

Abstract
Macrocyclic peptides have proven highly effective inhibitors of protein–protein interactions, but generally lack cell permeability to access intracellular targets. We show herein that macrocyclic peptides may be rendered highly cell-permeable and biologically active by conjugating with a cyclic cell-penetrating peptide (CPP). A previously reported cyclic peptidyl inhibitor against the Kelch-like ECH-associated protein 1 (Keap1)-nuclear factor erythroid-2 (Nrf2) interaction (KD = 18 nM) was covalently attached to a cyclic CPP through a flexible linker. The resulting bicyclic peptide retained the Keap1-binding activity, resisted proteolytic degradation, readily entered mammalian cells, and activated the transcriptional activity of Nrf2 at nanomolar to low micromolar concentrations in cell culture. The inhibitor provides a useful tool for investigating the biological function of Keap1-Nrf2 and a potential lead for further development into a novel class of anti-inflammatory and anticancer agents. Our data suggest that other membrane-impermeable cyclic peptides may be similarly rendered cell-permeable by conjugation with a cyclic CPP.
Keywords: Cell-penetrating peptide, Cyclic peptide, Drug delivery, Oxidative stress, Protein-protein interaction
Graphical Abstract
INTRODUCTION
Macrocyclic peptides have recently emerged as an exciting class of drug modality because of their capacity of targeting proteins that are challenging for current drugs (e.g., small molecules and biologics), such as those involved in intracellular protein-protein interactions (PPIs).1,2 Moreover, the advent of several powerful combinatorial library technologies has enabled the rapid discovery of macrocyclic peptide ligands with antibody-like affinity and specificity against essentially any protein target.3–10 A major limitation of macrocyclic peptides, however, is their impermeability to the cell membrane, preventing access to intracellular targets.11 As such, although an increasing number of highly potent, specific cyclic peptidyl ligands have been developed against intracellular targets, through library screening or other approaches, their therapeutic potential has not been realized. To date, therapeutic applications of macrocyclic peptides have largely been limited to extracellular targets.12
One approach to improving the cell-permeability of macrocyclic peptides is reduction of the number of peptide backbone-associated hydrogen bond donors and acceptors through Nα-methylation, formation of intramolecular hydrogen bonds, and/or steric occlusion with bulky side chains.11,13 However, this method is generally limited to relatively small macrocycles (≤11 amino acids) of predominantly hydrophobic residues.14 An alternative approach involves conjugation with cell-penetrating peptides (CPPs).15 Unfortunately, the first-generation CPPs had low cytosolic delivery efficiencies as well as poor bioavailability and biodistribution. Several investigators recently discovered small, amphipathic cyclic CPPs with greatly improved cytosolic delivery efficiencies, bioavailability and broad tissue distribution.16–23 We had previously improved the cell-permeability of cyclic peptide ligands by fusing them with cyclic CPPs to form bicyclic peptides.24 We further demonstrated that cell-permeable bicyclic peptides containing a target-binding sequence in one ring and a CPP motif in the second ring can be combinatorially synthesized and screened for binding to intracellular targets to identify cell-permeable, biologically active ligands.25,26 An advantage of the bicyclic approach is that, when properly designed, the CPP ring may perform the dual function of cell penetration and target binding.27 However, the close proximity of the CPP and target-binding motifs may also result in mutual interference of their individual functions. In principle, an operationally simpler approach is to covalently attach a macrocyclic peptide ligand of interest to a cyclic CPP through a flexible linker. The latter approach is especially attractive when a potent cyclic peptide ligand is already available, and the attached CPP is less likely to perturb the biological function of the cargo molecule (and vice versa). In this work, we apply this approach to develop a potent, cell-permeable inhibitor against Kelch-like ECH-associated protein 1 (Keap1).
Nuclear factor erythroid-2 (Nrf2) is a transcription factor involved in the cellular response to stress and its cellular level is regulated by Kelch-like ECH-associated protein 1 (Keap1).28 Under normal conditions, Keap1 associates with Nrf2, causing the ubiquitination and subsequent degradation of Nrf2. Oxidative stress perturbs the Nrf2-Keap1 interaction, leading to Nrf2 accumulation and translocation into the nucleus. Activated Nrf2 upregulates a series of antioxidant response genes, including heme-oxygenase-1 (HO-1), NADPH quinone oxidoreductase I (NQO1), and superoxide dismutase (SOD).28 Therefore, Nrf2 is considered a significant regulator of inflammation and a promising target for treatment of inflammatory diseases.29 In addition, Nrf2 protects cells and tissues against various toxins and carcinogens.30 As a result, there has been great interests in developing small-molecule31–33 and peptidyl inhibitors34–39 against the Keap1-Nrf2 interaction. Most of the reported peptidyl inhibitors target the Kelch domain of Keap1 and contain a key Kelch-binding motif of Nrf2, E-T-G-E. However, peptides containing the E-T-G-E motif are negatively charged and impermeable to the cell membrane. Other investigators have previously conjugated the peptidyl Keap1 inhibitors to cell-penetrating peptides (CPPs) such as the trans-activating transcriptional activator of HIV (Tat).36,38 As mentioned above, conventional CPPs (e.g., Tat) have low cytosolic delivery efficiencies and high peptide concentrations (≥10 μM) are usually required to generate significant biological activities in cellular assays. Additionally, conventional CPPs such as Tat have poor bioavailability and biodistribution properties, hampering their clinical application. Interestingly, a recent study by Lu et al.40 described a potent cyclic peptidyl inhibitor, cyclo(GQLDPETGEFL) (KD = 18 nM), which was reported to penetrate the mammalian cell membrane and exhibit robust activity in cellular assays, despite carrying a −3 charge under the physiological condition.
RESULTS AND DISCUSSION
Design and Synthesis of CPP9-Cyclic Peptide Conjugate.
This study was motivated by several factors. First, a highly potent and specific inhibitor against the Keap1-Nrf2 interaction would provide a useful tool for investigating the biological functions of Keap1 and Nrf2 proteins. Although potent small-molecule inhibitors have been developed against the Keap1-Nrf2 interaction,31–33 small molecules often have off-target activities, making them less ideal as chemical probes. On the other hand, peptidyl inhibitors engage larger surface areas on target proteins and as such, are often highly specific for their intended targets. Second, we were interested in testing whether our cyclic CPPs could be used to deliver macrocyclic peptides into mammalian cells by simply conjugating the two with a flexible linker. Finally, we were intrigued by the previous report40 that cyclo(GQLDPETGEFL) (Scheme 1, compound 1) had the capacity to penetrate the eukaryotic cell membrane; based on our current understanding of the mechanisms of cell penetration by cyclic peptides,11 highly polar peptides such as cyclo(GQLDPETGEFL) are not expected to have significant cellular entry activity.
Scheme 1. Design and Synthesis of CPP9-Cyclic Peptide Conjugate.
