

Abstract
IMPORTANCE
Pathogenic DNA variants associated with familial hypercholesterolemia, hereditary breast and ovarian cancer syndrome, and Lynch syndrome are widely recognized as clinically important and actionable when identified, leading some clinicians to recommend population-wide genomic screening.
OBJECTIVES
To assess the prevalence and clinical importance of pathogenic or likely pathogenic variants associated with each of 3 genomic conditions (familial hypercholesterolemia, hereditary breast and ovarian cancer syndrome, and Lynch syndrome) within the context of contemporary clinical care.
DESIGN, SETTING, AND PARTICIPANTS
This cohort study used gene-sequencing data from 49 738 participants in the UK Biobank who were recruited from 22 sites across the UK between March 21, 2006, and October 1, 2010. Inpatient hospital data date back to 1977; cancer registry data, to 1957; and death registry data, to 2006. Statistical analysis was performed from July 22, 2019, to November 15, 2019.
EXPOSURES
Pathogenic or likely pathogenic DNA variants classified by a clinical laboratory geneticist.
MAIN OUTCOMES AND MEASURES
Composite end point specific to each genomic condition based on atherosclerotic cardiovascular disease events for familial hypercholesterolemia, breast or ovarian cancer for hereditary breast and ovarian cancer syndrome, and colorectal or uterine cancer for Lynch syndrome.
RESULTS
Among 49 738 participants (mean [SD] age, 57 [8] years; 27 144 female [55%]), 441 (0.9%) harbored a pathogenic or likely pathogenic variant associated with any of 3 genomic conditions, including 131 (0.3%) for familial hypercholesterolemia, 235 (0.5%) for hereditary breast and ovarian cancer syndrome, and 76 (0.2%) for Lynch syndrome. Presence of these variants was associated with increased risk of disease: for familial hypercholesterolemia, 28 of 131 carriers (21.4%) vs 4663 of 49 607 noncarriers (9.4%) developed atherosclerotic cardiovascular disease; for hereditary breast and ovarian cancer syndrome, 32 of 116 female carriers (27.6%) vs 2080 of 27 028 female noncarriers (7.7%) developed associated cancers; and for Lynch syndrome, 17 of 76 carriers (22.4%) vs 929 of 49 662 noncarriers (1.9%) developed colorectal or uterine cancer. The predicted probability of disease at age 75 years despite contemporary clinical care was 45.3% for carriers of familial hypercholesterolemia, 41.1% for hereditary breast and ovarian cancer syndrome, and 38.3% for Lynch syndrome. Across the 3 conditions, 39.7% (175 of 441) of the carriers reported a family history of disease vs 23.2% (34 517 of 148 772) of noncarriers.
CONCLUSIONS AND RELEVANCE
The findings suggest that approximately 1% of the middle-aged adult population in the UK Biobank harbored a pathogenic variant associated with any of 3 genomic conditions. These variants were associated with an increased risk of disease despite contemporary clinical care and were not reliably detected by family history.
Introduction
Identification of individuals at high risk for cardiovascular disease or cancer remains a major public health need.1 Because these diseases have a genetic component, one approach is to use inherited DNA variation to stratify the population. The United States Centers for Disease Control and Prevention has identified 3 tier-1 genomic conditions in which genetic testing to identify carriers of a pathogenic variant may be particularly useful: familial hypercholesterolemia, hereditary breast and ovarian cancer syndrome, and Lynch syndrome.2–4
Pathogenic variants in any of 9 genes associated with these 3 genomic conditions perturb key driving pathways in disease pathogenesis.5–7 Familial hypercholesterolemia variants accelerate development of atherosclerotic cardiovascular disease by preventing clearance of atherogenic low-density lipoproteins (LDLs) from the circulation.8 Those associated with hereditary breast and ovarian cancer syndrome confer increased risk of breast, ovarian, and other malignant tumors by disrupting tumor suppressor genes integral to DNA repair.9 Lynch syndrome variants are associated with colorectal, uterine, and other cancers through perturbation of DNA mismatch repair pathways.10
These genomic conditions remain underdiagnosed and undertreated in current clinical practice; if identified, evidence-based screening or therapies to attenuate risk are available. Such interventions include lipid-lowering therapy, which has been associated with up to a 76% reduced risk for cardiovascular disease among patients with familial hypercholesterolemia11,12; prophylactic mastectomy, which has been associated with up to a 90% reduced risk for breast cancer among patients with hereditary breast and ovarian cancer syndrome13; and screening colonoscopies, which have been associated with up to a 62% reduced risk for colorectal cancer among patients with Lynch syndrome.14
The traditional approach, which is currently recommended by clinical guidelines, has focused on genetic testing for individuals affected by disease and their family members.15–19 However, this phenotype-first past may give way to a genotype-first future. In this paradigm, population-based genomic screening may identify high-risk individuals before disease onset, enabling targeted screening or prevention.20,21
Population genomic screening has become increasingly feasible because of rapid decreases in the cost of gene sequencing and is already underway or planned in several health care systems and national biobanks.22–24 However, several areas of uncertainty remain. First, the prevalence of pathogenic variants assessed using systematic gene sequencing and clinical-grade variant classification in a large population of otherwise unascertained adults has not been fully characterized. Previous studies have either been small (performing clinical-grade variant classification of up to 1640 individuals, noting a prevalence ranging from 0.9% to 1.5%)23,25,26 or restricted to a single disease entity.5–7 Second, contemporary clinical care has expanded efforts to prevent or screen for cardiovascular diseases and cancer; the relative and absolute risks of disease among pathogenic variant carriers within the context of such care warrants further study. Third, whether family history of disease can serve as a reliable proxy for pathogenic variant status and the incremental risk of a pathogenic variant among individuals stratified by family history has not been fully explored.
We analyzed gene sequencing data from participants of the UK Biobank and used clinical-grade variant classification to assess the prevalence of pathogenic or likely pathogenic variants for the 3 tier-1 genomic conditions. Next, we assessed the association of such variants with family history of disease and the clinical importance of such variants within the context of contemporary clinical care.
Methods
Study Design, Setting, and Participants
This cohort study used data from the UK Biobank, which enrolled more than 500 000 individuals between the ages of 40 and 69 years from 22 sites across the UK between March 21, 2006, and October 1, 2010.27,28 Analysis of data from the UK Biobank data was approved by the Mass General Brigham institutional review board in Boston, Massachusetts, and was performed under UK Biobank application #7089. All participants provided electronic informed consent at their initial visit. This study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline.29
All 49 738 participants with whole exome sequencing data available were included in this analysis. Family history of disease in parents and siblings was reported by participants using a structured assessment tool at the time of enrollment. The LDL cholesterol concentrations and medication lists were assessed at time of enrollment as part of the study protocol, enabling estimation of untreated levels as described previously (eTable 1 in the Supplement).30 At the initial study visit, participants completed questionnaires about health history and lifestyle and underwent physical assessment and phlebotomy.28
Exome Sequencing and Variant Classification
Whole exome sequencing, which enables identification of genetic variants affecting the protein-coding region of each gene, was performed for UK Biobank participants as described previously.31–33 Additional details with respect to sequencing and quality control are available in the eMethods in the Supplement. Either of 2 laboratory geneticists (H.M-S. and M.L.), who were certified by the American Board of Medical Genetics and Genomics and who were blinded to any phenotype information, classified the pathogenicity of observed variants in 9 genes known to be associated with any of 3 genomic conditions according to current clinical standards.34 Variants for which classification was not straightforward because of limitations in available evidence were reviewed by both geneticists to arrive at a consensus opinion. These 9 genes included 3 associated with familial hypercholesterolemia (APOB [OMIM:107730], LDLR [606945], and PCSK9 [607786]), 2 associated with hereditary breast and ovarian cancer syndrome (BRCA1 [113705], BRCA2 [600185]), and 4 associated with Lynch syndrome (MLH1 [120436], MSH2 [609309], MSH6 [600678], and PMS2 [600259]).
