50 min at 50 °C, and an additional 15 min at 70 °C. Gene expression levels were quantified by real-time quantitative PCR (RT-qPCR) using a QuantStudio System and TaqMan Gene Expression Master Mix and Assays (Thermo Fisher Scientific). Oligonucleotides were purchased from Thermo Fisher Scientific. Primer sequences are provided in Table S3. qRT-PCR data were analyzed by the 2−ΔΔCt method, and expression levels for each gene were normalized to Ppia (peptidylprolyl isomerase A-cyclophilin A).
2.10. Quantification and statistical analysis
Data are represented as the mean ± SD or as the mean ± SEM, as indicated. Statistical comparisons were made by one- or two-way ordinary ANOVA followed by Bonferroni post hoc or by unpaired two-tailed Student's t-test (only when two conditions) using GraphPad Prism version 8 software (San Diego, CA, USA). The Log-rank (Mantel–Cox) test was used to evaluate survival rates. A P value ≤ 0.05 was considered statistically significant.
3. Results
3.1. Toll-like receptor and notch genes are dysregulated in BM from HFD-fed Gipr−/− mice
TLRs are expressed in a variety of cell populations, including immune cells and nonimmune cells, such as HSPCs and endothelial cells [[21], [22], [23]]. When engaged, they regulate the proliferation, mobilization, and differentiation of HSC (Hematopoietic Stem Cells) and committed progenitors [21,[24], [25], [26]]. Recent studies reported a reduction in bone marrow progenitors and circulating myeloid cells in Gipr−/− mice [20]. To determine whether this defect may reflect alterations in TLR signaling, we studied the expression of TLRs and downstream effectors, MyD88 and Ticam1, in BM cells from 8- to 13-week-old RCD-fed Gipr−/− mice. Only the levels of Tlr8 and Tlr13 mRNA were downregulated in RNA from Gipr−/− mice (Figure 1A). In contrast, the BM from HFD-fed Gipr−/− mice exhibited reduced levels of Tlr4, Tlr5, Tlr6, Tlr7, Tlr9, Tlr13, MyD88, and Ticam1 (Figure 1B).
Figure 1.

TLR and Notch expression is downregulated in bone marrow cells of HFD-fed Gipr−/− mice. mRNA levels of the indicated TLR and Notch signaling-related genes, relative to Ppia gene expression, were assessed in isolated BM cells from 8- to 13-week-old RCD-fed (n = 6–17/group) (A, C) and from 30- to 34-week-old HFD-fed (n = 8–14/group) (B, D) WT and Gipr−/− male mice. (E) Gipr mRNA levels, relative to Ppia, in the indicated cell populations isolated by flow cytometry from peripheral blood, spleen, and bone marrow of 8-week-old Tie2-cre and GiprTie2−/- female mice. mRNA levels of the indicated TLR and Notch signaling-related genes, relative to Ppia gene expression, were assessed in isolated BM cells from 8- to 13-week-old RCD-fed (n = 6–17/group) (F,G) and from 30- to 34-week-old HFD-fed (n = 8–14/group) (H, I) Tie2-cre and GiprTie2−/− male mice. Data are presented as the mean ± SD. ∗P ≤ 0.05, ∗∗P ≤ 0.01, ∗∗∗P ≤ 0.001, and ∗∗∗∗P ≤ 0.0001. RCD: regular chow diet; HFD: high-fat diet; wks: weeks; BM: bone marrow; PB: peripheral blood; SPL: spleen; M cells: myeloid cells; HSC: hematopoietic stem cells; ST-HSC: short-term hematopoietic stem cells; LT-HSC: long-term hematopoietic stem cells; MPP: multipotent progenitor; CLP: common lymphoid progenitor; CMP: common myeloid progenitor; GMP: granulocyte-monocyte progenitor; MEP: megakaryocyte-erythroid progenitors.
We next analyzed the BM expression of Notch receptors (Notch1, Notch2, Notch3, and Notch4), ligands (Jag1 and Dkk), and targets (Hes1 and Hes3), genes important for hematopoiesis [31]. Only the levels of Notch2 were reduced in the BM from RCD-fed mice (Figure 1C). However, the levels of Notch1, Notch2, Notch3, Jag1, and Hes1 mRNA transcripts were lower in BM from HFD-fed Gipr−/− mice (Figure 1D). Hence, HFD-fed, but not RCD-fed Gipr−/− mice, display an impaired expression of multiple BM genes within the TLR and Notch pathways.
3.2. Gipr expression is downregulated in bone marrow cells from GiprTie2−/− mice
To further delineate the contribution of Gipr to hematopoietic cell lineages, we crossed Tie2-cre mice with GiprFlox/Flox to generate GiprTie2−/− mice (Figure S1A). Tunica intima endothelial kinase 2 (Tie2/Tek) is expressed in endothelial cells, within all progenitor and several differentiated hematopoietic cells, but not in the bone marrow stromal compartment [32]. Gipr mRNA transcripts were not reduced in the jejunum, epididymal, or mesenteric WAT of GiprTie2−/− mice (Figure S1B). Moreover, Gipr levels were very low and unchanged in lung, spleen, thymus, and lymph nodes from GiprTie2−/− mice, tissues with substantial contributions from hematopoietic cell lineages. Although GIPR expression has been reported in some endothelial cell lines [33], Gipr mRNA transcripts were not reduced in major blood vessels, specifically in the thoracic aorta and aortic arch, from GipTie2−/− mice (Figure S1B). In contrast, Gipr mRNA levels were markedly downregulated (by about 90%) in BM cells from GiprTie2−/− mice (Figure S1B).
Given our detection of abnormal BM expression of TLR and Notch genes in HFD-fed Gipr−/− mice (Figure 1B,D), we assessed GiprTie2−/− mice after 25 weeks of 45% HFD feeding. Although body weight and adiposity trended higher in GiprTie2−/− mice, glucose, lipid, and insulin tolerance were not different (Figures S1C–S1K), apart from a modest increase in glucose excursion after intraperitoneal glucose challenge (Figure S1I). Plasma GLP-1 levels were not different, but GIP levels were slightly higher after oral glucose in HFD-fed GiprTie2−/− mice, corresponding to increased insulin levels (Figures S1F–S1H).
The levels of Gipr mRNA were low or undetectable in circulating white blood cells (Figure 1E), relatively higher within the BM and spleen, and markedly reduced within the BM, including BM T cells and myeloid cells of GiprTie2−/− mice (Figure 1E). Strikingly, Gipr was not detectable in early bone marrow progenitor cells (LT-ST HSC, MPP), or in CLP. However, Gipr was expressed at low levels in the CMP lineage and in the more differentiated myeloid progenitor populations, GMP and MEP. Gipr expression in these cell populations was markedly reduced in GiprTie2−/− mice (Figure 1E). Tissue weights from spleen, iWAT, eWAT, mWAT, and BAT were not different between Tie2-cre and GiprTie2−/− mice (Figure S1L).
3.3. TLR and notch gene expression and hematopoiesis are not dysregulated in GiprTie2−/− mice
Body weight, spleen and femur weights, femur length, and spleen and femur cellularity were not different in Gipr−/− and GiprTie2−/− mice (Figures S2A–S2F). Flow cytometry analysis (Figures S3A–S3D and Tables S1-S2) revealed similar frequencies of CD45+ cells, and no differences were observed in populations of T cells, B cells, or myeloid cells in peripheral blood, spleen, or BM from Gipr−/− and GiprTie2−/− mice (Figures S2G–S2I). BM RNA from RCD-fed and HFD-fed GiprTie2−/− mice did not reveal differences in the expression of TLR or Notch pathway genes (Figure 1F–I).
