

Abstract
The ability of enzymes, including ribozymes, to catalyze side reactions is believed to be essential to the evolution of novel biochemical activities. It has been speculated that the earliest ribozymes, whose emergence marked the origin of life, were low in activity but high in promiscuity, and that these early ribozymes gave rise to specialized descendants with higher activity and specificity. Here, we review the concepts related to promiscuity and examine several cases of highly promiscuous ribozymes. We consider the evidence bearing on the question of whether de novo ribozymes would be quantitatively more promiscuous than later evolved ribozymes or protein enzymes. We suggest that while de novo ribozymes appear to be promiscuous in general, they are not obviously more promiscuous than more highly evolved or active sequences. Promiscuity is a trait whose value would depend on selective pressures, even during prebiotic evolution.
1. Introduction
Catalytic RNA sequences, or ribozymes, are widely accepted to have been central to the origin of life.1,2 Their dual capacity for information storage and catalytic activity is the basis for the RNA world theory,3−5 that an RNA-based metabolism could have preceded the more complex DNA-RNA-protein system that we observe in biology today. Regardless of whether an RNA world existed on early Earth, ribozymes represent an excellent laboratory model system for molecular evolution. Beginning with a pool of random sequences, strategies can be devised to select for particular activities. Cycles of selection and amplification by PCR allow enrichment and eventually isolation of active sequences. A prerequisite of successful in vitro evolution is the presence of one or more molecules with some activity, however slight, in the initial pool or early rounds. Once this kernel of activity exists, the active sequences can be selected and activity possibly improved by mutation during the evolutionary process. In addition to developing new ribozymes, in vitro evolution of RNA allows well-controlled experiments to observe and analyze the de novo emergence of biochemical functions.6−8
Promiscuous catalytic activities have been invoked as being particularly significant for the origin of enzymes,9,10 as one might intuit that early, simple ribozymes or enzymes would have little specificity and therefore might catalyze many reactions, albeit with slow rates. These sequences might possess kernels of activity for many different substrates or reactions. One landmark study of such a ribozyme is a sequence which was engineered to adopt two possible folds, one of which acted as a ligase and one of which acted as a self-cleaving ribozyme.11 This sequence had very low activity for each function, but a relatively small number of mutations would increase function to near wild-type in both directions. Such promiscuity would promote evolutionary innovation by poising sequences at a nonzero fitness for multiple activities, each of which could be potentially optimized by natural selection. This idea also raises the interesting question of whether ribozymes are intrinsically more promiscuous than protein enzymes. From extensive work on the directed evolution of enzymes, it has become clear that much of the success of the field as a whole is due to the presence of low levels of apparently promiscuous activity in extant protein enzymes. This surprising degree of promiscuity in highly evolved enzymes suggests that promiscuity is actually the rule rather than an exception for protein enzymes.
Here, we review what is known about the specificity and promiscuity of ribozymes. We first introduce major concepts and definitions in specificity and promiscuity, including how to quantify them, which were originally developed in the enzyme literature. An interesting concept is the relationship between activity and specificity, which underlies the intuition that early, relatively low-activity ribozymes would be more promiscuous. We then review several cases of ribozymes in which studies have demonstrated promiscuity in some way. When possible, we calculate a promiscuity index from what is known about these ribozymes, a first step toward rigorous comparisons of the promiscuity of ribozymes and protein enzymes. We end with a discussion of the implications of these comparisons for the hypothesis that early ribozymes were particularly promiscuous.
2. Promiscuity and Specificity: Concepts and Definitions
2.1. Defining Specificity
Specificity is the ability of an enzyme to discriminate between two different substrates assuming both are present. The question of how to measure enzyme specificity has been a matter of debate in the past (see refs (12 and 13) and references therein), but it is generally agreed that specificity in the presence of two different substrates should be compared based on the discrimination factor,14 defined as the ratio of the catalytic efficiencies (kcat/KM) for the corresponding reactions. According to transition-state theory, the logarithm of the catalytic efficiency (kcat/KM) is proportional to the free energy difference between the free enzyme and substrate vs the transition-state complex (ΔG⧧).15 When comparing the cognate with an alternate substrate, the discrimination factor is also called the accuracy A. Thus, A is exponentially dependent on the difference ΔΔG⧧ between the cognate and alternate substrates.
In some cases (e.g., polymerases), the use of an error ratio (the rate of incorrect product formation divided by the rate of correct product formation) is more appropriate. To gain an intuition about the possible scale of this ratio, one may note that the theoretical maximum discrimination between alternative substrates undergoing analogous reactions occurs when the formation of the enzyme–substrate complex is much faster than product conversion and release (as assumed in Michaelis–Menten kinetics). In this case, the theoretical minimum error ratio is equal to the ratio of KM values.16,17
2.2. General Mechanisms for Specificity and the Possible Trade-off with Rate
Discrimination among substrates can arise from different affinities in the initial enzyme–substrate complexes (ground-state discrimination) or in the transition-state complexes (catalytic or transition-state discrimination).18 The accuracy (A) for a cognate vs alternative substrate can be increased by three scenarios: (a) higher rate of substrate association (ground-state discrimination with koncog > konalt), (b) lower rate of substrate dissociation (ground-state discrimination with koffcog < koffalt), or (c) higher rate of conversion of the enzyme–substrate complex into the transition state (transition-state discrimination with kcatcog > kcatalt).
In ground-state discrimination, lowering the energy of the enzyme–substrate complex has two effects, namely decreasing KM as well as decreasing kcat. In other words, although selectivity may be improved via increased substrate affinity, the reaction rate suffers. Examples of enzymes exhibiting ground state discrimination include DNA methyltransferases and the ribosome.18 The trade-off between accuracy and rate might impose an evolutionary constraint limiting selectivity.18 Indeed, selection for activity on one substrate does not seem to induce high selectivity by itself,19 and therefore, negative selection against undesired substrates is used when engineering new enzymes.20,21 Interestingly, a trade-off between rate and accuracy created by ground-state discrimination would contradict the idea that early, less optimized ribozymes or enzymes were more promiscuous.
On the other hand, in transition-state discrimination, which tends to apply to relatively small substrates (e.g., DNA polymerases18,22−25), lowering the activation barrier increases kcat without necessarily affecting KM. Thus, in principle, transition-state discrimination might achieve higher selectivity at high activity because there is not necessarily a trade-off between accuracy and rate. In addition, nonequilibrium mechanisms driven by release of chemical energy may improve selectivity with or without a trade-off between accuracy and rate.26 Furthermore, such mechanisms can allow accuracy to surpass the theoretical thermodynamic limit based on binding energies. For example, in kinetic proofreading,16,27 discrimination between two possible substrates is achieved by the presence of one or more irreversible steps in the reaction pathway, whose rate(s) are biased by the identity of the substrate. These steps are made irreversible by consumption of chemical energy, and concatenation of such steps could be used to achieve arbitrarily small error ratios, in principle. Some biological processes can afford high specificity by using this mechanism.16 For example, although the valine concentration in vivo is ∼5-fold higher than that of isoleucine, and isoleucyl-tRNA synthetase favors the reaction with isoleucine over valine by only ∼100-fold, the rapid hydrolysis of misincorporated valine-tRNA decreases the error ratio to 1 in 3000. While kinetic proofreading can increase reaction specificity substantially, this comes with a relatively high energetic cost.18,28
However, in the absence of proofreading mechanisms, substrate specificity is inherently limited due to physicochemical reasons. Indeed, a recent survey of the BRENDA database (The Comprehensive Enzyme Information System29) suggests discrimination is usually much lower than the theoretical maximum.14 In the case of substrates differing by a single methyl group, discrimination was found to be lower than the theoretical maximum, for 23 out of the 25 enzymes surveyed, by typically 1–2 orders of magnitude. Interestingly, a similar discrepancy is found in nonenzymatic, template-directed polymerization of activated nucleotides,17 suggesting that this phenomenon is not specific to enzymes. A discrimination level lower than the expected theoretical maximum might reflect prioritization of increased rate during evolution, if the enzyme is subject to an accuracy-rate trade-off; in other words, the marginal fitness benefit of increased specificity may come with a larger fitness decrement due to slower rate. Thus, in general, specificity tends to be lower than the theoretical maximum, possibly because of the costs associated with accuracy.18
Specificity may appear to be quite suboptimal even for presumably highly evolved enzymes. For example, the carboxylase enzyme Rubisco plays an essential role in fixing atmospheric carbon dioxide into sugars during photosynthesis. However, considering its biomass and critical role, it is surprisingly slow and nonspecific, as oxygenation constitutes a major side reaction. Trade-off models have been proposed to explain the observed correlations between specificity and other kinetic parameters,30,31 which were recently revisited using an extended data set.32 A strong correlation was found between the catalytic efficiencies for carboxylation and oxygenation, indicating that lowering the effective CO2 addition energy barrier (i.e., faster carboxylation) entails a similar reduction in the effective O2 addition energy barrier (i.e., faster oxygenation). Therefore, the accuracy of Rubisco appears to be highly constrained.
2.3. Promiscuous vs Multispecific Enzymes
The term “catalytic promiscuity” was originally used to refer to enzymes known to catalyze more than one type of reaction.9,33 However, in practice, “promiscuity” has not been well-defined and thus has been used to refer to fundamentally different phenomena.14,34 Generally, catalytic promiscuity refers to the capability of enzymes to catalyze reactions mechanistically different from the primary biological reaction,33 and substrate promiscuity refers to the capability of enzymes to transform different substrates.35 These terms warrant additional consideration here, as their usage varies and can depend on incomplete knowledge.
The native function of an enzyme refers to the physiologically relevant chemical transformation and substrate for which an enzyme has evolved. Native function is selected for and contributes to organismal fitness. In this context, any physiological functions for which an enzyme has evolved are considered native, even if they are not the enzyme’s primary function. For example, while the primary function of aminoacyl-tRNA synthetases is to catalyze the attachment of tRNAs to their respective amino acids, some also catalyze generation of 5′,5′-diadenosine tetraphosphate in a reaction that appears to be physiologically relevant,36 and thus this additional function would be considered native. In practice, whether a particular function contributes to organismal fitness may be difficult to assess.
It is nowadays well-accepted that many, if not most, enzymes have multiple side activities.10,37,38 However, such side activities may or may not be a product of evolution. In the evolutionary biochemistry literature, promiscuity refers to side activities that are non-native (i.e., not evolved), so the alternative transformation or substrate is fortuitous. By definition, there is no evolutionary pressure on non-native activities, as they do not impact organismal fitness (e.g., alternative substrates are not available in the cell).37,39,40 For biologically evolved enzymes, promiscuity (as defined by evolutionary biochemists) is nearly impossible to ascertain in practice because we do not know what past environments and selective pressures might have applied to the protein. If promiscuity of a naturally evolved enzyme is suspected, we suggest use of the term apparent promiscuity, in contrast to true promiscuity, to acknowledge this uncertainty.
Interestingly, there are two special scenarios in which true promiscuity can indeed be characterized unequivocally. First, if the enzyme transforms a man-made compound not present in nature, the enzyme could not have evolved this activity, and the activity must be non-native. Examples are the atrazine chlorohydrolase and melamine deaminase enzymes, which degrade the man-made compounds atrazine and melamine, respectively. Despite very high similarity (98% identity), both enzymes show little activity on the alternative substrate.12 While it is likely that their host strains evolved the atrazine or melamine degradation function in response to environmental exposure (both strains were isolated from areas contaminated by the substrate12,41), it may be presumed that neither strain experienced both contaminants simultaneously. If so, these enzymes can be considered as lacking in true promiscuity.42,43 The second scenario in which true promiscuity might be determined is in the case of in vitro evolved enzymes and ribozymes, in which the different environments and selective pressures applied to the sequences are known.
An important contrast to promiscuous enzymes, whose side reactions are non-native, is multispecific enzymes (or broad-specificity enzymes), which evolved to perform many native transformations, such as on a broad range of available substrates. These enzymes are characterized by small accuracy values, with different substrates having similar kcat/KM. For example, theta class glutathione transferases from various species can catalyze the conjugation of the tripeptide glutathione to a variety of electrophilic substrates.44 The enzyme family of cytochromes P450 metabolizes a variety of different substrates, with activities including biosynthesis of steroids, fatty acids, or fat-soluble vitamins as well as the degradation of herbicides and insecticides. In particular, cytochrome P450 3A4 contributes to the metabolism of approximately 50% of marketed drugs.45−47 Most of the terpene cyclase enzymes (a.k.a. terpene synthases) are also multispecific. For example, the class I sesquiterpene cyclase γ-humulene synthase generates 52 different products, of which γ-humulene constitutes less than 30% in abundance.48,49 Methane monooxygenase oxidizes more than 150 different substrates.50 The RecBCD nuclease, originally named Exonuclease V, accepts both linear double-stranded DNA and single-stranded DNA with very low specificity.51,52 The distinction drawn between promiscuous and multispecific enzymes hinges on whether the additional substrates represented selective pressures on the enzyme. While this is an important conceptual distinction, assessing whether an enzyme is promiscuous vs multispecific may be difficult in practice due to lack of knowledge of the evolutionary and environmental history of the enzyme.
It should be noted that additional usages of the term “promiscuous” also exist. Promiscuity is sometimes used to refer to the capacity of an enzyme to transform different physiologically relevant substrates (see ref (14) and references therein), to be contrasted with “multifunctional enzymes” whose side activities may be either physiologically useful or detrimental.53 Unfortunately, this definition of promiscuity can be contradictory to the one given earlier, in which promiscuity refers to the capacity to perform non-native reactions. In addition, the determination of physiological relevance is difficult to make and again raises questions of evolutionary history. A third usage of the term promiscuity refers to enzymes whose catalytic domain executes multiple functions.53−56 In this review, we have favored the definition from the evolutionary biochemistry literature, because ribozymes are often evolved under known conditions in vitro, allowing true promiscuity to be characterized, while physiological relevance is unspecified and multiple domains are relatively uncommon.
