

Graphical Abstract
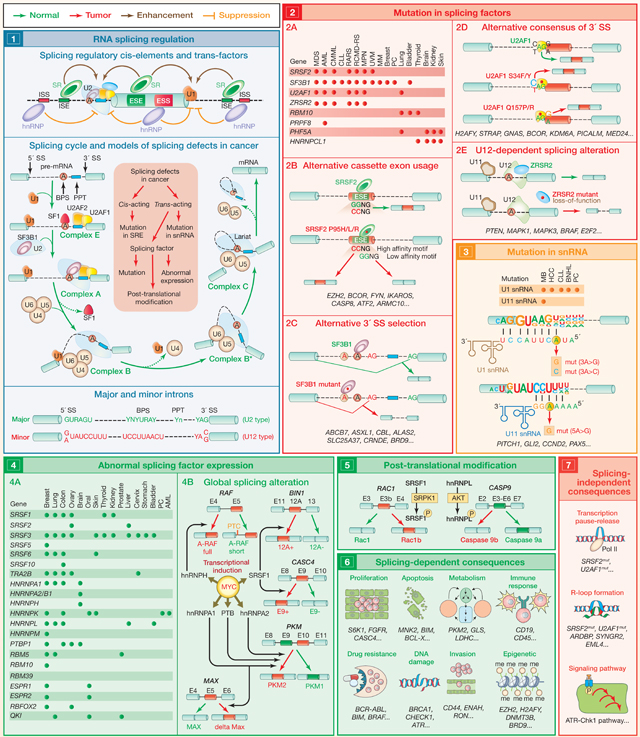
Splicing is the process by which introns are removed from pre-mRNA and exons joined to produce mature mRNA (1). Splicing is carried out by large macromolecular machineries, composed of small RNAs and proteins, known as the major (which recognizes ~99.5% of introns) and minor spliceosome (responsible for recognition of ~0.5% of introns) (Wahl et al., 2009). Splicing is coordinated by cis-elements within genes known as splicing enhancers and silencers (which can be located in exons or introns) and trans-acting splicing factors comprised of RNA-binding proteins (such as SR proteins and hnRNPs) (1) (Anczuków and Krainer, 2016). Compared to normal cells, cancer cells often display alterations in splicing, many of which contribute to disease. Some of these changes are caused by mutations in splicing-regulatory elements, whereas others result from alterations in splicing factors themselves, by either abnormal expression, mutation, or post-translational modification (1) (Dvinge et al., 2016; Zhang and Manley, 2013). This SnapShot provides a summarized view of splicing-factor alterations in cancer described to date.
The discovery of mutations in genes encoding RNA-splicing factors provided genetic evidence of a direct link between splicing misregulation and cancer pathogenesis (2A) (Wang et al., 2011; Yoshida et al., 2011). Spliceosome gene mutations are highly recurrent in myeloid malignancies, CLL, and non-cutaneous melanoma, but they also occur in lung, bladder, and breast cancers. These occur most commonly in SF3B1, SRSF2, U2AF1, and ZRSR2, where they affect highly restricted amino acids (except in ZRSR2), and they occur in a mutually exclusive manner. It is now clear that these mutations alter the normal RNA-recognition preferences of these proteins. For example, mutations in SRSF2 change its RNA-binding specificity, promoting its recognition of C-rich (CCNG) sequences while impairing its ability to bind G-rich sequences (GGNG), leading to global changes in cassette-exon recognition (2B) (Kim et al., 2015). In contrast, mutations in SF3B1 promote usage of aberrant branchpoint sequences, resulting in the global selection of cryptic 3′ splice sites (2C) (Darman et al., 2015). Mutations in U2AF1 are concentrated at S34 and Q157 residues and alter its normal RNA-binding preference and splicing properties for a subset of “AG-dependent” 3′ splice sites (2D) (Ilagan et al., 2015). In contrast, mutations in ZRSR2 are distributed throughout the gene and impair splicing of U12-type introns by the minor spliceosome (2E). Recurrent mutations have also been reported for a number of additional RNA-splicing factors in cancer, including PRPF8, RBM10, SFPQ, PHF5A, HNRNPCL1, PCBP1, PCBP2, FUBP1, FUBP3, and QKI. However, there has been little functional characterization of these mutations to date.
Mutations in the RNA components of the spliceosome (snRNAs) have been recently reported in cancer, mostly in the U1 snRNA, which is responsible for 5′-splice-site recognition (3) (Suzuki et al., 2019). Two prominent mutations at base position 3 in U1 snRNA were identified (3A>C and 3A>G), within the 5′-splice-site-binding region. The 3A>C mutation was identified in MB, CLL, HCC, B-NHL, and PC, whereas the 3A>G mutation was restricted to MB (most prominently in Sonic hedgehog MB). Interestingly, hotspot mutations in the fifth nucleotide of the U11 snRNA (responsible for 5′-splice-site recognition of minor introns) have also been described in MB (Suzuki et al., 2019). snRNA mutant tumors display significant aberrant splicing, with an excess of cryptic 5′-splice-site events.
