

ABSTRACT
Objective:
Heatstroke can induce serious physiological dysfunction in the intestine. However, the underlying mechanisms of this condition are unknown, and therapeutic strategies are not available. In this study, we explored the role of endoplasmic reticulum (ER) stress signaling in this process and assessed whether pretreating mice with an inhibitor of ER stress could alleviate intestinal damage.
Methods:
A heatstroke model was established in male mice. Mice were pretreated with 4-phenylbutyrate (4-PBA) before exposure to heat stress. Intestinal morphological changes were observed by hematoxylin and eosin (H&E) staining and transmission electron microscopy. The TUNEL assay was used to detect intestinal apoptosis. The expression of the ER stress-related proteins and apoptosis-related proteins was investigated by the Western blot assay.
Results:
Compared with control group, mice with heatstroke exhibited evidence of intestinal injury and epithelial apoptosis, accompanied by significantly increased expression of ER stress-related proteins in the intestines. The intestinal injury score and level of intestinal epithelial apoptosis were significantly reduced after administration of 4-PBA. Furthermore, the levels of the intestinal ER stress-related proteins GRP78, PERK, p-eIF2α, ATF4, and CHOP were decreased after 4-PBA treatment.
Conclusions:
Our results indicate that the ER stress-mediated apoptosis pathway is activated during heat stress-induced intestinal injury. 4-PBA can inhibit heatstroke-induced intestinal ER stress and attenuate intestinal injury. We provide evidence that the beneficial effect of 4-PBA is closely related to the inhibition of ER stress-mediated apoptosis. These findings suggest that ER stress may be a novel therapeutic target in patients with heatstroke.
Keywords: 4-Phenylbutyrate, apoptosis, endoplasmic reticulum stress, heatstroke, intestinal injury, pyroptosis
INTRODUCTION
Because of the global warming trend, heatstroke has become a serious threat to public health in recent years (1). There is no effective treatment for heatstroke, and the 28-day and 2-year mortality rates are reported to be 58% and 71%, respectively (2). Heatstroke has been defined as a core body temperature of greater than 40°C in humans, and it is accompanied by a systemic inflammatory response that can lead to multiorgan dysfunction syndrome (3). Currently, it is thought that the pathophysiological process of heatstroke is a “sepsis-like” reaction secondary to heat injury that triggers the process of multiorgan dysfunction. The intestine plays an important role not only in the digestion and absorption of nutrients, but also as an immune defense barrier (4). Because the gut is considered to be the largest reservoir of bacteria and endotoxins in the body, heat stress-induced intestinal injury can lead to bacterial translocation and endotoxemia (5). It has been reported that the local inflammatory response and degree of injury in the intestine are closely related to the severity of the systemic inflammatory response in heatstroke (6). Intestinal injury is a common complication of heatstroke, and, in turn, exacerbates the process of heatstroke injury. The underlying mechanisms of intestinal injury in heatstroke have not been fully investigated. Apoptosis triggered by heat stress has been reported to cause intestinal dysfunction (7), but the mechanisms are not clear and need further study.
The endoplasmic reticulum (ER) is an important intracellular organelle commonly known as a protein-folding factory. The ER provides the location for calcium storage, lipid synthesis, and carbohydrate metabolism. Certain pathological conditions, including sepsis, burns, trauma, and ischemia, can lead to the accumulation of unfolded proteins that alter the homeostasis of the ER and cause ER stress (8). It has been reported that the level and duration of ER stress can significantly affect the function, adaptation, or apoptosis of stress cells (9). Glucose-regulated protein 78 (GRP78) is a marker for ER stress that plays a critical role in the regulation of the unfolded protein response (UPR) signaling network (10). Under nonstress conditions, GRP78 binds to ER transmembrane proteins, including the membrane-embedded proteins inositol-requiring protein 1 (IRE1), protein kinase RNA-like endoplasmic reticulum kinase (PERK), and activating transcription factor 6 (ATF6) (11). As a response to ER stress, GRP78 dissociates from these transmembrane proteins, which leads to their activation and triggers three types of UPR, subsequently controlling the expression of specific transcription factors and UPR downstream pathways (12). Of the three UPR pathways, the protein kinase RNA-like endoplasmic reticulum kinase/activating transcription factor 4/C/EBP-homologous protein (PERK/ATF4/CHOP) signaling pathway has been explored to determine whether it plays a crucial role in cell death (13). Accumulated evidence shows that CHOP is a critical mediator responsible for ER stress-induced apoptosis through different mechanisms (14).
The intestinal epithelium is rich in developed ER structure. Exogenous or endogenous risk factors capable of impairing ER function can induce intestinal damage in the host. Upregulation of CHOP expression exacerbates the development of colitis, and a larger number of apoptotic cells in the intestinal mucosa of wild-type mice than in CHOP-null mice were observed to accompany the development of colitis (15). These results indicate that the CHOP signaling pathway is associated with apoptotic cell death in inflammatory bowel disease. Whether this signaling is involved in heatstroke-induced intestinal damage has not yet been investigated.
4-Phenylbutyrate (4-PBA), which is a chemical chaperone that alleviates ER stress and acts as an ER stress inhibitor, has been approved by the US Food and Drug Administration. It is mainly used for the treatment of urea cycle disorders as an ammonia scavenger (16). Recent research has found that 4-PBA can treat severe sepsis and septic shock in rat models and can significantly improve vital organ function and overall treatment outcome without apparent side effects (17). Therefore, our experiment was designed to investigate the effects of ER stress in heatstroke-associated intestinal injury and explore whether 4-PBA would prevent intestinal injury via the inhibition of the ER stress pathway.