2.4. Promiscuity and Evolutionary Innovation
Fortuitous side reactions of a promiscuous enzyme are believed to be central to evolutionary innovation, as an initial kernel of activity for a side reaction is a starting point for optimization of the new activity by evolution.9 In addition, an enzyme might exhibit new side activities under new environmental conditions (e.g., temperature, pH), and such enzymes are called condition-promiscuous enzymes. Conditional promiscuity is a possible path for adaptation in new environments.57−59 However, in the absence of selective pressure, side activities would be subject to neutral drift and may be lost if they are uncorrelated to native functions of the enzyme.
Interestingly, contrary to native functions, which are usually tolerant of mutations, directed evolution of enzymes has shown that the non-native functions can be greatly optimized by just a small number of mutations.19,55 The flip side of this so-called plasticity is that newly evolved non-native functions are typically not tolerant to mutations. One might therefore suspect that evolutionary robustness, if it is observed for native functions, likely evolved as a trait or correlate of a selected trait. In addition, the duality of plasticity for non-native functions and robustness for native functions implies that evolutionary optimization of non-native functions might not always lead to a significant decrease of the original native function. However, in the absence of continued selection pressure on the original function, specialization has been shown to occur due to trade-offs during selection of the secondary function, even without negative selection against the original function.60
The idea that enzyme evolution and promiscuity are connected goes back to the mid-1970s, when Yčas and Jensen proposed, independently, the first model for enzyme evolution.9,61 This general model hypothesizes that primitive life had minimal gene content and the number of available enzymes was limited. It posits that primordial enzymes may have been less specific, being able to catalyze broad classes of reactions on a variety of substrates. Gene duplication and mutation would have then increased genetic diversity, leading to the divergence of new enzymes whose secondary activities might give an evolutionary advantage under newly encountered selective pressures.62,63 This hypothesis is widely accepted, although direct evidence is scant.14,55 The molecular processes and evolutionary forces involved in the biological evolution of enzymes are very difficult to reconstruct, and hence the mechanisms under which duplication and specialization events shape enzyme evolution have been the subject of much debate.64 Nevertheless, understanding how specificity and promiscuity arise during in vitro selection and evolution of ribozymes, recapitulating an origin of life, can address this problem experimentally.
2.5. Promiscuity and the Fitness Landscape
The fitness landscape is a well-studied conceptualization of evolution through the space of all possible sequences (sequence space).65,66 Each point of sequence space is specified by a sequence and its associated fitness (e.g., activity on a given substrate), giving the fitness “landscape” in sequence space. At each point in sequence space, one might also imagine the large chemical space of possible substrates, and an activity profile for that sequence over substrate space, which reflects the promiscuity of the sequence.
Properties of the fitness landscape are not necessarily expected to correlate with properties of the promiscuity profile. Different fitness landscapes over sequence space can give rise to the same promiscuity profile in substrate space (Figure 1). In general, optimization for higher activity need not correspond to increased specificity. However, specific mechanisms, such as a trade-off between rate and specificity, could produce correlations between the fitness landscape and the associated promiscuity profiles. The idea that early, nonoptimized ribozymes were particularly promiscuous would translate into a correlation in which highly active sequences on the fitness landscape have lower promiscuity compared to less active sequences. Whether the specificity of a ribozyme can be improved through mutation or evolution would depend not only on the specificity of that individual ribozyme, but also on the fitness landscapes for the cognate and alternative substrates (Figure 1).
Figure 1.

Fitness landscapes and promiscuity profiles. Different fitness landscapes (A–D) can correspond to the same promiscuity profile (E–H). Vertical dashed lines (a–g) correspond to different ribozyme sequences. Ribozyme fitness landscapes (A–D) for two substrates may differ (blue and red) with or without overlap. The promiscuity profile (E–H), depicted here for two substrates (1: blue and 2: red) depends on the sequence tested, as seen in the comparison among sequences a–c in panel E. In addition, similar promiscuity profiles can be derived from qualitatively different fitness landscapes. Compare sequence a from (A, E) with sequence d from (B, F), sequence b from (A, E) with sequence e from (C, G), and sequence c from (A, E) with sequence g from (D, H). While ribozymes a and d have similar promiscuity profiles, their evolutionary potential is strikingly different. Ribozyme a could evolve through mutations to specialized activity, but ribozyme d is already at a local maximum and has no evolutionary potential for increasing activity. Similarly, ribozymes b and e have the same promiscuity profile, but only ribozyme b has the possibility to evolve into a sequence of higher activity and selectivity. Ribozymes c, f, and g are highly specific, but unlike ribozymes f and g, ribozyme c has increased potential to evolve into a promiscuous ribozyme.
2.6. Quantifying Promiscuity: The Promiscuity Index
Several possible methods exist to quantify substrate specificity. Here, we describe the promiscuity index (I) proposed by Nath and Atkins, which is a metric similar to a normalized information entropy67 (eq 1):
| 1 |
where N is the number of substrates that can be transformed and ei corresponds to their individual associated catalytic efficiencies. Due to the normalization, this metric goes from 0 (only uses one substrate) to 1 (equally efficient on all N substrates).
While this promiscuity index is simple and intuitive, it might be strongly influenced by the experimenter’s choice of substrates to test. In particular, when comparing promiscuity indices for different ribozymes or enzymes, one sequence might appear more promiscuous only because many chemically similar substrates were assayed. To account for this problem, a weighted promiscuity index (J) factoring in substrate similarity can be calculated (eq 2).67 Chemical similarity can be calculated using a bitwise dissimilarity metric between a pair of substrates (δ), which is based on the presence or absence of a number of different functional groups.
| 2 |
Any method for quantifying promiscuity from experimental data is likely to be biased in at least two ways. First, there is an experimental bias in the selection of substrates (e.g., synthetically accessible, similar to known substrates). For comparisons among enzymes, differences in these biases might affect the promiscuity index calculated, even when using the weighted value. Second, these metrics do not consider the chemical context in which an enzyme functions. If the environment never provides a certain substrate, it may not be justifiable to include such a substrate in the calculation even if the enzyme has nonzero activity on it in vitro. Additionally, the relationship between chemical similarity and promiscuity has not been well-established, and often little difference between unweighted (I) and weighted (J) values has been observed.67,68 Other metrics for promiscuity also exist, such as a measure based on structural information on the catalytic residues.69,70
Despite these limitations, the promiscuity index serves as a starting point for characterization and comparison of substrate specificities. In this review, we calculate promiscuity indices for ribozymes for which sufficient data is available in the literature.71,72 However, when necessary, catalytic rates were used in place of catalytic efficiency when the catalytic efficiency was inappropriate or unknown. Quantitative metrics like the promiscuity index provide the opportunity to compare the specificity of different molecules and potentially study the relationship between promiscuity and other measurable characteristics, such as activity.
3. Ribozymes Illustrating Promiscuity
In this section, we first describe substrate promiscuity using aminoacylation ribozymes, for which different substrates have been studied in some depth. Then, to gain mechanistic insight into a specific case, we turn to the hammerhead ribozyme, where specificity can be understood in terms of RNA annealing. An important consequence of this mechanism is that promiscuity is dependent on environmental conditions, such as temperature. The expression of promiscuity under new conditions (conditional promiscuity) is a possible mechanism for uncovering latent side activities. We then follow a series of in vitro evolution experiments seeking an RNA replicase, in which the presumption of promiscuous activity was essential to the design and success of the experiments. We next describe a different kind of promiscuity, catalytic promiscuity, in a case of a nucleotide synthase ribozyme that unexpectedly possesses two distinct catalytic mechanisms. Then, we end this section with a brief discussion of the ribosome, a proteinaceous ribozyme whose promiscuity appears to be unparalleled.
3.1. Substrate Promiscuity: Aminoacylation Ribozymes
Aminoacylation of tRNA is a key step in protein synthesis, and high selectivity for tRNA-amino acid pairs is crucial for the stability of the genetic code.73 It is presumed that ribozymes carried out aminoacylation reactions in the earliest stages of the evolution of the translation apparatus. Indeed, several aminoacyl-RNA synthase ribozymes have been identified through in vitro selection, which use a variety of activated amino acid substrates74−77 (Figure 2). These aminoacylating ribozymes show a range of specificities for the substrate side chain. For example, selection using a phenylalanine adenylate substrate 1 produced ribozymes that showed little discrimination (i.e., promiscuous ribozymes) as well as ribozymes showing a strong preference for aromatic amino acids.78 Although they are derived from the same selection, these ribozymes have quite different promiscuity profiles and indices (Figure 3, Table 1). Selection for aminoacylation with coenzyme A (CoA) thioester 2 produced ribozymes that could function with other CoA thioesters, but required the presence of a free α-amino group.76 None of these ribozymes match the specificity of the aminoacyl-tRNA synthetase enzymes found in modern biochemistry. This discrepancy cannot be the result of a trade-off between activity and specificity, because the ribozymes are generally much less efficient (∼1000-fold) than the corresponding enzymes.79,80 Instead, the general finding of promiscuity and the variation of specificities found among these ribozymes are consistent with the understanding that newly evolved sequences are not necessarily specific if they have not been selected for specificity.
Figure 2.

Substrates for aminoacylation ribozymes. Phenylalanyl-adenosine monophosphate74,78 (1), biocytinCoA76 (2), biotinyl-Tyr(Me)-oxazolone77 (3), amino acid cyanomethyl ester,90 (4) and amino acid 3,5-dinitrobenzyl ester90 (5). Substrates 4 and 5 are flexizyme substrates. The amino acid backbone is depicted in green; side chains are depicted in blue, and leaving groups are depicted in red. R indicates possible chemical variation in the side chain.
Figure 3.
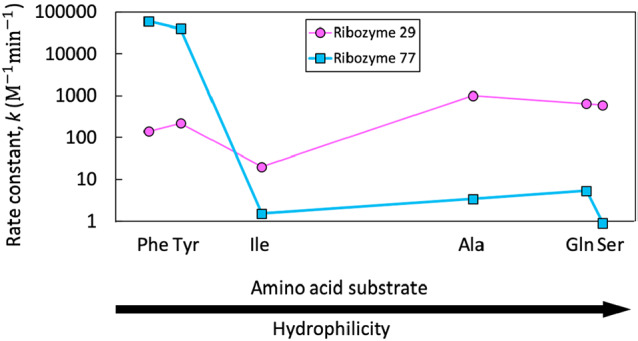
Promiscuity profiles for two aminoacylation ribozymes. Promiscuity profiles for ribozyme 77 (blue squares) and ribozyme 29 (pink circles) show catalytic rates for each tested amino acid substrate,78 ordered by hydrophilicity as defined by Hopp and Woods91,92 (Phe = −2.5, Tyr = −2.3, Ile = −1.8, Ala = −0.5, Gln = 0.2, and Ser = 0.3). Also see Table 1.
Table 1. Promiscuity Indices Calculated for Two Aminoacylation Ribozymes (77 and 29).
| substrate side chaina | CIDa | k (M–1 min–1)b | Ic | Jc |
|---|---|---|---|---|
| ribozyme 77 | ||||
| phenylalanine | 6140 | 60 000 | 0.376 | 0.439 |
| tyrosine | 6057 | 40 000 | ||
| isoleucine | 6306 | 1.5 | ||
| alanine | 5950 | 3.4 | ||
| glutamine | 5961 | 5.3 | ||
| serine | 5951 | 0.9 | ||
| ribozyme 29 | ||||
| phenylalanine | 6140 | 140 | 0.810 | 0.807 |
| tyrosine | 6057 | 220 | ||
| isoleucine | 6306 | 20 | ||
| alanine | 5950 | 1000 | ||
| glutamine | 5961 | 650 | ||
| serine | 5951 | 600 | ||
Amino acids (CID: PubChem Compound Identifier) were used to determine similarities for calculation of J.67,71,71 The substrates used are aminoacyl adenylates with the side chain indicated.
Rate constants are from Illangasekare et al.78
Both the unweighted (I) and weighted (J) promiscuity indices were calculated from the rate constants (k) shown.
The aminoacylating ribozymes discussed above have also been observed to catalyze reactions using alternative nucleophilic substrates to generate amide bonds in addition to esterification. In particular, a minimized, 29 nucleotide version of an aminoacylating ribozyme that utilizes Phe-AMP was found to catalyze successive reactions: aminoacylation of the RNA and the subsequent amide bond formation to generate a conjugated peptide.81,82 The rate of peptide formation was approximately 13-fold less than that for aminoacylation, but this difference could be tuned. Extending the 3′ tail of the RNA by three nucleotides resulted in a 3-fold reduction in the rate of aminoacylation and a 2-fold increase in the rate of peptide formation, presumably by increasing the flexibility around the active site.
The potential promiscuity of aminoacylating ribozymes is highlighted by the “flexizymes” developed by Suga and colleagues, so named for their flexibility in accommodating a variety of substrates. These ribozymes were generated over a series of selections with the ultimate goal of producing catalysts capable of charging tRNAs with a wide variety of both natural and non-natural substrates. The starting point of the selection was a library containing a 5′ random region and a 3′ tRNA. Ribozymes were selected for their ability to aminoacylate the 3′ terminus of the conjugated tRNA. This first selection produced ribozymes with a high level of specificity to both the tRNA and phenylalanine substrates.75,83,84 To broaden the tRNA substrate range, further design and selection was performed with an alternative tRNA sequence, which resulted in ribozymes capable of accepting a variety of tRNAs.85 These early flexizymes exhibited high affinity for the aromatic side chains. Using the ribozyme’s affinity to the aromatic group to broaden the side chain specificity, the initial substrate 4 was redesigned to substrate 5, which contains a 3,5-dinitrobenzyl ester as the leaving group in the aminoacylation reaction. The idea was that this leaving group could be kept constant, ensuring affinity to the ribozyme, while the side chain itself was varied. This substrate necessitated an altered reaction mechanism, but nevertheless, the strategy was successful, with further selection resulting in ribozymes capable of charging tRNAs without regard to amino acid side chain.85 More recently, flexizymes have been used to charge tRNAs with various non-natural amino acids, including d-amino acids, β-amino acids, and α-hydroxy acids, and 3′-aminoacyl-NH-tRNA can also be charged.86−89 Although the flexizyme does exhibit a minor degree of side chain specificity, yields with the non-natural analogues often rival those for the l-amino acids used in the initial selections. The additional substrates represent both promiscuous (non-native) as well as native activities. Overall, the flexizyme demonstrates the surprisingly broad substrate specificity that can be evolved and designed when substrate generality is a desired goal.