We next analyzed HSPCs using a panel of lineage markers (TER-119, CD11b, Ly-6G/Ly-6C (Gr-1), CD3e, and CD45R/B220) to discriminate between differentiated and nondifferentiated cells in the BM (Figure S3D and Tables S1-S2). Lineage-negative (Lin-) cells were not different in Gipr−/− or GiprTie2−/− animals (Figure S2J), and no differences were observed in Lin-cKit + Sca1+ (LKS) or in Lin-cKit + Sca1- (LK) stem cell populations (Figure S2K). Similarly, the LT-ST HSC population and the multipotent progenitor (MPP) cell frequencies were comparable (Figure S2L), and no differences were found in CLP, CMP cells, or the differentiated myeloid progenitor (GMP and MEP) cell populations (Figures S2M-S2N).
To discern the importance of GIPR signaling for hematopoiesis, we performed colony-forming unit (CFU) assays, a widely used technique to assess the ability of HSPC cells to proliferate and differentiate in vitro, using BM cells from femurs of 8-week-old female Gipr−/− and GiprTie2−/− mice. No major differences were observed in cell populations within colonies established from both Gipr−/− and GiprTie2−/− mice (Figures S2O–S2S). However, at 10 days, the granulocyte-macrophage (GM) population was increased in colonies originating from Gipr−/− compared to WT mice, without differences in the proportion of the other assessed populations (Figure S2P). In contrast, no changes were observed in colonies propagated from GiprTie2−/− mice (Figure S2P). A decrease in the granulocyte (G) population was observed in both Gipr−/− and GiprTie2−/− mice at 14 days (Figure S2Q). However, this decrease had no impact on other cell populations, nor were abnormalities observed at subsequent replating time points. Taken together, these results indicate that, despite the reduction of Gipr expression in different BM cell populations, loss of GIPR signaling within the Tie2+ lineage did not impact hematopoiesis.
3.4. Reconstitution of hematopoiesis after bone marrow transplant does not require the GIPR
To further probe the importance of the GIPR for hematopoiesis, we performed a noncompetitive BMT to assess HSPC repopulation capacity. BM cells from WT or Gipr−/− female donors (CD45.2) were transplanted into irradiated WT male recipients (CD45.1). Mice were maintained on RCD or HFD, and the frequency of cell populations and gene expression were examined 16 weeks after BMT (Figure S4A). Gipr expression was reduced in BM and spleen cells, but not in fat depots of RCD-fed Gipr−/− BM recipients, consistent with successful BMT (Figure S4B). Body weights were not different between genotypes in transplanted mice (Figure S4C). Repopulation capacity in peripheral blood (Figure S4D) was also similar between groups. Tissue weights were not different in Gipr−/− BM recipients, and no differences in femur length or femur and spleen cellularity were observed (Figure S4E).
To determine if prolonged high-fat feeding, a condition associated with dysregulation of TLR and Notch gene expression in Gipr−/− mice (Figure 1A–D), as well as increased circulating levels of GIP [1], could modify hematopoiesis after BMT, mice were placed on a 45% HFD for 14 weeks, starting two weeks following BMT (Figure S4A). Gipr expression was reduced in BM and spleen cells of HFD-fed Gipr−/− BM recipients, but not in adipose tissues (Figure S4F). Body weight gain was not different between groups (Figure S4G). The repopulating capacity of BM assessed through the analysis of peripheral blood was not different (Figure S4H), and tissue weights were similar in BM recipients (Figure S4I). Interestingly, spleen cellularity, but not weight, was decreased in Gipr−/− BM-transplanted mice (Figure 2A).
Figure 2.

Hematopoietic responses after RCD or HFD feeding in mice transplanted with Gipr−/−donor BM. (A) Spleen weight, relative to body weight, and spleen cellularity from 26-week-old WT male mice that received WT (BMT-WT) or Gipr−/− (BMT-Gipr−/−) bone marrow at the age of 8 weeks and were fed an HFD for 14 weeks. mRNA levels of the indicated TLR and Notch signaling-related genes, relative to Ppia gene expression, were assessed in isolated BM cells from 26-week-old WT male mice that received WT (BMT-WT) or Gipr−/− (BMT-Gipr−/−) bone marrow at the age of 8 weeks and were fed an RCD (B, C) (n = 5–7/group) or HFD (D, E) (n = 4–5/group) for 14 weeks. Examination of B cells, T cells, and M cells and monocyte lineage cells (neutrophils and monocytes) at 16 weeks after BMT as a percentage of donor repopulated cells (CD45.2) in peripheral blood (F), spleen (G), and BM (H), and (I) frequency of CMP, GMP, and MEP cells in bone marrow at 16 weeks after BMT from male mice that received WT (BMT-WT) or Gipr−/− (BMT-Gipr−/−) bone marrow at the age of 8 weeks and were fed an RCD (n = 5–7/group). Examination of B cells, T cells, and myeloid cell populations (neutrophils and monocytes) at 16 weeks after BMT as a percentage of donor repopulated cells (CD45.2) in peripheral blood (J) and spleen (K), and (L) frequency of LT-ST HSC and MPP cells in bone marrow at 16 weeks after BMT from male mice that received WT (BMT-WT) or Gipr−/− (BMT-Gipr−/−) bone marrow at the age of 8 weeks and were fed an HFD for 14 weeks (n = 4–5/group). Data are presented as the mean ± SD. ∗P ≤ 0.05 and ∗∗P ≤ 0.01. BMT: bone marrow transplant; RCD: regular chow diet; HFD: high-fat diet; BM: bone marrow; SPL: spleen; PB: peripheral blood; M cells: myeloid cells; BM-HPSC: bone marrow hematopoietic progenitor stem cells; Lin-: lineage negative; ST-HSC: short-term hematopoietic stem cells; LT-HSC: long-term hematopoietic stem cells; MPP: multipotent progenitor; CMP: common myeloid progenitor; GMP: granulocyte-monocyte progenitor; MEP: megakaryocyte-erythroid progenitors.
The analysis of BM mRNA transcripts corresponding to genes important for TLR and Notch signaling revealed only a few differences in Gipr−/− BM-transplanted mice (Figure 2B–E). The levels of Tlr11 and Hes1 were reduced in RCD-fed mice (Figure 2B and C), while Tlr1 and Tlr5 mRNA transcripts were decreased in Gipr−/− BM recipient mice fed an HFD (Figure 2D and E). The distributions of lymphocytes versus myeloid cell populations were not different in peripheral blood of RCD-fed mice; however, the proportion of CD11b + cells was increased (Figure 2F). Ly6C- cells were increased and circulating proinflammatory monocytes (Ly6C++) were decreased in Gipr−/− BM recipients (Figure 2F).
No differences in proportions of CD45+ cells or population frequency distribution of lymphocytes and myeloid cells in spleen or BM were observed (Figure 2G and H). However, monocytes (Ly6C+) were decreased in both spleen and BM cells of Gipr−/− BM recipients, Ly6C- cells were increased in the spleen, while the numbers of CD11b+ and proinflammatory monocytes (Ly6C++) were increased in BM (Figure 2G and H). No differences were found in the lineage-negative HSC population (Figure S5A), LKS and LK cell frequencies, LT-ST HSC or MPP progenitors (Figures S5B-S5C), or the CLPs (Figure S5D). The GMP cell population was increased in Gipr−/− BM recipients, but CMP and MEP populations were not perturbed (Figure 2I).
