

Abstract
Macrophages play a pivotal role in the early stages of atherosclerosis development; they excessively accumulate cholesterol in the cytosol in response to modified Low Density Lipoprotein (mLDL). The mLDL are incorporated through scavenger receptors. CD36 is a high-affinity cell surface scavenger receptor that facilitates the binding and uptake of long-chain fatty acids and mLDL into the cell. Numerous structurally diverse ligands can initiate signaling responses through CD36 to regulate cell metabolism, migration, and angiogenesis. Nitro-fatty acids are endogenous electrophilic lipid mediators that react with and modulate the function of multiple enzymes and transcriptional regulatory proteins. These actions induce the expression of several anti-inflammatory and cytoprotective genes and limit pathologic responses in experimental models of atherosclerosis, cardiac ischemia/reperfusion, and inflammatory diseases. Pharmacological and genetic approaches were used to explore the actions of nitro-oleic acid (NO2-OA) on macrophage lipid metabolism. Pure synthetic NO2-OA dose-dependently increased CD36 expression in RAW264.7 macrophages and this up-regulation was abrogated in BMDM from Nrf2-KO mice. Ligand binding analysis revealed that NO2-OA specifically interacts with CD36, thus limiting the binding and uptake of mLDL. Docking analysis shows that NO2-OA establishes a low binding energy interaction with the alpha helix containing Lys164 in CD36. NO2-OA also restored autophagy flux in mLDL-loaded macrophages, thus reversing cholesterol deposition within the cell. In aggregate, these results indicate that NO2-OA reduces cholesterol uptake by binding to CD36 and increases cholesterol efflux by restoring autophagy.
Keywords: Nitro-fatty acid, Atherosclerosis, CD36, Macrophages, Foam cell, Nitro-oleic acid
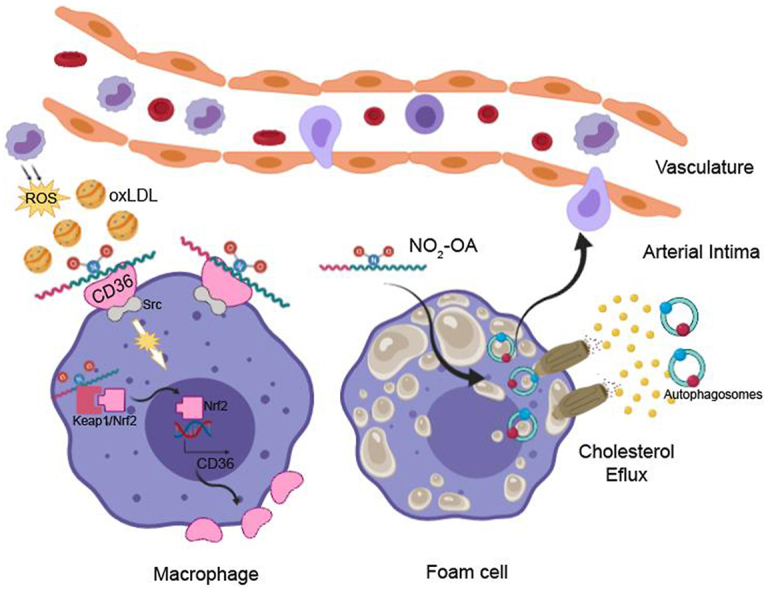
Graphical abstract
At the onset of atherosclerosis, inflammation, and ROS production in the arterial intima favors the redox modification of LDL, which is incorporated via CD36 receptor into macrophages. In the presence of NO2-OA, the binding and incorporation of mLDL to CD36 in macrophages is limited, decreasing intracellular lipid stores, despite the induction of CD36 expression via Keap1/Nrf2 pathway activation. In contrast, once the accumulation of lipids has taken place and the macrophages differentiate to foam cells, NO2-OA favors lipid efflux by a mechanism dependent on activation of AMPK and stimulation of the autophagic flux. In the autophagosomes, the red and blue balls represent the LC3II and p62 proteins, respectively.
Highlights
-
•
NO2-OA induces CD36 expression via Keap1/Nrf2 .
-
•
NO2-OA is a new ligand for CD36 .
-
•
NO2-OA decreases the uptake of mLDL in macrophages .
-
•
NO2-OA increases the cholesterol efflux in lipid-loaded macrophages .
1. Introduction
Hyperlipidemia exacerbates oxidative stress and increases inflammatory responses in the arterial intima, leading to atherogenesis [1]. In this context, monocyte-derived macrophages play a critical role in the uptake of modified Low Density Lipoprotein (mLDL) through the expression and activity of scavenger receptors [2]. The scavenger receptor CD36 recognizes, binds and internalizes mLDL, oxidized (oxLDL) and acetylated (acLDL) [3], to initiate a positive feedback loop enhancing the receptor expression [4,5].
CD36 or fatty acid translocase (FAT) is a member of the scavenger receptor class B family. This multi-ligand receptor has a hairpin-like topology with two transmembrane domains and cytoplasmic -NH2 and -COOH termini [6]. The extracellular domain is heavily glycosylated and contains the binding sites for diverse ligands such as long-chain fatty acids (LCFA) [[7], [8], [9]], oxLDL [10], phospholipids [11], Plasmodium falciparum-infected erythrocytes [12], and thrombospondin [13] among others. The structure of CD36 facilitates the access of LCFA to a binding pocket rich in hydrophobic amino acids, also named entrance 1, that channels fatty acids to the cell membrane or the cytoplasm.
The ionic interactions between Lys164, in the binding pocket of CD36, and fatty acid carboxylates induce the necessary conformational changes in the receptor to support internalization [6]. Both lipid uptake and CD36–dependent signaling responses are inhibited by targeting Lys164 with site-directed mutagenesis or by reaction with the amine-reactive sulfo-N-succinimidyl oleate (SSO) [[14], [15], [16]]. In macrophages, the regulation of CD36 expression is mainly controlled by activation of the nuclear receptor PPARγ and Nrf2-regulated antioxidant gene expression. Rosiglitazone, a full agonist for PPARγ, and nitro-linoleic acid, a partial PPARγ agonist bind PPARγ with high affinity (Rosiglitazone) and covalently (nitro-linoleic acid) to increase CD36 expression on RAW264.7 macrophages, a response partially inhibitable by PPARγ antagonists [17]. Nevertheless, these ligands recruit different sets of co-regulators that define their responses in downstream events such as adipogenesis, lipid metabolism, or renal fluid regulation [17].
Nitro-fatty acids (NO2-FA) are endogenous signaling mediators formed upon reaction of nitric oxide- and nitrite-derived nitrogen dioxide with unsaturated fatty acids, preferentially with those containing conjugated double bonds [18,19]. NO2-FA are normally present in plasma and urine of healthy individuals (~1–3 nM and 20 pg/mg creatinine respectively), and their local formation in tissues increases during inflammation and ischemia-reperfusion events [[20], [21], [22], [23], [24]]. The electronegativity of the nitro group present on the nitroalkene double bond renders the β-carbon electron-deficient and chemically reactive (electrophilic). In cells and tissues, NO2-FA react with nucleophilic moieties found in proteins and small molecules through Michael addition reactions. These reactions are key to the anti-inflammatory and antioxidant responses induced by NO2-FA in different preclinical disease models, including atherosclerosis, ischemia and reperfusion injury, diabetes and metabolic syndrome. These reactions are involved in the activation of PPARγ-dependent metabolic responses [25], Keap1/Nrf2 antioxidant gene expression [26], and downregulation of pro-inflammatory signaling through NF-κB and STING inhibition [25,27,28]. Despite advances in understanding the roles of NO2-FA in the modulation of inflammatory responses, the impact on macrophage lipid metabolism remains undefined. The subcutaneous administration of NO2-OA to ApoE-KO mice fed a high-fat diet (HFD) reduced the development of atherosclerotic plaque in the arterial intima and decreased inflammation, lipid content and plaque volume [29]. The mechanisms accounting for this protective effect remain poorly understood and a role for CD36 modulation or an impact on macrophage responses has not been addressed.
Numerous mechanisms have been proposed to participate in plaque reversion, with intracellular lipid removal emerging as a significant regulator of lipid metabolism in different cell types, including macrophage foam cells, hepatocytes, adipocytes, and others [[30], [31], [32]]. In particular, the inflammatory profile of foam cells is promoted by increased lipid uptake and cholesterol and cholesteryl ester accumulation in lipid droplets (LD). While a reduction of LD content is desirable, lipophagy, the autophagic flux that is key to lipid droplet clearance, is impaired in foam cells [31,33,34]. Thus, mechanisms that restore autophagy to reduce cholesterol content in foam cells are highly desirable. Herein, we reveal that NO2-OA restores the autophagy flux of macrophage lipids.
In this work, we also describe the regulation of CD36 expression by NO2-OA via activation of Keap1/Nrf2 and provide biochemical and in silico evidence that NO2-OA binds to CD36, reducing mLDL uptake and both cholesterol and cholesteryl ester accumulation. We show that this mechanism is operative in foam cells, where NO2-OA reduces the lipid load by restoring the autophagy flux. These findings provide a molecular mechanism for the reduction of cholesterol content in lipid-loaded macrophages and this can account for the athero-protective effects of NO2-OA in animal models [36].