3.2. Conditional Promiscuity: The Hammerhead Ribozyme
Due to their historical importance in the discovery of ribozymes, much is known about the self-cleaving ribozymes, which function through general acid–base catalysis.175 The ribozyme fold brings the reactant nucleotides to the vicinity of the cleavage site, with the catalytic strand acting as the general base or acid to activate the nucleophile or stabilize the leaving group, respectively (Figure 4). Many of these ribozymes can also catalyze the same transesterification reaction in reverse, using nucleophilic attack from a 5′-hydroxyl to ligate two substrate strands,93 which represents a possible case of catalytic promiscuity, a phenomenon discussed in Section 3.5 in the context of a different ribozyme. In this section, we focus on the substrate promiscuity of a self-cleaving ribozyme and how it arises. Although these ribozymes are cis-acting in vivo, they can be engineered to accept oligonucleotide substrates in trans with multiple turnover. While there are numerous self-cleaving ribozymes, here we confine our discussion to the case of the hammerhead ribozyme, a naturally occurring ribozyme found in plant viroid transcripts,94 for which the specificity of trans-acting variants has been extensively investigated.
Figure 4.

Proposed mechanism of RNA self-scission by general acid–base catalysis. A general base promotes deprotonation of the 2′-hydroxyl of the nucleophile, initiating formation of the cyclic intermediate. A general acid stabilizes the 5′-hydroxyl leaving group, allowing resolution of the intermediate to generate the cleavage products.
The trans-acting hammerhead ribozyme can be engineered from the cis-acting ancestor by removing a nucleotide loop of one helical arm, thereby creating a cleavable substrate strand and a catalytic strand.95−99 In such constructs, the catalytic strand can bind and cleave substrate strands with multiple turnover. In particular, separation of stem I from stem III of the ribozyme (the I/III construct) is most widely studied100−102 because this construction places most of the conserved nucleotides in the catalytic strand (Figure 5). This allows substrate specificity in the substrate strand to be probed.
Figure 5.
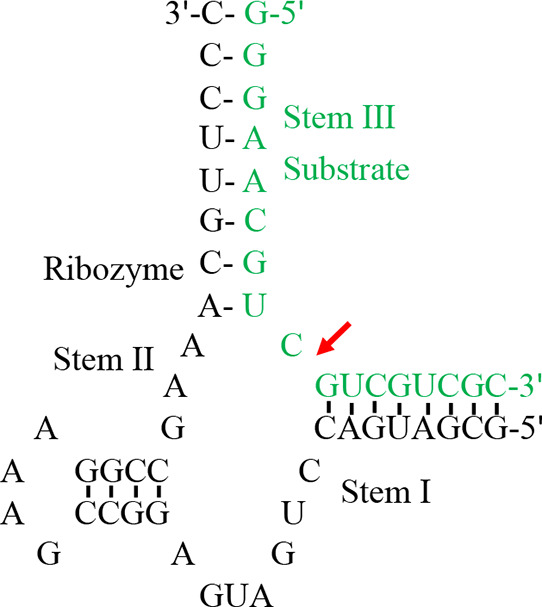
Structure of a trans-acting I/III hammerhead ribozyme HH16. The catalytic strand is shown in black, and substrate is shown in green.103 The red arrow indicates the cleavage site.
There are two main expectations for sequence specificity of the substrate in a trans-acting construct. First, residues critical for the catalytic mechanism are expected to be relatively intolerant to mutations, which would primarily affect kcat.104 Second, aside from critical residues, promiscuity for the substrate is expected to be determined by binding interactions (Km) between enzyme and substrate, namely base-pairing, which can lead to large variation in dissociation rates among different substrates. In the HH16 ribozyme (Figure 5), substrate affinity, which was dominated by stem III, was very high, implying a low dissociation rate, such that truncation from the 3′ end, down to a 2-nucleotide version of stem I, had little effect on the overall rate of cleavage.103 Specificity in either stem I or III of the substrate was therefore only observed when stem III was destabilized to give a dissociation rate that was on par with or slower than the overall cleavage rate. Conversely, extending the recognition sequence reduced specificity,105 in keeping with the idea that, if substrate dissociation is slow relative to cleavage, mutations in the substrate are tolerated because the bound complex is sufficiently populated. In terms of the active site itself, the hammerhead ribozyme has limited substrate promiscuity; substitution of the reactive phosphate with thiophosphate greatly reduces kcat.106
The example of the hammerhead ribozyme, particularly the sequence dependence of the substrate, illustrates, at a molecular level, the property of conditional substrate promiscuity, in which the apparent promiscuity depends on the environmental condition. Variants having longer binding regions or higher substrate affinity can tolerate weakening (or strengthening) of binding without much change in population of the bound state, and therefore are relatively insensitive to mutations and have high apparent promiscuity. On the other hand, variants that exist on the threshold of binding can display high specificity as they are sensitive to small changes in dissociation rate. Thus, exhibition of promiscuity depends on conditions such as substrate concentration, pH, ionic strength, and temperature. Conditional promiscuity can be the basis for cryptic genetic variation, in which an altered phenotype is uncovered in new environments. Thus, it is likely to be underappreciated in the literature due to observational bias, because most experimental studies tend to focus on a small set of reaction or environmental conditions. This is an area ripe for future research given the likely importance of conditional promiscuity for evolutionary innovation.
3.3. Convergent Mechanism, Convergent Promiscuity: A Tale of Two “Capping” Ribozymes
The influence of mechanism on promiscuity, illustrated by the hammerhead ribozyme, is exemplified in a comparison of two independently derived ribozymes that share a common mechanism. These ribozymes, isolated under different selection conditions, promote the formation of a phosphate-phosphate anhydride bond between the terminal phosphate of a nucleotide and the 5′-α-phosphate of RNA. The final product is similar to the 5′ cap found on eukaryotic mRNAs.
These two RNA capping ribozymes, the Iso6 and 6.17 ribozymes, were discovered in the Yarus and Unrau groups, respectively.107,108 Interestingly, both ribozymes were isolated from different selections for which this capping reaction was not the desired function. Iso6 was recovered from a selection originally designed to identify ribozymes that could produce aminoacyl adenylates through reaction between amino acids and triphosphorylated RNA. Instead, pyrophosphate release was observed in the absence of amino acids, and the selection pool even developed labeling with PPi. Selection for capping activity using UTP instead of PPi quickly resulted in high activity in the pool and the identification of Iso6.107 On the other hand,the 6.17 ribozyme derived from a selection initially designed to identify polymerase activity by incorporating labeled UMP into a primer annealed to a poly(A) template. The resultant ribozyme with the fastest kinetics, 6.17, instead was found to act on the 5′ end of the RNA, forming a 5′–5′ cap.108
Iso6 and 6.17 display no apparent sequence similarities and are expected to adopt different secondary structures, consistent with their unique origins. Despite these differences, the molecular mechanisms for these two ribozymes appear to be surprisingly similar. Both ribozymes are predicted to have helices that terminate at the site of capping, with the terminal 5′ nucleotide retaining some flexibility; this position is unpaired in Iso6 and requires wobble pairing in 6.17. Both ribozymes also display increased activity at lower pH and require divalent cations for activity, although Iso6 prefers Ca2+ while 6.17 tolerates Mg2+, Mn2+, and Ca2+. The ribozymes even possess similar substrate binding affinities.108,109 Finally, both ribozymes appear to have minimal substrate requirements. The identities of the sugar and base have little impact on activity, despite possible hydrogen bonding interactions with these moieties. However, decreasing the length of the phosphate chain results in a large decrease in substrate binding,108,110,111 indicating that the phosphate itself is responsible for most substrate interactions.
Thus, these ribozymes suggest a common molecular mechanism for RNA capping that permits a high degree of substrate promiscuity, provided a small number of key features is present. The fact that two independently evolved, structurally dissimilar ribozymes have the same requirements supports the idea that substrate promiscuity is determined by mechanism. In this case, evolutionary convergence on the same mechanism resulted in convergence to similar promiscuity as well.112
3.4. Relying on Promiscuity: Searching for an RNA Replicase
For those interested in the origin of life, one of the most sought-after de novo ribozyme functions is catalysis of template-directed RNA polymerization (an “RNA replicase”), which is thought to be important, if not essential, to a self-replicating RNA system. One of the major avenues for this search has relied heavily on the promiscuity of newly evolved ribozymes. The first ribozymes developed by use of in vitro selection from a large pool of random sequences were RNA ligase ribozymes, including the “class I ligase” (Figure 6). The class I ligase was selected to catalyze the ligation between a 5′-triphosphate on the ribozyme and a 3′-hydroxyl on an RNA oligonucleotide substrate, which caused the ribozyme to tag itself with a sequence on the substrate that was necessary for purification and amplification.113,114 The 3′,5′-phosphodiester bond formed during ligation is identical to that formed during template-directed polymerization. Modification of the original class I ligase to bind a primer-template complex generated a ribozyme that possessed some polymerization activity, being able to extend a primer through the incorporation of mononucleotide triphosphates.115,116 This reaction occurred with 92% fidelity, though activity decreased with successive nucleotide additions, topping out at six nucleotides added after a six-day incubation. Nevertheless, this initial finding signaled that catalytic promiscuity of the class I ligase could potentially lead to an RNA replicase.
Figure 6.
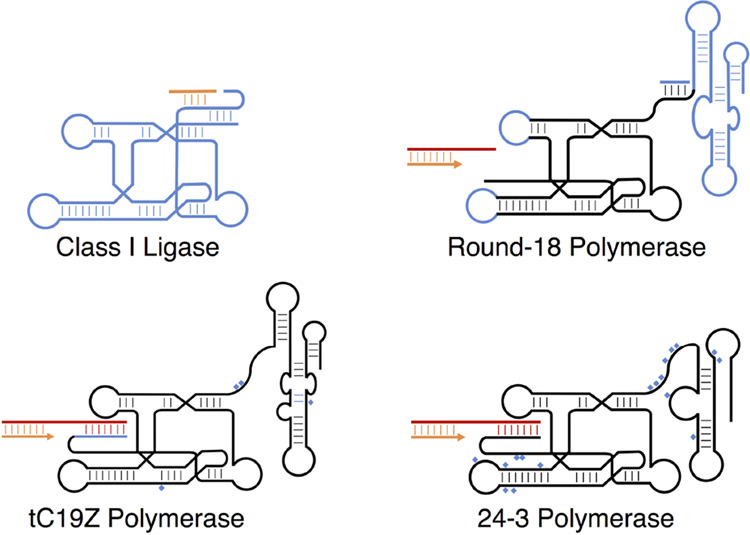
The class I ligase ribozyme and its descendants. Structures for the class I ligase;133,134 the round-18 polymerase introducing the new 3′ accessory domain (blue), which is known to interact with the loop depicted on the lower right;117,122 the tC19Z polymerase, introducing a new 5′ accessory domain;123 and the 24–3 polymerase.124 Blue regions denote new additions to the ribozyme with point mutations marked by blue diamonds. Primer and template oligonucleotides are shown in orange and red.
Subsequent work with the class I ligase and its derivatives aimed to increase its processivity, fidelity, and template generality. Important progress was made through attachment of an accessory domain to the 3′ end of the ribozyme, which was selected from a 76-nt random sequence with the idea that this domain could facilitate interaction of the ribozyme with the primer-template complex.117 Polymerization activity was selected for through incorporation of tagged nucleotides opposite an attached primer. To increase the sequence generality of polymerase activity, shorter templates were used to reduce hybridization with the ribozyme, and different primer-template sequences and lengths were used in different rounds of selection. One ribozyme, isolate 10.2, was found to function without attachment to the primer, and without recognition of a specific sequence. This feature was conferred by the new accessory domain, which increased binding of the primer-template complex, but polymerization activity itself occurred with minimal change to the ligase domain. Mutagenized versions of the 10.2 ribozyme were further selected for function on longer single-stranded templates and in the presence of higher concentrations of untagged nucleotides to improve fidelity. After eight more rounds of selection, the best resultant ribozyme, termed the round-18 polymerase (Figure 6), functioned much better with longer templates, and allowed for the extension of up to 14 nucleotides.
While catalytic promiscuity was key to the discovery of an RNA polymerase ribozyme, substrate promiscuity at a given template base is highly undesirable. That is, fidelity is important for an RNA replicase, because error rates represent a serious limit in the transmission of information.118 The round-18 polymerase copied templates with a per-base fidelity of 96.7%, which corresponds to relatively low promiscuity values (0.01–0.22; Table 2). One of the major determinants of this fidelity is misincorporation resulting from G:U wobble pairs, which is reflected by their higher promiscuity values compared to A and C (Table 2). While extension across a template A or C resulted in the correct addition (U or G, respectively) in over 99% of cases, G templated with an incorporation fidelity of 95.7%, and U templated with a fidelity of 92.1%, with the vast majority of mismatches resulting in a G:U mispair. This type of mispairing also appears to be the major limitation on the fidelity of nonenzymatic replication, and may be an echo of the thermodynamic limit on specificity.17,119
Table 2. Promiscuity Index (I) and Weighted Promiscuity Index (J) for the Round-18 Polymerase.
| substrate | CID | kA (M–1min–1)a | kC (M–1min–1)a | kG (M–1min–1)a | kU (M–1min–1)a |
|---|---|---|---|---|---|
| ATP | 5957 | 0.30 | 0.057 | 0.023 | 5.3 |
| CTP | 6176 | 0.02 | 0.008 | 5.4 | 0.0002 |
| GTP | 135 398 633 | 0.02 | 41 | 0.003 | 0.23 |
| UTP | 6133 | 87 | 0.004 | 0.46 | 0.001 |
| fidelity | 0.991 | 0.9996 | 0.957 | 0.921 | |
| I | 0.020 | 0.010 | 0.219 | 0.126 | |
| J | 0.020 | 0.010 | 0.220 | 0.125 |
Rate constants (kN, for N = A, C, G, U) are from Johnston et al.117
Despite the 3′ accessory domain, a major limitation of the round-18 polymerase continued to be low binding affinity for the primer-template complex, which was the primary contributor to the low processivity of the class I polymerase. The affinity also had a high degree of variability with regard to primer-template sequence, suggesting that further reduction of sequence specificity was still needed.120 Further progress was achieved by selecting directly for activity in trans using a water-in-oil emulsion. The first product of this method was the B6.61 polymerase, which was capable of generating sequences 20 nucleotides long.121,122 B6.61 showed a much faster polymerization rate than its predecessor, with an extension rate over 75 times faster for longer sequences. While there was no significant improvement in binding to the primer-template, this rate increase was accompanied by increased fidelity, including a minimization of G:U wobble insertions. As with the aminoacylation ribozymes (Table 1), this trend is a counterexample to the idea of a general trade-off between activity and specificity.