MATERIALS AND METHODS
Animals
Because estrogen can improve survival during heatstroke by ameliorating inflammatory responses and cardiovascular dysfunction (18), only pathogen-free, 6- to 8-week-old, male BALB/c mice weighing 20.0 ± 2.0 g were used. All animal procedures were approved by the Medical Ethics Committee of the General Hospital of Southern Theatre Command of PLA. Mice were housed in controlled environmental conditions (12-h light/dark cycle, 35% ± 5% humidity, temperature 23°C) and received water ad libitum.
Experimental design and heat stress protocol
In the pilot experiment, the heatstroke murine model and survival time were assessed. Eighteen mice were randomly divided into three groups: control group (n = 6), no heat exposure; heatstroke (HS) group (n = 6); and 4-PBA group (n = 6).
In the main experiment, intestinal injury and endoplasmic reticulum stress were assessed. Thirty mice were randomly divided into three groups: control group (n = 10), no heat exposure; HS group (n = 10); and 4-PBA group (n = 10).
In the 4-PBA group, each mouse received an intraperitoneal injection of 120 mg/kg 4-PBA (Sigma, Calif) (19) before heat exposure. Mice in the HS group and 4-PBA group were placed in an incubator that had been prewarmed and was maintained at a temperature of 35.5 ± 0.5°C with a relative humidity of 60% ± 5%, whereas mice in the control group were kept at an ambient temperature of 25 ± 0.5°C with a humidity of 35% ± 5%. The rectal core temperature (Tc) was continuously monitored with a rectal thermometer. When the core temperature (Tc) reached 42°C, the heatstroke model was established successfully. This murine model of heatstroke was used in our previous studies (6, 20). Mice received cooling treatment after the Tc reached 42°C. The treatment strategies were as follows: mice were removed from the incubator and allowed to cool at an ambient temperature of 25 ± 0.5°C with a humidity of 35% ± 5%; in addition, mice received water to drink. Our previous study showed that the average survival time of mice with a Tc above 42°C is approximately 6 h, even with immediate application of cooling treatment (6). Thus, this time point was selected for our follow-up experiment. Six hours after the cooling treatment, all animals in the three groups were sacrificed by cervical dislocation. The ileum was harvested, flushed with ice-cold phosphate-buffered saline (PBS), and stored for further study.
Histopathological analysis
Distal ileum tissues were fixed in neural-buffered formalin. Fixed samples were embedded in paraffin, cut into sections that were 4 to 5 μm thick, and stained with H&E. Morphological changes were assessed and graded in a blinded manner by two certified veterinary pathologists using the intestinal injury score developed by Chiu (21). According to changes in the villi and glands of the intestinal mucosa, the Chiu scoring system includes five subdivisions: grade 0, normal mucosa; grade 1, development of a subepithelial Gruenhagen space at the villus tip; grade 2, extension of the space with moderate epithelial lifting; grade 3, extensive epithelial lifting with a few denuded villi; grade 4, denuded villi with exposed capillaries; and grade 5, disintegration of the lamina propria, ulceration, and bleeding. Images were acquired by using a Nikon microscope (Eclipse Ci, Nikon Corporation, Tokyo, Japan).
Transmission electron microscopy
Ileum tissue samples for transmission electron microscopy were cut to a size of approximately 1 to 2 mm3 and fixed with 2.5% glutaraldehyde at 4°C for 4 h. After fixation, samples were dehydrated with a graded series of ethanol treatments and were embedded in Epon overnight. Then, ultrathin sections were cut and stained with uranyl acetate and lead citrate (22). Images were acquired by transmission electron microscopy (Hitachi, Tokyo, Japan).
Immunohistochemistry
For the immunohistochemical study, after deparaffinization, dehydration, antigen retrieval, quenching of endogenous peroxidase activity, and blocking in normal goat serum, sections were incubated overnight at 4°C with primary antibodies against CHOP, BAX, and Bcl-2, which were obtained from Proteintech Group, Inc. (Rosemont, Ill). After washing with PBS, sections were incubated with secondary antibodies (Zhongshanjinqiao, Beijing, China) for 1 h at room temperature and rinsed in PBS. Immunoreactivity was detected using diaminobenzidine (DAB), followed by counterstaining with hematoxylin. Sections were rinsed in water, dehydrated in a graded alcohol series, cleared with xylene, mounted with neutral balsam, and coverslipped. Proteins were visualized as brown pigment according to a standard diaminobenzidine protocol. Images were acquired by using a Nikon microscope (Eclipse Ci,Nikon Corporation, Tokyo, Japan).
Apoptosis assay of intestinal tissue
To detect apoptotic cell death in ileum tissues, a TUNEL assay (In Situ Cell Death Detection Kit, Roche, Mannheim, Germany) was performed according to the manufacturer's instructions. Ileum tissue sections were incubated with proteinase K for 20 min at room temperature and washed with PBS. After that sections were incubated in TdT buffer containing FITC-conjugated dUTP at 37°C for 60 min. Morphological changes in nuclei were observed by counterstaining with DAPI (Beyotime, Shanghai, China). Sections were analyzed under a Nikon fluorescence microscope (Eclipse Ti-SR,Nikon Corporation, Tokyo, Japan).