The distribution of lymphocytes versus myeloid cell populations was similar in peripheral blood of HFD-fed WT and Gipr−/− BM recipients (Figure 2J). Nevertheless, the levels of circulating neutrophils (CD115-) and inflammatory monocytes (LyC6++) were reduced and the numbers of anti-inflammatory monocytes (LyC6-) were increased in Gipr−/− BM recipients (Figure 2J). The analysis of the spleen revealed small differences in lymphoid and myeloid cell populations, including a slight increase in B cells and M cells, and reduced numbers of T cells in Gipr−/− BM recipients fed in an HFD (Figure 2K). Ly6C- cell populations were increased, while inflammatory monocytes (Ly6C+) were reduced (Figure 2K). BM cell populations were not different (Figure S5E). The BM lineage-negative population and LKS frequencies were comparable, and LK cell numbers were unchanged (Figures S5F-S5G). The numbers of MPP progenitors were lower; however, the proportions of LT-ST HSC cells were similar in Gipr−/− BM recipient mice (Figure 2L). Common lymphoid and myloid progenitors (CLP, CMP) populations were similar (Figure S5H,I).
3.5. GIPR agonism modifies TLR and notch pathway responses to 5-FU and Pam3CSK4
We next asked whether the GIPR was important for adaptive hematopoiesis in response to (i) the chemotherapeutic agent 5-FU and (ii) two distinct TLR ligands, LPS acting through TLR4 and Pam3CSK4 acting via TLR1/2. 5-FU is a chemotherapeutic agent that activates HSPCs while eliminating proliferative myeloid cells [34], whereas LPS and Pam3CSK4 engage TLR receptors to regulate HSPC proliferation and myelopoiesis [25,35]. Interestingly, plasma GIP levels were increased after 5-FU and trended higher after LPS administration in WT mice (Figure S6A). Baseline GIP levels were higher in Gipr−/− mice and did not increase further after 5-FU or LPS (Figure S6A), whereas GIP levels were not different after the Pam3CSK4 administration (Figure S6A).
Subsequently, we examined whether genes important for TLR signaling were dysregulated in isolated BM cells from WT and Gipr−/− mice following the administration of 5-FU, LPS, or Pam3CSK4 (Figure S6B). The majority of mRNA transcripts examined did not exhibit genotype-dependent regulation after 5-FU, LPS, or Pam3CSK4, with the exception of MyD88, which was markedly upregulated by Pam3CSK4 only in BM from Gipr-deficient mice, and Tlr6, which was upregulated in BM from WT but not Gipr−/− BM, whereas Tlr1 was induced in Gipr−/− mice BM after 5-FU (Figure S6B).
To assess whether the activation of GIPR signaling modified the BM gene expression profile response to 5-FU, LPS, or Pam3CSK4, we coadministered the degradation-resistant GIPR agonist, [DAla2]-GIP [10,36]. Interestingly, BM expression of Tlr2, Tlr4, Tlr5, Tlr8, Tlr9, Tlr13, Notch1, Jag1 and Notch2 and the downstream target, Ticam1, were differentially regulated by the coadministration of [DAla2]-GIP in 5-FU-treated mice (Figure 3A). Specifically, 5-FU and [DAla2]-GIP cotreatment increased the levels of Tlr4, Tlr5, Tlr8, and Notch1 expression relative to basal levels seen with [DAla2]-GIP alone. Coadministration of LPS and [DAla2]-GIP had relatively little effect on BM gene expression profiles, relative to LPS alone (Figure S6C). Conversely, cotreatment with [DAla2]-GIP attenuated changes in BM levels of Tlr1, Tlr8, Tlr13, MyD88, and Ticam1 mRNA transcripts observed with Pam3CSK4 alone (Figure 3B). Interestingly, Tlr9 expression was not affected by Pam3CSK4 alone, yet mRNA levels decreased with concomitant [DAla2]-GIP administration (Figure 3B).
Figure 3.

Bone marrow Tlr and Notch gene expression in response to 5-FU or Pam3CSK4 and [DAla2]-GIP coadministration. mRNA levels of the indicated TLR- and Notch-related genes, relative to Ppia gene expression, in bone marrow from 7-week-old WT male mice treated with PBS or [DAla2]-GIP and/or 5-FU (A) or Pam3CSK4 (B) as indicated (n = 6/group). Data are presented as the mean ± SD. ∗P ≤ 0.05, ∗∗P ≤ 0.01, ∗∗∗P ≤ 0.001, and ∗∗∗∗P ≤ 0.0001. 5-FU: 5-fluorouracil; Pam3CSK4: Pam3CysSerLys4; PBS: phosphate-buffered saline; GIP: glucose-dependent insulinotropic polypeptide; BM: bone marrow.
3.6. Gipr is dispensable for adaptive hematopoiesis in response to 5-FU
We next examined whether the loss of Gipr impaired adaptive hematopoiesis. Interestingly, Gipr−/− but not GiprTie2−/− mice displayed better survival following 5-FU treatment (Figure S7A), consistent with the findings of improved survival of stressed Gipr−/− mice in response to myocardial ischemia [37]. Hence, WT and Gipr−/− mice were examined in more detail after 5-FU administration (Figure S7B). No differences in spleen or femur weight or cellularity were detected in 5-FU-treated WT versus Gipr−/− mice; however, body weight trended lower in 5-FU-treated Gipr−/− mice (Figures S7C–S7G). Peripheral blood myeloid cell populations were reduced after 5-FU, whereas T cells were increased (Figure S7H). Splenic B and T cell numbers, as well as myeloid cell numbers, were not different in 5-FU-treated mice (Figure S7I). B and T cell numbers were also increased after 5-FU in WT and Gipr−/− BM (Figure S7J), whereas BM myeloid cell numbers trended lower or were reduced in 5-FU-treated WT and Gipr−/− mice, respectively (Figure S7J).
No differences were observed in the proportions of BM Lin- cells (Figure S7K); however, LK and LKS cell numbers were reduced in the BM of 5-FU-treated mice, independent of the genotype (Figure S7L). LT-ST HSC cells were decreased while MPP cells were increased after 5-FU; however, no differences were observed in Gipr−/− versus WT mice (Figure S7M). The CLP population decreased similarly in both 5-FU treated groups (Figure S7N), whereas the levels of CMP, GMP, and MEP cells were not significantly different (Figure S7O).
We next treated mice with [DAla2]-GIP and examined the hematopoietic response to 5-FU (Figure S8A). Body weight was reduced after 5-FU, but no differences in spleen or femur weights or femur cellularity were observed in mice treated with [DAla2]-GIP (Figure S8B-F). Administration of [DAla2]-GIP for 8 days did not alter the populations of B cells, T cells, or myeloid cells in peripheral blood (Figures S8G-S8H) or BM (Figure 4A) of mice treated with 5-FU. Intriguingly, the reduction in the CD115- BM population (neutrophils) was blunted in [DAla2]-GIP-treated mice (Figure 4B), whereas the majority of BM-HPSC lineages were not different (Figures S8I–S8M).
Figure 4.

Hematopoietic responses to 5-FU, LPS, and Pam3CSK4 in Gipr−/−mice and WT mice treated with [DAla2]-GIP. Frequencies of B cells, T cells, M cells (A), and monocyte lineage cells (neutrophils and monocytes) (B) in bone marrow from 7-week-old WT males treated with [D-Ala]-GIP and/or 5-FU and controls (n = 6–8/group). (C) Frequency of CLP cells in bone marrow from 7-week-old WT and Gipr−/- male mice that were treated with LPS or vehicle (PBS) (n = 6/group). Frequency of LKS and LK populations (D) and CLP (E) cells in bone marrow from 7-week-old WT male mice treated with [D-Ala]-GIP and/or LPS and controls (n = 6/group). (F) Frequency of CMP, GMP, and MEP cells in bone marrow from 7-week-old WT and Gipr−/- male mice that were treated with Pam3CSK4 or vehicle (PBS) (n = 6/group). Frequencies of B cells, T cells, M cells, and monocyte lineage cells (neutrophils and monocytes) in peripheral blood (G, H) and bone marrow (I, J) from 7-week-old WT males treated with [D-Ala]-GIP and/or Pam3CSK4 and controls (n = 6/group). Data are presented as the mean ± SD. ∗P ≤ 0.05, ∗∗P ≤ 0.01, ∗∗∗P ≤ 0.001, and ∗∗∗∗P ≤ 0.0001. PBS: phosphate-buffered saline; 5-FU: 5-fluorouracil; LPS: Lipopolysaccharide; Pam3CSK4: Pam3CysSerLys4; GIP: glucose-dependent insulinotropic polypeptide; PB: peripheral blood; BM: bone marrow; BM-HPSC: bone marrow hematopoietic progenitor stem cells; LK: Lin-cKit + Sca1-; LKS: Lin-cKit + Sca1+; CLP: common lymphoid progenitor; CMP: common myeloid progenitor; GMP: granulocyte-monocyte progenitor; MEP: megakaryocyte-erythroid progenitors.
