

Abstract
Proteases are fundamental to successful parasitism, including for the schistosome flatworm parasite, which causes the disease schistosomiasis in 200 million people worldwide. The proteasome is receiving attention as a potential drug target for treatment of a variety of infectious parasitic diseases, but has been understudied in the schistosome. Adult Schistosoma mansoni was incubated with 1 μM of the proteasome inhibitors bortezomib, carfilzomib and MG132. After 24 h, bortezomib and carfilzomib decreased worm motility by more than 85%, endogenous proteasome activity by >75%, and, after 72 h, increased caspase activity by >4.5-fold. The association between the engagement of the proteasome target, and the phenotypic and biochemical effects recorded, encouraged the chromatographic enrichment of the S. mansoni proteasome (Sm20S). Activity assays with fluorogenic proteasome substrates revealed that Sm20S contains caspase-type (β1), trypsin-type (β2) and chymotrypsin-type (β5) activities. Sm20S was screened with 11 peptide epoxyketone inhibitors derived from the marine natural product, carmaphycin B. Analog 17 was 27.4-fold less cytotoxic to HepG2 cells than carmaphycin B and showed equal potency for the β5 subunits of Sm20S, human constitutive proteasome and human immunoproteasome. However, this analog was 13.2-fold more potent at targeting Sm20S β2 compared to the equivalent subunits of the human enzymes. Furthermore, 1 μM of 17 decreased both worm motility and endogenous Sm20S activity by more than 90% after 24 h. We provide direct evidence of the proteasome’s importance to schistosome viability and identify a lead for which future studies will aim to improve the potency, selectivity and safety.
Keywords: Proteasome, Schistosoma, schistosomiasis, bortezomib, carfilzomib, carmaphycin, MG132
Graphical Abstract
Among the ‘neglected’ tropical diseases1, schistosomiasis, caused by the Schistosoma blood fluke, infects approximately 200 million worldwide, principally in sub-Saharan Africa2,3. Parasite eggs trapped in the viscera induce an inflammatory response that progresses to tissue fibrosis and portal vein hypertension or occlusion (intestinal schistosomiasis caused by S. mansoni and S. japonicum) or hydronephrosis and the possibility of squamous bladder cancer (urinary schistosomiasis caused by S. haematobium)4. The disease is most often chronic and painful, decreasing school attendance and worker productivity, and undermining economic development5,6. Treatment and control rely on the expanding use of just one drug, praziquantel (PZQ)6. PZQ is administered orally as a single-dose, has few side effects, and is active against the three main schistosome species. However, the drug is rarely curative7 and has a number of pharmacological and pharmaceutical weaknesses, including poorer efficacy against developing schistosomes compared to adult worms8,9. This potentially increases the risk for resistance by exposing parasites to sub-curative doses10. There is a clear need to identify novel drugs and drug targets to treat schistosomiasis.
Schistosome proteolytic enzymes (proteases) facilitate invasion of the mammalian host and digestion of host proteins, and are implicated in modulating the host’s physiology and immune response11–14. The anti-parasitic activities of protease inhibitors against this flatworm in culture and in vertebrate animal infection models have been demonstrated15,16. One protease system that has received little attention as a potential drug target for schistosomiasis is the proteasome. The proteasome is a multi-subunit protein complex that regulates normal protein turnover and degradation of misfolded proteins17. In eukaryotes, the 26S proteasome comprises one core particle (20S) flanked by two regulatory particles (19S). Proteins destined for degradation are covalently linked to one or more ubiquitin chains on exposed lysine residues. The regulatory particles remove the ubiquitin chains and unfold the substrate proteins, which are then threaded into the barrel-shaped proteolytic core and hydrolyzed into short peptide sequences with the released ubiquitin tags being recycled18. The core of the proteasome is formed by two stacked rings of seven β subunits sandwiched between two rings of seven α subunits. Three of the β subunits have catalytic activity. In humans, the constitutive 20S proteasome (c20S) is expressed in most cell types whereas immune cells such as antigen-presenting cells, also express an immunoproteasome 20S (i20S) that has alternative catalytic subunits19.
Recent studies have established the validity of the proteasome as a drug target for infectious organisms20, including parasites21–27. For example, a single intravenous dose of a rationally designed proteasome inhibitor reduced Plasmodium chabaudi burden by >95% in a mouse model of malaria infection without apparent toxicity28. Also, Khare et al. reported the clearance of Trypanosoma and Leishmania infections in rodents following oral dosing with an allosteric proteasome inhibitor29.
Indirect evidence points to the schistosome proteasome as a possible drug target. Mice exposed to Schistosoma mansoni infectious larvae (cercariae) that had been pre-treated with a high concentration (50 μM) of the proteasome inhibitor, MG132, had a 96% reduction in parasite establishment compared to control-treated mice30. Also, exposure in vitro of S. mansoni post-infective larvae (schistosomula) to short-interfering RNAs targeting the proteasome regulatory protein, SmRPN11, resulted in a 78% decrease in parasite viability31. Though encouraging, little biochemical characterization has been performed on the enzyme, and no studies have assessed whether chemical inhibition of the proteasome in the parasite is associated with decreased viability. An understanding of both would be required to consider initiating a program to develop and optimize schistosome-specific proteasome inhibitors.
In this study, we measured the phenotypic and biochemical responses of adult S. mansoni to proteasome inhibitors of diverse chemical warheads and evaluated whether those responses are associated with on-target engagement of the proteasome. We then enriched the parasite proteasome from parasite lysates and identified potent irreversible inhibitors from a natural product, peptide epoxyketone library. One analog preferentially targets the parasite proteasome over the human proteasome and demonstrated decreased mammalian cytotoxicity.
Results
Human proteasome inhibitors induce phenotypic changes, including decreased motility, in Schistosoma mansoni.
MG132 is a reversible, tripeptide-aldehyde proteasome inhibitor that induces apoptosis in mammalian cells32. Previous studies have shown that this inhibitor causes global gene expression changes when co-incubated in culture with S. mansoni33. Using a quantitative motility assay, known as WormAssay34, incubation of the mixed-sex adults with 1 μM MG132 for up to 24 h did not alter motility compared to DMSO-exposed controls (Figure 1A). When viewed under the microscope at 72 h, male and female worms were paired, were attached to the well surface by their oral and/or ventral suckers, and appeared normal in shape, movement and color (Supplementary Video 1) compared to DMSO controls (Supplementary Video 2). In contrast, treatment with 1 μM of bortezomib, a covalent, reversible, dipeptide boronic acid inhibitor of human proteasome35, significantly decreased parasite motility by 45% and 87% after 6 h and 24 h, respectively (Figure 1A). By 72 h, the parasites were relatively motionless, unable to extend or flex their bodies and darkened, and had detached from the surface of the well (Supplementary Video 3). Bortezomib can react with serine hydrolases18, which leaves open the question of whether the phenotypic responses recorded were in fact due to proteasome inhibition alone. Therefore, we also tested the tetrapeptide epoxyketone inhibitor carfilzomib, an agent used to treat multiple myeloma36 that exclusively reacts with the active site threonine residue found only in the proteasome18. At 1 μM, carfilzomib decreased worm motility by 47% and 89% after 6 h and 24 h, respectively. By 72 h, phenotypic changes were similar to those observed for bortezomib.
Fig 1. Proteasome inhibitors decrease worm motility and induce caspase activity.

Experiments employed 24 well plates with 5–6 adult S. mansoni pairs per well. A. At each time point, WormAssay recordings were taken over 45 – 60 sec and the mean motility value per well noted. For bortezomib (BTZ) and carfilzomib (CFZ), the decrease in motility at 6 and 24 h relative to the 1 h time point is significant (*p < 0.05; ** p < 0.005) by the Student’s t-test. B. Protein lysates were generated from S. mansoni worms following 24 h treatment with vehicle (Veh; 0.0001% DMSO) or 1 μM proteasome inhibitors. Caspase activity was quantified using the fluorogenic peptide substrate, Ac-DEVD-AFC. Caspase activity (relative fluorescent units/min/μg) was significantly (*p < 0.05) increased in lysates from worms treated with bortezomib and carfilzomib, but not MG132, compared to vehicle control. Data shown are from experiments performed in triplicate: in one experiment, duplicate wells were used per treatment, whereas in the second and third experiments, quadruplicate wells were used, thus yielding a total of 10 wells per treatment.
In mammalian cells, inhibition of the proteasome induces apoptosis including increased caspase activity37,38. Previously, our group measured an increase in caspase activity that was associated with death of S. mansoni adult worms following in vitro exposure to statin drugs39. Therefore, we prepared lysates from worms that had been incubated for 24 h in the presence of 1 μM MG132, bortezomib or carfilzomib and measured activity of the executioner caspases of apoptosis with the fluorogenic substrate, Ac-DEVD-AFC. Caspase activity in worms exposed to MG132 was not significantly different from the DMSO control (Figure 1B). In contrast, bortezomib and carfilzomib induced a significant increase in caspase activity (7- and 4.5-fold, respectively) compared to the DMSO control. Thus, the consistency noted between the appearance of phenotypic changes, decreased motility and increased caspase activity suggested that the schistosome proteasome was being engaged by bortezomib and carfilzomib, a hypothesis that encouraged further biochemical characterization of the putative enzyme target.
Human proteasome inhibitors engage the proteasome in S. mansoni.
In the human proteasome, the subunits responsible for peptide hydrolysis are β1, β2 and β5, and each subunit has a distinct substrate preference40. The β1 subunit generally cleaves on the carboxyl terminal side of acidic residues whereas the β2 subunit preferentially cleaves after basic residues. Most proteasome inhibitors, including MG132, bortezomib and carfilzomib, target the β5 subunit. This subunit generally cleaves on the carboxyl terminal side of hydrophobic residues41 and is typically monitored using the fluorogenic substrate, Succinyl-Leu-Leu-Val-Tyr-7-amido-4-methylcoumarin (Suc-LLVY-AMC). Under the same assay conditions, Suc-LLVY-AMC may also be cleaved by cysteine proteases such as calpains42 that are present in S. mansoni43. To eliminate the cysteine protease activity, protein lysates from mixed-sex adult worms were prepared in a buffer containing 100 μM of E-64, a broad acting cysteine protease inhibitor. When the worm lysate was then assayed with Suc-LLVY-AMC, proteolytic activity was reduced by 92%, 99% and 100% in the presence 1 μM of MG132, bortezomib or carfilzomib, respectively (Figure 2A). As each of these inhibitors have different chemical reactive groups and all are predicated to target the β5 proteasome subunit, we can conclude that all of the proteolytic activity being measured with Suc-LLVY-AMC in parasite extracts is due to proteasome activity.