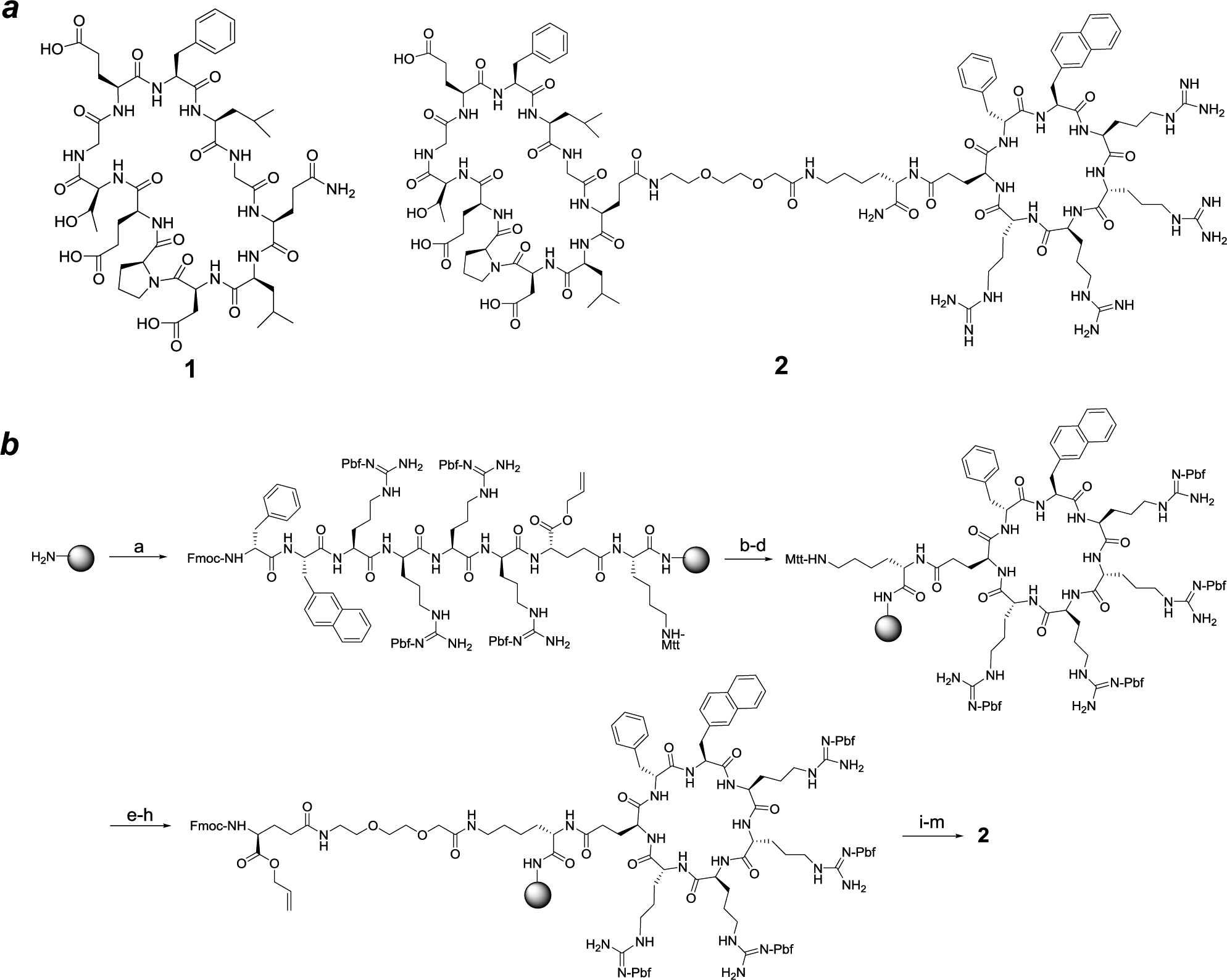
Reagents and Conditions: a) solid-phase peptide synthesis; b) Pd(PPh3)4; c) piperidine; d) PyBOP; e) 2% TFA; f) Fmoc-miniPEG/HATU; g) piperidine; h) Fmoc-Glu-OAll/HATU; i) solid-phase peptide synthesis; j) Pd(PPh3)4; k) piperidine; l) PyBOP; and m) TFA.
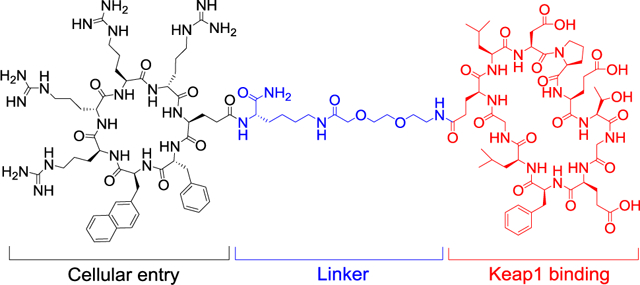
We conjugated cyclic peptide 1 to CPP9, a cyclic CPP of high cytosolic delivery efficiency,17 through a flexible polyethyleneglycol (miniPEG)-lysine linker to give peptide 2 (Scheme 1a). The Gln residue of peptide 1 was chosen as the site of attachment, as previous structural studies revealed that this Gln residue makes minimal interaction with Keap1.40,41 Peptide 2 was synthesized on solid phase, starting with a methoxytrityl (Mtt)-protected lysine residue (Scheme 1b). Following removal of the N-Fmoc group, the α-amine of the lysine residue was acylated with the side-chain carboxyl of Oα-allyl N-Fmoc-glutamate. Subsequent coupling of the CPP motif (phe-Nal-Arg-arg-Arg-arg, where phe is D-phenylalanine; Nal is 2-L-naphthylalanine, and arg is D-arginine) was carried out on the solid phase by using standard peptide chemistry. Cyclization of the CPP motif was effected by sequential treatment of the resin with Pd(PPh3)4 (deallylation of the C-terminal Glu), piperidine (deprotection of the N-terminal Fmoc group), and benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate (PyBOP). Next, the Mtt group was selectively removed from the lysine residue by treatment with 2% trifluoroacetic acid (TFA) and a Fmoc-miniPEG linker was added to the lysine side chain by using 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) as the coupling agent. Synthesis of the second, Keap1-binding macrocycle was similarly carried out. Finally, peptide 2 was cleaved off the resin and deprotected by treatment with TFA and purified to ≥95% purity by reversed-phase HPLC (Figure S1 in Supporting Information).
Biochemical Characterization of Peptides 1 and 2.
Peptides 1 and 2 were labeled with fluorescein (see Figure S1 for detailed structures) and tested for binding to the Kelch domain of Keap1 by a fluorescence polarization (FP) assay. Peptides 1 and 2 showed KD values of 82 ± 5 and 48 ± 3 nM, respectively (Figure 1a), which are somewhat higher than the previously reported KD value of 18 nM.40 To determine whether the dye molecule affected protein binding, we also performed an FP-based competition assay, in which unlabeled peptides 1 and 2 were used to compete with the fluorescein-labeled peptide 2 for binding to Keap1. Peptides 1 and 2 gave IC50 values of 19 ± 3 and 153 ± 7 nM, respectively (Figure 1b). These data indicate that labeling of peptide 1 with fluorescein or conjugation with CPP9 slightly reduces its Keap1-binding affinity. Since CPP9 and the Keap1-binding sequence carry +4 and −3 charges at the physiological pH, respectively, electrostatic interaction between the two macrocycles (either intra- or intermolecularly) presumably attenuated the potency of the Keap1 ligand. Nevertheless, competition between peptides 1 and 2 suggests that both peptides bind to the canonical Nrf2-binding site on Keap1.
Figure 1.