3.7. Modulation of GIPR signaling does not impair the hematopoietic response to LPS or Pam3CSK4
To determine the hematopoietic response to TLR engagement, we treated WT and Gipr−/− mice with the TLR ligands LPS or Pam3CSK4. Body weight was modestly lower in Gipr−/− mice treated with LPS, but not different in WT animals (Figures S9A-S9B). Spleen weight and splenic and femur cellularity were not different between genotypes (Figures S9C–S9F). LPS increased the frequency of myeloid populations in peripheral blood (Figure S9G), spleen (Figure S9H), and bone marrow (Figure S9I). In contrast, lymphoid cells (both B and T cells) were decreased in all tissues, except in the spleen where only T cells were reduced (Figures S9G–S9I). No differences in the response to LPS were detected between genotypes. Similarly, Lin-, LK, or LKS cell populations were not different (Figures S9J-S9K). The numbers of LT-ST HSCs were reduced, whereas MPPs were increased after LPS, to a similar extent in WT versus Gipr−/− mice (Figure S9L). Intriguingly, CLP populations were reduced by LPS only in the absence of the Gipr (Figure 4C), whereas proportions of CMPs, GMPs, and MEPs were not different in LPS-treated WT versus Gipr−/− mice (Figure S9M).
We next examined whether the activation of the GIPR impacted the hematopoietic response to LPS administration (Figure S10A). Body weight was reduced after LPS treatment and lower with [DAla2]-GIP administration (Figure S10B). No differences in spleen or femur weights or femur length were observed in [DAla2]-GIP-treated mice (Figures S10C–S10E). Femur cellularity was reduced in WT mice treated with [DAla2]-GIP alone (Figure S10F). Similarly, T cells, B cell, myeloid cell, neutrophil (CD115-), and monocyte (CD115+) cell populations in peripheral blood or BM were not different in [DAla2]-GIP-treated mice exposed to LPS (Figures S10G–S10J). [DAla2]-GIP administration increased LK cell proportions in BM (Figure 4D), while reducing CLP population (Figure 4E), without affecting other progenitor cell populations (Figures S10K–S10M).
Treatment with the TLR1/2 agonist Pam3CSK4 (Figure S11A) revealed no genotype-dependent differences in body weight, spleen, and femur weights, or femur and splenic cellularity (Figures S11B–S11F). Spleen weight was increased in Pam3CSK4-treated WT and Gipr−/− mice (Figure S11C); however, the numbers of B cells, T cells, and myeloid cells were not different in Pam3CSK4-treated WT versus Gipr−/− mice (Figures S11G–S11I). Myeloid cell populations were increased by Pam3CSK4 in peripheral blood, spleen and BM, without differences between genotypes (Figure S11G–I). Pam3CSK4 reduced B cell numbers in peripheral blood and BM, and T cell numbers in spleen and bone marrow (Figure S11G–I), however, no differences were observed between WT and Gipr-/- mice. Pam3CSK4 had a minimal genotype-dependent effect on BM-HPSC cell populations (Figure 4F, Figure S11J–S11M), with no differences being observed between WT and Gipr−/− mice. Similarly, the proportions of total lineage-negative populations were not different, LT-ST HSCs were decreased, and MPP frequencies were increased in both genotypes following Pam3CSK4 treatment (Figure S11J–L). CLP populations were similar (Figure S11M), however, CMP frequencies were increased in Pam3CSK4-treated WT but not Gipr-/- mice (Figure 4F). No genotype-dependent differences were observed in GMP and MEP progenitor cell populations (Figure 4F).
Coadministration of [DAla2]-GIP and Pam3CSK4 (Figure S12A) produced no differences in body weight (Figure S12B), spleen or femur weights, or femur cellularity compared to Pam3CSK4 alone (Figures S12C–S12F). B cell and T cell numbers were not different; however, myeloid cell numbers were higher in peripheral blood from mice treated with Pam3CSK4 plus [DAla2]-GIP versus [DAla2]-GIP alone (Figure 4G). CD11b + cells were increased with [DAla2]-GIP and Pam3CSK4 cotreatment, but the numbers of neutrophils (CD115-) and monocytes (CD115+) were not different (Figure 4H). [DAla2]-GIP treatment attenuated the induction of the inflammatory monocyte cell population (Ly6C+) by Pam3CSK4 (Figure 4H). BM T cells and B lymphocytes were reduced, whereas myeloid cells were increased by Pam3CSK4, without any impact of [DAla2]-GIP administration (Figure 4I). Interestingly, neutrophil levels (CD115-) were modestly increased in mice receiving [DAla2]-GIP and Pam3CSK4 versus mice treated with Pam3CSK4 alone (Figure 4J). No differences in Lin-cells, LKS, LK, LT-ST HSCs, MPPs, CLP, CMP, GMP, or MEPs were detected in the BM following [DAla2]-GIP administration in Pam3CSK4-treated mice (Figures S12G–S12K).
3.8. Gipr−/− BMT recipients are protected from HFD-induced adipose tissue inflammation
As GIPR + myeloid cells contribute to the control of WAT inflammation [38], we assessed mRNA transcripts relevant to inflammation in different WAT depots from Gipr−/− BM recipients fed an RCD or HFD for 16 weeks after BMT. In eWAT, the levels of S100a8 and S100a9 alarmin mRNA transcripts were increased after HFD feeding in WT but not in Gipr−/− BM-transplant recipients (Figure 5A). Similarly, S100a8, but not S100a9, was differentially expressed in iWAT of Gipr−/− BM-transplanted recipient mice (Figure 5B). Moreover, the HFD induction of Il1b, Il6, Tnf, Ccl2, AdgreI (F4/80), and Cxcl2 mRNA expression in eWAT of WT BM recipients was markedly attenuated in mice receiving Gipr−/− BM (Figure 5A). Conversely, Cxcl2 expression in iWAT was increased by HFD feeding only in Gipr−/− BM-transplanted mice (Figure 5B). Interestingly, the levels of S100a9 and Il6 in mWAT were increased in the recipients of Gipr−/− BM under RCD, but not HFD feeding (Figure S13A).
Figure 5.

Modification of adipose tissue inflammation in HFD-fed mice after transplantation of Gipr−/−BM. Tissue mRNA expression of S100a8, S100a9, cytokines, and chemokines in eWAT (A) and iWAT (B) from 26-week-old WT male mice that received WT (BMT-WT) or Gipr−/− (BMT-Gipr−/−) bone marrow at the age of 9 weeks and were fed an RCD (n = 4–7/group) or HFD (n = 4–5/group) for 12 weeks. Data are presented as the mean ± SD. ∗P ≤ 0.05, ∗∗P ≤ 0.01, ∗∗∗P ≤ 0.001, and ∗∗∗∗P ≤ 0.0001. BMT: bone marrow transplant; RCD: regular chow diet; HFD: high-fat diet; eWAT: epididymal white adipose tissue; iWAT: inguinal white adipose tissue.