Clinical End Points
Primary end points were composites specific to each genomic condition: coronary artery disease, ischemic stroke, and peripheral artery disease for familial hypercholesterolemia; breast or ovarian cancer for hereditary breast and ovarian cancer syndrome; and colorectal and uterine cancer for Lynch syndrome. Case definitions for each primary end point were defined in the UK Biobank using a combination of self-reported data confirmed by trained health care professionals, hospitalization records, and national procedural, cancer, and death registries. End points were assessed based on hospital inpatient data dating back to 1977, cancer data dating back to 1957, and death registry data available from time of initial enrollment from 2006 onward.35 Additional details with respect to disease ascertainment are available in the eMethods in the Supplement.
Statistical Analysis
Statistical analysis was performed from July 22, 2019, to November 15, 2019. Comparison of baseline characteristics between carriers of pathogenic or likely pathogenic variants and noncarriers was performed with the χ2 test for categorical variables and analysis of variance for continuous variables. Missing data were excluded. Hazard ratios for disease comparing carriers with noncarriers were calculated using Cox proportional hazards regression models with the covariates of enrollment age, sex, and genetic ancestry, as quantified by the first 4 genetic principal components.36 The age-dependent probability of disease in variant carriers and noncarriers was quantified using a Cox proportional hazards regression model adjusted for sex and the first 4 genetic principal components, with model standardized to the mean of each of the covariates. This model was extended to include family history and, when statistically significant, its interaction with carrier status. Statistical analyses were performed using R software, version 3.5 (R Project for Statistical Computing). Statistical significance was set at P < .05, and 2-sided P values were used.
Results
Sequencing of genetic samples from 49 738 participants (mean [SD] age, 57 [8] years; 27 144 female [54.6%]) identified 8777 total variants present in the 9 genes known to be associated with any of the 3 genomic conditions, including 3543 (40.4%) variants in genes associated with familial hypercholesterolemia, 2269 (25.9%) in genes associated with hereditary breast and ovarian cancer syndrome, and 2965 (33.8%) in genes associated with Lynch syndrome. Initial bioinformatic filtering was performed on this list of variants, restricting to those predicted to result in loss of function, rare missense variants (the maximum population allele frequency <0.005 in Genome Aggregation Database, a publicly available genetic variant frequency database),37–39 or variants that were previously classified as pathogenic or likely pathogenic in the ClinVar database (Figure 1).40 This filtering resulted in 3599 candidate variants for subsequent classification, performed according to current clinical criteria by clinical laboratory geneticists blinded to any phenotype information.34 A total of 232 variants met stringent clinical criteria to be classified as pathogenic or likely pathogenic, including 50 associated with familial hypercholesterolemia, 133 with hereditary breast and ovarian cancer syndrome, and 49 associated with Lynch syndrome. Details regarding evidence used to support pathogenicity assertions are provided for each variant in eTable 2 in the Supplement.
Figure 1.
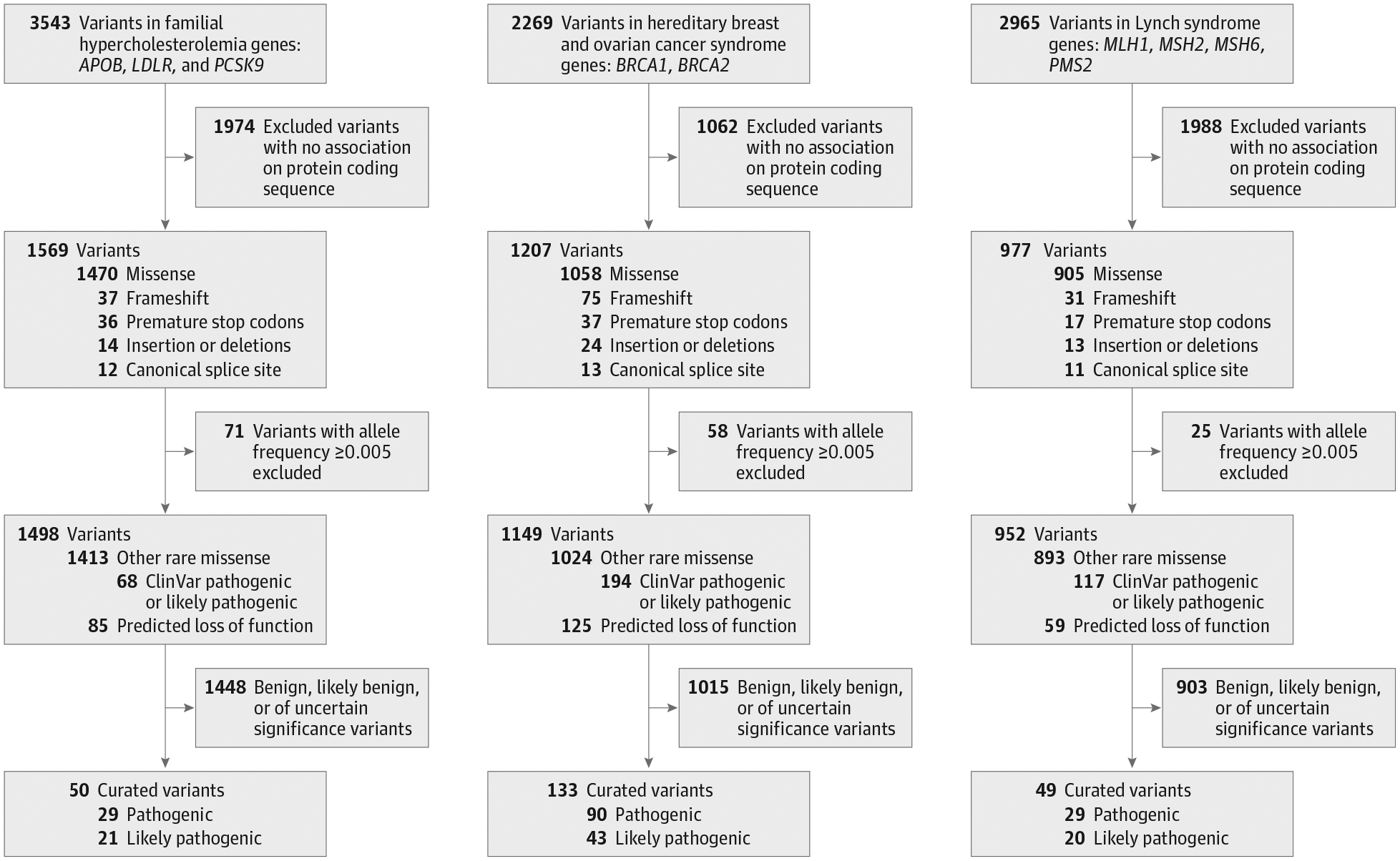
Filtration and Classification of Variants Identified by Whole-Exome Sequencing
Variants excluded (allele frequency [H11350]0.005) in the Genome Aggregation Database.37 Predicted loss-of-function variants do not necessarily meet all current standards to be classified as pathogenic or likely pathogenic.
A total of 441 individuals harbored any of these 232 pathogenic or likely pathogenic variants, corresponding to a prevalence of 0.89% (95% CI, 0.80%–0.98%). These individuals included 131 (0.26%; 95% CI, 0.22%–0.31%) with a variant associated with familial hypercholesterolemia, 235 participants (0.47%; 95% CI, 0.41%–0.54%) with a variant associated with hereditary breast and ovarian cancer syndrome, and 76 participants (0.15%; 95% CI, 0.12%–0.19%) with a variant associated with Lynch syndrome (eFigure 1 in the Supplement). Baseline characteristics of carriers and noncarriers of pathogenic or likely pathogenic variants are provided in eTables 3–5 in the Supplement.