Western blot analysis
Frozen ileum tissue was ground into powder in liquid nitrogen, lysed in ice-cold RIPA buffer mixed with 1 mM protease inhibitor and protein phosphatase inhibitor (Roche Applied Science, Rockford, Ill), and centrifuged at 12,000 × g for 30 min at 4°C. The protein concentration was determined using a BCA protein assay kit (Applygen Technologies, Inc., Beijing, China). Equal amounts of protein (80 μg) were separated on 12% SDS–PAGE gels and transferred to nitrocellulose membranes (Bio-Rad Laboratories, Inc., Hercules, Calif). Membranes were blocked in a 5% skim milk solution at room temperature for 1 h and were then incubated with diluted primary antibody overnight at 4°C. Antibodies against GRP-78, PERK, eIF2α, p-eIF2α, and CHOP were obtained from Cell Signaling Technology (Danvers, Mass). Antibodies against ATF4, Bcl-2, and Bax were obtained from Proteintech Group, Inc. (Rosemont). Blots were washed in PBS/Tween 20 (T20) for 5 min with shaking. Blots were incubated with the secondary antibody (Zhongshanjinqiao, Beijing, China) for 1 h at room temperature. The chemiluminescence signal was detected by using ImmobilonWestern (Millipore Corporation, Billerica, Mass). Protein bands were detected using GE ImageQuant LAS 500 (GE Healthcare, United States). Quantification of the digitized images of the Western blot bands was performed using ImageJ software (National Institutes of Health, Bethesda, Md).
Statistical analyses
All data were presented as mean ± SD and were analyzed using one-way ANOVA followed by Duncan's post hoc test using SPSS 13.0 software (SPSS, Chicago, Ill). A value of P < 0.05 was considered statistically significant.
RESULT
Inhibition of endoplasmic reticulum stress increased the survival time after heatstroke
The survival time is shown in Table 1. In the HS group, the survival time was 384 min. Pretreatment with 4-PBA significantly increased the survival time by 136 min (P < 0.01). Mice in the control group were monitored for 720 min, and no death was observed.
Table 1.
Survival time after heatstroke
| Group | Survival time, min |
| Control | >720 |
| HS | 384 ± 25 |
| HS+PBA | 520 ± 33# |
The results are presented as mean ± SD. Significant differences are indicated as follows: #P < 0.01 vs. the heatstroke group.
Inhibition of endoplasmic reticulum stress reduced ileum injury in animals affected by heatstroke
Gross morphological changes in the small intestine following heatstroke were evident, including edema, hyperemia, and petechial hemorrhages in the mesentery, and 4-PBA attenuated this intestinal damage (Fig. 1A). Histopathological findings are shown in Figure 2B. The morphology of the intestinal mucosa was approximately normal in the control groups. Heatstroke-induced acute intestinal mucosal damage and inflammation was manifested by extensive destruction of the villi and infiltration of inflammatory cells into the lamina propria. 4-PBA significantly attenuated heatstroke-induced pathological alterations, observed as reduced inflammatory cell infiltration and basically intact glands and intestinal villi. The intestinal injury scores in the heatstroke groups were significantly higher than those in the control group (P < 0.01), whereas the injury scores in the 4-PBA group were significantly lower than those in the heatstroke group (P < 0.01) (Fig. 1C).
