

Abstract
Stapled peptides recapitulate the binding affinity and specificity of α-helices in proteins, resist proteolytic degradation, and may provide a novel modality against challenging drug targets such as protein-protein interactions. However, most of the stapled peptides have limited cell-permeability or are impermeable to the cell membrane. We show herein that stapled peptides can be rendered highly cell-permeable by conjugating a cyclic cell-penetrating peptide to their N-terminus, C-terminus, or stapling unit. Application of this strategy to two previously reported, membrane-impermeable peptidyl inhibitors against the MDM2/p53 and β-catenin/TCF interactions resulted in the generation of potent proof-of-concept anti-proliferative agents against key therapeutic targets.
Keywords: Anticancer, cell-penetrating peptide, drug delivery, protein-protein interaction, stapled peptide
Graphial Abstract
Introduction
Stapled peptides have emerged as a novel class of therapeutic agents for targeting intracellular protein-protein interactions (PPIs), which are challenging targets for conventional small molecules and biologics.1,2 When appropriately designed, stapled peptides have demonstrated the capacity to resist proteolytic degradation, enter the cytosol and nucleus of mammalian cells, and inhibit intracellular PPIs mediated by α-helices. A variety of stapling methodologies have been described in the literature3, with hydrocarbon-stapled α-helices receiving considerable attention. The first demonstration of cellular activity by a stapled peptide was made by Walensky et al., who showed that a hydrocarbon-stapled α-helix mimicking the BCL-2 homology 3 (BH3) domain of the pro-apoptotic protein BID had the capacity for cellular uptake by endocytosis, resulting in apoptotic cell death.4 Further development of the stapling strategy has led to a stapled-peptide drug in clinical trials for targeting an intracellular PPI between HDM2/HDMX and p53 (clinicaltrials.gov identifier: NCT02264613). However, despite their remarkable promise as therapeutics, designing stapled peptides with predictable cell-permeability remains a difficult task. Comprehensive analyses of hundreds of stapled peptides by the Verdine5 and Walensky groups6 suggested that optimal hydrophobicity, positive charge, α-helicity, and staple composition and placement are the key drivers of cellular uptake, whereas excess hydrophobicity and positive charge can trigger membrane lysis at elevated peptide dosing. These studies revealed that while therapeutically relevant cell-permeability is feasible for certain stapled peptides, the vast majority of them are either impermeable to the cell membrane or of very limited cell-permeability. Thus, a more general method is needed to enhance the cytosolic entry of stapled peptides.
We previously discovered a novel family of cyclic cell-penetrating peptides (CPPs), which are capable of efficiently delivering membrane-impermeable small molecules, linear and cyclic peptides, and proteins into the cytosol and nucleus of mammalian cells by endocytic mechanisms.7–9 Other examples of cyclic CPPs (e.g. cyclic Tat10,11 and Arg-rich bicyclic peptides12) have also been reported in the literature, but generally lack significant cytosolic uptake except at high concentrations (≥25 μM) when alternative cellular entry mechanisms (e.g., direct translocation) occur. We envisioned that cyclic CPPs should also be able to improve the cell-permeability of stapled peptides. In this study, we tested this hypothesis by conjugating a cyclic CPP to the N-terminus, C-terminus, or stapling unit of a panel of stapled peptides of different intrinsic cell permeability. We show that conjugation to a cyclic CPP renders all of the stapled peptides tested highly cell-permeable.
RESULTS AND DISCUSSION
Stapled Peptidyl Inhibitors of MDM2-p53 Interaction.
We first chose to generate a cell-permeable stapled peptide inhibitor against the MDM2-p53 interaction. Intracellular accumulation of tumor suppressor protein p53 leads to a plethora of cellular effects, including apoptosis, to guard against the propagation of cells that carry damaged DNA with potentially oncogenic mutations. In healthy cells, a proper p53 level is maintained through synthesis and proteasomal degradation mediated by MDM2, a p53-specific E3 ubiquitin ligase.13 In many cancers, overexpression of MDM2 results in excessive degradation of p53 and loss of its growth-suppressive function.14 Therefore, inhibition of the MDM2-p53 interaction has emerged as a promising therapeutic approach to treating p53WT cancers.15,16 Multiple small molecules have entered clinical trials with limited success while displaying severe dose-limiting toxicity due to off-target effects.17 Researchers have since turned to stapled peptides as a promising therapeutic modality to target the MDM2-p53 interaction because of their improved target selectivity. Several highly potent stapled peptides have been developed against MDM2, but they generally suffer from poor cellular uptake.18,19 Efforts to rationally optimize cellular uptake by employing alternative stapling methodologies did yield some promising compounds, but they still suffer from low cellular potency.20 Improving uptake for hydrocarbon-stapled peptides through incorporation of cationic residues by Quach et al. resulted in considerable off-target toxicity21, similar to what was observed with direct conjugation to a linear cationic CPP.22–24 Therefore, generation of a cell-permeable stapled peptide with robust on-target activity would be able to ameliorate many of the issues observed with previous MDM2-p53 inhibitors.
We conjugated CPP9, a highly active cyclic CPP,9 to a previously reported MDM2 ligand, Ac-LTFEHYWAQLTS (PDI; Table 1, peptide 1).18 To staple PDI, we replaced Glu-4 and Ala-8 with aspartic acid and lysine, respectively, and crosslinked their side chains through an amide bond (Table 1, peptide 2). Fairlie and coworkers25 had previously shown that a similar lactam linker formed by the side chains of a lysine/aspartate pair at the i and i+4 positions (KD linker) results in consistently higher α-helicity for stapled peptides than other commonly used staples. The lactam linker is also more hydrophilic than other staples (e.g., all hydrocarbon staple), improving the aqueous solubility of the stapled peptides. Conjugation of CPP9 to the N- or C-terminus of peptide 2 via a miniPEG linker gave peptides 3 and 4, respectively (Figure 1a,b; see Figure S1 in Supporting Information for detailed structures). The binding affinity of peptides 2–4 for MDM2 was determined by a fluorescence polarization (FP)-based competition assay by using tetramethylrhodamine (TMR)-labeled PDI as probe. Peptides 2–4 showed IC50 values of 10.5 ± 0.8, 24 ± 3, and 11.9 ± 0.3 nM, respectively (Figure 2a). Peptide 4 was selected for further studies because of its higher binding affinity. Affinity differences observed between N- and C-terminal conjugation likely result from unfavorable steric interactions between the transporter sequence and MDM2. Computational modeling reveals that C-terminal conjugation results in the transporter sequence extending into the solvent without perturbing the native binding mode (Figure 1d). As a negative control, we synthesized peptide 5 by replacing a key binding residue of peptide 4, phenylalanine at position 3, with an alanine. Peptide 5 bound MDM2 with >1000-fold lower affinity than peptide 4 (IC50 > 20 μM, Figure 2a). For comparison, we also conjugated two prototypical linear CPPs, Tat and nonaarginine (R9), to peptide 2 to give peptides 6 and 7, respectively, which bound MDM2 with similar affinities to peptide 4 (Figure S2).
Table 1.
Sequences of peptides used in this work
| Peptide. | Sequence[a] | Staple |
|---|---|---|
| 1 | Ac-LTFEHYWAQLTS-miniPEG-K(TMR)-NH2 | none |
| 2 | Ac-LTFDHYWKQLTS-miniPEG-K-NH2 | Asp-Lys |
| 3 | CPP9-miniPEG-LTFDHYWKQLTS-NH2 | Asp-Lys |
| 4 | Ac-LTFDHYWKQLTS-miniPEG-K(CPP9) | Asp-Lys |
| 5 | Ac-LTADHYWKQLTS-miniPEG-K(CPP9) | Asp-Lys |
| 6 | Ac-LTFDHYWKQLTS-miniPEG-K(Tat) | Asp-Lys |
| 7 | Ac-LTFDHYWKQLTS-miniPEG-K(R9) | Asp-Lys |
| 8 | FAM-GGYPECILDCHLQRVIL-NH2 | xylene |
| 9 | Ac-GGYPEDILDKHLQRVIL-(miniPEG)2-Dap(CPP9)-NH2 | Asp-Lys |
| 10 | Ac-GGYPEDILDKHLQRVIL-(miniPEG)2-Dap(Tat)-NH2 | Asp-Lys |
| 11 | Ac-GGYPEDILDKHLQRVIL-(miniPEG)2-Dap(R9)-NH2 | Asp-Lys |
| 12 | NF-βA-RKFCRLFC-NH2 | xylene |
| 13 | NF-βA-RKFCRLFC-NH2 | BBA-CPP9 |
| 14 | NF-βA-ENPECILDCHVQRVM-NH2 | xylene |
| 15 | NF-βA-ENPECILDCHVQRVM-NH2 | BBA-CPP9 |
| 16 | NF-βA-NPECILDCHVQRVM-NH2 | xylene |
| 17 | NF-βA-NPECILDCHVQRVM-NH2 | BBA-CPP9 |
| 18 | NF-βA-TYRGAAQCAAQCVREV-NH2 | xylene |
| 19 | NF-βA-TYRGAAQCAAQCVREV-NH2 | BBA-CPP9 |
| 20 | Ac-LTFCHYWCQLTS-miniPEG-K(FAM) | xylene |
| 21 | Ac-LTFCHYWCQLTS-miniPEG-K(FAM) | BBA-CPP9 |
| 22 | Ac-ETFCHYWCQLTS-miniPEG-K(FAM) | xylene |
| 23 | Ac-ETFCHYWCQLTS-miniPEG-K(FAM) | BBA-CPP9 |
| 24 | Ac-EETFCHYWCQLTS-miniPEG-K(FAM) | xylene |
| 25 | Ac-EETFCHYWCQLTS-miniPEG-K(FAM) | BBA-CPP9 |
Underlined residues indicate the positions of stapling. βA, β-alanine; Dap, L-2,3-diaminopropionic acid; FAM, fluorescein; NF, naphthofluorescein. See Figure S1 for detailed structures.
Figure 1.
