

Summary
We report a comprehensive molecular analysis of 34 cases of small cell carcinoma (SCC) and 84 cases of conventional urothelial carcinoma (UC), with The Cancer Genome Atlas cohort of 408 conventional UC bladder cancers used as the reference. SCCs showed mutational landscapes characterized by nearly uniform inactivation of TP53 and were dominated by Sanger mutation signature 3 associated with loss of BRCA1/2 function. SCCs were characterized by downregulation of luminal and basal markers and were referred to as double-negative. Transcriptome analyses indicated that SCCs displayed lineage plasticity driven by a urothelial-to-neural phenotypic switch with a dysregulated epithelial-to-mesenchymal transition network. SCCs were depleted of immune cells, and expressed high levels of the immune checkpoint receptor, adenosine receptor A2A (ADORA2A), which is a potent inhibitor of immune infiltration. Our observations have important implications for the prognostication and development of more effective therapies for this lethal bladder cancer variant.
Subject Areas: Biological Sciences, Genomics, Cancer Systems Biology, Cancer, Transcriptomics
Graphical Abstract
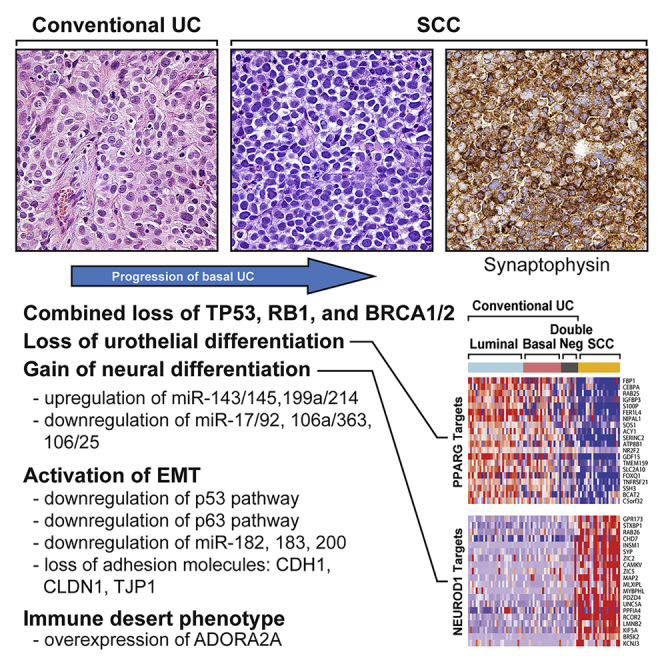
Highlights
-
•
SCCs show TP53/RB1 loss with mutational signature of BRCA1/2 loss of function
-
•
SCCs are driven by neural phenotypic switch with dysregulated EMT network
-
•
SCCs show depleted immune phenotype with upregulation of ADORA2A
Biological Sciences; Genomics; Cancer Systems Biology; Cancer; Transcriptomics
Introduction
The ability of cells to acquire alternative differentiation-associated phenotypes, referred to as lineage plasticity, is a common feature of embryogenesis and is also intrinsic to malignant transformation. Mild to moderate deviation from the tissue-of-origin terminal differentiation profile as a consequence of either stem cell transformation or dedifferentiation is a general feature of tumor initiation, but major changes in lineage identity can occur in tumor progression (Yuan et al., 2019). Such changes are associated with dramatic, microscopically evident phenotypic switches coupled with the acquisition of clinically aggressive behaviors. Here we show that the progression of urothelial bladder cancer to the clinically aggressive small cell carcinoma (SCC) variant is driven by lineage plasticity signified by the loss of urothelial differentiation and acquisition of neural phenotype and epithelial-to-mesenchymal transition (EMT).
Bladder cancer develops along two distinct tracks, referred to as papillary and non-papillary, that represent clinically and molecularly different forms of the disease (Czerniak et al., 2016). The vast majority of papillary tumors are of luminal molecular subtype characterized by gene expression patterns that are similar to normal intermediate and terminal urothelial differentiation (Choi et al., 2014b). Papillary tumors frequently recur but rarely progress to high-grade invasive carcinomas. On the other hand, non-papillary carcinomas are clinically aggressive, exhibiting a high propensity for invasive growth. A large proportion of them are lethal because of metastatic spread (Kamat et al., 2016). Many of them are of a basal molecular subtype and express genes characteristic of the normal basal urothelial layer and EMT. In addition to conventional urothelial carcinomas (UCs), many microscopically distinct bladder cancer variants have been described and in general are thought to develop via progression of conventional disease (Amin, 2009). The most frequent of these variants are sarcomatoid, small cell, micropapillary, and plasmacytoid. These variants are clinically more aggressive than conventional UCs and require uniquely tailored therapeutic management, which is often unavailable (Amin, 2009, Kamat et al., 2016).
In this report we focus on SCC, which comprises less than 1% of all bladder cancer and often coexists with conventional UC (Amin, 2009). SCC is a highly aggressive disease characterized by early progression to metastasis and shorter survival compared with conventional UC (Veskimae et al., 2019). We report on the genome-wide characterization of bladder SCC, including microRNA (miRNA), gene expression, and whole-exome mutational profiles of 34 paraffin-embedded SCC and 84 invasive conventional bladder UC samples from an MD Anderson Cancer Center (MDACC) cohort. A cohort of 408 muscle-invasive bladder cancers in The Cancer Genome Atlas (TCGA) was used as a reference (Table S1). These analyses revealed molecular features associated with its aggressive nature that may be relevant for the early detection and treatment of this highly lethal form of bladder cancer.
