

Abstract
BACE1 (Beta-site Amyloid Precursor Protein (APP) Cleaving Enzyme 1) is a promising therapeutic target for Alzheimer’s Disease (AD). However, efficient expression, purification, and crystallization systems are not well described or detailed in the literature nor are approaches for treatment of enzyme kinetic data for potent inhibitors well described. We therefore developed a platform for expression and purification of BACE1, including protein refolding from E.coli inclusion bodies, in addition to optimizing a reproducible crystallization procedure of BACE1 bound with inhibitors. We also report a detailed approach to the proper analysis of enzyme kinetic data for compounds that exhibit either rapid-equilibrium or tight-binding mechanisms. Our methods allow for the purification of ~15 mg of BACE1 enzyme from 1 L of culture which is higher than reported yields in the current literature. To evaluate the data analysis approach developed here, a well-known potent inhibitor and two of its derivatives were tested, analyzed, and compared. The inhibitory constants (Ki) obtained from the kinetic studies are in agreement with dissociation constants (Kd) that were also determined using isothermal titration calorimetry (ITC) experiments. The X-ray structures of these three compounds in complex with BACE1 were readily obtained and provide important insight into the structure and thermodynamics of the BACE1-inhibitor interactions.
Graphical Abstract
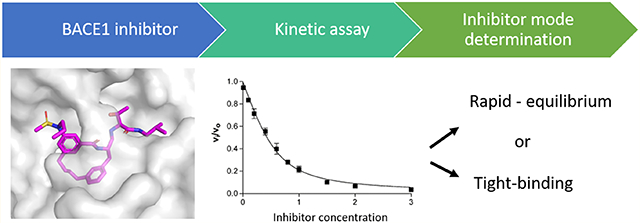
Alzheimer’s disease (AD) is a neurodegenerative disorder and it is the most common cause of dementia. Currently, it affects more than 5 million people in the United States. According to the Alzheimer’s Association, the estimated cost of caregiving for Alzheimer’s patients in the U.S. was around $277 billion in 2018. The cost of AD treatment is estimated to reach $1.1 trillion by 2050.1 Although a vast amount of research has been performed on AD, current treatments are still limited. So far, only symptomatic treatments are available and no disease-modifying therapies are on the market. Thus, effective treatments are greatly needed.
One of the strategies for current drug development for AD is driven by the amyloid hypothesis, which postulates that the accumulation of amyloid-β (Aβ) peptides in the brain of AD patients is a critical factor leading to the AD pathology.2 Based on this hypothesis, elimination of the Aβ peptides from the brain of AD patients could be a potential therapeutic approach. Aβ peptide is one of the proteolysis products from the Amyloid Precursor Protein (APP), resulting from sequential cleavage by two enzymes, β-secretase and γ-secretase BACE1. β-site APP Cleaving Enzyme 1, is an aspartic acid protease that has been established as the major β-secretase through the generation of BACE1 knockout mice.3 The generation of Aβ peptide is completely abolished in BACE1-deficient mice. It has also been reported that overexpression of BACE1 results in an increase in the production of Aβ.4–6 Furthermore, no apparently adverse effects were noted in BACE1-deficient mice,3,7 indicating that BACE1 might be a viable target for the treatment of AD. Several BACE1 inhibitors have been tested in mouse models and have shown promising results in rescuing age-related cognitive decline.8–10 In contrast, BACE1 inhibitors have yet to show any improvement in slowing cognitive decline in patients with advanced Alzheimer’s disease. Currently, a result, few BACE1 small molecular inhibitors are under clinical investigation. Some of the clinical trials on BACE1 inhibitors have been terminated due in part to severe adverse effects.11 In addition, a recently concluded phase 3 clinical trial Verubescestat (MK-8931) did not provide proof-of-concept for the amyloid hypothesis.12 It appears that multiple factors may be responsible for clinical failures including the dose of BACE1 inhibitor administered, the disease stage of the patient, the drug selectivity, and the intervention timeline.
BACE1 is a membrane protein containing 501 amino acids. The protein is organized with a signal sequence (pre), a pro peptide (pro), a catalytic, a transmembrane (tm), and a cytosolic (c) domain (Figure S1). Numerous studies have been conducted to understand the biological functions of each domain in BACE1. The signal peptide in the N-terminus of BACE1 protein directs the newly synthesized BACE1 proteins to the endoplasmic reticulum (ER). The pro domain has been demonstrated to assist the folding of the BACE catalytic domain from an in vitro study.13 However, with the pro domain at the N-terminus, the BACE1 protein is less active than the mature form. In cells, the pre and pro domains are removed by furin, or a furin-like protease in the Golgi apparatus,14,15 to convert BACE1 to the mature protein. The catalytic Asp residues are located in the canonical active site motifs, DTGS and DSGT, in the catalytic domain. The transmembrane domain anchors the BACE1 protein to the membrane, and the cytosolic domain regulates its cellular trafficking.14,16
Although there are more than 10 000 BACE1 inhibitors that have been reported in the patent and open literature,17 currently no BACE1 inhibitors have been developed for the treatment of Alzheimers. The field is still active in identifying potent leads and improving their pharmaceutical properties to facilitate the drug development of BACE1 inhibitors. Although the drug development process has been heavily investigated for over a decade, there are still some difficulties in performing rational drug design for BACE1. One of the challenges is to express and purify BACE1 protein using an E. coli expression system or other expression systems for that matter. E. coli is one of the most popular expression systems for the production of recombinant proteins because of its low-cost and convenience. However, due to the large size of BACE1 and the requirement for pro-domain cleavage and proper disulfide-bond formation for function, BACE1 is mainly produced in the inclusion-body fraction when expressing in E. coli.18,19 Solubilization and refolding from inclusion bodies makes protein production and purification more challenging. According to the current literature, purifying BACE1 using an E. coli expression system requires either multiple, pH-adjusted refolding steps18 or a particular affinity column for purification.19 Overall, the protein yield ranges from 1 to 8 mg from 1 L of culture. Here, a relatively simple refolding and purification method. We have developed a strategy and decision-making process for analysis of enzyme kinetic data (rapid-equilibrium or tight-binding), including the utility of using a dose–response titration curve for determination of active enzymes concentration, is also demonstrated. Finally, a reproducible method for BACE1 crystallization is also demonstrated. Together, the aforementioned approaches provide for a simplified platform for structure-based drug design studies of BACE1 that is amenable to both academic laboratories and the pharmaceutical industry.
EXPERIMENTAL PROCEDURES
Construct Design
The expression plasmid containing BACE1 pre, pro, and catalytic domains (AlaP14 to Thr393, the numbering of residues used here starts at Glu1 in the sequence of EMVDN- as shown in Figure S2A) was obtained from Prof. Jordan Tang (construct-A, Figure S2B).20 The codon-optimized construct (construct-B, Figure S2C) encoding BACE1 pre, pro, and catalytic domains (AlaP14 to Thr393) was synthesized for optimal expression in E. coli (Bio Basic Inc.). The codon-optimized gene was subcloned into a pET-11a expression vector with an N-terminal His8-tag. In addition, a TEV cleavage recognition sequence is engineered between the pro and catalytic domains for the N-terminal truncation to create a mature BACE1. Both constructs are verified by DNA sequencing at Purdue University Genomics Core Facility.