Familial hypercholesterolemia is unique among the 3 genomic conditions because of availability of a circulating biomarker (LDL cholesterol) that might serve as an additional proxy for pathogenic variant carrier status. Both observed LDL cholesterol levels at time of enrollment and estimated untreated LDL cholesterol levels were significantly higher among carriers compared with noncarriers: 24 mg/dL higher (95% CI, 15–33 mg/dL; P < .001) for observed values and 53 mg/dL higher (95% CI, 44–62 mg/dL; P < .001) for estimated untreated values (to convert LDL cholesterol to millimoles per liter, multiply by 0.0259). However, the broad overlap in LDL cholesterol values between carriers and noncarriers underscores the limitations in the ability to identify variant carriers without access to genetic data (eFigure 2 in the Supplement).
Individuals who harbored a pathogenic or likely pathogenic variant experienced substantially increased risk of associated diseases. For familial hypercholesterolemia, 28 of 131 carriers (21.4%) vs 4663 of 49 607 noncarriers (9.4%) developed atherosclerotic cardiovascular disease, corresponding to a 3.03-fold (95% CI, 2.09–4.40) increased risk. For hereditary breast and ovarian cancer syndrome, 32 of 116 female carriers (27.6%) vs 2080 of 27 028 female noncarriers (7.7%) developed associated cancers, corresponding to a 4.11-fold (95% CI, 2.90–5.83) increased risk. For Lynch syndrome, 17 of 76 carriers (22.4%) vs 929 of 49 662 noncarriers (1.9%) developed colorectal or uterine cancer, corresponding to a 12.77-fold increased risk (95% CI, 7.90–20.64). Results stratified by gene end points are given in Figure 2 and by individual disease end points in eTables 6–9 in the Supplement. Risk estimates were similar across the 3 genes associated with familial hypercholesterolemia and 2 genes associated with hereditary breast and ovarian cancer syndrome. For Lynch syndrome, consistent with previous studies,41–44 risk was greatest among those with variants in the MLH1 gene (eTable 9 in the Supplement).
Figure 2.
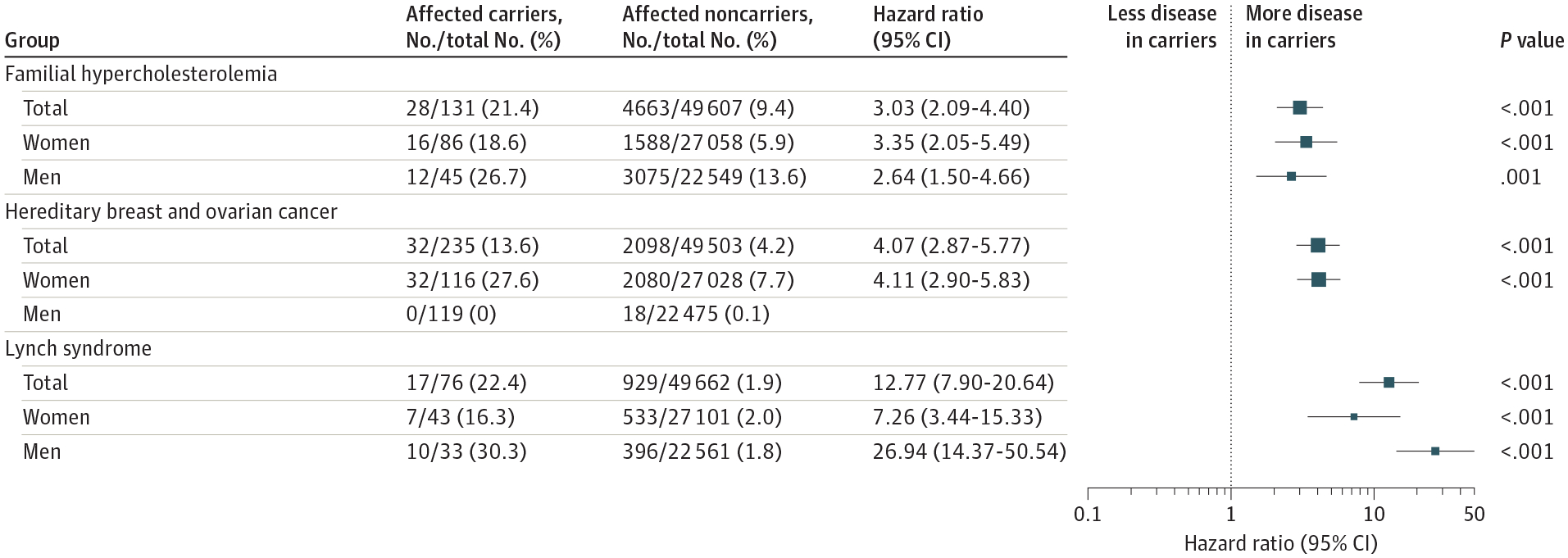
Prevalence and Clinical Importance of Pathogenic or Likely Pathogenic Variants for 3 Genomic Conditions
Risk calculated using Cox proportional hazards regression models with covariates of enrollment age, sex, and genetic ancestry as quantified by the first 4 genetic principal components for women, men, and total populations.
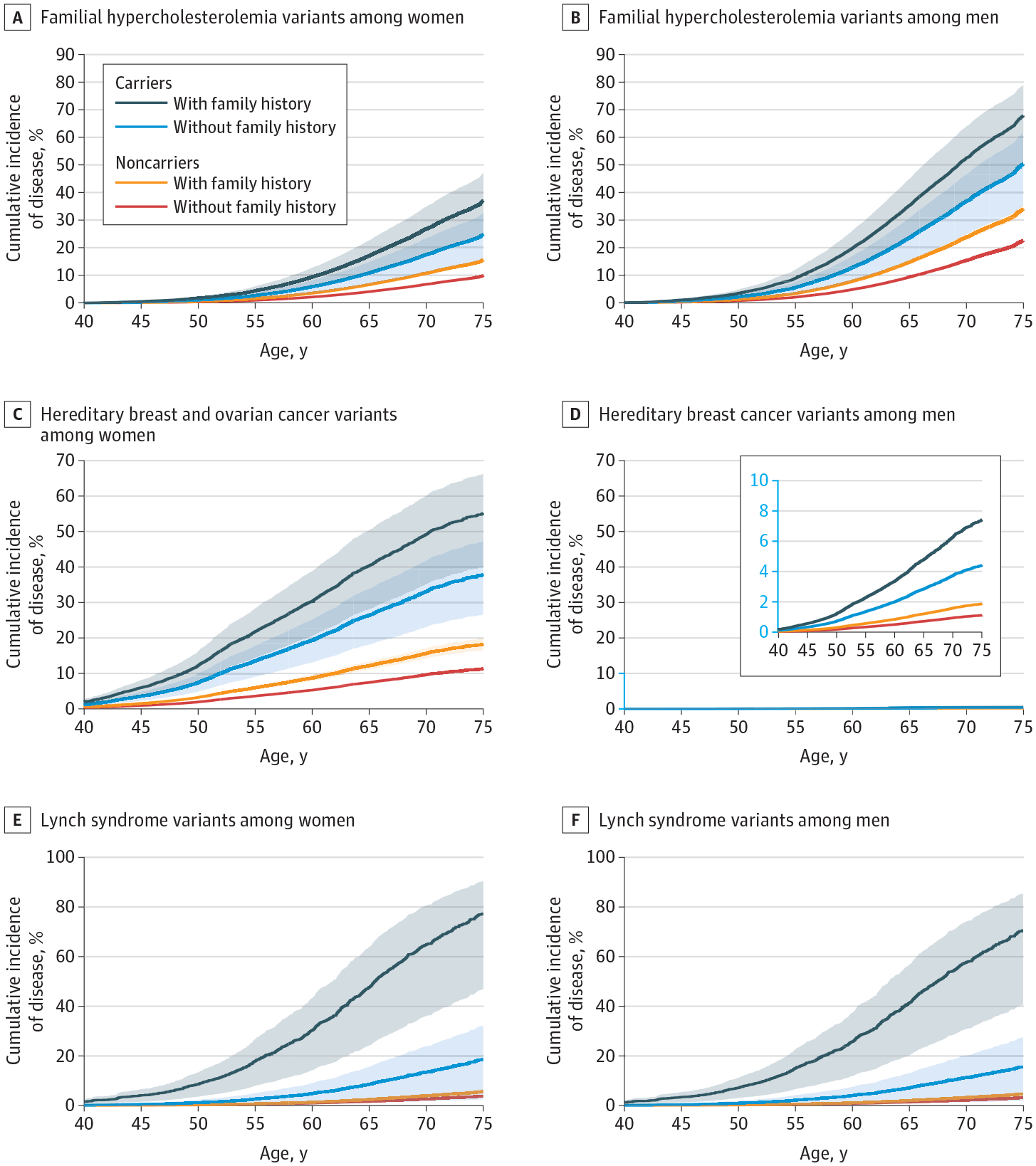
