

Using direct-write 3D printing, we tune structural color of bottlebrush block copolymer photonics via kinetic trapping.
Abstract
Additive manufacturing of functional materials is limited by control of microstructure and assembly at the nanoscale. In this work, we integrate nonequilibrium self-assembly with direct-write three-dimensional (3D) printing to prepare bottlebrush block copolymer (BBCP) photonic crystals (PCs) with tunable structure color. After varying deposition conditions during printing of a single ink solution, peak reflected wavelength for BBCP PCs span a range of 403 to 626 nm (blue to red), corresponding to an estimated change in d-spacing of >70 nm (Bragg- Snell equation). Physical characterization confirms that these vivid optical effects are underpinned by tuning of lamellar domain spacing, which we attribute to modulation of polymer conformation. Using in situ optical microscopy and solvent-vapor annealing, we identify kinetic trapping of metastable microstructures during printing as the mechanism for domain size control. More generally, we present a robust processing scheme with potential for on-the-fly property tuning of a variety of functional materials.
INTRODUCTION
Highly branched, ultrahigh–molecular weight block copolymers (BCPs) offer an attractive route toward bottom-up fabrication of organic photonic materials (1). In photonic crystals (PCs), the dielectric function (index of refraction) varies with spatial periodicity approaching optical wavelengths (approximately hundreds of nanometers), creating a photonic bandgap, i.e., constructive reflection of light of specific wavelengths (2). BCPs comprising covalently linked, but chemically incompatible, homopolymer segments have been widely used to generate well-ordered nanomaterials with domain d-spacings on the order of 5 to 100 nm (3). In general, chain entanglements and synthetic challenges impede the assembly of linear BCPs to the larger domain d-spacings (>100 nm) required for reflection spanning the visible spectrum (1). Recent reports of ultrahigh–molecular weight BCPs have achieved this (4, 5); however, long-range ordering may require extended solvent or thermal annealing (~1 week).
By contrast, numerous reports have shown that dense grafting of linear polymeric arms [bottlebrush BCPs (BBCPs)] (6–9) or pendant (10, 11) groups onto a common backbone suppresses chain entanglement to enable more rapid formation at large domain d-spacings. Over the past two decades, much attention has been paid to synthesis and near-equilibrium processing of BBCPs to explore their self-assembly behavior and fundamental chain dynamics (1, 7, 12). It is now well established that BBCP of sufficient grafting density (13, 14) can be described as semiflexible, worm-like chains with some degree of shape persistency (bending rigidity) (7, 15). Recent attention has also focused on the conformation of BBCP chains within lamellar domains formed in the solid phase. Many groups have reported scaling exponents (v) for the domain d-spacing (D0) versus degree of polymerization (DP) of greater than 0.8 [compared to v ~ 2/3 for strongly segregated linear BCP; (16)] as evidence that BBCPs adopt highly extended conformations in the solid state (6, 17–19), although more recent works by Dalsin et al. (9) and Sunday et al. (20) have clarified that there is markedly increased backbone flexibility within the core of lamellar domains. In each of the above references, efforts were taken to access near-equilibrium structures for comparison with the predictions of equilibrium field theories (9, 18, 20, 21).
The use of BBCP for photonic applications has attracted substantial research interest in the past decade, motivated by a wealth of potential industrial applications as environmentally friendly alternatives to dyes in decorative coatings, as low-cost radiant barriers, and (for particularly well-ordered crystals) in telecommunications. Sveinbjörnsson et al. in 2012 (6) presented a poly(lactic-acid)-block-poly(styrene) chemistry, which forms PC with molecular weight–dependent reflectivity spanning the visible/near-infrared spectrum for films prepared under slow evaporation and annealing between glass slides. Soon after, modulation of the peak reflected wavelength for BBCP-based PC was demonstrated by slow evaporation of BBCP blends with linear homopolymers [McFarlane et al. (22)] and BBCP of differing molecular weights [Miyake et al. (23)]. More recent reports have demonstrated multifunctional photonic bottlebrush-derived materials with behaviors such as strain-adaptive stiffening (24) and robust mechanical performance (25). Promising work by Boyle et al. (11) has demonstrated melt three-dimensional (3D) printing of dendritic (bulky pendant group) BCPs with more rapid microphase separation during filament drawing, but again, the process relies on changing the filament chemistry for modifying photonic properties. The materials and insights gleaned from these works are impressive; however, their industrial relevance is limited by the reliance on synthetic variation to modify optical properties and use of lengthy thermal annealing processes that are not suited for high–production volume printing applications. In this work, we address this challenge through a rapid, scalable, and highly customizable additive manufacturing approach toward BBCP deposition, which can achieve spatial, microstructural, and functional patterning of PCs.
3D printing (additive manufacturing) technologies have achieved widespread commercialization for melt extrusion of thermoplastic structural polymers [fused deposition modeling (FDM)], thanks to their low cost, reliability, and ease of use (26). Expanding the material palette to functional materials is a revolutionary next step that has been recently reported for cell-laden gels (27), electronic materials (28), and even pharmaceutical drugs (29), although the overwhelming focus has been on determining suitable ink formulations for material delivery, with less attention paid toward the molecular assembly process during deposition. In this work, we leverage the core advantage of 3D printing—the high level of hardware/software integration—to precisely direct materials assembly during the nonequilibrium printing process and to generate patterned PCs with tunable domain size and color. The incorporation of color into 3D printing has significant pedagogical and cosmetic advantages but has so far been demonstrated only for single colors through dyed filament stock or the use of complex and time-consuming multi-nozzle, multi-material methods for multicolored prints. By depositing BBCP from the solution phase with a volatile solvent, we force molecular assembly (microphase segregation) to compete with evaporation and demonstrate on-the-fly tuning of nanoscale morphology and structural color for vibrant, multicolored prints from a single stock ink.
In the following sections, we first detail the well-controlled synthesis of poly(dimethylsiloxane)-block-poly(lactic acid) (PDMS-b-PLA) BBCPs at the multigram scale. Then, we demonstrate tuning of optical properties for bottlebrush PCs by modulating printing speed and bed temperature during direct-write 3D printing. We show precise and programmable patterning of PCs with peak reflected wavelengths spanning a range of 223 nm (blue to red). Via ex situ scanning electron microscopy (SEM) and synchrotron small-angle x-ray scattering (SAXS) analysis of printed films, we then clarify the microstructural underpinning of these phenomena: tuning of domain d-spacing across a range of greater than 70 nm. Last, we confirm the role of kinetic trapping of metastable microstructures as the mechanism for domain size control based on solution SAXS, in situ optical microscopy, and solvent-vapor annealing experiments.
RESULTS AND DISCUSSION
Synthesis of PDMS-b-PLA BBCP
Well-defined PDMS-b-PLA BBCP were synthesized by graft-through polymerization of macromonomers (Fig. 1, A to C). PDMS macromonomers [Mn (number-average molecular weight) = 6200 g/mol; Mw (weight-average molecular weight)/Mn = 1.05] were synthesized via a seeded anionic ring opening polymerization of hexamethylcyclotrisiloxane (30, 31). PLA macromonomers (Mn = 5100 g/mol; Mw/Mn = 1.05) were synthesized by an 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) organocatalyzed ring opening polymerization of lactide (30, 32). Sequential graft-through ring-opening metathesis polymerization (ROMP) was used to synthesize the 50% mole fraction of PDMS and 50% mole fraction of PLA BBCP.
Fig. 1. PDMS-b-PLA bottlebrush synthesis, solution preparation, and dropcast film.
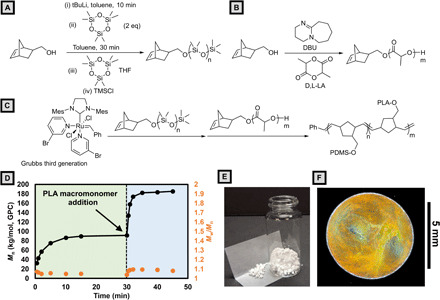
(A) Seed anionic ring opening polymerization of hexamethylcyclotrisiloxane to produce PDMS macromonomers. (B) 8-diazabicyclo[5.4.0]undec-7-ene (DBU) catalyzed ring opening polymerization of lactide to produce PLA macromonomers. (C) Sequential graft-through ring-opening metathesis polymerization (ROMP) of PDMS and PLA macromonomers. (D) Molecular weight versus time plot for the synthesis of PDMS-b-PLA bottlebrush. PDMS macromonomers are polymerized first (t < 30 min), and then PLA macromonomers are added and polymerized (t > 30 min). (E) Image of dried, as-synthesized bottlebrush stock material. (F) Microscope camera image of a drop-cast film taken at normal incidence under a ring light. Photograph courtesy of Bijal Patel, University of Illinois.