2. Materials and methods
Materials: RAW264.7 macrophages were purchased from American Type Culture Collection (ATCC®). Nitro-oleic acid (NO2-OA) and biotin-labeled NO2-OA (B-NO2-OA) were synthesized as previously [37,38]. Macrophage colony-stimulating factor (M-CSF) was purchased from Gibco. CDDO-Me (Nrf2 activator), GW9662 (PPAR-γ antagonist), rosiglitazone (PPARγ agonist), Compound C (AMPK inhibitor), SSO or sulfo-N-succinimidyl oleate (CD36 inhibitor), were purchased from Cayman Chemical Company. Bicinchoninic acid protein reaction kit (BCA) was sourced from Thermo Scientific. The lipophilic and fluorescent dye, DiI or 1,1′-dioctadecyl-3,3,3′,3′-tetramethyl indocarbocyanine perchlorate was purchased from Sigma-Aldrich, and recombinant CD36 (rCD36) was obtained from Abcam. Solvents used for extractions and mass spectrometric analyses were of HPLC grade or higher from Burdick and Jackson (Muskegon, MI).
2.1. Cell culture and BMDM differentiation
RAW264.7 macrophages were maintained in complete media Dulbecco's Modified Eagle's Medium (DMEM) high glucose containing 10% fetal bovine serum (FBS), streptomycin/penicillin 100 U/ml (Invitrogen) at 37 °C and 5% CO2. All murine model studies were performed using protocols approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Nrf2−/− male mice on a BALB/c background (provided by M. Yamamoto) and wild type BALB/c male mice were maintained under specific pathogen-free conditions. Bone marrow cells were obtained according to Ref. [39]. For Western blot and RT-qPCR analyses, 1 × 106 cells/well were seeded on 6-well plates after 7 days of differentiation with macrophage colony-stimulating factor (M-CSF, Gibco).
2.2. Isolation and modification of LDL
LDL were isolated from plasma of healthy volunteers (signed consent and approval) by ultracentrifugation [40]. After blood collection using EDTA as anticoagulant, KBr was added to reach a concentration of 0.29 g/ml plasma. Then, an equal volume of NaCl 0.15 M was added and samples centrifuged at 65.000 g for 1 h at 4 °C in a Beckman Coulter UltraCentrifuge (Optima DE-80K). After centrifugation, the middle band (orange color) was recovered, kept under nitrogen at 4 °C and protected from light. LDL protein content was determined using BCA. Native-LDL was obtained by adding 20 μM of BHT per each mg/ml of LDL and oxidation was performed with CuSO4 (10 μM per each 0.2 mg/ml of LDL) to obtain the oxLDL/mLDL fraction for 8 h at 25 °C. Oxidation was confirmed by LDL mobility changes in agarose gel. Native- and oxLDL were labeled with the fluorescent lipophilic dye, DiI. Briefly, 1 mg of LDL protein was mixed with 4 μL of DiI (30 mg/ml diluted in DMSO) for 16 h at 37 °C, filtered through 0.45 μm filters, and purified on a C-10 desalting column to eliminate unbound DiI. For cell culture studies, all LDL were sterilized using a 0.22 μm filter.
2.3. Flow cytometry
For cell surface antigen staining, cells were washed with cold PBS 3 times and then re-suspended in cold Accutase Enzyme Cell Detachment Medium (Invitrogen). Single-cell suspensions were washed twice with FACS buffer (PBS containing 2% calf serum, 1 mM EDTA, 0.1% sodium azide) and incubated with fluorochrome-labeled antibodies during 20 min at 4 °C. Data was collected on a FACSCanto II (BD Biosciences) and analyzed with FlowJo software (ThreeStar).
2.4. Immunoblotting
Cells (1 × 106/well) were seeded on a 6-well plate in complete media and serum-deprived for 2 h prior to treatment with 5 μM of NO2-OA, 5 μM oleic acid (OA), 300 nM CDDO-Me, 10 μM GW9662, 10 μM rosiglitazone, or 3.3 μM Compound C. All incubations were performed in DMEM containing 2% FBS. To evaluate signaling responses downstream of CD36, cells were serum-deprived in DMEM for 5 h and pretreated for 30 min with 50 μM SSO. Cells were stimulated with 5 μM NO2-OA, or 300 μM PA. Cells were lysed in 1% Triton-X-100 in PBS supplemented with 1 mM phenylmethylsulfonyl fluoride (PMSF), 10 mM sodium ortho-vanadate and protease inhibitor cocktails (Sigma-Aldrich). BCA protein assay kits were used for protein quantification. Total cell lysates were resolved on 10 or 15% SDS-PAGE and transferred onto nitrocellulose membranes (Bio-Rad). Membranes were probed with specific primary antibodies according to the antibody manufacturer's specifications and normalized using housekeeping proteins (actin or GAPDH). The following primary antibodies were used: rabbit monoclonal anti-CD36 (1/500; Abcam), anti–HO–1(1/1000; Enzo Life Science), anti-NQO1 (1/2000; Cell Signaling), anti-GAPDH (1/2000; Cell Signaling), rabbit monoclonal anti-βActin (1/2000; Sigma Aldrich), anti-pAMPK (1/1000; Cell Signaling), anti-pSrc (1/2000; Cell Signaling), anti-p62 (1/1000; Abcam), anti-LC3 (1/1000; Sigma Aldrich). Secondary antibodies were: IRDye 800 CW donkey anti-rabbit IgG or IRDye 800 CW donkey anti-mouse IgG antibodies (1:15000 in 1% BSA TBST). Membranes were visualized and quantified using the Odyssey Infrared Imaging System (LI-COR, Inc., Lincoln, NE, USA).
2.5. Real time qPCR
Cells were seeded in 6-well plates 24 h before analysis. After treatments, cells were washed 3 times with cold PBS and total RNA was extracted using TRIzol® (Invitrogen), according to the manufacturer's instructions [41]. Briefly, 1 μg of total RNA was reverse-transcribed in a total volume of 20 μL using random primers (Invitrogen) and 50 U of M-MLV reverse transcriptase (Promega Corp). For qPCR, cDNA was mixed with 1x SYBR Green PCR Master Mix (Applied Biosystems) and forward and reverse primers: CD36 forward: TCCTCTGACATTTGCAGGTCTATC/reverse: AAAGGCATTGGCTGGAAGAA, MAP1 forward: CGCTTGCAGCTCAATGCTAAC/reverse: TCGTACACTTCGGAGATGGG, ABCA1 forward: AGTGATAATCAAAGTCAAAGGCACAC/reverse: AGCAACTTGGCACTAGTAACTCTG, ABCG1 forward: TCACCCAGTTCTGCATCCTCTT/reverse: GCAGATGTGTCAGGACCGAGT. qPCR was carried out on an Applied Biosystems 7500 real-time PCR System with Sequence Detection Software v1.4. The cycling conditions included a hot start at 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s and 60 °C for 1 min. Specificity was verified by melting curve analysis. Relative gene expression was calculated according to the 2-ΔΔCt method with GAPDH as the housekeeping gene (forward: AAATGGTGAAGGTCGGTGTG/reverse: TGAAGGGGTCGTTGATGG). Each sample was analyzed in triplicate. No amplification was observed in PCRs using diethylpirocarbonate (DEPC) treated water as template or RNA samples incubated without reverse transcriptase during the cDNA synthesis.
2.6. Immunoprecipitation of CD36
RAW264.7 macrophages were incubated with 0.1 μM B-NO2-OA for 1 h at 4 °C to avoid endocytosis in the presence or absence of 0.1 μM SSO. After incubation, cells were washed 3x with cold PBS and then lysed with 1% Triton-X-100 in PBS supplemented with 1 mM PMSF, PhosSTOPTM phosphatase inhibitor, and a protease inhibitor cocktail (Sigma-Aldrich). Next, total protein was quantified with BCA, and 500 μg of protein were incubated overnight at 4 °C with rabbit monoclonal anti-CD36 antibody (1/50, Abcam). Then, 20 μL of Protein AG (Santa Cruz) was added and incubated during 2 h at 4 °C. Samples were centrifuged 5 min at 2500 rpm and washed 3 times with 1% TritonX-100. Pellets were re-suspended in Sample Buffer (1x) containing 2 mM TCEP (tris-2-carboxyethyl-phosphine). β-mercaptoethanol was replaced by TCEP as reducing agent to reduce loss of B-NO2-OA-CD36 adducts [26]. After heating for 5 min at 40 °C, samples were electrophoresed, transferred to a nitrocellulose membrane, incubated with HRP-Streptavidin (Thermo-Fisher) 1 h at RT and analyzed by a ChemiDoc MP System (Bio-Rad).
2.7. In vitro CD36 interaction analysis
rCD36 (0.1 μg) was incubated with B-NO2-OA in the presence of NO2-OA, OA, or GSH at molar ratios ranging from 1:1, 1:10 and 1:100 for 30 min at 37 °C. After incubation, samples were treated with 1x sample buffer containing TCEP as reducing agent to avoid the loss of protein–NO2–OA adducts and incubated 5 min at 40 °C before western blot analysis. To evaluate reversibility, rCD36 and B-NO2-OA were incubated for 30 min, and then the interaction was competed with SSO for 30 min at 37 °C and assessed by western blotting.