Cox proportional hazards regression modeling of cumulative age-specific probabilities of disease reinforced the clinical importance of these increased relative risk estimates. The estimated probability of developing atherosclerotic cardiovascular disease by age 75 years was 34.2% (95% CI, 24.0%–44.5%) in female familial hypercholesterolemia variant carriers vs 12.8% (95% CI, 12.1%–13.4%) in female noncarriers and 60.9% (95% CI, 47.3%–74.6%) in male carriers vs 26.4% (95% CI, 25.4%–27.4%) in male noncarriers. The estimated risk of developing breast or ovarian cancer syndrome by age 75 years in female hereditary breast and ovarian cancer variant carriers was 41.1% (95% CI, 30.2%–52.0%) in female carriers vs 12.2% (95% CI, 11.5%–12.8%) in female noncarriers, whereas the risk of developing male breast cancer by age 75 years was 0.5% (95% CI, 0.2%–0.8%) in male carriers and 0.1% (0.1%–0.2%) in male noncarriers. The estimated risk of developing colorectal or uterine cancer by age 75 years was 40.8% (95% CI, 25.9%–55.7%) in female Lynch syndrome variant carriers vs 4.1% (95% CI, 3.7%–4.5%) in female noncarriers, whereas the risk of developing colorectal cancer by age 75 years was 35.5% (95% CI, 21.7%–49.3%) in male carriers and 3.4% (95% CI, 3.1%–3.8%) in male noncarriers. Risk estimates appeared similar among male vs female participants, with no evidence of statistical interaction of carrier status by sex for any of the conditions (Figure 3) (eFigure 3 in the Supplement).
Figure 3.
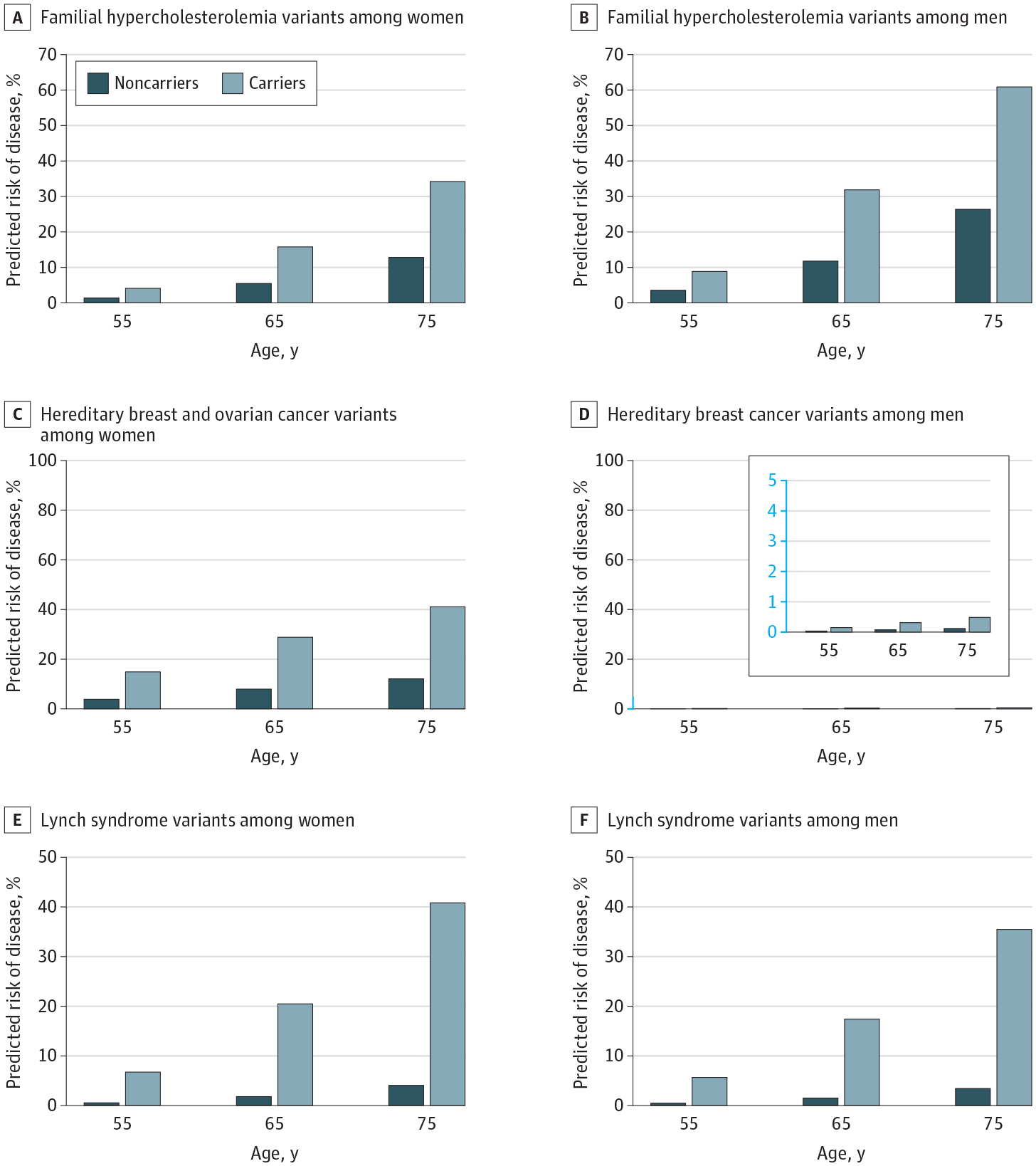
Predicted Risk of Disease by Ages 55, 65, and 75 Years by Pathogenic or Likely Pathogenic Variant Carrier Status
Risk of disease estimated using a Cox proportional hazards regression model standardized to the mean of the first 4 genetic principal components and sex and stratified by variant carrier status. All P < .001.
A family history of associated diseases as assessed in the UK Biobank by surveys completed at the time of enrollment would have failed to identify most of the individuals harboring a pathogenic or likely pathogenic variant. A first-degree relative with disease was reported by 66.4% (87 of 131) of those carrying a familial hypercholesterolemia variant, 25.5% (60 of 235) of those carrying a hereditary breast and ovarian cancer syndrome variant, and 36.8% (28 of 76) of those carrying a Lynch syndrome variant. Taken together across the 3 genomic conditions, a family history was reported in 175 of 441 carriers (39.7%) vs 34 517 of 148 772 noncarriers (23.3%) (Figure 4).
