

Abstract
High-throughput chemical screens typically use coarse assays such as cell survival, limiting what can be learned about mechanisms of action, off-target effects, and heterogeneous responses. Here, we introduce “sci-Plex,” which uses “nuclear hashing” to quantify global transcriptional responses to thousands of independent perturbations at single-cell resolution. As a proof of concept, we applied sci-Plex to screen three cancer cell lines exposed to 188 compounds. In total, we profiled ~650,000 single-cell transcriptomes across ~5000 independent samples in one experiment. Our results reveal substantial intercellular heterogeneity in response to specific compounds, commonalities in response to families of compounds, and insight into differential properties within families. In particular, our results with histone deacetylase inhibitors support the view that chromatin acts as an important reservoir of acetate in cancer cells.
High-throughput screens (HTSs) are a cornerstone of the pharmaceutical drug-discovery pipeline (1, 2). However, conventional HTSs have at least two major limitations. First, the readout of most are restricted to gross cellular phenotypes, e.g., proliferation (3, 4), morphology (5, 6), or a highly specific molecular readout (7, 8). Subtle changes in cell state or gene expression that might otherwise provide mechanistic insights or reveal off-target effects are routinely missed.
Second, even when HTSs are performed in conjunction with more comprehensive molecular phenotyping such as transcriptional profiling (9–12), a limitation of bulk assays is that even cells ostensibly of the same “type” can exhibit heterogeneous responses (13, 14). Such cellular heterogeneity can be highly relevant in vivo. For example, it remains largely unknown whether the rare subpopulations of cells that survive chemotherapeutics are doing so on the basis of their genetic background, epigenetic state, or some other aspect (15, 16).
In principle, single-cell transcriptome sequencing (scRNA-seq) represents a form of high-content molecular phenotyping that could enable HTSs to overcome both limitations. However, the per-sample and per-cell costs of most scRNA-seq technologies remain high, precluding even modestly sized screens. Recently, several groups have developed “cellular hashing” methods, in which cells from different samples are molecularly labeled and mixed before scRNA-seq. However, current hashing approaches require relatively expensive reagents [e.g., antibodies (17) or chemically modified DNA oligos (18, 19)], use cell-type-dependent protocols (20), and/or use scRNA-seq platforms with a high per-cell cost.
To enable cost-effective HTSs with scRNA-seq–based phenotyping, we describe a new sample labeling (hashing) strategy that relies on labeling nuclei with unmodified single-stranded DNA oligos. Recent improvements in single-cell combinatorial indexing (sci-RNA-seq3) have lowered the cost of scRNA-seq library preparation to <$0.01 per cell, with millions of cells profiled per experiment (21). Here, we combine nuclear hashing and sci-RNA-seq into a single workflow for multiplex transcriptomics in a process called “sci-Plex.” As a proof of concept, we use sci-Plex to perform HTS on three cancer cell lines, profiling thousands of independent perturbations in a single experiment. We further explore how chemical transcriptomics at single-cell resolution can shed light on mechanisms of action. Most notably, we find that gene-regulatory changes consequent to treatment with histone deacetylase (HDAC) inhibitors are consistent with the model that they interfere with proliferation by restricting a cell’s ability to draw acetate from chromatin (22, 23).
Results
Nuclear hashing enables multisample sci-RNA-seq
Single-cell combinatorial indexing (sci-) methods use split-pool barcoding to specifically label the molecular contents of large numbers of single cells or nuclei (24). Samples can be barcoded by these same indices, e.g., by placing each sample in its own well during reverse transcription in sci-RNA-seq (21, 25), but such enzymatic labeling at the scale of thousands of samples is operationally infeasible and cost prohibitive. To enable single-cell molecular profiling of a large number of independent samples within a single sci-experiment, we set out to develop a low-cost labeling procedure.
We noticed that single-stranded DNA (ssDNA) specifically stained the nuclei of permeabilized cells but not intact cells (Fig. 1A and fig. S1A). We therefore postulated that a polyadenylated ssDNA oligonucleotide could be used to label populations of nuclei in a manner compatible with sci-RNA-seq (Fig. 1B and fig. S1B). To test this concept, we performed a “barnyard” experiment. We separately seeded human (HEK293T) and mouse (NIH3T3) cells to 48 wells of a 96-well culture plate. We then performed nuclear lysis in the presence of 96 well-specific polyadenylated ssDNA oligos (“hash oligos”) and fixed the resulting nuclear suspensions with paraformaldehyde. Having labeled or “hashed” the nuclei with a molecular barcode, we pooled nuclei and performed a two-level sci-RNA-seq experiment. Because the hash oligos were polyadenylated, they had the potential to be combinatorially indexed identically to endogenous mRNAs. As intended, we recovered reads corresponding to both endogenous mRNAs [median 4740 unique molecular identifiers (UMIs) per cell] and hash oligos (median 270 UMIs per cell).
Fig. 1. sci-Plex uses polyadenylated single-stranded oligonucleotides to label nuclei, enabling cell hashing and doublet detection.