2.8. Immunofluorescence studies
For uptake assays, RAW264.7 cells (5 × 105 cells) were seeded on glass covers and incubated or coincubated with 5 μM NO2-OA,50 μM SSO, or 50 μM OA according to what is indicated on each experiment, for 15 min on ice (4 °C) to avoid endocytosis followed by 30 min DiI-oxLDL (50 μg/ml) incubation for CD36 binding also at 4 °C. Excess of DiI-oxLDL was washed, and cells incubated at 37 °C for 30 min. For cholesterol efflux assays, RAW264.7 macrophages were seeded on glass covers and treated with 100 μg/ml of DiI-oxLDL for 24 h. After 16 h of recovery, cells were treated with vehicle, 5 μM NO2-OA or 5 μM OA (non-electrophile control) for 24 h. Cells were washed 3 times with PBS, fixed with 4% paraformaldehyde, and mounted on glass slides with Mowiol (Merck, Darmstadt, Germany). Fluorescent images were acquired using confocal microscopy (Olympus FV 1200).
2.9. In vitro CD36-oxLDL interaction analysis
rCD36 (10 μg) was adsorbed in a 96-well black plate in 200 μl of 50 mM NaCO3 pH 9.6. Plates were kept overnight at 4 °C, washed 3 times with PBS, and incubated with 12.5 μg/ml of NO2-OA, OA and SSO for 15 min at 37 °C. After incubation, 70 μg/mL of DiI-oxLDL were added for 2 h at 37 °C. Plates were washed 3 times with PBS 0.1% Triton-X100 and fluorescence (excitation 530/25, emission 590/35) measured using a Biotek Synergy HT plate reader. A reaction blank where rCD36 was incubated in buffer without DiI-oxLDL and a reagent blank using buffer in the absence of reagent and rCD36 were used as controls.
2.10. Lipid extraction for MS analysis
For uptake assays, RAW264.7 macrophages were seeded in a 6-well plate to evaluate LDL cholesterol uptake. Macrophages were incubated in complete media in the presence or absence of 50 μg/ml of mLDL for 2, 4, 8 and 24 h. After treatment, supernatants and cells were washed and collected. Before lipids extraction, internal standards for cholesterol (Cholesterol-d7; Avanti Polar Lipids, Inc.), cholesteryl esters (16:0-cholesteryl ester; Avanti Polar Lipids, Inc.) were added to a final concentration of 500 nM. Lipids were extracted by adding 500 μl of solvent mixture [1 mol/L formic acid/isopropanol/hexane (2:20:30, v/v/v)] followed by 1 ml of hexanes. The organic phase (upper) was transferred to a clean vial, dried under nitrogen and reconstituted in 100 μl of chloroform/methanol (20/80, v/v). Samples were dried under nitrogen, resuspended in chloroform/methanol, and subjected to LC-MS analysis.
Cells loaded for 24 h with 50 μg/ml of mLDL in DMEM 2% BSA, were preincubated with 5 μM NO2-OA and 50 μM SSO during 20 min at 37 °C in the presence of ACAT1 inhibitor to reduce cholesterol efflux and allow for influx analysis. Lipids were extracted and cholesterol and cholesteryl esters quantified by LC-MS.
For cholesterol efflux assays, cells were seeded in a 24 well plate the day before the experiment. RAW264.7 were treated with oxLDL for 24 h. After 16 h of recovery, 5 μM NO2-OA or vehicle were added, and supernatant aliquots obtained at different endpoints. At the end of the experiment, cells were harvested and lipids extracted and quantified by LC-MS.
2.11. LC-MS measurement of cholesterol and cholesteryl esters
Cholesterol and cholesteryl esters were chromatographically resolved on a Vanquish UHPLC (Thermo Scientific), the flow splitted post-column and simultaneously detected on a QExactive mass spectrometer (Thermo Scientific) and a triple quadrupole mass spectrometer (API 5000, Sciex). The samples were resolved using a C18 reverse-phase column (100 × 2,1 mm, 1,7 μm particle size; Thermo-Fisher Scientific) at a 200 μl/min flow rate with a gradient solvent system consisting of solvent A: 50% water/50% acetonitrile/0.1% formic acid and solvent B: 90% isopropanol/10% acetonitrile/0.1% formic acid. Samples were applied to the column at 65% B and eluted with a linear increase in solvent B over 6 min (65%–100% B). The gradient was held at 100% B for 9 min and then returned to starting conditions for 2 min.
The QExactive mass spectrometer was equipped with a HESI electrospray source and operated in positive ion mode using the following parameters: Aux gas heater temperature 300 °C, capillary temperature 380 °C, sheath gas flow 38, auxiliary gas flow 11, sweep gas flow 2, spray voltage 4.5 kV, S-lens RF level 70 (%), full MS scans were obtained between 300–950 m/z window. For the API 5000, the following parameters were used: electrospray ionization in the positive-ion mode with the collision gas set at 5 units, curtain gas at 40 units, ion source gas number 1 at 55 units and number 2 at 50 units, ion spray voltage at 5000 V, and temperature at 600 °C. The declustering potential was 90 eV, entrance potential 5 eV, collision energy 35 eV, and the collision exit potential 10 eV. The following MRM transitions were used: 369.3/147.3 and 376.3/147.3 for Cholesterol or Cholesteryl esters and d7-Cholesterol or d7-cholesteryl ester, respectively as ionization in the API5000 source hydrolyzes cholesterol esters (Fig. S1). The cholesterol and total cholesteryl esters were determined from analyte/internal standard area ratios and content normalized to proteins.
2.12. Molecular docking modeling
For in silico studies, ligand charges were obtained with antechamber module at am1-bbc level. The crystal structure for CD36 was retrieved from the Protein Data Bank database (PDB ID: 5LGD). The portion of CD36-binding PfEMP1 protein domain was removed as well as all the hetero-atoms present in the crystallized structure followed by all the appropriate modifications and charge addition to the protein as previously [42]. CD36 protein was considered as a rigid entity with no flexible chains and both water and glycerol molecules were eliminated followed by hydrogen addition with H++ server (biophysics.cs.vt.edu/H++). Smina (biophysicis.cs.vt.edu/H++), a fork of AutoDock Vina [43], was used for the docking studies with two different scoring functions; Autodock-4 [44] and Vinardo [45], both afforded similar results. For simplicity, results with Autodock-4 were included in the main text. An exhaustiveness criteria of 1000 was used. Three regions of the protein around Lys164 were explored at 4, 6 and 8 Å of this residue. VMD (www.ks.uiuc.edu/Research/vmd/), Maestro (Schrödinger Release 2019-2: LigPrep, Schrödinger, LLC, New York, NY, 2019.) and PLIP servers (PLIP web service, provided by TU Dresden/Redivia, last access 09/26/2019) were used for the analysis of the results.
Statistical analysis was performed using the GraphPad Prism 5.0 software. After confirmation of normality and homoscedasticity, data was analyzed using one-way or two-way ANOVA with a Bonferroni test for post hoc analysis of multiple comparisons. p-values < 0,05 were considered to be significant. All experiments were repeated at least three independent times. Figures show mean ± standard error (SEM) of experimental conditions replicated at least 3 times.
3. Results
3.1. NO2-OA regulates CD36 expression in macrophages
Since nitrated fatty acids activate PPARγ and Nrf2-dependent signaling, we examined the role of NO2-OA on CD36 expression in macrophages [4,46]. Flow cytometry analysis of NO2-OA treated RAW264.7 macrophages showed dose-dependent (0–5.0 μM), statistically-significant increase in the percentage of cells expressing CD36 from 25.7 ± 5%, in macrophage control, to 32.9 ± 10.9%, 46.6 ± 9.84% and 53.8 ± 5.14% in cells treated with 1.0; 2.5 or 5.0 μM, respectively; with OA (5.0 μM) having no effect on CD36 expression (21.9 ± 13.73%) after 8 h of treatment (Fig. 1A). The response of CD36 expression to NO2-OA was further confirmed in primary cell cultures of BMDM obtained from wild-type C57BL/6 mice. In these cells, the non-electrophilic OA slightly increased CD36 expression as previously (Fig. S2) [47,48].
Fig. 1.