Results
Mutational Signatures
The mutational profiles of conventional UC were characterized by the presence of statistically significant recurrent somatic mutations in 30 genes (Figure 1A). The 10 most frequently mutated genes were TP53 (47%), ARID1A (25%), KDM6A (22%), PIK3CA (22%), RB1 (17%), EP300 (15%), FGFR3 (14%), STAG2 (14%), ELF3 (12%), and CREBBP (11%). The overall mutational landscapes of luminal, basal, and double-negative UC were similar, but several mutated genes were enriched in each molecular subtype. Mutations in FGFR3, ELF3, CDKN1A, and TSC1 were enriched in luminal tumors, whereas mutations in TP53, RB1, and PIK3CA were enriched in basal tumors (Figure 1A). The mutational landscape of the double-negative subtype was, in general, similar to the basal tumors, which show increased p53 and RB1 mutations with a low frequency of FGFR3 mutations. In fact, only one case in the double-negative subtype had FGFR3 mutation. The most striking difference of double-negative tumors was enrichment for RB1 mutations (47%) and presence of combined mutational inactivation of p53 and RB1 (35%). SCC exhibited high overall mutational rates (median mutational frequency = 259, interquartile range = 174), and their significantly mutated genes were similar to those observed in conventional UC (Figure 1B; Table S2). However, the top two most frequently mutated genes in SCCs (TP53 [93%] and RB1 [47%]) were mutated at significantly higher frequencies in SCC than they were in conventional UCs of the TCGA cohort (p < 0.01). This suggests that SCC evolved from precursor conventional UC carrying these mutations, which may drive the progression process. Consistent with this hypothesis, in the paired cases containing both SCC and conventional UC, nearly all mutations in the conventional UCs were also present in SCCs, indicating that they were clonally related, e.g., the identical mutations of p53 (p.H179Y; p.C176F) and RB1 (p.R798fs) were present in both conventional and small cell components of the same tumor further confirming their clonal evolution (Table S2). Several of the chromatin-remodeling genes that are frequently mutated in conventional UC, including KDM6A, EP300, ARID1A, and CREBBP, were not mutated in SCC (Gui et al., 2011). Instead, SCCs carried mutations in FSCN3 (13%), BRD4 (13%), ISLR2 (13%), MAG (13%), MAMDC2 (13%), and TAF1D (13%), which are involved in cellular extension, chromatin regulation, cell cycle, and signaling (Abudureyimu et al., 2018, Edwards and Bryan, 1995, Filippakopoulos et al., 2012, Pijnappel et al., 2009, Wierer et al., 2018). The functional significance of mutations in these genes for small cell progression remains unclear, but they are attractive candidates for future mechanistic studies. Interestingly, FGFR3 mutations, which were present in 14% of conventional UCs, were not present in SCCs.
Figure 1.

Mutational Landscape of SCC
(A) Mutational landscapes among the molecular subtypes of 408 muscle-invasive bladder cancers from the TCGA cohort showing the frequency of mutations in individual tumors and somatic mutations for significantly mutated genes. The frequencies of mutations of individual genes in the luminal, basal, and double-negative subtypes are shown on the left. Bars on the right show the numbers of specific substitutions for individual genes.
(B) Mutational landscapes of 13 cases of SCC and 2 paired samples of precursor conventional UC showing the frequency of mutations in individual genes and somatic mutations for significantly mutated genes. The frequencies of mutations of individual genes are shown on the left. Bars on the right show the numbers of specific substitutions for individual genes.
(C) Composite bar graphs showing the distributions of all nucleotide substitutions in two sets of samples corresponding to the TCGA cohort and SCC.
(D) Proportion of single-nucleotide variants (SNVs) in specific nucleotide motifs for each category of substitution in two sets of samples as shown in (C).
(E) False discovery rate (FDR) for specific nucleotide motifs in two sets of samples as shown in (C).
(F) Average weight scores of mutagenesis patterns in three sets of samples corresponding to the TCGA cohort, paired precursors conventional UC, and SCC.
(G) Weight scores of mutagenesis patterns in individual tumor samples of the TCGA cohort.
(H) Weight scores of mutagenesis patterns in SCCs and paired precursors conventional UCs.
(I) Statistical significance of mutagenesis patterns (p value) in SCC compared with conventional UC. For (E) and (I), p value was calculated using Wilcoxon rank-sum and Kruskal-Wallis tests, respectively.
Mechanisms of Mutagenesis
To further characterize the mutational process associated with progression from conventional UC to SCC, we examined six single-based substitutions (C>A, C>G, C>T, T>A, T>C, and T>G) in all cancer samples (Alexandrov et al., 2013). The results revealed that SCCs were enriched with C>T mutations compared with conventional UCs (Figures 1C–1E). Analyses of Sanger mutational signatures (Faltas et al., 2016) revealed six dominant signatures in the conventional UCs in the TCGA cohorts: signatures 1, 2, 3 (BRCA1/2 mutagenesis), 13 (APOBEC), 19, and 30 (Figures 1F and 1G). Clustering separated the conventional tumors into two subsets (α and β) that were characterized by different levels of signature 13 (APOBEC) prevalence. In contrast, SCCs and the paired precursor conventional UCs were characterized by the uniform dominance of signature 3 (Figure 1H). In addition, clustering segregated SCCs into two subsets that were also characterized by different levels of APOBEC activity. Finally, mutagenesis signatures 16, 21, and 22 were significantly enriched in SCCs compared with conventional UCs (Figure 1I). Overall, these data suggest that SCCs evolve from a distinct subset of conventional UCs.
RNA Expression
Whole-transcriptome expression profiling and unsupervised hierarchical clustering in a combined cohort of SCCs and UCs separated the tumors into two clusters, one dominated by SCCs and the other by conventional UCs (Figure 2A). We recognized that 28% top upregulated and downregulated genes in SCCs were involved in neural differentiation. Similarly, four of the top 10 upregulated genes in the SCC cluster controlled neural development (Figure 2B).
Figure 2.

Whole-Genome mRNA Expression Profile of SCC and Conventional UC
(A) The top 50 upregulated and the top 50 downregulated genes in SCC (n = 22) compared with conventional UC (n = 84).
(B) Hierarchical cluster analysis of the cohort shown in (A) using the top 10 upregulated and top 10 downregulated genes identified in SCC.
(C) Intrinsic molecular subtypes identified by the expression of luminal and basal markers in conventional UC (n = 84) and SCC (n = 22).
(D) The top 10 canonical pathways dysregulated in SCC, as revealed by Ingenuity Pathway Analysis (IPA) and Gene Set Enrichment Analysis (GSEA).