Figure 2. S. mansoni proteasome β5 subunit activity and its inhibition in living parasites.
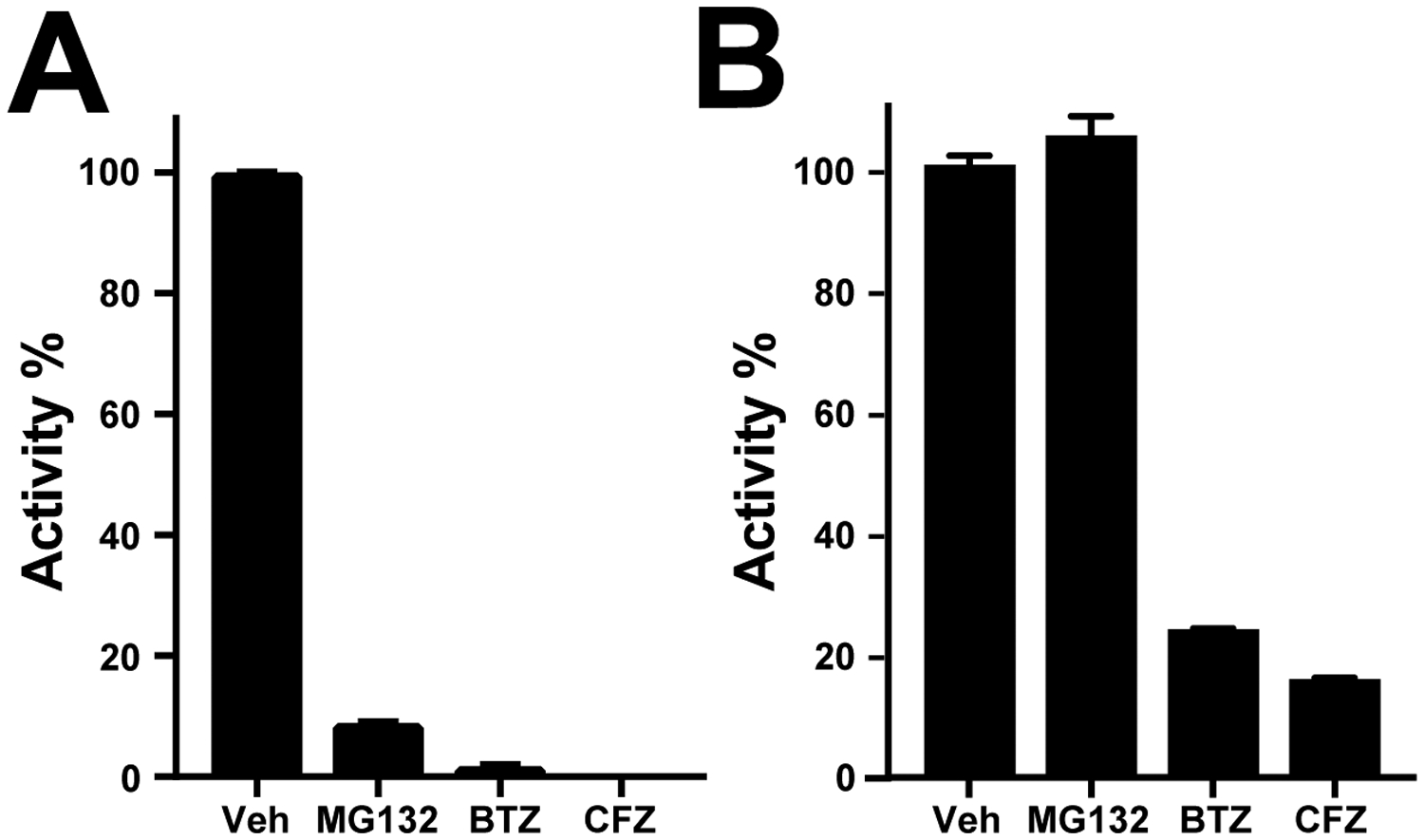
A. Proteolytic activity in S. mansoni adult lysate was measured with the fluorogenic substrate, Suc-LLVY-AMC. Proteasome inhibitors (1 μM) were added to the lysate and the rate of substrate cleavage quantified and compared to lysate treated with 0.0001% DMSO-. B. Adult S. mansoni worms (5–6 pairs per well) were incubated with 1 μM of MG132, BTZ or CFZ for 24 h. After extensive washing, protein extracts were prepared and assayed with Suc-LLVY-AMC. Proteasome activity was normalized to protein concentration across all groups and activity expressed as a percentage relative to vehicle (Veh) control (0.05% DMSO). Data are presented as the mean ± SD from triplicate experiments each performed in duplicate.
Having confirmed the assay conditions to specifically measure parasite β5 activity, we evaluated whether MG132, bortezomib and carfilzomib engage the proteasome in living S. mansoni. Parasites were exposed to 1 μM of each inhibitor for 24 h. After extensive washing, worm extracts were prepared and assayed for proteasome activity using Suc-LLVY-AMC. Activity was normalized to protein concentration. Bortezomib and carfilzomib decreased proteasome activity by 76% and 84%, respectively (Figure 2B). In contrast, MG132 had no effect on proteasome activity. These data demonstrate that bortezomib and carfilzomib but not MG132, engage the intended target in living worms. Furthermore, their inhibition of the target is associated with the major phenotypic and biochemical changes described above.
Enrichment of active proteasome from S. mansoni lysates.
To enrich for the S. mansoni proteasome (Sm20S), we developed a protocol using two chromatographic separation steps. Adult mixed-sex worm lysates were fractionated via gel filtration chromatography followed by DEAE-anion exchange column chromatography and activity was monitored using Suc-LLVY-AMC (Figure 3A). Attempts to enrich the proteasome further using cation exchange or hydrophobic interaction chromatography did not yield active enzyme in the eluted fractions. Therefore, fractions from the DEAE-anion exchange column containing the most abundant activity were pooled and resolved by SDS-PAGE followed by silver staining (Figure 3B).
Figure 3. Enrichment of Sm20S from S. mansoni protein extracts.
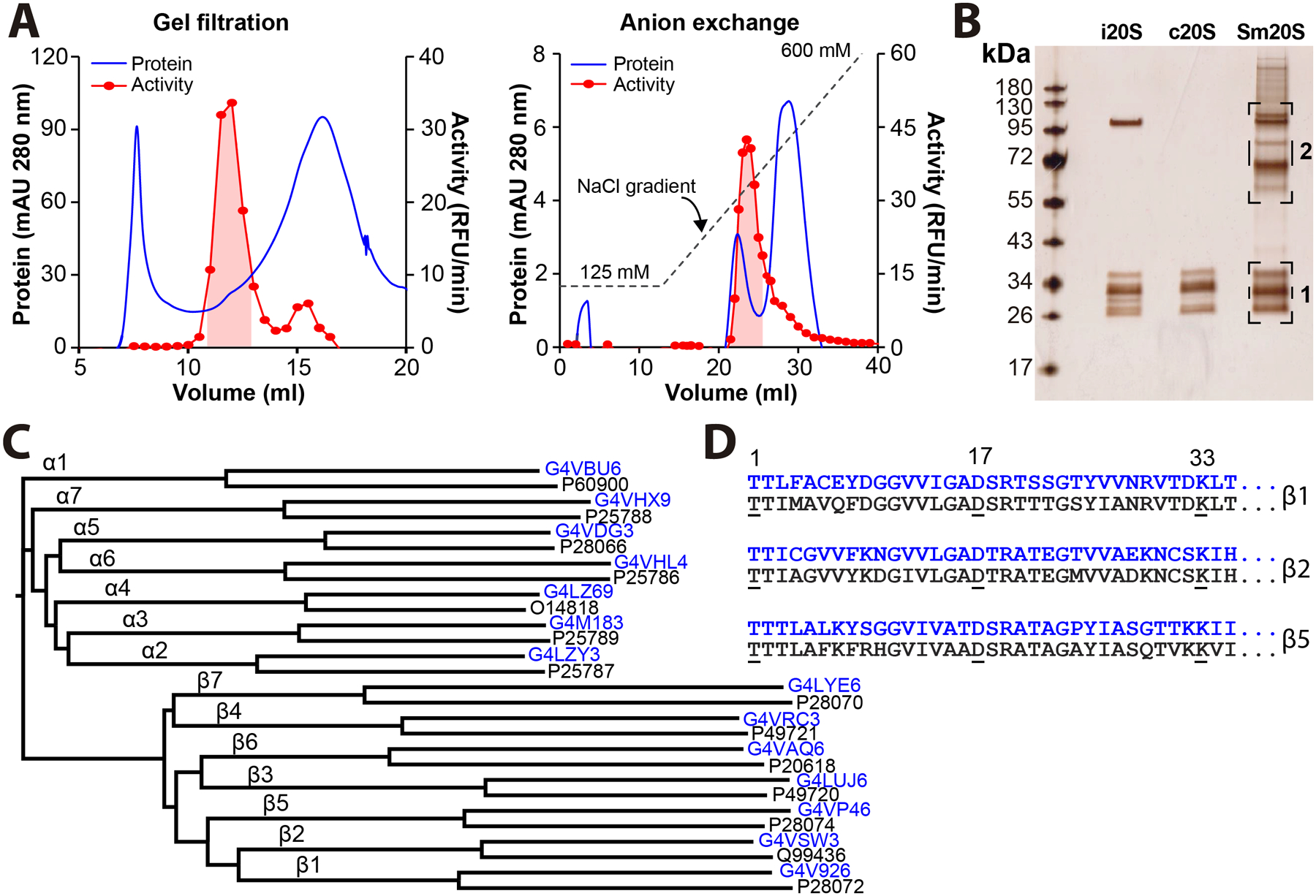
A. A two-step protocol involving gel filtration followed by DEAE-anion exchange chromatography was employed to enrich for Sm20S from S. mansoni lysates. Protein was separated on a Superose-6 10/300 gel filtration column and fractions with activity against Suc-LLVY-AMC were pooled (red shaded area). The pooled fractions were separated on a DEAE column and active fractions were again pooled (red shaded area). B. Pooled fractions containing Sm20S were resolved by SDS-PAGE beside the human immunoproteasome (i20S) and constitutive proteasome (c20S). Following silver staining, two gel slices, indicated in brackets, were excised for proteomic analysis. C. Dendogram showing the protein sequence homology between the human c20S proteasome subunits and those identified for Sm20S in gel slice 1. The UniProt identifiers for the Sm20S and c20S subunits are indicated in blue and black, respectively. D. Protein sequence alignment of residues 1 to 35 of the mature β1, β2 and β5 subunits from Sm20S (blue) and c20S (black). The amino acids involved in catalytic activity of c20S are underlined and conserved in Sm20S.