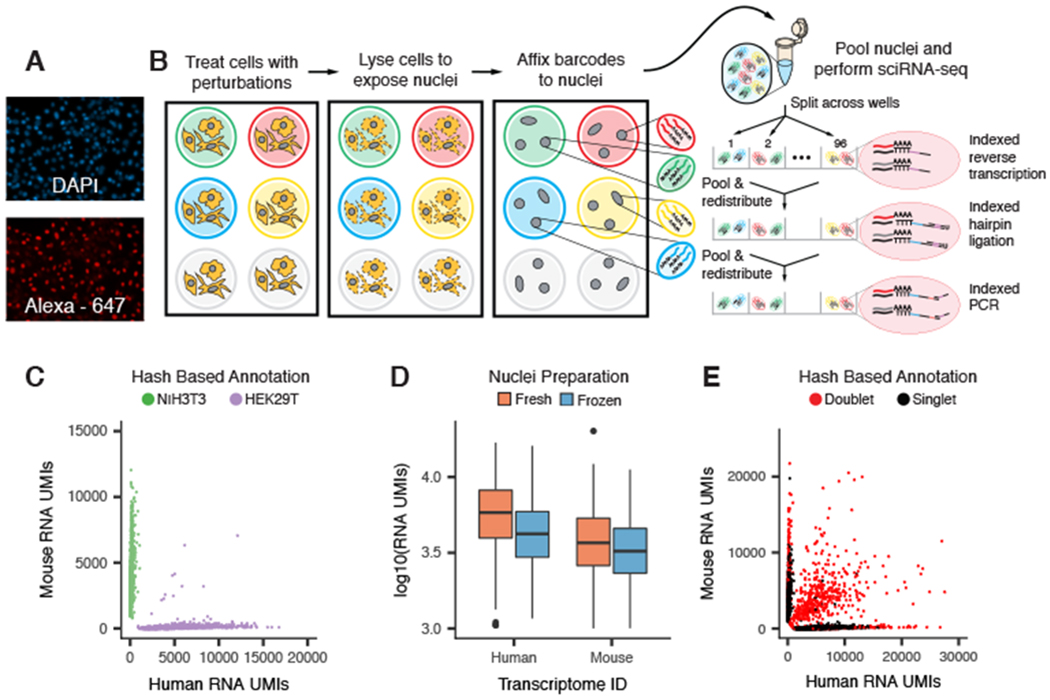
(A) Fluorescent images of permeabilized nuclei after incubation with DAPI (top) and an Alexa Fluor-647–conjugated single-stranded oligonucleotide (bottom). (B) Overview of sci-Plex. Cells corresponding to different perturbations are lysed in-well, their nuclei labeled with well-specific “hash” oligos, followed by fixation, pooling, and sci-RNA-seq. (C) Scatter plot depicting the number of UMIs from single-cell transcriptomes derived from a mixture of hashed human HEK293T cells and murine NIH3T3 cells. Points are colored on the basis of hash oligo assignment. (D) Boxplot depicting the number of mRNA UMIs recovered per cell for fresh versus frozen human and mouse cell lines. (E) Scatter plot of overloading experiment; axes are as in (C). Identified hash oligo collisions (red) identify cellular collisions with high sensitivity.
We devised a statistical framework to identity the hash oligos associated with each cell at a frequency exceeding background (table S1). We observed 99.1% concordance between species assignments on the basis of hash oligos versus endogenous cellular transcriptomes (Fig. 1C and fig. S1, C to F). Additionally, the association of hash oligos and nuclei was stable to a freeze–thaw cycle, highlighting the opportunity to label and store samples (Fig. 1D and fig. S1, G and H). These results demonstrate that hash oligos stably label nuclei in a manner that is compatible with sci-RNA-seq.
In sci-experiments, “collisions” are instances in which two or more cells are labeled with the same combination of barcodes by chance (24). To evaluate hashing as a means of detecting doublets resulting from collisions, we varied the number of nuclei loaded per polymerase chain reaction well, resulting in a range of predicted collision rates (7 to 23%) that was well matched by observation (fig. S1I). Hash oligos facilitated the identification of the vast majority of interspecies doublets (95.5%) and otherwise undetectable within-species doublets (Fig. 1E and fig. S1, J and K).
sci-Plex enables multiplex chemical transcriptomics at single-cell resolution
We next evaluated whether nuclear hashing could enable chemical screens by labeling cells that had undergone a specific perturbation, followed by single-cell transcriptional profiling as a high-content phenotypic assay. We exposed A549, a human lung adenocarcinoma cell line, to one of four compounds: dexamethasone (a corticosteroid agonist), nutlin-3a (a p53-Mdm2 antagonist), BMS-345541 (an inhibitor of nuclear factor κB–dependent transcription), or vorinostat [suberoylanilide hydroxamic acid (SAHA), an HDAC inhibitor], for 24 hours across seven doses in triplicate for a total of 84 drug–dose–replicate combinations and additional vehicle controls (Fig. 2A and fig. S2A). We labeled nuclei from each well and subjected them to sci-RNA-seq2 (fig. S2, B to D, and table S1).
Fig. 2. sci-Plex enables multiplex chemical transcriptomics at single-cell resolution.