To assess the effectiveness of ROMP for producing the desired diblock bottlebrushes, aliquots were taken at various time points during the polymerization and analyzed by gel permeation chromatography (GPC) using polystyrene standard (30). First-order consumption of both PDMS and PLA macromonomers was observed while maintaining narrow molecular weight dispersities for the growing bottlebrush. A doubling of molecular weight occurred upon completion of the addition of the second block, supporting a well-controlled synthesis (Fig. 1D). This experiment was followed up with the large-scale synthesis (5.5 g) of PDMS-b-PLA with a total backbone DP of 400 repeat units with narrow molecular weight dispersity (Mw/Mn = 1.09). The absolute molecular weight Mn(t-GPC) = 1930 kg/mol is in good agreement with the theoretical molecular weight (Mn = 2260 kg/mol). Further synthetic details can be found in section S2.
The as-synthesized BBCP was isolated as a coarse white powder (Fig. 1E) exhibiting glass transitions at −125.5° and 53.2°C (section S3). Upon the addition to tetrahydrofuran (THF) at a concentration of 100 mg/ml [10.1 weight % (wt %)], BBCP readily dissolves with stirring to form a weakly ordered micellar phase, as determined by SAXS and SEM (section S4). When drop-cast, the solution dries to form microphase-separated, colored films with significant stochastic color variation both within and between samples (Fig. 1F and movie S1).
Establishing a platform for direct-write 3D printing of BBCP
3D printing provides an ideal platform for efficient study of nonequilibrium assembly behavior due to its high flexibility with regard to pattern complexity, ease of programming (G-Code), printing speed (mm/min − m/min), and substrate temperature while maintaining moderate spatial resolution (~micrometers) and total system cost (~$10,000). In our work, a consumer 3D printer was chosen as the core element on the basis of its large and unenclosed build volume and open software and hardware. The key challenge in adapting consumer 3D printers to research on printing of functional polymers is that the common melt-extrusion (FDM) schemes are not material-agnostic. That is, they demand relatively large quantities (greater than tens of grams) of material ductile enough for filament spooling, exhibiting suitable thermoplasticity for extrusion and rapid hardening. By contrast, functional (optoelectronic) materials are rarely available or required in bulk, and applications are much more suited for thin-film fabrication. In this work, we choose to deposit from the solution phase, which also adds an additional dimension to the BCP phase diagram; the use of volatile solvents both imparts significantly enhanced molecular mobility at low temperature and provides a critical lever for control of the highly accelerated assembly process. The key advantage of our 3D printing scheme is the incorporation of a highly precise pneumatic extrusion system capable of applying a wide range of applied pressures (100 Pa to 680 kPa), at high speed (<0.1-s switching time), while preserving the high level of software and hardware integration that makes precise patterning and deposition possible.
To print well-defined patterns and to achieve on-the-fly modulation of photonic properties, it is critical that the dispensing and motion control systems achieve synchronization and can handle flow rate adjustment during printing and travel moves. To this end, we have developed and released an open-source program (PolyChemPrint; section S5), which handles bidirectional communication with the consumer 3D printer’s motion axes and substrate heater and the pneumatic dispenser controller. The software was written in Perl, with new versions available in Python 3.x, and runs on Windows/Linux distributions. It provides a simple text-based user interface and can be readily modified to accommodate any modern 3D printer operating the common Marlin firmware and any extrusion system capable of serial communication. The software offers three main operating modes to handle prints of increasing complexity: (i) direct hardware control, (ii) execution of user-defined code sequences (e.g., meanderlines, plates, and cubes) with adjustable parameters (length, width, printing speed, pressure, etc.), and (iii) execution of arbitrarily complex externally generated G-Code files. There are also a host of research-friendly features such as automatic data logging and combinatorial parameter tuning that may fill a critical need in the 3D printing/bioprinting research community. Last, the scheme allows for on-the-fly modification (scaling) of printing speed and applied pressure via the hardware controls on the 3D printer (Taz 6) and dispenser controller during the printing process. A rendering of the system is shown in Fig. 2A.
Fig. 2. Direct-write 3D printing scheme.
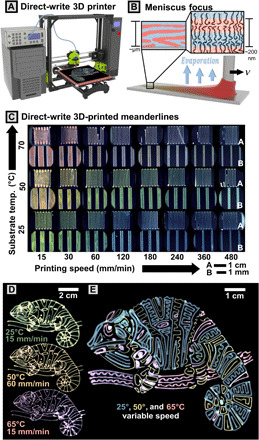
(A) Render of the printing setup comprising 3D motion axes, pneumatic dispenser, and computer control. (B) Cartoon of molecular assembly during the solution-casting process. Pressure is applied to cause outflow of polymer solution during translation at velocity, v. Microphase separation occurs simultaneously with solvent evaporation to form lamellae. (C) Programmatic variation of optical properties via modulation of printing speed and temperature. Optical microscopy images of printed meanderline patterns on bare silicon are shown in the figure. At each temperature (pair of rows), images at low magnification (inset A) and high magnification (inset B) are shown. (D) Chameleon patterns printed as continuous prints under constant printing conditions (pressure, printing speed, and bed temperature). (E) Complex pattern printed in three layers at three bed temperatures. Print speed was tweaked on the fly to tune line thickness, color, and intensity throughout the print, leading to intended variation seen in the green/blue 25°C lines.
In this work, concentrated solutions of PDMS-b-PLA BBCP (10.1 wt % in tetrahydrofuran) are loaded into a disposable piston-backed syringe, which is then connected to a nitrogen cylinder through the dispenser controller. Syringes are mounted onto a 3D-printed holder with attached pen camera for monitoring the meniscus during printing. After setting the end of the cylindrical nozzle 100 μm above the heated silicon substrate, gas pressure is applied on the fluid reservoir while translating the nozzle at a given speed. Figure 2B depicts a close-up of the solution-phase assembly process. As the nozzle moves, a concentration gradient develops between the leading edge of the liquid meniscus (at the nozzle exit) and the triple-phase contact line (where the film has solidified), accompanied by a morphology gradient between the solution-phase structure and the lamellar microphase-segregated state (Fig. 2B, inset).
Programmatic control of photonic properties via 3D printing
In this section, we demonstrate that 3D printing can be used to precisely control the photonic properties (color) of deposited films by varying printing speed, applied pressure, and substrate temperature. Samples prepared by the 3D printing process exhibit well-controlled, uniform, and highly tunable photonic properties (Fig. 2C and section S6). To prepare suitable samples for diffuse reflection measurements, we first defined a meanderline pattern in PolyChemPrint. A series of samples were prepared by varying printing speed systematically from 15 to 480 mm/min at three substrate temperatures (25°, 50°, and 70°C) with an applied pressure of 30 kPa (movie S2). Comparing printed samples, a very clear blueshift in reflected wavelength is observed as printing speed increases, while increasing temperature causes a pronounced redshift (detailed characterization and discussion follow). We also note that lines show a consistent coloration, spanning the majority of each 12-cm printed sample. Slight discolorations are visible near the beginning of the print and at corners (especially at high printing speeds); these are caused by interruptions in the constant velocity of the nozzle and could be mitigated by fine-tuning motion control and path design. At this relatively low applied pressure, it was possible to maintain a stable meniscus up to ~600 mm/min, after which discontinuous lines were printed because of insufficient ink supply (volume flow rate of solution dispensed). The relatively slow speeds and low pressure were chosen for ease of experimentation and high time resolution in in situ imaging. However, we will later show that the same phenomena are readily accessible at higher, industrially relevant printing speeds by scaling both the pressure and printing speed appropriately to maintain constant meniscus size.
We further demonstrate the versatility of the 3D printing process for depositing more complex patterns with a high degree of spatial and functional control (Fig. 2, D and E). To accomplish this goal, we incorporate two key functionalities into our control software: (i) the ability to import motion paths from common G-Code slicers and (ii) the ability to stop flow and perform travel moves. For the samples presented below, patterns were drawn in an image editor and exported as G-Code using a free plugin (see section S7). The result was a simple, user-friendly method to programmatically pattern PCs reflecting the desired color within patterns of arbitrary complexity. We first drew three separate chameleon patterns and printed them at fixed printing speed, pressure (35 kPa), and bed temperatures onto bare 4″ Si wafers (Fig. 2D and movie S3). To demonstrate control of not only spatial but also functional patterning, we then drew a much more complex chameleon pattern comprising different color PC printed as sequential layers. Figure 2E was printed in three steps, with each layer printed at a constant bed temperature (movie S4). Specifically, printing speeds for each layer were initially set to 60, 100, and 30 mm/min for printing green/blue, yellow, and red lines, respectively. To demonstrate on-the-fly tuning of photonic properties, we adjusted printing speed ±20% during printing of the green/blue layer, enabling fine-tuning of color and intensities across the sample. The three layers are printed with good registry, and the resulting image is an accurate reproduction of the original drawing (fig. S15), demonstrating that this workflow is highly applicable for printing complex functional patterns. We note that there is also no limitation to flat, planar substrates; in fig. S17, we demonstrate printing onto a spherical surface after generating motion code for PolyChemPrint by processing a 3D model in a commercial G-Code slicer (Cura).