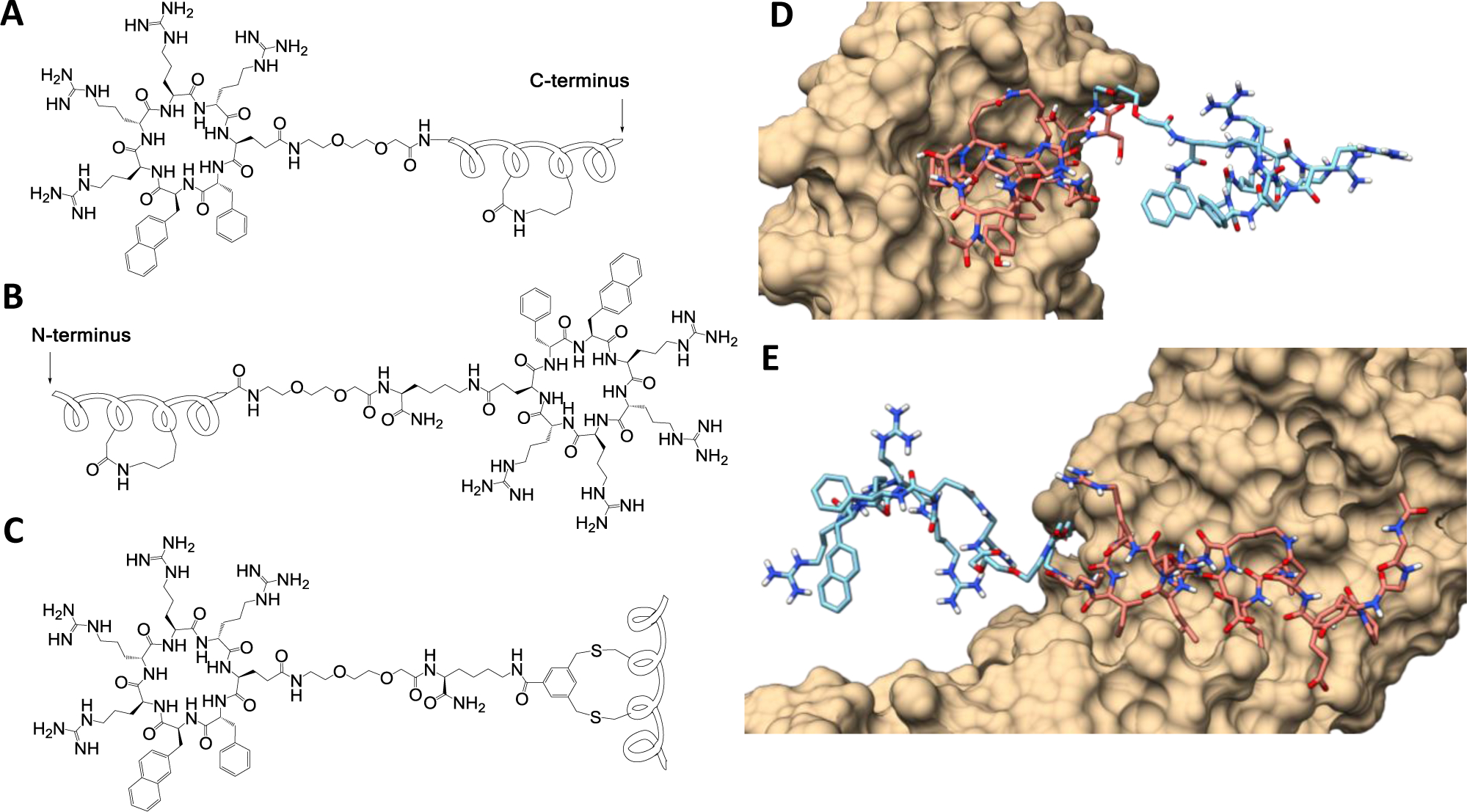
Cyclic CPP-stapled peptide conjugates. (A,B) Peptide stapling with a DK linker and conjugation to CPP at its N- or C-terminus. (C) Simultaneous stapling and conjugation to a CPP with a BBA staple. (D) Binding mode of stapled peptide-CPP conjugate 4 in complex with MDM2 as derived from MD simlations starting from PDBID:3EQS. (E) Binding mode of peptide 9 in complex with β-catenin derived from PDBID: 1QZ7. For (D) and (E), carbons in the stapled peptide are colored pink, while carbons in the transporter sequence are colored light blue, and the protein van der Waals surface is colored tan. Nitrogens, oxygens and polar hydrogens are depicted in blue, red, and white, respectively.
Figure 2.
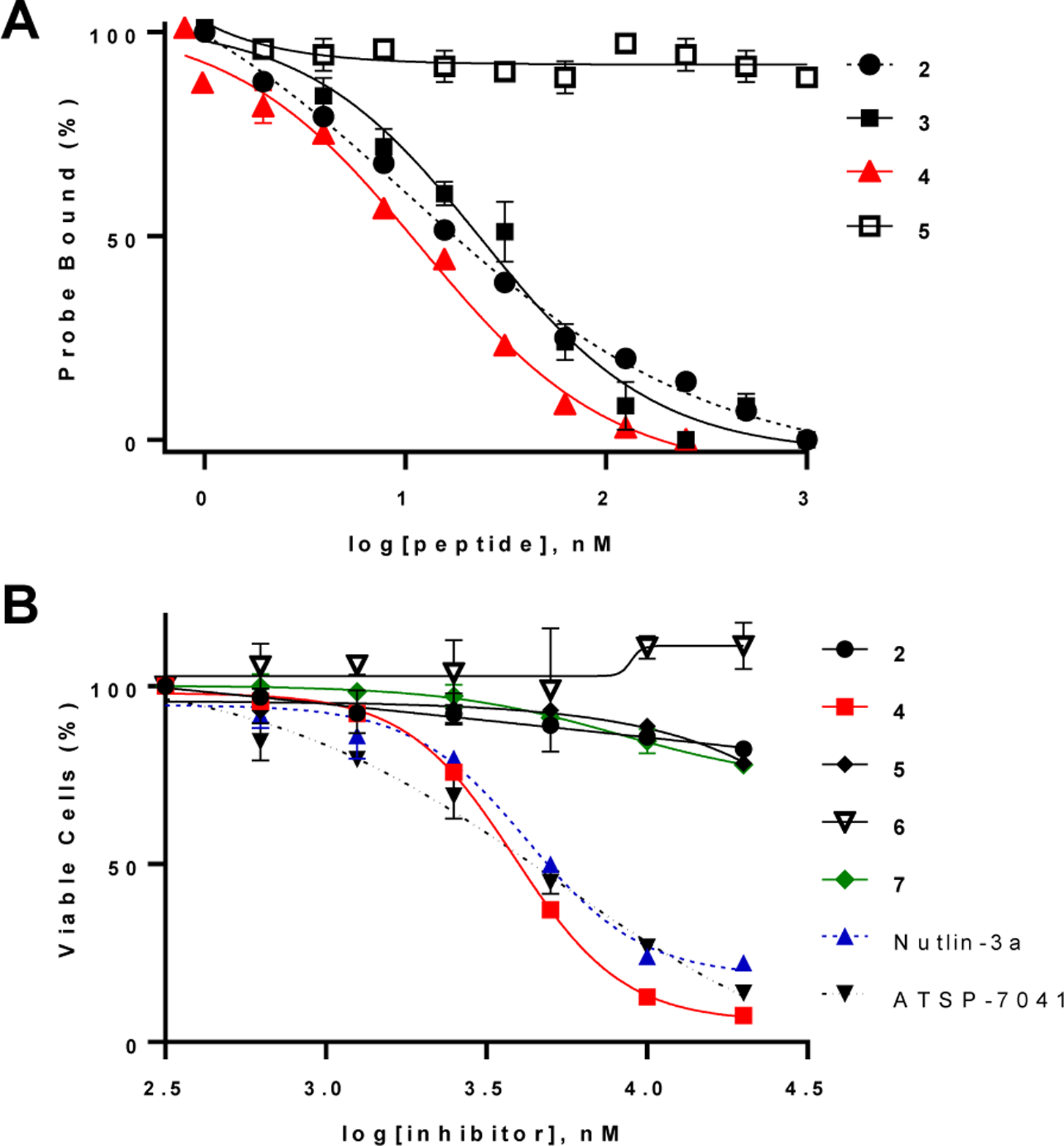
Anti-proliferative activity of MDM2 inhibitors. a) Competition of peptides 2–5 for binding to MDM2 (15 nM) with TMR-labeled PDI (probe; 15 nM). b) Effect of MDM2 inhibitors on the viability of SJSA-1 cells as determined by the MTT assay. Cells were serum starved for 24 h and then treated with indicated compounds for 72 h in complete growth media supplemented with 10% fetal bovine serum (FBS). Values reported represent the mean ± SD of ≥3 independent experiments.
Peptides 2 and 4–7 were evaluated for their ability to induce p53-dependent apoptosis of human osteosarcoma SJSA-1 cells, which express wild-type p53 and amplified levels of MDM2.26 Peptide 4 resulted in dose-dependent loss of viability of SJSA-1 cells, with an EC50 value of 3.8 ± 0.1 μM (Figure 2b). Under the same conditions, Nutlin-3a, a previously reported small-molecule inhibitor of MDM227 and a potent hydrocarbon-stapled peptidyl inhibitor (ATSP-7041)28 showed EC50 values of 4.2 ± 0.3 and 5.0 ± 1.4 μM, respectively. As expected, peptide 2 had no significant effect up to 20 μM concentration, given its poor cellular entry efficiency. Likewise, the “non-binding” peptide 5 showed no significant effect on SJSA-1 cells. Annexin V and propidium iodide (PI) staining indicated that SJSA-1 cells treated with peptide 4 underwent apoptotic cell death (Figure S3). To test whether peptide 4 disrupts the cell membrane, SJSA-1 cells were treated with increasing concentrations of peptide 4 (0–50 μM) for up to 4 h at 37 ᵒC and the amount of lactate dehydrogenase (LDH) activity released into the growth medium was quantified. Peptide 4 did not cause significant LDH release until at ≥25 μM concentration (Figure S4). Taken together, these results suggest that CPP9 effectively delivered PDI into the cytosol and peptide 4 induced apoptosis of SJSA-1 cells, possibly through the activation of p53, although we cannot rule out other off-target mechanism(s). The relatively high EC50 values of peptide 4, Nutlin-3a and ATSP-7041 may be caused by a number of factors, including the high intracellular MDM2 concentration in SJSA-1 cells, binding to serum proteins, and/or proteolytic degradation [in the case of peptide 4, which has a serum t1/2 of 13 h (Figure S5)]. Under the same assay condition, the Tat- and R9-conjugated peptides (peptides 6 and 7) did not cause significant reduction of the viability of SJSA-1 cells (Figure 2b), in agreement with earlier reports.22–24 Tat and R9 were previously shown to have 30- and 15-fold lower cytosolic delivery efficiencies, respectively, than cyclic CPP9.9
Peptide 4 also induced apoptosis of other cancer cells that express WT p53 protein, including MCF7 breast cancer cells (Figure S6), HCT116 colorectal carcinoma cells (Figure S7), and HeLa cervical cancer cells (Figure S7). However, these cells are less sensitive to MDM2 inhibition than SJSA-1 cells and treatment with ≤10 μM peptide 4 resulted in apoptosis of only a fraction of the cell population (5–50%). Higher concentrations of peptide 4 (≥20 μM) led to almost complete loss of cell viability, apparently through off-target mechanisms (e.g., membrane disruption), as suggested by the release of LDH (Figure S4).