(E) Expression patterns of cancer metastasis pathway genes in molecular subtypes of conventional UC and SCC.
(F) GSEA of metastasis-related genes in SCC compared with conventional UC. A p value <0.05 was considered statistically significant. A p value < 0.05 was considered statistically significant.
(G) The top 10 upstream regulators altered in SCC, as revealed by IPA and GSEA. Genes highlighted in red are involved in neural differentiation.
Conventional UCs segregated into 3 subtypes (basal, luminal, and double-negative), whereas the SCCs were uniformly double-negative for basal and luminal genes (Figure 2C). Ingenuity Pathway Analysis revealed that SCCs were highly enriched for genes that promote cancer metastasis, and more than half of the top activated canonical pathways were known to suppress adaptive (i.e., T-cell-mediated) immunity (Figures 2D–2F). Finally, several of the top downregulated pathways promoted inflammation (TNF, IFNG, JUN), stromal infiltration (TGFB1, PDGF BB), and basal bladder cancer biology (TP63, HIF1) (Figures 2D–2G, S1A, and S1B). Consistent with the presence of inactivating p53 mutations in virtually all SCCs, they showed widespread inactivation of the p53 target genes (Figure S1A). Kaplan-Meier analysis has shown that these phenotypic changes were associated with significantly shorter survival of patients with SCC when compared with conventional UC (Figure S2).
More in-depth analyses revealed that these effects were associated with increased expression of transcription factors that were also known to play important roles in neural crest development, pro-neural stem cell differentiation, and cell proliferation, and the Musashi RNA-binding protein family (MSI1 and MSI2) that functions as regulator of neural stem cell differentiation (Siddall et al., 2006) (Figures S3A and S3B). The maintenance of an embryonic/stem cell-like neural phenotype was further reinforced by the upregulation of two adhesion proteins, PROM1 and NCAM1 (Figures S3A and S3B) (Pruszak, 2015).
Comparative analyses of the miRNA expression profiles of conventional UC and SCC reinforced the conclusion that SCC progression involves urothelial-to-neural plasticity. Differential expression analysis revealed that over 200 miRNAs were selectively up- or downregulated in SCCs (Figure 3A), and in unsupervised clustering a subset of the top 20 up- and downregulated miRNAs was sufficient to distinguish SCCs from conventional UCs (Figure 3B). The upregulated miRNAs included the polycistronic miR-143/145 and miR-199a/214 clusters, which control neural crest development and pluripotent neural stem cell maintenance (Celia-Terrassa et al., 2017, Cordes et al., 2009). The downregulated miRNAs included three closely related polycistronic clusters: miR-17/92, miR-106a/363, and miR-106b/25, comprising over 10 individual miRNA species all involved in neural differentiation, including the regulation of neural stem cells (Rajman and Schratt, 2017) (Figure 3C). Interestingly, a subset of luminal miRNAs was downregulated in SCCs, whereas basal miRNAs were upregulated (Figure 3D) (Ochoa et al., 2016).
Figure 3.

miRNA Expression Profile in SCC and Conventional UC
(A) Top 50 upregulated and downregulated miRNAs in SCC compared with molecular subtypes of conventional UC.
(B) Hierarchical clustering analysis of the cohort shown in (A) using the top 10 up- and downregulated miRNA.
(C) Expression patterns of miRNAs involved in neural development in SCC and molecular subtypes of conventional UC.
(D) Expression patterns of luminal and basal miRNAs in SCC compared with molecular subtypes of conventional UC.
Analysis of Regulons and EMT
The analysis of both mRNA and miRNA expression profiles suggested that progression to SCC was driven by lineage plasticity characterized by the loss of urothelial differentiation and activation of neural transcriptional programs. To explore this relationship further, we performed additional analyses of regulons implicated in urothelial and neural differentiation (Tables S3 and S4). Most of 23 urothelial regulons displayed reduced enrichment scores in progression to SCC (Figures 4A and 4B). On the other hand, all 15 neural regulons exhibited activation with increases in their enrichment scores (Figures 4C and 4D).
Figure 4.

Enrichment Scores of Urothelial and Neural Regulons in Molecular Subtypes of Conventional UC (n = 84) and SCC (n = 22)
(A) Enrichment scores of urothelial regulons.
(B) The expression pattern of selected target genes of PPARG and TP63 regulons.
(C) Enrichment scores of neural regulons.
(D) The expression pattern of selected target genes of NCAM1 and NEUROD1 regulons.
As the development of the central nervous system is a canonical EMT program that is activated during embryogenesis, we hypothesized that dysregulation of EMT might represent an underlying mechanism driving the progression to SCC. In embryogenesis, an EMT-permissive state is activated by the developmental program. In solid tumors, at the core of this circuitry are TP53 and RB1, which negatively regulate EMT and their target genes, and they were coordinately downregulated in virtually all SCCs (Figure 5A). As the activation of EMT is driven by complex regulatory programs involving multiple pathways and their respective genes, we assessed its activation by calculating a quantitative EMT score based on a 76-gene signature developed by Byers et al. (Byers et al., 2013) (Figure 5B). We previously showed that TP63 controls the expression of basal high-molecular-weight keratins (KRT5, KRT6, and KRT14) and the EMT inhibitor, miR-205, in urothelial cells (Choi et al., 2014b, Tran et al., 2013). The central role of TP63 in the regulation of EMT was confirmed in several solid tumors (Stacy et al., 2017). Consistent with these observations SCCs showed downregulation of TP53 and TP63 target genes (Figure 5C). Correspondingly, E-cadherin (CDH1) and other homotypic adhesion molecules, including claudin-1 (CLDN1) and tight junction protein 1 (TJP1), were all downregulated in SCCs (Figure 5D). Surprisingly, none of the major transcriptional EMT regulators of the SNAIL, TWIST, and ZEB families was overexpressed in SCC (Figure 5E). However, several families of miRNAs typically involved in the activation of EMT such as miR-182, miR-183, and members of miR-200 family were downregulated in SCC (Figure 5A). The luminal and basal subtypes of conventional UC were characterized by positive EMT scores reflecting their epithelial phenotype (Figure 5F). The tumors in the double-negative category had intermediate scores indicating their partial EMT, whereas SCCs had the lowest scores confirming their fully activated EMT state. Similar results were obtained by Gene Set Enrichment Analysis using the 175 EMT gene signature developed by Yu et al. (Yu et al., 2013) (Figure 5G). These data confirm that the activation of EMT combined with urothelial-to-neural phenotypic switch is the driving mechanism of progression to SCC.