Fig. 1.
(A) Gross morphological changes in the ileum of mice.
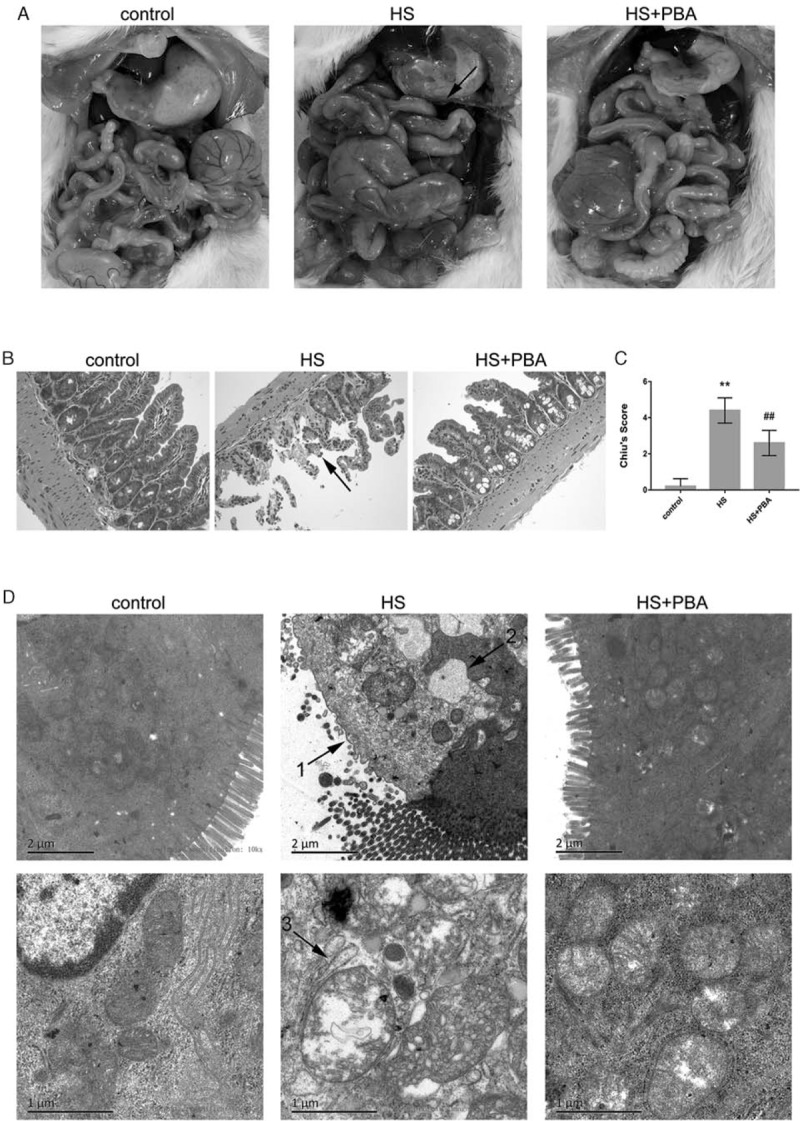
Edema, hyperemia, and petechial hemorrhages in the mesentery are clearly seen after heatstroke. 4-PBA attenuated intestinal damage. (B) Pathological changes in the ileum of mice (magnification ×200). Samples were harvested 6 h after heatstroke and stained with H&E. The arrow indicates inflammatory cell infiltration and loss of villi. (C) Histological scores for the ileum are presented as mean ± SD. Significant differences are indicated as follows: ∗∗P < 0.01 and ∗P < 0.05 vs. the control group; ##P < 0.01 and #P < 0.05 vs. the heatstroke group. (D) Changes in the mucosal ultrastructure of the small intestine at 10,000× magnification and 25,000× magnification by transmission electron microscopy. The arrows indicate the following: 1, loss of intestinal microvilli; 2, extreme expansion of the endoplasmic reticulum; 3, vacuole-like structure and mitochondrial swelling.
Fig. 2.
Immunohistochemical staining of CHOP, Bax, and Bcl-2 expression in the small intestine of mice.
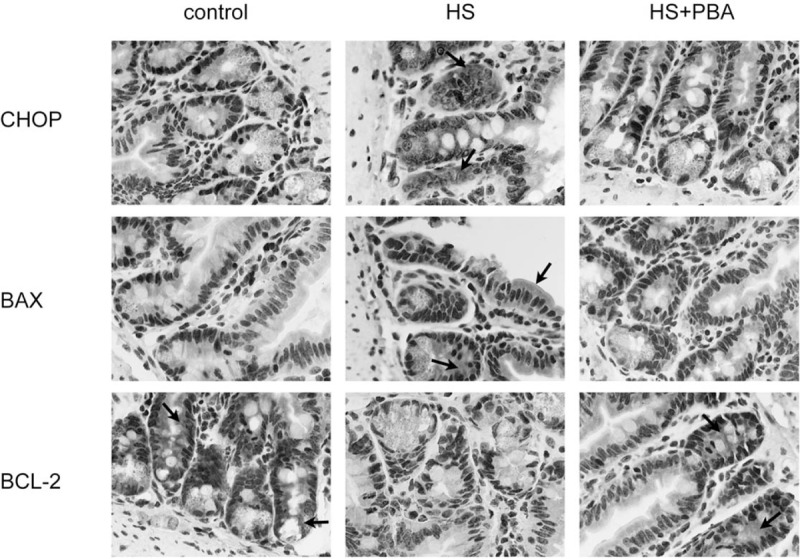
The arrows indicate cells positive by immunohistochemistry.
Findings from electron microscopic observation are shown in Figure 1D. The microvilli on the surface of intestinal mucosal epithelial cells in the control group were neatly arranged, the epithelial cells were structurally intact, and the organelle structures were normal. After heatstroke, microvilli were lost, and epithelial cells were bare. ER structures exhibited extreme expansion, and mitochondria were swollen and vacuole-like. As seen via transmission electron microscopy, treatment with 4-PBA appreciably ameliorated the heat-related changes in intestinal mucosal morphology, including providing a more orderly arrangement of the intestinal microvilli, although the villi were shorter than those in the control group; restoring the ER structure nearly to normal; and reducing mitochondrial swelling.
Inhibition of endoplasmic reticulum stress reduced intestinal epithelial apoptosis in animals affected by heatstroke
It has been reported that the CHOP-dependent pathway can lead to apoptotic cell death. Immunohistochemical images of ileum samples showed that the expression of the CHOP, Bcl-2, and Bax proteins was changed at the ileal epithelium. As shown in Figure 2, heatstroke increased the expression of CHOP and Bax, and decreased the expression of Bcl-2. Interestingly, 4-PBA significantly increased the expression of Bcl-2 and decreased the expression of the CHOP and Bax proteins.
The TUNEL assay results, shown in Figure 3, show that there were no obvious apoptotic cells in the control group. However, a large number of apoptotic cells at the ileal epithelium were seen in the heatstroke group, and the apoptosis and tissue damage were milder in the 4-PBA group than in the heatstroke group.
Fig. 3.