Figure 4.
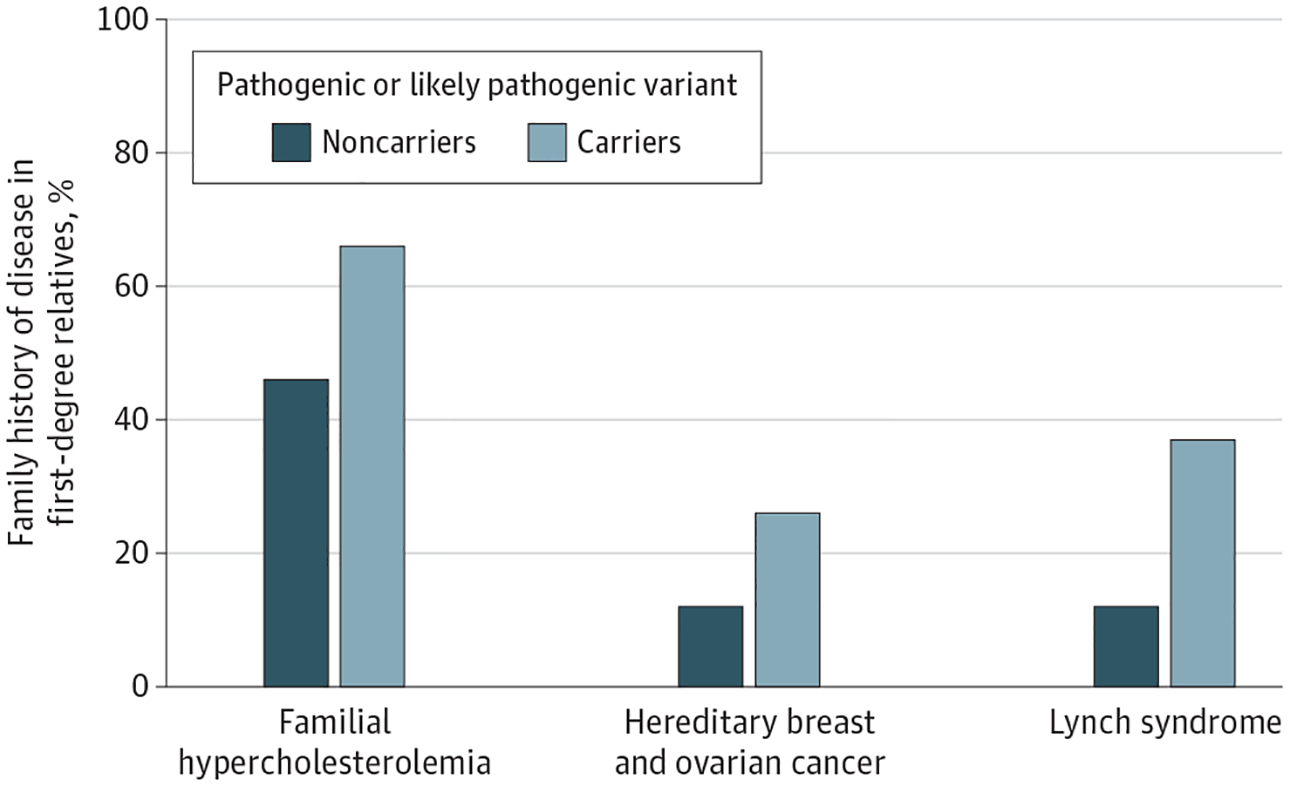
Family History of Disease in a First-Degree Relative by Presence of a Pathogenic or Likely Pathogenic Variant
First-degree relative with coronary artery disease for familial hypercholesterolemia, breast cancer for hereditary breast and ovarian cancer syndrome, and colorectal cancer for Lynch syndrome. All P < .001 designate statistical significance of differences in means of family history of disease based on variant carrier status.
Both family history of disease and pathogenic variant carrier status were independently associated with risk for each of the 3 genomic conditions studied (Figure 5). Within Cox proportional hazards regression models, the relative risk associated with a pathogenic variant was similar for those with and without a family history of disease. For Lynch syndrome, the association of family history with risk of disease was greater among those who reported a family history (eFigure 4 in the Supplement). For example, a woman’s risk for developing colorectal cancer or uterine cancer by age 75 years was estimated to be 3.9% (95% CI, 3.5%–4.3%) in a noncarrier without family history and 5.7% (95% CI, 4.7%–6.6%) in a noncarrier with family history and 18.7% (95% CI, 3.9%–33.4%) in a Lynch syndrome variant carrier without family history and 77.3% (95% CI, 58.0%–96.6%) in a variant carrier with family history (eFigure 4 in the Supplement).
Figure 5.
Age-Dependent Probability of Disease by Pathogenic or Likely Pathogenic Variant Carrier Status and Family History
Results estimated using a Cox proportional hazards regression model standardized to the mean of the first 4 genetic principal components and stratified by sex, variant carrier status, and family history for familial hypercholesterolemia (coronary artery disease, stroke, or peripheral artery disease), hereditary breast and ovarian cancer, and Lynch syndrome (colorectal and uterine cancer). The interaction of family history with carrier status was included in the models for Lynch syndrome variants, in which the interaction term was statistically significant.
The risk estimates presented reflect disease rates despite contemporary clinical practice. Many individuals identified with pathogenic or likely pathogenic variants did not receive recommended measures for screening and prevention of cardiovascular disease and cancer.15–17,19,45,46 For example, at the time of enrollment, only 75 of 131 individuals (57.3%) with familial hypercholesterolemia variants reported use of LDL cholesterol-lowering medications, only 12 of 25 female hereditary breast and ovarian cancer syndrome variant carriers younger than 50 years (48.0%) reported having ever undergone mammography screening, and only 4 of 15 of those with Lynch syndrome variants younger than 50 years (26.7%) reported undergoing screening for bowel cancer.
Discussion
In this study, we observed a rare pathogenic or likely pathogenic variant associated with any of 3 genomic conditions (familial hypercholesterolemia, hereditary breast and ovarian cancer syndrome, and Lynch syndrome) in 441 of 49 738 UK Biobank participants (0.9%). These individuals were at 3.03- to 12.77-fold relative risk for developing associated diseases despite contemporary clinical care, and their conditions were not reliably detected using self-reported family history.
These results build on previous efforts to understand the prevalence and clinical importance of monogenic risk variants in several key ways. First, blinded classification of genetic variants identified using systematic gene sequencing was performed by clinical laboratory geneticists. Second, estimates of increased risk were provided within the context of clinical care, including lipid-lowering therapy, mammography, and endoscopic screening in many participants. Third, all participants provided information about family history of disease in first-degree relatives using a structured survey assessment tool. Fourth, we analyzed a large cohort of individuals from a national biobank.
Although explicit demonstration that population genomic screening improves outcomes in a randomized clinical trial may not be feasible, identification of pathogenic variant carriers before disease onset may provide a clinically actionable opportunity for targeted screening, early therapy, and cascade testing of first-degree relatives.47 Previous studies have suggested that population-wide genomic screening of young adults may be cost-effective, particularly if the costs of genetic testing continue to decrease, although this topic warrants additional consideration.48 Moreover, although studies to date have not suggested psychologic harm associated with genetic risk disclosure, additional study is needed, particularly across a diverse set of diseases and individuals.49,50
Individuals with high-risk DNA variants may be difficult to reliably identify without direct gene sequencing, even for a condition like familial hypercholesterolemia, which has a measurable circulating biomarker with higher levels in carriers. In this study, family history of disease was reported in only 39.7% of those who harbored a pathogenic or likely pathogenic variant across all 3 genomic conditions, and even when present, clinicians often can not distinguish risk associated with shared DNA vs shared environment. Taken together, these results indicate an important opportunity to increase recognition and guideline-supported care of these individuals.