A substantial improvement to processivity came using a similar compartmentalization technique with the addition of a 5′ random region with the aim of improving interactions between the ribozyme and the primer-template complex. This yielded a 5′ accessory domain that forms stabilizing interactions with downstream portions of the template, thus increasing binding of the ribozyme to the template. Randomization and selection of the template sequence strengthened these interactions. With the optimized template, the new ribozyme, named tC19, ultimately yielded up to 95 nucleotide extensions with a per-base fidelity of 97.3%. However, the new interactions were largely intermolecular base-pairing, such that activity was strongly dependent on sequence. Selection on different templates identified four point mutations which, when introduced into tC19 to make tC19Z (Figure 6), improved the sequence generality. These new mutations further increased the measured per-base fidelity to 99.1%, largely due to a decrease in G insertion across template U. The tC19Z polymerase was shown to be capable of transcribing a functional 24 nt variant of the hammerhead ribozyme.123
Consideration of the promiscuity of the RNA polymerase ribozyme raises an interesting irony: while substrate promiscuity of the incoming monomer across a given template base is undesirable because it leads to copying errors, substrate promiscuity with respect to the template itself is highly desirable to obtain a ribozyme capable of copying many different, and ideally any, sequences. Sequences of particular concern are those with a high degree of structure that would need to be locally melted for ribozyme access, including sequences that comprise the ribozyme itself. Recent selections based on the RNA polymerase ribozyme focus on improving its sequence generality. One such study selected for the polymerase’s ability to synthesize complex folded RNA molecules, with selection tied to the creation of two functional aptamers, imposing pressure for sequence generality and high fidelity.124 The most active selected ribozyme, 24–3 (Figure 6), showed a ∼100-fold increased incorporation rate through structured sequences compared to the parent ribozyme. Likely as a result of selection for functional molecules instead of sequence fidelity, the 24–3 ribozyme displayed a higher error rate than its predecessors, in particular an increased tolerance for G-U wobble pairing. Despite this limitation, 24–3 was able to synthesize functional RNAs up to 76 nt long and could perform exponential amplification of an RNA template.
A different approach to overcoming the substrate generality problem takes advantage of plasticity, which occurs when non-native functions can be found through a relatively small number of mutations. In this case, it was hypothesized that copying via ligation of oligonucleotides could improve copying through structured sequences, because base-pairing to the oligonucleotide would mitigate some of the free energy cost of melting the template. Knowing that the RNA polymerase ribozyme was originally derived from the promiscuous activity of an RNA ligase, Attwater et al. engineered and evolved an ancestor of the tC19Z ribozyme to copy templates using trinucleotide triphosphates instead of NTPs.125 The triplet oligonucleotides use strand invasion to unfold structured RNA sequences for improved copying. The atavistic ribozyme t5+1 displayed reduced fidelity compared to its NTP-using counterpart, but selection for fidelity yielded an improved variant able to synthesize its own catalytic subunit. Interestingly, the t5+1 ribozyme consists of a heterodimer of the catalytic subunit and an RNA “cofactor” that assists interaction with the primer-template complex. Both subunits are descended from the same ancestral pool, illustrating how specialized descendants originated from distinct domains of the ancestor.
An ingenious orthogonal strategy to overcome the problem of sequence generality was developed by Joyce and coworkers, who reasoned that base-pairing between ribozyme and template was the major contributor to the energetics determining template specificity. Base-pairing is essentially absent between d-RNA and l-RNA sequences,126,127 and thus, a d-ribozyme is expected to have little base-pairing interaction with l-substrates. Selection for ligase activity indeed discovered d-ribozymes that could ligate l-RNA oligonucleotides.128 As expected, the non-natural, mirror-image l-ribozyme could perform the complementary reaction using d-RNA substrates and template. Furthermore, as with the class I ligase, these cross-chiral ligases also possessed polymerization activity. Unlike nonenzymatic templated RNA replication,129 these ribozymes displayed very little chiral inhibition, showing a high specificity for substrates of the desired chirality. A cross-chiral ligase was efficient enough to produce its mirror image enantiomer, which could then produce the original enantiomer. The cross-chiral ribozymes were not entirely sequence-general, as some substrates, such as those with 3′-terminal C or G residues, were more efficient than others. Nevertheless, given the precedent of the evolutionary strides demonstrated by the promiscuous class I ligase, the cross-chiral ligases represent an intriguing starting point for further development.
Polymerase (and ligase) ribozymes present a unique challenge in simultaneously requiring broad template accommodation and strict fidelity. Although this work was not undertaken for the purpose of studying promiscuity in ribozymes, the advances made with the class I ligase, spanning more than two decades of work by multiple groups, rely heavily on the promiscuity and plasticity of the ribozyme. Because this lineage of RNA polymerase ribozymes has only been selected on RNA substrates, true promiscuity can be clearly identified if the ribozymes accept different nucleic acids. One ribozyme displays some activity for incorporation of non-natural sugars and nucleobases, although it often stalls if modified nucleotides are present at specific positions.130 Unlike other ribozymes in its lineage, the 24–3 ribozyme, perhaps as a consequence of its selection for tolerance of different RNA aptamer templates, was observed to polymerize DNA on an RNA template (i.e., reverse transcription), permitting extension by up to 32 deoxyribonucleotides.131 A later generation of this ribozyme, 38–6, shows remarkable promiscuity, with activity on templates or nucleotides composed of multiple combinations of RNA, DNA, threose nucleic acid (TNA), or arabinose nucleic acids (ANA), though with reduced activity compared to an RNA-only system.132 38–6 also performs DNA-templated RNA synthesis and RNA-templated DNA synthesis more effectively than synthesis involving TNA or ANA, likely due to the lower structural similarities. These results further demonstrate the promiscuity of this ribozyme lineage. While the class I ligase and its descendants constitute a fascinating case study, it is unknown whether other ribozymes could exhibit similar versatility.
3.5. Catalytic Promiscuity: The Nucleotide Synthase Ribozyme
While substrate promiscuity appears to be commonly found among ribozymes,108,135,136 one may ask whether true catalytic promiscuity is also observed. Indeed, an interesting case was found in the pR1 nucleotide synthase.137 Selected to catalyze a reaction between ribose 5-phosphate (PR) and 6-thioguanosine (6SGua), this ribozyme was found to also be capable of catalyzing the reaction between 6SGua and 5-phosphoribosyl 1-pyrophospate (PRPP), an intermediate in the biological synthesis of nucleotides. These two reactions appear to have distinct reaction mechanisms and resultant products, depending on the substrate provided. Reaction with PRPP generates a glycosidic bond, resulting in the corresponding nucleotide, 6SGMP. However, the reaction with PR appears to require acyclization of ribose, allowing 6SGua to react with the corresponding aldehyde and generate a Schiff base (Figure 7). Each reaction has a unique dependence on magnesium ion concentration, supporting the existence of two different mechanisms. Interestingly, ribozymes selected for reactivity with PRPP instead of PR did not exhibit analogous activity on PR. Thus, not all ribozymes with the same function possess catalytic promiscuity. Despite the catalytic promiscuity of pR1, the ribozyme still displays a high degree of substrate specificity toward 6SGua, as analogous sulfur-containing purines are not recognized, in contrast to purine synthase ribozymes selected independently.138 The pR1 ribozyme demonstrates that catalytic promiscuity may differ in important ways from substrate promiscuity. While substrate promiscuity might be readily evolved through a relaxed binding mode, catalytic promiscuity requires a new reaction mechanism whose spontaneous emergence might be relatively unusual.
Figure 7.

Reactions catalyzed by the pR1 nucleotide synthase ribozyme. (A) Given a ribose 5-phosphate substrate, the acyclic form of ribose is stabilized and 6-thioguanosine (6SGua) reacts to form a Schiff base, which can then undergo an Amadori rearrangement. (B) Reaction with 5-phosphoribosyl 1-pyrophospate produces the desired nucleotide, 6SGMP. Adapted with permission from ref (137). Copyright 2009 Elsevier Ltd. Cell Press.
3.6. A Highly Promiscuous Ribozyme: The Ribosome
The ribosome is a ribozyme that translates genetic information on mRNA into protein sequences and is conserved across all the domains of life. The ribosome core consists of catalytic RNA, but farther from the catalytic center, both proteins and RNA are found.139 In eukaryotes, four rRNAs (rRNAs) associate with about 70 proteins, while in E. coli, the ribosome consists of 3 rRNAs and 52 proteins.140,141 Because the ribosome is a ribozyme and conserved across all domains, it is presumed to have existed in the last universal common ancestor (LUCA), and is also taken as circumstantial evidence of the RNA World.142 Protein translation necessitates high fidelity, with error rates of the overall process on the order of 10–4 per codon. Fidelity is primarily maintained through factors other than the ribosome, such as aminoacyl-tRNA synthetase editing and EF-Tu binding.143,144 While the peptidyl transferase center of the ribosome provides some steric selectivity (e.g., preferring l- rather than d-amino acids145), the ribosome itself is a surprisingly promiscuous molecule overall, permitting a wide assortment of substrates so long as there is a correct codon-anticodon match.
The ribosome accepts two aminoacyl-tRNAs at a time and catalyzes the formation of a peptide bond between the amino acids, releasing an uncharged tRNA and retaining a peptidyl-tRNA.141 The ribosome must accommodate a large variety of substrates: there are 20 canonical amino acids that can be associated with 50 or more different tRNAs, depending on the species.146 Even if one only considers two canonical amino acids coming together to be joined by a peptide bond, there are 400 possible substrate permutations that the ribosome must accommodate and catalyze. This level of multispecificity is essential for the production of all extant proteins in the organism. In addition to accommodating different amino acids and peptides in the active site, the ribosome must also accommodate different peptides in the exit tunnel. Interestingly, the exit tunnel is lined primarily by RNA and lacks significant patches of hydrophobicity, creating a “nonstick” character that allows peptides through regardless of sequence.139
The ribosome is similarly multispecific with respect to the mRNA templates, on which there are minimal sequence restrictions. Following initiation, which does involve sequence-specific interactions in some organisms, ribosome binding to mRNA is primarily facilitated through interactions with the mRNA backbone.147 However, the ribosome does display some slight substrate preferences. Early research on the ribosome, for example, discovered roughly 2-fold higher reactivity with leucine than phenylalanine.148 Additionally, ribosomes display codon preferences that can alter the elongation rate,149 a property which is used for regulation of gene expression. Still, the degree to which ribosomes are capable of utilizing a wide variety of substrates, including many noncanonical amino acids,150 representing promiscuous activity, is truly striking.
As with the RNA polymerase ribozymes, the substrate promiscuity of the ribosome must coexist with a requirement for high fidelity of information transfer. The promiscuity of the ribosome is tolerated by the cell in part because translation fidelity is handled during aminoacylation of tRNAs, including proofreading processes.151 In the ribosome, cognate and noncognate tRNAs can be distinguished through minor differences in base-pairing to mRNA. Recognition of the cognate tRNA leads to a structural change that is identified by elongation factor proteins which permit translation to proceed.152 Another restriction imposed on the incoming tRNA is the 3′ terminal CCA sequence, which forms specific interactions with the ribosome.139,147 This CCA sequence is required, and occasionally sufficient, for peptidyl transfer to occur.148,153 A small number of important interactions between tRNAs and the ribosome provide high fidelity of translation while permitting minimal restrictions on the mRNA or protein sequences.
The innate promiscuity of the ribosome is occasionally exploited by nature. One such example is puromycin, an antibiotic produced by the bacteria Streptomyces alboniger. Puromycin is an aminonucleoside, containing nucleoside and amino acid analogues, linked through an amide bond instead of the conventional ester. This structure mimics the 3′ terminus of a charged tRNA, which allows it to enter the ribosome and be irreversibly incorporated into the nascent polypeptide, terminating translation.154,155 The efficacy of this molecule suggests that evolutionary escape from this promiscuous activity has been difficult despite the selective pressure engendered by the antibiotic.
Synthetic biologists have also taken advantage of the substrate promiscuity of the ribosome, fundamentally altering the genetic code itself. tRNAs recognizing the amber stop codon can be charged with noncanonical amino acids. Because the ribosome is, to a first approximation, essentially agnostic with respect to the side chain of the incoming monomer, the amber codon is translated into the new amino acid.156,157 Amino acids with a remarkably diverse set of unnatural functional groups have been successfully incorporated by this method, including alkanes, polybenzenes, sugars and phosphate-containing species.158,159 The ribosome can even catalyze the formation of ester bonds, yielding mRNA-encoded polyesters, without mutation in the ribosome itself.86 Further evolution can push this versatility further, as seen with the ribosome variants ribo-Q1 and ribo-X, which translate quadruplet codons and thus introduce many “blank” codons to the genetic code.160−162 Although it was postulated early on that the genetic code might be a “frozen accident”,4 it now seems clear that the code itself has been the subject of evolution, as evidenced by the different version of the code found in mitochondria163 as well as statistical analyses suggesting that the code has evolved to minimize the biophysical impact of mutations.164 The evolvability and malleability of the genetic code attests to the remarkable combination of substrate promiscuity and informational fidelity in the ribosome.
4. Primordial Ribozymes: More Promiscuous?
We now return to a question posed at the beginning of this review: would primordial ribozymes be particularly promiscuous? There are two reasons why one might hypothesize this. First, given the importance of promiscuity for evolutionary innovation, one may suppose that primordial ribozymes might have been more promiscuous than highly evolved enzymes due to evolutionary pressure for greater specificity. Second, given the wider chemical diversity of functional groups available to proteins, one may suppose that proteins will have both superior specificity and activity, in general, compared to ribozymes, due to their ability to engage in more types of interactions. Although there is insufficient data in the literature to answer these questions definitively, here we consider two comparisons that bear on these issues. First, we consider whether newly evolved ribozymes are more promiscuous than highly evolved ribozymes. Second, we ask whether ribozymes are more promiscuous than proteins by examining a head-to-head comparison of a ribozyme and a protein enzyme, both of which were evolved de novo.