Optical characterization reveals systematic variation of photonic properties
To quantitatively analyze the photonic properties of our printed films, we measured normally incident diffuse reflection spectra using an integrating sphere geometry (Fig. 3A). Diffuse reflection mode was chosen to eliminate the contribution of the (nearly totally) specular reflection from the bare Si substrate. Curves are vertically shifted for clarity but share a common scale. It is readily apparent that as printing speed increases, there is a continual blueshift in the peak reflected wavelength and significant peak broadening, whereas increasing temperature leads to a redshift. This was validated by quantitative peak fitting of the diffuse reflection peaks. The peak center and full width at half maximum (FWHM) of each spectrum are displayed in Fig. 3B and discussed below.
Fig. 3. Optical properties of printed lines correlated to printing speed and substrate temperature.
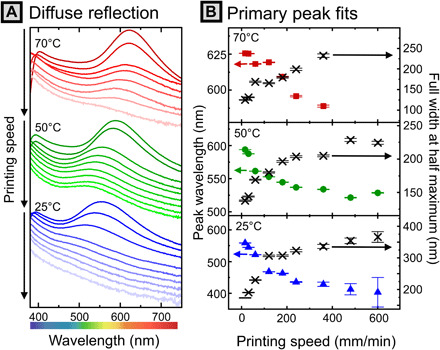
(A) Diffuse reflection spectra obtained using the integrating sphere geometry, vertically shifted for clarity. Y axis represents reflectivity (% versus spectralon standard). (B) Lorentzian peak fits describing peak position and FWHM as a function of printing speed. Error bars denote SE of the fit. Raw spectra can be found in fig. S18.
Peak position and FWHM are strongly correlated to the printing speed. In interpreting these results, it is useful to relate peak reflected wavelength to microstructural parameters (domain d-spacing). For a perfect 1D lamellar PC (Bragg stack), the governing equation for the first-order reflection can be simplified to the following combination of Bragg’s law and Snell’s law (for normally incident light) (1)
| ( 1) |
where λ represents the peak reflected wavelength, d1, d2 represent the thickness of the PDMS and PLA layers, respectively, and n1, n2 represent the refractive indices of each layer. While the as-cast material is by no means a perfect Bragg stack, this equation does allow us to make an estimate of the domain d-spacing and to infer the internal structure. For the estimates reported below, we use the bulk refractive indices of PDMS (1.40) (33) and PLA (1.46) (34) and weight domain sizes based on a volume fraction (ϕPDMS) of 0.61 (see calculation in section S9). As we will show, these estimates are in qualitative agreement with the values obtained more rigorously via SAXS. From the equation, it is apparent that dispersity in domain size can clearly lead to peak broadening, but domain orientation also plays a role, complicating interpretation of the dispersity of real peaks.
For samples printed at a substrate temperature at 25°C, the largest variation in peak wavelength was observed. By increasing printing speed from 15 to 600 mm/min, the peak reflected wavelength was modulated continuously from 403 to 558 nm (range of 155 nm). This corresponds to an estimated change in domain d-spacing (dopt) from 141 to 196 nm. As speed increases at low temperature, peaks become extremely broad, with the FWHM ranging from 200 to nearly 350 nm across the same speed range. Samples printed at 50°C demonstrate a smaller variation of wavelengths, with a redshifted window. By increasing printing speed from 15 to 600 mm/min, the peak reflected wavelength is adjusted from 521 to 594 nm (range of 73 nm), corresponding to estimated dopt of 183 to 209 nm. In this case, peaks are better resolved than at 25°C, with peak widths ranging from 177 to 230 nm. After further increasing the substrate temperature to 70°C (above the solvent boiling point), meniscus instabilities and stochastic boiling of the solvent begin to be apparent at low printing speeds. Nonetheless, there is still apparent modulation of peak position from 588 to 626 nm (range of 38 nm) across a printing speed range of 15 to 360 mm/min, corresponding to estimated dopt from 206 to 220 nm. In this case, the reflectivity of the samples printed at higher speed was too low for meaningful fitting of the peak location. Peak widths are comparable to the 50°C samples, ranging from 125 to 232 nm. In summary, the peak reflected wavelength of PCs of a single BBCP chemistry can be modulated continuously over a wide range from 403 to 626 nm by modulating both printing speed and substrate temperature. This corresponds to a domain size change of ~78 nm predicted by Eq. 1, which we will show in the following section is in good agreement with the value measured by x-ray scattering.
Substrate temperature and printing speed both can affect assembly in complex ways. As the temperature increases, (i) segregation strength χN decreases, (ii) the solvent removal rate increases, and (iii) the chain mobility increases. From the observed trend of increasing domain size with temperature, we suggest that factor (iii) outweighs the thermodynamic and kinetic considerations (i) and (ii), which we predict would both reduce domain size. We also observe a strong correlation of peak width with printing speed, which we attribute to a significant increase in microstructural disorder. Increasing temperature serves to decrease FWHM at the same printing speed. From this, we speculate that increasing molecular mobility aids in the elimination of the orientational and positional defects. We note that the correlation of optical morphology with printing speed does not provide insight into the molecular mechanism or driving force for domain size modulation. Changes in printing speed necessarily convolve changes in the fluid flow field, film thickness (potential confinement effects), and the kinetics of solvent removal. In following sections, we first link the optical properties of our printed films to their microstructure and address each factor, in turn, to elucidate the mechanism of printing speed–modulated nanoscale morphology.
Microstructural analysis indicates that domain size control drives photonic variation
Cross-sectional scanning electron micrographs (Fig. 4A) of cryo-fractured films unambiguously reveal lamellar morphologies. Images taken over large areas (~100 μm2) for samples deposited at all three temperatures under a range of printing speeds (figs. S20 to S23) also demonstrate lamellar morphologies throughout the films. Across different samples, however, the correlation length for lamellar domains (grain size) and the range of orientations observed appear to vary significantly. In the dropcast case, domains appear to be quite large (exceeding 50 layers), although there are significant fractions of lamellae that are not aligned parallel to the substrate (some even perpendicular). Thus, we believe that orientation dispersity is a significant contributor to the large variability in color for dropcast samples (Fig. 1F). In printed films, however, there are many smaller grains (approximately 10 layers), which adopt orientations at a smaller distribution of angles, preferably parallel with the substrate. Printed films are, however, still highly defective 1D PCs, with misoriented grains likely a major contribution to the breadth of the measured reflection peaks.
Fig. 4. Microstructural characterization.
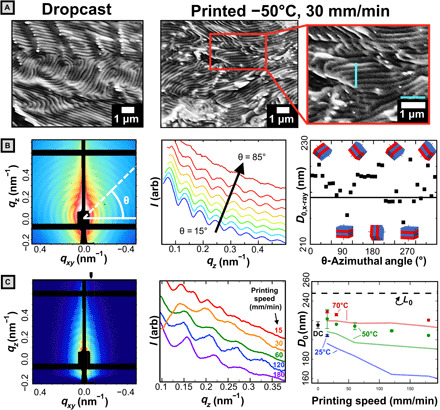
(A) Representative cross-sectional SEM of dropcast and printed films with 1-μm scale and guidelines. (B) Left: 2D SAXS data for dropcast sample with sample azimuthal line-cut (white). Middle: 1D linecuts plotted for various azimuthal angles (θ). Right: d-spacing determined from the q interval between adjacent peaks. Solid black line reflects azimuthally averaged data. (C) Left: 2D SAXS data for a sample printed at 50°C and 120 mm/min. The black tick mark indicates the region integrated for 1D profiles. Middle: 1D SAXS profiles for samples printed at 50°C. Deviation at low q (orange curve) caused by positioning the integrating region at an offset from qxy = 0 to avoid diffuse background intensity. Right: Domain d-spacing calculated from SAXS (solid points) versus printing speed. Error bars on colored points represent the range of two scans. Error bars for dropcast (DC) sample represent the SD of nine measurements across three samples. The dashed line represents the contour length of the bottlebrush estimated with a fixed backbone contour length of 0.62 nm per norbornene repeat unit. Faded lines connect the domain size estimates obtained by application of the Bragg-Snell law to optical peaks reported in Fig. 3.
We do not attempt to make any quantitative measure of domain d-spacing via SEM, however, other than to note that all samples measured appear to show domain d-spacings (dSEM) of approximately 200 nm. The absolute value of the lamellar thickness observed is highly dependent on factors such as fracture angle and distortion of the structure during the rapid cooling and fracture processes, particularly considering the very low glass transition temperature of PDMS (−125.5°C; fig. S5). Furthermore, the alternating bright and dark bands observed are due to both chemical and topographical differences, and in many images, roughness of the fracture plane can easily be mistaken for lamellar domain grain boundaries.