Expression and Refolding of BACE1 Catalytic Domain
The expression plasmid encoding BACE1 was transformed into E. coli BL21 (DE3) electro-competent cells using a Bio-Rad Gene Pulser electroporator. Transformants were plated out on a Luria–Bertani (LB) agar plate supplemented with 100 μg/mL carbenicillin at 37 °C in an incubator and grown overnight. To prepare a starter culture, a single colony was plucked out from the plate and used to inoculate 100 mL of LB supplemented with 100 μg/mL ampicillin. The starter culture was then shaken at 200 rpm and incubated at a 37 °C shaker (ATR Biotech Multitron HT Infors Dual-Stack Incubator-Shaker) overnight. The next day, 10 mL of the starter culture were added into 1 L of LB broth supplemented with 100 μg/mL ampicillin. The culture was grown at 37 °C until the optical density at 600 nm (O.D.600) reached a value of ~1 OD. Then, isopropyl-D-1-thiogalactopyranoside (IPTG) was added to the culture to a final concentration of 0.2 mM to induce the BACE1 protein expression. The culture was again shaken at 200 rpm for 3 h at 37 °C to overexpress BACE1. After growth, cells were harvested by centrifugation (12 006g, 10 min at 4 °C). The harvested cell pellet was frozen at −80 °C until use.
To lyse the cells, frozen cells were thawed on ice and were then resuspended using a ratio of 1 g of cell pellet to 3 mL of lysis buffer containing 50 mM Tris-HCl at pH 7.2 and 150 mM NaCl. Resuspended cells were homogenized with a homogenizer and then lysed by passing three times through a French press at 1500 p.s.i.. The cell lysate was further incubated with 750 mL lysis buffer supplemented with 100 mM β-mercaptoethanol at 4 °C overnight. The insoluble fraction was collected the next day by centrifuging at 6770g for 40 min. The supernatant was discarded, and the pellet was resuspended in wash buffer (50 mM Tris-HCl pH 7.5, 100 mM NaCl, 1 mM EDTA, 0.5% Triton X-100), homogenized, incubated at 4 °C for 15 min, and pelleted by centrifugation (6770g, 20 min at 4 °C). This step was repeated twice and then again with the wash buffer without 0.5% Triton X-100. The washed pellet was stored at −80 °C until further use.
The frozen inclusion bodies were thawed on ice and dissolved with solubilization buffer (50 mM CAPs, pH 10.7, 50 mM β-mercaptoethanol, 8 M urea). The protein concentration was adjusted to 3.6 mg/mL with solubilization buffer. The solubilized solution was stirred at room temperature (rt) for 3 h. To achieve refolding, the solubilized protein solution was diluted 80-fold into cold deionized water and stirred at 4 °C for 4–5 days. The refolded protein sample was concentrated to less than 5 mL for size exclusion chromatography using a stirred ultrafiltration cell (Amicon stirred cell, 400 mL for 76 mm 10K MWCO membrane).
Size-Exclusion Chromatography (SEC)
The refolded and concentrated protein was loaded onto a gel filtration column (Superdex 75 26/60, Amersham Biosciences) equilibrated with Buffer A (20 mM Tris-HCl, pH 7.5, 80 mM Urea). The protein was eluted at a flow rate of 1 mL/min. The fractions were pooled based on the BACE1 activity assay and SDS-PAGE analysis.
Mono Q Anion-Exchange Chromatography
The pooled protein sample from SEC was loaded onto an 8 mL Mono Q 10/100 column (Amersham Biosciences) equilibrated in Buffer A. The column was then washed with a two column volume of Buffer A and then washed with 30% Buffer B (20 mM Tris-HCl, pH 7.5, 80 mM Urea, 0.3 M NaCl) until absorbance values at UV280 were minimized and stable. The BACE1 protein was eluted using a linear gradient from 30% to 55% Buffer B. Fractions were collected and pooled based on specific activity and purity as judged by SDS-PAGE. Purified BACE1 protein was concentrated to 3–5 mg/mL using an Amicon ultracentrifugal device (3K MWCO filter) and stored at 4 °C until further use.
Fluorescence-Based Kinetic Assays
A FRET-based (Forster Resonance Energy Transfer) assay was used to monitor the BACE1 enzymatic activity. The assay was performed using a fluorogenic 8-mer peptide substrate (Mca-S-E-V-N-L-D-A-E-F-K-Dnp).21 The sequence of the peptide substrate is derived from the β-secretase cleavage site of the Swedish APP mutation. The Swedish APP mutation was found in familial AD and will lead to an increased cleavage rate of APP by the β-secretase.22 Assays were conducted in black, half-area, 96-well plates (Corning Glass) with a final assay volume of 100 μL in each well. To start the reaction, a final concentration of 100 nM BACE1 was mixed with the assay buffer (0.1 M acetic acid, pH 4). A final concentration of 1 μM substrate was added into the above mixture to initiate the reaction. Upon substrate cleavage, the increasing fluorescence signal (excitation at 328 nm and emission at 393 nm) was measured using a Synergy-H1 Hybrid Multi-Mode Microplate reader (BioTek). The initial slope of each reaction in relative fluorescence units per minute (RFU/min) was measured and converted to units of micromolar of product produced per minute (μM/min) using experimentally determined values of the fluorescence extinction coefficient of the peptide substrate. All reactions were carried out in triplicate.
Steady-State Kinetic Assays
The Km and kcat. values were determined using the fluorescence-based kinetics assay described above. Each assay contained 100 nM of BACE1 enzyme and various substrate concentrations (0 μM, 1 μM, 1.25 μM, 2 μM, 2.5 μM, 3.75 μM, 5 μM, 7.5 μM, 10 μM, 15 μM, 20 μM, 30 μM, and 40 μM) in the reaction buffer containing 0.1 M acetic acid (pH 4). All reactions were initiated by the addition of the peptide substrate. The initial slope of each reaction (RFU/min) was measured and converted to specific activity (μM/min/mg) using the experimentally derived fluorescence extinction coefficient and the enzyme concentration used in the assay. The specific activity values were plotted as a function of substrate concentration, and the data were then fit to the Michaelis–Menten equation using nonlinear regression and the program Graphpad Prism 6.
Determination of Inhibition Constant (Ki) Values of Compounds
The test compounds were synthesized in the laboratory of Prof. A. K. Ghosh, Department of Chemistry at Purdue University. The details of the synthesis will be reported elsewhere. Each of the test compounds was dissolved with 100% DMSO to a stock concentration of 50 mM. To obtain the inhibitory constant (Ki) values, varying concentrations of compound were prepared and tested using the fluorescence-based kinetics assay described above. The test compounds, or DMSO (100%) as a control, were first mixed with the assay buffer (0.1 M acetic acid, pH 4) and incubated at 37 °C for 5 min. The final concentration of DMSO in each well is 1%. Then, the BACE1 enzyme was added into the solution to a final concentration of 100 nM, and the enzyme–inhibitor mixture was further incubated at 37 °C for 10 min. All of the reactions were initiated by the addition peptide substrates to a final concentration of 1 μM. The initial velocity, Vi (presence of inhibitor) or Vo (no inhibitor), of each reaction (RFU/min) was measured. The Vi/Vo ratio and % inhibition were calculated using eq 1 and eq 2, respectively.
| (1) |
| (2) |
In the equations, Vsample is the rate of the enzyme with inhibitor, Vbackground is the rate with no enzyme, and VDMSO is the rate of enzyme without inhibitor. The data were then analyzed as described in the flowchart (see Figure 2).