The analysis of eWAT from HFD-fed Gipr−/− mice revealed a reduction in Tnf, AdgreI, Mgl2, and Cxcl1 gene expression, while only Mgl2 was reduced in iWAT (Figures S13B-S13C). An increase in alarmin S100a9 was observed in eWAT from Gipr−/− animals, whereas Il1b and Tnf mRNAs were increased in iWAT. In contrast, the levels of mRNA transcripts for Il6, Ccl2, AdgreI, and Cxcl1 were reduced in Gipr−/− mWAT (Figure S13D). Taken together, these results reveal depot-specific differences in the contribution of BM-derived cells to HFD-associated changes in adipose tissue inflammation.
4. Discussion
The availability of nutrients is a key determinant of hematopoiesis. Nutrient depletion or starvation reduces monocyte BM mobilization [39], enhances migration of memory T cells from peripheral organs to the BM [40], and shifts B cells from Peyer's patches to the BM, actions that are reversed upon refeeding [41]. Conversely, energy excess, as evident in mice exposed to an HFD, impairs hematopoiesis via reduction of BM HSPCs [42] or altered myelopoiesis, depending on the specific experimental context [43]. Although signals conveying nutrient status to BM populations are poorly understood, roles for leptin, 5′ AMP-activated protein kinase, CD36, and TLR4 as nutrient-sensitive regulators of hematopoiesis and adipose tissue inflammation have been proposed [[44], [45], [46], [47]]. Here, we extend these concepts by highlighting new roles for the nutrient-sensitive GIP-GIPR axis in the control of hematopoiesis.
Previous studies have shown that HSCs from HFD-fed mice increase the number of proinflammatory macrophages in adipose tissue, via a hematopoietic MyD88-dependent process [48]. Moreover, experimental and clinical obesity has been linked to the dysregulation of hematopoiesis [42,47,48], mediated in part via gut-derived mechanisms including nutrient signaling via fatty acids, as well as microbial-derived metabolites and TLR ligands [49,50]. In turn, BM TLR signaling regulates the extent of obesity-associated insulin resistance [46,51]. As circulating GIP levels are upregulated in the context of high-fat feeding, as well as experimental and clinical obesity [1,[52], [53], [54]], we hypothesized that GIPR signaling links energy availability to the control of hematopoiesis.
In this study, we showed that the absence of the GIPR in young healthy animals does not translate to a dysregulation of TLR or Notch gene expression within BM cells or any differences in hematopoiesis. In contrast, BM from older Gipr−/− mice fed an energy-rich diet displayed reduced expression of genes important for the TLR and Notch signaling pathways (Figure 6). Intriguingly, when BM cells were stressed using 5-FU or TLR agonists (LPS or Pam3CSK4), no differences in the expression of TLR- and Notch-related genes in Gipr−/− BM cells were observed. However, the activation of GIPR signaling in the context of concomitant 5-FU or Pam3CSK4 administration modified the expression of different TLR and Notch signaling members.
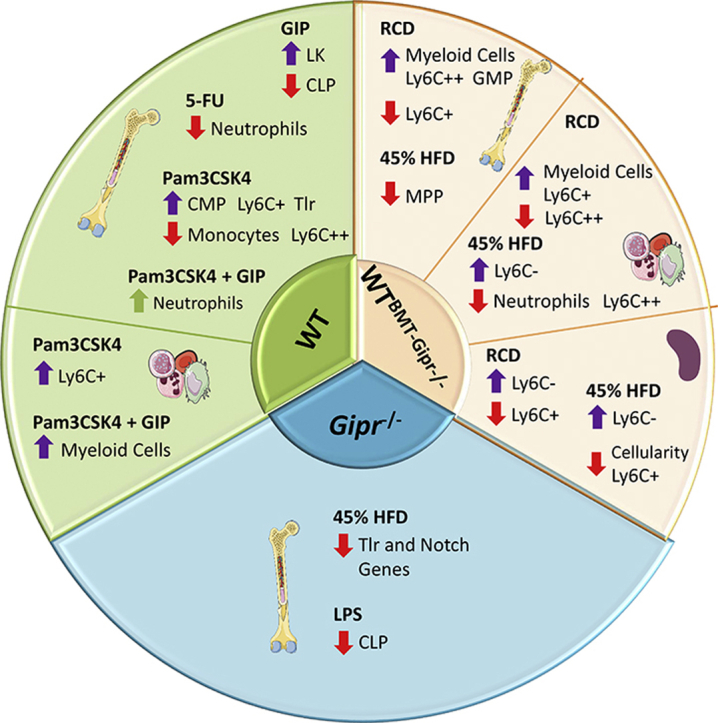
Figure 6.

Gain and loss of GIPR signaling impact hematopoiesis. Bone marrow, peripheral blood, and splenic cells were assessed to investigate the role of the GIPR in the control of hematopoiesis in different stress situations. HFD: high-fat diet; RCD: regular chow diet; BMT: bone marrow transplant; 5-FU: 5-fluorouracil; LPS: Lipopolysaccharide; Pam3CSK4 = Pam3CysSerLys4; GIP: glucose-dependent insulinotropic polypeptide; LK: Lin-cKit + Sca1-; MPP: multipotent progenitor; CLP: common lymphoid progenitor; CMP: common myeloid progenitor; GMP: granulocyte-monocyte progenitor.
Gipr expression was localized to myeloid progenitors and within subsets of differentiated myeloid cells in the BM. Moreover, a marked reduction of Gipr mRNA within these cell lineages was observed within the BM of 8-week-old GiprTie2−/− mice. Nevertheless, the loss of GIPR within Tie2+ cells or globally within all tissues of Gipr−/− mice did not result in alteration of progenitor or differentiated lymphoid or myeloid cell populations in circulating blood, BM, or spleen. Hence, our current data do not support a critical role for Tie2-GIPR + cells in the basal control of hematopoiesis.
Similarly, we did not detect dysregulation of TLR or Notch gene expression in the BM of GiprTie2−/− mice, despite the marked reduction of Gipr expression within total BM RNA and in major Gipr + cell lineages within the BM. These results imply that signals arising from one or more GIPR + cell types not directly targeted by Tie2-cre contribute to the dysregulation of Tlr and Notch expression evident in Gipr−/− mice. Although developmental adaptation to germline deletion of Gipr may contribute to these different observations, the reduced expression of a subset of these genes in the BM of mice transplanted with Gipr−/− BM demonstrates that these findings reflect a BM-intrinsic process. Hence, it seems unlikely that the compensation for developmental loss of Gipr expression underlies sustained dysregulation of BM Tlr and Notch expression following the loss of the Gipr.
The reduced expression of TLR and Notch family members raised the possibility that Gipr−/− mice might exhibit defective mobilization of hematopoietic cells in response to TLR ligands. Nevertheless, we did not detect major differences in the acute hematopoietic responses to the TLR agonists LPS or Pam3CSK4. Intriguingly, the representation of the CLP population was reduced following the LPS administration in Gipr−/− mice, raising the possibility that the loss of the GIPR differentially impacts the biology of the lymphoid differentiating cells, findings which merit further exploration.
Interestingly, we detected reduced eWAT expression of the alarmins S100a8 and S100a9, as well as attenuated WAT expression of genes encoding cytokines, chemokines, and F4/80, a marker of macrophage infiltration, following selective deletion of the GIPR within the BM. Hence, these findings, taken together with our recent studies [20,38], add further support for the BM GIPR as a determinant of the extent of adipose tissue inflammation in the context of nutrient excess.