Stapled Peptidyl Inhibitors of β-Catenin-TCF Interaction.
To test the generality of intracellular delivery of stapled peptides by cyclic CPPs, we next applied the above strategy to generate a cell-permeable peptidyl inhibitor against the β-catenin-TCF interaction. The Wnt signaling pathway has emerged as a key regulator for cancer stemness, metastasis, and self-regeneration. β-Catenin is an integral part of the canonical Wnt signaling cascade and is directly responsible for driving expression of Wnt target genes through interaction with transcription factor TCF.29 In the absence of Wnt, β-catenin resides in a “destruction complex” and is phosphorylated, ubiquitinated, and degraded. Wnt pathway activation induces dissociation of β-catenin from the complex, nuclear translocation and downstream gene expression. Mutations in the Wnt pathway lead to aberrant β-catenin activation in virtually all colorectal cancers, suggesting that inhibitors against the β-catenin-TCF interaction may provide a novel anticancer treatment.30 Therapeutic intervention in the periphery of the Wnt signaling pathway has raised concerns for off-target toxicity, while direct small-molecule inhibitors of the β-catenin-TCF interaction have been met with limited translational success owing to the challenge of targeting a PPI.31 Hydrocarbon- and thioether-stapled peptides have been developed against this interaction, but they lack potent biological activity due to their limited membrane permeability.32–34 Efforts by Dietrich et al. to incorporate a small cationic CPP sequence in the context of a hydrocarbon-stapled peptide resulted in modest biological activity.35 More recently, linear as well as macrocyclic peptidomimetic inhibitors of β-catenin have also been reported.36,37
To this end, we chose a previously reported m-xylene-stapled peptidyl inhibitor, FAM-GGYPECILDCHLQRVIL-NH2 (Table 1, peptide 8), which has high affinity for β-catenin (KD = 18 nM) but is membrane-impermeable.34 We replaced the two cysteine residues with aspartic acid and lysine and the m-xylene staple with a DK linker. The N-terminal FAM dye was removed to increase the aqueous solubility and the resulting peptide was conjugated to cyclic CPP9 at its C-terminus via a (miniPEG)2 linker (Table 1, peptide 9). Analysis of the binding mode through molecular dynamics indicated that C-terminal conjugation resulted in minimal disruption of the productive binding conformation with CPP9 extended into the solvent (Figure 1e). In an FP-based competition assay, peptide 9 bound to β-catenin with an IC50 value of 152 ± 8 nM (Figure S8). We also replaced the CPP9 moiety of peptide 9 with Tat and R9, to give peptides 10 and 11, respectively. Peptides 8–11 were tested for anti-proliferative activity against SW480 cells, a colorectal cancer cell line which has elevated levels of β-catenin due to an APC mutation and requires the Wnt signaling pathway for proliferation.38 Peptide 9 potently and dose-dependently reduced the viability of SW480 cells by up to ~60% after 72 h of incubation, with an EC50 value of 1.2 ± 0.4 μM (Figure 3a). In comparison, peptide 8 resulted in only a slight decrease in the viability of SW480 cells at the highest concentration tested (20 μM). Peptides 10 and 11 showed weak activities with IC50 values exceeding 20 μM (Figure 3b). To test whether the peptides exerted anti-proliferative effects through specific inhibition of Wnt signaling or as a result of general cytotoxicity, we tested the same set of peptides against Wnt-independent MCF7 cells. Peptide 9 up to 20 μM showed negligible effect on MCF7 cell viability (Figure 3a), consistent with the notion that peptide 9 inhibits SW480 cell proliferation through inhibition of β-catenin. On the other hand, peptides 10 and 11 also reduced the viability of MCF7 cells, albeit to a lesser extent compared to SW480 cells, indicating that peptides 10 and 11 cause cytotoxicity due to as yet unidentified off-target effects (Figure 3b). Peptide 9 reduced the mRNA and protein levels of Myc, Axin2, and LGR5 (which are genes upregulated by β-catenin) in Wnt-dependent cells (SW480 and HCT-116) but not Wnt-independent cells (data not shown). Annexin V and PI staining revealed that peptide 9 did not induce apoptosis or necrosis of SW480 cells or another Wnt-addicted colorectal cancer cell line, DLD-1 cells, until 25 μM concentration, when ~10% cells underwent apoptotic cell death (Figures S9 and S10). Further, treatment of SW480 cells with up to 50 μM peptide 9 did not cause significant LDH release (Figure S4). Our results are in agreement with an earlier report that knockdown of β-catenin results in activation of apoptosis among ~20% of the cell population,39 indicating that peptide 9 reduces the viability of SW480 and DLD-1 cells primarily by inhibiting their rate of proliferation. Peptide 9 has a serum t1/2 of ~2 h (Figure S5); proteolytic degradation of the peptide likely contributed to the relatively large difference between the in vitro IC50 and cellular EC50 values (8-fold).
Figure 3.
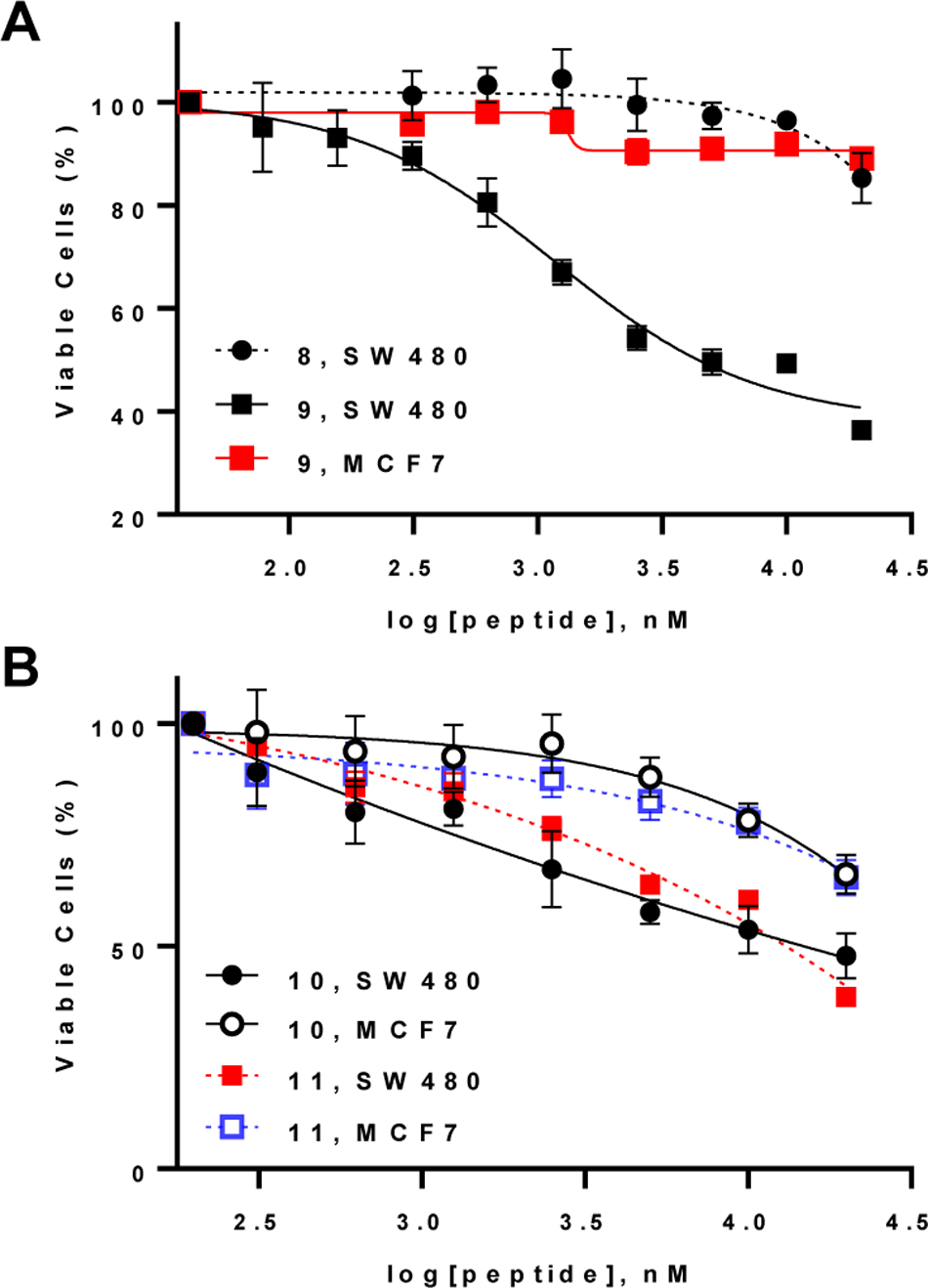
Effect of β-catenin-TCF inhibitors on the viability of SW480 (Wnt-addicted) and MCF7 (Wnt-independent) cells as determined by the MTT assay. Cells were incubated with serially diluted compounds for 72 h in the presence of 10% FBS. Data reported represent the mean ± SD of ≥3 independent experiments.
Intracellular Delivery of Other Stapled Peptides by Cyclic CPP9.
The generality of our approach was further tested by exploring alternative stapling and conjugation methods as well as additional peptide sequences. Other investigators have previously explored bifunctional stapling and conjugation strategies in the context of linear CPP’s with modest success.40,41 We envisioned that 3,5-bis(bromomethyl)benzoic acid (BBA) would be a particularly appealing stapling agent, as stapling by BBA results in concomitant conjugation to a CPP (Figure 4). A structurally similar compound, m-xylene dibromide, reacts rapidly with two spatially proximal cysteines at a low reagent/peptide stoichiometry and has been widely used to staple α-helical peptides in high yields.42 We first synthesized CPP9 containing a miniPEG-Lys(Mtt) linker by standard solid-phase peptide chemistry (Figure 4). While still on resin, the methyltrityl (Mtt) group on the lysine side chain was selectively removed with 2% trifluoroacetic acid (TFA) and the exposed amine was acylated with BBA. The CPP9-BBA adduct was released from the resin and side-chain deprotected by treatment with TFA and purified by HPLC. For stapling, a linear peptide of interest is modified to contain two cysteine residues at i and i+4 positions and mixed with CPP9-BBA to generate CPP9-stapled peptide conjugates. Note that once prepared, CPP9-BBA is stable upon storage and can be used to staple/cyclize any peptide containing two thiol groups.