Figure 2.

Kinetic data analysis flowchart.
Determination of BACE1 Protein Concentration with a Titration Experiment
A well-characterized compound, GRL-8234, was used as a standard compound for kinetic experiments to determine the concentration of active BACE1. The synthesis of GRL-8234 and its Ki value (1.8 nM) against BACE1 were reported previously.8,23 The inhibition assay of GRL-8234 against BACE1 was run as described above. The inhibitory data were fit to the Morrison equation (see eqs 3–5 in the Results section) using nonlinear regression and the program Graphpad Prism 6. For curve fitting, the Ki value was initially constrained to 1.8 nM, the substrate concentration was constrained to 1000 nM, and the Km value was constrained to 13820 nM. The [E]o value returned from the curve fitting process is the active enzyme concentration in the assay that is capable of being inhibited.
Determination of the Cellular EC50 on the Reduction of Aβ1–40 with an Enzyme-Linked Immuno Sorbent Assay (ELISA)
Cellular production of Aβ1–40 was determined in BE(2)-M17 human neuroblastoma cells (ATCC CRL-2267TM). Cells were seeded overnight in 96-well plates with the medium volume of 100 μL [1:1 mixture of Eagle’s minimum essential medium (EMEM) and F12 medium plus 10% fetal bovine serum (FBS)] in each well. After 24 h, the medium was replaced with 70 μL of serum-limited medium [1:1 mixture of Eagle’s minimum essential medium (EMEM) and F12 medium plus 5% fetal bovine serum (FBS)] containing different concentrations of inhibitors. Cells were then returned to the incubator and incubated for an additional 24 h. The media from each well were then harvested and assayed for the presence of Aβ1–40 using an Aβ1–40 ELISA kit (Novex ELISA kits, Invitrogen Corporation). Aβ1–40 concentration values were normalized for cell viability, which was determined using the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) tetrazolium reduction assay. The production of Aβ1–40 for each concentration were plotted to get EC50 values using the four-parameter logistic program in Graphpad Prism 6.
Isothermal Titration Calorimetry (ITC)
Prior to the ITC experiments, purified BACE1 protein was dialyzed overnight against dialysis buffer containing 50 mM sodium acetate (pH 5.2) and 200 mM NaCl. The protein and inhibitor concentrations were adjusted to the desired concentrations using the dialysis buffer supplemented with 0.3% DMSO. ITC experiments were performed at 25 °C using a GE/MicroCal iTC200 calorimeter. For the ITC experiments, 150 μM of protein in the syringe was titrated into a 15 μM solution of inhibitor in the sample cell after thermal equilibrium at 25 °C was achieved. The injection schedule started with an initial 60 s delay and a single 0.4 μL injection of BACE1 that was then followed by 21 serial injections of 1.8 μL (or 29 serial injections of 1.2 μL) of BACE1 using an interval of 180 s between each injection. Baseline correction was performed by NITPIC,24 and data were analyzed and fit using a one-site model from SEDPHAT.25
Co-crystallization and X-ray Structure Determination of BACE1 in Complex with Inhibitors
Crystals of BACE1 in complex with different inhibitors were obtained using the hanging-drop, vapor-diffusion method. Freshly purified BACE1 protein was concentrated to 3.5 mg/mL using an ultracentrifugal device (3K MWCO filter, Amicon). For setting up crystallization trays, a solution of BACE1 protein in buffer containing 80 mM urea, 20 mM Tris-HCl pH 7.5, and 0.3 M NaCl was mixed with 500 μM inhibitor and then incubated for at least 1 h to form the BACE1–inhibitor complex. Crystallization drops were formed by mixing 2 μL of the BACE1–inhibitor complex with 1 μL of the reservoir solution that contained 0.2 M MgSO4, 0.1 M sodium citrate (pH varied from 5.0 to 6.0), and 14% to 20% PEG4000. Crystals suitable for X-ray data collection grew within 2 weeks. Crystals were retrieved with a nylon loop and then quickly dragged through well solution that was supplemented with 30% glycerol. The crystals were immediately flash-cooled by plunging into liquid nitrogen. Crystals were stored in cryo-shipping dewars containing liquid nitrogen until X-ray data collection. All X-ray diffraction data were collected at beamline 31-ID-D at the Lilly Research Laboratories Collaborative Access Team (LRL-CAT), at the Advanced Photon Source (APS), Argonne National Laboratories.
X-ray data were processed and scaled using the program HKL2000.26 Scaled and merged intensity data were converted to structure factor amplitudes using CCP4.27 BACE1–inhibitor complexes crystallized in space group P 1 21 1 with three BACE1 molecules in an asymmetric unit. Molecular library files and coordinates for the inhibitors were built using the program Phenix.28 Inhibitors were manually modeled into electron density using the program Coot.29 Fourier maps were calculated and visualized using the program Coot, and the structure was refined using the program Phenix. Iterative rounds of manual building and refinement were continued until Rwork and Rfree values reached their lowest values. Electron density maps presented in the figures were calculated using Phenix, and the figures were generated using the program PyMoL molecular graphics system, Version 1.7.2.1 Schrodinger, LLC.
RESULTS
BACE1 Catalytic Domain: Construct Design, Expression, Solubilization, and Purification
The extracellular catalytic domain (AlaP14 to Thr393) was readily expressed and purified from E. coli. Two different BACE1 expression constructs were investigated. The first was a wild-type BACE1 construct that was cloned from a mammalian cell line (construct-A, Figure S2B). The second was a wild-type, codon-optimized construct with a N-terminal His8-tag and a TEV recognition sequence between the pro and the catalytic domains (construct-B, Figure S2C). Each of these BACE1 expression constructs was individually overexpressed in E. coli and then subjected to the purification steps.
Inclusion bodies were solubilized with 8 M urea, and the protein solution was quickly poured and diluted into a container of stirring cold water at 4 °C. A dilution factor of 80:1 cold water to protein was found to be optimal. Rapid dilution was used to promote proper refolding which occurred over a period of 4 days at 4 °C with continual stirring. The resulting refolded and dilute protein solution was then concentrated and injected onto a Superdex 75 size-exclusion column (SEC) to separate high molecular mass misfolded protein aggregates from folded monomeric BACE1 protein. Active BACE1 protein fractions from SEC were pooled and then further purified using a Mono-Q, strong-anion-exchange column. Using this expression and purification procedure, a total of 5 to 6 mg of pure and active BACE1 enzyme can be purified from 1 L of E. coli culture using construct-A, or up to 16 mg of pure using codon-optimized construct-B.