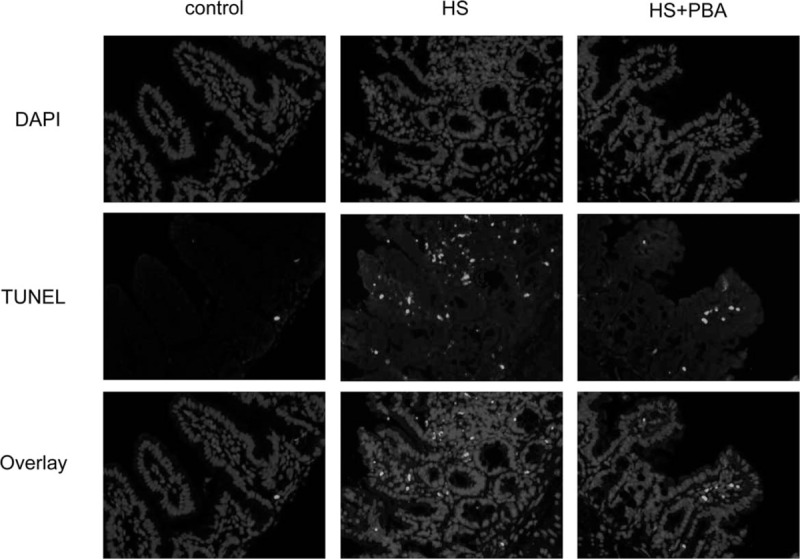
Representative photographs showing TUNEL immunofluorescence in the small intestine of mice.
Inhibition of heatstroke-induced endoplasmic reticulum stress by 4-PBA
As shown in Figure 4, the levels of ER stress-related proteins, that is, GRP78, PERK, p-eIF2α, eIF2α, ATF4, and CHOP, in the intestinal tissue were assessed by Western blot analysis. The protein levels of intestinal GRP78, PERK, p-eIF2α, ATF4, and CHOP in the heatstroke group were significantly higher than those in the control group (P < 0.01). The application of 4-PBA significantly attenuated the expression of the GRP78, PERK, p-eIF2α, ATF4, and CHOP proteins compared with that in the heatstroke group (P < 0.01).
Fig. 4.
(A) The levels of the ER stress-related proteins GRP78, PERK, p-eIF2α, eIF2α, ATF4, and CHOP were measured using the Western blot method. p-eIF2α, phosphorylated eIF2α.
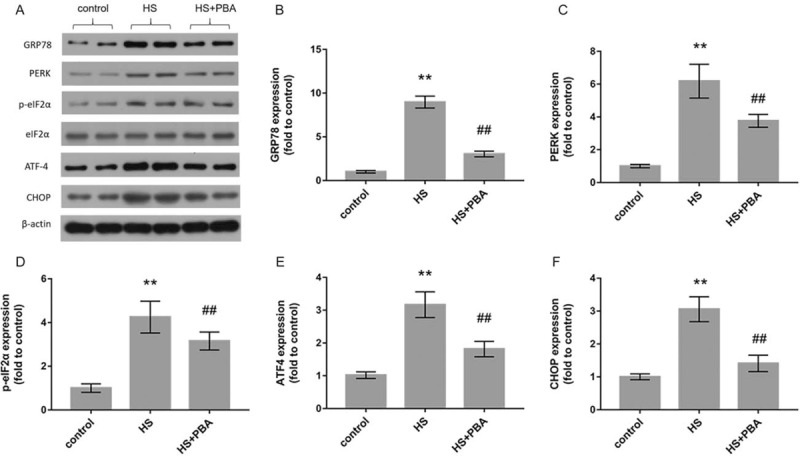
(B)–(F) Relative protein levels of GRP78, PERK, p-eIF2α, ATF4, and CHOP. The graphs show the relative band densities of the target protein to β-actin, normalized against the control group. The results are presented as mean ± SD. Significant differences are indicated as follows: ∗∗P < 0.01 and ∗P < 0.05 vs. the control group; ##P < 0.01 and #P < 0.05 vs. the heatstroke group.
PBA attenuated heatstroke-induced apoptosis by upregulation of Bcl-2 protein expression and downregulation of bax protein expression
In Figure 5, we can see that the proapoptotic protein BAX was more highly activated in the intestine of heat-exposed mice than in control group mice (P < 0.01). In contrast, the expression of the antiapoptotic protein Bcl-2 was downregulated compared with that in control group mice (P < 0.01), whereas 4-PBA attenuated heatstroke-induced apoptosis by upregulating Bcl-2 protein expression and downregulating Bax protein expression compared with that in heatstroke group mice (P < 0.01).
Fig. 5.
(A) The levels of Bax and Bcl-2 protein expression were measured by the Western blot method.
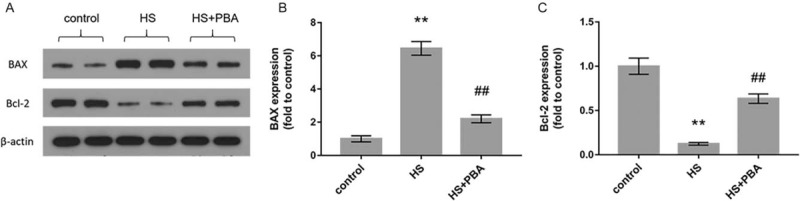
(B)–(C) The graphs show the relative band densities of BAX and Bcl-2 to β-actin, normalized against the control group. The results are presented as mean ± SD. Significant differences are indicated as follows: ∗∗P < 0.01 and ∗P < 0.05 vs. the control group; ##P < 0.01 and #P < 0.05 vs. the heatstroke group.