Two gel slices were excised for mass spectrometry analysis (Figure 3B). Gel Slice 1 contained numerous proteins in the range of 25–40 kDa that correspond in size to the α and β subunits of the human constitutive proteasome (c20S) and the human immunoproteasome (i20S). Fourteen putative 20S subunits were identified and each of these shared more than 46% sequence homology with a single S. mansoni proteasome subunit. The close alignment of proteasome subunits allowed us to assign a number to each S. mansoni subunit using the human proteasome nomenclature (Figure 3C). The active site triad consisting of Thr-1, Asp-17 and Lys-33 found in the c20S β1, β2 and β5 subunits44 is conserved within the subunits of the Sm20S counterparts indicating that the parasite proteasome has three catalytically active β subunits (Figure 3D).
Gel slice 2 contained many high molecular weight proteins in the range of 60–110 kDa. Among a number of diverse proteins identified by mass spectrometry, six regulatory subunits of the S. mansoni proteasome were found (Supplementary File 1). Taken together, these data confirm that the core 20S proteasome and its associated regulatory subunits are enriched from adult worm lysates using a two-step column chromatography protocol.
Subunit characterization of the S. mansoni proteasome.
Sm20S was enriched from S. mansoni lysates by monitoring for β5 chymotrypsin-type activity using the substrate, Suc-LLVY-AMC. The concentration of this enzyme complex was determined by active site titration with carfilzomib. Using 5 nM of Sm20S, we then compared the β5 activity with an equi-molar concentration of c20S and i20S. The human enzymes cleaved Suc-LLVY-AMC at 35- to 40-fold faster rates than Sm20S. Proteomic data indicated that the caspase-type (β1: G4V926) and trypsin-type (β2: G4VSW3) catalytic subunits were also present and likely enzymatically active due to the presence of a conserved nucleophilic threonine residue (Figure 3D). Therefore, we predicted that the enriched Sm20S could cleave the human β1 caspase-type substrate, z-LLE-AMC, and the human β2 trypsin-type substrate, z-LRR-AMC45. Using an equal concentration of Sm20S, c20S and i20S, we compared their activities with the β1 and β2 fluorogenic substrates and demonstrated that the parasite proteasome can hydrolyze these peptides albeit at a slower rate than the human proteasomes (Figure 4A).
Figure 4. Substrate and inhibitor profiling of the catalytic subunits of Sm20S.
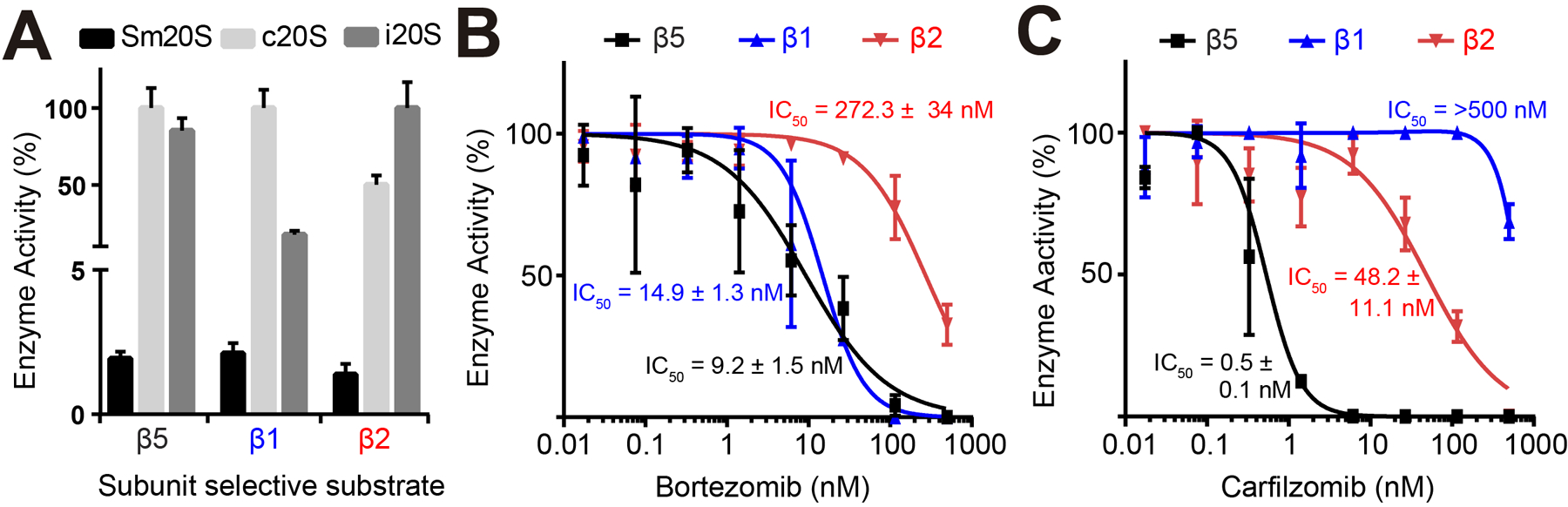
A. Activity assays with subunit-selective proteasome substrates for β1 (z-LLE-AMC), β2 (z-LRR-AMC) and β5 (Suc-LLVY-AMC). B. Determination of bortezomib potency with Sm20S using subunit-selective substrates. C. Determination of carfilzomib potency with Sm20S using the same substrates. All assays were performed in triplicate. IC50 values ± SD are shown.
We next measured the potency of bortezomib and carfilzomib against the three catalytic subunits of Sm20S. Bortezomib inhibited the β1, β2 and β5 subunits with IC50 values of 15 nM, 272 nM and 9 nM, respectively (Figure 4B). In contrast, carfilzomib was more than 33-fold less potent against the β1 subunit (IC50 = >500 nM) when compared to bortezomib, however, it was 5.6-fold more potent against the β2 subunit (IC50 = 48 nM) and 18-fold more potent against the β5 subunit (IC50 = 0.5 nM) (Figure 4C). Although bortezomib and carfilzomib are potent inhibitors of Sm20S activity and have different selectivity for each of the subunits, they are generally cytotoxic to mammalian cells, which limits their potential use as anthelmintics. Therefore, we evaluated other proteasome inhibitors that have been developed for non-human targets.
The carmaphycin B scaffold as a platform to develop novel Sm20S inhibitors.
Carmaphycin B is a marine peptide isolated from cyanobacterium comprising of an N-hexanoyl amino terminus capping group, Val, Met sulfone and Leu at P1, P2 and P3, respectively, and a carboxyl terminal epoxyketone group46 (Figure 5A). Carmaphycin B inhibited Sm20S β2 and β5 activity with IC50 values of 0.6 nM and 9.8 nM, respectively, and did not inhibit β1 activity up to 500 nM (Supplementary Figure 1). This compound also potently inhibited the β5 subunits of c20S and i20S with IC50 values of less than 5 nM, and also inhibited the β2 subunit of c20S (IC50 = 12 nM) and i20S (IC50 = 62 nM) (Figure 5A). Like Sm20S, no inhibition of c20S and i20S β1 activity was detected up to 500 nM.
Figure 5. A screen of carmaphycin B and 11 analogs identifies a compound that preferentially inhibits Sm20S with decreased cytotoxicity to HepG2 cells.