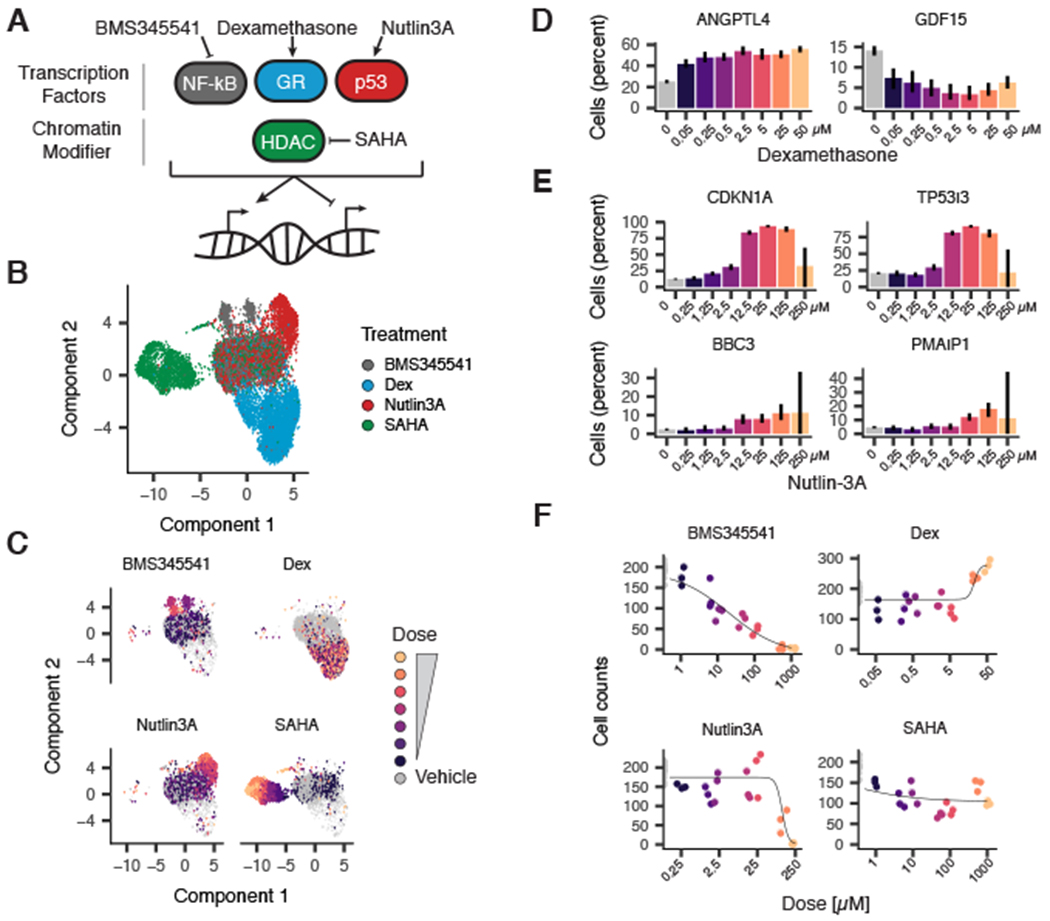
(A) Diagram depicting compounds and corresponding targets assayed within the pilot sci-Plex experiment. A549 lung adenocarcinoma cells were treated with either vehicle [dimethylsulfoxide (DMSO) or ethanol] or one of four compounds (BMS345541, dexamethasone, nutlin-3a, or SAHA). (B) UMAP embedding of chemically perturbed A549 cells colored by drug treatment. (C) UMAP embedding of chemically perturbed A549 cells faceted by treatment with cells colored by dose. (D and E) Expression of a canonical (D) glucocorticoid receptor activated (ANGPTL4) and repressed (GDF15) target genes as a function of dexamethasone dose or (E) p53 target genes as a function of nutlin-3a dose. y-axes indicate the percentage of cells with at least one read corresponding to the transcript. (F) Dose–response viability estimates for BMS345541-, dexamethasone-, nutlin-3a-, and SAHA-treated A549 cells on the basis of the relative number of cells recovered at each dose.
We used Monocle 3 (21) to visualize these data using Uniform Manifold Approximation and Projection (26) (UMAP) and Louvain community detection to identify compound-specific clusters of cells, which were distributed in a dose-dependent manner (Fig. 2, B and C, and fig. S2, E and F). To quantify the “population average” transcriptional response of A549 cells to each of the four drugs, we modeled each gene’s expression as a function of dose through generalized linear regression. A total of 7561 genes were sensitive to at least one drug, and 3189 genes were differentially expressed in response to multiple drugs (fig. S3A and table S2). These included canonical targets of dexamethasone (Fig. 2D) and nutlin-3a (Fig. 2E). Gene ontology analysis of differentially expressed genes revealed the involvement of drug-specific pathways (e.g., hormone signaling for dexamethasone; p53 signaling for nutlin-3a; fig. S3B). Additionally, we evaluated whether the number of cells recovered at each concentration could be used to infer toxicity akin to traditional screens. After fitting a response curve to the recovered cellular counts, we inferred a “viability score” from sci-Plex data, a metric that was concordant with “gold standard” measurements (Fig. 2F and fig. S2, G to I).
sci-Plex scales to thousands of samples and enables HTS
To assess how sci-Plex scales for HTS, we performed a screen of 188 compounds targeting a diverse range of enzymes and molecular pathways (Fig. 3A). Half of this panel was chosen to target transcriptional and epigenetic regulators. The other half was chosen to sample diverse mechanisms of action. We exposed three well-characterized human cancer cell lines, A549 (lung adenocarcinoma), K562 (chronic myelogenous leukemia), and MCF7 (mammary adenocarcinoma), to each of these 188 compounds at four doses (10 nM, 100 nM, 1 μM, and 10 μM) in duplicate, randomizing compounds and doses across well positions in replicate culture plates (table S3). These conditions, together with vehicle controls, accounted for 4608 of 4992 independently treated cell populations in this experiment. After treatment, we lysed cells to expose nuclei, hashed them with a specific combination of two oligos (fig. S4A), and performed sci-RNA-seq3 (21). After sequencing and filtering based on hash purity (fig. S4, B to F), we obtained transcriptomes for 649,340 single cells, with median mRNA UMI counts of 1271,1071, and 2407 for A549, K562, and MCF7, respectively (fig. S5A). The aggregate expression profiles for each cell type were highly concordant between replicate wells (Pearson correlation = 0.99) (fig. S5B).