DISCUSSION
The most important finding in our study is that ER stress participates in the occurrence of heatstroke-induced intestinal epithelial apoptosis by the activation of the PERK/eIF2α/ATF4/CHOP unfolded protein response pathway, which subsequently upregulates the protein expression of Bax and downregulates the protein expression of Bcl-2. Inhibition of ER stress by 4-PBA ameliorated intestinal damage and reduced intestinal epithelial apoptosis, thus indicating a potential therapeutic approach for the treatment of heatstroke.
Heatstroke, defined as severe heat stress, has been reported to induce gastrointestinal dysfunction, which is thought to be of great importance in the pathophysiological process of heatstroke (23). Intact epithelial barrier function is important for maintaining intestinal homeostasis. The intestinal mucus layer is located on the outermost surface of the intestinal barrier, which participates in protection and lubrication. Goblet cells are a major secretory cell lineage in the intestinal epithelium that produces mucus. The intestinal mucosal surface area and goblet cell area are greatly reduced after heat stress (24). Tight junctions are significantly opened at high temperatures and thus increase the permeability of the intestinal epithelium (25). Heatstroke also causes small intestinal epithelial cell apoptosis and increases oxidative stress (22). Other studies have indicated that endotoxins from the gut cause an inflammatory response, fuel the progression from heat stress to heatstroke, and contribute to multiple-organ dysfunction syndrome, often causing death (26). In our study, the mouse model of heatstroke was established according to our previous report. Pathological sections showed that heatstroke induced the destruction of villi and inflammatory cell infiltration. Moreover, the pathological score was greatly increased, suggesting that serious intestinal damage is induced by heatstroke. Transmission electron micrographs revealed that heatstroke caused severe damage to the ultrastructure of the intestinal epithelium. Intriguingly, the ER stress inhibitor 4-PBA reduced intestinal congestion and neutrophil infiltration and protected important organelles such as the ER and mitochondria, suggesting that 4-PBA effectively prevents the progression of intestinal damage in heatstroke.
Thermal tissue injury due to cell death can be caused in different ways. Apoptosis and necrosis are the most described mechanisms by which different types of stress can cause cell death. Apoptosis produces a form of cell debris called apoptotic bodies, which are rapidly cleared by phagocytosis before the contents of the cell spill onto the surrounding cells and cause damage. It has been reported that apoptosis induced by hyperthermia is dependent on the dose and duration of heat stress (27). Extreme heat stress (41°C–45°C) can induce apoptosis through different mechanisms, such as the death receptor, mitochondria, and reactive oxygen species (28, 29). In our study, a large number of apoptotic cells in the intestinal epithelium were observed in the heatstroke group by TUNEL. Application of the ER stress inhibitor 4-PBA protected the intestinal epithelium against heatstroke-induced apoptosis, which reveals that ER stress may be involved in intestinal epithelial apoptosis.
As we mentioned before, ER stress can be triggered by many conditions, such as inflammatory stimulation, oxidative stress, and ischemia–reperfusion injury. Thus, ER stress is a regulated cell process that plays a crucial role in regulating a large number of physiological processes. The UPR is a double-edged sword. The primary aim of the three UPR signaling responses is to reduce the accumulation of unfolded proteins in the ER and restore cellular homeostasis. When these events are excessive or sustained, however, apoptosis, autophagy, and other types of cell death are initiated and eventually lead to disease (30). In our study, we found that intestinal damage was accompanied by ER stress. Our findings showed that the protein levels of GRP78, PERK, p-eIF2α, ATF4, and CHOP, which have been found to be involved in one of the ER stress-mediated UPR signaling pathways, were significantly increased in intestinal tissues under conditions of heat stress. In this UPR pathway, PERK activates eIF2α phosphorylation to promote the translation of ATF4, which increases the transcription of specific UPR target genes, including CHOP, and CHOP is thought to induce apoptosis (31). In addition, 4-PBA suppressed the UPR in the intestine, as evidenced by the decreased protein levels of GRP78, PERK, p-eIF2α, ATF4, and CHOP.
In addition, in the heatstroke group, BAX, which is a proapoptotic protein, was upregulated. In contrast, BCL-2, which is an antiapoptotic protein, was downregulated. The Bcl-2 family consists of a number of proteins that share Bcl-2 homology (BH) domains. The proteins in the Bcl-2 family are located in the cytoplasm and outer mitochondrial membrane (32). They are most notable for their regulation of apoptosis, a form of programmed cell death, in mitochondria (33). Under prolonged ER stress, PERK signaling can trigger cell death, and CHOP is crucial in downregulating the expression of Bcl-2, sensitising the cell to apoptosis (34). Our results are consistent with this point of view. Heat treatment activated ER stress and stimulated the proapoptotic protein Bax, whereas inhibition of the antiapoptotic protein Bcl-2 was observed. The ER stress inhibitor 4-PBA reversed this condition and alleviated intestinal epithelial apoptosis.
Some interesting phenomena were observed in recent studies, that is, ER stress was found to be involved in the regulation of NLRP3 inflammasome activation, which mediates cysteine-like proteolytic enzyme-1 (caspase-1)-dependent pyroptosis (35). Our previous study demonstrated heat stress-induced NLRP3 inflammasome activation and consequent IL-1 release and hepatocyte pyroptosis in the rat liver (36). Whether ER stress can mediate heat stress-induced pyroptosis needs further study. In addition to forming the intestinal physical barrier, intestinal epithelial cells also contribute to maintaining intestinal homeostasis by regenerating itself every 3 to 5 days. The histone deacetylase inhibitor Givinostat can promote intestinal epithelial regeneration via autocrine TGFβ1 signaling in inflammation (16). Current findings suggest that 4-PBA can also inhibit histone deacetylases (37), but whether it can promote intestinal epithelial regeneration in heatstroke requires further investigation. In addition, the ATF6 and XBP1 UPR pathways also facilitate ER stress (11), and we need to confirm whether they participate in heat stress. Further work also needs to be accomplished in studying the postheatstroke treatment strategy because we only performed 4-PBA pretreatment in mice with heatstroke, and it thus remains unknown whether postheatstroke treatment confers any protective effects.