Figure 4.
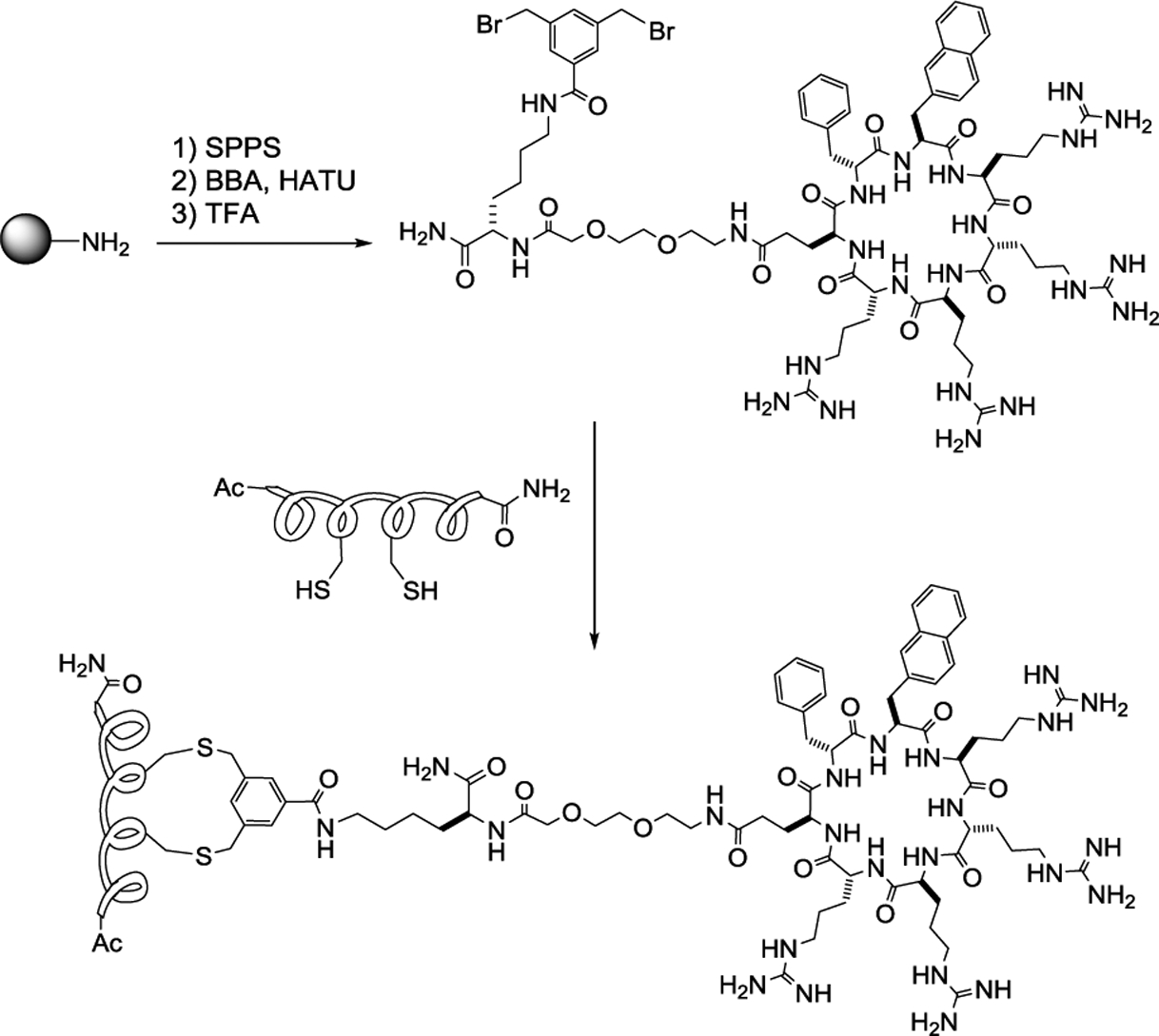
Simultaneous stapling and conjugation of Cys-containing peptides to a cyclic CPP. SPPS, solid-phase peptide synthesis; BBA, 3,5-bis(bromomethyl)benzoic acid; HATU, O-Benzotriazole-N,N,N’,N’-tetramethyluronium hexafluorophosphate; TFA, trifluoroacetic acid.
We synthesized four pairs of xylene/BBA-stapled peptides, with and without conjugation to CPP9, labeled them with naphthofluorescein (NF) at the N-termini, and compared their cellular entry efficiencies into HeLa cells by flow cytometry analysis (Table 1, peptides 12–19). NF is nonfluorescent inside the acidic endosome and lysosome and, therefore, any intracellular fluorescence should reflect the amount of peptide that had successfully escaped from the endosomal/lysosomal pathway into the cytosol.43 According to a recent study by Verdine and co-workers,5 peptide 12 was one of the most cell-permeable stapled peptides, out of more than 200 peptides tested. On the other hand, peptides 14, 16, and 18 were among the least permeable sequences. In agreement with Verdine’s finding, xylene-stapled peptide 12 (no CPP9) showed good cell-permeability (47% of CPP9), while xylene-stapled peptides 14 and 16 did not (2.5% and 8.9%, respectively) (Figure 5A). The cellular entry efficiency of peptide 18 could not be determined due to limited solubility. After conjugation with CPP9, all four peptides (13, 15, 17, and 19) became highly cell-permeable, exhibiting 11- to 152-fold higher cytosolic entry than their unconjugated counterparts. Even for the most permeable peptide 12, conjugation to CPP9 further increased its cell-permeability by 11-fold. To ascertain that the observed cellular fluorescence was not due to peptides bound to the cell surface, we resuspended the cells after treatment with NF-labeled peptides in a pH 6.5 buffer (which should quench the fluorescence of any cell-surface associated peptide) and repeated the flow cytometry analysis. Similar permeability enhancements were observed for the four peptides after conjugation to CPP9 (Figure S11).
Figure 5.
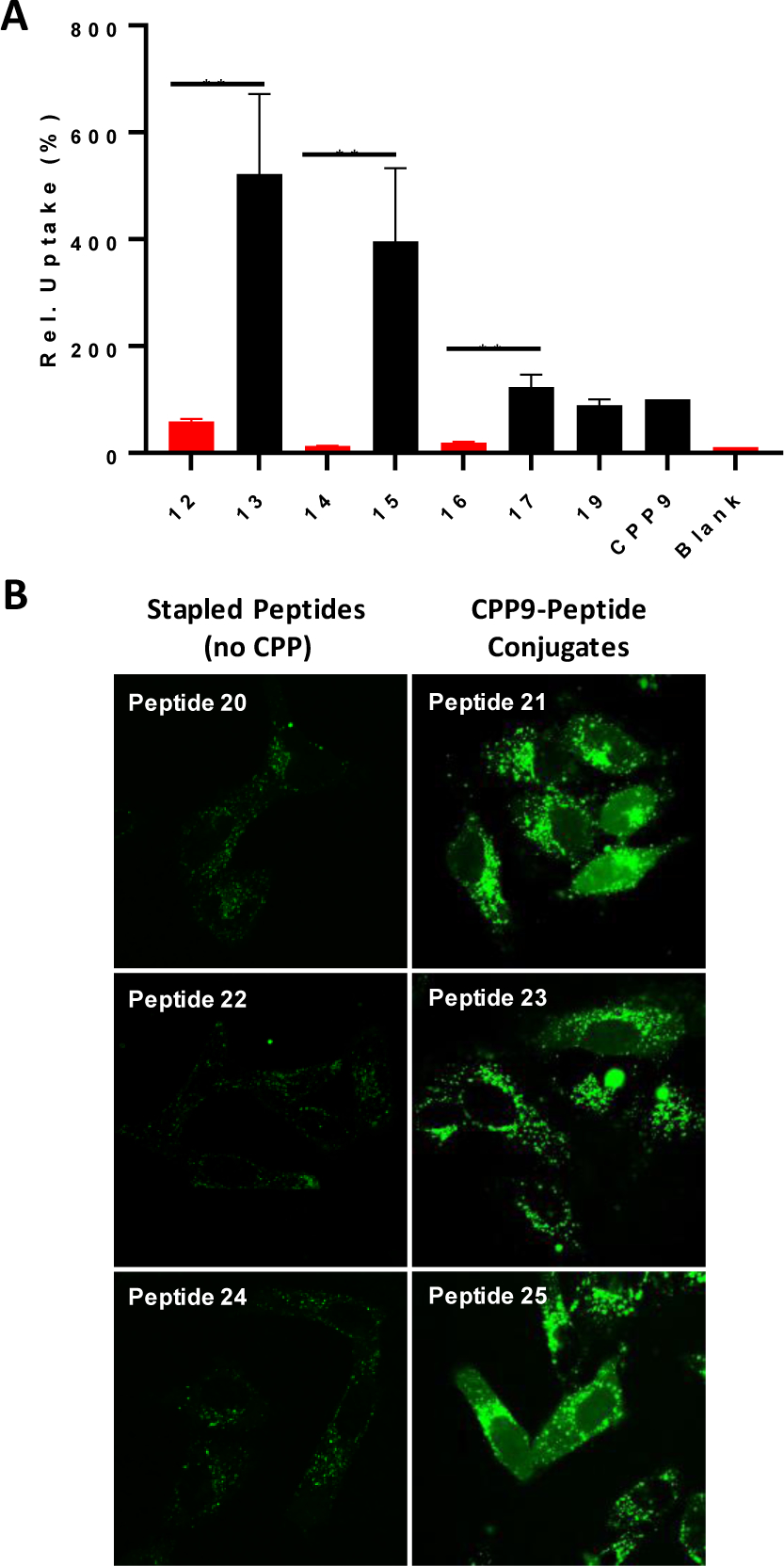
Effect of CPP9 on the cellular entry efficiencies of stapled peptides. (A) Comparison of the cytosolic entry efficiencies of NF-labeled stapled peptides, without (peptides 12, 14, and 16) and with CPP9-conjugation (peptides 13, 15, 17, and 19) into HeLa cells as measured by flow cytometry. Cells were incubated with 5 μM peptide in the presence of 10% FBS for 2 h before flow cytometry analysis. All values are relative to that of CPP9 (100%) and represent the mean ± SD of three independent experiments. Blank, no peptide added; **, p ≤ 0.01. (B) Representative live-cell confocal microscopic images of HeLa cells (~5000 cells) after treatment with FAM-labeled peptides 20-25 (5 μM) for 2 h in the presence of 1% FBS.