Of potential translational relevance, exogenous GIPR agonism had no major deleterious consequences on the BM hematopoietic responses to experimental stressors such as 5-FU, LPS, or Pam3CSK4. Interestingly, circulating myeloid numbers were higher in [DAla2]-GIP-treated mice after Pam3CSK4. As a complementary approach to identify the functional importance of BM GIPR + cell populations, we used noncompetitive BM repopulation to examine cell lineages within the BM and peripheral blood. A reduction in circulating and splenic monocyte cell populations was evident as early as 4 weeks after the transplantation in Gipr−/− BM recipients. Although RCD-fed Gipr−/− BM recipients showed a decrease in circulating proinflammatory monocytes (Ly6C++), this same population was increased in isolated BM cells, accompanied by a decrease in monocytes (LyC6+). Similarly, HFD-fed Gipr−/− BM recipient mice had a decrease in circulating proinflammatory monocytes (Ly6C++) and neutrophils (CD115-) compared to WT BM-transplanted mice. Taken together with the dysregulated expression of inflammatory genes within adipose tissue, the transplantation experiments illustrate an important biological role of the BM GIPR in the formation of hematopoietic lineages and the response to HFD feeding (Figure 6).
Our studies have several limitations. First, several analyses used mice with germline inactivation of the Gipr gene, and hence developmental adaptations may have masked the importance of the loss of the GIPR in the hematopoietic system of adult mice. Although Gipr expression has been difficult to detect in normal endothelial cells [33] and was not reduced within major blood vessels of GiprTie2−/− mice, it remains possible that low-level Gipr expression within subsets of endothelial cells, when extinguished, may contribute to the phenotypes observed. Moreover, BMT may confer partial resistance to diet-induced obesity [55], potentially attenuating metabolic phenotypes arising in recipients of Gipr−/− BM. We did not study the importance of the GIPR for hematopoiesis in markedly obese older mice with severe insulin resistance, metabolic features likely to be found in human populations targeted therapeutically for manipulation of the GIPR signaling system. Similarly, it will be interesting to assess the importance of the GIPR for hematopoiesis under conditions characterized by bacterial and viral infections, cancer, and additional immune challenges, which may unmask new roles for the hematopoietic GIPR in these contexts. Additionally, we analyzed the gene expression in RNA from the whole BM and adipose tissue, potentially obscuring meaningful changes in cellular subsets within the tissue microenvironment.
Our current findings reveal that the gain and loss of GIPR signaling produce dysregulation of hematopoiesis and myeloid lineages and regulate the expression of multiple BM TLR and Notch genes (Figure 6), as well as the control of adipose tissue inflammation. Collectively, these observations are consistent with a role for GIP as a gut-derived signal communicating changes in nutrient intake to the BM compartment. However, our analyses do not support an important role for the GIPR in the control of basal or adaptive hematopoiesis. As GIP-based coagonists such as tirzepatide [5,56] are in phase 3 clinical trials and GIPR antagonism continues to be explored therapeutically [6,7,13], our findings support the hematopoietic safety of translational studies targeting the GIPR for the treatment of metabolic disorders.
Authors’ contributions
Conceptualization was contributed by G. P., and D. J. D.; investigation, G. P., E. M. V., L. L. B., E. E. M., J. A. K., D. M., and K. W. A. B.; formal analysis and visualization, G. P.; writing the original draft, G. P. D. J. D; reviewing and editing the manuscript, G. P., E. M. V., L. L. B., E. E. M., J. A. K., and D. J. D.; funding acquisition and project administration, D. J. D.; supervision, D. J. D. In addition, D. J. D. takes the primary responsibility for the data described in this manuscript.
Acknowledgments
G. P. has received a postdoctoral fellowship from the Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS). E. M. V. received fellowship funding from Diabetes Canada. E. E. M. has received fellowship funding from the Canadian Diabetes Association and the Canadian Institutes of Health Research. D. J. D. was supported by a CIHR Foundation Grant 154321, an investigator-initiated operating grant from Novo Nordisk Inc., the Novo Nordisk Foundation-Sinai Health-University of Toronto Fund in Incretin biology, the Canada-Israel Health Research Initiative, jointly funded by the Canadian Institutes of Health Research 154321, the Israel Science Foundation, the International Development Research Centre, and the Azrieli Foundation, and a Banting and Best Diabetes Centre-Novo Nordisk Chair in Incretin biology. Some of the equipment used in this study was supported by the 3D (Diet, Digestive Tract, and Disease) Centre funded by the Canadian Foundation for Innovation and Ontario Research Fund, project numbers 19442 and 30961. We thank J. E. Dick, S. Xie, and J. F. Woolley for their scientific guidance and assistance.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.molmet.2020.101008.
Conflict of interest
D. J. D. has served as an advisor or consultant or speaker within the past 12 months to Forkhead Biotherapeutics, Intarcia Therapeutics, Merck Research Laboratories, Novo Nordisk Inc., and Pfizer Inc. None of the other authors has conflicts of interest. Investigator-initiated research for studies of GLP-1 and GIP is supported in part by grants from Novo Nordisk Inc. To Mt. Sinai Hospital and D. J. D.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Campbell J.E., Drucker D.J. Pharmacology physiology and mechanisms of incretin hormone action. Cell Metabolism. 2013;17(4):819–837. doi: 10.1016/j.cmet.2013.04.008. [DOI] [PubMed] [Google Scholar]
- 2.Muller T.D., Finan B., Bloom S.R., D'Alessio D., Drucker D.J., Flatt P.R. Glucagon-like peptide 1 (GLP-1) Mol Metab. 2019:3072–3130. doi: 10.1016/j.molmet.2019.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Drucker D.J. The cardiovascular biology of glucagon-like peptide-1. Cell Metabolism. 2016;24(1):15–30. doi: 10.1016/j.cmet.2016.06.009. [DOI] [PubMed] [Google Scholar]
- 4.Nauck M.A., Meier J.J., Cavender M.A., Abd El Aziz M., Drucker D.J. Cardiovascular actions and clinical outcomes with glucagon-like peptide-1 receptor agonists and dipeptidyl peptidase-4 inhibitors. Circulation. 2017;136(9):849–870. doi: 10.1161/CIRCULATIONAHA.117.028136. [DOI] [PubMed] [Google Scholar]
- 5.Frias J.P., Nauck M.A., Van J., Kutner M.E., Cui X., Benson C. Efficacy and safety of LY3298176, a novel dual GIP and GLP-1 receptor agonist, in patients with type 2 diabetes: a randomised, placebo-controlled and active comparator-controlled phase 2 trial. Lancet. 2018;392(10160):2180–2193. doi: 10.1016/S0140-6736(18)32260-8. [DOI] [PubMed] [Google Scholar]
- 6.Finan B., Muller T.D., Clemmensen C., Perez-Tilve D., DiMarchi R.D., Tschop M.H. Reappraisal of GIP pharmacology for metabolic diseases. Trends in Molecular Medicine. 2016;22(5):359–376. doi: 10.1016/j.molmed.2016.03.005. [DOI] [PubMed] [Google Scholar]