4.1. Newly Evolved vs Highly Evolved Ribozymes
An interesting comparison can be made between the promiscuity of ribozymes from in vitro selection, which have very short evolutionary histories, to highly evolved ribozymes, in particular, the ribosome. The first ribozymes to catalyze amide bond formation were initially selected for a different activity, to catalyze the transfer of an aminoacyl group from the 3′-hydroxyl of a short tRNA mimic to the 5′-hydroxyl of the ribozyme.165 However, like the ribosome, one of the selected ribozymes was able to use an alternative nucleophilic substrate. When the 5′-hydroxyl was substituted with an amino group, amide bond formation was observed at a similar rate. In both cases, the ribozyme accelerated the respective noncatalyzed reaction by over 1000-fold. Later, ribozymes were selected to perform peptide bond formation by linking a phenylalanine to the 5′ end of the RNA and selecting for the ability to attach a biotinylated methionine from a 3′ acylated AMP substrate.166 The best ribozymes from this selection displayed a rate enhancement of ∼106. This reaction was inhibited by the presence of AMP, but not other nucleotides or methionine, suggesting that the ribozyme functions primarily through specific interactions with AMP. Consistent with this, activity was also observed with leucine, phenylalanine, and lysine substrates, with methionine and leucine being the best peptidyl donors.
These peptide synthase ribozymes, much like the flexizyme, illustrate that if there are sufficient interactions with other parts of the small molecule substrate, the amino acid side chain may be quite variable. In addition, the fact that one of the ribozymes is capable of both ester and amide bond formation, much like the ribosome, further corroborates its promiscuity of function. (In this case, these non-native activities represent true promiscuity because we know the complete environmental history of the ribozyme.) However, while it may be possible to evolve increased promiscuity in these ribozymes, it seems a hard task indeed to match or exceed the promiscuity of the ribosome. This comparison at least suggests that newly evolved ribozymes are not necessarily more promiscuous than highly evolved ones. Instead, specificity or promiscuity itself may be a selectable trait, and natural selection may favor either greater or lesser promiscuity.
4.2. De Novo Ribozyme vs de Novo Protein Enzyme: The Diels–Alderases
Are protein enzymes superior to ribozymes, such that ribozymes emerging in the RNA world would be worse than their protein counterparts? While it seems clear that proteins have greater activity in general, nearly all protein enzymes have much longer evolutionary histories compared to ribozymes, most of which have been evolved in vitro. To avoid this confounding factor, one may compare a de novo ribozyme with a de novo protein enzyme. Such a comparison can be made with the Diels–Alderase ribozyme and protein enzymes, which both catalyze a reaction (Figure 8) previously not known to occur in biology.
Figure 8.

Diels–Alder cycloaddition. A concerted reaction between a conjugated diene and an alkene (dienophile) results in the formation of a cyclized product.172 The dienophile substituents shown (R3 and R4) are added to the same face of the cyclohexane ring.
Interestingly, the first biochemical catalyst discovered for the Diels–Alder reaction was a ribozyme, not a protein. While most ribozyme reactions involve RNA or amino acid modifications and often involve base-pairing interactions, an early discovery that demonstrated the catalytic versatility of ribozymes was carbon–carbon bond formation by Diels–Alderase ribozymes.167,168 The specificity of one such ribozyme was extensively characterized by the Jäschke laboratory through testing a series of potential substrates.169,170 The initial experiments selected for cycloaddition of a biotin-maleimide to anthracene, which was conjugated to the RNA via a polyethylene glycol linker. The ribozyme produced from this selection could catalyze this reaction on free substrate with a high degree of enantioselectivity. Additionally, a synthesized mirror image of this ribozyme composed of l-nucleotides produced the opposite enantiomer. This enantioselectivity was the result of a “tail” group on the anthracene substrate (e.g., the PEG linker), which restricted the molecule’s orientation in the binding pocket. Important structural features of both substrates include: the diene must contain three linearly annellated rings, the dienophile must be a five-membered maleimide ring with a hydrophobic tail, and both substrates must be arranged in a stacked, coplanar manner.170 These results present one of the most rigorous characterizations of ribozyme specificity on a non-nucleotide substrate.
One decade later, a Diels–Alderase protein enzyme was developed by the Baker lab.171 This enzyme was created de novo using computational design and site-directed mutagenesis to catalyze the reaction between 4-carboxybenzyl trans-1,3-butadiene-1-carbamate and N,N-dimethylacrylamide. Like the ribozyme, this protein enzyme demonstrated a high level of product stereoselectivity (>97%). The best Diels–Alderase enzymes possessed higher catalytic activity than the Diels–Alderase ribozymes, but were still markedly slower than natural enzymes.
Although these catalysts were discovered through different means (in vitro selection vs computational design), both were created in a laboratory setting independent of natural evolutionary influences, and therefore they are an interesting test comparison to understand the promiscuity of de novo functions. While the reactions catalyzed by these molecules use different substrates, the promiscuity indices can be compared between them (Table 3 vs Table 4). Note that different values are calculated for the diene and dienophile when possible, and that I and J varies depending on whether they are calculated from kcat or from kcat/kuncat (catalytic power). Despite these differences, in general, the promiscuity indices are not very different; all values for the dienophiles lie in the range of 0.66–0.83, with the values for the protein enzyme lying in the middle of this range. Therefore, comparing these two de novo catalysts, it does not appear that the ribozyme is more promiscuous than the protein enzyme.
Table 3. Unweighted (I) and Weighted (J) Promiscuity Indices for Diels–Alderase Ribozymes, Calculated Separately for the Diene and Dienophile Substrates.

HEG indicates hexa(ethylene glycol).
kcat and kuncat values from Stuhlmann and Jäschke.170
Dash indicates kuncat not reported; these values could not be included in the calculation of I or J.
Table 4. Unweighted (I) and Weighted (J) Promiscuity Indices for the Protein Diels–Alderase, Calculated for Dienophile Substrates.

Rate values were estimated from Siegel et al.171
It is of practical interest to note that the weighted and unweighted promiscuity indices (Tables 1–4) are often not very different from one another, as the difference between these values ranges from 0 to 0.1. This may reflect ruggedness in the promiscuity profile over the chemical space of the substrates. The motivation for creating a weighted index was to account for the expectation that chemically similar substrates would have similar activity. While this must be true to some extent, over the substrates that were tested and reported in these examples, the additional accounting did not alter the overall calculation by much.
5. Concluding Remarks
Ribozymes identified by in vitro selection or evolution represent an ideal model system for studying true promiscuity because the selective pressures on these ribozymes are controlled by the experimenter and their entire evolutionary history is available for study. In addition, the promiscuity of ribozymes in particular is a fascinating question relating to the origin of living systems. An attractive but untested hypothesis is that the earliest ribozymes emerging from the prebiotic milieu of random polymers would be highly promiscuous, presenting a kernel of activity across many functions that could be optimized by evolution individually (e.g., after duplication events). Although a rigorous test of this hypothesis is currently lacking, we may consider how current knowledge informs this hypothesis of promiscuous ribozymes.
What are the likely properties of a ribozyme selected de novo, i.e., a primordial ribozyme? It is clear that the activity is likely to be low initially, simply because there are more sequences of low activity compared to high activity (i.e., the frequency of sequences is a decreasing function of activity),173 leaving room for optimization of activity by natural selection. What about promiscuity? While it might seem intuitive that simple, low activity ribozymes would have high promiscuity, we do not see solid evidence for this so far in the literature. As discussed above, a de novo peptide synthase ribozyme is less promiscuous than its highly evolved counterpart (the ribosome). While there are some examples of ribozymes where in vitro evolution resulted in both improved activity and specificity (discussed above), it is not clear that there would be a positive correlation between activity and specificity in general. Indeed at least one mechanism (discussed in Section 2.2) has the opposite effect, causing a negative correlation, i.e., a trade-off, between activity and specificity.
The intuition that there should be a positive correlation between activity and specificity is based on the general idea that increased molecular interactions give both increased activity and increased specificity. This seems to be reasonable, but in one rigorous study of RNA aptamers, activity was found to be uncorrelated to specificity.174 It is even less clear that increased interactions should increase specificity in ribozymes, particularly cis-acting ribozymes, because the entire reaction pathway would be stabilized. How the ground state, transition state, and product would be affected in relative terms is not clear. Therefore, it should not be assumed that the earliest emerging ribozymes were particularly promiscuous. Empirical data is required to resolve the relationship between activity and specificity of ribozymes.
The reason why the hypothesis of promiscuous primordial ribozymes is attractive, despite the current lack of evidence to support it, is that it solves an important problem in prebiotic evolution. If the first ribozyme to emerge by chance possesses the ability to catalyze many reactions, albeit at low activity, this ribozyme could serve as the ancestral catalyst to a suite of different reactions, rapidly forming a metabolic network of ribozymes. In evolutionary terms, a network of ribozymes might then arise from exaptation (or preadaptation) of a small number of ancestral ribozymes. However, it may be that promiscuity, rather than being an automatic property of a low-activity primordial ribozyme, should be considered as an evolvable or fortuitous trait itself, possibly uncorrelated to activity. In this case, the selective pressures on the RNA world would play an important role in shaping ribozyme evolvability.
Acknowledgments
The authors thank Sabine Muller, John Sutherland, Dieter Braun, Abhinav Nath, and William Atkins for their insights. Funding from the Simons Foundation (Grant 290356FY18), NASA (NNX16AJ32G), NIH (DP2 GM123457-01), and the Camille Dreyfus Teacher-Scholar Program is gratefully acknowledged.
Biographies
Evan Janzen is a PhD candidate in the Biomolecular Science and Engineering Program at the University of California, Santa Barbara. He received a BS in biochemistry and molecular biology from Emporia State University and an MS in molecular and integrative physiology from the University of Kansas.
Celia Blanco is an Otis Williams postdoctoral fellow at the Chemistry and Biochemistry department at the University of California, Santa Barbara. Her research focuses on analyzing deep sequencing data to understand de novo evolution of functional molecules. Previously, she was a Calvo-Rodes graduate student fellow at the Astrobiology Center in the National Institute of Aerospace Technology in Madrid (Spain), where she engaged in research related to the origin of biological homochirality, under the supervision of Dr. David Hochberg. She received her BS, MS, and PhD in Theoretical Physics from the Complutense University of Madrid.
Huan Peng is currently a postdoctoral fellow in the Chemistry and Biochemistry department at the University of California, Santa Barbara. He works on small self-cleaving ribozymes and development of chimeric phage functionalized with nanomaterials for bacterial detection. In 2016 he received a PhD under the supervision of Prof. Andrij Pich and Prof. Ulrich Schwaneberg from RWTH Aachen University, where he worked on water-soluble reactive macromolecules for protein encapsulation and delivery. He worked as a joint postdoctoral fellow in Maastricht University and SABIC Europe B.V. on functional porous polymer membranes prior to moving to California in 2017.
Josh Kenchel is a PhD candidate in the Biomolecular Sciences and Engineering Program at the University of California, Santa Barbara. He received a BS in biochemistry and an MS in biology from the University of California, San Diego.
Irene A. Chen is an associate professor of Chemistry and Biochemistry at the University of California, Santa Barbara. She received a BA in chemistry and an MD/PhD in biophysics from Harvard, where she worked with Jack Szostak on protocell membranes. She studied nucleic acid replication and RNA evolution as a Bauer Fellow in systems biology at Harvard. She has been named a Searle Scholar, NIH New Innovator, and Camille Dreyfus Teacher-Scholar. Since 2013, she has been an Investigator of the Simons Collaboration on the Origin of Life.
The authors declare no competing financial interest.