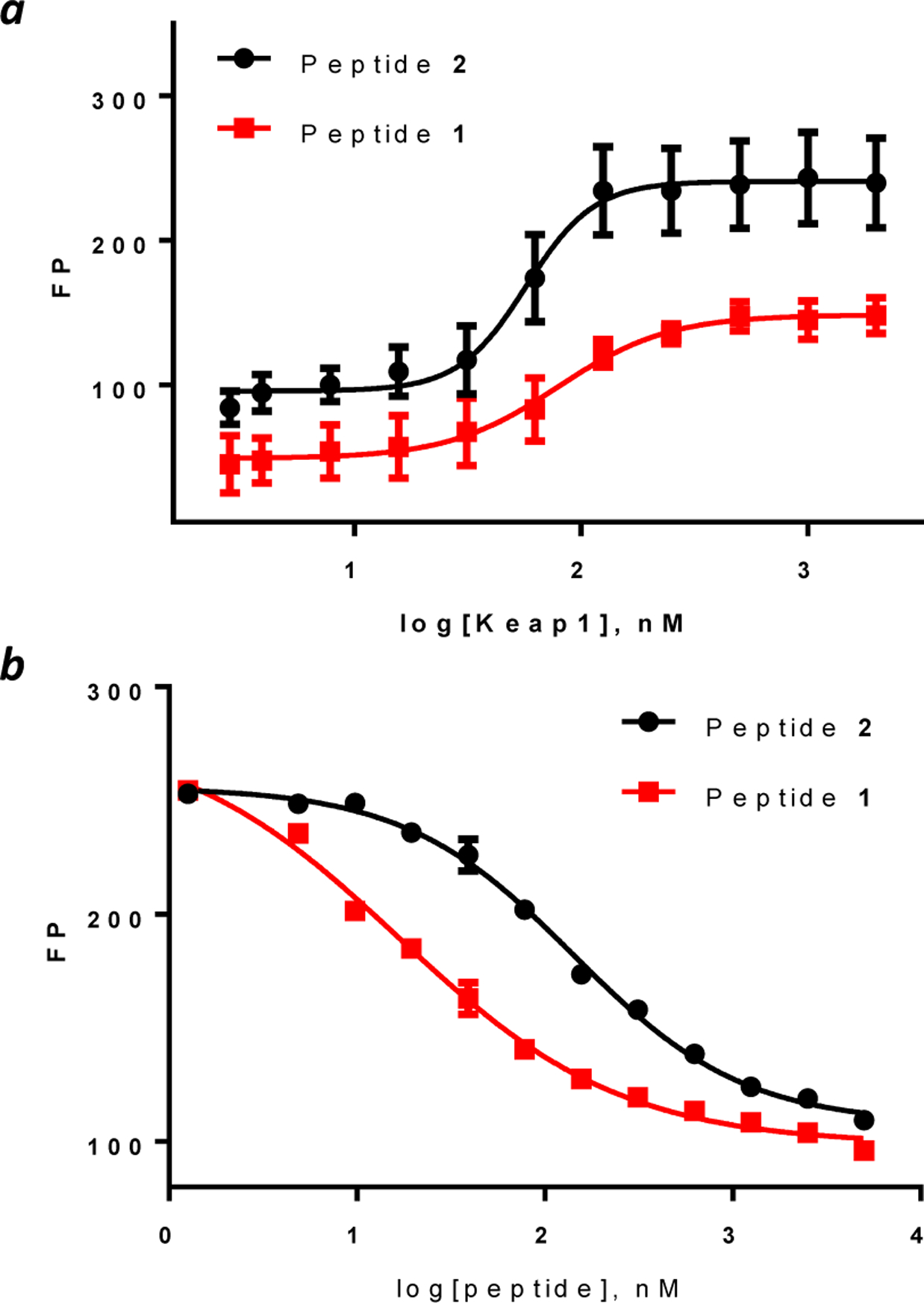
Binding of peptides 1 and 2 to Keap1 as monitored by fluorescence polarization (FP). (a) Binding of fluorescein-labeled peptides (20 nM) as a function of Keap1 concentration; (b) Competition for binding to Keap1 (40 nM) between fluorescein-labeled peptide 2 (20 nM) and increasing concentrations of unlabeled peptide 1 or 2. Data shown represent the mean ± SD of three independent experiments.
The metabolic stability of peptide 2 was assessed by incubating it in 25% human serum and examining the amounts of remaining intact peptide at various time points (0–24 h) by reversed-phase HPLC. As expected, peptide 2 is highly stable against proteolysis; ~80% of the peptide remained intact after 24 h of incubation at 37 °C (Figure S2). Under the same condition, a stapled control peptide, Ac-GGYPED*ILDK*HLQRVIL-(miniPEG)2-Dap-CPP9 (where the side chains of D* and K* were crosslinked via an amide bond and Dap is L-2,3-diaminopropionic acid), was rapidly degraded with a half-life of 109 min.
Cellular Entry Efficiency of Peptides 1 and 2.
The cellular uptake efficiency of peptides 1 and 2 was first assessed by live-cell confocal microscopy. Human cervical cancer (HeLa) cells were treated with 5 μM fluorescein-labeled peptide for 2 h, washed, and immediately imaged. Cells treated with peptide 1 (no CPP) showed weak, punctate fluorescence, consistent with a small amount of peptide 1 being entrapped inside the endosomal/lysosomal compartments (Figures 2a and S3a). Under the same condition, cells treated with peptide 2 produced much stronger fluorescence (Figures 2b and S3b). Although the fluorescence pattern of peptide 2 was also predominantly punctate, its intracellular distribution was dramatically different from that of peptide 1. First, the peptide fluorescence was present throughout the cytoplasmic space, but not in the nucleus. Second, the fluorescence signals appear to fall into two categories – the first being bright, discrete spots and the other less intense, more diffuse and fibrous structures (Figure S3). The former appears to be peptides that remained entrapped inside the endosomes and lysosomes, whereas the latter is consistent with peptide 2 that reached the cytosol and became bound to Keap1 (or another cytosolic component(s)). It was previously reported that the intracellular distribution of Keap1 is exclusively cytosolic and often associated with the cytoskeleton of the cell.42
Figure 2.
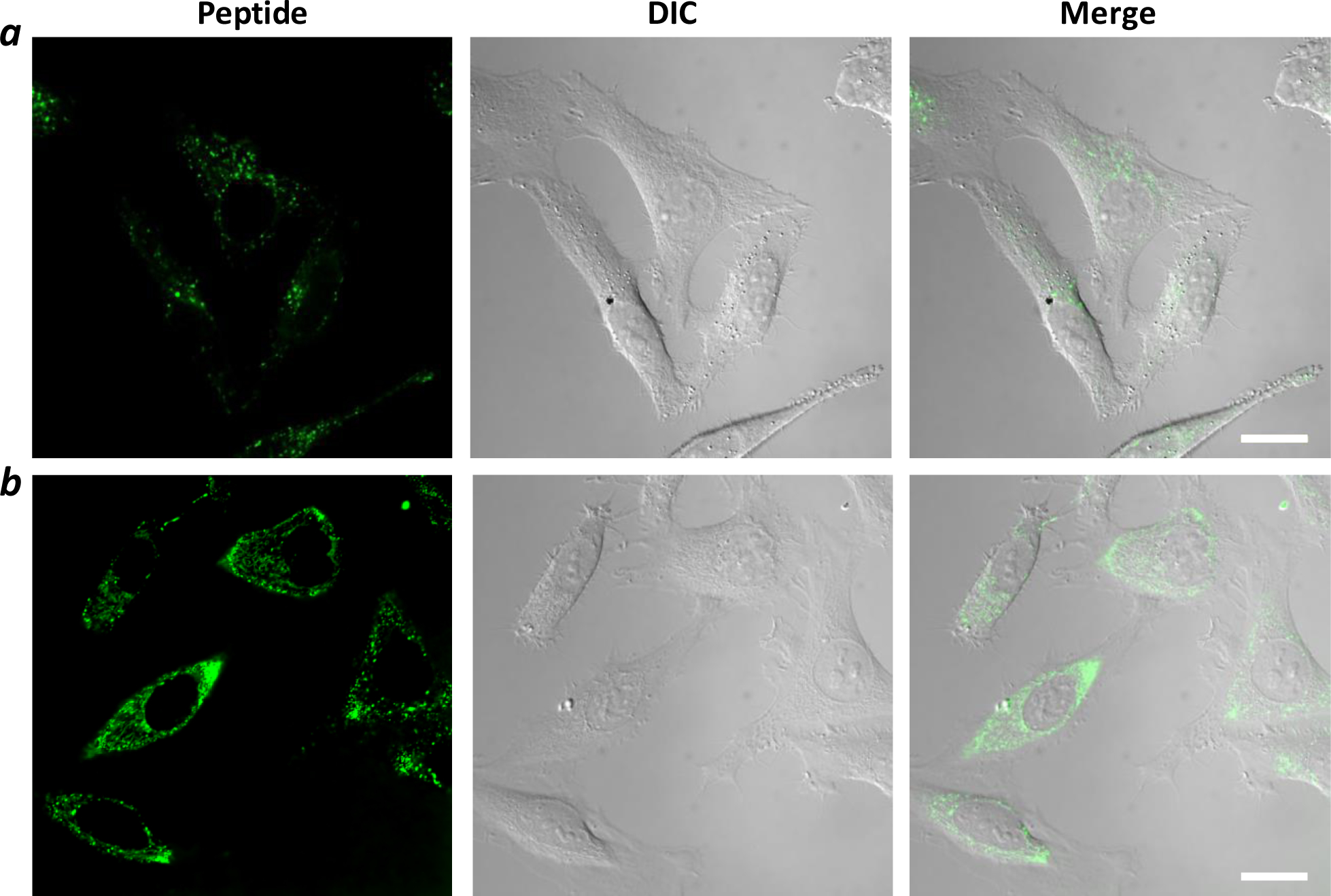
Live-cell confocal images of HeLa cells after treatment with 5 μM fluorescein-labeled peptide 1 (a) or 2 (b) for 2 h in the presence of 1% fetal bovine serum (FBS). Scale bar, 20 μm.