Fig. 3. sci-Plex enables global transcriptional profiling of thousands of chemical perturbations in a single experiment.
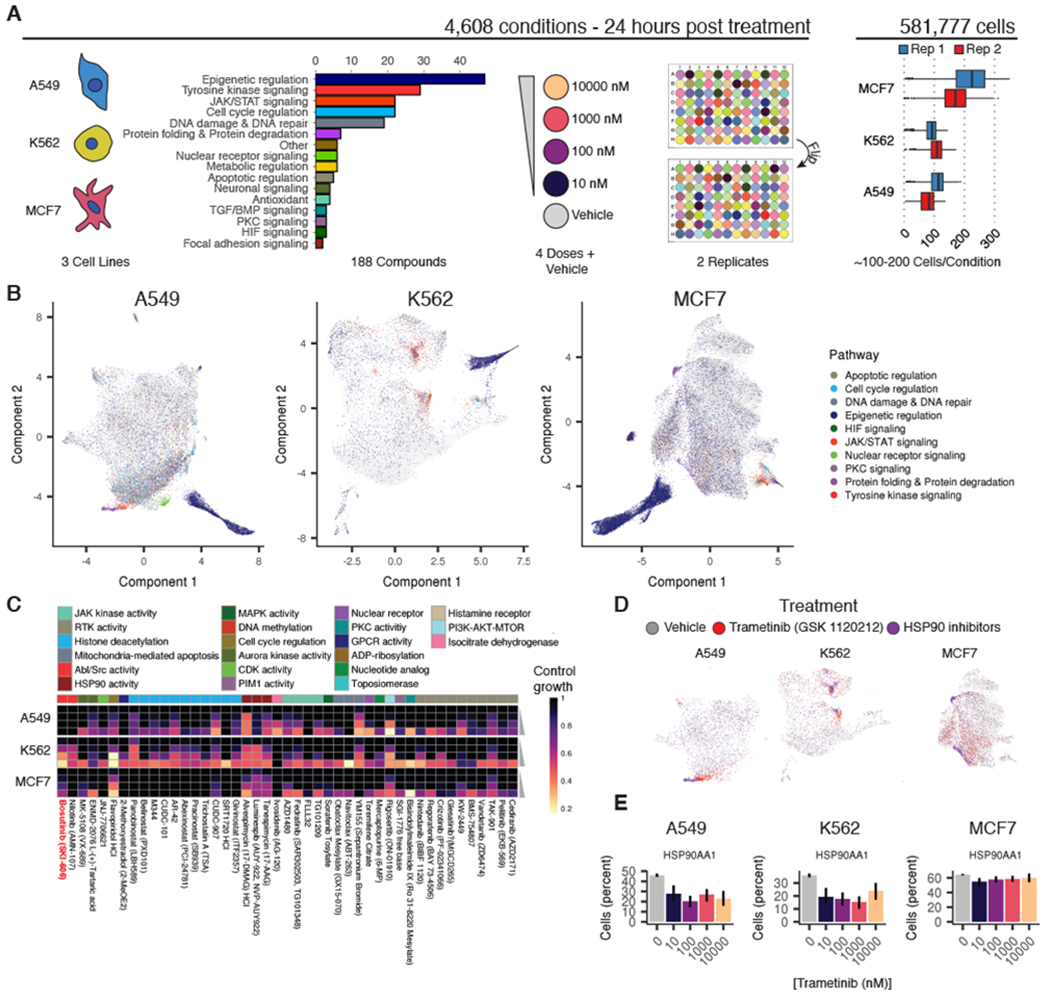
(A) Schematic of the large-scale sci-Plex experiment (sci-RNA-seq3). A total of 188 small molecules were tested for their effects on A549, K562, and MCF7 human cell lines, each at four doses and in biological replicate, after 24 hours of treatment. The plate positions of doses and drugs were varied between replicates, and a median of 100 to 200 cells were recovered per condition. Colors demarcate cell line, compound pathway, and dose. (B) UMAP embeddings of A549, K562, and MCF7 cells in our screen with each cell colored by the pathway targeted by the compound to which a given cell was exposed. To facilitate visualization of significant molecular phenotypes, we added transparency to cells treated with compound or dose combinations that did not appreciably alter the corresponding cells’ distribution in UMAP space compared with vehicle controls (Fisher’s exact test, FDR < 1%). (C) Viability estimates obtained from hash-based counts of nuclei at each dose of selected compounds (bosutinib is highlighted in red text). Rows represent compound doses increasing from top to bottom, and columns represent individual compounds. Annotation bar at top depicts the broad cellular activity targeted by each compound. (D) UMAP embeddings highlighted by treatment with the MEK inhibitor trametinib (red), an HSP90 inhibitor (purple), or vehicle control (gray). (E) HSP90AA1 expression levels in cells exposed to increasing doses of trametinib. y-axes indicate the percentage of cells with at least one read corresponding to the transcript.