References
- Robertson M. P.; Joyce G. F. The Origins of the RNA World. Cold Spring Harbor Perspect. Biol. 2012, 4, a003608. 10.1101/cshperspect.a003608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pressman A.; Blanco C.; Chen I. A. The RNA World as a Model System to Study the Origin of Life. Curr. Biol. 2015, 25, R953–963. 10.1016/j.cub.2015.06.016. [DOI] [PubMed] [Google Scholar]
- Woese C. R. On the Evolution of the Genetic Code. Proc. Natl. Acad. Sci. U. S. A. 1965, 54, 1546–1552. 10.1073/pnas.54.6.1546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. H. The Origin of the Genetic Code. J. Mol. Biol. 1968, 38, 367–379. 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- Orgel L. E. Evolution of the Genetic Apparatus. J. Mol. Biol. 1968, 38, 381–393. 10.1016/0022-2836(68)90393-8. [DOI] [PubMed] [Google Scholar]
- Ellington A. D.; Szostak J. W. In Vitro Selection of RNA Molecules That Bind Specific Ligands. Nature 1990, 346, 818–822. 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- Robertson D. L.; Joyce G. F. Selection In Vitro of an RNA Enzyme That Specifically Cleaves Single-Stranded DNA. Nature 1990, 344, 467–468. 10.1038/344467a0. [DOI] [PubMed] [Google Scholar]
- Ellington A. D.; Chen X.; Robertson M.; Syrett A. Evolutionary Origins and Directed Evolution of RNA. Int. J. Biochem. Cell Biol. 2009, 41, 254–265. 10.1016/j.biocel.2008.08.015. [DOI] [PubMed] [Google Scholar]
- Jensen R. A. Enzyme Recruitment in Evolution of New Function. Annu. Rev. Microbiol. 1976, 30, 409–425. 10.1146/annurev.mi.30.100176.002205. [DOI] [PubMed] [Google Scholar]
- Khersonsky O.; Tawfik D. S. Enzyme Promiscuity: a Mechanistic and Evolutionary Perspective. Annu. Rev. Biochem. 2010, 79, 471–505. 10.1146/annurev-biochem-030409-143718. [DOI] [PubMed] [Google Scholar]
- Schultes E. A.; Bartel D. P. One Sequence, Two Ribozymes: Implications for the Emergence of New Ribozyme Folds. Science 2000, 289, 448–452. 10.1126/science.289.5478.448. [DOI] [PubMed] [Google Scholar]
- Seffernick J. L.; de Souza M. L.; Sadowsky M. J.; Wackett L. P. Melamine Deaminase and Atrazine Chlorohydrolase: 98% Identical but Functionally Different. J. Bacteriol. 2001, 183, 2405–2410. 10.1128/JB.183.8.2405-2410.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornish-Bowden A.; Cardenas M. L. Specificity of Non-Michaelis-Menten Enzymes: Necessary Information for Analyzing Metabolic Pathways. J. Phys. Chem. B 2010, 114, 16209–16213. 10.1021/jp106968p. [DOI] [PubMed] [Google Scholar]
- Peracchi A. The Limits of Enzyme Specificity and the Evolution of Metabolism. Trends Biochem. Sci. 2018, 43, 984–996. 10.1016/j.tibs.2018.09.015. [DOI] [PubMed] [Google Scholar]
- Yagisawa S. Enzyme Kinetics Based on Free-Energy Profiles. Biochem. J. 1995, 308, 305–311. 10.1042/bj3080305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopfield J. J. Kinetic Proofreading: a New Mechanism for Reducing Errors in Biosynthetic Processes Requiring High Specificity. Proc. Natl. Acad. Sci. U. S. A. 1974, 71, 4135–4139. 10.1073/pnas.71.10.4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leu K.; Obermayer B.; Rajamani S.; Gerland U.; Chen I. A. The Prebiotic Evolutionary Advantage of Transferring Genetic Information from RNA to DNA. Nucleic Acids Res. 2011, 39, 8135–8147. 10.1093/nar/gkr525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tawfik D. S. Accuracy-Rate Tradeoffs: How Do Enzymes Meet Demands of Selectivity and Catalytic Efficiency?. Curr. Opin. Chem. Biol. 2014, 21, 73–80. 10.1016/j.cbpa.2014.05.008. [DOI] [PubMed] [Google Scholar]
- Tokuriki N.; Jackson C. J.; Afriat-Jurnou L.; Wyganowski K. T.; Tang R.; Tawfik D. S. Diminishing Returns and Tradeoffs Constrain the Laboratory Optimization of an Enzyme. Nat. Commun. 2012, 3, 1257. 10.1038/ncomms2246. [DOI] [PubMed] [Google Scholar]
- Brustad E. M.; Arnold F. H. Optimizing Non-Natural Protein Function with Directed Evolution. Curr. Opin. Chem. Biol. 2011, 15, 201–210. 10.1016/j.cbpa.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson J. C.; Badran A. H.; Guggiana-Nilo D. A.; Liu D. R. Negative Selection and Stringency Modulation in Phage-Assisted Continuous Evolution. Nat. Chem. Biol. 2014, 10, 216–222. 10.1038/nchembio.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beard W. A.; Shock D. D.; Vande Berg B. J.; Wilson S. H. Efficiency of Correct Nucleotide Insertion Governs DNA Polymerase Fidelity. J. Biol. Chem. 2002, 277, 47393–47398. 10.1074/jbc.M210036200. [DOI] [PubMed] [Google Scholar]
- Ram Prasad B.; Warshel A. Prechemistry Versus Preorganization in DNA Replication Fidelity. Proteins: Struct., Funct., Genet. 2011, 79, 2900–2919. 10.1002/prot.23128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caglayan M.; Bilgin N. Temperature Dependence of Accuracy of DNA Polymerase I from Geobacillus anatolicus. Biochimie 2012, 94, 1968–1973. 10.1016/j.biochi.2012.05.019. [DOI] [PubMed] [Google Scholar]
- Azpurua J.; Ke Z.; Chen I. X.; Zhang Q.; Ermolenko D. N.; Zhang Z. D.; Gorbunova V.; Seluanov A. Naked Mole-Rat Has Increased Translational Fidelity Compared with the Mouse, as Well as a Unique 28S Ribosomal RNA cleavage. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 17350–17355. 10.1073/pnas.1313473110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee K.; Kolomeisky A. B.; Igoshin O. A. Elucidating Interplay of Speed and Accuracy in Biological Error Correction. Proc. Natl. Acad. Sci. U. S. A. 2017, 114, 5183–5188. 10.1073/pnas.1614838114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninio J. Kinetic Amplification of Enzyme Discrimination. Biochimie 1975, 57, 587–595. 10.1016/S0300-9084(75)80139-8. [DOI] [PubMed] [Google Scholar]
- Murugan A.; Huse D. A.; Leibler S. Speed, Dissipation, and Error in Kinetic Proofreading. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 12034–12039. 10.1073/pnas.1119911109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schomburg I.; Jeske L.; Ulbrich M.; Placzek S.; Chang A.; Schomburg D. The BRENDA Enzyme Information System-From a Database to an Expert System. J. Biotechnol. 2017, 261, 194–206. 10.1016/j.jbiotec.2017.04.020. [DOI] [PubMed] [Google Scholar]
- Tcherkez G. G.; Farquhar G. D.; Andrews T. J. Despite Slow Catalysis and Confused Substrate Specificity, All Ribulose Bisphosphate Carboxylases May Be Nearly Perfectly Optimized. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 7246–7251. 10.1073/pnas.0600605103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savir Y.; Noor E.; Milo R.; Tlusty T. Cross-Species Analysis Traces Adaptation of Rubisco Toward Optimality in a Low-Dimensional Landscape. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 3475–3480. 10.1073/pnas.0911663107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flamholz A. I.; Prywes N.; Moran U.; Davidi D.; Bar-On Y. M.; Oltrogge L. M.; Alves R.; Savage D.; Milo R. Revisiting Trade-offs between Rubisco Kinetic Parameters. Biochemistry 2019, 58, 3365–3376. 10.1021/acs.biochem.9b00237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Brien P. J.; Herschlag D. Catalytic Promiscuity and the Evolution of New Enzymatic Activities. Chem. Biol. 1999, 6, R91–R105. 10.1016/S1074-5521(99)80033-7. [DOI] [PubMed] [Google Scholar]
- Khersonsky O.; Roodveldt C.; Tawfik D. S. Enzyme Promiscuity: Evolutionary and Mechanistic Aspects. Curr. Opin. Chem. Biol. 2006, 10, 498–508. 10.1016/j.cbpa.2006.08.011. [DOI] [PubMed] [Google Scholar]
- Hult K.; Berglund P. Enzyme Promiscuity: Mechanism and Applications. Trends Biotechnol. 2007, 25, 231–238. 10.1016/j.tibtech.2007.03.002. [DOI] [PubMed] [Google Scholar]
- Brevet A.; Plateau P.; Cirakoglu B.; Pailliez J. P.; Blanquet S. Zinc-Dependent Synthesis of 5′,5′-Diadenosine Tetraphosphate by Sheep Liver Lysyl- and Phenylalanyl-tRNA Synthetases. J. Biol. Chem. 1982, 257, 14613–14615. [PubMed] [Google Scholar]
- Copley S. D. Shining a Light on Enzyme Promiscuity. Curr. Opin. Struct. Biol. 2017, 47, 167–175. 10.1016/j.sbi.2017.11.001. [DOI] [PubMed] [Google Scholar]
- Newton M. S.; Arcus V. L.; Gerth M. L.; Patrick W. M. Enzyme Evolution: Innovation is Easy, Optimization is Complicated. Curr. Opin. Struct. Biol. 2018, 48, 110–116. 10.1016/j.sbi.2017.11.007. [DOI] [PubMed] [Google Scholar]
- James L. C.; Tawfik D. S. Conformational Diversity and Protein Evolution--a 60-Year-Old Hypothesis Revisited. Trends Biochem. Sci. 2003, 28, 361–368. 10.1016/S0968-0004(03)00135-X. [DOI] [PubMed] [Google Scholar]
- Khersonsky O.; Tawfik D. S. In Comprehensive Natural Products II; Liu H.-W., Mander L., Eds.; Elsevier: Oxford, 2010. [Google Scholar]
- Seffernick J. L.; Johnson G.; Sadowsky M. J.; Wackett L. P. Substrate Specificity of Atrazine Chlorohydrolase and Atrazine-Catabolizing Bacteria. Appl. Environ. Microbiol. 2000, 66, 4247–4252. 10.1128/AEM.66.10.4247-4252.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibert C. M.; Raushel F. M. Structural and Catalytic Diversity within the Amidohydrolase Superfamily. Biochemistry 2005, 44, 6383–6391. 10.1021/bi047326v. [DOI] [PubMed] [Google Scholar]
- Scott C.; Jackson C. J.; Coppin C. W.; Mourant R. G.; Hilton M. E.; Sutherland T. D.; Russell R. J.; Oakeshott J. G. Catalytic Improvement and Evolution of Atrazine Chlorohydrolase. Appl. Environ. Microbiol. 2009, 75, 2184–2191. 10.1128/AEM.02634-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griswold K. E.; Aiyappan N. S.; Iverson B. L.; Georgiou G. The Evolution of Catalytic Efficiency and Substrate Promiscuity in Human Theta Class 1–1 Glutathione Transferase. J. Mol. Biol. 2006, 364, 400–410. 10.1016/j.jmb.2006.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt R. Cytochromes P450 as Versatile Biocatalysts. J. Biotechnol. 2006, 124, 128–145. 10.1016/j.jbiotec.2006.01.026. [DOI] [PubMed] [Google Scholar]
- Ekroos M.; Sjogren T. Structural Basis for Ligand Promiscuity in Cytochrome P450 3A4. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 13682–13687. 10.1073/pnas.0603236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt R.; Urlacher V. B. Cytochromes P450 as Promising Catalysts for Biotechnological Application: Chances and Limitations. Appl. Microbiol. Biotechnol. 2014, 98, 6185–6203. 10.1007/s00253-014-5767-7. [DOI] [PubMed] [Google Scholar]
- Steele C. L.; Crock J.; Bohlmann J.; Croteau R. Sesquiterpene Synthases from Grand Fir (Abies grandis). Comparison of Constitutive and Wound-Induced Activities, and cDNA Isolation, Characterization, and Bacterial Expression of Delta-Selinene Synthase and Gamma-Humulene Synthase. J. Biol. Chem. 1998, 273, 2078–2089. 10.1074/jbc.273.4.2078. [DOI] [PubMed] [Google Scholar]
- Hammer S. C.; Syren P. O.; Seitz M.; Nestl B. M.; Hauer B. Squalene Hopene Cyclases: Highly Promiscuous and Evolvable Catalysts for Stereoselective CC and CX Bond Formation. Curr. Opin. Chem. Biol. 2013, 17, 293–300. 10.1016/j.cbpa.2013.01.016. [DOI] [PubMed] [Google Scholar]
- Pilkington S. J.; Dalton H. Soluble Methane Monooxygenase from Methylococcus-Capsulatus Bath. Methods Enzymol. 1990, 188, 181–190. 10.1016/0076-6879(90)88032-6. [DOI] [Google Scholar]
- Dillingham M. S.; Kowalczykowski S. C. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol. Mol. Biol. Rev. 2008, 72, 642–671. 10.1128/MMBR.00020-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett S. T.The DNA Exonucleases of Escherichia coli. EcoSal Plus 2011, 4. 10.1128/ecosalplus.4.4.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X. Y.; Huang W. J.; Hu S. C.; Zhang H. L.; Wang H.; Zhang J. X.; Lin H. H.; Chen Y. Z.; Zou Q.; Ji Z. L. A Global Characterization and Identification of Multifunctional Enzymes. PLoS One 2012, 7, e38979 10.1371/journal.pone.0038979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffery C. J. Moonlighting Proteins: Old Proteins Learning New Tricks. Trends Genet. 2003, 19, 415–417. 10.1016/S0168-9525(03)00167-7. [DOI] [PubMed] [Google Scholar]
- Aharoni A.; Gaidukov L.; Khersonsky O.; Gould S. M.; Roodveldt C.; Tawfik D. S The ‘Evolvability’ of Promiscuous Protein Functions. Nat. Genet. 2005, 37, 73–76. 10.1038/ng1482. [DOI] [PubMed] [Google Scholar]
- Copley S. D. Enzymes with Extra Talents: Moonlighting Functions and Catalytic Promiscuity. Curr. Opin. Chem. Biol. 2003, 7, 265–272. 10.1016/S1367-5931(03)00032-2. [DOI] [PubMed] [Google Scholar]
- James L. C.; Tawfik D. S. Catalytic and Binding Poly-Reactivities Shared by Two Unrelated Proteins: The Potential Role of Promiscuity in Enzyme Evolution. Protein Sci. 2001, 10, 2600–2607. 10.1110/ps.14601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James L. C.; Roversi P.; Tawfik D. S. Antibody Multispecificity Mediated by Conformational Diversity. Science 2003, 299, 1362–1367. 10.1126/science.1079731. [DOI] [PubMed] [Google Scholar]
- Yang K.; Metcalf W. W. A New Activity for an Old Enzyme: Escherichia coli Bacterial Alkaline Phosphatase is a Phosphite-Dependent Hydrogenase. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 7919–7924. 10.1073/pnas.0400664101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasan R.; Meharenna Y. T.; Snow C. D.; Poulos T. L.; Arnold F. H. Evolutionary History of a Specialized p450 Propane Monooxygenase. J. Mol. Biol. 2008, 383, 1069–1080. 10.1016/j.jmb.2008.06.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ycas M. On Earlier States of the Biochemical System. J. Theor. Biol. 1974, 44, 145–160. 10.1016/S0022-5193(74)80035-4. [DOI] [PubMed] [Google Scholar]
- Amitai G.; Gupta R. D.; Tawfik D. S. Latent Evolutionary Potentials Under the Neutral Mutational Drift of an Enzyme. HFSP J. 2007, 1, 67–78. 10.2976/1.2739115/10.2976/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wroe R.; Chan H. S.; Bornberg-Bauer E. A Structural Model of Latent Evolutionary Potentials Underlying Neutral Networks in Proteins. HFSP J. 2007, 1, 79–87. 10.2976/1.2739116/10.2976/1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espinosa-Cantu A.; Ascencio D.; Barona-Gomez F.; DeLuna A. Gene Duplication and the Evolution of Moonlighting Proteins. Front. Genet. 2015, 6, 227. 10.3389/fgene.2015.00227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Visser J. A.; Krug J. Empirical Fitness Landscapes and the Predictability of Evolution. Nat. Rev. Genet. 2014, 15, 480–490. 10.1038/nrg3744. [DOI] [PubMed] [Google Scholar]
- Blanco C.; Janzen E.; Pressman A.; Saha R.; Chen I. A. Molecular Fitness Landscapes from High-Coverage Sequence Profiling. Annu. Rev. Biophys. 2019, 48, 1–18. 10.1146/annurev-biophys-052118-115333. [DOI] [PubMed] [Google Scholar]
- Nath A.; Atkins W. M. A Quantitative Index of Substrate Promiscuity. Biochemistry 2008, 47, 157–166. 10.1021/bi701448p. [DOI] [PubMed] [Google Scholar]
- Nath A.; Zientek M. A.; Burke B. J.; Jiang Y.; Atkins W. M. Quantifying and Predicting the Promiscuity and Isoform Specificity of Small-Molecule Cytochrome P450 Inhibitors. Drug Metab. Dispos. 2010, 38, 2195–2203. 10.1124/dmd.110.034645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S.; Rao B. J. A measure of the promiscuity of proteins and characteristics of residues in the vicinity of the catalytic site that regulate promiscuity. PLoS One 2012, 7, e32011 10.1371/journal.pone.0032011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S.; Asgeirsson B.; Rao B. J. A measure of the broad substrate specificity of enzymes based on ’duplicate’ catalytic residues. PLoS One 2012, 7, e49313 10.1371/journal.pone.0049313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honaker M. T.; Acchione M.; Zhang W.; Mannervik B.; Atkins W. M. Enzymatic Detoxication, Conformational Selection, and the Role of Molten Globule Active Sites. J. Biol. Chem. 2013, 288, 18599–18611. 10.1074/jbc.M112.445767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Promiscuity indices were calculated using online tools available at http://hetaira.herokuapp.com (accessed on August 1, 2019).