Figure 5.

Dysregulation of EMT Network in Molecular Subtypes of Conventional UC and SCC
(A) Expression patterns of representative genes of the EMT regulatory network in molecular subtypes of conventional UC and SCC.
(B) EMT scores in molecular subtypes of conventional UC and SCC.
(C) Gene Set Enrichment Analysis (GSEA) analysis of TP53 (left) and TP63 (right) target genes in SCC compared with conventional UC.
(D) Boxplot analysis of expression levels of CDH1, TJP1, and CLDN1 in molecular subtypes of conventional UC and SCC.
(E) Boxplot analysis of expression levels of SNAI2, SNAI3, TWIST1, ZEB1, and ZEB2 in molecular subtypes of conventional UC and SCC.
(F) Boxplot analysis of EMT scores in molecular subtypes of conventional UC and SCC.
(G) GSEA analysis of 175 EMT target genes in SCC compared with conventional UC. In panels C, D, E, F, and G, a p value < 0.05 was considered statistically significant.
Immune Infiltrate
Immune checkpoint blockade is clinically active in approximately 20% patients with bladder cancer. The response is positively associated with high mutational burden, infiltration with activated cytotoxic lymphocytes, and specific molecular subtypes (van Dijk et al., 2019). Given the contradictory presence of high mutational rates and immunosuppressive gene expression signatures observed in SCC, we characterized their expression of immune-related genes. In general, conventional carcinomas in the basal subtype were characterized by increased immune gene expression signatures (Figure 6A). In contrast, SCCs were characterized by decrease of immune gene expression. The null immune signature of SCCs was confirmed by significantly reduced immune scores (Figure 6B). More in-depth analysis of immune infiltrate status was performed using the CIBERSORT algorithm, which provided quantitative assessment of expression signatures related to 22 immune cell types (Newman et al., 2015). CIBERSORT confirmed that SCCs were characterized by an immune-null signature when compared with conventional UCs (Figures 6C and 6D). The analysis of the expression signature of immune checkpoint ligands and their receptors (Sharma and Allison, 2015) included CD70 and CD27, CD80 and CTLA4, TNFSF9 and TNFRSF18, ADA and ADORA2A, and PDCD1G2 and CD274 (Figure 6E). SCCs were also characterized by downregulation of nearly all immune checkpoint ligands and their receptors with the exception of ADORA2A, a major suppressor of immune infiltration, which was overexpressed (Figure 6F). This unique feature of SCC potentially explains their null immune phenotype, and ADORA2A may therefore represent an unexplored therapeutic target.
Figure 6.

Expression Pattern of Immune Cell Infiltrate and Immune Checkpoint Genes in Molecular Subtypes of Conventional UC and SCC
(A) Expression pattern of immune cell infiltrate. Top to bottom: B cell, T cell, CD8, MacTH1, and dendritic cell expression clusters.
(B) Boxplot of immune scores calculated using the gene expression profile shown in (A) for molecular subtypes of conventional UC and SCC.
(C) Heatmap of CIBERSORT scores for 22 immune cell types in molecular subtypes of conventional UC and SCC. A p value < 0.05 was considered statistically significant.
(D) Proportion of cases with significant CIBERSORT scores in conventional UC and SCC (left). Proportion of cases with significant CIBERSORT scores in molecular subtypes of conventional UC and SCC (right).
(E) Expression of immune checkpoint genes in conventional UC and SCC in relation to their molecular subtypes.
(F) Boxplot of ADORA2A mRNA expression levels in molecular subtypes of conventional UC and SCC. In panels B, D, and F, a p value < 0.05 was considered statistically significant.
Validation Studies
We verified the expression patterns of signature luminal markers (GATA3), basal markers (KRT5/6, KRT14), and neural markers (Synaptophysin, Chromogranin, and INSM1) by immunohistochemistry (IHC). The SCCs were all negative for GATA3, KRT5/6, and KRT14. In contrast, SCCs were IHC-positive for neural marker such as Synaptophysin, Chromogranin, and INSM1, consistent with the RNA expression data (Figure 7A). IHC stains for CD3 and CD8 T cells confirmed the immune-null phenotype of SCCs, which showed practically no T cell infiltrate (Figure 7B). In contrast, many double-negative conventional UCs showed pronounced infiltrate of CD3 and CD8 T cells. On the other hand, only a sparse T cell infiltrate was present in luminal and basal conventional UC subtypes. Overexpression of synpatophysin, MSI1, E2F1, and ADORA2A with downregulation of E-cadherin in SCCs was confirmed on several fresh frozen tissues by western blotting (Figure 7C).
Figure 7.

Immunohistochemical Validation Studies of Molecular Features of SCC when Compared with Conventional UC
(A) The immunohistochemical expression signatures of luminal, basal, and neuroendocrine markers in representative luminal and basal cases of conventional UC and SCC on a tissue microarray (TMA) composed of conventional UC (n = 76) and SCC (n = 14). Scale bars, 40 μm.
(B) The immunohistochemical expression of CD3 and CD8 T cells in representative luminal, basal, and double-negative cases of conventional UC and SCC on a TMA composed of conventional UC (n = 76) and SCC (n = 14). Scale bars, 40 μm.
(C) Western blot documenting the overexpression of synaptophysin, MSI1, E2F1, and ADORA2A with loss of E-cadherin in fresh frozen tissue of conventional UC and SCC. All cases of conventional UC are of the basal subtype.