Instead, SAXS experiments were conducted to quantitatively evaluate lamellar domain size (d-spacing). Samples were run in transmission mode but parallel to the substrate at a shallow negative angle (−3 ° < θ < 0°). In this configuration [akin to grazing-transmission SAXS; (35)], long-range lamellar stacking manifests as a series of peaks present at regular intervals in q-space. As before, we first treat the dropcast system and then compare to the printed films. For dropcast samples, the ring-like SAXS pattern (Fig. 4B, left) indicates close to isotropic distribution in lamellar domain orientations. A slight preference for lamellae oriented parallel to the substrate was observed, evidenced by the larger number of higher-order peaks along qz. These observations are consistent with the SEM imaging. Furthermore, there is evidence for a small domain size variation as a function of stacking angle (as reported elsewhere for solvent-cast BCPs) (36). By taking linecuts at 10° increments and evaluated domain d-spacing as plotted in Fig. 4B (middle and right), we determine that domain d-spacings range between 213 and 226 nm. Thus, for dropcast samples, the uncontrolled color variation is confirmed to be due to both domain size and orientation dispersity.
By contrast, for 3D-printed films, the color change can primarily be attributed to domain size variation, while change in domain orientation plays only a minor role. For 3D-printed samples, we observe a much stronger signal from lamellae oriented parallel to the substrate as evidenced by the significantly higher intensity of peaks that center on the qz axis in Fig. 4C (left). Calculation of the 2D orientation parameter (S2D; section S11) (37), for the film printed at 15 mm/min at a substrate temperature of 50°C, yields a value of −0.96, compared to −0.73 for the dropcast case. A value of −1 indicates that lamellae are oriented parallel to the substrate, and a value of 0 indicates isotropy. This suggests that while both cases have lamellae oriented primarily parallel to the substrate, the printed films are more strongly oriented. To calculate domain d-spacing, a vertical integrating region was taken near the qz axis to obtain the 1D line profiles as in Fig. 4C (middle). Domain d-spacings evaluated by averaging the interpeak distance in q-space are shown as symbols in Fig. 4C (right). We note that peak fittings accounted for the systematic absence of even-numbered peaks, indicating that thicknesses of both blocks are nearly identical (see Materials and Methods). For the most systematically examined series (50°C), there is a variation of domain d-spacing from 221 to 204 nm across a range of printing speeds from 15 to 180 mm/min. For samples printed at 70°C, domain sizes at 15 and 30 mm/min are increased by ~10 nm from the 50°C counterparts to 229 and 226 nm, respectively. Similarly, the sample printed at 25°C, 15 mm/min has a domain spacing of 204 nm, 20 nm smaller than that at 50°C. We compare these results with the domain sizes estimated from the optical peak wavelength, as shown by the red, green, and blue lines in Fig. 4C and find generally good agreement. We note that there is a constant offset of ~10 nm between the values of domain d-spacing determined by x-ray and predicted by the simplified optical equation. The discrepancy is likely due to disorder such as misoriented lamellae and d-spacing dispersity that is not captured by the optical equation. As the lamellar normal shifts away from the incident angle of light, the wavelength reflected blueshifts, leading to the observed underestimate of domain size as compared to SAXS measurements (38). Nonetheless, the good qualitative agreement between the optical estimates and x-ray measurements allows us to conclude that domain size change of >70 nm underpins the full range of the color change in 3D-printed films, primarily driven by substrate temperature.
Last, we speculate as to the molecular architecture within domains and the molecular origin of domain spacing modulation. We propose that bottlebrush diblock polymers take on “head-to-head” configurations within lamellar domains, meaning that each chemical domain is composed of a “bilayer,” as depicted in Fig. 2B (inset). From our x-ray data, we find that measured domain sizes (maximum of ~230 nm) are comparable to the length of a single contour length of our bottlebrush polymer [248 nm, based on 0.62 nm per poly(norbornene) repeat unit (9, 19), indicated by the dotted line in Fig. 4C, right]. This is consistent with recent reports of head-to-head configurations for similar BBCP under near-equilibrium conditions, which was reported to be both interface-minimizing and entropically favorable (9). As the domain size spans two molecular layers, this picture suggests significant compression/flexibility of the backbone in the lamella structure, i.e., domains are roughly half as large as they would be if the bottlebrush was fully extended. With this proposed structure, it is possible to rationalize domain size variation as marginal extension/compression of the backbone within domains rather than further extension of nearly rod-like bottlebrush backbones, as would be the case for domains comprising single, interdigitated bottlebrushes.
Topographical characterization and evidence against confinement/flow effects
Having established the microstructural underpinning of the observed color change, we dedicate the rest of this manuscript to discussing the mechanism of domain size modulation. Numerous driving forces for the directed assembly of BCPs have been reported, commonly including confinement (39) and fluid flow gradients (40). In this section, we briefly present our topographical analysis, which suggests that confinement does not play a major role in the domain size selection, and then introduce experimental results which suggest that fluid flow plays only a weak role. Further details are provided in section S12.
The as-printed line patterns were characterized via laser confocal scanning microscopy to measure film thickness profiles. Printed lines exhibit significant surface roughness and a characteristic “bowed” shape (fig. S36A) indicative of strong capillary flow toward the edges during drying. The film thickness, width, and cross-sectional area vary as a function of printing speed (fig. S36B), although all three measures of topography vary similarly and exhibit consistent trends across printing temperatures. We find that across the range of conditions tested, film thicknesses range from 1.5 to 50 μm, and linewidths vary from 118 to 870 μm, with the thickest lines always at lowest speed, as expected.
To evaluate the role of thin-film confinement effects on optical properties, we plot the correlation between peak reflected wavelength and FWHM of printed films and their thickness in fig. S36C. It is quite evident that within each temperature series as film thickness increases, so does the peak reflected wavelength. We furthermore note a strong correlation in the FWHM of the reflection (dispersity in domain size/orientation) with thickness; as thickness increases, FWHM decreases markedly. However, we believe that these correlations belie the true mechanism of domain size selection. We first note that the data show that samples of similar thickness can exhibit peak wavelengths spanning a range of up to 200 nm by modifying printing speed and substrate temperature (fig. S36C). This suggests that film thickness does not play a dominant role on assembly outcomes. It has been frequently reported that maintaining commensurability (integer number of layers) while changing film thickness may lead to domain stretching/compression in ultrathin films (nL < 10) (41). In this study, however, films are significantly thicker, with approximately nL = 15 to 250 layers, and the greatest peak shift present in thicker films. Therefore, we believe that the effect of commensurability on domain size is not significant in this case. Instead, we propose that the correlation between optical properties and film thickness is due to modified drying kinetics. Thinner films are printed at higher speeds and undergo more rapid drying, as we discuss in detail in the following section.
We also assert that flow gradients are unlikely to be the cause of the observed trend. For BCPs, a wealth of literature has demonstrated that flow gradients can align lamellar domains in complex ways (40). Thus, it is reasonable to expect that as we change printing speed over a very wide range, the accompanying increase in strain rate in the meniscus would lead to variation in assembly outcomes. Unexpectedly, we find that this is not the case. By keeping meniscus size and film thickness nearly constant while scaling both printing speed and pressure proportionately, we can isolate the impact of fluid flow while removing the influence of confinement or drying rate. By printing in this way, we observe negligible domain color variation (fig. S43). That is, even after increasing strain rates in both the nozzle (extension) and meniscus (mixed shear and extension) by increasing pressure from 20 to 58.6 kPa and printing speed from 50 to 278.7 mm/min, we find that the shift in the reflection peak value is less than 10 nm. Qualitatively, we find that the color stays remarkably constant even over a range of pressures from 12.5 to 254 kPa and printing speed of 15 to 1841.4 mm/min (fig. S42). Thus, flow gradients are unlikely to be the origin of our color (domain size) selection phenomena, although they quite possibly affect lamellar orientation and the extent of long-range order during printing.
Solution SAXS and in situ optical microscopy support a kinetic trapping mechanism
Kinetic trapping is frequently used to control nonequilibrium structure for solvent-cast films, because the time scale of rapid drying naturally limits the time scale of molecular assembly. Essentially, by controlling processing conditions, it is possible to arrest the evolution toward the equilibrium solid-state structure at an intermediate, nonequilibrium stage. In this section, we first use in situ optical microscopy to demonstrate that the drying time as modulated by the printing speed sets the time scale for microstructural evolution, which is a necessary condition for kinetic trapping. We then combine our previous microstructural analysis of printed films with solution SAXS of the ink to demonstrate that the obtained films do, in fact, exhibit morphology (domain size) intermediate between the equilibrium solution and solid-state structures and thus are likely the result of kinetic trapping.