To quantitate the cytosolic entry of peptides 1 and 2, the peptides were next labeled with naphthofluorescein (NF), a pH sensitive dye (pKa = 7.8) which is fluorescent at neutral and basic pH, but essentially nonfluorescent inside the acidic endosome (pH 5.5–6.5) and lysosome (pH 4.5–5.5).43 HeLa cells were treated with the NF-labeled peptides (5 μM) for 2 h and analyzed by flow cytometry. Cells treated with CPP9 (positive control) and peptides 1 and 2 produced mean fluorescence intensity (MFI) values of 100%, 10%, and 980%, respectively (Figure 3). Thus, conjugation with CPP9 increased the cytosolic entry efficiency of peptide 1 by 98-fold. To test whether the increased fluorescence was the result of peptide binding to the outer surface of the cell, the flow cytometry experiment was repeated by suspending the peptide treated cells in a pH 6.5 buffer (which would quench the fluorescence of any surface-bound peptides) immediately before flow cytometry. Similar results were obtained (Figure S4), indicating that the observed cell-associated fluorescence was not due to peptide binding to the cell surface. Interestingly, a small amount of peptide 1 (10% relative to CPP9) reached the cytosol of mammalian cells, despite having a −3 charge and no recognizable cell-penetrating motif. We hypothesize that this population of peptide 1 likely traverses the plasma membrane by “hijacking” the organic anion transporting polypeptide (OATP) on the plasma embrane,11 whereas the endocytosed population largely remains entrapped inside the endosomes and lysosomes (Figures 2a and S3a). Two factors may contribute to the greater cellular entry efficiency of peptide 2 than CPP9 alone. The first is the impact of protein binding. Binding of cyclic CPPs (e.g., CPP9) to serum proteins substantially slows their cellular entry kinetics and reduces the uptake efficiency as measured by the flow cytometry assay. Conjugation to a hydrophilic cargo likely reduces the extent of serum protein binding. The cargo moiety may also interact with the plasma and/or endosomal membranes and enhance the total cellular uptake and/or endosomal escape, respectively.17
Figure 3.
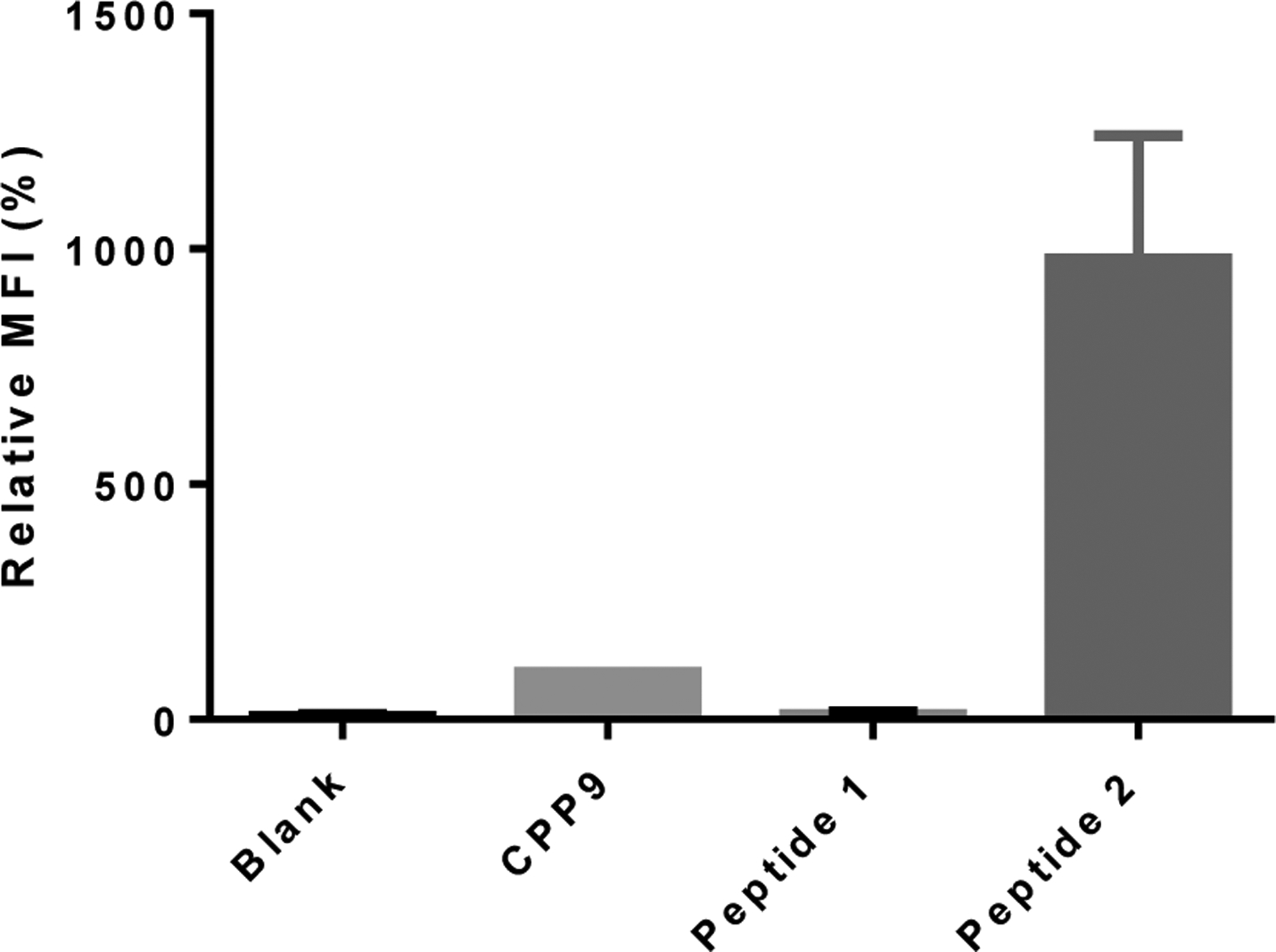
Cytosolic entry efficiencies of peptides 1 and 2. HeLa cells were treated with 5 μM NF-labeled peptide for 2 h in the presence of 10% FBS, washed, and analyzed by flow cytometry. All MFI values are relative to that of CPP9-NF (100%) and represent the mean ± SD of four independent experiments. Blank, cells without peptide treatment.
Biological Activity of Peptides 1 and 2.
The capability of peptides 1 and 2 to inhibit the intracellular Keap1-Nrf2 interaction was first evaluated on an engineered cell line, ARE reporter–HepG2 cell, which carries a firefly luciferase gene under the transcriptional control of Nrf2.44 In the absence of any inhibitor, Nrf2 is bound to Keap1 and sequestered inside the cytosol, and the cells produced a low, basal level of luciferase activity (Figure 4a). Treatment of the cells with peptide 2 dose-dependently increased the luciferase expression, with an EC50 value of ~1.1 μM and a maximum effect of ~6-fold at 5 μM concentration. Note that a significant increase in luciferase activity was already evident at 160 nM peptide 2, a concentration similar to the IC50 value for inhibition of the Keap1-Nrf2 interaction by peptide 2 in vitro (Figure 1b). Under the same condition, peptide 1 (no CPP) did not result in significant increase in luciferase expression until 5 μM, when the luciferase activity was increased by 1.6-fold (EC50 >10 μM). Our results are in qualitative agreement with the previous report,40 in that peptide 1 has the unusual capacity to gain access to the cytosol of mammalian cells and activate Nrf2. However, in our hands, the cytosolic entry efficiency of peptide 1 is substantially lower than was previously reported (i.e., 4-fold induction in luciferase activity at 5 μM peptide 1).40
Figure 4.