As the miR17 family is considered to be a regulator of neural differentiation and was consistently downregulated in SCC, we hypothesized that this downregulation played a role in neural switch (Mogilyansky and Rigoutsos, 2013). To verify this hypothesis, we first analyzed the mRNA expression signatures of 30 UC cell lines and found that, similar to patient samples, they could be separated into luminal and basal subtypes. However, a large proportion (33%) of these cell lines was negative for the basal and luminal markers and was considered to be double-negative. Consistent with our observation on patient samples, double-negative UC cell lines showed upregulation of neural markers. As SCCs appeared to be related to basal subtype, we ablated miR17-5p by locked nucleic acid in the basal UC cell line UC6 and confirmed it by RT-qPCR (Figures 8A and S4). The ablation of miR17-5p caused dysregulation of multiple genes enriched for neural markers (Figures 8B and 8C). This included activation of several regulons (TP63, FOXM1, EGFR, PROM1, SOX2, and E2F1) implicated in neural differentiation (Figures 8D–8F). In summary, ablation of miR17 validated the functional role of the miRNA in the induction of neural phenotype.
Figure 8.

Induction of Neural-like Phenotype by Locked Nucleic Acid (LNA) Ablation of miR-17-5p in UC6 Cell Line
(A) Detection of miR17-5p ablation by RT-qPCR induced by miR17-5p LNA with NegCtl LNA as control and miR-125a as an external reference.
(B) Top 50 up- and downregulated genes induced by miR17-5p LNA by RNA sequencing (RNA-seq). Genes highlighted in red are involved in neural differentiation.
(C) Representative miR17-5p target genes upregulated induced by miR17-5p LNA transfection revealed by RNA-seq.
(D) Representative upregulated urothelial and neural regulons induced by miR17-5p LNA transfection revealed by RNA-seq.
(E) Upregulation of E2F1 expression induced by miR17-5p LNA quantified by RT-qPCR.
(F) Representative upregulated E2F1 targets induced by miR17-5p LNA revealed by RNA-seq. In panels A and E, a p value < 0.05 was considered statistically significant.
Discussion
Bladder cancer is the ninth most common cancer worldwide, affecting 430,000 people and resulting in 165,000 deaths per year (Antoni et al., 2017). In the United States it is the fourth most common cancer in men with an estimated incidence of approximately 80,000 cases in 2019 (Siegel et al., 2019). More than 90% bladder cancers are UCs that originate from precursor lesions in the epithelial urothelial layer of the bladder. Progression of conventional UC to the so-called variant UCs such as SCC, sarcomatoid, micropapillary, or plasmacytoid is associated with increased clinical aggressiveness (Amin, 2009). In the following we summarize the key findings concerning molecular characterization of SCCs and suggest how the findings contribute to our understanding of its clinically aggressive behavior as well as how to open more effective therapeutic opportunities.
Similar to conventional UC, SCCs have high mutational rates comparable to those observed in melanomas and non-small cell lung cancers (Hoadley et al., 2018). In SCC, these high mutational rates are associated with mutation signature 3, characteristic of loss of BRCA1/2 function. SCCs can be separated into two subgroups, i.e., with and without mutation signature of the endogenous mutagenic enzyme, APOBEC cytidine deaminase. When compared with conventional UC, SCCs are enriched for specific mutations. Practically all of them have inactivating mutations of p53 and many of them show combined inactivation of p53 and RB1, resulting in the combined loss of TP53- and RB1-associated transcriptional pathway activities seen in these tumors. Consistent with their basal origin, none of the SCCs contained the mutations that are typically enriched in the luminal subset of conventional UCs. Another feature of SCCs is the absence of mutations in chromatin-remodeling genes, which are common in conventional UC (Gui et al., 2011), reinforcing the concept that SCCs evolve from a unique subset of conventional UCs.
SCCs show a genome-wide change in their expression profiles affecting nearly 40% of the protein-encoding genome. Much of this expressional change resulted in lineage plasticity, exhibited by global downregulation of genes involved in urothelial differentiation and upregulation of neural differentiation programs. The urothelial-to-neural phenotypic switch was further confirmed by the in-depth analysis of the urothelial and neural regulons, which revealed widespread downregulation of practically all targets controlled by master regulators of the urothelial phenotype and activation of genes in neural regulons. This dramatic phenotypic switch occurred in a background of dysregulated EMT. Although the transcription factors that serve as master regulators of EMT (i.e., the SNAIL, TWIST, and ZEB families) were not activated, SCCs showed downregulation of epithelial micro RNAs (i.e., the miR-200 family and miR-182/183) and homotypic epithelial adhesion molecules such as E-cadherin (CDH1), claudin-1 (CLDN1), and tight junction protein 1 (TJP1). Our quantitative assessment of EMT showed that SCC had the lowest EMT scores, whereas the double-negative subtype of conventional UC had intermediate EMT scores corresponding to their respective complete and partial EMT states. At the core of EMT circuitry was the combined loss of p53 and RB complemented by the downregulation of p63, which positively regulates basal markers and negatively regulates EMT. These features not only activate EMT but also correspond to the essential component of lineage plasticity representing the fundamental biologic feature of progression to SCC.
The neural signature based on limited number of markers was originally identified by the TCGA and Lund groups and was further confirmed on several multi-institutional cohorts (Batista da Costa et al., 2019, Marzouka et al., 2018, Robertson et al., 2017, Sjodahl et al., 2017, Tan et al., 2019). The neural expression profile of these tumors overlap with those of SCC, but the coexpression of urothelial markers and the retention or loss of urothelial phenotype varies in different cohorts. Therefore, it is uncertain whether the tumors in these cohorts represent conventional UCs with upregulation of neural/neuroendocrine genes or they are SCC neuroendocrine variants exhibiting loss of urothelial features with gain of neural differentiation.