Visualizing sci-RNA-seq profiles separately for each cell line revealed compound-specific transcriptional responses and patterns that were common to multiple compounds. For each of the cell lines, UMAP projected most cells into a central mass, flanked by smaller clusters (Fig. 3B). These smaller clusters were largely composed of cells treated with compounds from only one or two compound classes (figs. S6 and S7, A to C). For example, A549 cells treated with triamcinolone acetonide, a synthetic glucocorticoid receptor agonist, were markedly enriched in one such small cluster, comprising 95% of its cells [Fisher’s exact test, false discovery rate (FDR) < 1%; fig. S7, D and E]. Although many drugs were associated with a seemingly homogeneous transcriptional response, we also identified cases in which distinct transcriptional states were induced by the same drug. For example, in A549, the microtubule-stabilizing compounds epothilone A and epothilone B were associated with three such focal enrichments, each composed of cells from both compounds at all four doses (fig. S7, F and G). The cells in each focus were distinct from one another, but transcriptionally similar to other treatments: a recently identified microtubule destabilizer, rigosertib (27); the SETD8 inhibitor UNC0397; or untreated proliferating cells (fig. S7H).
We next assessed the effects of each drug on the “population average” transcriptome of each cell line. In total, 6238 genes were differentially expressed in a dose-dependent manner in at least one cell line (FDR < 5%; fig. S8 and tables S4 and S5). Bulk RNA-seq measurements collected for five compounds across four doses and vehicle agreed with averaged gene expression values and estimated effect sizes across identically treated single cells, although correlations between small effect sizes were diminished (fig. S9). Moreover, sci-Plex dose-dependent effect profiles correlated with compound-matched L1000 measurements (11) (fig. S10).
Genes associated with the cell cycle were highly variable across individual cells, and many drugs reduced the fraction of cells that expressed proliferation marker genes (figs. S11 and S12). In principle, scRNA-seq should be able to distinguish shifts in the proportion of cells in distinct transcriptional states from gene-regulatory changes within those states. By contrast, bulk transcriptome profiling would confound these two signals (fig. S13A) (14). We therefore tested for dose-dependent differential expression on subsets of cells corresponding to the same drug but expressing high versus low levels of proliferation marker genes (fig. S13B). Correlation between the dose-dependent effects on the two fractions of each cell type varied across drug classes (fig. S13C), with some frankly discordant effects for individual compounds (fig. S13D). Viability analysis performed as in the pilot experiment revealed that after drug exposure at the highest dose, only 52 (27%) compounds caused a decrease in viability of 50% or more (Fig. 3C and fig. S5C). Among the drugs that reduced viability, we observed a higher sensitivity of K562 to the Src and Abl inhibitor bosutinib (Fig. 3C), a result that we confirmed by cell counting (fig. S14A). This result is consistent with K562 cells harboring a constitutively active BCR-ABL fusion kinase (28) and an observed increased sensitivity of hematopoietic and lymphoid cancer cell lines to Abl inhibitors (29) (fig. S14B).
To assess whether each compound elicited similar responses across the three cell lines, we clustered compounds using the effect sizes for dose-dependent genes as loadings in each cell line (figs. S15 to S18). Joint analysis of the three cell lines revealed common and cell-type–specific responses to different compounds (figs. S19 and S20). For example, trametinib, a mitogen-activated protein kinase kinase (MEK) inhibitor, induced a transcriptionally distinct response in MCF7 cells. Inspection of UMAP projections revealed trametinib-treated MCF7 cells interspersed among vehicle controls, reflecting limited effects. By contrast, trametinib-treated A549 and K562 cells, which harbor activating KRAS and ABL mutations (30), respectively, were tightly clustered, consistent with a strong, specific transcriptional response to inhibition of MEK signaling by trametenib (Fig. 3D). Further, we observed that these A549 and K562 cells appeared proximal to clusters enriched with inhibitors of HSP90, a key chaperone for protein folding (Fig. 3D). This observation was corroborated by concordant changes in HSP90AA1 expression in trametinib-treated cells (Fig. 3E). Analysis of Connectivity Map data (11,12) revealed further evidence that MEK inhibitors do indeed induce highly similar gene expression signatures to HSP90 perturbations (fig. S14C), especially in A549 but not in MCF7 (fig. S14, D and E). These results are concordant with previous observations of the regulation of HSP90AA1 downstream of MEK signaling (31) and suggest that similarity in single-cell transcriptomes treated with distinct compounds can highlight drugs that target convergent molecular pathways.
Inference of chemical and mechanistic properties of HDAC inhibitors
For each of the three cell lines, the most prominent compound response was composed of cells treated with one of 17 HDAC inhibitors (Fig. 3B, dark blue, and table S6). To assess the similarity of the dose–response trajectories between cell lines, we aligned HDAC-treated cells and vehicle-treated cells from all three cell lines using a mutual-nearest neighbor (MNN) matching approach (32) to produce a consensus HDAC inhibitor trajectory, which we call “pseudodose” [analogous to “pseudotime” (33)] (Fig. 4A and fig. S21). We observed that some HDAC inhibitors induced homogeneous responses, with nearly all cells localized to a relatively narrow range of the HDAC inhibitor trajectory at each dose (e.g., pracinostat in A549), whereas other drugs induced much greater cellular heterogeneity (Fig. 4B and fig. S22).