A summary of the purification procedure using the codon-optimized BACE1 construct (construct-B) is presented in Table 1, since this construct produced the highest yield and purity. SDS-PAGE analysis for the purification steps is shown in Figure 1A. Size-exclusion chromatography (Figure 1B) was used to confirm that only the monomeric species was obtained after refolding and purification. The estimated molecular mass from the sixe-exclusion chromatography was around 40 kDa, which is the expected molecular mass for a monomer (44 kDa). The specific activity of BACE1 purified from both constructs is comparable to commercial BACE1 tagged at the N-terminus with (His)6. BACE1 protein produced from both constructs also produces enzyme that readily crystallizes under the same crystallization conditions; e.g., construct-A produced crystals that diffract to 2.1 Å resolution (I/σ is greater than 2 in the last shell).
Table 1.
BACE1 Purification from 2 L E. coli Culture Using Codon-Optimized Construct
| Purification steps | Volume (mL) | Protein (mg) | Specific activity (μM/min/mg) | Fold purification | Total units |
|---|---|---|---|---|---|
| Solubilize | 180 | 642 | – | – | – |
| Refold and concentrate | 10 | 321 | 24 | 1 | 7704 |
| Size exclusion | 65 | 85 | 79 | 3.3 | 6715 |
| Anion exchanger | 1.7 | 32 | 126 | 5.3 | 4032 |
Figure 1.

SDS-PAGE and size-exclusion chromatography analysis of BACE1 purification. (A) SDS-PAGE analysis throughout the purification process of BACE1 protein. 1, Molecular weight ladder; 2, protein sample after solubilization; 3, BACE1 protein sample after refold (After refolding, the active protein underwent autocleaving process to remove its pro-domain and the molecular weight went down to 44 kDa); 4, Size-exclusion chromatography pool; 5, Anion exchange elute pool. (B) Size-exclusion chromatography of pure BACE1.
The kinetic response of BACE1 to increasing concentrations of the FRET-based substrate described in Experimental Procedures was determined at 37 °C and pH 4 (the optimal pH for BACE1 activity, Figure S3). The initial rates were plotted versus substrate concentrations, and a fit of the data to the Michaelis–Menten equation yields values for kcat. = 8.0 ± 0.4 min−1 and Km = 13.8 ± 1.6 μM. The calculated catalytic efficiency (kcat./Km) is therefore 0.58 min−1 ywM−1.
Kinetic Analysis of BACE1 Inhibition by Tight-Binding Inhibitors
Based on the accumulated knowledge from a number of structure–activity relationship (SAR) studies on BACE1, many of the recently reported compounds are potent BACE1 inhibitors with Ki values in the low nanomolar range (nM). As a result, the concentrations of inhibitors must be varied into the low nM which approaches the concentration of BACE1 used in the assays (~100 nM). Detection limitations make it difficult to perform assays on BACE1 below 50 nM. Under these conditions, the assays are being performed under tight-binding conditions where free [I] ≈ [I]t cannot be met. Therefore, the classic steady-state or rapid-equilibrium rate equations for inhibition mechanisms are no longer applicable.30 A number of reports in the literature fit inhibition data using simple dose–response curves to estimate IC50 values.31,32 However, IC50 values can vary significantly depending on the assay conditions and analysis methods used. Therefore, it is better to determine the Ki values for compounds since they are true equilibrium dissociation constants instead of determining IC50 values which are less reliable.
To determine Ki values under tight-binding conditions, the Morrison equation (eq 3) should be applied.33 However, only limited information is found in the current literature regarding the analysis of BACE1 inhibitory data with the Morrison equation. Therefore, data analysis was performed using the Morrison equation according to the decision making flowchart shown in Figure 2. The flowchart is useful for treating the inhibitory data for BACE1 inhibitors with different potency. The Morrison equation is a five-parameter equation, where [E]o is the active enzyme concentration, is the apparent inhibitory constant, [I]o is the initial inhibitor concentration, Vi is the initial velocity at certain inhibitor concentrations, and Vo is the control velocity observed in the absence of inhibitor.
The relationship between the true Ki value and is described by eq 4 (for competitive inhibitors only). Rearranging eq 4 to solve for and substituting this equation into eq 3 yields eq 5 which is then used for curve fitting data to the Morrison equation using Graphpad Prism 6.
| (3) |
| (4) |
| (5) |
In order to fit eq 5 to experimental data, three parameters—the active enzyme concentration [E]o, the substrate concentration [S], and the Michaelis–Menten constant Km—have to be known and constrained during curve fitting. These three parameters are all experimentally determined and therefore subject to experimental error that will influence Ki value determination. To examine the effect of experimental error in each of these parameters on the resulting Ki values, the Ki value for a well-characterized and potent BACE1 inhibitor, GRL-8234, was used.8 The Ki of GRL-8234 was first determined with the enzymatic assay. The Ki value obtained from the assay is consistent with the reported value (1.8 nM). An error of 20% was intentionally introduced into each parameter while computing the Ki values (Table 2). The simulated results indicate that an inaccurate [E]o value has a significant impact on the Ki value determination. It also suggests that an accurate enzyme concentration is a crucial factor when plotting the data with the Morrison equation (eq 5).
Table 2.
Ki Determination of GRL-8234 against BACE1 Using the Morrison Plot
 | ||||
|---|---|---|---|---|
| Parameters | Experimental data | Simulated [E]o | Simulated [S] | Simulated Km |
| Vo | 0.9635 | 0.9606 | 0.9365 | 0.9891 |
| [E]o | = 100 | = 80 | = 100 | = 100 |
| [S] | = 1000 | = 1000 | = 800 | = 1000 |
| Km | = 13 820 | = 13 820 | = 13 820 | = 11 056 |
| Ki | 1.676 | 5.664 | 1.699 | 1.648 |
To assess whether the rapid-equilibrium model or tight-binding inhibition model should be applied for curve fitting depending on the data, the inhibitory data obtained from assays are first fit to the Morrison equation (eq 5). The [E]o can be initially determined using a Bradford assay. If the approximate Ki value obtained from the curve fitting is 10-fold greater than the estimated [E]o, the inhibitor is classified as a classical inhibitor and the inhibitory data can be analyzed with the rapid-equilibrium model (eq 6) to get the IC50 values.
| (6) |
In eq 6, the % inhibition values are calculated from eq 2 and are then plotted as a function of inhibitor concentration, [I]o. The data are then fit to eq 6, which describes a typical dose–response curve, and the IC50 and % inhibition max values and their associated errors are obtained from the fit. The IC50 values resulting from eq 6 can then be used to calculate Ki values using eq 7 (for competitive inhibitors only).
| (7) |
If the approximate Ki value is less than or similar to the estimated [E]o, then the Morrison equation (eq 5) should be applied for Ki determination. To obtain a more accurate Ki value, a titration experiment as described in the Experimental Procedures has to be performed to determine the active enzyme concentration. When the Ki/[E]o value is between 1 and 10, tight-binding begins to show up. In this case, both rapid equilibrium and tight binding models can be applied for data analysis. Theoretically, the Ki values computed from each equation are similar under this situation. However, if the inhibitor concentrations used in most of the data points are greater than the estimated [E]o, then the rapid-equilibrium model is preferred for data fitting.