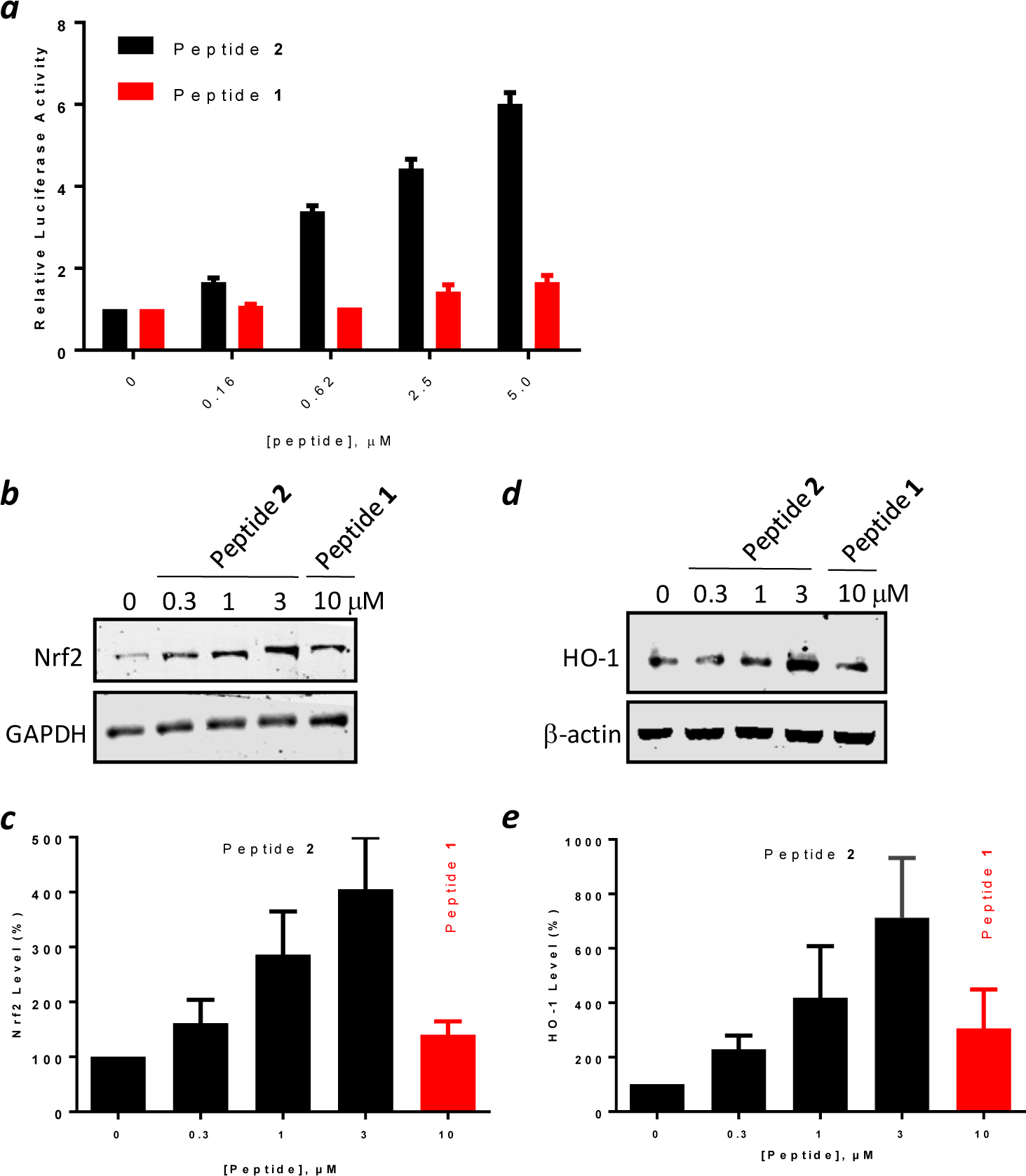
Biological activity of peptides 1 and 2. (a) Induction of luciferase expression by HepG2-ARE (Luc) cells by increasing concentrations of peptides 1 and 2. (b) Representative anti-Nrf2 western blot showing the effect of peptides 1 and 2 on the Nrf2 level in HEK293T cells. (c) Quantitation of western blots from (b). (d) Representative anti-HO-1 western blot showing the effect of peptides 1 and 2 on the HO-1 level in HEK293T cells. (e) Quantitation of western blots from (d). Data reported in (a), (c), and (e) represent the mean ± SD of at least three independent sets of experiments.
We next examined the effect of peptides 1 and 2 on the expression levels of proteins involved in the Nrf2 signaling pathway by western blotting analysis. Treatment of HEK293T cells with peptide 2 dose-dependently increased the Nrf2 protein level, by 4-fold at 3 μM peptide concentration (Figure 4b,c). In comparison, treatment with 10 μM peptide 1 only increased the Nrf2 level by 1.4-fold. Similarly, peptide 2 dose-dependently increased the expression of heme-oxygenase 1 (HO-1), a gene under the transcriptional control of Nrf2, by up to 7-fold at 3 μM peptide 2 (Figure 4d,e). Again, peptide 1 was less effective, showing 3-fold induction of HO-1 expression at 10 μM concentration.
Finally, peptides 1 and 2 were tested for potential cytotoxicity with HeLa and HEK293T cells by using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) viability assay. Incubation with up to 20 μM peptide 2 for 72 h reduced the viability of HeLa cells by ~20%, but not that of HEK293T cells (Figure S5). Peptide 1 showed essentially the same profile (data not shown). These results indicate that peptides 1 and 2 do not cause significant cytotoxicity.
CONCLUSION
This work results in a potent, cell-permeable, and metabolically stable cyclic peptidyl inhibitor against the Keap1-Nrf2 interaction, which should provide a useful chemical probe for investigating the biological functions of Keap1 and Nrf2 proteins. Peptide 2 may be further developed into therapeutic agents for treatment of inflammatory diseases and certain cancers. The success of peptide 2 suggests that other membrane-impermeable cyclic peptidyl ligands may similarly be rendered cell-permeable and biologically active by conjugation with a cyclic CPP, thus overcoming hitherto one of the major limitations of cyclic peptides as a drug modality. Our study demonstrates that cyclic peptide 1 has weak cell-penetrating activity, but does not have robust cytosolic entry efficiency as previously claimed.
EXPERIMENTAL SECTION
Materials.
Reagents for peptide synthesis and Rink amide resin (100–200 mesh, 0.27 mmol/g) were purchased from Chem-Impex (Wood Dale, IL). Rink amide resin LS (100–200 mesh, 0.27 mmol/g) was purchased from Advanced ChemTech (Louisville, KY). DMEM and RPMI cell culture media, fetal bovine serum, penicillin-streptomycin, 0.25% trypsin-EDTA, and DPBS were purchased from Invitrogen (Carlsbad, CA). Isopropyl β-D-1-thiogalactopyranoside (IPTG), protease inhibitor cocktail tablets, and ampicillin were purchased from Sigma-Aldrich (St. Louis, MO). All solvents and other chemical reagents were obtained from Sigma-Aldrich, Fisher Scientific (Pittsburgh, PA), or VWR (West Chester, PA) and were used without further purification unless noted otherwise. The ARE reporter (Luc)-HepG2 cell line and One-Step™ luciferase assay system were purchased from BPS Bioscience (San Diego, CA). The HEK293T as well as HeLa cells were obtained from ATCC (Manassas, VA). The cell proliferation kit (MTT) was purchased from Roche (Indianapolis, IN). Recombinant human Keap1 protein (aa 322–624) was expressed in Escherichia coli and purified as previously described,45 by using a prokaryotic expression vector (pET15b) containing the coding sequence for the Kelch domain of Keap1 (aa 322–624), which was kindly provided by Professor Gaya K. Amarasinghe (Washington University, St Louis, MO).
Peptide Synthesis and Labeling.