- 7.Killion E.A., Lu S.C., Fort M., Yamada Y., Veniant M.M., Lloyd D.J. Glucose-dependent insulinotropic polypeptide receptor therapies for the treatment of obesity, do agonists = antagonists? Endocrine Reviews. 2020;41(1) doi: 10.1210/endrev/bnz002. [DOI] [PubMed] [Google Scholar]
- 8.Kaneko K., Fu Y., Lin H.Y., Cordonier E.L., Mo Q., Gao Y. Gut-derived GIP activates central Rap1 to impair neural leptin sensitivity during overnutrition. Journal of Clinical Investigation. 2019:1303786–1303791. doi: 10.1172/JCI126107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adriaenssens A.E., Biggs E.K., Darwish T., Tadross J., Sukthankar T., Girish M. Glucose-dependent insulinotropic polypeptide receptor-expressing cells in the hypothalamus regulate food intake. Cell Metabolism. 2019;30(5):987–996. doi: 10.1016/j.cmet.2019.07.013. e986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Campbell J.E., Ussher J.R., Mulvihill E.E., Kolic J., Baggio L.L., Cao X. TCF1 links GIPR signaling to the control of beta cell function and survival. Nature Medicine. 2016;2284–90 doi: 10.1038/nm.3997. [DOI] [PubMed] [Google Scholar]
- 11.Thondam S.K., Daousi C., Wilding J.P., Holst J.J., Ameen G.I., Yang C. Glucose-dependent insulinotropic polypeptide promotes lipid deposition in subcutaneous adipocytes in obese type 2 diabetes patients: a maladaptive response. American Journal of Physiology. Endocrinology and Metabolism. 2017;312(3):E224–E233. doi: 10.1152/ajpendo.00347.2016. [DOI] [PubMed] [Google Scholar]
- 12.Miyawaki K., Yamada Y., Ban N., Ihara Y., Tsukiyama K., Zhou H. Inhibition of gastric inhibitory polypeptide signaling prevents obesity. Nature Medicine. 2002;8(7):738–742. doi: 10.1038/nm727. [DOI] [PubMed] [Google Scholar]
- 13.Killion E.A., Wang J., Yie J., Shi S.D., Bates D., Min X. Anti-obesity effects of GIPR antagonists alone and in combination with GLP-1R agonists in preclinical models. Science Translational Medicine. 2018;10(472) doi: 10.1126/scitranslmed.aat3392. [DOI] [PubMed] [Google Scholar]
- 14.Svendsen B., Capozzi M.E., Nui J., Hannou S.A., Finan B., Naylor J. Pharmacological antagonism of the incretin system protects against diet-induced obesity. Mol Metab. 2020:3244–3255. doi: 10.1016/j.molmet.2019.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhong Q., Itokawa T., Sridhar S., Ding K.H., Xie D., Kang B. Effects of glucose-dependent insulinotropic peptide on osteoclast function. American Journal of Physiology. Endocrinology and Metabolism. 2007;292(2):E543–E548. doi: 10.1152/ajpendo.00364.2006. [DOI] [PubMed] [Google Scholar]
- 16.Bollag R.J., Zhong Q., Phillips P., Min L., Zhong L., Cameron R. Osteoblast-derived cells express functional glucose-dependent insulinotropic peptide receptors. Endocrinology. 2000;141(3):1228–1235. doi: 10.1210/endo.141.3.7366. [DOI] [PubMed] [Google Scholar]
- 17.Nagashima M., Watanabe T., Terasaki M., Tomoyasu M., Nohtomi K., Kim-Kaneyama J. Native incretins prevent the development of atherosclerotic lesions in apolipoprotein E knockout mice. Diabetologia. 2011;54(10):2649–2659. doi: 10.1007/s00125-011-2241-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nogi Y., Nagashima M., Terasaki M., Nohtomi K., Watanabe T., Hirano T. Glucose-dependent insulinotropic polypeptide prevents the progression of macrophage-driven atherosclerosis in diabetic apolipoprotein E-null mice. PloS One. 2012;7(4) doi: 10.1371/journal.pone.0035683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pivovarova O., Hornemann S., Weimer S., Lu Y., Murahovschi V., Zhuk S. Regulation of nutrition-associated receptors in blood monocytes of normal weight and obese humans. Peptides. 2015:6512–6519. doi: 10.1016/j.peptides.2014.11.009. [DOI] [PubMed] [Google Scholar]
- 20.Mantelmacher F.D., Fishman S., Cohen K., Pasmanik Chor M., Yamada Y., Zvibel I. Glucose-dependent insulinotropic polypeptide receptor deficiency leads to impaired bone marrow hematopoiesis. The Journal of Immunology. 2017;198(8):3089–3098. doi: 10.4049/jimmunol.1601441. [DOI] [PubMed] [Google Scholar]
- 21.Nagai Y., Garrett K.P., Ohta S., Bahrun U., Kouro T., Akira S. Toll-like receptors on hematopoietic progenitor cells stimulate innate immune system replenishment. Immunity. 2006;24(6):801–812. doi: 10.1016/j.immuni.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sioud M., Floisand Y., Forfang L., Lund-Johansen F. Signaling through toll-like receptor 7/8 induces the differentiation of human bone marrow CD34+ progenitor cells along the myeloid lineage. Journal of Molecular Biology. 2006;364(5):945–954. doi: 10.1016/j.jmb.2006.09.054. [DOI] [PubMed] [Google Scholar]
- 23.Takizawa H., Regoes R.R., Boddupalli C.S., Bonhoeffer S., Manz M.G. Dynamic variation in cycling of hematopoietic stem cells in steady state and inflammation. Journal of Experimental Medicine. 2011;208(2):273–284. doi: 10.1084/jem.20101643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rodriguez S., Chora A., Goumnerov B., Mumaw C., Goebel W.S., Fernandez L. Dysfunctional expansion of hematopoietic stem cells and block of myeloid differentiation in lethal sepsis. Blood. 2009;114(19):4064–4076. doi: 10.1182/blood-2009-04-214916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Esplin B.L., Shimazu T., Welner R.S., Garrett K.P., Nie L., Zhang Q. Chronic exposure to a TLR ligand injures hematopoietic stem cells. The Journal of Immunology. 2011;186(9):5367–5375. doi: 10.4049/jimmunol.1003438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Megias J., Yanez A., Moriano S., O'Connor J.E., Gozalbo D., Gil M.L. Direct Toll-like receptor-mediated stimulation of hematopoietic stem and progenitor cells occurs in vivo and promotes differentiation toward macrophages. Stem Cells. 2012;30(7):1486–1495. doi: 10.1002/stem.1110. [DOI] [PubMed] [Google Scholar]
- 27.Miyawaki K., Yamada Y., Yano H., Niwa H., Ban N., Ihara Y. Glucose intolerance caused by a defect in the entero-insular axis: a study in gastric inhibitory polypeptide receptor knockout mice. Proceedings of the National Academy of Sciences of the U S A. 1999;96(26):14843–14847. doi: 10.1073/pnas.96.26.14843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Lange W.J., Halabi C.M., Beyer A.M., Sigmund C.D. Germ line activation of the Tie2 and SMMHC promoters causes noncell-specific deletion of floxed alleles. Physiological Genomics. 2008;35(1):1–4. doi: 10.1152/physiolgenomics.90284.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koehler J.A., Baggio L.L., Yusta B., Longuet C., Rowland K.J., Cao X. GLP-1R agonists promote normal and neoplastic intestinal growth through mechanisms requiring Fgf7. Cell Metabolism. 2015;21(3):379–391. doi: 10.1016/j.cmet.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 30.Mulvihill E.E., Varin E.M., Gladanac B., Campbell J.E., Ussher J.R., Baggio L.L. Cellular sites and mechanisms linking reduction of dipeptidyl peptidase-4 activity to control of incretin hormone action and glucose homeostasis. Cell Metabolism. 2017;25(1):152–165. doi: 10.1016/j.cmet.2016.10.007. [DOI] [PubMed] [Google Scholar]