Representative key challenges to widespread implementation of population-based genomic screening into clinical practice exist. First, even for genes with known disease associations such as those included in this study, rigorous classification of the individual variants using current clinical guidelines is necessary.34 This requires manual curation of evidence, which relies heavily on a given variant co-segregating with disease within a family-based study, absence of presence at an appreciable frequency in diverse population databases of genomic variation, and rare allele frequency in global populations. Within our study, only 232 of 8777 observed genetic variants (2.6%) were ultimately classified as pathogenic or likely pathogenic in blinded review by clinical laboratory geneticists.
Second, few health systems have the necessary personnel and infrastructure to implement genomic medicine. Most current electronic health records do not allow for structured integration of genetic testing results into a patient’s medical record, which can lead to inefficiencies and duplicative testing. Moreover, few clinicians have a nuanced understanding of various forms of genetic testing. An instructive recent example involves testing for 3 known pathogenic hereditary breast and ovarian cancer variants by 23andMe (23andMe Inc), a direct-to-consumer genetic testing company. Although each of these 3 variants is associated with disease, this test failed to identify more than 90% of pathogenic variants.51 Anecdotal evidence suggests that some individuals or clinicians may be falsely reassured by a negative (but not comprehensive) test result.52
Third, carriers of monogenic risk variants may infer that they are predestined to develop disease. However, despite a several-fold increased risk associated with such variants, we estimated that 54.7% of familial hypercholesterolemia variant carriers, 59.0% of hereditary breast and ovarian cancer variant carriers, and 61.7% of Lynch syndrome variant carriers remain unaffected at age 75 years. This observation was consistent with recent results in a US health care system, in which more than 75% of female hereditary breast and ovarian cancer variant carriers remained free of disease well into middle age but was different from other mendelian diseases in which high-risk variants were associated with disease in a nearly 1:1 fashion.6 This overlap of disease-associated phenotypes was additionally noted when examining the distributions of LDL cholesterol levels in carriers vs noncarriers of familial hypercholesterolemia variants; consistent with multiple previous studies,5,53,54 we found a substantial right shift but overlapping distributions. Additional efforts are needed to integrate genetic and nongenetic factors into integrated risk estimation tools that better facilitate shared decision-making for patients and their health care teams. Moreover, these observations suggest a potential need to alter the current framework for clinical risk disclosure in which risk is described in a probabilistic manner rather than as pathogenic vs not pathogenic.55
Limitations
This study has limitations. First, UK Biobank participants were recruited at age 40 to 69 years, raising the possibility of survivorship or selection bias that limits generalizability to younger patients. Second, disease end points were ascertained through participant self-report, diagnosis codes from inpatient admissions, and national procedure, cancer, and death registries rather than manual review of records by a clinician. Third, participants in research studies tend to be healthier than the general population; recalibration of disease risk models for a given target population may be needed before clinical deployment.27,28,31 Fourth, we aggregated all pathogenic and likely pathogenic variants for each monogenic condition together, but even among this group, heterogeneity in risk for each specific variant may be present.34 Fifth, we analyzed whole-exome sequencing to identify pathogenic variants, but some classes of genetic variation, including cryptic splice sites, copy number variants, or structural variants, were not reliably detected using this technology.32,33 Previous work suggests that such variants account for a relatively small proportion of pathogenic variants, but this topic warrants additional study, particularly as algorithms to detect such variants continue to improve and whole genome sequencing data become available.56
Conclusions
The findings suggest that 0.9% of the middle-aged adult population in the UK Biobank harbored a pathogenic variant associated with familial hypercholesterolemia, hereditary breast or ovarian cancer syndrome, or Lynch syndrome. These individuals were at increased risk of disease, even within the context of routine clinical care, and carrier status was not reliably detected based on family history. Population genomic screening efforts may enable identification of these high-risk individuals before disease onset so that established risk mitigation strategies to overcome inherited disease susceptibility can be implemented.
Supplementary Material
Key Points.
Question
What is the prevalence and clinical importance of pathogenic DNA variants in genes associated with familial hypercholesterolemia, hereditary breast and ovarian cancer syndrome, and Lynch syndrome in a national biobank?
Findings
In this cohort study of 49 738 participants in the UK Biobank, a pathogenic or likely pathogenic variant associated with the 3 genomic conditions was identified in 0.9% of participants. These individuals had an increased risk of disease identified by gene sequencing that was not found through self-reported family history.
Meaning
The findings suggest that some participants in the UK Biobank harbored a pathogenic variant associated with familial hypercholesterolemia, hereditary breast and ovarian cancer syndrome, or Lynch syndrome and may have an increased risk of disease even within the context of routine clinical care; genomic screening efforts may enhance detection and treatment for these individuals.
Conflict of Interest Disclosures:
Dr Fahed reported being a consultant and reported holding equity and receiving personal fees from GoodPath outside the submitted work. Dr Philippakis reported receiving personal fees from GV outside the submitted work. Dr Cassa reported receiving grants from the National Institutes of Health during the conduct of the study. Dr Ellinor reported receiving a grant from Bayer AG to the Broad Institute focused on the genetics and therapeutics of cardiovascular diseases and receiving personal fees from serving on advisory boards or consulting for Bayer AG, Novartis AG, and Quest Diagnostics Inc during the conduct of the study. Dr Kathiresan reported being an employee of Verve Therapeutics and having equity in Catabasis, Maze Therapeutics, San Therapeutics, and Verve Therapeutics; being a member of the scientific advisory boards for Corvidia Therapeutics and Regeneron Genetics Center; reported serving as a consultant for Acceleron, Aegerion, Alnylam, Bayer Healthcare, Blackstone, Color Genomics Inc, Haug Partners, Illumina, Ionis, Leerink Partners, Eli Lilly and Company, MedGenome, Medscape, Merck & Company Inc, Noble Insights, Novartis AG, Novo Nordisk, Novo Ventures, Pfizer, and Quest Diagnostics outside the submitted work; and receiving patents related to a method of identifying and treating a person having a predisposition to or afflicted with cardiometabolic disease and a genetics risk predictor. Dr Ng reported being an employee of IBM Research. Dr Lebo reported receiving grant funding from the National Institutes of Health. Dr Khera reported serving as a consultant and receiving personal fees from Color Genomics, Maze Therapeutics, Medicines Company, Navitor Pharmaceuticals, and Sanofi Pharmaceuticals outside the submitted work; receiving speaking fees from Illumina; receiving grants and personal fees from the Novartis Institute for Biomedical Research; receiving sponsored research agreements from IBM Research and the Novartis Institute for Biomedical Research; receiving grants from the Broad Institute of MIT and Harvard, Massachusetts General Hospital, and the National Human Genome Research Institute (NHGRI) during the conduct of the study; and having a patent related to a genetic risk predictor. No other disclosures were reported.
Funding/Support: Funding support was provided by grant T32HL007208 from the National Heart, Lung, and Blood Institute (Drs Patel and Fahed); grant 14CVD01 from the Fondation Leducq (Dr Ellinor); grants 1RO1HL092577, R01HL128914, and K24HL105780 from the National Heart, Lung, and Blood Institute (Dr Ellinor); grant 18SFRN34110082 from the American Heart Association (Dr Ellinor); an institutional grant from the Broad Institute of MIT and Harvard (Merkin Institute Fellowship, Dr Khera); grants 1K08HG010155 and 5UM1HG008895 (Dr Khera) and R01HG010372 (Drs Cassa and Lebo) from the NHGRI; a Hassenfeld Scholar Award from Massachusetts General Hospital (Dr Khera); and a sponsored research agreement from IBM Research (Drs Philippakis and Khera).