Finally, the capacity of CPP9 to enhance the cellular uptake of stapled peptides was examined by confocal microscopy. We initially labeled peptides 4–7 at their N-termini with fluorescein (FAM) and treated HeLa cells with the dye-labeled peptides. Unexpectedly, the dye-labeled peptides (but not the unlabeled peptides) were cytotoxic and caused significant membrane damage to mammalian cells. We therefore modified the PDI peptide to install a pair of cysteines at positions 4 and 8 and stapled it with m-xylene or BBA to give peptides 20 and 21, respectively. Peptides 20 and 21 were labeled with FAM at their C-termini via a miniPEG-Lys linker (see Figure S1 for structures). To improve the solubility of peptide 20, we also replaced the N-terminal leucine with a glutamate to give peptide 22, and added a second glutamate residue to produce peptide 24 (Table 1). Conjugation of peptides 22 and 24 with CPP9 generated peptides 23 and 25, respectively. As expected, HeLa cells treated with peptide 20, 22, or 24 (5 μM) for 2 h showed only weak, punctate fluorescence in the cytoplasmic region (Figure 5B). In contrast, cells treated with peptide 21, 23, or 25 were brightly fluorescent, showing a combination of punctate and diffuse fluorescence signals in HeLa cells and with the latter being throughout the entire cell volume including the nucleus. Thus, conjugation to a cyclic CPP (e.g., CPP9) has rendered all of the stapled peptides tested so far (including ones not featured in this study) cell-permeable. Interestingly, 6 out of the 7 CPP9-peptide conjugates tested in this work showed greater cytosolic entry efficiency than CPP9 alone (Figure 5A). The cargo moiety can potentially affect the cellular entry efficiency by several different mechanisms. It may bind to the plasma and/or endosomal membrane and enhance the endocytic uptake and/or endosomal escape, respectively. The cargo may also affect the CPP activity via protein binding, since the flow cytometry experiments were conducted in the presence of 10% fetal bovine serum (FBS). Binding of a CPP to serum proteins would inhibit its interaction with the cell membrane and reduce its cellular entry kinetics and/or efficiency. Depending on the nature of the cargo, it may either increase or decrease the extent of protein binding by CPP9. Intramolecular (or intermolecular) interaction between the cyclic CPP and the cargo moiety may affect the function of the CPP (and the cargo) as well.
CONCLUSION
We have demonstrated that conjugation with a cyclic CPP offers a potentially general approach to endowing stapled peptides with greatly enhanced cell-permeability, regardless of their intrinsic membrane permeability, thus overcoming a key limitation of stapled peptides as a novel drug modality. Compared to conventional linear CPPs (e.g., Tat and R9), cyclic CPPs offer superior cytosolic delivery efficiency as well as improved metabolic stability, bioavailability, and biodistribution.9 The resulting cyclic CPP-stapled peptide conjugates may be leveraged to target previously challenging proteins such as those involved in intracellular PPIs.
While this work was under review, Soudah et al. reported the effective delivery of peptide nucleic acids into glioblastoma cells by using one of our cyclic CPPs (CPP12).44
EXPERIMENTAL SECTION
Materials.
Fmoc-protected amino acids for peptide synthesis were purchased from Advanced ChemTech (Louisville, KY), NovaBiochem (La Jolla, CA), or Aapptec (Louisville, KY). Fmoc-protected Asp-(O-2-PhiPr) was purchased from Sigma Aldrich (St. Louis, MO). Rink amide resin (100–200 mesh, 0.3–0.6 meq/g) was purchased from Chem-Impex (Wood Dale, IL). O-Benzotriazole-N,N,N’,N’-tetramethyluronium hexafluorophosphate (HATU), 1-hydroxybenzotriazole hydrate (HOBt), and (Benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP) were purchased from Aapptec. Fluorescein isothiocyanate (FITC), 5-(and-6)-carboxynaphthofluorescein succinimidyl ester (NF), Nutlin-3a, phenylsilane (PhSiH3), human serum, and tetrakis(triphenylphosphine)palladium(0) [Pd(PPh3)4] were purchased from Sigma Aldrich (St. Louis, MO), and 5(6)-carboxy-tetramethylrhodamine was purchased from Chem-Impex (Wood Dale, IL). Cell culture media, fetal bovine serum, penicillin-streptomycin, Dulbecco’s phosphate-buffered saline (DPBS) (2.67 mM potassium chloride, 1.47 mM potassium phosphate monobasic, 137 mM sodium chloride, 8.06 mM sodium phosphate dibasic), and 0.25% trypsin-EDTA were purchased from Invitrogen (Carlsbad, CA). Cell proliferation kit (MTT) was purchased from Roche (Indianapolis, IN). Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit and CyQUANT LDH Release Kit was purchased from ThermoFisher Scientific (Waltham, MA). ATSP-7041 was custom synthesized by WuXi AppTec and used without further purification. All solvents and other chemical reagents were obtained from Sigma-Aldrich, VWR (West Chester, PA), or Fisher Scientific and were used without further purification unless noted otherwise. Mammalian cell lines were purchased from American Type Culture Collection (ATCC).
General Peptide Synthesis.
Peptides were manually synthesized by SPPS on Rink amide resin by using Fmoc chemistry and 2-(7-aza-1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (HATU) as the coupling agent. Coupling reactions typically involved Fmoc-amino acids (5 equiv), HATU (5 equiv), and diisopropylethylamine (DIPEA; 10 equiv) and were carried out at room temperature (RT) for 45 min. The peptides were cleaved off the resin and deprotected by treatment with 92.5% TFA, 2.5% water, 2.5% triisopropylsilane (TIPS), and 2.5% 1,3-dimethoxybenzene for 3 h at RT. The solvents were removed by flowing a stream of N2 over the solution and the residue was triturated three times with cold Et2O. The crude peptides were purified by reversed-phase HPLC equipped with a Waters XBridge C18 column, which was eluted with linear gradients of acetonitrile (containing 0.05% TFA) in ddH2O (containing 0.05% TFA).
For peptides containing a side-chain lactam crosslink, Lys(Mtt) and Asp(O-2-PhiPr) were incorporated at their designated positions during manual SPPS using the previously indicated coupling reagents. Following completion of the linear sequence, the N-terminal Fmoc group was removed and acylated with Ac2O (10 equiv), DIPEA (10 equiv) in DCM for 10 min twice. Acid-labile side-chain protecting groups were removed by incubating the resin with 2% TFA, 1% TIPS in DCM, three times for 5 min. Lactam formation was performed using PyBOP (5 equiv), DIPEA (5 equiv) in 1:1 (v/v) DMF/DCM for 2 h, followed by overnight incubation. Peptides were washed and any remaining amine was acylated using Ac2O (10 equiv) and DIPEA (10 equiv) in DCM 2 × 10 min. Peptides were cleaved from the resin by the addition of 92.5% TFA, 2.5% water, 2.5% TIPS, and 2.5% 1,3-dimethoxybenzene for 3 h at RT, triturated with diethyl ether, and purified by HPLC as described above.
For peptides containing a C-terminal cell-penetrating peptide, the linear precursor peptides were synthesized via manual SPPS containing a C-terminal allyloxycarbonyl (Alloc)-protected Lys or Dap residue. Following general synthesis and side-chain cyclization as mentioned previously, the C-terminal Alloc group was removed using Pd(PPh3)4 (0.3 equiv) and PhSiH3 (15 equiv) in dry DCM (3 × 15 min). Following deprotection, the resin was incubated in 10% sodium dimethyldithiocarbamate (SDDC) in DMF (w/v) and washed thoroughly with DMF/DCM. Cell-penetrating sequences were then synthesized by manual SPPS as mentioned previously.
For BBA- or xylene-stapled peptides, linear peptides were first synthesized as previously described and purified. After lyophilization, 2 mg of each peptide was dissolved in 3 mL of DMF and 7 mL of 100 mM NH4HCO3 (pH 8.1) to a final concentration of 0.1 mM peptide. To this mixture, tris(carboxyethyl)phosphine (TCEP; 1.1 equiv) was added and mixed for 1 h at RT. CPP9-BBA (2 equiv) or m-xylene dibromide was freshly prepared in DMF and added to the reduced peptide solution and mixed for 3 h at RT. Reaction progress was monitored by MALDI-TOF mass spectrometry. Upon completion, the reaction mixture was quenched with TFA and purified by RP-HPLC to obtain the crosslinked peptides.
For peptides containing a fluorescent label, precursor peptides were first synthesized and purified as previously described. Approximately 1 mg of lyophilized peptide was incubated with 5 equiv. of an activated fluorescent labeling reagent [e.g., FITC, 5(6)-carboxyfluorescein succinimidyl ester, or 5(6)-carboxynaphthofluorescein succinimidyl ester) and 5 equiv. of DIPEA in 150 μL of 1:1 (v/v) DMF/150 mM sodium bicarbonate (pH 8.5) for 2 h. The reaction was quenched by TFA and the labeled peptides were purified again by HPLC and their authenticity was confirmed by MALDI-TOF mass spectrometry. To generate TMR-labeled peptides, an Nε−4-methyltrityl-L-lysine was added to the C-terminus. The lysine side chain was selectively deprotected using 2% (v/v) TFA in DCM. The resin was incubated with 5(6)-carboxy-tetramethylrhodamine (5 equiv), DIC (5 equiv) and DIPEA (5 equiv) in DMF overnight. The resin was washed and subjected to deprotection by 92.5% TFA, 2.5% water, 2.5% TIPS, and 2.5% 1,3-dimethoxybenzene for 3 h at RT, triturated with diethyl ether, and purified by HPLC as described above.
The purity of each peptide was assessed by reversed-phase HPLC equipped with a Waters XBridge C18 analytical column and its authenticity was confirmed by high-resolution mass spectrometry using a custom Bruker 15-Tesla MALDI-FT-ICR instrument. The detailed structures of peptides used in this study and their analytical data are provided in Figure S1. All peptides used in this study had ≥95% as judged by analytical HPLC (Table S1).
Protein-Ligand Binding Assays.