- 31.Pajcini K.V., Speck N.A., Pear W.S. Notch signaling in mammalian hematopoietic stem cells. Leukemia. 2011;25(10):1525–1532. doi: 10.1038/leu.2011.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tang Y., Harrington A., Yang X., Friesel R.E., Liaw L. The contribution of the Tie2+ lineage to primitive and definitive hematopoietic cells. Genesis. 2010;48(9):563–567. doi: 10.1002/dvg.20654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pujadas G., Drucker D.J. Vascular biology of glucagon receptor superfamily peptides: mechanistic and clinical relevance. Endocrine Reviews. 2016;37(6):554–583. doi: 10.1210/er.2016-1078. [DOI] [PubMed] [Google Scholar]
- 34.Randall T.D., Weissman I.L. Phenotypic and functional changes induced at the clonal level in hematopoietic stem cells after 5-fluorouracil treatment. Blood. 1997;89(10):3596–3606. [PubMed] [Google Scholar]
- 35.Herman A.C., Monlish D.A., Romine M.P., Bhatt S.T., Zippel S., Schuettpelz L.G. Systemic TLR2 agonist exposure regulates hematopoietic stem cells via cell-autonomous and cell-non-autonomous mechanisms. Blood Cancer Journal. 2016;6e437 doi: 10.1038/bcj.2016.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lamont B.J., Drucker D.J. Differential anti-diabetic efficacy of incretin agonists vs. DPP-4 inhibition in high fat fed mice. Diabetes. 2008;57(1):190–198. doi: 10.2337/db07-1202. [DOI] [PubMed] [Google Scholar]
- 37.Ussher J.R., Campbell J.E., Mulvihill E.E., Baggio L.L., Bates H.E., McLean B.A. Inactivation of the glucose-dependent insulinotropic polypeptide receptor improves outcomes following experimental myocardial infarction. Cell Metabolism. 2018;27(2):450–460. doi: 10.1016/j.cmet.2017.11.003. [DOI] [PubMed] [Google Scholar]
- 38.Mantelmacher F.D., Zvibel I., Cohen K., Epshtein A., Pasmanik-Chor M., Vogl T. GIP regulates inflammation and body weight by restraining myeloid-cell-derived S100A8/A9. Nature Metabolism. 2019;1(1):58–69. doi: 10.1038/s42255-018-0001-z. [DOI] [PubMed] [Google Scholar]
- 39.Jordan S., Tung N., Casanova-Acebes M., Chang C., Cantoni C., Zhang D. Dietary intake regulates the circulating inflammatory monocyte pool. Cell. 2019;178(5):1102–1114. doi: 10.1016/j.cell.2019.07.050. e1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Collins N., Han S.J., Enamorado M., Link V.M., Huang B., Moseman E.A. The bone marrow protects and optimizes immunological memory during dietary restriction. Cell. 2019;178(5):1088–1101. doi: 10.1016/j.cell.2019.07.049. e1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nagai M., Noguchi R., Takahashi D., Morikawa T., Koshida K., Komiyama S. Fasting-refeeding impacts immune cell dynamics and mucosal immune responses. Cell. 2019;178(5):1072–1087 e1014. doi: 10.1016/j.cell.2019.07.047. [DOI] [PubMed] [Google Scholar]
- 42.van den Berg S.M., Seijkens T.T., Kusters P.J., Beckers L., den Toom M., Smeets E. Diet-induced obesity in mice diminishes hematopoietic stem and progenitor cells in the bone marrow. The FASEB Journal. 2016;30(5):1779–1788. doi: 10.1096/fj.201500175. [DOI] [PubMed] [Google Scholar]
- 43.Christ A., Gunther P., Lauterbach M.A.R., Duewell P., Biswas D., Pelka K. Western diet triggers NLRP3-dependent innate immune reprogramming. Cell. 2018;172(1–2):162–175. doi: 10.1016/j.cell.2017.12.013. e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Galic S., Fullerton M.D., Schertzer J.D., Sikkema S., Marcinko K., Walkley C.R. Hematopoietic AMPK beta1 reduces mouse adipose tissue macrophage inflammation and insulin resistance in obesity. Journal of Clinical Investigation. 2011;121(12):4903–4915. doi: 10.1172/JCI58577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nicholls H.T., Kowalski G., Kennedy D.J., Risis S., Zaffino L.A., Watson N. Hematopoietic cell-restricted deletion of CD36 reduces high-fat diet-induced macrophage infiltration and improves insulin signaling in adipose tissue. Diabetes. 2011;60(4):1100–1110. doi: 10.2337/db10-1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Saberi M., Woods N.B., de Luca C., Schenk S., Lu J.C., Bandyopadhyay G. Hematopoietic cell-specific deletion of toll-like receptor 4 ameliorates hepatic and adipose tissue insulin resistance in high-fat-fed mice. Cell Metabolism. 2009;10(5):419–429. doi: 10.1016/j.cmet.2009.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu A., Chen M., Kumar R., Stefanovic-Racic M., O'Doherty R.M., Ding Y. Bone marrow lympho-myeloid malfunction in obesity requires precursor cell-autonomous TLR4. Nature Communications. 2018;9(1):708. doi: 10.1038/s41467-018-03145-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singer K., DelProposto J., Morris D.L., Zamarron B., Mergian T., Maley N. Diet-induced obesity promotes myelopoiesis in hematopoietic stem cells. Mol Metab. 2014;3(6):664–675. doi: 10.1016/j.molmet.2014.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yan H., Baldridge M.T., King K.Y. Hematopoiesis and the bacterial microbiome. Blood. 2018;132(6):559–564. doi: 10.1182/blood-2018-02-832519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Griffin C., Eter L., Lanzetta N., Abrishami S., Varghese M., McKernan K. TLR4, TRIF, and MyD88 are essential for myelopoiesis and CD11c(+) adipose tissue macrophage production in obese mice. Journal of Biological Chemistry. 2018;293(23):8775–8786. doi: 10.1074/jbc.RA117.001526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Razolli D.S., Moraes J.C., Morari J., Moura R.F., Vinolo M.A., Velloso L.A. TLR4 expression in bone marrow-derived cells is both necessary and sufficient to produce the insulin resistance phenotype in diet-induced obesity. Endocrinology. 2015;156(1):103–113. doi: 10.1210/en.2014-1552. [DOI] [PubMed] [Google Scholar]
- 52.Creutzfeldt W., Ebert R., Willms B., Frerichs H., Brown J.C. Gastric inhibitory polypeptide (GIP) and insulin in obesity: increased response to stimulation and defective feedback control of serum levels. Diabetologia. 1978;14(1):15–24. doi: 10.1007/BF00429703. [DOI] [PubMed] [Google Scholar]
- 53.Salera M., Giacomoni P., Pironi L., Cornia G., Capelli M., Marini A. Gastric inhibitory polypeptide release after oral glucose: relationship to glucose intolerance, diabetes mellitus, and obesity. Journal of Clinical Endocrinology & Metabolism. 1982;55(2):329–336. doi: 10.1210/jcem-55-2-329. [DOI] [PubMed] [Google Scholar]
- 54.Hampton S.M., Kwasowski P., Tan K., Morgan L.M., Marks V. Effect of pretreatment with a high fat diet on the gastric inhibitory polypeptide and insulin responses to oral triolein and glucose in rats. Diabetologia. 1983;24(4):278–281. doi: 10.1007/BF00282713. [DOI] [PubMed] [Google Scholar]
- 55.Katiraei S., Hoving L.R., van Beek L., Mohamedhoesein S., Carlotti F., van Diepen J.A. BMT decreases HFD-induced weight gain associated with decreased preadipocyte number and insulin secretion. PloS One. 2017;12(4) doi: 10.1371/journal.pone.0175524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Frias J.P., Nauck M.A., Van J., Benson C., Bray R., Cui X. Efficacy and tolerability of tirzepatide, a dual glucose-dependent insulinotropic peptide and glucagon-like peptide-1 receptor agonist in patients with type 2 diabetes: a 12-week, randomized, double-blind, placebo-controlled study to evaluate different dose-escalation regimens. Diabetes, Obesity and Metabolism. 2020 doi: 10.1111/dom.13979. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.