Several groups of investigators proposed various classification schemes of bladder cancer based on their molecular profile (Choi et al., 2014b, Damrauer et al., 2014, Robertson et al., 2017, Sjodahl et al., 2012). The original classification proposed by the Lund group divided bladder cancers into five categories (Sjodahl et al., 2012). Similarly the TCGA group identified five molecularly distinct classes of bladder cancer as luminal-papillary, luminal-infiltrated, luminal, basal/squamous, and neuronal (Robertson et al., 2017). Damrauer et al. and our group proposed classifiers dividing bladder cancer into major groups referred to as luminal and basal with a third small subset referred to as double-negative (Choi et al., 2014b, Dadhania et al., 2016, Damrauer et al., 2014). Additional meta- and consensus analyses of bladder cancer were recently published (Kamoun et al., 2020, Tan et al., 2019). Although various groups of investigators propose a different terminology, the set of markers used to classify bladder cancers among the groups is overlapping, and it appears that the top level of dichotomy is between luminal and basal categories. These two major subsets of bladder cancer show features of undifferentiated basal and more differentiated intermediate luminal urothelial cell layers (Choi et al., 2014a, Choi et al., 2014b). A small subset of tumors, referred to as double-negative, appears to be negative for both luminal and basal markers (Dadhania et al., 2016). They can be perceived as a poorly differentiated variant of the basal subtype as they retain the expression of a signature basal marker, CD44. They also show overlapping mutational signature with the basal subtype (Dadhania et al., 2016). Several animal model studies confirmed the distinct cellular origin of basal and luminal tumors. These studies suggest that basal tumors originate from undifferentiated uroprogenitor cells (Shin et al., 2014, Van Batavia et al., 2014). In contrast, luminal tumors are derived from intermediate uroplakin-expressing cells (Saito et al., 2018). Our combined analyses of the mutational landscape and expression signatures suggest that SCC evolves from basal subtype with double-negative tumors representing an intermediate step in the progression.
We found that SCCs are depleted of immune cell infiltration and are characterized by the so-called immune desert phenotype. In contrast to closely related sarcomatoid carcinomas of the bladder, which show overexpression of the immune checkpoint receptor ligand PD-L1, SCCs express a unique immune checkpoint receptor adenosine receptor A2A (ADORA2A) that has been shown to be a potent inhibitor of immune infiltration and especially of CD8+ cytotoxic lymphocytes (Ma et al., 2017). The overexpression of ADORA2A in the majority of SCCs may explain their overall immune-null phenotype but most importantly represents as of yet unexplored therapeutic target for patients affected by this highly lethal variant of bladder cancer. A phase I clinical trial of A2AR antagonists (PBF-509 and CPI-444) alone or in combination with the immune checkpoint inhibitor atezolizumab is currently recruiting participants to study the efficiency of A2AR blockade for solid tumor including bladder cancer (NCT02655822).
Limitations of the Study
-
•
The number of SCCs analyzed in our study is a limiting factor, and our observations should be validated on larger numbers of cases possibly from the inter-institutional cohorts.
-
•
Immune desert phenotype of SCC and the putative role of ADORA2A may represent therapeutic targets and should be validated in preclinical models.
Resource Availability
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, B. Czerniak (bczernia@mdanderson.org).
Materials Availability
Requests for materials related to this study should be directed to the Lead Contact of this study.
Data and Code Availability
Data were deposited on SRA (SRA SUB6960718) and GEO (GEO: GSE128192, GSE145260, GSE145259, and GSE128277).
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This study was supported by NCI Genitourinary Bladder SPORE Grant P50CA 91846 (Project 1 and Core C to B.C.). G.Y. is a visiting scientist from China, and his permanent institutional affiliation is Department of Urology, Renji Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
Author Contributions
B.C. conceived and supervised the whole project and wrote the manuscript. G.Y., D.C., J.B., S.L., J.G.L., S.Z., W.C., and D.J.M. performed the experiments and generated data. Z.W., Y.W., and H.Y. performed data analysis. M.K., P.W., and J.N.W. designed and supervised data analysis. J.B., C.C.G., V.D., Y.L., G.W., and Y.W. organized and analyzed pathologic data. A.K., C.D., D.T., A.S.-R., C.L., and J.G. provided and analyzed clinical data.
Declaration of Interests
The authors declare no conflict of interest.
Published: June 26, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101201.
Contributor Information
Charles C. Guo, Email: ccguo@mdanderson.org.
Bogdan Czerniak, Email: bczernia@mdanderson.org.
Supplemental Information
References
- Abudureyimu S., Asai N., Enomoto A., Weng L., Kobayashi H., Wang X., Chen C., Mii S., Takahashi M. Essential role of Linx/Islr2 in the development of the forebrain anterior commissure. Sci. Rep. 2018;8:7292. doi: 10.1038/s41598-018-24064-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov L.B., Nik-Zainal S., Wedge D.C., Aparicio S.A., Behjati S., Biankin A.V., Bignell G.R., Bolli N., Borg A., Borresen-Dale A.L. Signatures of mutational processes in human cancer. Nature. 2013;500:415–421. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin M.B. Histological variants of urothelial carcinoma: diagnostic, therapeutic and prognostic implications. Mod. Pathol. 2009;22:S96–S118. doi: 10.1038/modpathol.2009.26. [DOI] [PubMed] [Google Scholar]