Peptide 1:
Its binding affinity was determined using FP. TMR-labeled peptide 1 (50 nM) was incubated with serial dilutions of GST-MDM2 in PBS (pH 7.4) containing 5 mM dithiothreitol (DTT) and 0.01% Triton-X100. After 1 h, aliquots from each serial dilution were transferred into black-on-black 384-well non-binding plates (Greiner) and FP was measured using a Tecan Infinite M1000 Pro plate reader. KD values were calculated using KaleidaGraph v. 3.6 using the equation:
Peptides 2–7:
Binding affinity was determined using a FP-based competition assay. TMR-labeled peptide 1 (KD ~ 12 nM, 15 nM) was incubated with 15 nM GST-MDM2 in PBS (pH 7.4) containing 5 mM DTT and 0.01% Triton-X100 for 1 h at RT. Serial dilutions of a competitor peptide were prepared in PBS (pH 7.4) containing 5 mM DTT and 0.01% Triton-X100. After 1 h, aliquots of the equilibrated peptide 1-MDM2 solution were added to each compound serial dilution and incubated for 1 h at RT. Samples were transferred into black-on-black 384-well non-binding microplates (Greiner) and FP was measured using a Tecan M1000 Infinite plate reader. The data were analyzed using GraphPad Prism v. 8.0 using log[inhibitor] vs. response (four parameters) and beginning from the model equation:
where Top is the highest FP value observed and Bottom is the lowest and normalized to FP values corresponding to fully bound/unbound probe.
Peptide 8:
Binding affinity was determined using FP. Peptide 8 (10 nM) was added to serial dilutions of GST-β-catenin in 20 mM Tris, 300 mM NaCl, pH 8.8, 0.01% Triton-X100 as reported previously.34 After 1 h, aliquots were transferred from the reaction mixtures into black-on-black 384-well non-binding plates (Greiner) and FP was measured using a Tecan Infinite M1000 Pro plate reader. The KD value was calculated using KaleidaGraph v. 3.6 using the above equation.
Peptides 9–11:
Binding affinity was determined using a FP-based competition assay. FAM-labeled peptide 8 (10 nM) was incubated with 50 nM GST-β-catenin in 20 mM Tris, 300 mM NaCl, pH 8.8, 0.01% Triton-X100 for 1 h as reported previously.34 Serial dilutions of a competitor peptide were prepared in 20 mM Tris, 300 mM NaCl, pH 8.8, 0.01% Triton-X100. After 1 h, aliquots of the equilibrated peptide 8-β-catenin solution were added to serially diluted peptide solutions and incubated for 1 h at RT. Samples were transferred into black-on-black 384-well non-binding microplates (Greiner) and FP was measured using a Tecan M1000 Infinite plate reader. The data were analyzed using GraphPad Prism v. 8.0 and normalized to FP values corresponding to fully bound/unbound probe. All binding values reported are the mean ± SD of ≥3 independent experiments.
Cell Viability Assay.
Cell viability was determined using the MTT assay.45 For SJSA-1, cells were maintained in RPMI-1640 supplemented with 10% FBS and 1% penicillin/streptomycin in an atmosphere of 5% CO2 at 37 °C. Cells were harvested by first washing with DPBS and trypsinized using 0.025% porcine trypsin-EDTA for 5 min at 37 °C. Trypsin was neutralized with the addition of warm DPBS and the cells were pelleted at 200 g for 5 min at 4 °C. Cell density was determined using a hemocytometer and cells were resuspended in fresh FBS-free RPMI-1640 supplemented with 1% penicillin/streptomycin, pipetted into clear culture-treated 96-well plates (Sigma-Aldrich) at a final density of 5.0 × 103 cells/well, and incubated for 24 h at 37 °C in an atmosphere containing 5% CO2. Compounds were serially diluted in DPBS with standardized DMSO concentration (0.5% v/v) and added to each well along with 10% FBS (final concentration). Cells were incubated at 37 °C for 72 h, after which 10 μL of a MTT solution was added to each well, followed by incubation for 4 h at 37 °C, 5% CO2. Following MTT incubation, the formazan crystals formed were solubilized by the addition of 100 μL of a solubilization buffer and allowed to stand overnight at 37 °C. The next day, A565 was determined using a Tecan M1000 Infinite plate reader and absorbance values were standardized against cell-free wells and normalized to untreated control cells. Data were analyzed using GraphPad Prism v. 8.0.
SW480 and MCF7 cells were maintained as described above. Cell density was determined using a hemocytometer and cells were resuspended in fresh RPMI-1640 or MEM supplemented with 10% FBS and 1% penicillin/streptomycin, pipetted into clear culture-treated 96-well plates (Sigma-Aldrich) at a final density of 5.0 × 103 cells/well, and incubated for 24 h at 37 °C, 5% CO2. Compounds were serially diluted in DPBS with standardized DMSO concentration (0.5% v/v) and added to each well. Treated cells were incubated at 37 °C for 72 h. After that, 10 μL of MTT solution was added to each well, followed by incubation for 4 h at 37 °C, 5% CO2. The formazan crystals formed were solubilized through the addition of 100 μL of a solubilization buffer and allowed to stand overnight at 37 °C. The next day, A565 was determined using a Tecan M1000 Infinite plate reader and absorbance values were standardized against cell-free wells and normalized to untreated control cells. Data was analyzed using GraphPad Prism v. 8.0. Values reported are the mean ± SD of three independent experiments.
Annexin V/PI Staining.
For SJSA-1, cells were maintained in RPMI-1640 supplemented with 10% FBS and 1% penicillin/streptomycin in an atmosphere of 5% CO2 at 37 °C. Cells were harvested by first washing with DPBS, then trypsinized using 0.025% porcine trypsin-EDTA for 5 min at 37 °C. Trypsin was neutralized with the addition of warm DPBS and cells were pelleted down at 200 g for 5 min at 4 °C. Cell density was determined using a hemocytometer and cells were resuspended in fresh FBS-free RPMI-1640 supplemented with 1% penicillin/streptomycin, transferred into clear culture-treated 12-well plates (Sigma-Aldrich) at a final density of 1.0 × 105 cells/well, and incubated for 24 h at 37 °C in the presence of 5% CO2. Compounds were prepared in DPBS, and added to each well along with FBS to a final concentration of 10%. Treated cells were incubated at 37 °C for 48 h. After that, cells were washed with cold DPBS and harvested as mentioned previously. The cell pellet was resuspended and washed with cold DPBS twice, followed by resuspension in 100 μL of Annexin V binding buffer containing 5 μL of Annexin V-Alexa 488 and 1 μL of 100 μg/mL propidium iodide and incubated at RT for 15 min. After incubation, 400 μL of Annexin V binding buffer was added to each sample, mixed gently on ice and immediately analyzed on a BD Fortessa flow cytometer. Data was analyzed using FlowJo.
For DLD1, SW480, and MCF7, cells were maintained in RPMI-1640 or MEM supplemented with 10% FBS and 1% penicillin/streptomycin in an atmosphere of 5% CO2 at 37 °C. Cells were harvested by first washing with DPBS, then trypsinized using 0.025% trypsin-EDTA for 5 min at 37 °C. Trypsin was neutralized with the addition of warm DPBS and cells were pelleted down at 200 g for 5 min at 4 °C. Cell density was determined using a hemocytometer and cells were resuspended in RPMI-1640 or MEM supplemented with 10% FBS and 1% penicillin/streptomycin, pipetted into clear culture-treated 12-well plates (Sigma-Aldrich) at a final concentration of 1.0 × 105 cells/well, and incubated for 24 h at 37 °C, 5% CO2. Compounds were prepared in DPBS and added to each well. Treated cells were incubated at 37 °C for 48 h. After that, cells were washed with cold DPBS and harvested as mentioned previously. The cell pellet was resuspended and washed with cold DPBS twice, followed by resuspension in 100 μL of Annexin V binding buffer containing 5 μL of Annexin V-Alexa 488 and 1 μL of 100 μg/mL propidium iodide and incubated at RT for 15 min. After incubation, 400 μL of Annexin V binding buffer was added to each sample, mixed gently on ice and immediately analyzed on a BD Fortessa flow cytometer. Data was analyzed using FlowJo.
Flow Cytometry.
HeLa cells were seeded in 12-well plates (1.5 × 105 cells/well) for 24 h. On the day of the experiment, the cells were incubated with 5 μM NF-labeled peptide in DMEM with 10% FBS at 37 °C for 2 h in the presence of 5% CO2. At the end of incubation, the peptide-containing medium were removed, and the cells were washed with DPBS twice, detached from the plate with 0.025% trypsin, diluted into DPBS, pelleted at 200 g for 5 min at 4 °C, washed twice with DPBS, resuspended in 200 μL of DPBS, and analyzed on a BD FACS LSR II flow cytometer. For lower pH experiments, glycine-HCl buffer, pH 2.0, was added immediately before analysis to obtain a final pH of 6.5. For NF-labeled peptides, 633-nm laser was used for excitation and the fluorescence emission was analyzed in the APC channel. Data was analyzed using FlowJo and plotted using GraphPad Prism v. 8.0. Values provided are the mean ± SD of three independent experiments.
Human Serum Stability.
Whole human serum was diluted 1:4 in sterile DPBS and equilibrated at 37 °C for 15 min. Compound was added to the diluted serum to a final concentration of 100 μM and incubated at 37 °C with gentle mixing. At varying time points, 100-μL aliquots were withdrawn and combined with 100 μL of 15% trichloroacetic acid in MeOH (w/v), 100 μL of MeCN and stored at 4 °C for 24 h. After protein precipitation was complete, each aliquot was centrifuged (15000 g, 5 min, 4 °C) and analyzed by RP-HPLC.
Confocal Microscopy.
HeLa cells (1 mL, 5 × 103 cells/mL) suspended in DMEM supplemented with 10% FBS and 1 % penicillin/streptomycin were seeded into glass-bottomed culture dishes (MatTek) and cultured overnight at 37 °C, 5% CO2. The next day, cells were washed with DPBS (3x) and then compounds were added in fresh DMEM supplemented with 1% FBS and 1% penicillin/streptomycin and incubated at 37 °C for 2 h. After 2 h, cells were washed with phenol red-free DMEM containing 1% FBS before imaging in the same media using a Nikon AIR live-cell confocal microscope equipped with a 100X oil objective. Images were processed using NIS-Elements AR.
Molecular Dynamics.