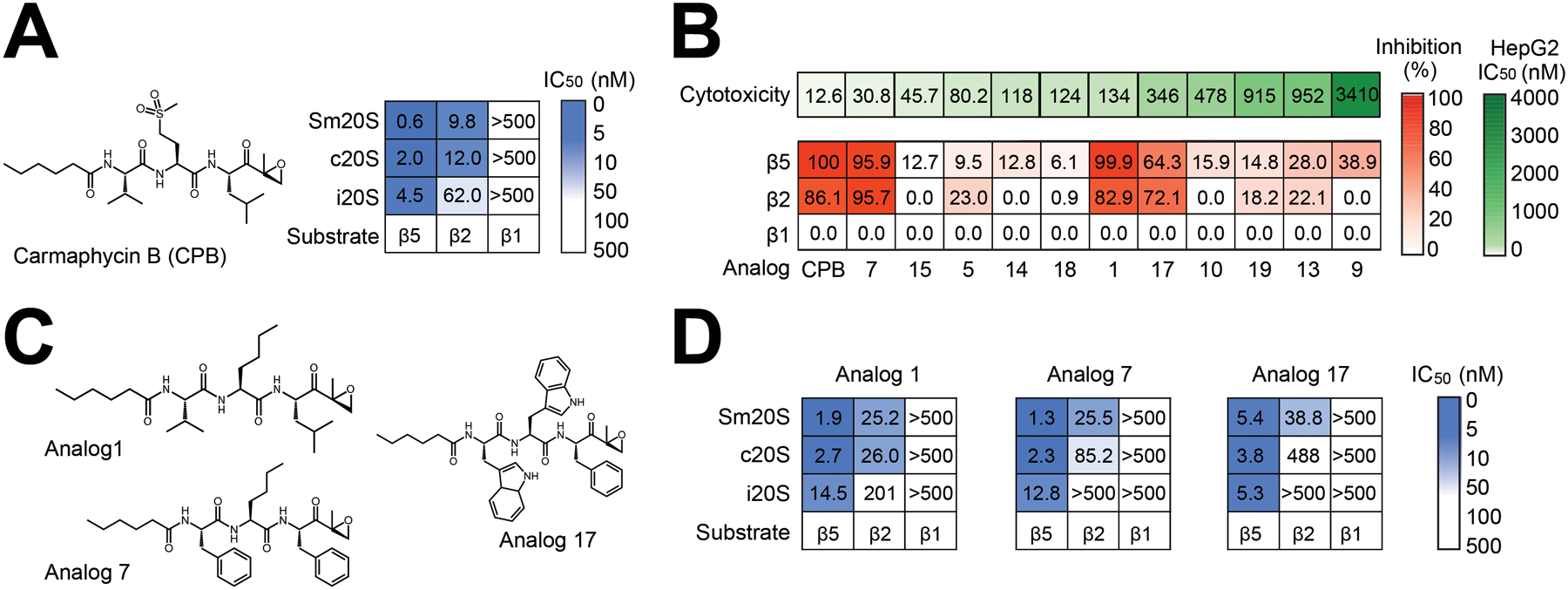
A. Structure of carmaphycin B and IC50 values for this compound against each catalytic subunit of Sm20S, c20S and i20S B. Inhibition of Sm20S by 11 carmaphycin B analogs (red heatmap) that have lower HepG2 cytotoxicity than the parent molecule (toxicity data and numbering system taken from LaMonte et al47). Activity of Sm20S was quantified in the presence of 10 nM inhibitor for β5 and β1 assays and 50 nM inhibitor for β2 assays. Mean % inhibition values relative to vehicle (DMSO) are shown . C. Structures of carmaphycin B analogs 1, 7 and 17. D. Mean IC50 values for carmaphycin B analogs 1, 7 and 17 for each catalytic subunit of the Sm20S, c20S and i20S.
Carmaphycin B is toxic to HepG2 cells with a 24 h EC50 value of 12.6 nM47 and as part of a campaign to target the Plasmodium falciparum proteasome we developed 20 carmaphycin B analogs of which 11 were less cytotoxic (EC50 values of 31 nM to 3410 nM)47. Sm20S was pre-incubated with either 10 nM (for β5 & β1 assays) or 50 nM (for β2 assays) of these compounds and the percentage inhibition was calculated. Like the parent compound, none of these carmaphycin B analogs inhibited the β1 subunit and only compounds 1, 7, and 17 decreased the β5 and β2 subunit activity by more than 60% (Figure 5B). The remaining compounds weakly inhibited the β5 subunit and/or the β2 subunit.
The IC50 values of compounds 1, 7 and 17 were calculated because they strongly inhibited both the β5 and β2 subunits of Sm20S. In parallel, selectivity was determined by comparing these IC50 values with those generated for c20S and i20S. Analog 1 differs from carmaphycin B by substitution of a P2 norleucine (Nle) for a Met sulfone (Figure 5C) that results in an 11-fold reduction in HepG2 cytotoxicity (134 nM vs. 12.6 nM) (Figure 5B). This compound showed no difference in potency between c20S and Sm20S for all of the subunits although it was at least 5-fold less potent towards the i20S β5 and β2 subunits (Figure 5D). Analog 7 also contains Nle in P2, but has substitutions of Phe for Leu in P1 and Phe for Val in P3 (Figure 5C). This compound is had a similar potency for the β5 subunits of c20S and Sm20S and was at least 5-fold less potent against i20S β5. However, for the Sm20S β2 subunit, 7 was 3.3-fold and at least 19-fold more potent relative to c20S and i20S, respectively (Figure 5D). Finally, analog 17, which comprises Trp-Trp-Phe at the P3–P2–P1 positions, was similar to the other analogs in not providing specificity for the β5 subunit over the human proteasomes. However, for the β2 subunit, analog 17 was at least 12.5-fold more potent for Sm20S compared to c20S and i20S (Figure 5D). Of the three analogs evaluated in detail, 7 and 17 showed the greatest selectivity for the parasite proteasome and therefore were further characterized in vitro with adult S. mansoni.
Activity of carmaphycin analogs against S. mansoni in vitro.
The anti-schistosomal activity of carmaphycin B and analogs 7 and 17, was evaluated at 1 μM using adult mixed-sex S. mansoni in culture. These compounds were compared to analogs 5 and 10 which were weaker inhibitors of Sm20S subunits (Supplementary Figure 2). Bortezomib was included as a positive control. After 24 h, bortezomib, carmaphycin B, 7 and 17 decreased worm movement by at least 80% (Figure 6A) and generated the same deleterious phenotypic changes previously noted for bortezomib. These changes were associated with inhibition of the Sm20S β5 activity by 80% or more (Figure 6B). In contrast, analogs 5 and 10 decreased motility by just 22% and 40%, respectively, and no obvious phenotypic changes were observed. Also, β5 subunit activity was reduced by 52% and 70% for analogs 5 and 10, respectively. Overall, there was strong correlation between proteasome inhibition and worm motility (Pearson correlation coefficient = 0.922), indicating that engagement of the schistosome proteasome target is responsible for the phenotypic responses measured and observed. When comparing analogs 7 and 17, the latter had a stronger phenotypic performance (Figure 6A), increased selectivity for Sm20S over c20S and i20S (Figure 5D) and a lower HepG2 cytotoxicity (Figure 5B). Therefore 17 was identified as the best starting point for the future design of more potent and selective inhibitors of Sm20S.
Figure 6. Decreased parasite motility in the presence of carmaphycin B and its analogs correlates with inhibition of the proteasome.
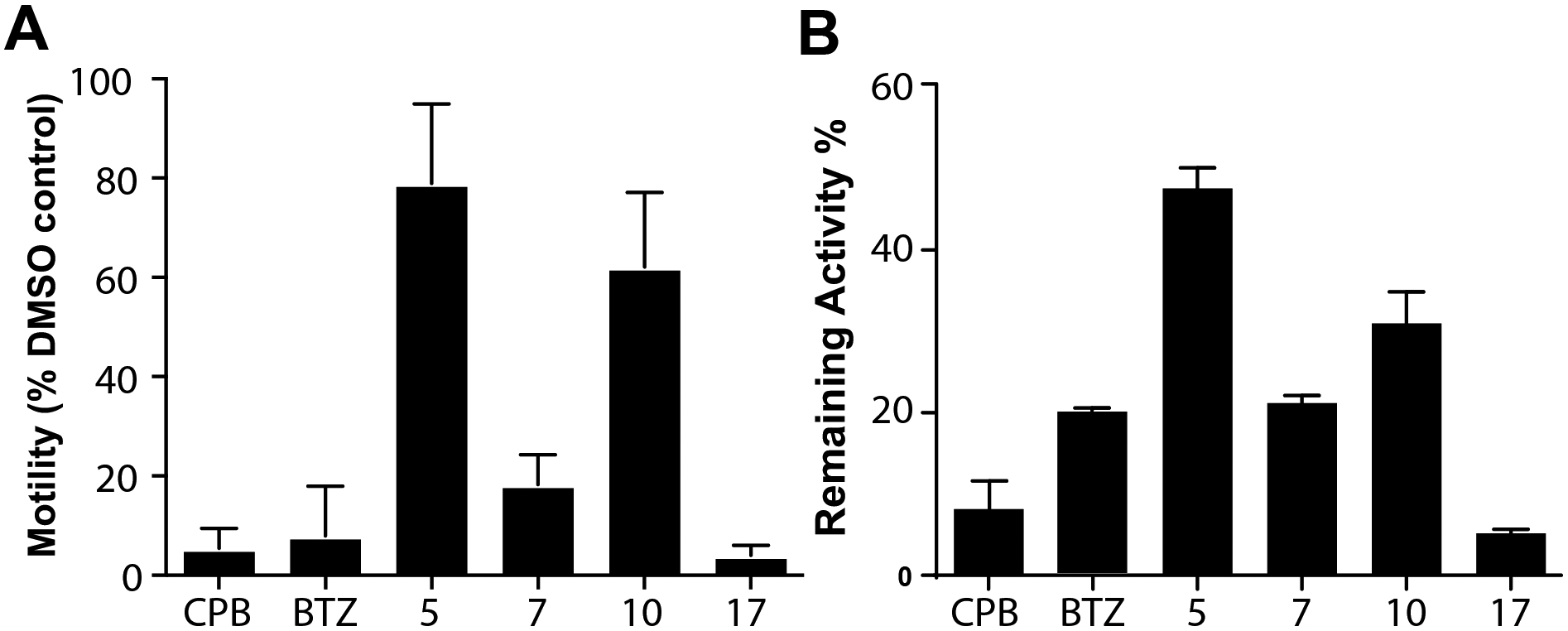
A. Experiments employed 24 well plates with 5–6 adult S. mansoni pairs per well and 1 μM of carmaphycin B (CPB), bortezomib (BTZ) and the indicated analogs. After 24 h, WormAssay recordings were taken over 45–60 sec and the mean motility value per well noted. B. The same worms used in A were extensively washed and protein extracts prepared in the presence of 100 μM E-64 for assay with Suc-LLVY-AMC. Proteasome activity was normalized to protein concentration across all groups and activity expressed as the percentage remaining activity relative to DMSO controls. Data are presented as means ± SD from three experiments each in triplicate.