We first focus on demonstrating that our direct-write 3D printing scheme is, in fact, able to induce kinetic trapping in printed films by precisely controlling drying time. Through in situ microscopy of printed films (Fig. 5, A to C), we confirm this by demonstrating (i) that drying time decreases with increasing printing speed and (ii) that assembly time has a corresponding decrease. To accomplish the first task, we captured a series of side-view videos of the printing meniscus in the transmission geometry (movie S5 and section S14) to track wet film thickness over time with good spatial and temporal resolutions (~10 μm and 0.03 s, respectively). Videos were analyzed in MATLAB to yield the meniscus height profiles depicted in Fig. 5A. Starting from the time the nozzle passes (t = 0), the height of the meniscus falls before reaching a baseline value. This time interval is taken to be the drying time and is plotted in the inset for a range of different printing speeds. The results clearly indicate that faster printing speeds did, in fact, reduce the drying time of the films.
Fig. 5. In situ optical monitoring of the 3D printing process.
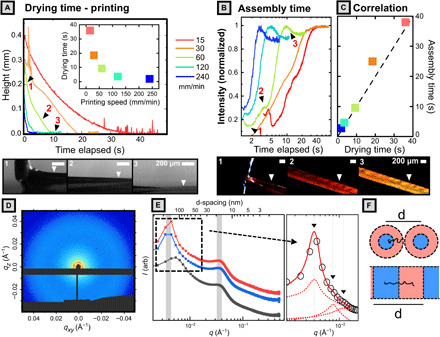
Full analysis details and code are provided in section S14. Images beside plot correspond to the numbered ticks. All data shown in (A) to (C) were obtained at a substrate temperature of 50°C and applied pressure of 30 kPa. (A) Meniscus height profile versus time for samples 3D printed at various printing speed. Images below are snapshots of transmission-mode video taken for sample printed at 60 mm/min. Inset contains plots of drying time (x intercept) versus printing speed. (B) Intensity plotted against elapsed time for 3D-printed samples. Images below are snapshots from the reflection-mode video corresponding to labeled tick marks for sample (60 mm/min) within plot. (C) Assembly time (peak intensity) plotted against drying time, showing close matching between the two. Dashed line indicates a slope of one. (D) 2D SAXS pattern for solution (100 mg/ml) of BBCP in THF. (E) 1D azimuthally averaged profiles for backbone DP of 400 (top two curves) and 200 (lower curve). Inset depicts fitting of the low-q peak to a lamellar structure factor. (F) Cartoon of bottlebrush conformation in micellar and lamellar assemblies.
We then paired this analysis with a series of reflection geometry videos (movie S6 and section S14) where we track the intensity of reflection of an area of the film during 3D printing as a means to obtain assembly time (Fig. 5B). For a range of printing speeds, we observe that intensity is initially low, goes through a rapid rise, and then plateaus. While the color is dictated by domain size only, the reflected intensity is governed by two parameters: refractive index contrast between domains and the number of domains. As the solvent dries, the refractive index contrast increases, while unimers and disordered micelles progressively add to the long-range–ordered lamella to grow lamellar domains, leading to the observed saturation of reflected intensity. As a result, the location of the peak in intensity can conceptually represent upper bounds for the “assembly time” of the system and is plotted in Fig. 5C against the drying time. We find that there is a close matching of assembly time and drying time, fulfilling a critical requirement of kinetic trapping. We further note that in all cases, the assembly times (<40 s) for printed samples are an order of magnitude lower than those observed for dropcast samples (~200 s), providing indirect support for the presence of metastable domains.
Through SAXS experiments on the initial ink solution, we can clarify the initial state of polymer chains in solution. The 1D azimuthally averaged data for BBCP with backbone DP of 400 (red and blue curves) and DP of 200 (gray curve) are shown in Fig. 5E. The high-q peak for both samples occurs at q ~ 0.035 Å−1, corresponding to a correlation length of 18.0 nm. We attribute this structure factor peak to the correlation between individual bottlebrush polymer chains across their short axis (radial direction), as reported elsewhere (42). This conclusion is supported by the observation that for two BBCP with identical arm lengths, the correlation peak occurs at the same q value and is insensitive to backbone length. By contrast, the low-q peak is very sensitive to backbone length and provides information on the PLA/PDMS compositional fluctuations along the backbone direction. SEM imaging of the freeze-dried solution (fig. S8) indicates that at 100 mg/ml, the solution is largely composed of disordered spherical micelles that have begun to fuse/overlap into possibly lamella structures. Supporting this, close inspection of the scattering profile for BBCP with backbone DP of 400 repeat units (Fig. 5E, inset) reveals the presence of two very weak diffraction orders suggesting lamella structures. From the very broad primary peak and weak diffraction orders, we infer that BBCP in the initial ink primarily form disordered/liquid-like micelles, which have begun to fuse to form lamellae. Fitting of the primary low-q peak gives a correlation length of 157 ± 8 nm, while the locations of the two diffraction peaks indicate weak lamellar stacking with almost the same d-spacing. Figure 5F relates this correlation length to intermicellar spacing/lamellar spacing.
We can now use this result to evaluate whether printed films do exhibit a morphology intermediate to the equilibrium solution and solid states. Bottlebrushes appear to adopt a less extended conformation in micellar solution than in the printed films, as indicated by the increase in d-spacing from 157 nm in solution to greater than 200 nm in the printed films. By comparison, the equilibrium domain spacing in thermally annealed films is approximately 212 nm, exhibiting a far more extended polymer conformation. If the kinetic trapping mechanism holds, then samples that should exhibit the closest morphology to the solution state are those printed at room temperature at high speeds; the estimate domain spacing of ~160 nm at this printing condition is in good agreement with the measured solution d-spacing. These results are consistent with the existing BCP solution theory: During drying from a weakly selective solvent, BCP microphase separation is promoted by the rapidly increasing segregation strength of the mixture (43). This process can only continue, however, for as long as there remains sufficient solvent to suppress the glass transition temperature (Tg) and afford molecular mobility (43, 44). We speculate that the formation of domains with larger d-spacing and more extended chain conformation is thermodynamically favored in that it reduces both interfacial area and the bending energy of constituent BBCP. Thus, in situ optical microscopy of the printing process and examination of the solution-state structure both indicate that kinetic trapping is the mechanism for domain size control.
SVA further validates the kinetic trapping mechanism
We have so far proposed that printing at higher speeds leads to the observed blueshift in color by trapping metastable, smaller lamellar d-spacings. To test this hypothesis, we use solvent vapor annealing (SVA) to “relax” the metastable conformation of BBCP in printed films. If kinetic trapping is the cause of d-spacing modulation, then exposing a series of samples printed at different speeds to solvent vapor and allowing them to equilibrate before the removal of solvent should eliminate the impact of different solvent removal rates and so cause d-spacings to converge. To test this, we printed two sets of BBCP samples at substrate temperatures of 25° and 50°C and at printing speeds of 30, 90, and 150 mm/min (for 12 total samples). As expected, as-printed samples show differences in peak reflected wavelength as a function of printing speed and temperature. Within each set, these samples show good dimensional and optical reproducibility (figs. S47 to S50). These conditions were chosen to cover a large range of film thicknesses (~3 to 30 μm) and initial color range (blue-green to orange).
Snapshots of the annealing process are shown in Fig. 6A. Upon exposure to solvent vapor, printed thin films showed a gradual redshift in color over a period of ~1 min, before becoming largely colorless (even under very high light exposure). The system was held for a total solvent exposure time of 300 s to allow for molecular rearrangement in the highly mobile solvent-laden state before the removal of solvent vapor, with the film blueshifting to its stable color over ~10s of seconds. Figure 6B (Tprint = 25°C) and Fig. 6C (Tprint = 50°C) show the ultraviolet-visible (UV-Vis) reflection spectra for the line patterns printed at 30 mm/min (solid), 90 mm/min (dashed), and 150 mm/min (dashed-dotted) before annealing (lower curves), after annealing at the print temperature (middle curves), and after annealing at higher temperatures (top curves). These spectra were then fit, and the peak reflectance is plotted in Fig. 6D. Comparison of samples printed and annealed at the same temperature (labeled 25/25 and 50/50 in Fig. 6D) clearly shows that after SVA, the d-spacing for samples printed at different speeds converges to a single value. That is, by eliminating variations in processing time, we eliminate the difference in optical properties, supporting our kinetic trapping hypothesis.
Fig. 6. SVA experiments.
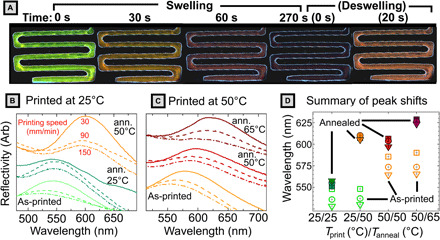
(A) Microscopy images during the SVA process for a sample printed at 30 mm/min at 25°C and annealed at 50°C. At time = 0 s, solvent was introduced to the chamber, and the sample was allowed to swell for 300 s before the cover was removed and the sample deswelled. (B and C) Ultraviolet-visible (UV-Vis) reflection spectra for the line patterns printed at 30 mm/min (solid), 90 mm/min (dashed), and 150 mm/min (dashed-dotted) before annealing (ann.) (lower curves), after annealing at the printing temperature (middle curves), and a higher temperature (top curves). For reflectivity measurements, the detector spot size encompasses the entire printed sample. (D) Plot of peak reflected wavelength for the 12 samples before (hollow symbol) and after (filled symbols) annealing for each printing speed: 30 mm/min (triangles), 90 mm/min (circles), and 150 mm/min (squares). Vertical error bars denote the SE from fitting peaks to Lorentzian profiles.