NO2-OA regulates CD36 expression in RAW 264.7 macrophages and BMDM. A. Density plots of CD36 in RAW264.7 macrophages stimulated with increasing concentrations of NO2-OA (0–5.0 μM) for 8 h (Side Scatter-A vs CD36-APC fluorescence). B. Time course effect of NO2-OA or OA (5 μM) on CD36 mRNA expression in RAW264.7 macrophages. C. CD36 and HO-1 expression in macrophages incubated with NO2-OA (0–5 μM) or CDDO-Me (300 nM) for 8 h. GAPDH was used as a loading control. D. NO2-OA induction of CD36 expression. Macrophages were pre-incubated with 10 μM of the PPARγ antagonist GW9662 or 10 μM of the agonist rosiglitazone during 10 min or 20 min with the Nrf2 activator CDDO-Me (300 nM) before NO2-OA addition. E and F. CD36 expression in BMDM from Nrf2-KO mice. Protein (HO-1, NQO1 and CD36) and mRNA levels (CD36) of BMDM obtained from WT and Nrf2-KO mice were treated with 5 μM of NO2-OA, 5 μM OA or 300 nM of CDDO-Me (8 h for Western blot analysis and 4 h for RT-qPCR analysis). The data is a representative experiment selected from at least 3 independent experiments, n = 3- for each determination showing mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
To gain insight into the mechanisms of action, NO2-OA induction of CD36 expression was evaluated at both the transcriptional and translational levels. Upon incubation of RAW264.7 cells with 5.0 μM NO2-OA, CD36 mRNA expression significantly increased at 2 and 4 h and returned to basal levels at 6 h after treatment (Fig. 1B). OA had no significant impact on CD36 transcript levels at the evaluated concentration. This dose-dependent increase in CD36 transcript levels in RAW264.7 macrophages stimulated with NO2-OA correlated with increases in CD36 and HO-1 protein expression, in agreement with previous results (Fig. 1C) [26]. CDDO-Me, a potent Nrf2 activator, also induced the expression of CD36 and its target gene HO-1 (Fig. 1C). To evaluate the role of PPARγ in CD36 expression, the PPARγ agonist rosiglitazone was used. No significant changes in CD36 expression were observed in RAW264.7 macrophages (Fig. 1D), and the up-regulation induced by NO2-OA was not inhibited by PPARγ antagonist GW9662. This suggests that the activation of the PPARγ receptor is not involved in CD36 expression (Fig. 1D). To further validate the role of NO2-OA on Keap1/Nrf2 activation of CD36 expression, BMDM from Nrf2-KO and wild type mice were evaluated. The induction of CD36 expression by NO2-OA and CDDO-Me was significantly reduced in Nrf2-KO macrophages when compared to those derived from wild type mice. Similar observations were made for other Nrf2 target genes such as HO-1 and NQO1 (Fig. 1E). These effects observed at the transcriptional level were also confirmed at the protein level (Fig. 1F). In aggregate, these results provide new evidence for the role of Nrf2 signaling in NO2-OA-dependent CD36 expression.
3.2. NO2-OA inhibits cellular uptake of mLDL
The fatty acid translocase CD36 is a key receptor involved in the cellular uptake of mLDL and LCFA. The increased CD36 expression induced by NO2-OA motivated the evaluation of the role of NO2-OA in CD36-dependent mLDL uptake in macrophages. LC-MS-based quantification of cholesterol and cholesteryl ester accumulation in mLDL treated RAW264.7 cells showed that NO2-OA had an inhibitory effect (Fig. 2A). Fluorescence microscopy using DiI-labeled mLDL showed that 15 min pre-incubation of cells with either NO2-OA or the CD36 antagonist SSO inhibited mLDL binding and uptake by macrophages (Fig. 2B and C). These results were further confirmed using an ELISA-like assay where pre-treatment with NO2-OA or SSO for 15 min significantly decreased the binding of DiI-labeled mLDL to rCD36 (Fig. 2D). In addition, we tested the effect of CCDO-Me on lipid uptake, and in agreement with the activation of the Keap1/Nrf2/CD36 axis, CCDO increased DiI-mLDL accumulation. In contrast, NO2-OA treatment counterbalanced the activation of Keap1/Nrf2/CD36 axis by reducing DiI-mLDL incorporation by macrophages (Fig. S3).Thus, the reduced incorporation and accumulation of cholesterol in macrophages indicate an inhibitory role for NO2-OA in mLDL binding to CD36.
Fig. 2.

NO2-OA inhibits mLDL incorporation by macrophages. A. Cholesterol and cholesteryl ester levels were reduced in RAW264.7 macrophages co-incubated with NO2-OA (5 μM) and mLDL for 24 h as assessed by LC-MS-MS. Analyses were performed using deuterated cholesterol and cholesteryl palmitate as internal standards. B and C. Immunofluorescence images and quantification of fluorescence from RAW264.7 cells incubated with NO2-OA (5 μM) and SSO (50 μM) for 15 min follow by treatment with DiI-labeled oxLDL (50 μg/uL) for 2 h at 4 °C and then washed and incubated at 37 °C for 30 min. Quantification of fluorescence was performed on at least 20 cells per condition. D. NO2-OA but not OA preincubation (15 min) with recombinant CD36 protein inhibits binding of DiI-labeled oxLDL (50 μg/μL). Binding was assessed after incubation for 2 h at 37 °C. The data is obtained from a representative experiment selected from at least 3 independent experiments. Mean ± SEM are represented in graphs (*p < 0.05, **p < 0.01, ***p < 0.001).
3.3. NO2-OA interacts with CD36
Since NO2-OA interferes with CD36-dependent binding and uptake of mLDL, we further investigated a potential direct interaction between NO2-OA and CD36. The co-immunoprecipitation of CD36 in RAW264.7 macrophages incubated with B-NO2-OA revealed a molecular association between CD36 and B-NO2-OA (Fig. 3A). Furthermore, this interaction was disrupted when macrophages were pretreated with SSO (Fig. 3A), suggesting that SSO and NO2-OA were competing for the same binding site. Analysis of the in vitro reaction of rCD36 and B-NO2-OA showed a dose-dependent association at mol ratios ranging from 1:0.5 to 1:10 (Fig. 3B). Given the robust interaction observed at the 1:2 mol ratio, this condition was used for subsequent experiments. To compete for the binding of B-NO2-OA to CD36, non-biotinylated NO2-OA and OA were incubated at ratios ranging from 1 to 100 fold excess over B-NO2-OA. Both NO2-OA and OA effectively competed with B-NO2-OA for binding to CD36 (Fig. 3C). Strikingly, a 10 fold excess of OA demonstrated a better competition than higher concentrations of OA (100 fold) [49,50]. Finally, GSH inhibited B-NO2-OA binding with rCD36 in a dose-dependent manner by inhibiting nitroalkene electrophilic reactivity (Fig. 3D). To evaluate the reversibility of the alkylation reaction between B-NO2-OA and rCD36, displacement assays were performed with SSO. After allowing the B-NO2-OA/rCD36 complex formation for 20 min, SSO was added to assess displacement. A molar ratio of 1:10 was necessary to induce a significant displacement of B-NO2-OA from rCD36 (Fig. 3E). These findings support our previous results with RAW264.7 macrophages and confirm the reversible nature of the binding of NO2-OA to CD36, which may involve the critical residue Lys164 [14]. Consistent with rCD36 binding studies showing competition of NO2-OA by OA, GSH, and SSO, excess of OA restored binding and uptake of DiI-oxLDL in the presence of NO2-OA in RAW264.7 macrophages. On the other hand, SSO, as expected, abolished binding and uptake of DiI-oxLDL independently of NO2-OA (Fig. S4). However, GSH, per se, interfered with the fluorescence measurement independent of NO2-OA treatment (Fig. S4).
Fig. 3.

NO2-OA binds reversibly and covalently to CD36. A. RAW264.7 macrophages were incubated with B-NO2-OA for 15 min at 4 °C to limit receptor endocytosis. Immunoprecipitation of CD36 was blotted for B-NO2-OA using HRP-Streptavidin. B. Recombinant CD36 was incubated with different mole ratios of B-NO2OA and the complexes B-NO2-OA-rCD36 revealed by SDS-PAGE using HRP-Streptavidin. C. Competition of B-NO2-OA with NO2-OA or OA for rCD36 binding. D. GSH inhibits B-NO2OA interaction with rCD36. E. SSO displacement of B-NO2-OA from B-NO2-OA-rCD36 adduct. The data is from a representative experiment selected from at least 3 independent experiments, n = 3 for each determination showing mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
3.4. NO2-OA interaction with the CD36 binding pocket is revealed by molecular docking analysis
Based on experimental data showing that SSO blocks the interaction of NO2-OA with CD36, we proposed that both ligands could share the same binding site (containing the Lys164). Hence, the region around Lys164 was modeled to explore the formation of a complex between CD36 and NO2-OA. OA was used as a non-electrophilic fatty acid control. The search included three regions at 4, 6 and 8 Å around Lys164. The most stable complexes were obtained at 6 Å, analysis of bigger exploration boxes (>8 Å) found no new complexes in other areas of the protein. Both ligands, NO2-OA and OA bind to the same region of the protein (Fig. 4A and Fig. S5A), although the complexes formed with the nitroalkene present lower energies than those of OA (Table 1). Fig. 4A displays all the geometries obtained for NO2-OA and Fig. 4B and C shows the representation of the two most stable geometries. The main difference between these two structures is the position of the nitro group, with both structures showing the aliphatic chains located in the same region (Fig. 4B and C) and the carboxylate group forming an H-bond with the hydroxyl group of Ser168 (2.63 and 3.62 Å respectively). In the first geometry, the nitro group forms a strong H-bond with Lys166 (~2.55 Å) (Fig. 4B), and the second geometry with the amide group of Asn163 (3.19 Å) (Fig. 4C). The 3D-view of these interactions is represented in Fig. 4D, left and right panel, respectively. No strong interactions were established with the amine of Lys164, instead this group moved freely and became available for nucleophilic reaction with the activated double bond in the nitroalkene (at 7.8 and 8.1 Å of the C-C double bond). Thus, the H-bond may be working as an anchor for the nitroalkene and Lys164 would move as a scorpion tail to reach the electrophilic β-carbon. It should be noted that while the geometries obtained for OA were similar to those for NO2-OA (Figs. S5B and S5C), the OA complex can not be further stabilized with a covalent bond (Fig. S5D). This binding dissimilarity may explain the functional differences between OA and NO2-OA in mLDL binding and the subsequent uptake through CD36.