Fig. 4. HDAC inhibitor trajectory captures cellular heterogeneity in drug response and biochemical affinity.
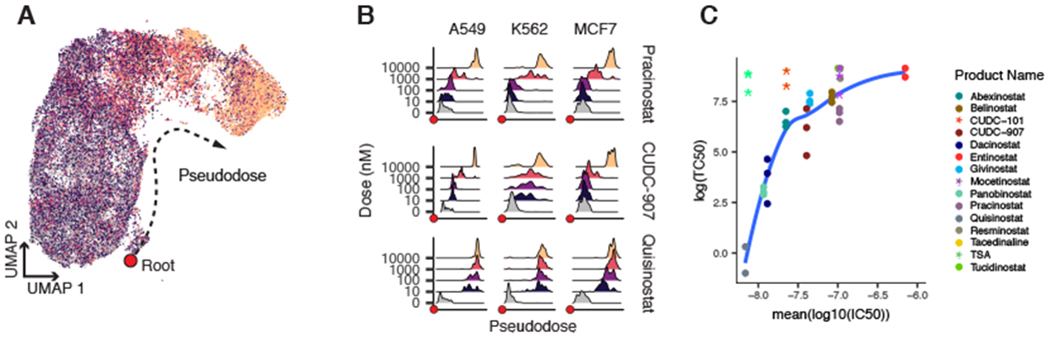
(A) MNN alignment and UMAP embedding of transcriptional profiles of cells treated with one of 17 HDAC inhibitors. Pseudodose root is displayed as a red dot. (B) Ridge plots displaying the distribution of cells along pseudodose by dose shown for three HDAC inhibitors with varying biochemical affinities. (C) Relationship between TC50 and average log10(IC50) from in vitro measurements. Asterisks indicate compounds with a solubility <200 μM (in DMSO) that were not included in the fit.
Such heterogeneity could be explained by cells executing a defined transcriptional program asynchronously, with the dose of drug that the cells are exposed to modulating the rates of their progression through it. To test this hypothesis, we sequenced the transcriptomes of 64,440 A549 cells that were treated for 72 hours with one of 48 compounds, including many of the HDAC inhibitors from the large sci-Plex screen. Upon accounting for confluency-dependent cell-cycle effects and MNN alignment (figs. S23 and S24), the coembedded UMAP projection revealed new focal concentrations of cells at 72 hours that were not evident at the 24-hour time point, e.g., SRT1024 (fig. S25). However, for the majority of HDAC inhibitors tested, we did not observe that cells at a given dose moved farther along an aligned HDAC trajectory at 72 hours (fig. S26). This suggests that the dose of many HDAC inhibitors governs the magnitude of a cell’s response rather than its rate of progression and that any observed heterogeneity cannot be attributed solely to asynchrony (fig. S26).
Next, we assessed whether a given HDAC inhibitor’s target affinity explained its global transcriptional response to the compound. We used dose-response models to estimate each compound’s transcriptional median effective concentration (TC50), i.e., the concentration needed to drive a cell halfway across the HDAC inhibitor pseudodose trajectory (fig. S27A and table S6). To compare the transcriptionally derived measures of potency with the biochemical properties of each compound, we collected published median inhibitory concentration (IC50) values for each compound from in vitro assays performed on eight purified HDAC isoforms (table S7). With the exception of two relatively insoluble compounds, our calculated TC50 values increased as a function of compound IC50 values (Fig. 4C and fig. S27, B and C).
To assess the components of the HDAC inhibitor trajectory, we performed differential expression analysis using pseudodose as a continuous covariate. Of the 4308 genes that were significantly differentially expressed over this consensus trajectory, 2081 (48%) responded in a cell-type–dependent manner and 942 (22%) exhibited the same pattern in all three cell lines (fig. S28, A and B, and table S8). One prominent pattern shared by the three cell lines was an enrichment for genes and pathways indicative of progression toward cell-cycle arrest (figs. S28C and S29, A and B). DNA content staining and flow cytometry confirmed that HDAC inhibition resulted in the accumulation of cells in the G2/M phase of the cell cycle (34) (fig. S29, C and D).
The shared response to HDAC inhibition included not only cell-cycle arrest but also the altered expression of genes involved in cellular metabolism (fig. S28C). Histone acetyltransferases and deacetylases regulate chromatin accessibility and transcription factor activity through the addition or removal of charged acetyl groups (35–37). Acetate, the product of HDAC class I-, II-, and IV-mediated histone deacetylation and a precursor to acetyl-coenzyme A (acetyl-CoA), is required for histone acetylation but also has important roles in metabolic homeostasis (23, 38, 39). Inhibition of nuclear deacetylation limits recycling of chromatin-bound acetyl groups for both catabolic and anabolic processes (39). Accordingly, we observed that HDAC inhibition led to sequestration of acetate in the form of markedly increased acetylated lysine levels after exposure to a 10 μM dose of the HDAC inhibitors pracinostat and abexinostat (fig. S30).