- Antoni S., Ferlay J., Soerjomataram I., Znaor A., Jemal A., Bray F. Bladder cancer incidence and mortality: a global overview and recent trends. Eur. Urol. 2017;71:96–108. doi: 10.1016/j.eururo.2016.06.010. [DOI] [PubMed] [Google Scholar]
- Batista da Costa J., Gibb E.A., Bivalacqua T.J., Liu Y., Oo H.Z., Miyamoto D.T., Alshalalfa M., Davicioni E., Wright J., Dall'Era M.A. Molecular characterization of neuroendocrine-like bladder cancer. Clin. Cancer Res. 2019;25:3908–3920. doi: 10.1158/1078-0432.CCR-18-3558. [DOI] [PubMed] [Google Scholar]
- Byers L.A., Diao L., Wang J., Saintigny P., Girard L., Peyton M., Shen L., Fan Y., Giri U., Tumula P.K. An epithelial-mesenchymal transition gene signature predicts resistance to EGFR and PI3K inhibitors and identifies Axl as a therapeutic target for overcoming EGFR inhibitor resistance. Clin. Cancer Res. 2013;19:279–290. doi: 10.1158/1078-0432.CCR-12-1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celia-Terrassa T., Liu D.D., Choudhury A., Hang X., Wei Y., Zamalloa J., Alfaro-Aco R., Chakrabarti R., Jiang Y.Z., Koh B.I. Normal and cancerous mammary stem cells evade interferon-induced constraint through the miR-199a-LCOR axis. Nat. Cell Biol. 2017;19:711–723. doi: 10.1038/ncb3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W., Czerniak B., Ochoa A., Su X., Siefker-Radtke A., Dinney C., McConkey D.J. Intrinsic basal and luminal subtypes of muscle-invasive bladder cancer. Nat. Rev. Urol. 2014;11:400–410. doi: 10.1038/nrurol.2014.129. [DOI] [PubMed] [Google Scholar]
- Choi W., Porten S., Kim S., Willis D., Plimack E.R., Hoffman-Censits J., Roth B., Cheng T., Tran M., Lee I.L. Identification of distinct basal and luminal subtypes of muscle-invasive bladder cancer with different sensitivities to frontline chemotherapy. Cancer Cell. 2014;25:152–165. doi: 10.1016/j.ccr.2014.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordes K.R., Sheehy N.T., White M.P., Berry E.C., Morton S.U., Muth A.N., Lee T.H., Miano J.M., Ivey K.N., Srivastava D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature. 2009;460:705–710. doi: 10.1038/nature08195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czerniak B., Dinney C., McConkey D. Origins of bladder cancer. Annu. Rev. Pathol. 2016;11:149–174. doi: 10.1146/annurev-pathol-012513-104703. [DOI] [PubMed] [Google Scholar]
- Dadhania V., Zhang M., Zhang L., Bondaruk J., Majewski T., Siefker-Radtke A., Guo C.C., Dinney C., Cogdell D.E., Zhang S. Meta-analysis of the luminal and basal subtypes of bladder cancer and the identification of signature immunohistochemical markers for clinical use. EBioMedicine. 2016;12:105–117. doi: 10.1016/j.ebiom.2016.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damrauer J.S., Hoadley K.A., Chism D.D., Fan C., Tiganelli C.J., Wobker S.E., Yeh J.J., Milowsky M.I., Iyer G., Parker J.S. Intrinsic subtypes of high-grade bladder cancer reflect the hallmarks of breast cancer biology. Proc. Natl. Acad. Sci. U S A. 2014;111:3110–3115. doi: 10.1073/pnas.1318376111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards R.A., Bryan J. Fascins, a family of actin bundling proteins. Cell Motil. Cytoskeleton. 1995;32:1–9. doi: 10.1002/cm.970320102. [DOI] [PubMed] [Google Scholar]
- Faltas B.M., Prandi D., Tagawa S.T., Molina A.M., Nanus D.M., Sternberg C., Rosenberg J., Mosquera J.M., Robinson B., Elemento O. Clonal evolution of chemotherapy-resistant urothelial carcinoma. Nat. Genet. 2016;48:1490–1499. doi: 10.1038/ng.3692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P., Picaud S., Mangos M., Keates T., Lambert J.P., Barsyte-Lovejoy D., Felletar I., Volkmer R., Muller S., Pawson T. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149:214–231. doi: 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gui Y., Guo G., Huang Y., Hu X., Tang A., Gao S., Wu R., Chen C., Li X., Zhou L. Frequent mutations of chromatin remodeling genes in transitional cell carcinoma of the bladder. Nat. Genet. 2011;43:875–878. doi: 10.1038/ng.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoadley K.A., Yau C., Hinoue T., Wolf D.M., Lazar A.J., Drill E., Shen R., Taylor A.M., Cherniack A.D., Thorsson V. Cell-of-Origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell. 2018;173:291–304.e6. doi: 10.1016/j.cell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamat A.M., Hahn N.M., Efstathiou J.A., Lerner S.P., Malmstrom P.U., Choi W., Guo C.C., Lotan Y., Kassouf W. Bladder cancer. Lancet. 2016;388:2796–2810. doi: 10.1016/S0140-6736(16)30512-8. [DOI] [PubMed] [Google Scholar]
- Kamoun A., de Reynies A., Allory Y., Sjodahl G., Robertson A.G., Seiler R., Hoadley K.A., Groeneveld C.S., Al-Ahmadie H., Choi W. A consensus molecular classification of muscle-invasive bladder cancer. Eur. Urol. 2020;77:420–433. doi: 10.1016/j.eururo.2019.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma S.R., Deng W.W., Liu J.F., Mao L., Yu G.T., Bu L.L., Kulkarni A.B., Zhang W.F., Sun Z.J. Blockade of adenosine A2A receptor enhances CD8(+) T cells response and decreases regulatory T cells in head and neck squamous cell carcinoma. Mol. Cancer. 2017;16:99. doi: 10.1186/s12943-017-0665-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marzouka N.A., Eriksson P., Rovira C., Liedberg F., Sjodahl G., Hoglund M. A validation and extended description of the Lund taxonomy for urothelial carcinoma using the TCGA cohort. Sci. Rep. 2018;8:3737. doi: 10.1038/s41598-018-22126-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogilyansky E., Rigoutsos I. The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013;20:1603–1614. doi: 10.1038/cdd.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman A.M., Liu C.L., Green M.R., Gentles A.J., Feng W., Xu Y., Hoang C.D., Diehn M., Alizadeh A.A. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods. 2015;12:453–457. doi: 10.1038/nmeth.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ochoa A.E., Choi W., Su X., Siefker-Radtke A., Czerniak B., Dinney C., McConkey D.J. Specific micro-RNA expression patterns distinguish the basal and luminal subtypes of muscle-invasive bladder cancer. Oncotarget. 2016;7:80164–80174. doi: 10.18632/oncotarget.13284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijnappel W.P., Kolkman A., Baltissen M.P., Heck A.J., Timmers H.M. Quantitative mass spectrometry of TATA binding protein-containing complexes and subunit phosphorylations during the cell cycle. Proteome Sci. 2009;7:46. doi: 10.1186/1477-5956-7-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruszak J. Elsevier; 2015. Neural Surface Antigens from Basic Biology towards Biomedical Applications. [Google Scholar]