It is also immediately apparent that SVA at the higher temperature markedly increases domain size for all samples regardless of printing speed, even exceeding the peak reflectance obtained via long thermal annealing (605 nm). This phenomenon is also observed for dropcast samples (fig. S51). By solvent annealing even at 25°C, there is a pronounced redshift in reflectance, and further annealing at higher temperatures homogenizes droplet appearance indicating relaxation of defects. Thus, direct-write 3D printing followed by rapid SVA has utility as a method for accessing higher domain sizes in a consistent, macroscopically patterned way. There is, in fact, no need for a closed SVA chamber at all—if combined with a rastering solvent vapor nozzle as recently reported by Epps et al. (45), then this method could readily be used to create spatially localized regions of large domain size that are not accessible via the original 3D printing process due to solvent boiling at high temperature.
CONCLUSIONS
In conclusion, we demonstrate a direct-write 3D printing approach for both spatial and functional patterning of PDMS-b-PLA BBCP PCs. We show that by systematic variation of printing speed and substrate temperature during 3D printing, programmatic variation of film color can be achieved—tuning peak reflected wavelength continuously across a range of 403 to 626 nm. Via SEM and synchrotron SAXS analysis, we show that printed films comprise lamellar structures with periodicity on the order of 200 nm and that the microstructural underpinning of the observed optical variation is tuning of domain d-spacing over a range of greater than 70 nm. Last, we investigate the mechanism of domain size selection. After excluding the roles of volume confinement and fluid flow, we use in situ optical microscopy to provide evidence for kinetic trapping as the mechanism for microstructural tunability. Subsequent SVA experiments support this conclusion by showing that metastable domains can relax and converge to larger, equilibrium domain spacings when allowed more time for assembly.
BBCPs are a new and exciting class of materials whose hierarchical structure grants them great applicability to a variety of fields including photonics, surfactants, and organic electronics. Significant progress has been and is being made in growing understanding of their chemistry, fundamental physics, and equilibrium self-assembly behavior. We hope that this work can stimulate more investigation into and highlight the importance of nonequilibrium assembly techniques for bottlebrushes and other advanced functional materials. Using kinetic trapping to optimize materials properties is not a new principle; for at least two millenia (46), materials scientists have trapped metastable phases of metal alloys and oxides via quenching (47). In this work, we show that a modern integrated hardware and software approach to direct-write 3D printing can leverage this technique to unlock functional property modulation and present a versatile testbed for the nonequilibrium study of other functional materials.
MATERIALS AND METHODS
[(H2IMes)(3-Br-py)2(Cl)2Ru = CHPh], G3 was synthesized according to literature (48). 5-Norbornene-2-methanol was synthesized according to literature (mixture of 20% exo/endo used) (49). Full details on the synthesis of PDMS-b-PLA BBCPs and extended methods can be found in the Supplementary Materials.
Direct-write 3D printing apparatus
The apparatus depicted in Fig. 2A of the main text was designed and constructed for this work. Fully documented software source files and schematics of custom hardware are available or linked in section S5. The 3D printer hardware is built around the Aleph Objects Lulzbot Taz 6 printer with a custom 3D-printed mount for disposable syringes. A glass plate was mounted on top of the supplied Polyetherimide-coated glass heating bed, and a thermocouple was attached to the top surface for independent temperature verification. The 3D printer firmware (Marlin) was modified to shrink the motion command buffer to one value, to send an “end-of-motion” confirmation, and to use a baud rate of 115,200. A link to the modified firmware (Marlinv1.2BP) can be found in the Supplementary Materials. The Ultimus V pneumatic extruder, disposable Amber 3-ml polypropylene syringe barrels, white polyethylene pistons, and 32-gauge standard nozzles were acquired from Nordson EFD. Gas was supplied via an external dry nitrogen cylinder. A portable fume snorkel (Sentry Air Systems) was placed several inches from the nozzle to protect the user (very mild forced convection). The open-source program PolyChemPrint v2.2 was written to control both extruder and printer axes. Software was run on a low-spec, salvaged PC running the free Linux variant Debian 9 “Stretch.” The system is highly flexible and has been used successfully within our group for patterned deposition of a variety of materials including organic conductors/semiconductors, particle-laden hydrogels, and chocolate.
Direct-write 3D printing process
In the present study, the PDMS-b-PLA BBCP was dissolved in THF (Macron) at 100 mg/ml. It was found that for concentrations that were significantly lower, (1 to 10 mg/ml), deposited films were extremely thin and entirely colorless. Increasing concentration up to 50 mg/ml resulted in colored films with significant nonuniformities. At 100 mg/ml, continuous, vibrantly colored films could be printed over a wide range of printing speeds (as reported in the main text). After stirring at room temperature for 2 hours, a homogeneous solution with a faint angle-independent purple color was formed. Upon aging up to ~2 hours without stirring, the color intensified. We note that the samples printed before 2 hours of aging exhibited nonuniformities, with spots of the same color as the aged samples alternating with colorless regions. There was no apparent impact upon aging the solution past 2 hours. This may indicate that the formation of “ripened” solution-phase micellular structures is a critical component of successful solution deposition, but further study is needed.
After loading solutions into the disposable amber syringe barrels and capping with a piston, air was removed from the syringe by sealing the end and applying pressure to the piston back before installing the 32-gauge syringe cap. Approximately 2 ml of solution (maximum) was loaded at a time to avoid evaporative loss during extended printing sessions. Samples were printed onto bare silicon wafer segments (University Wafers no. 1113) that had been sonicated for 5 min sequentially in toluene, acetone, and isopropanol, followed by plasma treatment (Harrick Plasma PDC-001, 6 min at 250 mtorr in dry air, 30 W) for no more than 30 min before use. To set printing height, the nozzle was positioned between an inexpensive light-emitting diode backlight and a microscope camera and moved into contact with the substrate before being raised to a height of 100 μm. Before execution of printing code, the nozzle was “primed” at low pressure and cleaned with a foam swab coated in acetone to remove any blockage at the tip due to solidified polymer. It was found that modifying printing speed or applied pressure has essentially the same effect on color; thus, applied pressure was held fixed at 30 kPa.
For meanderline patterns, a parameterized script was written with user-adjustable printing speed, dimensions, pressure, etc. For complex “chameleon” patterns, we have incorporated the ability to import XYZ motion paths from G-Code files prepared by common slicing programs (e.g., slic3r and Cura) or (as here) using the free plugin “G-CodeTools” for open-source vector image editing program Inkscape. Upon importing a G-Code file, PolyChemPrint strips away all information except the motion and extruder commands (if present) and allows the user to specify printing speed, travel speed, temperature, the XYZ home position, and other relevant printing parameters. If extruder commands are present, then the software can either convert from extruder length units to pressure commands with a user-supplied scaling function or simply ignore them and use a fixed pressure specified by the user. The system fully supports stopping flow for travel moves and can emulate filament retraction by the application of vacuum. Further details are available in section S5.
Optical and topographical characterization
All static images of dropcast/printed films are taken by microscope cameras under diffuse (ring) lights at low magnification. Diffuse reflection spectra were collected using a Varian Cary 5G instrument with the integrating sphere attachment located at the Illinois Materials Research Laboratory (MRL). Before measurement, the “start bead” and corners of meanderlines were cut and delaminated with a clean razor blade to leave only steady-state regions. Reference spectra were taken with respect to a Spectralon standard.
Optical profiling over large regions (>1 mm by 1 mm) was performed using a Keyence VK-X1000 3D laser scanning confocal microscope in “film top” mode at the Illinois MRL. Measurements were validated against values obtained using a Sloan Dektak3ST contact profilometer at the Illinois MRL.
In situ optical microscopy was taken using a XIMEA xiQ USB3 camera connected to a high-magnification Navitar lens system purchased from W. Nuhsbaum Inc. We note that top-view videos were taken at ~20° from the substrate normal to track the evolution of structure color over time. As a result of the viewing angle and the use of strong lighting at ~45°, the color seen in the videos does not match the color apparent during normally incident diffuse reflection; however, the evolution of the color intensity is consistent. Videos were obtained at moderate frame rate (~45 fps) and analyzed frame by frame using custom MATLAB (The MathWorks Inc., Natick, MA, USA) scripts. Further software details are provided in section S14.