Fig. 4.

In silico CD36-NO2-OA docking studies reveal the interaction of NO2-OA in the region of Lys164. A. Superimposed conformations obtained from docking simulation for searches performed at 4 (red), 6 (blue) and 8 Å (yellow) around Lys164. B and C. Representation of the two most stable complexes obtained for NO2-OA around Lys164. Ligand atoms are represented with balls and sticks and Lys164 and 166 of the receptor with bold sticks. The non-polar hydrogens of the ligand were included in the figures for a better representation. D. Three-dimensional (3D) representation of the CD36-NO2-OA complexes nearby Lys164 showed interactions with relevant residues of CD36. Left panel: CD36-NO2-OA complex represented in Fig. 5B and Right panel: CD36-NO2-OA complex represented in Fig. 5C. The carbons of the ligand are represented in orange while the carbons of the amino acids from the receptor are in cyan. Blue is used for nitrogen atoms, red for oxygen and white for hydrogen. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Table 1.
Binding energies values for the two most stable complexes of each ligand at different regions.
|
Ligand |
Binding Energy (kcal/mol) of the complexes at different regions |
||
|---|---|---|---|
| 4 Å | 6 Å | 8 Å | |
| NO2-OA | −23.7 | −27.1 | −25.7 |
| −23.2 | −27.1 | −25.3 | |
| OA | −22.6 | −23.7 | −22.4 |
| −22.0 | −23.7 | −22.3 | |
3.5. NO2-OA modulates lipid metabolism via activation of autophagy in RAW264.7
Given that lipid storage in macrophage foam cells has been associated with the deregulation of cholesterol efflux, we evaluated whether NO2-OA could alter lipid metabolism in lipid-loaded macrophages. Hence, macrophages were incubated with mLDL and sampling at different endpoints was used to determine that 24 h was, in our model, the best experimental condition for loading macrophages with lipids (Fig. S6A). Interestingly, lipid-loaded macrophages with DiI-labeled mLDL showed decreased immunofluorescence in NO2-OA treated cells compared to OA or vehicle (Fig. 5A). The expression of ABCA1 and ABCG1 transporters by qPCR did not show any significant changes in the presence or absence of NO2-OA (Fig. 5B). Cholesterol and cholesteryl esters were measured in the supernatant of lipid-loaded macrophages and efflux rate was calculated for the first 4 h as this timeframe presented the highest cholesterol release (Fig. S6B). Therefore, assessment of cholesterol export from lipid-loaded macrophages using HDL (50 μg/ml) as a cholesterol acceptor showed increased export for NO2-OA treated cells when compared to vehicle (Fig. 5C). These results suggest that the efflux of cholesterol and cholesterol esters in macrophages is regulated by NO2-OA independently of the transcriptional expression of fatty acid transporters ABC. To confirm this finding, we explored whether NO2-OA impacted other mechanisms involved in the regulation of cellular lipid storage and cholesterol efflux. The autophagosomal markers LC3II and p62 were evaluated in lipid-loaded macrophages as a measurement of autophagic flux. An increase in LC3II and p62 protein was observed in lipid-loaded macrophages, supporting previous reports indicating an inhibition of the autophagic flux (Fig. 5D). In contrast, incubation with 5 μM of NO2-OA for 8 h, significantly decreased levels of LC3II and p62, denoting restoration of a defective autophagic flux (Fig. 5D). Neither oxLDL nor NO2-OA changed the transcriptional regulation of MAP1 (the gene encoding LC3 protein) (Fig. S7). The action of the inducer of autophagic flux rapamycin on depletion of autolysosome markers LC3II paralleled the effect of NO2-OA (Fig. S8), while chloroquine, an inhibitor of the autophagy flux, showed accumulation of LC3II caused by the fusion failure of autophagosomes and lysosomes (Fig, S8). In aggregate, these results indicate that in lipid-loaded macrophages, NO2-OA enhances the efflux of cholesterol and recovery of the autophagic flux.
Fig. 5.

NO2-OA regulates cholesterol metabolism. A. RAW264.7 macrophages loaded with DiI-oxLDL (100 μg/ml/24 h) were incubated with vehicle, NO2-OA or OA during 24 h. Images were obtained in a Leica DMi8 microscope. At least 20 cells per condition were randomly analyzed. The data is from a representative experiment selected from at least 3 independent experiments. B. ABCG1 and ABCA1 mRNA expression in RAW264.7 macrophages treated with 5 μM of NO2-OA during 2, 4 and 8 h. At least 3 independent experiments were carried out. C. Cholesterol efflux in RAW 264.7 macrophages loaded with oxLDL 100 μg/ml/24 h. Macrophages were treated with vehicle or NO2-OA (5 μM), cholesterol release was evaluated in the cell culture supernatant by MS. The graph shows the data obtained from at least 3 independent experiments. D. RAW 264.7 macrophages loaded with oxLDL 100 μg/ml for 24 h. Then cells were treated with vehicle or NO2-OA (5 μM) for 8 h in presence or absence of AMPK inhibitor CompC (3,3 μM). Macrophages were harvested in lysis buffer and samples analyzed by Western blot. The data is from a representative experiment selected from at least 3 independent experiments. Each quantification shows mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001.
Given that a link between AMPK and mTOR inhibition had been previously established and that NO2-OA induced the phosphorylation of Src and AMPK (Fig. S9), we evaluated the role of AMPK activation in the restoration of autophagic flux [51,52]. Treatment with the AMPK inhibitor Compound C abolished the beneficial effects on autophagy flux by NO2-OA (Fig. 5D). Thus, the regulation of the lipid metabolism by NO2-OA in lipid-loaded macrophages is, at least, partially mediated by AMPK phosphorylation and activation of autophagy.
4. Discussion
The present study shows that the electrophilic fatty acid nitroalkene NO2-OA limits macrophage mLDL incorporation and cholesterol accumulation despite increasing CD36 expression. Macrophages are important mediators in the pathogenesis of atherosclerosis because of a central role in the propagation of chronic inflammation, lipid accumulation, and differentiation to atherogenic foam cells [1]. Upon loss of endothelial barrier function, the extravasation of humoral and cellular pro-inflammatory components into the arterial intima leads to enhanced rates of nitro-oxidative reactions that further exacerbate inflammation and oxidative stress in the local intima. Modified lipoproteins are incorporated by macrophages through cell surface scavenger receptors such as CD36, SR-A1, LOX-1, and LRP-1, disrupting macrophage lipid metabolism and promoting foam cell formation that propagates inflammation, lipid accumulation, and allied atherogenic responses. A strong correlation has been established both clinically and in experimental models between the expression levels of CD36 in vascular cells and atherosclerosis [[53], [54], [55]].
Relevant to the present study, the administration of NO2-OA in ApoE-/- mice fed a high-fat diet (HFD) protected aorta from atherosclerotic plaque development, reduced inflammatory cell infiltration, lipid deposition and both plaque size and stability [29]. The anti-inflammatory actions of NO2-FA inhibit cytokine production (IL-1 and IL-6), expression of vascular adhesion molecules (VCAM) and TLR4 activation in inflammatory macrophages, thus both limiting and facilitating the resolution of inflammatory processes [56]. While NO2-FA modulate inflammatory responses in macrophages in vitro and in vivo, an impact on macrophage lipid uptake and metabolism has not been previously explored.
NO2-OA strongly modulated macrophage lipid metabolism via the activation of CD36-mediated mLDL incorporation and cholesterol efflux. While NO2-OA induced the expression of CD36 via activation of Nrf2-dependent gene expression, this effect was counterbalanced by covalent binding of NO2-OA with CD36, the net effect being a reduction of total macrophage cholesterol and cholesteryl ester accumulation. These findings are consistent with responses to other CD36 ligands that modulate mLDL binding and internalization [[57], [58], [59]]. For NO2-OA, covalent binding with CD36 via Michael addition is reversible, with NO2-OA competing for binding with other non-covalent ligands such as OA. This indicates well-integrated mechanisms of post-translational protein modification and signaling responses by NO2-OA that both regulate cholesterol uptake and CD36 expression.