- Pang Y. L.; Poruri K.; Martinis S. A. tRNA Synthetase: tRNA Aminoacylation and Beyond. Wiley Interdiscip. Rev. RNA 2014, 5, 461–480. 10.1002/wrna.1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illangasekare M.; Sanchez G.; Nickles T.; Yarus M. Aminoacyl-RNA Synthesis Catalyzed by an RNA. Science 1995, 267, 643–647. 10.1126/science.7530860. [DOI] [PubMed] [Google Scholar]
- Saito H.; Kourouklis D.; Suga H. An In Vitro Evolved Precursor tRNA with Aminoacylation Activity. EMBO J. 2001, 20, 1797–1806. 10.1093/emboj/20.7.1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N.; Huang F. Ribozyme-Catalyzed Aminoacylation from CoA Thioesters. Biochemistry 2005, 44, 4582–4590. 10.1021/bi047576b. [DOI] [PubMed] [Google Scholar]
- Pressman A. D.; Liu Z.; Janzen E.; Blanco C.; Muller U. F.; Joyce G. F.; Pascal R.; Chen I. A. Mapping a Systematic Ribozyme Fitness Landscape Reveals a Frustrated Evolutionary Network for Self-Aminoacylating RNA. J. Am. Chem. Soc. 2019, 141, 6213–6223. 10.1021/jacs.8b13298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illangasekare M.; Yarus M. Specific, Rapid Synthesis of Phe-RNA by RNA. Proc. Natl. Acad. Sci. U. S. A. 1999, 96, 5470–5475. 10.1073/pnas.96.10.5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xin Y.; Li W.; First E. A. The ’KMSKS’ Motif in Tyrosyl-tRNA Synthetase Participates in the Initial Binding of tRNA(Tyr). Biochemistry 2000, 39, 340–347. 10.1021/bi991675l. [DOI] [PubMed] [Google Scholar]
- Francklyn C. S.; First E. A.; Perona J. J.; Hou Y. M. Methods for Kinetic and Thermodynamic Analysis of Aminoacyl-tRNA Synthetases. Methods 2008, 44, 100–118. 10.1016/j.ymeth.2007.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illangasekare M.; Yarus M. A Tiny RNA that Catalyzes Both Aminoacyl-RNA and Peptidyl-RNA Synthesis. RNA 1999, 5, 1482–1489. 10.1017/S1355838299991264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turk R. M.; Chumachenko N. V.; Yarus M. Multiple Translational Products from a Five-Nucleotide Ribozyme. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 4585–4589. 10.1073/pnas.0912895107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito H.; Suga H. A Ribozyme Exclusively Aminoacylates the 3′-Hydroxyl Group of the tRNA Terminal Adenosine. J. Am. Chem. Soc. 2001, 123, 7178–7179. 10.1021/ja015756s. [DOI] [PubMed] [Google Scholar]
- Saito H.; Watanabe K.; Suga H. Concurrent Molecular Recognition of the Amino Acid and tRNA by a Ribozyme. RNA 2001, 7, 1867–1878. [PMC free article] [PubMed] [Google Scholar]
- Murakami H.; Ohta A.; Ashigai H.; Suga H. A Highly Flexible tRNA Acylation Method for Non-Natural Polypeptide Synthesis. Nat. Methods 2006, 3, 357–359. 10.1038/nmeth877. [DOI] [PubMed] [Google Scholar]
- Ohta A.; Murakami H.; Higashimura E.; Suga H. Synthesis of Polyester by Means of Genetic Code Reprogramming. Chem. Biol. 2007, 14, 1315–1322. 10.1016/j.chembiol.2007.10.015. [DOI] [PubMed] [Google Scholar]
- Fujino T.; Goto Y.; Suga H.; Murakami H. Reevaluation of the d-Amino Acid Compatibility with the Elongation Event in Translation. J. Am. Chem. Soc. 2013, 135, 1830–1837. 10.1021/ja309570x. [DOI] [PubMed] [Google Scholar]
- Fujino T.; Goto Y.; Suga H.; Murakami H. Ribosomal Synthesis of Peptides with Multiple Beta-Amino Acids. J. Am. Chem. Soc. 2016, 138, 1962–1969. 10.1021/jacs.5b12482. [DOI] [PubMed] [Google Scholar]
- Katoh T.; Suga H. Flexizyme-Catalyzed Synthesis of 3′-Aminoacyl-NH-tRNAs. Nucleic Acids Res. 2019, 47, e54 10.1093/nar/gkz143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohuchi M.; Murakami H.; Suga H. The Flexizyme System: a Highly Flexible tRNA Aminoacylation Tool for the Translation Apparatus. Curr. Opin. Chem. Biol. 2007, 11, 537–542. 10.1016/j.cbpa.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Hopp T. P.; Woods K. R. Prediction of Protein Antigenic Determinants from Amino Acid Sequences. Proc. Natl. Acad. Sci. U. S. A. 1981, 78, 3824–3828. 10.1073/pnas.78.6.3824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez R. M.; Polanco J. A.; Lupták A. Chemistry and biology of self-cleaving ribozymes. Trends Biochem. Sci. 2015, 40 (11), 648–661. 10.1016/j.tibs.2015.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco C.; Bayas M.; Yan F.; Chen I. A. Analysis of Evolutionarily Independent Protein-RNA Complexes Yields a Criterion to Evaluate the Relevance of Prebiotic Scenarios. Curr. Biol. 2018, 28, 526–537. 10.1016/j.cub.2018.01.014. [DOI] [PubMed] [Google Scholar]
- Ferre-D’Amare A. R.; Scott W. G. Small Self-Cleaving Ribozymes. Cold Spring Harbor Perspect. Biol. 2010, 2, a003574. 10.1101/cshperspect.a003574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prody G. A.; Bakos J. T.; Buzayan J. M.; Schneider I. R.; Bruening G. Autolytic Processing of Dimeric Plant-Virus Satellite Rna. Science 1986, 231, 1577–1580. 10.1126/science.231.4745.1577. [DOI] [PubMed] [Google Scholar]
- Uhlenbeck O. C. A. Small Catalytic Oligoribonucleotide. Nature 1987, 328, 596–600. 10.1038/328596a0. [DOI] [PubMed] [Google Scholar]
- Haseloff J.; Gerlach W. L. Simple RNA Enzymes with New and Highly Specific Endoribonuclease Activities. Nature 1988, 334, 585–591. 10.1038/334585a0. [DOI] [PubMed] [Google Scholar]
- Jeffries A. C.; Symons R. H. A. Catalytic 13-Mer Ribozyme. Nucleic Acids Res. 1989, 17, 1371–1377. 10.1093/nar/17.4.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odai O.; Hiroaki H.; Tanaka T.; Uesugi S. Properties of a Hammerhead-Type Rna Enzyme-System That Consists of 3 Rna Oligomer Strands. Nucleosides Nucleotides 1994, 13, 1569–1579. 10.1080/15257779408012172. [DOI] [Google Scholar]
- Clouet-D’Orval B.; Uhlenbeck O. C. Kinetic Characterization of Two I/II Format Hammerhead Ribozymes. RNA 1996, 2, 483–491. [PMC free article] [PubMed] [Google Scholar]
- Ruffner D. E.; Stormo G. D.; Uhlenbeck O. C. Sequence Requirements of the Hammerhead Rna Self-Cleavage Reaction. Biochemistry 1990, 29, 10695–10702. 10.1021/bi00499a018. [DOI] [PubMed] [Google Scholar]
- Perriman R.; Delves A.; Gerlach W. L. Extended Target-Site Specificity for a Hammerhead Ribozyme. Gene 1992, 113, 157–163. 10.1016/0378-1119(92)90391-2. [DOI] [PubMed] [Google Scholar]
- Zoumadakis M.; Tabler M. Comparative-Analysis of Cleavage Rates after Systematic Permutation of the Nux-Down-Arrow Consensus Target Motif for Hammerhead Ribozymes. Nucleic Acids Res. 1995, 23, 1192–1196. 10.1093/nar/23.7.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hertel K. J.; Herschlag D.; Uhlenbeck O. C. Specificity of Hammerhead Ribozyme Cleavage. EMBO J. 1996, 15, 3751–3757. 10.1002/j.1460-2075.1996.tb00745.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimayama T.; Nishikawa S.; Taira K. Generality of the Nux Rule - Kinetic-Analysis of the Results of Systematic Mutations in the Trinucleotide at the Cleavage Site of Hammerhead Ribozymes. Biochemistry 1995, 34, 3649–3654. 10.1021/bi00011a020. [DOI] [PubMed] [Google Scholar]
- Herschlag D. Implications of Ribozyme Kinetics for Targeting the Cleavage of Specific RNA Molecules In Vivo: More isn’t Always Better. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 6921–6925. 10.1073/pnas.88.16.6921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koizumi M.; Ohtsuka E. Effects of Phosphorothioate and 2-Amino Groups in Hammerhead Ribozymes on Cleavage Rates and Mg2+ Binding. Biochemistry 1991, 30, 5145–5150. 10.1021/bi00235a005. [DOI] [PubMed] [Google Scholar]
- Huang F.; Yarus M. 5′-RNA Self-Capping from Guanosine Diphosphate. Biochemistry 1997, 36, 6557–6563. 10.1021/bi970475b. [DOI] [PubMed] [Google Scholar]
- Zaher H. S.; Watkins R. A.; Unrau P. J. Two Independently Selected Capping Ribozymes Share Similar Substrate Requirements. RNA 2006, 12, 1949–1958. 10.1261/rna.131306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F.; Yarus M. A Calcium-Metalloribozyme with Autodecapping and Pyrophosphatase Activities. Biochemistry 1997, 36, 14107–14119. 10.1021/bi971081n. [DOI] [PubMed] [Google Scholar]
- Huang F.; Yarus M. Versatile 5′ Phosphoryl Coupling of Small and Large Molecules to an RNA. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 8965–8969. 10.1073/pnas.94.17.8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F.; Yarus M. Kinetics at a Multifunctional RNA Active Site. J. Mol. Biol. 1998, 284, 255–267. 10.1006/jmbi.1998.2169. [DOI] [PubMed] [Google Scholar]
- Zaher H. S.; Unrau P. J. A General RNA-Capping Ribozyme Retains Stereochemistry During Cap Exchange. J. Am. Chem. Soc. 2006, 128, 13894–13900. 10.1021/ja0639822. [DOI] [PubMed] [Google Scholar]
- Bartel D. P.; Szostak J. W. Isolation of New Ribozymes from a Large Pool of Random Sequences. Science 1993, 261, 1411–1418. 10.1126/science.7690155. [DOI] [PubMed] [Google Scholar]
- Ekland E. H.; Szostak J. W.; Bartel D. P. Structurally Complex and Highly Active RNA Ligases Derived from Random RNA Sequences. Science 1995, 269, 364–370. 10.1126/science.7618102. [DOI] [PubMed] [Google Scholar]
- Ekland E. H.; Bartel D. P. The secondary structure and sequence optimization of an RNA ligase ribozyme. Nucleic Acids Res. 1995, 23, 3231–3238. 10.1093/nar/23.16.3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekland E. H.; Bartel D. P. RNA-Catalysed RNA Polymerization Using Nucleoside Triphosphates. Nature 1996, 382, 373–376. 10.1038/382373a0. [DOI] [PubMed] [Google Scholar]
- Johnston W. K.; Unrau P. J.; Lawrence M. S.; Glasner M. E.; Bartel D. P. RNA-Catalyzed RNA Polymerization: Accurate and General RNA-Templated Primer Extension. Science 2001, 292, 1319–1325. 10.1126/science.1060786. [DOI] [PubMed] [Google Scholar]
- Eigen M. Selforganization of Matter and the Evolution of Biological Macromolecules. Naturwissenschaften 1971, 58, 465–523. 10.1007/BF00623322. [DOI] [PubMed] [Google Scholar]
- Rajamani S.; Ichida J. K.; Antal T.; Treco D. A.; Leu K.; Nowak M. A.; Szostak J. W.; Chen I. A. Effect of Stalling after Mismatches on the Error Catastrophe in Nonenzymatic Nucleic Acid Replication. J. Am. Chem. Soc. 2010, 132, 5880–5885. 10.1021/ja100780p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M. S.; Bartel D. P. Processivity of Ribozyme-Catalyzed RNA Polymerization. Biochemistry 2003, 42, 8748–8755. 10.1021/bi034228l. [DOI] [PubMed] [Google Scholar]
- Zaher H. S.; Unrau P. J. Selection of an Improved RNA Polymerase Ribozyme with Superior Extension and Fidelity. RNA 2007, 13, 1017–1026. 10.1261/rna.548807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q. S.; Cheng L. K.; Unrau P. J. Characterization of the B6.61 Polymerase Ribozyme Accessory Domain. RNA 2011, 17, 469–477. 10.1261/rna.2495011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wochner A.; Attwater J.; Coulson A.; Holliger P. Ribozyme-Catalyzed Transcription of an Active Ribozyme. Science 2011, 332, 209–212. 10.1126/science.1200752. [DOI] [PubMed] [Google Scholar]