Role of the Funder/Sponsor: The funding organizations had no role in the design and conduct of the study; collection, management, analysis, and interpretation of the data; preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Additional Information: The Laboratory for Molecular Medicine is a not-for-profit reference molecular genetic testing laboratory performing fee-for-service testing. Work was performed under UK Biobank application #7089.
SUPPLEMENT.
eMethods. Study Populations, Gene Sequencing, Variant Quality Control, and Variant Classification.
eTable 1. Adjustment of Lipid Levels in the UK Biobank Based on Self-report of Lipid-Lowering Medications
eTable 2. Variant Characteristics and Evidence in Support of Pathogenic or Likely Pathogenic Classifications
eTable 3. Baseline Characteristics of Familial Hypercholesterolemia Variant Carriers and Noncarriers
eTable 4. Baseline Characteristics of Women Hereditary Breast and Ovarian Cancer Syndrome Variant Carriers and Noncarriers
eTable 5. Baseline Characteristics of Lynch Syndrome Variant Carriers and Noncarriers
eTable 6. Familial Hypercholesterolemia, Hazard Ratios for Each Component of the Composite End Point
eTable 7. Hereditary Breast and Ovarian Cancer Syndrome, Hazard Ratios for Primary Cancers
eTable 8. Lynch Syndrome, Hazard Ratios for Primary Cancers
eTable 9. Prevalence and Clinical Importance of Pathogenic or Likely Pathogenic Variants for 3 Genomic Conditions Stratified by Gene
eFigure 1. Frequency of Carriers of Pathogenic or Likely Pathogenic Variants for Each of 3 Genomic Conditions According to Gene
eFigure 2. Observed and Estimated Untreated Low-Density Lipoprotein Cholesterol Levels According to Familial Hypercholesterolemia Variant Status
eFigure 3. Age-Dependent Cumulative Probability of Disease According to Pathogenic or Likely Pathogenic Variant Carrier Status
eFigure 4. Predicted Risk of Disease at Ages 55, 65, and 75 According to Pathogenic or Likely Pathogenic Variant Carrier Status and Family History
eReferences.
REFERENCES
- 1.Heron MP. Deaths: leading causes for 2017. Natl Vital Stat Rep. 2019;68(6). Accessed September 17, 2019 https://stacks.cdc.gov/view/cdc/79488 [PubMed] [Google Scholar]
- 2.Murray MF, Evans JP, Khoury MJ. DNA-based population screening: potential suitability and important knowledge gaps. JAMA. 2020;323(4):307–308. doi: 10.1001/jama.2019.18640 [DOI] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention. Tier 1 genomics applications and their importance to public health. Published April 2, 2019. Accessed September 17, 2019 https://www.cdc.gov/genomics/implementation/toolkit/tier1.htm
- 4.George R, Kovak K, Cox SL. Aligning policy to promote cascade genetic screening for prevention and early diagnosis of heritable diseases. J Genet Couns. 2015;24(3):388–399. doi: 10.1007/s10897-014-9805-5 [DOI] [PubMed] [Google Scholar]
- 5.Abul-Husn NS, Manickam K, Jones LK, et al. Genetic identification of familial hypercholesterolemia within a single US health care system. Science. 2016;354(6319). doi: 10.1126/science.aaf7000 [DOI] [PubMed] [Google Scholar]
- 6.Manickam K, Buchanan AH, Schwartz MLB, et al. Exome sequencing–based screening for BRCA1/2 expected pathogenic variants among adult Biobank participants. JAMA Netw Open. 2018;1(5):e182140. doi: 10.1001/jamanetworkopen.2018.2140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haraldsdottir S, Rafnar T, Frankel WL, et al. Comprehensive population-wide analysis of Lynch syndrome in Iceland reveals founder mutations in MSH6 and PMS2. Nat Commun. 2017;8:14755. doi: 10.1038/ncomms14755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gidding SS, Champagne MA, de Ferranti SD, et al. ; American Heart Association Atherosclerosis, Hypertension, and Obesity in Young Committee of Council on Cardiovascular Disease in Young, Council on Cardiovascular and Stroke Nursing, Council on Functional Genomics and Translational Biology, and Council on Lifestyle and Cardiometabolic Health. The agenda for familial hypercholesterolemia: a scientific statement from the American Heart Association. Circulation. 2015;132(22):2167–2192. doi: 10.1161/CIR.0000000000000297 [DOI] [PubMed] [Google Scholar]
- 9.Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002;108(2):171–182. doi: 10.1016/S0092-8674(02)00615-3 [DOI] [PubMed] [Google Scholar]
- 10.Lynch HT, Lynch PM, Lanspa SJ, Snyder CL, Lynch JF, Boland CR. Review of the Lynch syndrome: history, molecular genetics, screening, differential diagnosis, and medicolegal ramifications. Clin Genet. 2009;76(1):1–18. doi: 10.1111/j.1399-0004.2009.01230.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luirink IK, Wiegman A, Kusters DM, et al. 20-Year follow-up of statins in children with familial hypercholesterolemia. N Engl J Med. 2019;381(16):1547–1556. doi: 10.1056/NEJMoa1816454 [DOI] [PubMed] [Google Scholar]
- 12.Versmissen J, Oosterveer DM, Yazdanpanah M, et al. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. BMJ. 2008;337:a2423. doi: 10.1136/bmj.a2423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ludwig KK, Neuner J, Butler A, Geurts JL, Kong AL. Risk reduction and survival benefit of prophylactic surgery in BRCA mutation carriers, a systematic review. Am J Surg. 2016;212(4):660–669. doi: 10.1016/j.amjsurg.2016.06.010 [DOI] [PubMed] [Google Scholar]
- 14.Järvinen HJ, Aarnio M, Mustonen H, et al. Controlled 15-year trial on screening for colorectal cancer in families with hereditary nonpolyposis colorectal cancer. Gastroenterology. 2000;118(5):829–834. doi: 10.1016/S0016-5085(00)70168-5 [DOI] [PubMed] [Google Scholar]
- 15.Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;139(25):e1082–e1143. doi: 10.1161/CIR.0000000000000625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Owens DK, Davidson KW, Krist AH, et al. ; US Preventive Services Task Force. Risk assessment, genetic counseling, and genetic testing for BRCA-related cancer: US Preventive Services Task Force recommendation statement [published correction in JAMA. 2019;322(18):1830. JAMA. 2019;322(7):652–665. doi: 10.1001/jama.2019.10987 [DOI] [PubMed] [Google Scholar]
- 17.Bevers TB, Helvie M, Bonaccio E, et al. Breast cancer screening and diagnosis, version 3.2018, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. 2018;16(11):1362–1389. doi: 10.6004/jnccn.2018.0083 [DOI] [PubMed] [Google Scholar]
- 18.Oeffinger KC, Fontham ETH, Etzioni R, et al. ; American Cancer Society. Breast cancer screening for women at average risk: 2015 guideline update from the American Cancer Society. JAMA. 2015;314(15):1599–1614. doi: 10.1001/jama.2015.12783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Giardiello FM, Allen JI, Axilbund JE, et al. Guidelines on genetic evaluation and management of Lynch syndrome: a consensus statement by the US Multi-society Task Force on Colorectal Cancer. Am J Gastroenterol. 2014;109(8):1159–1179. doi: 10.1038/ajg.2014.186 [DOI] [PubMed] [Google Scholar]
- 20.King M-C, Levy-Lahad E, Lahad A. Population-based screening for BRCA1 and BRCA2: 2014 Lasker Award. JAMA. 2014;312(11):1091–1092. doi: 10.1001/jama.2014.12483 [DOI] [PubMed] [Google Scholar]
- 21.Manchanda R, Gaba F. Population based testing for primary prevention: a systematic review. Cancers (Basel). 2018;10(11):424. doi: 10.3390/cancers10110424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Leitsalu L, Haller T, Esko T, et al. Cohort profile: Estonian Biobank of the Estonian Genome Center, University of Tartu. Int J Epidemiol. 2015;44(4):1137–1147. doi: 10.1093/ije/dyt268 [DOI] [PubMed] [Google Scholar]