Discussion
In spite of the strong conservation of the ubiquitin-proteasome system among eukaryotes, recent reports demonstrate that potent proteasome inhibitors can be designed with selectivity for pathogens over the human host. In particular, much effort has be made to target the Plasmodium proteasome with epoxyketone21,47, vinyl sulfone22,28, boronic acid27 and ethylenediamine24 inhibitors. From these studies, compounds with more than 380-fold selectivity for Plasmodium falciparum over mammalian cells have emerged24,47. Further, a large-scale library screening and medicinal chemistry effort led to the development of non-competitive proteasome inhibitors that selectively target the kinetoplastid parasites responsible for leishmaniasis, Chagas disease, and sleeping sickness29. Using these studies as a precedent, we anticipated that the proteasome in S. mansoni could also be a target for the development of potent and selective proteasome inhibitors.
Previous genetic31 and chemical evidence30,33 suggest that the S. mansoni proteasome is important for the establishment of infection in the mammalian host. Also, gene expression studies have communicated the biological importance of the schistosome ubiquitin proteasome system during oxidative stress and heat shock conditions48 and for egg development and morphology49. Here, we focused on adult parasites as they are ultimately responsible for disease morbidity via the eggs they produce. We showed that 1 μM of either bortezomib or carfilzomib inhibited proteasome activity in parasite lysates and caused pronounced phenotypic and biochemical changes to adult S. mansoni. MG132 also inhibited proteasome activity in the lysate but did not cause any phenotypic or biochemical changes to the worm. The lack of efficacy of MG132 may be due to poor penetration of the compound or the rapid dissociation half-life of peptide-aldehydes from the proteasome, compared to the much slower peptide-boronate (bortezomib) and the irreversible peptide epoxyketone (carfilzomib). The lack of potency may also be due to the aldehyde group being oxidized rapidly within the worm, as has been shown in mouse studies50. In addition, MG132 reacts with papain-fold cysteine proteases51,52 and these enzymes are abundantly expressed in S. mansoni adult worms13. Therefore, cysteine proteases may sequester much of the MG132 thereby reducing the inhibitor concentration that can interact with the proteasome.
The lack of efficacy of MG132 (at 1 μM) in our studies contrasts with the significant reduction in the survival of S. mansoni larvae in mice following exposure to a 50-fold higher concentration of the inhibitor30. Thus, the decrease in parasite survival after exposure to 50 μM MG132 may have been due to engaging other targets, and/or to the dual inhibition of the proteasome and papain-type cysteine proteases as was noted in studies with P. falciparum52. Using bortezomib and carfilzomib, we observed a clear association between the phenotypic changes in adult S. mansoni, the induction of caspase activity and engagement of the proteasome target. This encouraged us to isolate and biochemically characterize Sm20S.
Sm20S was enriched from adult worm lysates and all subunits were identified by proteomic analysis, and annotated based on sequencing similarity with the human proteasome. The protein sequence analysis revealed that the Sm20S complex has all of the amino acids associated with the catalytic triads of human β1, β2 and β5 subunits44. The schistosome enzyme complex readily hydrolyzed the standard β1 caspase-type, β2 trypsin-type and β5 chymotrypsin-type substrates albeit at lower rates to the human constitutive and immune proteasomes. These substrate specificity and turnover data indicate that there may be subtle differences between the S. mansoni and human proteasome that could be exploited for inhibitor development.
We found that bortezomib inhibits Sm20S β1 and β5 subunits with equal potency, whereas the epoxyketone inhibitor, carfilzomib, is approximately 100-fold and 1000-fold more selective for the Sm20S β5 subunit over the β2 and β1 subunits, respectively. These data reveal that the similar phenotypic and biochemical changes in adult S. mansoni that were recorded in the presence of bortezomib and carfilzomib, involve targeting of the β5 subunit. It has yet to be determined whether inhibition of β5 in combination with β1 or β2 is required to cause the phenotypic changes observed. Peptide vinyl sulfone inhibitors that were developed to target the Plasmodium proteasome inhibited both the β5 and β2 subunits22,28 but inhibition of β2 alone was insufficient to kill the parasite24. Also, peptide epoxyketone inhibitors that only targeted the P. falciparum β5 subunit47, killed the parasite as effectively as the β5/β2 vinyl sulfone inhibitors. None of the compounds tested to date preferentially target S. mansoni β1 or β2 and, thus, it is unclear whether targeting these subunits, alone or in combination, is sufficient for the anti-schistosomal activity measured.
In order to discover proteasome inhibitors that engage Sm20S but have reduced toxicity to host mammalian cells, we screened 11 peptide-epoxyketone analogs derived from the marine natural product, carmaphycin B46. These analogs are part of a library that previously yielded a tri-peptide epoxyketone inhibitor with a 379-fold selectivity for P. falciparum over HepG2 cells47. The lead anti-malarial compound, 18, consisted of D-valine, L-norleucine and L-leucine in the P3, P2 and P1 positions, respectively. This compound weakly inhibited Sm20S β5 and β2, thereby indicating clear differences in the subunit specificities of these parasite proteasomes. Our proteasome activity screen identified three hit compounds (1, 7 and 17) all of which contain L-amino acids in the P3 position. In the activity assays, analogs 7 and 17 targeted Sm20S β5 and β2 subunits and had selectivity for Sm20S over c20S in the β2 subunit. Of these two compounds, 17 was more selective for Sm20S with the least HepG2 toxicity. Our future studies will aim to improve the attributes of 17 regarding potency, selectivity and safety. With this in mind, the recent development of orally bioavailable peptide boronic acid53 proteasome inhibitors to treat multiple myeloma is encouraging and consistent with the target product profile for new anti-schistosomals (and other anthelmintics) that demands orally active drugs54,55.
Conclusion
We demonstrate that exposure of adult S. mansoni to FDA-approved proteasome inhibitors causes significant phenotypic and biochemical changes. As these anti-cancer drugs are broadly cytotoxic to mammalian cells, we screened 11 proteasome inhibitors that are derived from the marine natural product, carmaphycin B and are less cytotoxic. We identified a hit molecule, analog 17, with improved selectivity for the S. mansoni proteasome over the human proteasome. We show that treatment of adult S. mansoni with this inhibitor causes the same phenotypic changes as the FDA-approved drugs and that these changes are directly due to engagement of the target enzyme.
Experimental Section
Maintenance of S. mansoni
S. mansoni was maintained in male Golden Syrian hamsters and Biomphalaria glabrata (NMRI isolate) vector snails. Hamsters were infected at four weeks of age with 600–800 S. mansoni cercariae. At seven weeks post-infection, animals were euthanized by a lethal injection of sodium pentobarbital solution (Fatal-Plus; Vortech Pharmaceuticals Ltd) and adult worms harvested by reverse perfusion of the hepatic portal system56–59. Maintenance and handling of small vertebrate animals were carried out in accordance with a protocol approved by the Institutional Animal Care and Use Committee (IACUC) of the University of California San Diego. UCSD-IACUC derives its authority for these activities from the United States Public Health Service (PHS) Policy on Humane Care and Use of Laboratory Animals, and the Animal Welfare Act and Regulations (AWAR).
Quantitative phenotypic effects of proteasome inhibitors on adult S. mansoni in vitro.
After perfusion, parasites were washed extensively in Basch medium 16959,60.Worms were distributed into 24w plates at five-six adult pairs per well and allowed to acclimatize overnight at 37 °C and 5% CO2 in 1 ml Basch medium containing 4% heat-inactivated FBS. The next morning, a final concentration of 1 μM of proteasome inhibitor (dissolved in DMSO) was added and the volume made up to 2 ml (final DMSO concentration was 0.05%). After 1, 6 and 24 h, worm motility was measured using WormAssay34,61,62. WormAssay comprises a commodity digital movie camera connected to an Apple Mac computer that operates an open source software application to automatically process multiple wells in 6-, 12- or 24-well plate geometries. Using the “Consensus Voting Luminance Difference” option, the application detects the aggregate changes in the occupation and vacancy of pixels between frames that are due to worm movement. Parasites were also observed using an inverted microscope (Zeiss AxioVert A1, 1.25X objective) at the same time points and up to 72 h, and changes in motility, shape, color and pairing status recorded using a constrained nomenclature59,61,63,64.
Evaluation of caspase activation in worms exposed to proteasome inhibitors.
After recording the phenotypic responses in the presence of proteasome inhibitors, worms were transferred to 1.5 mL tubes and washed with 0.5 ml of ice-cold PBS every 30 min for 2 h. Worms were allowed to settle and subsequently homogenized in 130 μL of 40 mM PIPES, 20% sucrose, 200 mM NaCl, 2% CHAPS, 2 mM EDTA, pH 7.2, using a pestle connected to a hand-held motor. The lysate was centrifuged for 10 min at 15,000 ×g and 4 °C, and the supernatant was recovered for analysis. The protein concentration was quantified using the Pierce BCA kit (Thermo Scientific). Prior to assaying for caspase activity, the supernatant was pre-incubated at 37 °C for 1 h in the above buffer containing 10 mM DTT. Likewise, 200 μM of the caspase substrate, Ac-Asp-Glu-Val-Asp-7-amino-4-trifluoromethylcoumarin (Ac-DEVD-AFC; CPC Scientific) was pre-incubated at 37 °C for 10 min in the same buffer containing 10 mM DTT. Equal volumes of both the supernatant and substrate were then mixed in white-walled 96-well flat-bottomed plates and activity was measured using a CLARIOstar monochromator microplate reader (BMG Labtech) at excitation and emission wavelengths of 400 nm and 505 nm, respectively. Activity was quantified as relative fluorescence units (RFU) min−1 μg−1 protein.
Inhibition of proteasome activity in S. mansoni extracts.
To prepare S. mansoni lysates, mixed-sex adult worms were homogenized using a pestle connected to a handheld motor in 1.5-ml tubes containing 100 mM Tris-HCl, 100 μM E-64, pH 7.5. Lysates were then centrifuged for 15 min at 15,000 ×g and 4 °C. The upper lipid layer was discarded and the protein concentration of the remaining supernatant was quantified using the Pierce BCA kit. Supernatants were incubated with inhibitors for 1 h at room temperature and activity was assayed in 50 μl total volume reactions containing 7.5 μg of protein and 25 μM of the substrate, Succinyl-Leu-Leu-Val-Tyr-7-amido-4-methyl coumarin (Suc-LLVY-AMC), in 20 mM Tris-HCl, 0.02% SDS, pH 7.5, in black, round-bottomed 96w plates. The final concentration of inhibitors used was 10 μM E-64 and 1 μM for the proteasome inhibitors. Controls contained 0.0001% DMSO. The release of the AMC fluorophore was monitored at 24°C in a Synergy HTX multi-mode reader (BioTek Instruments, Winooski, VT) with excitation and emission wavelengths set to 340 nm and 460 nm, respectively. Activity was quantified as RFU min−1 μg−1 protein and normalized to the DMSO control reactions.