We had previously reported the PPARγ–dependent regulation of CD36 expression by nitroalkenes, specifically nitro-linoleic acid (NO2-LA), another member of the nitro-fatty acid class, but did not explore the participation of Keap1/Nrf2 signaling responses [17]. 4-Hydroxynonenal (4-HNE), another electrophilic lipid peroxidation byproduct, also up-regulates the expression of CD36; however, 4-HNE strongly promotes, rather than limits, macrophage foam cell formation [60]. These starkly contrasting actions of NO2-OA, 4-HNE and the Nrf2 agonist CDDO-Me may be attributed to the inhibition of CD36-dependent mLDL uptake induced by fatty acid nitroalkenes. These beneficial effects on lipid metabolism further translate to in vivo models, since the administration of NO2-FA to high fat diet-induced obese mice normalized hepatic triglyceride levels, limited indices of hepatic inflammation and reversed established hepatic steatosis [61]. Similarly, nitroalkene-containing α-tocopherol derivatives reduce inflammation and limit atherosclerotic lesion formation in ApoE-/- mice [36].
Oxidized LDL and FA bind to an α-helix at the membrane-exposed distal tip of CD36, a lipid binding pocket which contains the critical Lys164 to either initiate downstream signaling or facilitate FA binding and internalization [[62], [63], [64]]. Herein, a CD36-selective lysine alkylating agent (SSO) revealed that NO2-OA interacts with a region vicinal to Lys164 in CD36 (Fig. 4A), displaying lower binding energies than OA and forming strong H-bonds with the carboxylate and nitro groups (Fig. 4 and Table 1). Upon initial binding, the nucleophilic Lys164 is free to move and access the electrophilic β-carbon of the nitroalkene substituent to initiate Michael addition. For the most stable complexes modeled in silico, the distances from Lys164 to the β-carbon were 7.8 and 8.1 Å. This modeling reveals that the H-bonds are working as anchors for the nitroalkene and Lys164 can in turn access the β-carbon. This role for Lys164 is supported by our findings, including the inhibition of mLDL interaction with CD36 by NO2-OA, the inhibition of binding and downstream signaling by SSO, and a lack of regulation by OA.
In atherogenesis, the regulation of lipid homeostasis in macrophages is crucial to prevent the formation of foam cells. Thus, macrophages in early atherosclerotic lesions exhibit accumulation of CE in lipid droplets, which are degraded at lysosomal levels in a process associated with autophagy (lipophagy). However, in advanced lesions, accumulation of cholesterol and cholesteryl esters impair macrophage lipophagy, blocking cholesterol efflux. Here, we demonstrated that NO2-OA improves cholesterol efflux in oxLDL-loaded macrophages. Significant increases in cholesterol and cholesteryl ester efflux was observed during the first 4 h of treatment, a time frame corresponding to the highest rates of cholesterol release. This effect was unrelated to changes in expression levels of ATP-binding cassette transporter A1 or G1. Based on the finding that oxLDL impairs the autophagic pathway in lipid-loaded macrophages [33,65,66], these results provide multiple lines of evidence supporting that NO2-OA stimulates autophagy. NO2-OA reduced cholesterol and LD accumulation and reversed autophagic flux, as evidenced by reductions in LC3II and p62 protein levels.
Activation of AMPK-dependent signaling induces autophagic flux by inhibiting mTOR [51,52]. In macrophages, NO2-OA induced the activation Src and AMPK, an effect that can also lead to restoration of the autophagy flux.
Beneficial effects of NO2-OA on lipid metabolism have been extensively studied in the context of liver diseases, but a role in cardiovascular diseases and atherosclerosis is less well understood. NO2-OA protects the vasculature from neointimal hyperplasia induced by arterial injury and the development of atherosclerotic plaque [67]. Until now, the underlying metabolic reactions, signaling mechanisms and the modulation of macrophage function by NO2-OA have not been addressed. In this study, we reveal that NO2-OA not only reduces oxLDL uptake but also reduces macrophage foam cell formation through the regulation of the macrophage autophagy flux.
5. Conclusion
Our data reveals that NO2-OA is a novel ligand for CD36 that inhibits mLDL binding, uptake, and lipid droplet formation in macrophages. NO2-OA induces the Nrf2-dependent expression of CD36 in macrophages. This potentially pro-atherogenic effect is counterbalanced by the inhibition of mLDL incorporation by macrophages through binding and covalent modification of CD36 that results in a net increase in lipid efflux. The modulation of this pathway provides a novel target for therapeutic approaches that restore cell function and reduce pro-inflammatory status under pathological conditions.
Declaration of competing interest
BAF and FJS acknowledge an interest in Complexa, Inc. and Creegh Pharmaceuticals.
Acknowledgements
The authors thank Dra. María Cecilia Sanchez for her kind contributions. This work was supported by Fondo para la Investigación Científica y Tecnológica (FONCYT) del Ministerio de Ciencia, Tecnología e Innovación Argentina. PICT 3063-2013, and Secretaria de Ciencia y Técnica (SECyT) de la Universidad Nacional de Córdoba. Argentina (G.B.). National Institutes of Health, United State: Grants R01-HL64937, R01-HL132550 and P01-HL103455 (B.A.F.), R01-GM125944, R01-DK112854 and American Heart Association, United State, 17GRN33660955 (F.J.S.).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101591.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Moore K.J., Sheedy F.J., Fisher E.A. Macrophages in atherosclerosis: a dynamic balance. Nat. Rev. Immunol. 2013;13:709–721. doi: 10.1038/nri3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore K.J., Tabas I. The cellular biology of macrophages in atherosclerosis. Cell. 2011;145:341–355. doi: 10.1016/j.cell.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Freeman M.W., Febbraio M., Silverstein R., Koehn S., Hoff H.F., Andersson L., Moore K.J., Kunjathoor V.V., Rhee J.S., Podrez E.A. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J. Biol. Chem. 2002;277:49982–49988. doi: 10.1074/jbc.m209649200. [DOI] [PubMed] [Google Scholar]
- 4.Ishii T., Itoh K., Ruiz E., Leake D.S., Unoki H., Yamamoto M., Mann G.E. Role of Nrf2 in the regulation of CD36 and stress protein expression in murine macrophages: activation by oxidatively modified LDL and 4-hydroxynonenal. Circ. Res. 2004;94:609–616. doi: 10.1161/01.RES.0000119171.44657.45. [DOI] [PubMed] [Google Scholar]
- 5.Tontonoz P., Nagy L., Alvarez J.G.A., Thomazy V.A., Evans R.M. PPARγ promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93:241–252. doi: 10.1016/S0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- 6.Hsieh F.L., Turner L., Bolla J.R., Robinson C.V., Lavstsen T., Higgins M.K. The structural basis for CD36 binding by the malaria parasite. Nat. Commun. 2016;7:1–11. doi: 10.1038/ncomms12837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abumrad N.A., El-Maghrabi M.R., Ez-Zoubir A., Lopez E., Grimaldill P.A. Cloning of a rat adipocyte membrane protein implicated in binding or transport of long-chain fatty acids that is induced during preadipocyte differentiation. J. Biol. Chem. 1993;268:17665–17668. doi: 10.1002/pd.4617. [DOI] [PubMed] [Google Scholar]
- 8.Baillie A.G.S., Coburn C.T., Abumrad N.A. Reversible binding of long-chain patty acids to purified FAT, the adipose CD36 homolog. J. Membr. Biol. 1996;153:75–81. doi: 10.1007/s002329900111. [DOI] [PubMed] [Google Scholar]
- 9.Coburn C.T., Knapp J., Febbraio M., Beets A.L., Silverstein R.L., Abumrad N.A. Defective uptake and utilization of long chain fatty acids in muscle and adipose tissues of CD36 knockout mice. J. Biol. Chem. 2000;275:32523–32529. doi: 10.1074/jbc.M003826200. [DOI] [PubMed] [Google Scholar]
- 10.Endemann G., Stanton L.W., Madden K.S., Bryant C.M., White R.T., Protter A.A. CD36 is a receptor for oxidized low density lipoprotein. J. Biol. Chem. 1993;268:11811–11816. [PubMed] [Google Scholar]