Many cancer types exhibit alterations in the level of splicing factors, either through changes in gene copy number and/or expression (4A). For example, the transcription factor MYC, often amplified in cancers, induces expression of several splicing factors, some of which possess oncogenic properties (such as SRSF1, SRSF3, SRSF6, hnRNPA2/B1, and hnRNPH) (Anczuków and Krainer, 2016). Splicing factors often elicit changes in alternative splicing in a concentration-dependent manner and promote oncogenic transformation through genome-wide splicing alterations that affect diverse cellular processes. Representative examples are shown in the diagram (4B).
Post-translational modifications of splicing factors often alter their function or nuclear-cytoplasmic distribution. These modifications include phosphorylation, methylation, acetylation, sumoylation, and ubiquitination and are regulated by signaling pathways (5). For example, phosphorylation of SRSF1 by SRPK1 promotes a tumor-specific Rac1b isoform in colorectal cells.
The consequences of global splicing alterations in cancer can be divided into effects that are directly linked to altered mRNA splicing of protein-coding gene(s) (“splicing-dependent consequences”) (6) and effects that are not directly related to altered splicing of any individual RNA (“splicing-independent consequences”) (7). Many mRNA isoforms promoted by mutant splicing factors harbor a premature termination codon and are degraded by nonsense-mediated mRNA decay or may result in impaired nuclear export of the aberrant mRNA. In addition, splicing-independent effects of these mutations have been identified. The clearest example to date includes elevated DNA:RNA hybrid (“R-loop”) formation in the presence of SRSF2 and U2AF1 mutations and consequent activation of the ATR-Chk1 pathway. The growing evidence of splicing-factor alterations and perturbed splicing in tumors underscores this area as ripe for cancer therapeutic development.
Acknowledgments
DECLARATION OF INTERESTS
O.A.-W. is a member of the SAB for Envisagenics. A.R.K. is a founder, director, advisor, stockholder, and chair of the SAB of Stoke Therapeutics and receives compensation from the company; he is a paid consultant for Biogen; he is a member of the SABs of Skyhawk Therapeutics, Envisagenics BioAnalytics, and Autoimmunity Biologic Solutions and has received compensation from these companies in the form of stock; he is a research collaborator of Ionis Pharmaceuticals and has received royalty income from Ionis through his employer, Cold Spring Harbor Laboratory.
ABBREVIATIONS
- snRNA
small nuclear RNA
- SRE
splicing regulatory elements
- ESE
exonic splicing enhancer
- ESS
exonic splicing silencer
- ISE
intronic splicing enhancer
- ISS
intronic splicing silencer
- SS
splice site
- BPS
branch point sequence
- PPT
polypyrimidine tract
- R
purine (A/G)
- Y
pyrimidine (C/U)
- SR
serine/arginine-rich protein
- hnRNPs
heterogeneous nuclear ribonucleoproteins
- PTC
premature termination codon
- MDS
myelodysplastic syndromes
- AML
acute myeloid leukemia
- CMML
chronic myelomonocytic leukemia
- CLL
chronic lymphocytic leukemia
- RARS
refractory anemia with ringed sideroblasts
- RCMD-RS
refractory cytopenia with multilineage dysplasia with ringed sideroblasts
- MPN
myeloproliferative neoplasms
- UVM
uveal melanoma
- MM
mucosal melanoma
- PC
pancreatic adenocarcinoma
- GB
glioblastoma
- MB
medulloblastoma
- HCC
hepatocellular carcinoma
- B-NHL
B cell non-Hodgkin lymphoma
REFERENCES
- Anczuków O, and Krainer AR (2016). Splicing-factor alterations in cancers. RNA 22, 1285–1301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, et al. (2015). Cancer-Associated SF3B1 Hotspot Mutations Induce Cryptic 3′ Splice Site Selection through Use of a Different Branch Point. Cell Rep. 13, 1033–1045. [DOI] [PubMed] [Google Scholar]
- Dvinge H, Kim E, Abdel-Wahab O, and Bradley RK (2016). RNA splicing factors as oncoproteins and tumour suppressors. Nat. Rev. Cancer 16, 413–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, and Bradley RK (2015). U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res. 25, 14–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, et al. (2015). SRSF2 Mutations Contribute to Myelodysplasia by Mutant-Specific Effects on Exon Recognition. Cancer Cell 27, 617–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki H, Kumar SA, Shuai S, Diaz-Navarro A, Gutierrez-Fernandez A, De Antonellis P, Cavalli FMG, Juraschka K, Farooq H, Shibahara I, et al. (2019). Recurrent noncoding U1 snRNA mutations drive cryptic splicing in SHH medulloblastoma. Nature 574, 707–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, and Lührmann R. (2009). The spliceosome: design principles of a dynamic RNP machine. Cell 136, 701–718. [DOI] [PubMed] [Google Scholar]
- Wang L, Lawrence MS, Wan Y, Stojanov P, Sougnez C, Stevenson K, Werner L, Sivachenko A, DeLuca DS, Zhang L, et al. (2011). SF3B1 and other novel cancer genes in chronic lymphocytic leukemia. N. Engl. J. Med 365, 2497–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. (2011). Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478, 64–69. [DOI] [PubMed] [Google Scholar]
- Zhang J, and Manley JL (2013). Misregulation of pre-mRNA alternative splicing in cancer. Cancer Discov. 3, 1228–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]