Evaluation of proteasome inhibition in worms exposed to proteasome inhibitors.
After evaluation of the phenotypic responses in the presence of proteasome inhibitors, worms were transferred to 1.5 ml tubes and washed with 0.5 ml of ice-cold PBS every 30 min for 2 h. Protein lysates were prepared from homogenized worms as described above and assays were performed with 25 μM Suc-LLVY-AMC in 20 mM Tris–HCl, 0.02% SDS, pH 7.5. Activity was expressed as RFU min−1 μg−1 and normalized to the DMSO control reactions.
Enrichment of the S. mansoni proteasome.
The starting material for enrichment of the S. mansoni proteasome (Sm20S) was approximately 500 adult worm pairs that had been extensively washed in PBS and frozen at −80 °C. Worms were homogenized using a pestle connected to a motor in 1.5-ml tubes in ice cold 100 mM Tris-HCl, 100 μM E-64, pH 7.5. Lysates were centrifuged for 15 min at 10,000 ×g and 4 °C, and the supernatant subjected to two ammonium sulfate precipitation steps, each for 30 min on ice, at 30 and 60% saturation, respectively. After centrifugation for 15 min at 10,000 ×g and 4 °C, the supernatant was discarded and the precipitated proteins were re-suspended in 5 mL of ice cold 100 mM Tris-HCl, pH 7.5. Protein was concentrated and buffer exchange into 20 mM Tris-HCl, 10% glycerol, 0.125 M NaCl, pH 7.5 using a 100 kDa centrifugal filter unit (Amicon). Approximately 5 mg of soluble protein was loaded onto a Superose 6 10/300 gel filtration column under the control of an ÄKTA Pure instrument (GE Healthcare Life Sciences). Protein was eluted using 20 mM Tris-HCl, 10% glycerol, 0.125 M NaCl, pH 7.5. Fractions of 0.5 mL were collected, and assayed for proteasome activity with 25 μM Suc-LLVY-AMC in assay buffer (20 mM Tris, 0.02% SDS, pH 7.5). The AMC fluorophore release was monitored at 24°C with excitation and emission wavelengths of 340 nm and 460 nm, respectively, using a Synergy HTX multi-mode reader (BioTek Instruments).
Fractions containing proteasome activity were pooled and loaded onto a 5 ml anion exchange HiTrap DEAE FF column. Protein was eluted using 20 mM Tris-HCl, 10% glycerol, pH 7.5, and a linear gradient from 0.125 to 0.6 M NaCl. Fractions (1.5 ml) were collected and assayed with Suc-LLVY-AMC. Fractions containing proteasome activity were pooled and an aliquot was denatured and loaded into a 4–12% Bis-Tris Plus gel (Thermo Scientific) next to 70–130 ng of the human constitutive proteasome, c20S (Boston Biochem). The gel was stained using the Pierce silver stain kit (Thermo Scientific) and the concentration of Sm20S was estimated by comparing band density with the highly purified human c20S proteasome using Image J software.
Gel slice excision and proteomics.
Silver stained gel bands were excised with a clean scalpel, diced into 1 mm cubes and placed into 0.6 mL tubes (Axygen). An in-gel digestion was performed as described by Shevchenko and colleagues65. Following digestion and extraction, the peptides were desalted with C18 LTS tips (Rainin) and dried in a vacuum centrifuge. Peptides were resuspended in 12 μL 0.1% TFA and 5 μL was analyzed on a Q Exactive Mass Spectrometer (Thermo Scientific). Peptides were separated by reverse phase chromatography on an UltiMate 3000 HPLC system (Thermo Scientific) equipped with an in-house packed C18 column (1.7 μm ACQUITY UPLC BEH, 75 μm × 25 cm, heated to 65 °C) at a flow rate of 300 nL/min over a 76 min linear gradient from 5% Solvent A to 25% Solvent B, where solvent A and B correspond to 0.1% formic acid in water and 0.1% formic acid in acetonitrile, respectively. Survey scans were recorded at a resolution of 35,000 at 200 m/z over a range of 350–1500 m/z with the automatic gain control (AGC) at 1 × 106 and a maxIT of 300 ms. MS/MS was performed in data-dependent acquisition mode with higher-energy collision dissociation (HCD) fragmentation (28 normalized collision energy) on the 20 most intense precursor ions at a resolution of 17,500 at 200 m/z with the AGC at 5 × 106 and a maxIT of 50 ms.
Data analysis was performed using the PEAKS 8.5 software (Bioinformatics Solutions Inc.). RAW files were searched against the S. mansoni (Puerto Rican isolate; UP000008854) reference proteome from UniProt that contains 11,723 entries with the following settings: 15 ppm precursor mass tolerance, 0.01 Da MS/MS mass tolerance and an enzyme digest of trypsin with a maximum of three missed cleavages. Carbamidomethylation of cysteines was included as a fixed modification, and oxidation of methionine and protein N-terminal acetylation as variable modifications. The false discovery rate for protein identification was set to 1%. All mass spectrometry data can be accessed here: ftp://massive.ucsd.edu/MSV000083021. Annotation of the putative S. mansoni proteasome subunits with the α and β subunit nomenclature was performed by comparing sequence alignments against the human 20S proteasome subunits using BLAST 2.6.0+ command line tools.
Proteasome activity and inhibition assays.
Proteasome activity assays were performed using 5 nM of Sm20S, c20S or i20S and 25 μM of z-LLE-AMC or Suc-LLVY-AMC in assay buffer consisting of 20 mM Tris-HCl, pH 7.5, 0.02% SDS. For single concentration inhibition assays, Sm20S was preincubated with 10 nM inhibitor for 30 min at 4°C in assay buffer before the addition of z-LLE-AMC or Suc-LLVY-AMC. Assays with z-LRR-AMC were performed with 25 nM of enzyme in 20 mM Tris-HCl, pH 7.5. SDS was omitted from the z-LRR-AMC assays as it is was found to interfere with this substrate. For single concentration inhibition assays, Sm20S was preincubated with 50 nM inhibitor for 30 min at 4°C in assay buffer before the addition of z-LRR-AMC. The rate of AMC release was determined from 60 to 120 min and normalized to a DMSO control. For dose-response inhibition assays, an eight-point serial dilution of inhibitors in DMSO was acoustically transferred to 384 well assay plates using an ATS Gen 4 Plus (Biosero, San Diego, CA). After pre-incubation of enzyme with inhibitor for 30 min at 4°C, substrate was added to the assay plate using a Biomek FXP workstation equipped with a 96 multichannel pipetting head (Beckman Coulter, Brea, CA). IC50 values were calculated in GraphPad Prism 6 by normalizing activity to DMSO controls and interpolating the data using the log(inhibitor) – variable slope curve fitting algorithm.
Supplementary Material
Supplementary File 1 Total list of protein IDs with two unique peptides from mass spectrometry data.
Supplementary Figure 1 Dose response curves for Sm20S, c20S and i20S with carmaphycin B and analogs 1, 7 and 17.
Supplementary Figure 2 Dose response curves for Sm20S, c20S and i20S with carmaphycin B analogs 5 and 10.
Supplementary Video 1 Three-day incubation of S. mansoni adult worms with MG132.
Supplementary Video 2 Three-day incubation of S. mansoni adult worms with DMSO control.
Supplementary Video 3 Three-day incubation of S. mansoni adult worms with Bortezomib.
Acknowledgements
The research reported was supported by R21AI133393 to AJO and R21AI126296 to CRC. S. mansoni-infected hamsters were in part provided by the NIH-NIAID Schistosomiasis Resource Center for distribution through BEI Resources, NIH-NIAID Contract HHSN272201700014I. ZJ is supported by the UCSD Chancellor’s Research Excellence Scholarship and CL was supported by NIH T32MH019934 (awarded to D. Jeste, UC San Diego). JA is grateful to the University of Jordan and the Scientific Research Support Fund for financial support.