In Vitro and in Cellulo Evaluation of Macrocyclic BACE1 Inhibitors
Macrocyclilization has been proposed as a strategy to improve potency and drug-like properties of BACE1 inhibitors.34–36 Using the kinetic assays and the data analysis strategies described above, the inhibitory activity of three macrocyclic compounds was determined against BACE1. Inhibitor-1 (Table 3 and Figure S4) is a macrocyclic compound that has been characterized by Stachel and co-workers.34 It exhibits nanomolar potency against BACE1 with an IC50 value of 4 nM and a cellular EC50 value of 76 nM. In addition, inhibitor-1 shows in vivo efficacy when dosed in a murine model.34
Table 3.
Ki and EC50 Values of Macrocyclic Compounds against BACE1
 | ||||
|---|---|---|---|---|
| compound | R1 | Ki (nM) | computed Kia (nM) | EC50 (nM) |
| inhibitor-1 | pentane | 21 ± 34b | 28 ± 2 | 76b |
| inhibitor-2 | 2-propanol | 5 ± 3 | 15 ± 5 | 124 |
| inhibitor-3 | 2-pentanol | 7 ± 5 | 18 ± 6 | 8 |
The [E]o was constrained in a simulated value (1/3 lower than the experimental determined value) while computing the Ki value with the Morrison equation.
Values are obtained from J. Med. Chem. 2006, 49, 6147–6150.34
The inhibition of BACE1 by Inhibitor-1 was reassessed using the kinetic assays and analysis approaches described above and in Figure 2. To start the inhibitory data analysis, an accurate [E]o was first determined by performing a dose–response inhibition experiment using GRL-8234 as a control compound.8 The Ki value was set to a value of 1.8 nM8, and then [E]o was determined from a fit of the data to eq 5. To confirm the effect of [E]o on the determination of the Ki value for compound-1, the value of [E]o was constrained either to its experimentally determined value or to a value that is one-third lower than this value (computed Ki value, Table 3). The Ki values for the other two compounds (inhibitors-2–3) were also obtained this way.
The kinetic results are summarized in Table 3. The new Ki value for compound-1 (21 nM) compared to the previous value of 4 nM demonstrates that an inaccurate value of [E]o leads to an inaccurate determination of Ki. A similar conclusion has been drawn from previous studies using computational simulation.37,38 Calculations were done similarly for compound-2 and compound-3. The data show that although the overall trend in relative potency of the compounds remains the same, the Ki values are different if the [E]o is not accurately determined. The data also indicate that inaccurate [E]o values have a greater impact on more potent compounds than the less potent ones which can be problematic in SAR analysis.
The cellular efficacy of these compounds was also determined in a neuroblastoma cell line. All three compounds also exhibit strong potency in cellulo (Table 3).
Thermodynamic Parameters of Macrocyclic Inhibitors Interacting with BACE1
The thermodynamic parameters for inhibitors-1–3 were determined using isothermal titration calorimetry (ITC), and the results are shown and summarized in Figure 3 and Table 4. The dissociation constants (Kd) obtained from ITC are in agreement with the Ki values determined from the kinetic assays (Tables 3 and 4). Both inhibitor-2 and inhibitor-3 exhibit 2–3-fold improvement in potency compared to the parental compound, inhibitor-1. The observation that the Kd values derived from the ITC experiments closely match the Ki values determined from the kinetic studies supports the experimental strategy outlined in Figure 2 for more accurately determining the Ki values of the compounds binding to BACE1.
Figure 3.

ITC titration curves for determination of Kd values for (A) inhibitor-1, (B) inhibitor-2, and (C) inhibitor-3 to BACE1.
Table 4.
Thermodynamic Parameters of Macrocyclic Compounds for BACE1
| compound | ΔH (kcal/mol) | TΔS (kcal/mol) | Kd (nM) | ΔG (kcal/mol) |
|---|---|---|---|---|
| inhibitor-1 | −9.1 ± 0.2 | 1.46 ± 0.18 | 22.1 ± 9.5 | −10.56 ± 0.24 |
| inhibitor-2 | −13.9 ± 0.4 | −2.95 ± 0.28 | 12.1 ± 5.8 | −10.95 ± 0.27 |
| inhibitor-3 | −11.8 ± 0.1 | −0.49 ± 0.16 | 6.4 ± 2.9 | −11.31 ± 0.26 |
X-ray Structure of BACE1 in Complex with Macrocyclic Inhibitors
The X-ray structure of the BACE1 – inhibitor-1 complex was determined to a resolution of 2.3 A (Table S1). As shown in Figure 4A and 4B, inhibitor-1 sits nicely in the active site and is well described by the surrounding electron density difference map (Fo – Fc). As expected, the P2 sulfonamide group fits into the S2 subsite and makes extensive polar contacts with residues Ser325, Arg235, Asn233, and Thr232. The P1′ amine moiety forms hydrogen bonds with the catalytic residue (Asp228) (Figure 4C and 4D).23 The P1′ pentane in inhibitor-1 is mainly stabilized in the S1′ subsite through hydrophobic interactions (Figure S5). The X-ray structure of BACE1–inhibitor-1 complex reveals that the S1′ subsite could accommodate a polar moiety. Therefore, the P1′ functional group of inhibitor-1 has been optimized based on the structural observation. Here, the P1′ functionality was substituted with either 2-propanol (inhibitor-2) or 2-pentanol (inhibitor-3) as shown in Table 3 and Figure S4. These designed substitutions lead to compounds with a 2- to 3-fold increase in potency compared to inhibitor-1 as judged by the decrease in Ki and Kd values.
Figure 4.

X-ray crystal structure of BACE1-inhibitor-1 complex. Inhibitor-1 is shown in ball and stick and colored according to atom types. The binding orientation of inhibitor-1 is similar in the active sites of BACE1 in three molecules in the asymmetric unit, and therefore only one active site is shown for clarity. (A) Electron density map of inhibitor-1. Electron density omit map (Fo – Fc) is contoured to 3.0 σ and shown in gray mesh. (B) inhibitor-1 in the active site of BACE1. The active site of BACE1 is shown in gray surface representation and colored according to atom type. (C) 3D and (D) 2D representation of polar contact interactions of inhibitor-1 with residues in BACE1 active site. Active side residues are shown in lines and colored according to atom types. Water molecules are shown as solid spheres in red, and polar contacts are shown in black dashes. Distances are shown between heteroatoms.