- 23.Dewey FE, Murray MF, Overton JD, et al. Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science. 2016;354(6319):aaf6814. doi: 10.1126/science.aaf6814 [DOI] [PubMed] [Google Scholar]
- 24.Grzymski JJ, Coppes MJ, Metcalf J, et al. The Healthy Nevada Project: rapid recruitment for population health study. Preprint. Posted online January 19, 2018. bioRxiv 250274. doi: 10.1101/250274 [DOI] [Google Scholar]
- 25.Tang CS-M, Dattani S, So M-T, et al. Actionable secondary findings from whole-genome sequencing of 954 East Asians. Hum Genet. 2018;137(1):31–37. doi: 10.1007/s00439-017-1852-1 [DOI] [PubMed] [Google Scholar]
- 26.Haer-Wigman L, van der Schoot V, Feenstra I, et al. 1 in 38 Individuals at risk of a dominant medically actionable disease. Eur J Hum Genet. 2019;27(2):325–330. doi: 10.1038/s41431-018-0284-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bycroft C, Freeman C, Petkova D, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562(7726):203–209. doi: 10.1038/s41586-018-0579-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sudlow C, Gallacher J, Allen N, et al. UK Biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12(3):e1001779. doi: 10.1371/journal.pmed.1001779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.von Elm E, Altman DG, Egger M, Pocock SJ, Gøtzsche PC, Vandenbroucke JP; STROBE Initiative. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Lancet. 2007;370(9596):1453–1457. doi: 10.1016/S0140-6736(07)61602-X [DOI] [PubMed] [Google Scholar]
- 30.Benn M, Watts GF, Tybjærg-Hansen A, Nordestgaard BG. Mutations causative of familial hypercholesterolaemia: screening of 98 098 individuals from the Copenhagen General Population Study estimated a prevalence of 1 in 217. Eur Heart J. 2016;37(17):1384–1394. doi: 10.1093/eurheartj/ehw028 [DOI] [PubMed] [Google Scholar]
- 31.Van Hout CV, Tachmazidou I, Backman JD, et al. Whole exome sequencing and characterization of coding variation in 49,960 individuals in the UK Biobank. Preprint. Posted online March 9, 2019. bioRxiv 572347. doi: 10.1101/572347 [DOI] [Google Scholar]
- 32.Regier AA, Farjoun Y, Larson DE, et al. Functional equivalence of genome sequencing analysis pipelines enables harmonized variant calling across human genetics projects. Nat Commun. 2018;9(1):4038. doi: 10.1038/s41467-018-06159-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics. 2014;30(20):2843–2851. doi: 10.1093/bioinformatics/btu356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richards S, Aziz N, Bale S, et al. ; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.UK Biobank. Data providers and dates of availability. Accessed November 10, 2019 http://biobank.ndph.ox.ac.uk/showcase/exinfo.cgi?src=Data_providers_and_dates
- 36.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38(8):904–909. doi: 10.1038/ng1847 [DOI] [PubMed] [Google Scholar]
- 37.Karczewski KJ, Francioli LC, Tiao G, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding. Preprint. Posted online August 13, 2019. bioRxiv 531210. doi: 10.1101/531210 [DOI] [Google Scholar]
- 38.Lek M, Karczewski KJ, Minikel EV, et al. ; Exome Aggregation Consortium. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi: 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McLaren W, Gil L, Hunt SE, et al. The Ensembl Variant Effect Predictor. Genome Biol. 2016;17(1):122. doi: 10.1186/s13059-016-0974-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Landrum MJ, Lee JM, Benson M, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016;44(D1):D862–D868. doi: 10.1093/nar/gkv1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dowty JG, Win AK, Buchanan DD, et al. Cancer risks for MLH1 and MSH2 mutation carriers. Hum Mutat. 2013; 34(3):490–497. doi: 10.1002/humu.22262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Møller P, Seppälä T, Bernstein I, et al. ; Mallorca Group. Cancer incidence and survival in Lynch syndrome patients receiving colonoscopic and gynaecological surveillance: first report from the prospective Lynch syndrome database. Gut. 2017;66(3):464–472. doi: 10.1136/gutjnl-2015-309675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Senter L, Clendenning M, Sotamaa K, et al. The clinical phenotype of Lynch syndrome due to germ-line PMS2 mutations. Gastroenterology. 2008;135(2):419–428. doi: 10.1053/j.gastro.2008.04.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bonadona V, Bonaïti B, Olschwang S, et al. ; French Cancer Genetics Network. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA. 2011;305(22):2304–2310. doi: 10.1001/jama.2011.743 [DOI] [PubMed] [Google Scholar]
- 45.Gupta S, Provenzale D, Llor X, et al. NCCN guidelines insights: genetic/familial high-risk assessment: colorectal, version 2.2019. J Natl Compr Canc Netw. 2019;17(9):1032–1041. doi: 10.6004/jnccn.2019.0044 [DOI] [PubMed] [Google Scholar]
- 46.Arnett DK, Blumenthal RS, Albert MA, et al. 2019 ACC/AHA guideline on the primary prevention of cardiovascular disease: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;140(11):e596–e646. doi: 10.1161/CIR.0000000000000678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Knowles JW, Rader DJ, Khoury MJ. Cascade screening for familial hypercholesterolemia and the use of genetic testing. JAMA. 2017;318(4):381–382. doi: 10.1001/jama.2017.8543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhang L, Bao Y, Riaz M, et al. Population genomic screening of all young adults in a health-care system: a cost-effectiveness analysis. Genet Med. 2019;21(9):1958–1968. doi: 10.1038/s41436-019-0457-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Green RC, Roberts JS, Cupples LA, et al. ; REVEAL Study Group. Disclosure of APOE genotype for risk of Alzheimer’s disease. N Engl J Med. 2009;361(3):245–254. doi: 10.1056/NEJMoa0809578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bloss CS, Schork NJ, Topol EJ. Effect of direct-to-consumer genomewide profiling to assess disease risk. N Engl J Med. 2011;364(6):524–534. doi: 10.1056/NEJMoa1011893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murphy H Don’t count on 23andMe to detect most breast cancer risks, study warns. New York Times. April 16, 2019. Accessed October 18, 2019 https://www.nytimes.com/2019/04/16/health/23andme-brca-gene-testing.html [Google Scholar]
- 52.Esplin ED. Limitations of direct-to-consumer genetic screening for hereditary breast, ovarian, and colorectal cancer risk Abstract presented at: American Society of Human Genetics; October 17, 2019; Houston, TX. [Google Scholar]
- 53.Khera AV, Won H-H, Peloso GM, et al. Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J Am Coll Cardiol. 2016;67(22): 2578–2589. doi: 10.1016/j.jacc.2016.03.520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Benn M, Watts GF, Tybjaerg-Hansen A, Nordestgaard BG. Familial hypercholesterolemia in the Danish general population: prevalence, coronary artery disease, and cholesterol-lowering medication. J Clin Endocrinol Metab. 2012;97(11):3956–3964. doi: 10.1210/jc.2012-1563 [DOI] [PubMed] [Google Scholar]
- 55.Khera AV, Mason-Suares H, Brockman D, et al. Rare genetic variants associated with sudden cardiac death in adults. J Am Coll Cardiol. 2019;74(21):2623–2634. doi: 10.1016/j.jacc.2019.08.1060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Collins RL, Brand H, Karczewski KJ, et al. An open resource of structural variation for medical and population genetics. Preprint. Posted online October 21, 2019. bioRxiv 578674. doi: 10.1101/578674 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.