Abbreviations Used
- Sm20S
Schistosoma mansoni 20S proteasome
- c20S
human constitutive 20S proteasome
- i20S
human immune 20S proteasome
- AMC
7-amido-4-methylcoumarin
- AFC
7-Amino-4-trifluoromethylcoumarin
- Suc
Succinyl
- Ac
Acetyl
- DEAE
Diethylaminoethyl cellulose
- SDS-PAGE
Sodium Dodecyl Sulfate–Polyacrylamide gel electrophoresis
- RFU
Relative Fluorescent Units
References
- (1).Hotez PJ, Brindley PJ, Bethony JM, King CH, Pearce EJ, and Jacobson J (2008) Helminth infections: the great neglected tropical diseases. J. Clin. Invest 118, 1311–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Steinmann P, Keiser J, Bos R, Tanner M, and Utzinger J (2006) Schistosomiasis and water resources development: systematic review, meta-analysis, and estimates of people at risk. Lancet. Infect. Dis 6, 411–25. [DOI] [PubMed] [Google Scholar]
- (3).Hotez PJ (2018) Human Parasitology and Parasitic Diseases: Heading Towards 2050. Adv. Parasitol 100, 29–38. [DOI] [PubMed] [Google Scholar]
- (4).Gryseels B, Polman K, Clerinx J, and Kestens L (2006) Human schistosomiasis. Lancet (London, England) 368, 1106–1118. [DOI] [PubMed] [Google Scholar]
- (5).King CH, and Dangerfield-Cha M (2008) The unacknowledged impact of chronic schistosomiasis. Chronic Illn. 4, 65–79. [DOI] [PubMed] [Google Scholar]
- (6).Caffrey CR (2015) Schistosomiasis and its treatment. Future Med. Chem 7, 675–6. [DOI] [PubMed] [Google Scholar]
- (7).Doenhoff M, Cioli D, and Utzinger J (2008) Praziquantel: mechanisms of action, resistance and new derivatives for schistosomiasis. Curr Opin Infect Dis. 21, 659–67. [DOI] [PubMed] [Google Scholar]
- (8).Sabah AA, Fletcher C, Webbe G, and Doenhoff MJ (1986) Schistosoma mansoni: chemotherapy of infections of different ages. Exp. Parasitol 61, 294–303. [DOI] [PubMed] [Google Scholar]
- (9).Gönnert R, and Andrews P (1977) Praziquantel, a new board-spectrum antischistosomal agent. Z. Parasitenkd 52, 129–50. [DOI] [PubMed] [Google Scholar]
- (10).Doenhoff M, and Pica-Mattoccia L (2006) Praziquantel for the treatment of schistosomiasis: its use for control in areas with endemic disease and prospects for drug resistance. Expert Rev Anti Infect Ther. 4, 199–210. [DOI] [PubMed] [Google Scholar]
- (11).de Oliveira Fraga LA, Lamb EW, Moreno EC, Chatterjee M, Dvořák J, Delcroix M, Sajid M, Caffrey CR, and Davies SJ (2010) Rapid induction of IgE responses to a worm cysteine protease during murine pre-patent schistosome infection. BMC Immunol. 11, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Dvořák J, Fajtová P, Ulrychová L, Leontovyč A, Rojo-Arreola L, Suzuki BM, Horn M, Mareš M, Craik CS, Caffrey CR, and O’Donoghue AJ (2016) Excretion/secretion products from Schistosoma mansoni adults, eggs and schistosomula have unique peptidase specificity profiles. Biochimie 122, 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Caffrey CR, Goupil L, Rebello KM, Dalton JP, and Smith D (2018) Cysteine proteases as digestive enzymes in parasitic helminths. PLoS Negl. Trop. Dis 12, e0005840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Grote A, Caffrey CR, Rebello KM, Smith D, Dalton JP, and Lustigman S (2018) Cysteine proteases during larval migration and development of helminths in their final host. PLoS Negl. Trop. Dis 12, e0005919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Jílková A, Rezácová P, Lepsík M, Horn M, Váchová J, Fanfrlík J, Brynda J, McKerrow JH, Caffrey CR, and Mares M (2011) Structural basis for inhibition of cathepsin B drug target from the human blood fluke, Schistosoma mansoni. J. Biol. Chem 286, 35770–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Abdulla M-H, Lim K-C, Sajid M, McKerrow JH, and Caffrey CR (2007) Schistosomiasis mansoni: novel chemotherapy using a cysteine protease inhibitor. PLoS Med. 4, e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bhattacharyya S, Yu H, Mim C, and Matouschek A (2014) Regulated protein turnover: snapshots of the proteasome in action. Nat. Rev. Mol. Cell Biol 15, 122–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Kisselev AF, and Goldberg AL (2001) Proteasome inhibitors: from research tools to drug candidates. Chem. Biol 8, 739–758. [DOI] [PubMed] [Google Scholar]
- (19).Ferrington DA, and Gregerson DS (2012) Immunoproteasomes : Structure , Function , and Antigen Presentation. Prog. Mol. Biol. Transl. Sci 109, 75–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Bibo-Verdugo B, Jiang Z, Caffrey CR, and O’Donoghue AJ (2017) Targeting proteasomes in infectious organisms to combat disease. FEBS J. 284, 1503–1517. [DOI] [PubMed] [Google Scholar]
- (21).Li H, Ponder EL, Verdoes M, Asbjornsdottir KH, Deu E, Edgington LE, Lee JT, Kirk CJ, Demo SD, Williamson KC, and Bogyo M (2012) Validation of the proteasome as a therapeutic target in plasmodium using an epoxyketone inhibitor with parasite-specific toxicity. Chem. Biol 19, 1535–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Yoo E, Stokes BH, de Jong H, Vanaerschot M, Kumar T, Lawrence N, Njoroge M, Garcia A, Van der Westhuyzen R, Momper JD, Ng CL, Fidock DA, and Bogyo M (2018) Defining the Determinants of Specificity of Plasmodium Proteasome Inhibitors. J. Am. Chem. Soc 140, 11424–11437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Jalovecka M, Hartmann D, Miyamoto Y, Eckmann L, Hajdusek O, O’Donoghue AJ, and Sojka D (2018) Validation of Babesia proteasome as a drug target. Int. J. Parasitol. Drugs drug Resist 8, 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kirkman LA, Zhan W, Visone J, Dziedziech A, Singh PK, Fan H, Tong X, Bruzual I, Hara R, Kawasaki M, Imaeda T, Okamoto R, Sato K, Michino M, Alvaro EF, Guiang LF, Sanz L, Mota DJ, Govindasamy K, Wang R, Ling Y, Tumwebaze PK, Sukenick G, Shi L, Vendome J, Bhanot P, Rosenthal PJ, Aso K, Foley MA, Cooper RA, Kafsack B, Doggett JS, Nathan CF, and Lin G (2018) Antimalarial proteasome inhibitor reveals collateral sensitivity from intersubunit interactions and fitness cost of resistance. Proc. Natl. Acad. Sci. U. S. A 115, E6863–E6870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Zhan W, Visone J, Ouellette T, Harris J, Wang R, Zhang H, Singh P, Ginn J, Sukenick G, Wong T, Okoro J, Scales R, Tumwebaze P, Rosenthal P, Kafsack B, Cooper R, Meinke P, Kirkman L, and Lin G (2019) Improvement of asparagine ethylenediamines as anti-malarial Plasmodium-selective proteasome inhibitors. J. Med. Chem 10.1021/acs.jmedchem.9b00363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Wyllie S, Brand S, Thomas M, De Rycker M, Chung C, Pena I, Bingham RP, Bueren-Calabuig JA, Cantizani J, Cebrian D, Craggs PD, Ferguson L, Goswami P, Hobrath J, Howe J, Jeacock L, Ko E-J, Korczynska J, MacLean L, Manthri S, Martinez MS, Mata-Cantero L, Moniz S, Nühs A, Osuna-Cabello M, Pinto E, Riley J, Robinson S, Rowland P, Simeons FRC, Shishikura Y, Spinks D, Stojanovski L, Thomas J, Thompson S, Viayna Gaza E, Wall RJ, Zuccotto F, Horn D, Ferguson MAJ, Fairlamb AH, Fiandor JM, Martin J, Gray DW, Miles TJ, Gilbert IH, Read KD, Marco M, and Wyatt PG (2019) Preclinical candidate for the treatment of visceral leishmaniasis that acts through proteasome inhibition. Proc. Natl. Acad. Sci 116, 9318–9323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Xie SC, Gillett DL, Spillman NJ, Tsu C, Luth MR, Ottilie S, Duffy S, Gould AE, Hales P, Seager BA, Charron CL, Bruzzese F, Yang X, Zhao X, Huang SC, Hutton CA, Burrows JN, Winzeler EA, Avery VM, Dick LR, and Tilley L (2018) Target Validation and Identification of Novel Boronate Inhibitors of the Plasmodium falciparum Proteasome. J. Med. Chem 61, 10053–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Li H, O’Donoghue AJ, van der Linden WA, Xie SC, Yoo E, Foe IT, Tilley L, Craik CS, da Fonseca PCA, and Bogyo M (2016) Structure- and function-based design of Plasmodium-selective proteasome inhibitors. Nature 530, 233–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Khare S, Nagle AS, Biggart A, Lai YH, Liang F, Davis LC, Barnes SW, Mathison CJN, Myburgh E, Gao M-Y, Gillespie JR, Liu X, Tan JL, Stinson M, Rivera IC, Ballard J, Yeh V, Groessl T, Federe G, Koh HXY, Venable JD, Bursulaya B, Shapiro M, Mishra PK, Spraggon G, Brock A, Mottram JC, Buckner FS, Rao SPS, Wen BG, Walker JR, Tuntland T, Molteni V, Glynne RJ, and Supek F (2016) Proteasome inhibition for treatment of leishmaniasis, Chagas disease and sleeping sickness. Nature 537, 1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Guerra-Sá R, Castro-Borges W, Evangelista EA, Kettelhut IC, and Rodrigues V (2005) Schistosoma mansoni: Functional proteasomes are required for development in the vertebrate host. Exp. Parasitol 109, 228–236. [DOI] [PubMed] [Google Scholar]
- (31).Nabhan JF, El-Shehabi F, Patocka N, and Ribeiro P (2007) The 26S proteasome in Schistosoma mansoni: Bioinformatics analysis, developmental expression, and RNA interference (RNAi) studies. Exp. Parasitol 117, 337–347. [DOI] [PubMed] [Google Scholar]
- (32).He Q, Huang Y, and Sheikh MS (2004) Proteasome inhibitor MG132 upregulates death receptor 5 and cooperates with Apo2L/TRAIL to induce apoptosis in Bax-proficient and -deficient cells. Oncogene 23, 2554–2558. [DOI] [PubMed] [Google Scholar]