- 11.Kar N.S., Ashraf M.Z., Valiyaveettil M., Podrez E.A. Mapping and characterization of the binding site for specific oxidized phospholipids and oxidized low density lipoprotein of scavenger receptor CD36. J. Biol. Chem. 2008;283:8765–8771. doi: 10.1074/jbc.M709195200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Oquendo P., Hundt E., Lawler J., Seed B. CD36 directly mediates cytoadherence of Plasmodium falciparum parasitized erythrocytes. Cell. 1989;58:95–101. doi: 10.1016/0092-8674(89)90406-6. [DOI] [PubMed] [Google Scholar]
- 13.Asch A.S., Silbiger S., Heimer E., Nachman R.L. Thrombospondin sequence motif (CSVTCG) is responsible for CD36 binding. Biochem. Biophys. Res. Commun. 1992;182:1208–1217. doi: 10.1016/0006-291X(92)91860-S. [DOI] [PubMed] [Google Scholar]
- 14.Kuda O., Abumrad N.A., Pietka T.A., Cvacka J., Kopecky J., Demianova Z., Kudova E. Sulfo- N -succinimidyl oleate (SSO) inhibits fatty acid uptake and signaling for intracellular calcium via binding CD36 lysine 164. J. Biol. Chem. 2013;288:15547–15555. doi: 10.1074/jbc.m113.473298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coort S.L.M., Willems J., Coumans W.A., van der Vusse G.J., Bonen A., Glatz J.F.C., Luiken J.J.F.P. Sulfo-N-succinimidyl esters of long chain fatty acids specifically inhibit fatty acid translocase (FAT/CD36)-mediated cellular fatty acid uptake. Mol. Cell. Biochem. 2002;239:213–219. doi: 10.1023/A:1020539932353. [DOI] [PubMed] [Google Scholar]
- 16.Vögtle T., Sadou H., Chouabe C., Abdoul-Azize S., Khan N.A., Besnard P., Dramane G., Akpona S., Hichami A., Nieswandt B. STIM1 regulates calcium signaling in taste bud cells and preference for fat in mice. J. Clin. Invest. 2012;122:2267–2282. doi: 10.1172/jci59953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schopfer F.J., Lin Y., Baker P.R.S., Cui T., Garcia-Barrio M., Zhang J., Chen K., Chen Y.E., a Freeman B. Nitrolinoleic acid: an endogenous peroxisome proliferator-activated receptor ligand. Proc. Natl. Acad. Sci. Unit. States Am. 2005;102:2340–2345. doi: 10.1073/pnas.0408384102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beckman J., Beckman T., Chen J. Apparent hydroxyl radical production by peroxynitrite: implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. U.S.A. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schopfer F.J., Cipollina C., Freeman B.a. Formation and signaling actions of electrophilic lipids. Chem Rev. 2011;111:5997–6021. doi: 10.1021/cr200131e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bonacci G, Baker P.R., Salvatore S.R., Shores D., Khoo N.K.H, Koenitzer J.R., Vitturi D.A., Woodcock S.R, Gollin-Bisello F., Cole M.P., Watkins S., Croix St.C., Batthyany C.I., Freeman B.A., Schopfer F.J. Conjugated linoleic acid is a preferential substrate for fatty acid nitration. J. Biol. Chem. 2012;287:44071–44082. doi: 10.1074/jbc.m112.401356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nadtochiy S.M., Baker P.R.S., Freeman B.A., Brookes P.S. Mitochondrial nitroalkene formation and mild uncoupling in ischaemic preconditioning: implications for cardioprotection. Cardiovasc. Res. 2009;82:333–340. doi: 10.1093/cvr/cvn323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lima É.S., Di Mascio P., Rubbo H., Abdalla D.S.P. Characterization of linoleic acid nitration in human blood plasma by mass spectrometry. Biochemistry. 2002;41:10717–10722. doi: 10.1021/bi025504j. [DOI] [PubMed] [Google Scholar]
- 23.Salvatore S.R., Vitturi D.A., Fazzari M., Jorkasky D.K., Schopfer F.J. Evaluation of 10-nitro oleic acid bio-elimination in rats and humans. Sci. Rep. 2017;7:1–14. doi: 10.1038/srep39900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Batthyany C., Schopfer F.J., Baker P.R.S., Durán R., Baker L.M.S., Huang Y., Cerveñansky C., Branchaud B.P., Freeman B.A. Reversible post-translational modification of proteins by nitrated fatty acids in vivo. J. Biol. Chem. 2007;281:20450–20463. doi: 10.1074/jbc.M602814200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schopfer F.J., Cole M.P., Groeger A.L., Chen C.S., Khoo N.K.H., Woodcock S.R., Golin-Bisello F., Nkiru Motanya U., Li Y., Zhang J., Garcia-Barrio M.T., Rudolph T.K., Rudolph V., Bonacci G., Baker P.R.S., Xu H.E., Batthyany C.I., Chen Y.E., Hallis T.M., Freeman B.A. Covalent peroxisome proliferator-activated receptor γ adduction by nitro-fatty acids: selective ligand activity and anti-diabetic signaling actions. J. Biol. Chem. 2010;285:12321–12333. doi: 10.1074/jbc.M109.091512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kansanen E., Bonacci G., Schopfer F.J., Kuosmanen S.M., Tong K.I., Leinonen H., Woodcock S.R., Yamamoto M., Carlberg C., Yla S., Freeman B.A. Electrophilic nitro-fatty acids activate NRF2 by a KEAP1 cysteine 151-independent mechanism *. J. Biol. Chem. 2011;286:14019–14027. doi: 10.1074/jbc.M110.190710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cui T., Schopfer F.J., Zhang J., Chen K., Ichikawa T., Baker P.R., Batthyany C., Chacko B.K., Feng X., Patel R.P., Agarwal A., Freeman B.A., Chen Y.E. Nitrated fatty acids: endogenous anti-inflammatory signaling mediators. J. Biol. Chem. 2006;281:35686–35698. doi: 10.1074/jbc.M603357200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hansen A.L., Buchan G.J., Rühl M., Mukai K., Salvatore S.R., Ogawa E., Andersen S.D., Iversen M.B., Thielke A.L., Gunderstofte C., Motwani M., Møller C.T., Jakobsen A.S., Fitzgerald K.A., Roos J., Lin R., Maier T.J., Goldbach-Mansky R., Miner C.A., Qian W., Miner J.J., Rigby R.E., Rehwinkel J., Jakobsen M.R., Arai H., Taguchi T., Schopfer F.J., Olagnier D., Holm C.K. Nitro-fatty acids are formed in response to virus infection and are potent inhibitors of STING palmitoylation and signaling. Proc. Natl. Acad. Sci. U.S.A. 2018;115:E7768–E7775. doi: 10.1073/pnas.1806239115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rudolph T.K., Rudolph V., Edreira M.M., Cole M.P., Bonacci G., Schopfer F.J., Woodcock S.R., Franek A., Pekarova M., Khoo N.K.H., Hasty A.H., Baldus S., Freeman B.a. Nitro-fatty acids reduce atherosclerosis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2010;30:938–945. doi: 10.1161/ATVBAHA.109.201582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin C.Y., Duan H., Mazzone T. Apolipoprotein E-dependent cholesterol efflux from macrophages: kinetic study and divergent mechanisms for endogenous versus exogenous apolipoprotein E. J. Lipid Res. 1999;40:1618–1627. http://www.ncbi.nlm.nih.gov/pubmed/10484608 [PubMed] [Google Scholar]
- 31.Ouimet M., Franklin V., Mak E., Liao X., Tabas I., Marcel Y.L. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metabol. 2011;13:655–667. doi: 10.1016/j.cmet.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang S., Zhang X., Liu M., Luan H., Ji Y., Guo P., Wu C. Chrysin inhibits foam cell formation through promoting cholesterol efflux from RAW264.7 macrophages. Pharm. Biol. 2015;53:1481–1487. doi: 10.3109/13880209.2014.986688. [DOI] [PubMed] [Google Scholar]
- 33.Ning H., Liu D., Yu X., Guan X. Oxidized low-density lipoprotein-induced p62/SQSTM1 accumulation in THP-1-derived macrophages promotes IL-18 secretion and cell death. Exp. Ther. Med. 2017;14:5417–5423. doi: 10.3892/etm.2017.5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh R., Kaushik S., Wang Y., Xiang Y., Novak I., Komatsu M., Tanaka K., Cuervo A.M., Czaja M.J. Autophagy regulates lipid metabolism. Nature. 2009;458:1131–1135. doi: 10.1038/nature07976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rodriguez-Duarte J., Galliussi G., Dapueto R., Rossello J., Malacrida L., Kamaid A., Schopfer F.J., Escande C., López G.V., Batthyány C. A novel nitroalkene-α-tocopherol analogue inhibits inflammation and ameliorates atherosclerosis in Apo E knockout mice. Br. J. Pharmacol. 2019 doi: 10.1111/bph.14561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woodcock S.R., Bonacci G., Gelhaus S.L., Schopfer F.J. 2013. Nitrated Fatty Acids: Synthesis and Measurement. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schopfer F.J., Vitturi D.A., Jorkasky D.K., Freeman B.A. Nitro-fatty acids: new drug candidates for chronic inflammatory and fibrotic diseases. Nitric Oxide - Biol. Chem. 2018;79:31–37. doi: 10.1016/j.niox.2018.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ruotsalainen A.K., Lappalainen J.P., Heiskanen E., Merentie M., Sihvola V., Näpänkangas J., Raikaslehto L.L., Kansanen E., Adinolfi S., Kaarniranta K., Ylä-Herttuala S., Jauhiainen M., Pirinen E., Levonen A.L. Nuclear Factor E2-related Factor 2 Deficiency Impairs Atherosclerotic Lesion Development but Promotes Features of Plaque Instability in Hypercholesterolaemic Mice. Cardiovasc. Res. 2019;115:243–254. doi: 10.1093/cvr/cvy143. [DOI] [PubMed] [Google Scholar]