Scanning electron microscopy
Details on freeze-drying of solution samples are present in section S4. Solid samples were freeze-fractured in liquid nitrogen to preserve their internal structure. Best results were obtained by delaminating printed films from silicon substrates with a clean razor blade and placing between copper tape with a portion of the sample unsupported. After placing the sample in liquid nitrogen for an extended period (~20 min), the exposed portion of the film was scraped within the nitrogen bath to create a clean interface. Samples were then mounted in a mini vise holder and coated with ~10-nm Au-Pd using an Emitech K575 sputter coater at the Illinois MRL. Final micrographs were obtained using a JEOL JSM-7000F Analytical SEM at 3-kV accelerating voltage at the Illinois MRL and processed using the software package ImageJ2 (50).
Small-angle x-ray scattering
Experiments were performed at beamline 12-ID-B of the Advanced Photon Source (Lemont, IL) with a beam energy of 13.3 keV using the Pilatus 2M 2D detector at a sample-detector distance of 3.6 m. Q-calibration was performed against a silver behenate standard. 2D data reduction was performed using the Nika package for Igor Pro (WaveMetrics, Lake Oswego, OR, USA) developed by J. Ilavsky (51).
Because of their high surface roughness/curvature, printed films were unsuitable for running in grazing incidence configuration, which is more typical for thin films. Instead, SAXS experiments were performed in transmission mode at a shallow negative angle. Spectra were restricted to the region of q < ~0.4 nm−1 before peaks were deconvoluted and fit using Igor Pro’s built-in multi-peak fitting function. Peaks were fit sequentially from weakest to strongest as Voigt functions before refitting the entire curve. For lamellar morphology, constructive interference from successive layers causes peak spacing to follow Eq. 2.
| (2) |
where n is the order of the diffraction peak and dx is the domain d-spacing. When the lowest order of diffraction is clearly visible, n takes the value 1, and the equation can be solved directly for d-spacing. In our case, the predicted domain size (~230 nm) puts the estimated first-order peak at q = 0.0027 A−1, which is below the minimum q value we can detect. By rearranging Eq. 3 in series form, however, an alternate approach can be taken on the basis of the difference in peak location between successive peaks (Eq. 3).
| (3) |
Here, we present domain sizes obtained by averaging the predicted spacing from at least four diffraction orders to minimize the impact of diffuse scattering in biasing the low q peak positions. For lamellar domains of a real symmetric BCPs, peaks appear at integer multiples of the fundamental peak, with every other peak (n = 2, n = 4, and n = 6) significantly weakened (52). In our case, we predict that ϕPDMS ~ 0.61 is based on molecular weight, synthetic parameters, and the bulk density of the arm species (calculation is shown in the Supplementary Materials), and we find that for most of our spectra, we do not observe well-resolved even-numbered peaks (particularly at higher q). As a result, the peak spacing we obtain must be multiplied by 2 to reflect the domain d-spacing of the lamellar repeat unit. In addition, all samples manifest a large, diffuse signal along qz, requiring us to use a large vertical beamstop and further obscure the weaker even-numbered peaks. As a result, we choose a region of integration for peak fitting that is offset from the center line, further reducing the signal we observe. This source of noise complicates data analysis but is not so prevalent as to obscure the strong, periodic odd-numbered lamellar stacking peaks. Full SAXS analysis and spectra are provided in the Supplementary Materials.
Solvent vapor annealing
The apparatus for annealing during in situ microscopy comprised a small glass petri dish sitting (base down) within a larger petri dish (also base down) on a hot plate set to the target temperature underneath a microscope camera (fig. S46). In the absence of solvent vapor, structural rearrangement is not evident after several hours even at the highest temperature used (65°C). Into the small petri dish were placed a metal weight and the desired sample. An excess of THF solvent was injected into the larger petri dish, and the system was partially capped with a glass petri dish cover (preheated to 150°C to avoid condensation) and held closed for 300 s. Then, the dish is uncapped, and the remaining THF solvent was removed via syringe from the reservoir, and the sample could deswell. All experiments took place in a fume hood with strong forced convection.
Supplementary Material
Acknowledgments
We would like to thank C. Sing, S. Rogers, M. Wade, S. Dutta, J. Kurtz, and K. Park for intellectual discussions regarding this work. Experiments were carried out, in part, in the MRL Central Research Facilities, University of Illinois. PolyAnalytik Inc. (London, Canada) is acknowledged for performing triple-detection GPC. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by the Argonne National Laboratory under contract no. DE-AC02-06CH11357. We gratefully acknowledge the assistance and advice of A. Quental, J. Strzalka, and R. Zeigler at the X-ray Science Division beamlines 12-ID-B and 8-ID-E at the Advanced Photon Source, Argonne National Laboratory. Funding: This work was supported by the NSF under DMREF award no. DMR-1727605. J.K. and Y.D. acknowledge partial support by the NSF CAREER award under grant no. NSF DMR 18-47828. Major funding for the 500-MHz Bruker CryoProbe was provided by the Roy J. Carver Charitable Trust to the University of Illinois School of Chemical Sciences NMR Lab. The authors thank Umicore for the gift of Grubbs Catalyst. Author contributions: The manuscript was prepared through contributions of all authors. All synthetic procedures and chemical characterization were carried out by D.J.W. with guidance from D.G. J.K. assisted in equipment design, developing MATLAB code, and performing solution-state SAXS experiments mentioned in section S4. All other experiments were performed by B.B.P., assisted by D.H.K., with guidance from Y.D. B.L. of the Argonne National Laboratory provided invaluable insight and assistance with running and analyzing SAXS experiments. All authors have given approval to the final version of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/24/eaaz7202/DC1
REFERENCES AND NOTES
- 1.Liberman-Martin A. L., Chu C. K., Grubbs R. H., Application of bottlebrush block copolymers as photonic crystals. Macromol. Rapid Commun. 38, 10.1002/marc.201700058, (2017). [DOI] [PubMed] [Google Scholar]
- 2.J. D. Joannopoulos, S. G. Johnson, J. N. Winn, R. D. Meade, Photonic Crystals: Molding the Flow of Light (Princeton Univ. Press, ed. 2, 2008). [Google Scholar]
- 3.Bates C. M., Bates F. S., 50th Anniversary Perspective: Block polymers—Pure potential. Macromolecules 50, 3–22 (2017). [Google Scholar]
- 4.Appold M., Grune E., Frey H., Gallei M., One-step anionic copolymerization enables formation of linear ultrahigh-molecular-weight block copolymer films featuring vivid structural colors in the bulk state. ACS Appl. Mater. Interfaces 10, 18202–18212 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Mapas J. K. D., Thomay T., Cartwright A. N., Ilavsky J., Rzayev J., Ultrahigh molecular weight linear block copolymers: Rapid access by reversible-deactivation radical polymerization and self-assembly into large domain nanostructures. Macromolecules 49, 3733–3738 (2016). [Google Scholar]
- 6.Sveinbjörnsson B. R., Weitekamp R. A., Miyake G. M., Xia Y., Atwater H. A., Grubbs R. H., Rapid self-assembly of brush block copolymers to photonic crystals. Proc. Natl. Acad. Sci. U.S.A. 109, 14332–14336 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verduzco R., Li X., Pesek S. L., Stein G. E., Structure, function, self-assembly, and applications of bottlebrush copolymers. Chem. Soc. Rev. 44, 2405–2420 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Chae C.-G., Yu Y.-G., Seo H.-B., Kim M.-J., Grubbs R. H., Lee J.-S., Experimental formulation of photonic crystal properties for hierarchically self-assembled POSS–bottlebrush block copolymers. Macromolecules 51, 3458–3466 (2018). [Google Scholar]
- 9.Dalsin S. J., Rions-Maehren T. G., Beam M. D., Bates F. S., Hillmyer M. A., Matsen M. W., Bottlebrush block polymers: Quantitative theory and experiments. ACS Nano 9, 12233–12245 (2015). [DOI] [PubMed] [Google Scholar]