For peptide 4 in complex with MDM2, the system was prepared from a previously reported crystal structure of MDM2 in complex with a peptidyl inhibitor (PDBID: 3EQS).46 First, a cubic (10Å × 10Å × 10Å) protein grid for docking was produced using the Receptor Grid Generation47 tool in Maestro v. 10.248 centered on the geometric center of the existing ligand. Docking for PDI was carried out using Glide XP49 with the OPLS3 forcefield, with settings applied for enhanced planarity of conjugated π systems, 100,000 poses generated per ligand and 5,000 poses carried forward for energy minimization. To ensure that the docked peptides adhered to an α-helical conformation, a minimal core motif consisting of backbone C, N, and Cα from the Tyr-Trp-Ala region in the crystallized sequence was used as a constraint, with poses having higher than a 1.5-Å RMSD against this core sequence discarded. The final docked pose of PDI with the lactam staple installed was then subjected to a 50 ns production To ensure that the docked peptides adhered to an α-helical conformation, a minimal core motif consisting of backbone C, N, and Cα from the Ile-Leu-Asp region in the crystallized sequence was used as a constraint, with poses having higher than a 1.5-Å RMSD against this core sequence discarded. MD run using Desmond50 in an octahedral box, solvated with TIP3P water containing 0.15 M NaCl in addition to ions necessary to neutralize the system, and using the OPLS3 forcefield as configured in the Schrodinger Suite.51 From this final pose, the C-terminal transporter sequence was constructed in Maestro, appended and an additional 50 ns MD run using Desmond was performed using the previously mentioned conditions.
For peptide 9 in complex with β-catenin, the system was prepared from a previously reported crystal structure of β-catenin in complex with Axin (PDBID: 1QZ7).52 First, a cubic (10Å × 10Å × 10Å) protein grid for docking was produced using the Grid Generation tool in Maestro v. 10.2 centered on the geometric center of the existing ligand. Docking for Ax4 was carried out using Glide XP with the OPLS3 forcefield, with settings applied for enhanced planarity of conjugated π systems, 100,000 poses generated per ligand and 5,000 poses carried forward for energy minimization. The final docked pose of Ax4 with the lactam staple installed was then subjected to a 50 ns production MD run using Desmond in an octahedral box, solvated with TIP3P water containing 0.15 M NaCl in addition to ions necessary to neutralize the system, and using the OPLS3 forcefield as configured in the Schrodinger Suite. From this final pose, the C-terminal transporter sequence was constructed in Maestro, appended and an additional 50 ns MD run using Desmond was performed using the previously mentioned conditions.
Protein Purification.
Escherichia coli BL21 (DE3) cells harboring plasmid pGEX-MDM2(1–139)14 were grown in LB medium supplemented with 50 mg/L ampicillin at 37 °C until an OD600 = 0.7 and then induced by the addition of 0.3 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for 5 h at 30 °C before harvesting by centrifugation. Cells were lysed on ice in lysis buffer (20 mM Tris, 150 mM NaCl, 5 mM DTT, pH 8.0) containing lysozyme and HALT protease inhibitor for 20 min while stirring before the addition of 400 mg of protamine sulfate and another 10 min of stirring. Cells were homogenized by sonication (8 s off, 2 s on, 1 min total, performed twice at 70% amplitude), and the lysate was centrifuged at 13,000 g for 30 min at 4 °C. The clear lysate was loaded onto an equilibrated glutathione-agarose column (Pierce Scientific) and incubated for 1 h at 4 °C. The column was allowed to drain by gravity flow and washed exhaustively using a washing buffer (20 mM Tris, 150 mM NaCl, 5 mM DTT, pH 7.4). The bound protein was eluted using the washing buffer containing 10 mM reduced glutathione, concentrated, and stored at −80 °C in 25% glycerol.
For β-catenin, E. coli BL21 (DE3) cells harboring plasmid pGEX-β-catenin(133–665)53 were grown in LB medium supplemented with 50 mg/L ampicillin at 37 °C to an OD600 of 0.6 before induction with 0.2 mM IPTG overnight at 30 °C. Cells were harvested by centrifugation and lysed in lysis buffer (20 mM Tris, pH 8.0, 150 mM NaCl, 2 mM DTT, 1 % Triton-X100, 200 mM EDTA, 10 mg/mL PMSF, and 0.2 mg/mL lysozyme) on ice for 20 min, followed by sonication (8 s off, 2 s on, 1 min total, performed twice at 70% amplitude). The homogenate was centrifuged (13,000 g for 30 min at 4 °C and the clear supernatant was loaded onto an equilibrated glutathione-agarose column. After exhaustive washing with wash buffer (20 mM Tris, pH 7.4, 150 mM NaCl, 2 mM DTT), the bound GST-β-catenin was eluted with wash buffer supplemented with 10 mM glutathione, concentrated, and stored at −80 °C in 30% glycerol.
LDH Release.
SJSA-1 or SW480 cells were maintained as described above. Cells were harvested and seeded into clear 96-well plates at a final density of 5 × 103 cells/well in complete growth media and incubated overnight at 37 °C, 5% CO2. The next day, compounds serially diluted in DPBS were added to each well to give a constant final concentration of 0.5% DMSO (v/v), with control wells containing 10 μL of lysis buffer, cell-free complete growth media, or positive LDH control. The plates were incubated for 45 min to 4 h at 37 °C, 5% CO2. After that, 50 μL of growth medium was withdrawn from each well, transferred to a clear 96-well plate, mixed with 50 μL of LDH substrate mix, and incubated at RT for 30 min with gentle mixing. Finally, 50 μL of 1 N HCl was added to each well and the absorbances at 490 and 680 nm were immediately measured on a TECAN Infinite M1000 plate reader. The absorbance values, after subtraction of background signal at 680 nm, were plotted by using GraphPad PRISM v. 8.0. Values represent the mean ± SD of three replicates from two independent experiments (n = 6).
Supplementary Material
ACKNOWLEDGMENT
We thank Pei group members for helpful discussions and the Ohio State University CCIC for assistance with high-resolution mass spectrometry and flow cytometry.
Funding Sources
Financial support from the National Institutes of Health (GM122459 and CA234124) is gratefully acknowledged.
ABBREVIATIONS
- BBA
3,5-bis(bromomethyl)benzoic acid
- CPP
cell-penetrating peptide
- FAM
5(6)-carboxyfluorescein
- FP
fluorescence polarization
- MDM2
mouse double minute 2
- miniPEG
8-amino-3,6-dioxaoctanoic acid
- NF
5(6)-carboxynaphthofluorescein
- PPI
protein-protein interaction
- TMR
tetramethylrhodamine
Footnotes
Supporting Information
Peptide structures, additional experimental data, and quality control data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interests: D.P. is a co-founder and shareholder of Entrada Therapeutics, Inc.
REFERENCES
- [1].Verdine GL; Hilinski GL Stapled Peptides for Intracellular Drug Targets. Methods Enzymol. 2012, 503, 3–33. [DOI] [PubMed] [Google Scholar]
- [2].Walensky L D; Bird, G. H. Hydrocarbon-Stapled Peptides: Principles, Practice, and Progress. J. Med. Chem 2014, 57, 6275–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lau YH; de Andrade P; Wu Y; Spring DR Peptide Stapling Techniques Based on Different Macrocyclisation Chemistries. Chem. Soc. Rev 2015, 44, 91–102. [DOI] [PubMed] [Google Scholar]
- [4].Walensky LD; Kung AL; Escher I; Malia TJ; Barbuto S; Wright RD; Wagner G; Verdine GL; Korsmeyer SJ Activation of Apoptosis In Vivo by a Hydrocarbon-Stapled BH3 Helix. Science 2004, 305, 1466–1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chu Q; Moellering RE; Hilinski GJ; Kim Y; Grossman TN; Yeh JT-H; Verdine GL Towards Understanding Cell Penetration by Stapled Peptides. Med. Chem. Commun 2015, 6, 111–119. [Google Scholar]
- [6].Bird GH; Mazzola E; Opoku-Nsiah K; Lammert MA; Godes M; Neuberg DS; Walensky LD Biophysical Determinants for Cellular Uptake of Hydrocarbon-Stapled Peptide Helices. Nat. Chem. Biol 2016, 12, 845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Qian Z; Liu T; Briesewitz R; Barrios AM; Jhiang SM; Pei D Efficient Delivery of Cyclic Peptides into Mammalian Cells with Short Sequence Motifs. ACS Chem. Biol 2013, 8, 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Qian Z; LaRochelle JR; Jiang B; Lian W; Hard RL; Selner NG; Luechapanichkul R; Barrios AM; Pei D Early Endosomal Escape of a Cyclic Cell-Penetrating Peptide Allows Effective Cytosolic Cargo Delivery. Biochemistry 2014, 53, 4034–4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Qian Z; Martyna A; Hard RL; Wang J; Appiah-Kubi G; Coss C; Phelps MA; Rossman JS; Pei D Discovery and Mechanism of Highly Efficient Cyclic Cell-Penetrating Peptides. Biochemistry 2016, 55, 2601–2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nischan N; Herce HD; Natale F; Bohlke N; Budisa N; Cardoso MC; Hackenberger CPR Covalent Attachment of Cyclic TAT Peptides to GFP Results in Protein Delivery into Live Cells with Immediate Bioavailability. Angew. Chem. Int. Ed. Engl 2014, 54, 1950–1953 [DOI] [PubMed] [Google Scholar]
- [11].Lättig-Tünnemann G; Prinz M; Hoffman D; Behlke J; Palm-Apergi C; Morano I; Herce HD; Cardoso MC Backbone Rigidity and Static Presentation of Guanidinium Groups Increases Cellular Uptake of Arginine-Rich Cell-Penetrating Peptides. Nat. Comm 2011, 2, 453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Wallbrecher R; Depré L; Verdurmen WPR; Bovée-Geurts PH; van Duinkerken RH; Zekveld MJ; Timmerman P; Brock R Exploration of the Design Principles of a Cell-Penetrating Bicyclic Peptide Scaffold. Bioconjugate Chem. 2014, 25, 944–964. [DOI] [PubMed] [Google Scholar]