- (33).Morais ER, Oliveira KC, de Paula RG, Ornelas AMM, Moreira ÉBC, Badoco FR, Magalhães LG, Verjovski-Almeida S, and Rodrigues V (2017) Effects of the proteasome inhibitor MG-132 on the 1 parasite Schistosoma mansoni. PLoS One 12, e0184192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Marcellino C, Gut J, Lim KC, Singh R, McKerrow J, and Sakanari J (2012) WormAssay: a novel computer application for whole-plate motion-based screening of macroscopic parasites. PLoS Negl. Trop. Dis 6, e1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Adams J, Behnke M, Chen S, Cruickshank AA, Dick LR, Grenier L, Klunder JM, Ma YT, Plamondon L, and Stein RL (1998) Potent and selective inhibitors of the proteasome: Dipeptidyl boronic acids. Bioorganic Med. Chem. Lett 8, 333–338. [DOI] [PubMed] [Google Scholar]
- (36).Demo SD, Kirk CJ, Aujay MA, Buchholz TJ, Dajee M, Ho MN, Jiang J, Laidig GJ, Lewis ER, Parlati F, Shenk KD, Smyth MS, Sun CM, Vallone MK, Woo TM, Molineaux CJ, and Bennett MK (2007) Antitumor activity of PR-171, a novel irreversible inhibitor of the proteasome. Cancer Res. 67, 6383–6391. [DOI] [PubMed] [Google Scholar]
- (37).Concannon CG, Koehler BF, Reimertz C, Murphy BM, Bonner C, Thurow N, Ward MW, Villunger A, Strasser A, Kögel D, and Prehn JHM (2007) Apoptosis induced by proteasome inhibition in cancer cells: Predominant role of the p53/PUMA pathway. Oncogene 26, 1681–1692. [DOI] [PubMed] [Google Scholar]
- (38).Imajohohmi S, Kawaguchi T, Sugiyama S, Tanaka K, Omura S, and Kikuchi H (1995) Lactacystin, a specific inhibitor of the proteasome, induces apoptosis in human monoblast U937 cells. Biochem. Biophys. Res. Commun 217, 1070–1077. [DOI] [PubMed] [Google Scholar]
- (39).Rojo-Arreola L, Long T, Asarnow D, Suzuki BM, Singh R, and Caffrey CR (2014) Chemical and genetic validation of the statin drug target to treat the helminth disease, schistosomiasis. PLoS One 9, e87594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Kisselev AF, Garcia-Calvo M, Overkleeft HS, Peterson E, Pennington MW, Ploegh HL, Thornberry NA, and Goldberg AL (2003) The caspase-like sites of proteasomes, their substrate specificity, new inhibitors and substrates, and allosteric interactions with the trypsin-like sites. J. Biol. Chem 278, 35869–35877. [DOI] [PubMed] [Google Scholar]
- (41).Dick TP, Nussbaum AK, Deeg M, Heinemeyer W, Groll M, Schirle M, Keilholz W, Stevanović S, Wolf DH, Huber R, Rammensee HG, and Schild H (1998) Contribution of proteasomal β-subunits to the cleavage of peptide substrates analyzed with yeast mutants. J. Biol. Chem 273, 25637–25646. [DOI] [PubMed] [Google Scholar]
- (42).Giguere CJ, and Schnellmann RG (2008) Limitations of SLLVY-AMC in calpain and proteasome measurements. Biochem. Biophys. Res. Commun 371, 578–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Wang Q, Da’dara AA, and Skelly PJ (2017) The human blood parasite Schistosoma mansoni expresses extracellular tegumental calpains that cleave the blood clotting protein fibronectin. Sci. Rep 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Huber EM, Heinemeyer W, Li X, Arendt CS, Hochstrasser M, and Groll M (2016) A unified mechanism for proteolysis and autocatalytic activation in the 20S proteasome. Nat. Commun 7, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Kisselev AF, Akopian TN, Castillo V, and Goldberg AL (1999) Proteasome active sites allosterically regulate each other, suggesting a cyclical bite-chew mechanism for protein breakdown. Mol. Cell 4, 395–402. [DOI] [PubMed] [Google Scholar]
- (46).Pereira AR, Kale AJ, Fenley AT, Byrum T, Debonsi HM, Gilson MK, Valeriote FA, Moore BS, and Gerwick WH (2012) The Carmaphycins: New Proteasome Inhibitors Exhibiting an α,β-Epoxyketone Warhead from a Marine Cyanobacterium. ChemBioChem 13, 810–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).LaMonte GM, Almaliti J, Bibo-Verdugo B, Keller L, Zou BY, Yang J, Antonova-Koch Y, Orjuela-Sanchez P, Boyle CA, Vigil E, Wang L, M. Goldgof G, Gerwick L, J. O’Donoghue A, Winzeler Elizabeth A. Gerwick WH, and Ottilie S (2017) Development of a Potent Inhibitor of the Plasmodium Proteasome with Reduced Mammalian Toxicity. jounal Med. Chem 60, 6721–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).De Paula RG, De Magalhães Ornelas AM, Morais ER, De Castro Borges W, Natale M, Magalhães LG, and Rodrigues V (2014) Biochemical characterization and role of the proteasome in the oxidative stress response of adult Schistosoma mansoni worms. Parasitol. Res 113, 2887–2897. [DOI] [PubMed] [Google Scholar]
- (49).Mathieson W, Castro-Borges W, and Wilson RA (2011) The proteasome-ubiquitin pathway in the Schistosoma mansoni egg has development- and morphology-specific characteristics. Mol. Biochem. Parasitol 175, 118–125. [DOI] [PubMed] [Google Scholar]
- (50).Lindsten K, Menéndez-Benito V, Masucci MG, and Dantuma NP (2003) A transgenic mouse model of the ubiquitin/proteasome system. Nat. Biotechnol 21, 897–902. [DOI] [PubMed] [Google Scholar]
- (51).Kisselev AF, Van Der Linden W. a., and Overkleeft HS (2012) Proteasome inhibitors: An expanding army attacking a unique target. Chem. Biol 19, 99–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Prasad R, Atul, Kolla VK, Legac J, Singhal N, Navale R, Rosenthal PJ, and Sijwali PS (2013) Blocking Plasmodium falciparum development via dual inhibition of hemoglobin degradation and the ubiquitin proteasome system by MG132. PLoS One 8, e73530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Kupperman E, Lee EC, Cao Y, Bannerman B, Fitzgerald M, Berger A, Yu J, Yang Y, Hales P, Bruzzese F, Liu J, Blank J, Garcia K, Tsu C, Dick L, Fleming P, Yu L, Manfredi M, Rolfe M, and Bolen J (2010) Evaluation of the proteasome inhibitor MLN9708 in preclinical models of human cancer. Cancer Res. 70, 1970–1980. [DOI] [PubMed] [Google Scholar]
- (54).Caffrey CR (2007) Chemotherapy of schistosomiasis: present and future. Curr. Opin. Chem. Biol 11, 433–439. [DOI] [PubMed] [Google Scholar]
- (55).Olliaro P, Seiler J, Kuesel A, Horton J, Clark JN, Don R, and Keiser J (2011) Potential drug development candidates for human soil-transmitted helminthiases. PLoS Negl. Trop. Dis 5, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Duvall RH, and DeWitt WB (1967) An improved perfusion technique for recovering adult schistosomes from laboratory animals. Am. J. Trop. Med. Hyg 16, 483–6. [DOI] [PubMed] [Google Scholar]
- (57).Colley DG, and Wikel SK (1974) Schistosoma mansoni: simplified method for the production of schistosomules. Exp. Parasitol 35, 44–51. [DOI] [PubMed] [Google Scholar]
- (58).Abdulla M-H, Lim K-C, Sajid M, McKerrow JH, and Caffrey CR (2007) Schistosomiasis mansoni: novel chemotherapy using a cysteine protease inhibitor. PLoS Med. 4, e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Abdulla M-H, Ruelas DS, Wolff B, Snedecor J, Lim K-C, Xu F, Renslo AR, Williams J, McKerrow JH, and Caffrey CR (2009) Drug discovery for schistosomiasis: hit and lead compounds identified in a library of known drugs by medium-throughput phenotypic screening. PLoS Negl. Trop. Dis 3, e478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Basch PF, and Humbert R (1981) Cultivation of Schistosoma mansoni in vitro. III. implantation of cultured worms into mouse mesenteric veins. J. Parasitol 67, 191–5. [PubMed] [Google Scholar]
- (61).Long T, Rojo-Arreola L, Shi D, El-Sakkary N, Jarnagin K, Rock F, Meewan M, Rascón AA, Lin L, Cunningham KA, Lemieux GA, Podust L, Abagyan R, Ashrafi K, McKerrow JH, and Caffrey CR (2017) Phenotypic, chemical and functional characterization of cyclic nucleotide phosphodiesterase 4 (PDE4) as a potential anthelmintic drug target. PLoS Negl. Trop. Dis 11, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Weeks JC, Roberts WM, Leasure C, Suzuki BM, Robinson KJ, Currey H, Wangchuk P, Eichenberger RM, Saxton AD, Bird TD, Kraemer BC, Loukas A, Hawdon JM, Caffrey CR, and Liachko NF (2018) Sertraline, Paroxetine, and Chlorpromazine Are Rapidly Acting Anthelmintic Drugs Capable of Clinical Repurposing. Sci. Rep 8, 975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Glaser J, Schurigt U, Suzuki BM, Caffrey CR, and Holzgrabe U (2015) Anti-Schistosomal Activity of Cinnamic Acid Esters: Eugenyl and Thymyl Cinnamate Induce Cytoplasmic Vacuoles and Death in Schistosomula of Schistosoma mansoni. Molecules 20, 10873–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Long T, Neitz RJ, Beasley R, Kalyanaraman C, Suzuki BM, Jacobson MP, Dissous C, McKerrow JH, Drewry DH, Zuercher WJ, Singh R, and Caffrey CR (2016) Structure-Bioactivity Relationship for Benzimidazole Thiophene Inhibitors of Polo-Like Kinase 1 (PLK1), a Potential Drug Target in Schistosoma mansoni. PLoS Negl. Trop. Dis 10, e0004356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Shevchenko A, Tomas H, Havlis J, Olsen JV, and Mann M (2006) In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc 1, 2856–60. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary File 1 Total list of protein IDs with two unique peptides from mass spectrometry data.
Supplementary Figure 1 Dose response curves for Sm20S, c20S and i20S with carmaphycin B and analogs 1, 7 and 17.
Supplementary Figure 2 Dose response curves for Sm20S, c20S and i20S with carmaphycin B analogs 5 and 10.
Supplementary Video 1 Three-day incubation of S. mansoni adult worms with MG132.
Supplementary Video 2 Three-day incubation of S. mansoni adult worms with DMSO control.
Supplementary Video 3 Three-day incubation of S. mansoni adult worms with Bortezomib.