- 10.Boyle B. M., Heinz O., Miyake G. M., Ding Y., Impact of the pendant group on the chain conformation and bulk properties of norbornene imide-based polymers. Macromolecules 52, 3426–3434 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boyle B. M., French T. A., Pearson R. M., McCarthy B. G., Miyake G. M., Structural color for additive manufacturing: 3D-printed photonic crystals from block copolymers. ACS Nano 11, 3052–3058 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rzayev J., Molecular bottlebrushes: New opportunities in nanomaterials fabrication. ACS Macro Lett. 1146–1149 (2012). [DOI] [PubMed] [Google Scholar]
- 13.Lin T.-P., Chang A. B., Luo S.-X., Chen H.-Y., Lee B., Grubbs R. H., Effects of grafting density on block polymer self-assembly: From linear to bottlebrush. ACS Nano 11, 11632–11641 (2017). [DOI] [PubMed] [Google Scholar]
- 14.Sheiko S. S., Sumerlin B. S., Matyjaszewski K., Cylindrical molecular brushes: Synthesis, characterization, and properties. Prog. Polym. Sci. 33, 759–785 (2008). [Google Scholar]
- 15.Dutta S., Wade M. A., Walsh D. J., Guironnet D., Rogers S. A., Sing C. E., Dilute solution structure of bottlebrush polymers. Soft Matter 15, 2928–2941 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Fredrickson G. H., Bates F. S., Dynamics of block copolymers: Theory and experiment. Annu. Rev. Mater. Sci. 26, 501–550 (1996). [Google Scholar]
- 17.Rzayev J., Synthesis of polystyrene−polylactide bottlebrush block copolymers and their melt self-assembly into large domain nanostructures. Macromolecules 42, 2135–2141 (2009). [Google Scholar]
- 18.Gu W., Huh J., Hong S. W., Sveinbjornsson B. R., Park C., Grubbs R. H., Russell T. P., Self-assembly of symmetric brush diblock copolymers. ACS Nano 7, 2551–2558 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Xia Y., Olsen B. D., Kornfield J. A., Grubbs R. H., Efficient synthesis of narrowly dispersed brush copolymers and study of their assemblies: The importance of side chain arrangement. J. Am. Chem. Soc. 131, 18525–18532 (2009). [DOI] [PubMed] [Google Scholar]
- 20.Sunday D. F., Chang A. B., Liman C. D., Gann E., Delongchamp D. M., Thomsen L., Matsen M. W., Grubbs R. H., Soles C. L., Self-assembly of ABC bottlebrush triblock terpolymers with evidence for looped backbone conformations. Macromolecules 51, 7178–7185 (2018). [PMC free article] [PubMed] [Google Scholar]
- 21.Semenov A. N., Contribution to the theory of microphase layering in block-copolymer melts. Sov. Phys. JETP 61, 733–742 (1985). [Google Scholar]
- 22.Macfarlane R. J., Kim B., Lee B., Weitekamp R. A., Bates C. M., Lee S. F., Chang A. B., Delaney K. T., Fredrickson G. H., Atwater H. A., Grubbs R. H., Improving brush polymer infrared one-dimensional photonic crystals via linear polymer additives. J. Am. Chem. Soc. 136, 17374–17377 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Miyake G. M., Piunova V. A., Weitekamp R. A., Grubbs R. H., Precisely tunable photonic crystals from rapidly self-assembling brush block copolymer blends. Angew. Chem. Int. Ed. 51, 11246–11248 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Vatankhah-Varnosfaderani M., Keith A. N., Cong Y., Liang H., Rosenthal M., Sztucki M., Clair C., Magonov S., Ivanov D. A., Dobrynin A. V., Sheiko S. S., Chameleon-like elastomers with molecularly encoded strain-adaptive stiffening and coloration. Science 359, 1509–1513 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Song D.-P., Jacucci G., Dundar F., Naik A., Fei H.-F., Vignolini S., Watkins J. J., Photonic resins: Designing optical appearance via block copolymer self-assembly. Macromolecules 51, 2395–2400 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ngo T. D., Kashani A., Imbalzano G., Nguyen K. T. Q., Hui D., Additive manufacturing (3D printing): A review of materials, methods, applications and challenges. Compos. Part B Eng. 143, 172–196 (2018). [Google Scholar]
- 27.He Y., Yang F., Zhao H., Gao Q., Xia B., Fu J., Research on the printability of hydrogels in 3D bioprinting. Sci. Rep. 6, 29977 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewis J. A., Direct ink writing of 3D functional materials. Adv. Funct. Mater. 16, 2193–2204 (2006). [Google Scholar]
- 29.Norman J., Madurawe R. D., Moore C. M. V., Khan M. A., Khairuzzaman A., A new chapter in pharmaceutical manufacturing: 3D-printed drug products. Adv. Drug Deliv. Rev. 108, 39–50 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Walsh D. J., Guironnet D., Macromolecules with programmable shape, size, and chemistry. Proc. Natl. Acad. Sci. U.S.A. 116, 1538–1542 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maschke U., Wagner T., Coqueret X., Synthesis of high-molecular-weight poly(dimethylsiloxane) of uniform size by anionic polymerization, 1. Initiation by a monofunctional lithium siloxanolate. Die Makromolekulare Chemie 193, 2453–2466 (1992). [Google Scholar]
- 32.Lohmeijer B. G. G., Pratt R. C., Leibfarth F., Logan J. W., Long D. A., Dove A. P., Nederberg F., Choi J., Wade C., Waymouth R. M., Hedrick J. L., Guanidine and amidine organocatalysts for ring-opening polymerization of cyclic esters. Macromolecules 39, 8574–8583 (2006). [Google Scholar]
- 33.J. E. Mark, Polymer Data Handbook (Oxford Univ. Press, 1999). [Google Scholar]
- 34.Garlotta D., A literature review of poly(lactic acid). J. Polym. Environ. 9, 63–84 (2001). [Google Scholar]
- 35.Mahadevapuram N., Strzalka J., Stein G. E., Grazing-incidence transmission small angle x-ray scattering from thin films of block copolymers. J. Polym. Sci. B 51, 602–610 (2013). [Google Scholar]
- 36.Zhang J., Posselt D., Smilgies D.-M., Perlich J., Kyriakos K., Jaksch S., Papadakis C. M., Lamellar diblock copolymer thin films during solvent vapor annealing studied by GISAXS: Different behavior of parallel and perpendicular lamellae. Macromolecules 47, 5711–5718 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tao Y., Zohar H., Olsen B. D., Segalman R. A., Hierarchical nanostructure control in rod−coil block copolymers with magnetic fields. Nano Lett. 7, 2742–2746 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Edrington A. C., Urbas A. M., DeRege P., Chen C. X., Swager T. M., Hadjichristidis N., Xenidou M., Fetters L. J., Joannopoulos J. D., Fink Y., Polymer-based photonic crystals. Adv. Mater. 13, 421–425 (2001). [Google Scholar]
- 39.Albert J. N. L., Epps T. H., Self-assembly of block copolymer thin films. Mater. Today 13, 24–33 (2010). [Google Scholar]
- 40.M. Cloitre, D. Vlassopoulos, Block copolymers in external fields: Rheology, flow-induced phenomena, and applications, in Applied Polymer Rheology, M. Kontopoulou, Ed., (John Wiley & Sons Inc., 2011), pp 209–239. [Google Scholar]
- 41.Marencic A. P., Register R. A., Controlling order in block copolymer thin films for nanopatterning applications. Annu. Rev. Chem. Biomol. Eng. 1, 277–297 (2010). [DOI] [PubMed] [Google Scholar]
- 42. Bottle-brush macromolecules in solution: Comparison between results obtained from scattering experiments and computer simulations. Polymer 47, 7318–7327 (2006). [Google Scholar]
- 43.Lodge T. P., Pudil B., Hanley K. J., The full phase behavior for block copolymers in solvents of varying selectivity. Macromolecules 35, 4707–4717 (2002). [Google Scholar]
- 44.Sinturel C., Vayer M., Morris M., Hillmyer M. A., Solvent vapor annealing of block polymer thin films. Macromolecules 46, 5399–5415 (2013). [Google Scholar]
- 45.Luo M., Scott D. M., Epps III T. H., Writing highly ordered macroscopic patterns in cylindrical block polymer thin films via raster solvent vapor annealing and soft shear. ACS Macro Lett. 4, 516–520 (2015). [DOI] [PubMed] [Google Scholar]
- 46.Mackenzie D. S., History of quenching. Int. Heat Treatment Surface Eng. 2, 68–73 (2013). [Google Scholar]
- 47.Jones H., A perspective on the development of rapid solidification and nonequilibrium processing and its future. Mater. Sci. Eng. A 304–306, 11–19 (2001). [Google Scholar]
- 48.Love J. A., Morgan J. P., Trnka T. M., Grubbs R. H., A practical and highly active ruthenium-based catalyst that effects the cross metathesis of acrylonitrile. Angew. Chem. 114, 4207–4209 (2002). [DOI] [PubMed] [Google Scholar]
- 49.Radzinski S. C., Foster J. C., Chapleski R. C., Troya D., Matson J. B., Bottlebrush polymer synthesis by ring-opening metathesis polymerization: The significance of the Anchor Group. J. Am. Chem. Soc. 138, 6998–7004 (2016). [DOI] [PubMed] [Google Scholar]
- 50.Rueden C. T., Schindelin J., Hiner M. C., DeZonia B. E., Walter A. E., Arena E. T., Eliceiri K. W., ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18, 529 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ilavsky J., Nika: Software for two-dimensional data reduction. J. Appl. Cryst. 45, 324–328 (2012). [Google Scholar]
- 52.R. J. Roe, Methods of X-Ray and Neutron Scattering in Polymer Science; Topics in polymer science (Oxford Univ. Press, 2000). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/24/eaaz7202/DC1