Cyclic peptides were synthesized by utilizing standard Fmoc/HATU chemistry on Rink amide resin (0.27 mmol/g). The typical coupling reaction contained 5 equivalent of Fmoc-amino acid with HATU/HOBT/DIPEA (5, 5, and 10 equiv, respectively) and was allowed to proceed with mixing for 1 h. For peptide 2, Fmoc-Lys(Mtt)-OH was first added at the C-terminus, to serve as a linker for conjugation of CPP9 and cyclic peptide 1. The linear CPP sequence was then synthesized by standard peptide chemistry. The allyl group on the α-carboxyl group of the C-terminal L-Glu was removed by treatment with tetrakis(triphenylphosphine)palladium/phenylsilane (0.3 and 10 equiv, respectively) in DCM for 15 min (3 times). The resin was washed by sodium dimethyldithiocarbamate dihydrate (SDDNa, 0.5 M in DMF) twice and the N-terminal Fmoc group of D-Phe was removed by the addition of 20% piperidine in DMF. The resin was washed with DMF, DCM, and DMF (3 times each) and incubated with 1 M HOBt for 15 min. The CPP sequence was cyclized on resin by treatment with PyBOP/HOBt/DIPEA (5, 5, and 10 equiv, respectively) for 2 h (twice). Next, the Mtt group on the C-terminal Lys was removed by treatment with 2% TFA and 1% triisopropylsilane (TIPS) in DCM (6 × 5 min). Fmoc-miniPEG was coupled to Lys side chain followed by synthesis of the linear sequence of peptide 1. Finally, the allyl protecting group on the L-Glu and the Fmoc group of the N-terminal Gly residue were removed as described above. The linear sequence of peptide 1 was cyclized on resin by treatment with PyBOP/HOBt/DIPEA as described above. The peptide was cleaved from the resin and deprotected by treatment with 92.5:2.5:2.5:2.5 (vol/vol) TFA/H2O/1,4-dimethoxybenzene/TIPS for 2 h. The crude peptide was triturated three times with cold ethyl ether and purified by reversed-phase HPLC equipped with a Waters C18 column, which was eluted with a linear gradient of acetonitrile in ddH2O (containing 0.05% TFA).
To fluorescently label a peptide, a miniPEG-Lys (Boc protected) linker was added to the C-terminus of the peptide at the inception of peptide synthesis. For solution-phase labeling reaction, ~1 mg of lyophilized pure peptide was incubated with 5 equiv. of an activated fluorescent labeling reagent (e.g., FITC, 5(6)-carboxyfluorescein succinimidyl ester, or 5(6)-carboxynaphthofluorescein succinimidyl ester) and 5 equiv. of DIPEA in 150 μL of 1:1 (v/v) DMF/150 mM sodium bicarbonate (pH 8.5) for 2 h. The reaction was quenched by TFA and the labeled peptide was purified again by reversed-phase HPLC. The purity of each peptide was assessed by analytical reversed-phase HPLC using a Waters C18 analytical column (Figure S1). All peptides used had ≥95% purity. Peptide authenticity was confirmed by high-resolution MALDI FT-ICR mass spectrometry at Campus Chemical Instrumentation Center of The Ohio State University.
Peptide 1: HRMS (MALDI FT-ICR) m/z: [M + H]+ Calcd for C53H79N12O19 1187.5579; Found 1187.5567.
Peptide 2: HRMS (MALDI FT-ICR) m/z: [M + H]+ Calcd for C116H175N34O31 2540.3157; Found 2540.3048.
FAM-Peptide 1: HRMS (MALDI FT-ICR) m/z: [M + H]+ Calcd for C80H101N14O26 1673.6967; Found 1673.6957.
FITC-Peptide 2: HRMS (MALDI FT-ICR) m/z: [M + H]+ Calcd for C149H209N38O40S 3202.5035; Found 3202.5008.
NF-Peptide 1: HRMS (MALDI FT-ICR) m/z: [M + H]+ Calcd for C88H105N14O26 1773.7319; Found 1773.7289.
NF-Peptide 2: LRMS (MALDI TOF) m/z: [M + H]+ Calcd for C157H212N37O40 3271.5641; Found 3271.976.
NF-CPP9: HRMS (MALDI FT-ICR) m/z: [M + H]+ Calcd for C92H114N23O18 1828.8712; Found 1828.8686.
Protein-Ligand Binding Assays.
The binding affinity of peptides 1 and 2 was determined by FP. Fluorescein-labeled peptide (20 nM) was incubated with serial dilutions of Keap1 Kelch domain protein (aa 322–624) in 20 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM dithiothreitol, and 0.01% Triton-X for 1 h. The solutions were transferred to a black 384-well plate (Grenier) and FP values were measured on a Tecan Infinite M1000 Pro plate reader with excitation and emission wavelengths at 470 and 535 nm, respectively. Binding affinity was also assessed with an FP-based competition assay. Fluorescein-labeled peptide 2 (20 nM) was incubated for 1 h with 40 nM Keap1 protein in 20 mM HEPES, pH 7.5, 150 mM NaCl, 5 mM dithiothreitol, and 0.01% Triton-X. Serial dilutions of peptide 1 or 2 were prepared in the same buffer. Aliquots of the equilibrated peptide probe-protein mixture were mixed with each peptide dilution and incubated for 1 h. The samples were transferred to a 384-black microplate (Greiner) and FP values were measured on a Tecan Infinite M1000 Pro plate reader. The data was analyzed by GraphPad Prism with log [inhibitor] vs. response (four parameters).
Cell Culture.
HEK293T (human embryonic kidney) and HeLa (human cervical cancer) cells were purchased from the ATTC. The cells were cultured in Dulbecco’s modified eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin sulfate (Abs). Hep-G2 (human liver carcinoma) -antioxidant response element reporter cells were Purchased from BPS Bioscience, Inc. (San Diego, CA) and cultured in MEM medium supplemented with 10% FBS, 1% Abs, 1% non-essential amino acids, and 1 mM Na-pyruvate.
Luciferase Reporter Assay.
ARE reporter–HepG2 cells (5000 cells per well) were seeded in 100 μL of assay medium (MEM, 10% FBS, and 1% Abs) in a 96-well opaque microtiter plate and cultured overnight. Cells were treated with peptide inhibitor (which was dissolved in DMSO and serially diluted in 10 μL of the assay medium) for 18 h at 37 °C in the presence of 5% CO2, with constant DMSO concentration (0.5% v/v) in all samples. Finally, 100 μL of One-Step luciferase assay reagent was added to each well and after 15 min of shaking, the luminescence was determined using a Tecan Infinite M1000 Pro microplate reader. P values (< 0.001) was estimated using Student’s t test.
Immunoblotting.
HEK293T cells were cultured in a 6-well plate to 80–90% confluency in standard DMEM supplemented with 10% FBS and 1% Abs at 37 °C in the presence of 5% CO2. Cells were starved in serum free medium for 24 h and then treated with indicated concentrations of peptide inhibitor in 1 mL of the media. The cells were washed with cold PBS, treated with 0.25% trypsin-EDTA solution, centrifuged (5000 rpm, 5 min), and washed again with PBS. The cell pellets were lysed in 100 μL of Pierce RIPA Buffer (Thermo) containing protease and phosphatase inhibitors for 30 min on ice. Proteins were extracted by centrifugation at 15,000 rpm for 20 min. The total protein concentration in each sample was measured using the BCA Protein Assay Kit (Thermo). Equal amounts of protein were loaded in each lane of a 10% SDS-PAGE gel and separated by electrophoresis (120 V, 2.5 h). The proteins were electrophoretically transferred to a nitrocellulose membrane at 4 °C (90 V, 2.5 h). The membrane was blocked with 5% nonfat milk proteins (Bio-Rad) in TBST buffer (20 mM Tris, pH 7.5, 150 mM NaCl, 0.1% (v/v) Tween-20) at room temperature for 1 h. The membrane was incubated with the primary antibodies at 4 °C overnight, including anti-Nrf2 monoclonal antibody (1:1000 dilution, abcam, ab62352), anti-β-Actin (1:1000 dilution, Cell Signaling Technologies, 3700), anti-GAPDH (1:5000 dilution, Cell Signaling Technologies, 5174), and anti-HO-1 (1:1000 dilution, Enzo, ADI-SPA-895). The membrane was washed with TBST three times and incubated with fluorescently labeled secondary antibodies (Licor, 1:15,000 dilution) for 2 h at room temperature. The membrane was washed thrice with TBST and fluorescent signals were recorded using a LICOR Odyssey CLx instrument. Quantification of Nrf2 and HO-1 levels was performed by Image Studio. The ratio of Nrf2 or HO-1 signal to the loading control without peptide treatment was defined as 100%, and two-sided t tests were used to calculate P values.
Flow cytometry.
HeLa cells were seeded in a 24-well plate (7.5 × 104 cells/well) and cultured overnight. On the day of experiment, cells in DMEM media supplemented with 10% FBS and 1% Abs were incubated with 5 μM NF-labeled peptide 1, peptide 2, or CPP-9 for 2 h. The cells were washed with cold DPBS and harvested by trypsinization. The detached cells were washed twice with DPBS, resuspended in DPBS, and analyzed by flow cytometry (BD LSR II), with excitation at 633 nm and emission detected in the APC channel.