In order to get molecular insights into the increase in binding affinity of inhibitors-2 and −3, the X-ray structures of BACE1 cocrystallized with inhibitor-2 and inhibitor-3 were determined. The statistics for X-ray data collection and refinement are provided in Table S1. The X-ray structures of both complexes were determined to 2.1 Å resolution, and the electron density maps surrounding the ligands are clearly defined (Figure 5A and 5B). As shown in Figure 5C, inhibitor-1, inhibitor-2, and inhibitor-3 align almost perfectly in the BACE1 active site. The P1′ pentane, 2-propanol, and 2-pentanol from inhibitor-1, inhibitor-2, and inhibitor-3 fit nicely in the S1′ binding pocket. Importantly, the hydroxyl group from both P1′ 2-propanol (inhibitor-2) and 2-pentanol (inhibitor-3) forms a direct H-bond with Thr72 and two water-mediated H-bonds with Thr231 and Arg235 (Figure 5D and 5E). These additional interactions likely explain the 3-fold improvement in the potency of inhibitor-2 and inhibitor-3 as well as the 3–4 kcal/mol enthalpy gain of these two optimized compounds. The thermodynamic parameters also reveal that this new design causes a 2–3.5 kcal/mol entropy penalty and results in a total of only 0.5–1 kcal/mol net change in the Gibbs free energy (Table 4).
Figure 5.

X-ray crystal structures of BACE1 in complex with macrocyclic compounds. Ligands are shown in ball and stick and colored according to atom type. The binding orientation of inhibitor-2 and inhibitor-3 was the same in all active sites of the BACE1 trimer; therefore, only one active site is shown for clarity. Electron density map of (A) inhibitor-2 and (B) inhibitor-3. Electron density omit map (Fo – Fc) is contoured to 3.0 σ and shown in gray mesh. (C) Superposition of the X-ray structures of inhibitor-1 (shown in pink, ball and stick), inhibitor-2 (shown in green, ball and stick), and inhibitor-3 (shown in blue, ball and stick). The active site of BACE1 is shown in gray surface representation. The hydrogen bonding interactions of P1′ hydroxyl group from (D) inhibitor-2 and (E) inhibitor-3 with residues in the BACE1 active site. Active side residues are shown in lines and colored according to atom type. Water molecules are shown as solid spheres in red, and polar contacts are shown in black dashes. Distances are shown between heteroatoms.
DISCUSSION
BACE1 continues to be a promising therapeutic target for the treatment of Alzheimer’s disease. Over the past 18 years, numerous BACE1 inhibitors have been synthesized and tested. A number of BACE1 inhibitors have entered into clinical trials, and although there are encouraging accomplishments over the past decade, no clinically approved BACE1 inhibitors exist. Recent failures of BACE1 compounds such as Verubecestat and Lanabecestat in Phase III trials have dampened enthusiasm over the past few years, yet compounds such as Elenbecestat continue to move forward in Phase III. To achieve successful drug development against BACE1, reliable and efficient structure–activity relationships (SARs) and in-depth structural studies continue to be a prerequisite before compounds are taken to the clinic. Such studies require large amounts of functional BACE1 enzyme.
The production of BACE1 protein has been reported by several groups with different expression systems (Table S2). Among these different expression systems, E. coli produces the highest yield and is the simplest and most economical expression system. However, BACE1 protein is mainly expressed in the insoluble fractions and the refolding process has to be optimized. In 2004, Sardana et al.18 reported their BACE1 refolding and purification protocol. In their work, multiple pH-adjusted steps are required to achieve refolding. Compared to the methods reported by Tomasselli et al. in 2008,19 the refolding and purification protocol reported by Sardana et al. gives a relatively low protein yield. Tomasselli et al. refolded BACE1 protein in water, and they were able to yield 5–10 mg of pure BACE1 enzyme from a 1 L E. coli culture. However, a particular BACE1 affinity column (cross-linked to a BACE1 ligand) is required in their purification steps.
In this study, we developed and optimized a protocol for BACE1 refolding and purification from E. coli inclusion bodies without the need of an affinity column. Similar to Tomasselli et al., BACE1 protein was refolded in cold water. For purification, we used only two chromatographic steps, size exclusion and strong anion exchange, which are commonly used in laboratories. Using the refolding and purification protocol optimized in this study, the final protein yield is about 5–16 mg of pure BACE1 enzyme from a 1 L E. coli culture, depending on the construct used. The yield is higher than the values reported in the current literature (Table S2).
The steady-state kinetic parameters of BACE1 for the synthetic peptide substrate derived from Sweidish APP mutation were determined. Kinetic parameters have also been reported by Ermolieff et al.21 and Sardana et al.18 who used a similar substrate. Our observed kcat. and Km values are 8.0 ± 0.4 min−1 and 13.8 ± 1.6 μM (kcat./Km = 0.58 min−1 μM−1), respectively. Ermolieff et al. reported the kinetic parameters kcat 0.94 min−1 and Km 5.8 μM (kcat./Km = 0.16 min−1 μM−1). Sardana et al. published their kinetic parameters with a kcat. value of 9.3 min−1 and a Km value of 55 μM (kcat./Km = 0.17 min−1 μM−1). The different values of kinetic parameters reported from each group may differ due to the assay conditions or different refolded enzyme products.
Currently, there are many studies on designing potent BACE1 inhibitors in the literature. However, none of them report in detail the process they use to determine inhibition constants such as Ki. Here, we report a data analysis approach for evaluating inhibitors exhibiting markedly different inhibitory potencies against BACE1 ranging from the low nanomolar into the high micromolar range. The process outlined in Figure 2 requires that well-characterized standard compound be used in a titration experiment to first determine an accurate active enzyme concentration [E]o. Once this value is known, tight-binding inhibition constants of potent compounds can be determined using the Morrison equation. Fitting the Morrison equation with an inaccurate enzyme concentration would result in an inaccurate Ki value. The inaccurate enzyme concentration has a greater impact on the more potent inhibitors and thus will lead to inaccurate SARs. Running the titration experiment with a standard compound prior to the assay would not only allow one to obtain an accurate enzyme concentration but also make the comparison between different assays possible. This approach is applied to analyze the kinetic data of not only BACE1 inhibitors but also all other enzymes.
In this work, inhibitor-1 was tested as an example compound for evaluating the aforementioned approaches. Stachel and co-workers reported the same compound in 2006 with an IC50 value of 4 nM. In this study, the same compound has a Ki value of 21 nM which is close to its Kd value determined by ITC. The 5-fold difference is substantial when compared to the Ki values determined for two optimized derivatives that were synthesized. The Ki values of these two optimized compounds were determined using our approach to be near 5 nM. By comparing the results with the IC50 of inhibitor-1 against BACE1 reported previously, it would suggest that the optimization strategy introduced at the P1′ functional group does not improve the potency. The ITC results are also in agreement with the kinetic data obtained from this study, suggesting the data analysis approach outlined in Figure 2 leads to more accurate and reliable results.
The X-ray structures of BACE1 in complex with macrocyclic compounds were also determined and reveal that the P1′ substitution of 2-propanol or 2-pentanol is involved in a network of hydrogen bonding with the residues in the S1′ subsite. The thermodynamic parameters obtained from ITC experiments reveal a 3–4 kcal/mol enthalpy gain for the two optimized compounds. To correlate the ITC results with the structural observations, the 3–4 kcal/mol gain in enthalpy might be ascribed to the hydrogen bonding network between the P1′ substitution and the residues in the S1′ subsite. Furthermore, the hydroxyl group of 2-propanol or 2-pentanol in the P1′ moiety forms two water-mediated hydrogen bonds in the S1′ subsite. This interaction brings an ordered water molecular into the active site potentially explaining the 2–3.5 kcal/mol entropic penalty observed from ITC experiments. To avoid the entropy penalty, the P1′ moiety could be further optimized to make direct hydrogen bonds instead of water-mediated hydrogen bonds. The information gained from this study will help guide subsequent lead optimization studies.