Upon further inspection of pseudodose-dependent genes, we observed that enzymes critical for cytoplasmic acetyl-CoA synthesis from either citrate (ACLY) or acetate (ACSS2) were up-regulated (Fig. 5A). Genes involved in cytoplasmic citrate homeostasis (GLS, IDH1, and ACO1), citrate cellular import (SLC13A3), and mitochondrial citrate production and export (CS, SLC25A1) were also up-regulated. Up-regulation of SIRT2, which deacetylates tubulin, was also observed in response to HDAC inhibition.
Fig. 5. HDAC inhibitors shared transcriptional response indicative of acetyl-CoA deprivation.
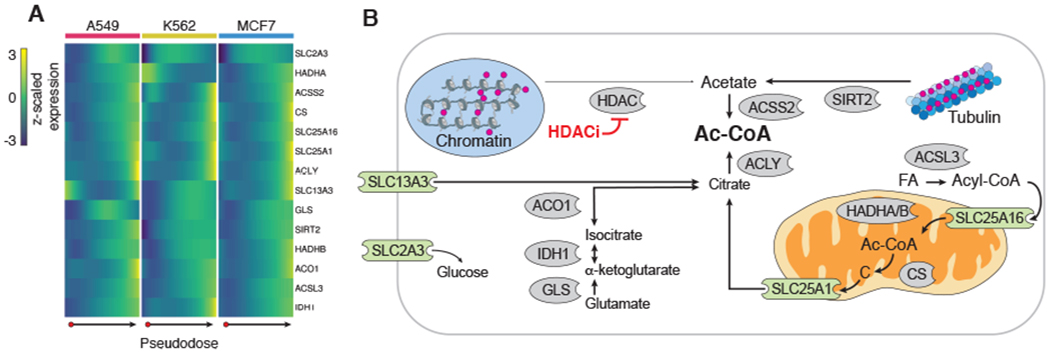
(A) Heatmap of row-centered and z-scaled gene expression depicting the up-regulation of pseudodose-dependent genes involved in cellular carbon metabolism. (B) Diagram of the roles of genes from (A) in cytoplasmic acetyl-CoA regulation. Red circles indicate acetyl groups. Enzymes are shown in gray. Transporters are shown in green (FA, fatty acid; Ac-CoA, acetyl-CoA; C, citrate).
Together with increases in chromatin-bound acetate, these transcriptional responses suggest a metabolically consequential depletion of cellular acetyl-CoA reserves in HDAC-inhibited cells (Fig. 5B). To validate this further, we sought to shift the distribution of cells along the HDAC inhibitor trajectory by modulating cellular acetyl-CoA levels. We treated A549 and MCF7 cells with pracinostat in the presence and absence of acetyl-CoA precursors (acetate, pyruvate, or citrate) or inhibitors of enzymes (ACLY, ACSS2, or PDH) involved in replenishing acetyl-CoA pools. After treatment, cells were harvested and processed using sci-Plex and trajectories constructed for each cell line (figs. S31 and S32). In both A549 and MCF7 cells, acetate, pyruvate, and citrate supplementation was capable of blocking pracinostat-treated cells from reaching the end of the HDAC inhibitor trajectory (fig. S31, F, J, H, and L). In MCF7 cells, both ACLY and ACSS2 inhibition shitted cells farther along the HDAC inhibitor trajectory, although no such shift was observed in A549 (fig. S31, G, K, I, and M). Taken together, these results suggest that a major feature of the response of cells to HDAC inhibitors, and possibly their associated toxicity, is the induction of an acetyl-CoA-deprived state.
Discussion
Here, we present sci-Plex, a massively multiplex platform for single-cell transcriptomics. sci-Plex uses chemical fixation to cost-effectively and irreversibly label nuclei with short, unmodified ssDNA oligos. In the proof-of-concept experiment described here, we applied sci-Plex to quantify the dose-dependent responses of cancer cells to 188 compounds through an assay that is both high content (global transcription) and high resolution (single cell). By profiling several distinct cancer cell lines, we distinguished between shared and cell-line-specific molecular responses to each compound.
sci-Plex offers some distinctive advantages over conventional HTS: it can distinguish a compound’s distinct effects on cellular subsets (including complex in vitro systems such as cellular reprogramming, organoids, and synthetic embryos); it can unmask heterogeneity in cellular response to a perturbation; and it can measure how drugs shift the relative proportions of transcriptionally distinct subsets of cells. Highlighting these features, our study provides insight into the mechanism of action of HDAC inhibitors. Specifically, we find that the main transcriptional responses to HDAC inhibitors involve cell-cycle arrest and marked shifts in genes related to acetyl-CoA metabolism. For some HDAC inhibitors, we observed clear heterogeneity in responses observed at the single-cell level. Although HDAC inhibition is conventionally thought to act through mechanisms directly involving chromatin regulation, our data support an alternative model, albeit not a mutually exclusive one, in which HDAC inhibitors impair growth and proliferation by interfering with a cancer cell’s ability to draw acetate from chromatin (22,23,39). As such, variation in cells’ acetate reservoirs is a potential explanation for their heterogeneous responses to HDAC inhibitors.