Confocal Microscopy.
HeLa cells (5 × 104 cells/mL) were seeded in a 35-mm glass-bottomed microwell dish (Grenier Bio-One) and cultured overnight. The next day, cells were treated with 5 μM fluorescein-labeled peptide 1 or 2 for 2 h in DMEM media containing 1% FBS and 1% Abs. After removal of the media, the cells were gently washed with DPBS twice and imaged on a Nikon A1R live-cell confocal equipped with a 100X oil objective. Data was analyzed using NIS-Elements AR.
MTT Cell Viability Assay.
HEK293T and HeLa cells were seeded in a 96-well plate at a density of 5000 cell/well (100 μL in each well) in DMEM supplemented with 10% FBS and 1% Abs and cultured overnight. The next day, peptide 1 or 2 was added to the cells and incubated for 72 h at 37 ᵒC in a 5% CO2 incubator. Ten μL of MTT solution was added to each well and incubated for 4 h. Finally, 100 μL of the SDS solubilizing buffer was added to each well and incubated overnight. The absorbance of the solubilized formazan was measured at 570 nm on a Tecan Infinite M1000 Pro plate reader.
Supplementary Material
ACKNOWLEDGMENTS
We thank Professor Gaya K. Amarasinghe (Washington University, St Louis, MO) for kindly providing the bacterial expression vector for the Keap1 Kelch domain. This work was supported by the National Institutes of Health (GM122459 and CA234124). J.S. was supported by a visiting professorship from China Scholarship Council.
Footnotes
Supporting Information
Additional data on compound characterization including HPLC chromatograms and HRMS spectra. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no conflict of interest.
REFERENCES
- 1.Cardote TA; Ciulli A, Cyclic and Macrocyclic Peptides as Chemical Tools to Recognise Protein Surfaces and Probe Protein-Protein Interactions. ChemMedChem 2016, 11 (8), 787–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Passioura T; Katoh T; Goto Y; Suga H, Selection-based Discovery of Druglike Macrocyclic Peptides. Annu Rev Biochem 2014, 83, 727–52. [DOI] [PubMed] [Google Scholar]
- 3.Tavassoli A; Benkovic SJ, Genetically Selected Cyclic-Peptide Inhibitors of AICAR Transformylase Homodimerization. Angew Chem Int Ed Engl 2005, 44 (18), 2760–3. [DOI] [PubMed] [Google Scholar]
- 4.Joo SH; Xiao Q; Ling Y; Gopishetty B; Pei D, High-throughput Sequence Determination of Cyclic Peptide Library Members by Partial Edman Degradation/Mass Spectrometry. J Am Chem Soc 2006, 128 (39), 13000–9. [DOI] [PubMed] [Google Scholar]
- 5.Lian W; Upadhyaya P; Rhodes CA; Liu Y; Pei D, Screening Bicyclic Peptide Libraries for Protein-Protein Interaction Inhibitors: Discovery of a Tumor Necrosis Factor-alpha Antagonist. J Am Chem Soc 2013, 135 (32), 11990–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Millward SW; Fiacco S; Austin RJ; Roberts RW, Design of Cyclic Peptides that Bind Protein Surfaces with Antibody-like Affinity. ACS Chem Biol 2007, 2 (9), 625–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heinis C; Rutherford T; Freund S; Winter G, Phage-Encoded Combinatorial Chemical Libraries based on Bicyclic Peptides. Nat Chem Biol 2009, 5 (7), 502–7. [DOI] [PubMed] [Google Scholar]
- 8.Sako Y; Morimoto J; Murakami H; Suga H, Ribosomal Synthesis of Bicyclic Peptides via Two Orthogonal Inter-Side-Chain Reactions. J Am Chem Soc 2008, 130 (23), 7232–7234. [DOI] [PubMed] [Google Scholar]
- 9.Kleiner RE; Dumelin CE; Tiu GC; Sakurai K; Liu DR, In vitro Selection of a DNA-Templated Small-molecule Library Reveals a Class of Macrocyclic Kinase Inhibitors. J Am Chem Soc 2010, 132 (33), 11779–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schlippe YV; Hartman MC; Josephson K; Szostak JW, In vitro Selection of Highly Modified Cyclic Peptides that Act as Tight Binding Inhibitors. J Am Chem Soc 2012, 134 (25), 10469–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dougherty PG; Sahni A; Pei D, Understanding Cell Penetration of Cyclic Peptides. Chem Rev 2019, 119, 10241–10287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zorzi A; Deyle K; Heinis C, Cyclic Peptide Therapeutics: Past, Present and Future. Curr Opin Chem Biol 2017, 38, 24–29. [DOI] [PubMed] [Google Scholar]
- 13.(a) Rezai T; Bock JE; Zhou MV; Kalyanaraman C; Lokey RS; Jacobson MP, Conformational Flexibility, Internal Hydrogen Bonding, and Passive Membrane Permeability: Successful In Silico Prediction of the Relative Permeabilities of Cyclic Peptides. J. Am. Chem. Soc 2006, 128, 14073–14080. [DOI] [PubMed] [Google Scholar]; (b) Bockus AT; Schwochert JA; Pye CR; Townsend CE; Sok V; Bednarek MA; Lokey RS, Going out on a Limb: Delineating the Effects of β-Branching, N-Methylation, and Side Chain Size on the Passive Permeability, Solubility, and Flexibility of Sanguinamide A Analogues. J. Med. Chem 2015, 58, 7409–7418. [DOI] [PubMed] [Google Scholar]; (c) Räder AFB; Weinmüller M; Reichart F; Schumacher-Klinger A; Merzbach S; Gilon C; Hoffman A; Kessler H Orally Active Peptides: Is There a Magic Bullet?. Angew. Chem., Int. Ed 2018, 57, 14414–14438. [DOI] [PubMed] [Google Scholar]
- 14.Nielsen DS; Shepherd NE; Xu W; Lucke AJ; Stoermer MJ; Fairlie DP, Orally Absorbed Cyclic Peptides. Chem. Rev 2017, 117, 8094–8128. [DOI] [PubMed] [Google Scholar]
- 15.(a) Sasaki Y; Minamizawa M; Ambo A; Sugawara S; Ogawa Y; Nitta K, Cell-penetrating Peptide-conjugated XIAP-inhibitory Cyclic Hexapeptides Enter into Jurkat Cells and Inhibit Cell Proliferation. FEBS J 2008, 275, 6011–6021. [DOI] [PubMed] [Google Scholar]; (b) Desimmie BA; Humbert M; Lescrinier E; Hendrix J; Vets S; Gijsbers R; Ruprecht RM; Dietrich U; Debyser Z; Christ F, Phage Display-Directed Discovery of LEDGF/P75 Binding Cyclic Peptide Inhibitors of HIV Replication. Mol Ther 2012, 20, 2064–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Qian Z; Liu T; Liu YY; Briesewitz R; Barrios AM; Jhiang SM; Pei D, Efficient Delivery of Cyclic Peptides into Mammalian Cells with Short Sequence Motifs. ACS Chem Biol 2013, 8 (2), 423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Qian Z; Martyna A; Hard RL; Wang J; Appiah-Kubi G; Coss C; Phelps MA; Rossman JS; Pei D, Discovery and Mechanism of Highly Efficient Cyclic Cell-Penetrating Peptides. Biochemistry 2016, 55 (18), 2601–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lattig-Tunnemann G; Prinz M; Hoffmann D; Behlke J; Palm-Apergi C; Morano I; Herce HD; Cardoso MC, Backbone Rigidity and Static Presentation of Guanidinium Groups Increases Cellular Uptake of Arginine-Rich Cell-Penetrating Peptides. Nat Commun 2011, 2, 453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mandal D; Nasrolahi Shirazi A; Parang K, Cell-Penetrating Homochiral Cyclic Peptides as Nuclear-Targeting Molecular Transporters. Angew Chem Int Ed Engl 2011, 50 (41), 9633–7. [DOI] [PubMed] [Google Scholar]