In summary, we demonstrated for the first time that heatstroke induced apoptosis through the PERK/eIF2α/ATF4/CHOP ER stress pathway in the intestines of mice. Inhibition of ER stress by 4-PBA ameliorated intestinal injury and intestinal epithelial apoptosis. Thus, ER stress is expected to be a novel therapeutic target for heatstroke.
Footnotes
This work was supported by grants from the National Natural Science Foundation of China [No. 81571940] and PLA Logistics Research Project of China [CWH17L020, 17CXZ008, 18CXZ030].
The authors report no conflicts of interest.
REFERENCES
- 1.Glazer JL. Management of heat stroke and heat exhaustion. Am Fam Physician 71 (11):2133–2140, 2005. [PubMed] [Google Scholar]
- 2.Argaud L, Ferry T, Le QH, Marfisi A, Ciorba D, Achache P, Ducluzeau R, Robert D. Short- and long-term outcomes of heatstroke following the 2003 heat wave in Lyon, France. Arch Intern Med 167 (20):2177–2183, 2007. [DOI] [PubMed] [Google Scholar]
- 3.Bouchama A, Knochel JP. Heat stroke. N Engl J Med 346 (25):1978–1988, 2002. [DOI] [PubMed] [Google Scholar]
- 4.Lambert GP. Intestinal barrier dysfunction, endotoxemia, and gastrointestinal symptoms: the ‘canary in the coal mine’ during exercise-heat stress? Med Sport Sci 53:61–73, 2008. [DOI] [PubMed] [Google Scholar]
- 5.Yang PC, He SH, Zheng PY. Investigation into the signal transduction pathway via which heat stress impairs intestinal epithelial barrier function. J Gastroenterol Hepatol 22 (11):1823–1831, 2007. [DOI] [PubMed] [Google Scholar]
- 6.Liu Z, Sun X, Tang J, Tang Y, Tong H, Wen Q, Liu Y, Su L. Intestinal inflammation and tissue injury in response to heat stress and cooling treatment in mice. Mol Med Rep 4 (3):437–443, 2011. [DOI] [PubMed] [Google Scholar]
- 7.Gao Z, Liu F, Yin P, Wan C, He S, Liu X, Zhao H, Liu T, Xu J, Guo S. Inhibition of heat-induced apoptosis in rat small intestine and IEC-6 cells through the AKT signaling pathway. BMC Vet Res 9:241, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mohammad MK, Yang WL, Wang P. Endoplasmic reticulum stress in sepsis. Shock 44 (4):294–304, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yoshino H, Kumai Y, Kashiwakura Effects of endoplasmic reticulum stress on apoptosis induction in radioresistant macrophages. Mol Med Rep 15 (5):2867–2872, 2017. [DOI] [PubMed] [Google Scholar]
- 10.Chen JC, Wu ML, Huang KC, Lin WW. HMG-CoA reductase inhibitors activate the unfolded protein response and induce cytoprotective GRP78 expression. Cardiovasc Res 80 (1):138–150, 2008. [DOI] [PubMed] [Google Scholar]
- 11.Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol 5 (3):a013169, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13 (2):89–102, 2012. [DOI] [PubMed] [Google Scholar]
- 13.Iurlaro R, Muñoz-Pinedo C. Cell death induced by endoplasmic reticulum stress. FEBS J 283 (14):2640–2652, 2016. [DOI] [PubMed] [Google Scholar]
- 14.Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ 11 (4):381–389, 2004. [DOI] [PubMed] [Google Scholar]
- 15.Namba T, Tanaka K, Ito Y, Ishihara T, Hoshino T, Gotoh T, Endo M, Sato K, Mizushima T. Positive role of CCAAT/enhancer-binding protein homologous protein, a transcription factor involved in the endoplasmic reticulum stress response in the development of colitis. Am J Pathol 174 (5):1786–1798, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kolb PS, Ayaub EA, Zhou W, Yum V, Dickhout JG, Ask K. The therapeutic effects of 4-phenylbutyric acid in maintaining proteostasis. Int J Biochem Cell Biol 61:45–52, 2015. [DOI] [PubMed] [Google Scholar]
- 17.Liu L, Wu H, Zang J, Yang G, Zhu Y, Wu Y, Chen X, Lan D, Li T. 4-Phenylbutyric acid reveals good beneficial effects on vital organ function via anti-endoplasmic reticulum stress in septic rats. Crit Care Med 44 (8):e689–e701, 2016. [DOI] [PubMed] [Google Scholar]
- 18.Chen SH, Chang FM, Niu KC, Lin MY, Lin MT. Resuscitation from experimental heatstroke by estrogen therapy. Crit Care Med 34 (4):1113–1118, 2006. [DOI] [PubMed] [Google Scholar]
- 19.Kusama H, Kon K, Ikejima K, Arai K, Aoyama T, Uchiyama A, Yamashina S, Watanabe S. Sodium 4-phenylbutyric acid prevents murine acetaminophen hepatotoxicity by minimizing endoplasmic reticulum stress. J Gastroenterol 52 (5):611–622, 2017. [DOI] [PubMed] [Google Scholar]