As the cost of single-cell sequencing continues to fall, the opportunities for leveraging sci-Plex for basic and applied goals in biomedicine may be substantial. The proof-of-concept experiments described here, consisting of nearly 5000 independent treatments with transcriptional profiling of >100 single cells per treatment, can potentially be scaled toward a comprehensive, high-resolution atlas of cellular responses to pharmacologic perturbations (e.g., hundreds of cell lines or genetic backgrounds, thousands of compounds, multichannel single-cell profiling, etc.). The ease and low cost of oligo hashing, coupled with the flexibility and exponential scalability of single-cell combinatorial indexing, would facilitate this goal.
Supplementary Material
ACKNOWLEDGMENTS
We thank members of the Shendure lab; the Trapnell lab; and others, particularly A. Adey, G. Booth, A. Hill, S. Henikoff, K. Cherukumilli, P. Selvaraj, and T. Zhou, for helpful suggestions, discussion, and mentorship; and D. Prunkard and A. Leith for assistance in flow sorting.
Funding: This work was funded by grants from the NIH (DP1HG007811 and R01HG006283 to J.S.; DP2 HD088158 to C.T.), the W. M. Keck Foundation (to C.T. and J.S.), the NSF (DGE-1258485 to S.R.S.), and the Paul G. Allen Frontiers Group (to J.S. and C.T.). J.S. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Competing interests: L.C., F.Z., and F.S. declare competing financial interests in the form of stock ownership and paid employment by Illumina, Inc. One or more embodiments of one or more patents and patent applications filed by Illumina and the University of Washington may encompass the methods, reagents, and data disclosed in this manuscript.
Data and materials availability: Processed and raw data can be downloaded from NCBI GEO (#GSE139944). Code used to perform analyses can be accessed on Zenodo and https://github.com/cole-trapnell-lab/sci-plex. All methods for making the transposase complexes used in this paper are described in Cao et al. (25); however, Illumina will provide transposase complexes in response to reasonable requests from the scientific community subject to a material transfer agreement.
science.sciencemag.org/content/367/6473/45/suppl/DC1
Materials and Methods
REFERENCES AND NOTES
- 1.Broach JR, Thorner J, Nature 384 (Suppl), 14–16 (1996). [PubMed] [Google Scholar]
- 2.Pereira DA, Williams JA, Br. J. Pharmacol 152, 53–61 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shum D et al. J. Enzyme Inhib. Med. Chem 23, 931–945 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yu C et al. Nat. Biotechnol 34, 419–423 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perlman ZE et al. Science 306, 1194–1198 (2004). [DOI] [PubMed] [Google Scholar]
- 6.Futamura Y et al. Chem. Biol 19, 1620–1630 (2012). [DOI] [PubMed] [Google Scholar]
- 7.Kang J et al. Nat. Biotechnol 34, 70–77 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huss KL, Blonigen PE, Campbell RM, J. Biomol. Screen 12, 578–584 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Ye C et al. Nat. Commun 9, 4307 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bush EC et al. Nat. Commun 8, 105 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Subramanian A et al. , Cell 171, 1437–1452.e17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lamb J et al. Science 313, 1929–1935 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Elowitz MB, Levine AJ, Siggia ED, Swain PS, Science 297, 1183–1186 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Trapnell C, Genome Res 25, 1491–1498 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shaffer SM et al. Nature 546, 431–435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spencer SL, Gaudet S, Albeck JG, Burke JM, Sorger PK, Nature 459, 428–432 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stoeckius M et al. Genome Biol 19, 224 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gehring J, Park JH, Chen S, Thomson M, Pachter L, Highly multiplexed single-cell RNA-seq for defining cell population and transcriptional spaces. bioRxiv 315333 [Preprint] 5 May 2018. 10.1101/315333. [DOI] [Google Scholar]
- 19.McGinnis CS et al. Nat. Methods 16, 619–626 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shin D, Lee W, Lee JH, Bang D, Sci. Adv 5, eaav2249 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cao J et al. Nature 566, 496–502 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McBrian MA et al. Mol. Cell 49, 310–321 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Comerford SA et al. Cell 159, 1591–1602 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cusanovich DA et al. Science 348, 910–914 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao J et al. Science 357, 661–667 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McInnes L, Healy J, UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv:1802.03426 [stat.ML] (9 February 2018). [Google Scholar]
- 27.Jost M et al. Mol. Cell 68, 210–223.e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grosveld G et al. Mol. Cell. Biol 6, 607–616 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Greuber EK, Smith-Pearson P, Wang J, Pendergast AM, Nat. Rev. Cancer 13, 559–571 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barretina J et al. Nature 483, 603–607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dai C et al. J. Clin. Invest 122, 3742–3754 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Haghverdi L, Lun ATL, Morgan MD, Marioni JC, Nat. Biotechnol 36, 421–427 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Trapnell C et al. Nat. Biotechnol 32, 381–386 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brazelle W et al. PLOS ONE 5, e14335 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roe J-S, Mercan F, Rivera K, Pappin DJ, Vakoc CR, Mol. Cell 58, 1028–1039 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brownell JE et al. Cell 84, 843–851 (1996). [DOI] [PubMed] [Google Scholar]
- 37.Taunton J, Hassig CA, Schreiber SL, Science 272, 408–411 (1996). [DOI] [PubMed] [Google Scholar]
- 38.Kurdistani SK, Curr. Opin. Genet. Dev 26, 53–58 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wellen KE et al. Science 324, 1076–1080 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.