Supplementary Material
ACKNOWLEDGMENTS
We would like to acknowledge Dr. Jordan Tang for sending us the expression plasmid (construct-A) and Dr. Zhongren Wu for sharing his experience on BACE1 purification. Also, we would like to thank the LRL-CAT beamline staff for their help in acquiring X-ray data. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli LillyCompany, which operates the facility. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Funding
Partial financial support by the National Institutes of Health (AG 18933) is gratefully acknowledged. Partial financial support by the Walther Cancer Foundation and the Purdue EVPRP were also provided to ADM. Crystallization and DNA sequencing were partially supported by the Purdue Center for Cancer Research Macromolecular Crystallography and DNA Sequencing Shared Resources which are partially supported by NIH grant P30 CA023168.
ABBREVIATIONS
- Aβ
Amyloid beta peptide
- AD
Alzheimer’s disease
- APP
amyloid precursor protein
- BACE1
Beta-site APP cleaving enzyme 1
- ITC
isothermal titration calorimetry
- SAR
structure–activity relationship
- TEV
tobacco etch virus
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.bio-chem.9b00714.
Figure S1. Protein domain scheme of BACE1; Figure S2. The amino acid sequence of BACE1 and the expression construct scheme; Figure S3. BACE1 activity dependence on pH; Figure S4. Chemical structure of test inhibitors; Figure S5. Ligand interaction plots of BACE1 and inhibitors; Table S1. Data collection and refinement statistics; Table S2. BACE1 expression systems (PDF)
Accession Codes
PDB codes: BACE1-inhibitor 1 complex, 6NW3; BACE1-inhibitor 2 complex, 6NV7; BACE1-inhibitor 3 complex, 6NV9.
REFERENCES
- (1).Alzheimer’s; Association 2018 Alzheimer’s disease facts and figures. Alzheimers Dement, 2018, 14, 63. [Google Scholar]
- (2).Karran E, Mercken M, and De Strooper B (2011) The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discovery 10 (9), 698–712. [DOI] [PubMed] [Google Scholar]
- (3).Luo Y, Bolon B, Kahn S, Bennett BD, Babu-Khan S, Denis P, Fan W, Kha H, Zhang J, Gong Y, Martin L, Louis JC, Yan Q, Richards WG, Citron M, and Vassar R (2001) Mice deficient in BACE1, the Alzheimer’s beta-secretase, have normal phenotype and abolished beta-amyloid generation. Nat. Neurosci. 4 (3), 231–2. [DOI] [PubMed] [Google Scholar]
- (4).Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson- Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, Seubert P, Suomensaari SM, Wang S, Walker D, Zhao J, McConlogue L, and John V (1999) Purification and cloning of amyloid precursor protein beta-secretase from human brain. Nature 402 (6761), 537–40. [DOI] [PubMed] [Google Scholar]
- (5).Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, Collins F, Treanor J, Rogers G, and Citron M (1999) Beta-secretase cleavage of Alzheimer’s amyloid precursor protein by the transmembrane aspartic protease BACE. Science 286 (5440), 735–41. [DOI] [PubMed] [Google Scholar]
- (6).Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashler JR, Stratman NC, Mathews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, and Gurney ME (1999) Membrane-anchored aspartyl protease with Alzheimer’s disease beta-secretase activity. Nature 402 (6761), 533–7. [DOI] [PubMed] [Google Scholar]
- (7).Roberds SL, Anderson J, Basi G, Bienkowski MJ, Branstetter DG, Chen KS, Freedman SB, Frigon NL, Games D, Hu K, Johnson-Wood K, Kappenman KE, Kawabe TT, Kola I, Kuehn R, Lee M, Liu W, Motter R, Nichols NF, Power M, Robertson DW, Schenk D, Schoor M, Shopp GM, Shuck ME, Sinha S, Svensson KA, Tatsuno G, Tintrup H, Wijsman J, Wright S, and McConlogue L (2001) BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer’s disease therapeutics. Hum. Mol. Genet. 10 (12), 1317–24. [DOI] [PubMed] [Google Scholar]
- (8).Chang WP, Huang X, Downs D, Cirrito JR, Koelsch G, Holtzman DM, Ghosh AK, and Tang J (2011) Beta-secretase inhibitor GRL-8234 rescues age-related cognitive decline in APP transgenic mice. FASEB J. 25 (2), 775–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Neumann U, Rueeger H, Machauer R, Veenstra SJ, Lueoend RM, Tintelnot-Blomley M, Laue G, Beltz K, Vogg B, Schmid P, Frieauff W, Shimshek DR, Staufenbiel M, and Jacobson LH (2015) A novel BACE inhibitor NB-360 shows a superior pharmacological profile and robust reduction of amyloid-β and neuroinflammation in APP transgenic mice. Mol. Neurodegener 10, 441–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Thakker DR, Sankaranarayanan S, Weatherspoon MR, Harrison J, Pierdomenico M, Heisel JM, Thompson LA, Haskell R, Grace JE, Taylor SJ, Albright CF, and Shafer LL (2015) Centrally Delivered BACE1 Inhibitor Activates Microglia, and Reverses Amyloid Pathology and Cognitive Deficit in Aged Tg2576 Mice. J. Neurosci. 35 (17), 6931–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Volloch V, and Rits S Results of Beta Secretase-Inhibitor Clinical Trials Support Amyloid Precursor Protein-Independent Generation of Beta Amyloid in Sporadic Alzheimer’s Disease. Med. Sci. (Basel) 2018, 6 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Egan MF, Kost J, Voss T, Mukai Y, Aisen PS, Cummings JL, Tariot PN, Vellas B, van Dyck CH, Boada M, Zhang Y, Li W, Furtek C, Mahoney E, Harper Mozley L, Mo Y, Sur C, and Michelson D (2019) Randomized Trial of Verubecestat for Prodromal Alzheimer’s Disease. N. Engl. J. Med. 380 (15), 1408–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shi XP, Chen E, Yin KC, Na S, Garsky VM, Lai MT, Li YM, Platchek M, Register RB, Sardana MK, Tang MJ, Thiebeau J, Wood T, Shafer JA, and Gardell SJ (2001) The pro domain of beta-secretase does not confer strict zymogen-like properties but does assist proper folding of the protease domain. J. Biol. Chem. 276 (13), 10366–73. [DOI] [PubMed] [Google Scholar]
- (14).Benjannet S, Elagoz A, Wickham L, Mamarbachi M, Munzer JS, Basak A, Lazure C, Cromlish JA, Sisodia S, Checler F, Chretien M, and Seidah NG (2001) Post-translational processing of beta-secretase (beta-amyloid-converting enzyme) and its ectodomain shedding. The pro- and transmembrane/cytosolic domains affect its cellular activity and amyloid-beta production. J. Biol. Chem. 276 (14), 10879–87. [DOI] [PubMed] [Google Scholar]