- [13].Kastenhuber E; Scott L Putting p53 in Context. Cell 2017, 170, 1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fang S; Jensen JP; Ludwig RL; Vousden KH; Weissman AM Mdm2 is a RING Finger-Dependent Ubiquitin Protein Ligase for Itself and p53. J. Biol. Chem 2000, 275, 8945–8951. [DOI] [PubMed] [Google Scholar]
- [15].Karni-Schmidt O; Lokshin M; Prives C The Roles of MDM2 and MDMX in Cancer. Annu. Rev. Pathol. Mech. Dis 2016, 11, 617–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wade M; Li YC; Wahl GM MDM2, MDMX and p53 in Oncogenesis and Cancer Therapy. Nat. Rev. Cancer 2013, 13, 83–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zhao Y; Aguilar A; Bernard D; Wang S Small-Molecule Inhibitors of the MDM2-p53 Protein-Protein Interaction (MDM2 Inhibitors) in Clinical Trials for Cancer Treatment. J. Med. Chem 2015, 58, 1038–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Phan J; Li Z; Kasprzak A; Li B; Sebti S; Guida W; Schönbrunn E; Chen J Structure-Based Design of High Affinity Peptides Inhibiting the Interaction of p53 with MDM2 and MDMX. J. Biol. Chem 2010, 285, 2174–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bernal F; Tyler AF; Korsmeyer SJ; Walensky LD; Verdine GL Reactivation of the p53 Tumor Suppressor Pathway by a Stapled p53 Peptide. J. Am. Chem. Soc 2007, 129, 2456–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wu Y; Kaur A; Fowler E; Wiedmann MM; Young R; Galloway WRJD; Olsen L; Sore HF; Chattopadhyay A; Kwan TT-L; Xu W; Walsh SJ; de Andrade P; Janecek M; Arumugam S; Itzhaki LS; Lau YH; Spring DR Toolbox of Diverse Linkers for Navigating the Cellular Efficacy Landscape of Stapled Peptides. ACS Chem. Biol 2019, 14, 526–533. [DOI] [PubMed] [Google Scholar]
- [21].Quach K; LaRochelle J; Li X -H.; Rhoades, E.; Schepartz, A. Unique Arginine Array Improves Cytosolic Localization of Hydrocarbon-Stapled Peptides. Bioorg. Med. Chem 2018, 26, 1197–1202. [DOI] [PubMed] [Google Scholar]
- [22].Liu M; Li C; Pazgier M; Li C; Mao Y; Lv Y; Gu B; Wei G; Yuan W; Zhan C; Lu WY; Lu W D-Peptide Inhibitors of the p53-MDM2 Interaction for Targeting Molecular Therapy of Malignant Neoplasms. Proc. Nat. Acad. Sci. U. S. A 2010, 107, 14321–14326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Do TN; Rosal RV; Drew L; Raffo AJ; Michl J; Pincus MR; Friedman FK; Petrylak DP; Cassai N; Szmulewicz J; Sidhu G; Fine RL; Brandt-Rauf PW; Preferential Induction of Necrosis in Human Breast Cancer Cells by a p53 Peptide Derived from the MDM2 Binding Site. Oncogene 2003, 22, 1431–1444. [DOI] [PubMed] [Google Scholar]
- [24].Yamada S; Kanno H; Kawahara N Trans-membrane Peptide Therapy for Malignant Glioma by Use of a Peptide Derived from the MDM2 Binding Site of p53. J. Neurooncol 2012, 109, 7–14. [DOI] [PubMed] [Google Scholar]
- [25].Shepherd NE; Hoang HN; Abbenante G; Fairlie DP Single Turn Peptide Alpha Helices with Exceptional Stability in Water. J. Am. Chem. Soc 2005, 127, 2974–2983. [DOI] [PubMed] [Google Scholar]
- [26].Okoro DR; Arva N; Gao C; Polotskaia A; Puente C; Rosso M; Bargonetti J Endogenous Human MDM2-C Is Highly Expressed in Human Cancers and Functions as a p53-Independent Growth Activator. PLoS One, 2013, 8, e77643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Vassilev LT; Vu BT; Carvajal D; Podlaski F; Filipovic Z; Kong N; Kammlott U; Lukacs C; Klein C; Fotouhi N; Liu EA In Vivo Activation of the p53 Pathway by Small-Molecule Antagonists of MDM2. Science 2004, 303, 844–848. [DOI] [PubMed] [Google Scholar]
- [28].Chang YS; Graves B; Guerlavais V; Tovar C; Packman K; To KH; Olson KA; Kesavan K; Gangurde P; Mukherjee A; Baker T; Darlak K; Elkin C; Filipovic Z; Qureshi FZ; Cai H,; Berry P; Feyfant E; Shi XE; Horstick J; Annis DA; Manning AM; Fotouhi N; Nash H; Vassilev LT; Sawyer TK Stapled α-Helical Peptide Drug Development: a Potent Dual Inhibitor of MDM2 and MDMX for p53-Dependent Cancer Therapy. Proc. Nat. Acad. Sci. U. S. A 2013, 110, 3445–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Clevers H; Nusse R Wnt/β-Catenin Signaling and Disease. Cell 2012, 149, 1192–1205. [DOI] [PubMed] [Google Scholar]
- [30].Kim JS; Crooks H; Foxworth A; Waldman T Proof-of-Principle: Oncogenic β-Catenin is a Valid Molecular Target for the Development of Pharmacological Inhibitors. Mol. Cancer Ther 2002, 1, 1355–1359. [PubMed] [Google Scholar]
- [31].Huan Tran F; Zheng JJ Modulating the Wnt Signaling Pathway with Small Molecules. Protein Sci. 2017, 26, 650–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Grossmann TN; Yeh JT; Bowman BR; Chu Q; Moellering RE; Verdine GL Inhibition of Oncogenic Wnt Signaling through Direct Targeting of beta-Catenin. Proc. Natl. Acad. Sci. U.S.A 2012, 109, 17942–17947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Cui HK; Zhao B; Li Y; Guo Y; Hu H; Liu L; Chen YG Design of Stapled alpha-Helical Peptides to Specifically Activate Wnt/beta-Catenin Signalling. Cell Res. 2013, 23, 581–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Diderich P; Bertoldo D; Dessen P; Khan MM; Pizzitola I; Held W; Huelsken J; Heinis C Phage Selection of Chemically Stabilized α-Helical Peptide Ligands. ACS Chem. Biol 2016, 11, 1422–1427. [DOI] [PubMed] [Google Scholar]
- [35].Dietrich L; Rathmer B; Ewan K; Bange T; Heinrichs S; Dale TC; Schade D; Grossman TN Cell Permeable Stapled Peptide Inhibitor of Wnt Signalling that Targets β-Catenin Protein-Protein Interactions. Cell Chem. Biol 2017, 24, 958–968. [DOI] [PubMed] [Google Scholar]
- [36].Schneider JA; Craven TW; Kasper AC; Yun C; Haugbro M; Briggs EM; Svetlov V; Nudler E; Knaut H; Bonneau R; Garabedian MJ; Kirshenbaum K; Logan SK Design of Peptoid-peptide Macrocycles to Inhibit the β-Catenin TCF Interaction in Prostate Cancer. Nat. Commun 2018, 9, 4396–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wang Z; Zhang M; Wang J; Ji H Optimization of Peptidomimetics as Selective Inhibitors for the β-Catenin/T-Cell Factor Protein–Protein Interaction. J. Med. Chem 2019, 62, 3617–3635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B, Kinzler KW Activation of β-Catenin-Tcf Signaling in Colon Cancer by Mutations in β-Catenin or APC. Science 1997, 275, 1787–1790. [DOI] [PubMed] [Google Scholar]
- [39].Su N; Wang P; Li Y Role of Wnt/β-Catenin Pathway in Inducing Autophagy and Apoptosis in Multiple Myeloma Cells. Oncol. Lett 2016, 12, 4623–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Iegre J; Ahmed NS; Gaynord JS; Wu Y; Herlihy KM; Tan YS; Lopes-Pires ME; Jha R; Lau YH; Sore HF; Verma C; O’Donovan DH; Pugh N; Spring DR Stapled Peptides as a New Technology to Investigate Protein-Protein Interactions in Human Platelets. Chem. Sci 2018, 9, 4638–4643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Assem N; Ferreira DJ; Wolan DW; Dawson PE Acetone-Linked Peptides: A Convergent Approach for Peptide Macrocyclization and Labeling. Angew. Chem. Int. Ed. Engl 2015, 54, 8665–8668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Fairlie DP; Dantas de Araujo A Stapling Peptides Using Cysteine Crosslinking. Pept. Sci 2016, 106, 843–852. [DOI] [PubMed] [Google Scholar]
- [43].Qian Z; Dougherty PG; Pei D Monitoring the Cytosolic Entry of Cell-Penetrating Peptides using a pH-Sensitive Fluorophore. Chem. Commun 2015, 51, 2162–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Soudah T; Khawaled S; Aqeilan RI; Yavin E AntimiR-155 Cyclic Peptide–PNA Conjugate: Synthesis, Cellular Uptake, and Biological Activity. ACS Omega 2019, 4, 13954–13961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Kumar P; Nagarajan A; Uchil PD Analysis of Cell Viability by the MTT Assay. Cold Spring Harb Protoc. 2018, 6, pdb.prot095505 [DOI] [PubMed] [Google Scholar]
- [46].Pazgier M; Liu M; Zou G; Yuan W; Li C; Li C; Li J; Monbo J; Zella D; Tarasov SG; Lu W; Structural Basis for High-Affinity Peptide Inhibition of p53 Interactions with MDM2 and MDMX. Proc. Natl. Acad. Sci. USA 2009, 106, 4665–4670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Schrödinger Release 2017–1: Receptor Grid Generation, Schrödinger, LLC, New York, NY, 2019. [Google Scholar]
- [48].Schrödinger Release 2017–1: Maestro, Schrödinger, LLC, New York, NY, 2019. [Google Scholar]
- [49].Friesner RA; Murphy RB; Repasky MP; Frye LL; Greenwood JR; Halgren TA; Sanschagrin PC; Mainz DT, Extra Precision Glide: Docking and Scoring Incorporating a Model of Hydrophobic Enclosure for Protein-Ligand Complexes. J. Med. Chem, 2006, 49, 6177–6196. [DOI] [PubMed] [Google Scholar]
- [50].Bowers KJ; Chow E; Xu H; Dror RO; Eastwood MP; Gregersen BA; Klepeis JL; Kolossváry I; Moraes MA; Sacerdoti FD; Salmon JK; Shan Y; Shaw DE Scalable Algorithms for Molecular Dynamics Simulations on Commodity Clusters. Proc. ACM/IEE Conf. Supercomputing 2006 [Google Scholar]
- [51].Schrödinger Release 2017–1: Desmond Molecular Dynamics System, Shaw DE Research, New York, NY, 2019. Maestro-Desmond Interoperability Tools, Schrödinger, New York, NY, 2019. [Google Scholar]
- [52].Xing Y; Clements WK; Kimelman D; Xu W Crystal Structure of a Beta-Catenin/Axin Complex Suggests a Mechanism for the β-Catenin Destruction Complex. Genes Dev. 2003, 17, 2753–2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Huber AH; Nelson WJ; Weis WI; Three-Dimensional Structure of the Armadillo Repeat Region of Beta-Catenin. Cell 1997, 90, 871–882. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.