- Horning D. P.; Joyce G. F. Amplification of RNA by an RNA Polymerase Ribozyme. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 9786–9791. 10.1073/pnas.1610103113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwater J.; Raguram A.; Morgunov A. S.; Gianni E.; Holliger P. Ribozyme-Catalysed RNA Synthesis Using Triplet Building Blocks. eLife 2018, 7, e35255 10.7554/eLife.35255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbesi A.; Capobinanco M. L.; Colonna F. P.; Tondelli L.; Arcamone F.; Manzini G.; hilbers C. W.; Aelen J. M.E.; Blommers M. J.J. l-DNAs as Potential Antimessenger Oligonucleotides: a Reassessment. Nucleic Acids Res. 1993, 21, 4159–4165. 10.1093/nar/21.18.4159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbesi A.; Capobianco M. L.; Colonna F. P.; Maflini M.; Niccoiai D.; Tondelli L. Chirally-Modified Oligonucleotides and the Control of Gene Expression. The Case of l-DNAs and -RNAs. Nucleosides Nucleotides 1998, 17, 1275–1287. 10.1080/07328319808004239. [DOI] [Google Scholar]
- Sczepanski J. T.; Joyce G. F. A Cross-Chiral RNA Polymerase Ribozyme. Nature 2014, 515, 440–442. 10.1038/nature13900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce G. F.; Visser G. M.; van Boeckel C. A.; van Boom J. H.; Orgel L. E.; van Westrenen J. Chiral Selection in Poly(C)-Directed Synthesis of Oligo(G). Nature 1984, 310, 602–604. 10.1038/310602a0. [DOI] [PubMed] [Google Scholar]
- Attwater J.; Tagami S.; Kimoto M.; Butler K.; Kool E. T.; Wengel J.; Herdewijn P.; Hirao I.; Holliger P. Chemical Fidelity of an RNA Polymerase Ribozyme. Chem. Sci. 2013, 4, 2804–2814. 10.1039/c3sc50574j. [DOI] [Google Scholar]
- Samanta B.; Joyce G. F. A Reverse Transcriptase Ribozyme. eLife 2017, 6, e31153 10.7554/eLife.31153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horning D. P.; Bala S.; Chaput J. C.; Joyce G. F. RNA-Catalyzed Polymerization of Deoxyribose, Threose, and Arabinose Nucleic Acids. ACS Synth. Biol. 2019, 8, 955–961. 10.1021/acssynbio.9b00044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekland E. H.; Bartel D. P. The Secondary Structure and Sequence Optimization of an Rna Ligase Ribozyme. Nucleic Acids Res. 1995, 23, 3231–3238. 10.1093/nar/23.16.3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shechner D. M.; Grant R. A.; Bagby S. C.; Koldobskaya Y.; Piccirilli J. A.; Bartel D. P. Crystal Structure of the Catalytic Core of an RNA-Polymerase Ribozyme. Science 2009, 326, 1271–1275. 10.1126/science.1174676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F.; Yang Z.; Yarus M. RNA Enzymes with Two Small-Molecule Substrates. Chem. Biol. 1998, 5, 669–678. 10.1016/S1074-5521(98)90294-0. [DOI] [PubMed] [Google Scholar]
- Forconi M.; Herschlag D. Promiscuous Catalysis by the Tetrahymena Group I Ribozyme. J. Am. Chem. Soc. 2005, 127, 6160–6161. 10.1021/ja050180i. [DOI] [PubMed] [Google Scholar]
- Lau M. W.; Unrau P. J. A Promiscuous Ribozyme Promotes Nucleotide Synthesis in Addition to Ribose Chemistry. Chem. Biol. 2009, 16, 815–825. 10.1016/j.chembiol.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Lau M. W.; Cadieux K. E.; Unrau P. J. Isolation of Fast Purine Nucleotide Synthase Ribozymes. J. Am. Chem. Soc. 2004, 126, 15686–15693. 10.1021/ja045387a. [DOI] [PubMed] [Google Scholar]
- Nissen P.; Hansen J.; Ban N.; Moore P. B.; Steitz T. A. The Structural Basis of Ribosome Activity in Peptide Bond Synthesis. Science 2000, 289, 920–930. 10.1126/science.289.5481.920. [DOI] [PubMed] [Google Scholar]
- Green R.; Noller H. F. Ribosomes and Translation. Annu. Rev. Biochem. 1997, 66, 679–716. 10.1146/annurev.biochem.66.1.679. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan V. Ribosome Structure and the Mechanism of Translation. Cell 2002, 108, 557–572. 10.1016/S0092-8674(02)00619-0. [DOI] [PubMed] [Google Scholar]
- Fox G. E. Origin and Evolution of the Ribosome. Cold Spring Harbor Perspect. Biol. 2010, 2, a003483. 10.1101/cshperspect.a003483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogle J. M.; Ramakrishnan V. Structural Insights into Translational Fidelity. Annu. Rev. Biochem. 2005, 74, 129–177. 10.1146/annurev.biochem.74.061903.155440. [DOI] [PubMed] [Google Scholar]
- Ling J.; Reynolds N.; Ibba M. Aminoacyl-tRNA Synthesis and Translational Quality Control. Annu. Rev. Microbiol. 2009, 63, 61–78. 10.1146/annurev.micro.091208.073210. [DOI] [PubMed] [Google Scholar]
- Kuncha S. K.; Kruparani S. P.; Sankaranarayanan R. Chiral Checkpoints During Protein Biosynthesis. J. Biol. Chem. 2019, 294, 16535–16548. 10.1074/jbc.REV119.008166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Z. M.; Bussema C. 3rd; Schmidt T. M. rrnDB: Documenting the Number of rRNA and tRNA Genes in Bacteria and Archaea. Nucleic Acids Res. 2009, 37, D489–493. 10.1093/nar/gkn689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selmer M.; Dunham C. M.; Murphy F. V. t.; Weixlbaumer A.; Petry S.; Kelley A. C.; Weir J. R.; Ramakrishnan V. Structure of the 70S Ribosome Complexed with mRNA and tRNA. Science 2006, 313, 1935–1942. 10.1126/science.1131127. [DOI] [PubMed] [Google Scholar]
- Monro R. E.; Cerna J.; Marcker K. A. Ribosome-Catalyzed Peptidyl Transfer: Substrate Specificity at the P-site. Proc. Natl. Acad. Sci. U. S. A. 1968, 61, 1042–1049. 10.1073/pnas.61.3.1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruan X. L.; Li S.; Geng P.; Zeng X. T.; Yu G. Z.; Meng X. Y.; Gao Q. P.; Ao X. B. Association Between TP53 Gene Codon 72 Polymorphism and Acute Myeloid Leukemia Susceptibility: Evidence Based on a Meta-Analysis. Med. Sci. Monit. 2015, 21, 3048–3053. 10.12659/MSM.894625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C. C.; Schultz P. G. Adding New Chemistries to the Genetic Code. Annu. Rev. Biochem. 2010, 79, 413–444. 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- Mohler K.; Ibba M. Translational Fidelity and Mistranslation in the Cellular Response to Stress. Nat. Microbiol. 2017, 2, 17117. 10.1038/nmicrobiol.2017.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogle J. M.; Murphy F. V.; Tarry M. J.; Ramakrishnan V. Selection of tRNA by the Ribosome Requires a Transition from an Open to a Closed Form. Cell 2002, 111, 721–732. 10.1016/S0092-8674(02)01086-3. [DOI] [PubMed] [Google Scholar]
- Moazed D.; Noller H. F. Sites of Interaction of the CCA End of Peptidyl-tRNA with 23S rRNA. Proc. Natl. Acad. Sci. U. S. A. 1991, 88, 3725–3728. 10.1073/pnas.88.9.3725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarmolinsky M. B.; Haba G. L. Inhibition by Puromycin of Amino Acid Incorporation into Protein. Proc. Natl. Acad. Sci. U. S. A. 1959, 45, 1721–1729. 10.1073/pnas.45.12.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathans D. Inhibition of Protein Synthesis by Puromycin. Fed. Proc. 1964, 23, 984–989. [PubMed] [Google Scholar]
- Bain J. D.; Diala E. S.; Glabe C. G.; Dix T. A.; Chamberlin A. R. Biosynthetic Site-Specific Incorporation of a Non-Natural Amino Acid into a Polypeptide. J. Am. Chem. Soc. 1989, 111, 8013–8014. 10.1021/ja00202a052. [DOI] [Google Scholar]
- Noren C. J.; Anthony-Cahill S. J.; Griffith M. C.; Schultz P. G. A General Method for Site-Specific Incorporation of Unnatural Amino Acids into Proteins. Science 1989, 244, 182–188. 10.1126/science.2649980. [DOI] [PubMed] [Google Scholar]
- Dougherty D. Unnatural Amino Acids as Probes of Protein Structure and Function. Curr. Opin. Chem. Biol. 2000, 4, 645–652. 10.1016/S1367-5931(00)00148-4. [DOI] [PubMed] [Google Scholar]
- Hohsaka T.; Sisido M. Incorporation of Non-Natural Amino Acids into Proteins. Curr. Opin. Chem. Biol. 2002, 6, 809–815. 10.1016/S1367-5931(02)00376-9. [DOI] [PubMed] [Google Scholar]
- Xie J.; Schultz P. G. A Chemical Toolkit for Proteins--an Expanded Genetic Code. Nat. Rev. Mol. Cell Biol. 2006, 7, 775–782. 10.1038/nrm2005. [DOI] [PubMed] [Google Scholar]
- Wang K.; Neumann H.; Peak-Chew S. Y.; Chin J. W. Evolved Orthogonal Ribosomes Enhance the Efficiency of Synthetic Fenetic Code Expansion. Nat. Biotechnol. 2007, 25, 770–777. 10.1038/nbt1314. [DOI] [PubMed] [Google Scholar]
- Neumann H.; Wang K.; Davis L.; Garcia-Alai M.; Chin J. W. Encoding Multiple Unnatural Amino Acids via Evolution of a Quadruplet-Decoding Ribosome. Nature 2010, 464, 441–444. 10.1038/nature08817. [DOI] [PubMed] [Google Scholar]
- Jukes T. H.; Osawa S. The Genetic Code in Mitochondria and Chloroplasts. Experientia 1990, 46, 1117–1126. 10.1007/BF01936921. [DOI] [PubMed] [Google Scholar]
- Freeland S. J.; Hurst L. D. The Genetic Code Is One in a Million. J. Mol. Evol. 1998, 47, 238–248. 10.1007/PL00006381. [DOI] [PubMed] [Google Scholar]
- Lohse P. A.; Szostak J. W. Ribozyme-Catalysed Amino-Acid Transfer Reactions. Nature 1996, 381, 442–444. 10.1038/381442a0. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Cech T. R. Peptide Bond Formation by In Vitro Selected Ribozymes. Nature 1997, 390, 96–100. 10.1038/36375. [DOI] [PubMed] [Google Scholar]
- Tarasow T. M.; Tarasow S. L.; Eaton B. E. RNA-Catalysed Carbon-Carbon Bond Formation. Nature 1997, 389, 54–57. 10.1038/37950. [DOI] [PubMed] [Google Scholar]
- Seelig B.; Jaschke A. A Small Catalytic RNA Motif with Diels–Alderase Activity. Chem. Biol. 1999, 6, 167–176. 10.1016/S1074-5521(99)89008-5. [DOI] [PubMed] [Google Scholar]
- Seelig B.; Keiper S.; Stuhlmann F.; Jaschke A. Enantioselective Ribozyme Catalysis of a Bimolecular Cycloaddition Reaction. Angew. Chem., Int. Ed. 2000, 39, 4576–4579. . [DOI] [PubMed] [Google Scholar]
- Stuhlmann F.; Jaschke A. Characterization of an RNA Active Site: Interactions Between a Diels–Alderase Ribozyme and Its Substrates and Products. J. Am. Chem. Soc. 2002, 124, 3238–3244. 10.1021/ja0167405. [DOI] [PubMed] [Google Scholar]
- Siegel J. B.; Zanghellini A.; Lovick H. M.; Kiss G.; Lambert A. R.; St. Clair J. L.; Gallaher J. L.; Hilvert D.; Gelb M. H.; Stoddard B. L.; Houk K. N.; Michael F. E.; Baker D.; et al. Computational Design of an Enzyme Catalyst for a Stereoselective Bimolecular Diels–Alder Reaction. Science 2010, 329, 309–313. 10.1126/science.1190239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouellette R. J.; Rawn J. D.. Organic Chemistry: Structure, Mechanism, Synthesis; 2nd ed.; Academic Press: London, San Diego, CA, 2018. [Google Scholar]
- Pressman A.; Moretti J. E.; Campbell G. W.; Muller U. F.; Chen I. A. Analysis of In Vitro Evolution Reveals the Underlying Distribution of Catalytic Activity Among Random Sequences. Nucleic Acids Res. 2017, 45, 10922. 10.1093/nar/gkx816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carothers J. M.; Oestreich S. C.; Szostak J. W. Aptamers Selected for Higher-Affinity Binding Are Not More Specific for the Target Ligand. J. Am. Chem. Soc. 2006, 128, 7929–7937. 10.1021/ja060952q. [DOI] [PMC free article] [PubMed] [Google Scholar]