- 40.Batthyány C., Santos C.X.C., Botti H., Cerveñansky C., Radi R., Augusto O., Rubbo H. Direct evidence for apo B-100-mediated copper reduction: studies with purified apo B-100 and detection of tryptophanyl radicals. Arch. Biochem. Biophys. 2000;384:335–340. doi: 10.1006/abbi.2000.2102. [DOI] [PubMed] [Google Scholar]
- 41.Ridano M.E, Racca A.C., Flores-Martín J., Camolotto S.A., Magnarelli de Potas G., Genti-Raimondi S., Panzetta-Dutari G.M. Chlorpyrifos modifies the expression of genes involved in human placental function. Reprod. Toxicol. 2012;33:331–338. doi: 10.1016/j.reprotox.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 42.Morris G.M., Huey R., Olson A.J. Wiley Brand; 2008. Using AutoDock for Ligand-Receptor Docking, Current Protocols in Bioinformatics. [DOI] [PubMed] [Google Scholar]
- 43.Trott O., Olson A. Autodock vina: improving the speed and accuracy of docking. J. Comput. Chem. 2010;31:455–461. doi: 10.1002/jcc.21334.AutoDock. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Forli S., Botta M. Lennard-Jones potential and dummy atom settings to overcome the AUTODOCK limitation in treating flexible ring systems. J. Chem. Inf. Model. 2007;47:1481–1492. doi: 10.1021/ci700036j. [DOI] [PubMed] [Google Scholar]
- 45.Quiroga R., Villarreal M.A. Vinardo: a scoring function based on autodock vina improves scoring, docking, and virtual screening. PloS One. 2016;11:1–18. doi: 10.1371/journal.pone.0155183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Moore K.J., Rosen E.D., Fitzgerald M.L., Randow F., Andersson L.P., Altshuler D., Milstone D.S., Mortensen R.M., Spiegelman B.M., Freeman M.W. The role of PPAR-γ in macrophage differentiation and cholesterol uptake. Nat. Med. 2001;7:41–47. doi: 10.1038/83328. [DOI] [PubMed] [Google Scholar]
- 47.Yang P., Su C., Luo X., Zeng H., Zhao L., Wei L., Zhang X., Varghese Z., Moorhead J.F., Chen Y., Ruan X.Z. Dietary oleic acid-induced CD36 promotes cervical cancer cell growth and metastasis via up-regulation Src/ERK pathway. Canc. Lett. 2018;438:76–85. doi: 10.1016/j.canlet.2018.09.006. [DOI] [PubMed] [Google Scholar]
- 48.Sfeir Z., Ibrahimi A., Amri E., Grimaldi P., Abumrad N. Regulation of FAT/CD36 gene expression: further evidence in support of a role of the protein in fatty acid binding/transport. Prostagl. Leukot. Essent. Fat. Acids. 1997;57:17–21. doi: 10.1016/S0952-3278(97)90487-7. [DOI] [PubMed] [Google Scholar]
- 49.Morigaki K., Walde P. Fatty acid vesicles. Curr. Opin. Colloid Interface Sci. 2007;12:75–80. doi: 10.1016/j.cocis.2007.05.005. [DOI] [Google Scholar]
- 50.Chen I.A., Szostak J.W. Membrane growth can generate a transmembrane pH gradient in fatty acid vesicles. Proc. Natl. Acad. Sci. U.S.A. 2004;101:7965–7970. doi: 10.1073/pnas.0308045101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Krishan S., Richardson D.R., Sahni S. Adenosine monophosphate-activated kinase and its key role in catabolism: structure, regulation, biological activity, and pharmacological activation. Mol. Pharmacol. 2015;87:363–377. doi: 10.1124/mol.114.095810. [DOI] [PubMed] [Google Scholar]
- 52.Ou H., Liu C., Feng W., Xiao X., Tang S., Mo Z. Role of AMPK in atherosclerosis via autophagy regulation. Sci. China Life Sci. 2018;61:1212–1221. doi: 10.1007/s11427-017-9240-2. [DOI] [PubMed] [Google Scholar]
- 53.Han J., Hajjar D.P., Febbraio M., Nicholson A.C. Native and modified low density lipoproteins increase the functional expression of the macrophage class B scavenger receptor, CD36. J. Biol. Chem. 1997;272:21654–21659. doi: 10.1074/jbc.272.34.21654. [DOI] [PubMed] [Google Scholar]
- 54.Nakata A., Nakagawa Y., Nishida M., Nozaki S., Miyagawa J.I., Nakagawa T., Tamura R., Matsumoto K., Kameda-Takemura K., Yamashita S., Matsuzawa Y. CD36, a novel receptor for oxidized low-density lipoproteins, is highly expressed on lipid-laden macrophages in human atherosclerotic aorta. Arterioscler. Thromb. Vasc. Biol. 1999;19:1333–1339. doi: 10.1161/01.ATV.19.5.1333. [DOI] [PubMed] [Google Scholar]
- 55.Febbraio M., Podrez E.A., Smith J.D., Hajjar D.P., Hazen S.L., Hoff H.F., Sharma K., Silverstein R.L. Targeted disruption of the class B, scavenger receptor CD36 protects against atherosclerotic lesion development in mice. J. Clin. Invest. 2000;105:1049–1056. doi: 10.1172/JCI9259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Villacorta L., Chang L., Salvatore S.R., Ichikawa T., Zhang J., Petrovic-Djergovic D., Jia L., Carlsen H., Schopfer F.J., Freeman B.A., Chen Y.E. Electrophilic nitro-fatty acids inhibit vascular inflammation by disrupting LPS-dependent TLR4 signalling in lipid rafts. Cardiovasc. Res. 2013;98:116–124. doi: 10.1093/cvr/cvt002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hung J.P., Paz M.A., Hamilton J.A., Jay A.G., Chen A.N. CD36 binds oxidized low density lipoprotein (LDL) in a mechanism dependent upon fatty acid binding. J. Biol. Chem. 2015;290:4590–4603. doi: 10.1074/jbc.m114.627026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jay A.G., Hamilton J.A. The enigmatic membrane fatty acid transporter CD36: new insights into fatty acid binding and their effects on uptake of oxidized LDL. Prostagl. Leukot. Essent. Fat. Acids. 2018;138:64–70. doi: 10.1016/j.plefa.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 59.Li J., Yu C., Wang R., Xu J., Chi Y., Qin J., Liu Q. The ω-carboxyl group of 7-ketocholesteryl-9-carboxynonanoate mediates the binding of oxLDL to CD36 receptor and enhances caveolin-1 expression in macrophages. Int. J. Biochem. Cell Biol. 2017;90:121–135. doi: 10.1016/j.biocel.2017.07.022. [DOI] [PubMed] [Google Scholar]
- 60.Yun M.R., Im D.S., Lee S.J., Park H.M., Bae S.S., Lee W.S., Kim C.D. 4-Hydroxynonenal enhances CD36 expression on murine macrophages via p38 MAPK-mediated activation of 5-lipoxygenase. Free Radic. Biol. Med. 2009;46:692–698. doi: 10.1016/j.freeradbiomed.2008.12.013. [DOI] [PubMed] [Google Scholar]
- 61.Shiva S., Khoo N.K.H., Fazzari M., Chatoumpekis D.V., Li L., Guimaraes D.A., Arteel G.E., Freeman B.A. Electrophilic nitro-oleic acid reverses obesity-induced hepatic steatosis. Redox Biol. 2019;22:101132. doi: 10.1016/j.redox.2019.101132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.El-Yassimi A., Hichami A., Besnard P., Khan N.A. Linoleic acid induces calcium signaling, Src kinase phosphorylation, and neurotransmitter release in mouse CD36-positive gustatory cells. J. Biol. Chem. 2008;283:12949–12959. doi: 10.1074/jbc.M707478200. [DOI] [PubMed] [Google Scholar]
- 63.Abumrad N.A., Goldberg I.J. CD36 actions in the heart: lipids, calcium, inflammation, repair and more? Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 2016;1861:1442–1449. doi: 10.1016/j.bbalip.2016.03.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Samovski D., Sun J., Pietka T., Gross R.W., Eckel R.H., Su X., Stahl P.D., Abumrad N.a. Regulation of AMPK activation by CD36 links fatty acid uptake to $β$-Oxidation. Diabetes. 2015;64:353–359. doi: 10.2337/db14-0582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ackermann K., Bonaterra G.A., Kinscherf R., Schwarz A. Growth differentiation factor-15 regulates oxLDL-induced lipid homeostasis and autophagy in human macrophages. Atherosclerosis. 2019;281:128–136. doi: 10.1016/j.atherosclerosis.2018.12.009. [DOI] [PubMed] [Google Scholar]
- 66.Ma Y., Huang Z., Zhou Z., He X., Wang Y., Meng C., Huang G., Fang N. A novel antioxidant Mito-Tempol inhibits ox-LDL-induced foam cell formation through restoration of autophagy flux. Free Radic. Biol. Med. 2018;129:463–472. doi: 10.1016/j.freeradbiomed.2018.10.412. [DOI] [PubMed] [Google Scholar]
- 67.Cole M.P., Rudolph T.K., Khoo N.K.H., Motanya U.N., Wertz J.W., Schopfer F.J., Rudolph V., Steven R., Bolisetty S., Ali M.S., Zhang J., Chen Y.E., Agarwal A., Freeman B.A., Bauer P.M. Nitro-fatty acid inhibition of neointima formation after endoluminal vessel injury. Circ. Res. 2010;105:965–972. doi: 10.1161/CIRCRESAHA.109.199075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.