- 20.Nischan N; Herce HD; Natale F; Bohlke N; Budisa N; Cardoso MC; Hackenberger CP, Covalent Attachment of Cyclic TAT Peptides to GFP Results in Protein Delivery into Live Cells with Immediate Bioavailability. Angew Chem Int Ed Engl 2015, 54 (6), 1950–3. [DOI] [PubMed] [Google Scholar]
- 21.Traboulsi H; Larkin H; Bonin MA; Volkov L; Lavoie CL; Marsault E, Macrocyclic Cell Penetrating Peptides: A Study of Structure-Penetration Properties. Bioconjug Chem 2015, 26 (3), 405–11. [DOI] [PubMed] [Google Scholar]
- 22.Horn M; Reichart F; Natividad-Tietz S; Diaz D; Neundorf I, Tuning the Properties of a Novel Short Cell-Penetrating Peptide by Intramolecular Cyclization with a Triazole Bridge. Chem Commun 2016, 52 (11), 2261–2264. [DOI] [PubMed] [Google Scholar]
- 23.Amoura M; Illien F; Joliot A; Guitot K; Offer J; Sagan S; Burlina F, Head to Tail Cyclisation of Cell-Penetrating Peptides: Impact on GAG-Dependent Internalisation and Direct Translocation. Chem. Commun 2019, 55, 4566–4569. [DOI] [PubMed] [Google Scholar]
- 24.Lian W; Jiang B; Qian Z; Pei D, Cell-permeable Bicyclic peptide Inhibitors against Intracellular Proteins. J Am Chem Soc 2014, 136 (28), 9830–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang B; Pei D, A Selective, Cell-Permeable Nonphosphorylated Bicyclic Peptidyl Inhibitor against Peptidyl–Prolyl Isomerase Pin1. J Med Chem 2015, 58 (15), 6306–6312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trinh TB; Upadhyaya P; Qian Z; Pei D, Discovery of a Direct Ras Inhibitor by Screening a Combinatorial Library of Cell-Permeable Bicyclic Peptides. ACS Comb Sci 2016, 18 (1), 75–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rhodes CA; Dougherty PG; Cooper JK; Qian Z; Lindert S; Wang QE; Pei D, Cell-Permeable Bicyclic Peptidyl Inhibitors against NEMO-IkappaB Kinase Interaction Directly from a Combinatorial Library. J Am Chem Soc 2018, 140 (38), 12102–12110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deshmukh P; Unni S; Krishnappa G; Padmanabhan B, The Keap1-Nrf2 Pathway: Promising Therapeutic Target to Counteract ROS-Mediated Damage in Cancers and Neurodegenerative Diseases. Biophys Rev 2017, 9 (1), 41–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahmed SMU; Luo L; Namani A; Wang XJ; Tang X, Nrf2 Signaling Pathway: Pivotal Roles in Inflammation. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease 2017, 1863 (2), 585–597. [DOI] [PubMed] [Google Scholar]
- 30.Jaramillo MC; Zhang DD, The Emerging Role of the Nrf2-Keap1 Signaling Pathway in Cancer. Genes Dev 2013, 27 (20), 2179–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tran KT; Pallesen JS; Solbak SMØ; Narayanan D; Baig A; Zang J; Aguayo-Orozco A; Carmona R; Garcia A; Bach A, A Comparative Assessment Study of Known Small‐Molecule Keap1‐Nrf2 Protein‐Protein Interaction Inhibitors: Chemical Synthesis, Binding Properties, and Cellular Activity. J Med Chem 2019, 62, 8028–8052. [DOI] [PubMed] [Google Scholar]
- 32.Davies TG; Wixted WE; Coyle JE; Griffiths-Jones C; Hearn K; McMenamin R; Norton D; Rich SJ; Richardson C; Saxty G; Willems HM; Woolford AJ; Cottom JE; Kou JP; Yonchuk JG; Feldser HG; Sanchez Y; Foley JP; Bolognese BJ; Logan G; Podolin PL; Yan H; Callahan JF; Heightman TD; Kerns JK, Monoacidic Inhibitors of the Kelch-like ECH-Associated Protein 1: Nuclear Factor Erythroid 2-Related Factor 2 (KEAP1:NRF2) Protein-Protein Interaction with High Cell Potency Identified by Fragment-Based Discovery. J Med Chem 2016, 59 (8), 3991–4006. [DOI] [PubMed] [Google Scholar]
- 33.Winkel AF; Engel CK; Margerie D; Kannt A; Szillat H; Glombik H; Kallus C; Ruf S; Gussregen S; Riedel J; Herling AW; von Knethen A; Weigert A; Brune B; Schmoll D, Characterization of RA839, a Noncovalent Small Molecule Binder to Keap1 and Selective Activator of Nrf2 Signaling. J Biol Chem 2015, 290 (47), 28446–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Inoyama D; Chen Y; Huang X; Beamer LJ; Kong AN; Hu L, Optimization of Fluorescently Labeled Nrf2 Peptide Probes and the Development of a Fluorescence Polarization Assay for the Discovery of Inhibitors of Keap1-Nrf2 Interaction. J Biomol Screen. 2012, 17(4), 435–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hancock R; Bertrand HC; Tsujita T; Naz S; El-Bakry A; Laoruchupong J; Hayes JD; Wells G, Peptide inhibitors of the Keap1-Nrf2 protein-protein interaction. Free Radic Biol Med. 2012, 52(2) 444–51. [DOI] [PubMed] [Google Scholar]
- 36.Steel R; Cowan J; Payerne E; O’Connell MA; Searcey M, Anti-inflammatory Effect of a Cell-Penetrating Peptide Targeting the Nrf2/Keap1 Interaction. ACS Med Chem Lett 2012, 3 (5), 407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Steel RJ; O’Connell MA; Searcey M, Perfluoroarene-Based Peptide Macrocycles that Inhibit the Nrf2/Keap1 Interaction. Bioorg Med Chem Lett 2018, 28 (16), 2728–2731. [DOI] [PubMed] [Google Scholar]
- 38.Tu J; Zhang X; Zhu Y; Dai Y; Li N; Yang F; Zhang Q; Brann DW; Wang R, Cell-Permeable Peptide Targeting the Nrf2-Keap1 Interaction: A Potential Novel Therapy for Global Cerebral Ischemia. J Neurosci. 2015, 35(44), 14727–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Georgakopoulos ND; Talapatra SK; Gatliff J; Kozielski F; Wells G, Modified Peptide Inhibitors of the Keap1-Nrf2 Protein-Protein Interaction Incorporating Unnatural Amino Acids. ChemBioChem. 2018, 19(17), 1810–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lu MC; Jiao Q; Liu T; Tan SJ; Zhou HS; You QD; Jiang ZY, Discovery of a Head-to-Tail Cyclic Peptide as the Keap1-Nrf2 Protein-Protein Interaction Inhibitor with High Cell Potency. Eur J Med Chem 2018, 143, 1578–1589. [DOI] [PubMed] [Google Scholar]
- 41.Lo SC; Li X; Henzl MT; Beamer LJ; Hannink M, Structure of the Keap1:Nrf2 Interface Provides Mechanistic Insight into Nrf2 Signaling. EMBO J 2006, 25 (15), 3605–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Velichkova M; Hasson T, Keap1 Regulates the Oxidation-Sensitive Shuttling of Nrf2 into and out of the Nucleus via a Crm1-Dependent Nuclear Export Mechanism. Mol Cell Biol 2005, 25 (11), 4501–4513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Qian Z; Dougherty PG; Pei D, Monitoring the Cytosolic Entry of Cell-Penetrating Peptides Using a pH-Sensitive Fluorophore. Chem Commun 2015, 51 (11), 2162–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee JM; Johnson JA, An Important Role of Nrf2-ARE Pathway in the Cellular Defense Mechanism. J Biochem Mol Biol. 2004, 37(2), 139–143. [DOI] [PubMed] [Google Scholar]
- 45.Johnson B; Li J; Adhikari J; Edwards MR; Zhang H; Schwarz T; Leung DW; Basler CF; Gross ML; Amarasinghe GK, Dimerization Controls Marburg Virus VP24-dependent Modulation of Host Antioxidative Stress Responses. J Mol Biol 2016, 428 (17), 3483–3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.