- Rajman M., Schratt G. MicroRNAs in neural development: from master regulators to fine-tuners. Development. 2017;144:2310–2322. doi: 10.1242/dev.144337. [DOI] [PubMed] [Google Scholar]
- Robertson A.G., Kim J., Al-Ahmadie H., Bellmunt J., Guo G., Cherniack A.D., Hinoue T., Laird P.W., Hoadley K.A., Akbani R. Comprehensive molecular characterization of muscle-invasive bladder cancer. Cell. 2017;171:540–556.e5. doi: 10.1016/j.cell.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito R., Smith C.C., Utsumi T., Bixby L.M., Kardos J., Wobker S.E., Stewart K.G., Chai S., Manocha U., Byrd K.M. Molecular subtype-specific immunocompetent models of high-grade urothelial carcinoma reveal differential neoantigen expression and response to immunotherapy. Cancer Res. 2018;78:3954–3968. doi: 10.1158/0008-5472.CAN-18-0173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P., Allison J.P. Immune checkpoint targeting in cancer therapy: toward combination strategies with curative potential. Cell. 2015;161:205–214. doi: 10.1016/j.cell.2015.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin K., Lim A., Odegaard J.I., Honeycutt J.D., Kawano S., Hsieh M.H., Beachy P.A. Cellular origin of bladder neoplasia and tissue dynamics of its progression to invasive carcinoma. Nat. Cell Biol. 2014;16:469–478. doi: 10.1038/ncb2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddall N.A., McLaughlin E.A., Marriner N.L., Hime G.R. The RNA-binding protein Musashi is required intrinsically to maintain stem cell identity. Proc. Natl. Acad. Sci. U S A. 2006;103:8402–8407. doi: 10.1073/pnas.0600906103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R.L., Miller K.D., Jemal A. Cancer statistics, 2019. CA Cancer J. Clin. 2019;69:7–34. doi: 10.3322/caac.21551. [DOI] [PubMed] [Google Scholar]
- Sjodahl G., Eriksson P., Liedberg F., Hoglund M. Molecular classification of urothelial carcinoma: global mRNA classification versus tumour-cell phenotype classification. J. Pathol. 2017;242:113–125. doi: 10.1002/path.4886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sjodahl G., Lauss M., Lovgren K., Chebil G., Gudjonsson S., Veerla S., Patschan O., Aine M., Ferno M., Ringner M. A molecular taxonomy for urothelial carcinoma. Clin. Cancer Res. 2012;18:3377–3386. doi: 10.1158/1078-0432.CCR-12-0077-T. [DOI] [PubMed] [Google Scholar]
- Stacy A.J., Craig M.P., Sakaram S., Kadakia M. DeltaNp63alpha and microRNAs: leveraging the epithelial-mesenchymal transition. Oncotarget. 2017;8:2114–2129. doi: 10.18632/oncotarget.13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan T.Z., Rouanne M., Tan K.T., Huang R.Y., Thiery J.P. Molecular subtypes of urothelial bladder cancer: results from a meta-cohort analysis of 2411 tumors. Eur. Urol. 2019;75:423–432. doi: 10.1016/j.eururo.2018.08.027. [DOI] [PubMed] [Google Scholar]
- Tran M.N., Choi W., Wszolek M.F., Navai N., Lee I.L., Nitti G., Wen S., Flores E.R., Siefker-Radtke A., Czerniak B. The p63 protein isoform DeltaNp63alpha inhibits epithelial-mesenchymal transition in human bladder cancer cells: role of MIR-205. J. Biol. Chem. 2013;288:3275–3288. doi: 10.1074/jbc.M112.408104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Batavia J., Yamany T., Molotkov A., Dan H., Mansukhani M., Batourina E., Schneider K., Oyon D., Dunlop M., Wu X.R. Bladder cancers arise from distinct urothelial sub-populations. Nat. Cell Biol. 2014;16:982–991. doi: 10.1038/ncb3038. 981–5. [DOI] [PubMed] [Google Scholar]
- van Dijk N., Funt S.A., Blank C.U., Powles T., Rosenberg J.E., van der Heijden M.S. The cancer immunogram as a framework for personalized immunotherapy in urothelial cancer. Eur. Urol. 2019;75:435–444. doi: 10.1016/j.eururo.2018.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veskimae E., Espinos E.L., Bruins H.M., Yuan Y., Sylvester R., Kamat A.M., Shariat S.F., Witjes J.A., Comperat E.M. What is the prognostic and clinical importance of urothelial and nonurothelial histological variants of bladder cancer in predicting oncological outcomes in patients with muscle-invasive and metastatic bladder cancer? A European Association of Urology muscle invasive and metastatic bladder cancer guidelines panel systematic review. Eur. Urol. Oncol. 2019;2:625–642. doi: 10.1016/j.euo.2019.09.003. [DOI] [PubMed] [Google Scholar]
- Wierer M., Prestel M., Schiller H.B., Yan G., Schaab C., Azghandi S., Werner J., Kessler T., Malik R., Murgia M. Compartment-resolved proteomic analysis of mouse aorta during atherosclerotic plaque formation reveals osteoclast-specific protein expression. Mol. Cell Proteomics. 2018;17:321–334. doi: 10.1074/mcp.RA117.000315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M., Bardia A., Wittner B.S., Stott S.L., Smas M.E., Ting D.T., Isakoff S.J., Ciciliano J.C., Wells M.N., Shah A.M. Circulating breast tumor cells exhibit dynamic changes in epithelial and mesenchymal composition. Science. 2013;339:580–584. doi: 10.1126/science.1228522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan S., Norgard R.J., Stanger B.Z. Cellular plasticity in cancer. Cancer Discov. 2019;9:837–851. doi: 10.1158/2159-8290.CD-19-0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data were deposited on SRA (SRA SUB6960718) and GEO (GEO: GSE128192, GSE145260, GSE145259, and GSE128277).