- 20.Ji J, Gu Z, Li H, Su L, Liu Z. Cryptdin-2 predicts intestinal injury during heatstroke in mice. Int J Mol Med 41 (1):137–146, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiu CJ, McArdle AH, Brown R, Scott HJ, Gurd FN. Intestinal mucosal lesion in low-flow states. I. A morphological, hemodynamic, and metabolic reappraisal. Arch Surg 101 (4):478–483, 1970. [DOI] [PubMed] [Google Scholar]
- 22.Yu J, Liu F, Yin P, Zhao H, Luan W, Hou X, Zhong Y, Jia D, Zan J, Ma W, et al. Involvement of oxidative stress and mitogen-activated protein kinase signaling pathways in heat stress-induced injury in the rat small intestine. Stress 16 (1):99–113, 2013. [DOI] [PubMed] [Google Scholar]
- 23.Yang PC, He SH, Zheng PY. Investigation into the signal transduction pathway via which heat stress impairs intestinal epithelial barrier function. J Gastroenterol Hepatol 4 (22):1823–1831, 2007. [DOI] [PubMed] [Google Scholar]
- 24.Abuajamieh M, Kvidera SK, Mayorga EJ, Kaiser A, Lei S, Seibert JT, Horst EA, Sanz Fernandez MV, Ross JW, Selsby JT, et al. The effect of recovery from heat stress on circulating bioenergetics and inflammatory biomarkers. J Anim Sci 96 (11):4599–4610, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiao G, Yuan F, Geng Y, Qiu X, Liu Z, Lu J, Tang L, Zhang Y, Su L. Eicosapentaenoic acid enhances heatstroke-impaired intestinal epithelial barrier function in rats. Shock 44 (4):348–356, 2015. [DOI] [PubMed] [Google Scholar]
- 26.Hall DM, Buettner GR, Oberley LW, Xu L, Matthes RD, Gisolfi CV. Mechanisms of circulatory and intestinal barrier dysfunction during whole body hyperthermia. Am J Physiol Heart Circ Physiol 280 (2):H509–H521, 2001. [DOI] [PubMed] [Google Scholar]
- 27.Li L, Tan HP, Gu ZT, Liu ZF, Geng Y, Liu YS, Tong HS, Tang YQ, Qiu JM, Su L. Heat stress induces apoptosis through a Ca2+-mediated mitochondrial apoptotic pathway in human umbilical vein endothelial cells. PLoS One 9 (12):e111083, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tran SE, Meinander A, Holmström TH, Rivero-Müller A, Heiskanen KM, Linnau EK, Courtney MJ, Mosser DD, Sistonen L, Eriksson JE. Heat stress downregulates FLIP and sensitizes cells to Fas receptor-mediated apoptosis. Cell Death Differ 10 (10):1137–1147, 2003. [DOI] [PubMed] [Google Scholar]
- 29.Slimen IB, Najar T, Ghram A, Dabbebi H, Mrad MB, Abdrabbah M. Reactive oxygen species, heat stress and oxidative induced mitochondrial damage. A review. Int J Hyperthermia 30 (7):513–523, 2014. [DOI] [PubMed] [Google Scholar]
- 30.Senft D, Ronai ZA. UPR, autophagy, and mitochondria crosstalk underlies the ER stress response. Trends Biochem Sci 40 (3):141–148, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Han J, Back SH, Hur J, Lin YH, Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M, et al. ER-stress-induced transcriptional regulation increases protein synthesis leading to cell death. Nat Cell Biol 15 (5):481–490, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hatok J, Racay P. Bcl-2 family proteins: master regulators of cell suvival. Biomol Concepts 7 (4):259–270, 2016. [DOI] [PubMed] [Google Scholar]
- 33.Logue SE, Cleary P, Saveljeva S, Samali A. New direction in ER stress-induced cell death. Apoptosis 18 (5):537–546, 2013. [DOI] [PubMed] [Google Scholar]
- 34.McCullough KD, Martindale JL, Klotz LO, Aw TY, Holbrook NJ. Gadd153 sensitizes cells to endoplasmic reticulum stress by down-regulating Bcl2 and perturbing the cellular redox state. Mol Cell Biol 21 (4):1249–1259, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lebeaupin C, Proics E, de Bieville CH, Rousseau D, Bonnafous S, Patouraux S, Adam G, Lavallard VJ, Rovere C, Le Thuc O, et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis 10 (6):e1879, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geng Y, Ma Q, Liu YN, Peng N, Yuan FF, Li XG, Li M, Wu YS, Li BL, Song WB, et al. Heatstroke induces liver injury via IL-1β and HMGB1-induced pyroptosis. J Hepatol 63 (3):622–633, 2015. [DOI] [PubMed] [Google Scholar]
- 37.Friedrich M, Gerbeth L, Gerling M, Rosenthal R, Steiger K, Weidinger C, Keye J, Wu H, Schmidt F, Weichert W, et al. HDAC inhibitors promote intestinal epithelial regeneration via autocrine TGFβ1 signalling in inflammation. Mucosal Immunol 12 (3):656–667, 2019. [DOI] [PubMed] [Google Scholar]