- (15).Creemers JW, Ines Dominguez D, Plets E, Serneels L, Taylor NA, Multhaup G, Craessaerts K, Annaert W, and De Strooper B (2001) Processing of beta-secretase by furin and other members of the proprotein convertase family. J. Biol. Chem. 276 (6), 4211–7. [DOI] [PubMed] [Google Scholar]
- (16).Vetrivel KS, Meckler X, Chen Y, Nguyen PD, Seidah NG, Vassar R, Wong PC, Fukata M, Kounnas MZ, and Thinakaran G (2009) Alzheimer disease Abeta production in the absence of S-palmitoylation-dependent targeting of BACE1 to lipid rafts. J. Biol. Chem. 284 (6), 3793–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Southan C, Varkonyi P, Boppana K, Jagarlapudi SA, and Muresan S (2013) Tracking 20 years of compound-to-target output from literature and patents. PLoS One 8 (10), No. e77142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sardana V, Xu B, Zugay-Murphy J, Chen Z, Sardana M, Darke PL, Munshi S, and Kuo LC (2004) A general procedure for the purification of human beta-secretase expressed in Escherichia coli. Protein Expression Purif. 34 (2), 190–6. [DOI] [PubMed] [Google Scholar]
- (19).Tomasselli AG, Paddock DJ, Emmons TL, Mildner AM, Leone JW, Lull JM, Cialdella JI, Prince DB, Fischer HD, Heinrikson RL, and Benson TE (2008) High yield expression of human BACE constructs in Eschericia coli for refolding, purification, and high resolution diffracting crystal forms. Protein Pept. Lett. 15 (2), 131–43. [DOI] [PubMed] [Google Scholar]
- (20).Lin X, Koelsch G, Wu S, Downs D, Dashti A, and Tang J (2000) Human aspartic protease memapsin 2 cleaves the beta-secretase site of beta-amyloid precursor protein. Proc. Natl. Acad. Sci. U. S. A. 97 (4), 1456–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Ermolieff J, Loy JA, Koelsch G, and Tang J (2000) Proteolytic activation of recombinant pro-memapsin 2 (Pro-beta-secretase) studied with new fluorogenic substrates. Biochemistry 39 (51), 16263. [DOI] [PubMed] [Google Scholar]
- (22).Haass C, Lemere CA, Capell A, Citron M, Seubert P, Schenk D, Lannfelt L, and Selkoe DJ (1995) The Swedish mutation causes early-onset Alzheimer’s disease by beta-secretase cleavage within the secretory pathway. Nat. Med. 1 (12), 1291–6. [DOI] [PubMed] [Google Scholar]
- (23).Ghosh AK, Kumaragurubaran N, Hong L, Kulkarni S, Xu X, Miller HB, Reddy DS, Weerasena V, Turner R, Chang W, Koelsch G, and Tang J (2008) Potent memapsin 2 (beta-secretase) inhibitors: design, synthesis, protein-ligand X-ray structure, and in vivo evaluation. Bioorg. Med. Chem. Lett. 18 (3), 1031–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Keller S, Vargas C, Zhao H, Piszczek G, Brautigam CA, and Schuck P (2012) High-precision isothermal titration calorimetry with automated peak-shape analysis. Anal. Chem. 84 (11), 5066–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Houtman JC, Brown PH, Bowden B, Yamaguchi H, Appella E, Samelson LE, and Schuck P (2007) Studying multisite binary and ternary protein interactions by global analysis of isothermal titration calorimetry data in SEDPHAT: application to adaptor protein complexes in cell signaling. Protein Sci. 16 (1), 30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Otwinowski Z, and Minor W (1997) [20] Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276, 307–326. [DOI] [PubMed] [Google Scholar]
- (27).Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, and Wilson KS (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr., Sect. D: Biol. Crystallogr. 67 (4), 235–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66 (2), 213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot. Acta Crystallogr., Sect. D: Biol. Crystallogr. 66 (4), 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Michaelis L, Menten ML, Johnson KA, and Goody RS (2011) The original Michaelis constant: translation of the 1913 Michaelis-Menten paper. Biochemistry 50 (39), 8264–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Atwal JK, Chen Y, Chiu C, Mortensen DL, Meilandt WJ, Liu Y, Heise CE, Hoyte K, Luk W, Lu Y, Peng K, Wu P, Rouge L, Zhang Y, Lazarus RA, Scearce-Levie K, Wang W, Wu Y, Tessier-Lavigne M, and Watts RJ (2011) A therapeutic antibody targeting BACE1 inhibits amyloid-β production in vivo. Sci. Transl. Med. 3 (84), 84ra43. [DOI] [PubMed] [Google Scholar]
- (32).Hattori Y, Kobayashi K, Deguchi A, Nohara Y, Akiyama T, Teruya K, Sanjoh A, Nakagawa A, Yamashita E, and Akaji K (2015) Evaluation of transition-state mimics in a superior BACE1 cleavage sequence as peptide-mimetic BACE1 inhibitors. Bioorg. Med. Chem. 23 (17), 5626–40. [DOI] [PubMed] [Google Scholar]
- (33).Copeland RA Enzymes: A practical Introduction to Structure Mechanism, and Data Analysis; Wiley-VCH: New York. [Google Scholar]
- (34).Stachel SJ, Coburn CA, Sankaranarayanan S, Price EA, Wu G, Crouthamel M, Pietrak BL, Huang Q, Lineberger J, Espeseth AS, Jin L, Ellis J, Holloway MK, Munshi S, Allison T, Hazuda D, Simon AJ, Graham SL, and Vacca JP (2006) Macrocyclic inhibitors of beta-secretase: functional activity in an animal model. J. Med. Chem. 49 (21), 6147–50. [DOI] [PubMed] [Google Scholar]
- (35).Ghosh AK, Devasamudram T, Hong L, DeZutter C, Xu X, Weerasena V, Koelsch G, Bilcer G, and Tang J (2005) Structure-based design of cycloamide-urethane-derived novel inhibitors of human brain memapsin 2 (beta-secretase). Bioorg. Med. Chem. Lett. 15 (1), 15–20. [DOI] [PubMed] [Google Scholar]
- (36).Machauer R, Veenstra S, Rondeau JM, Tintelnot-Blomley M, Betschart C, Neumann U, and Paganetti P (2009) Structure-based design and synthesis of macrocyclic peptidomimetic beta-secretase (BACE-1) inhibitors. Bioorg. Med. Chem. Lett. 19 (5), 1361–5. [DOI] [PubMed] [Google Scholar]
- (37).Kuzmic P, Elrod KC, Cregar LM, Sideris S, Rai R, and Janc JW (2000) High-throughput screening of enzyme inhibitors: simultaneous determination of tight-binding inhibition constants and enzyme concentration. Anal Biochem. 286 (1), 45–50. [DOI] [PubMed] [Google Scholar]
- (38).Murphy DJ (2004) Determination of accurate KI values for tight-binding enzyme inhibitors: an in silico study of experimental error and assay design. Anal Biochem. 327 (1), 61–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.