


Abstract
Translation fidelity relies essentially on the ability of ribosomes to accurately recognize triplet interactions between codons on mRNAs and anticodons of tRNAs. To determine the codon-anticodon pairs that are efficiently accepted by the eukaryotic ribosome, we took advantage of the IRES from the intergenic region (IGR) of the Cricket Paralysis Virus. It contains an essential pseudoknot PKI that structurally and functionally mimics a codon-anticodon helix. We screened the entire set of 4096 possible combinations using ultrahigh-throughput screenings combining coupled transcription/translation and droplet-based microfluidics. Only 97 combinations are efficiently accepted and accommodated for translocation and further elongation: 38 combinations involve cognate recognition with Watson-Crick pairs and 59 involve near-cognate recognition pairs with at least one mismatch. More than half of the near-cognate combinations (36/59) contain a G at the first position of the anticodon (numbered 34 of tRNA). G34-containing tRNAs decoding 4-codon boxes are almost absent from eukaryotic genomes in contrast to bacterial genomes. We reconstructed these missing tRNAs and could demonstrate that these tRNAs are toxic to cells due to their miscoding capacity in eukaryotic translation systems. We also show that the nature of the purine at position 34 is correlated with the nucleotides present at 32 and 38.
INTRODUCTION
In the three kingdoms of life, translation of genetic information into proteins takes place on the macromolecular machine called the ribosome. Using messenger RNA as a template, the ribosome catalyses the sequential addition of amino acids to the nascent polypeptide chain by recruiting cognate aminoacylated tRNAs according to the successive codons. Cell integrity requires a high fidelity rate in order to avoid the production of potentially toxic aberrant proteins. Nevertheless, it has been estimated both in bacteria and eukaryotes that 18% of the proteins (from a 400-amino acid long protein) contain at least one mis-incorporated amino acid under normal physiological conditions (1). Escherichia coli tolerates up to 10% of error-containing protein (2), while higher error rates, up to 50%, often lead to lethality by various mechanisms such as toxic protein production under stress conditions, for example, or protein misfolding (1,3). In E. coli, errors due to incorrect tRNA aminoacylations occur rarely (4–7). Another source of errors is aberrant decoding, also called miscoding, on the ribosome itself. Such errors are caused by abnormal frameshifting (10−5 in prokaryotes) (8) or more frequently by missense errors when the ribosome accommodates a near-cognate aminoacylated tRNA on a codon (10−3–10−4 both in E. coli and yeast) (1,9–13). Structural investigations using X-ray crystallography with several near-cognate codon–anticodon pairs in the A site of the ribosome demonstrated that the ribosomal decoding grip can accommodate near-cognate tRNAs when pairing with the codon adopts a ‘Watson–Crick-like geometry’ (14–17). After being accommodated in the ribosomal A site, the aminoacylated tRNA base-paired to the codon undergoes the translocation step that is defined by a concerted movement of the tRNA and the mRNA with respect to ribosomal subunits towards the P and later on the E sites (18,19). Importantly, to avoid frameshifting, base pairing in the codon/anticodon helix is maintained during the whole translocation process (20). The low in vivo misreading rate suggests that the ribosome discriminates against most potential errors by preventing their translocation (10). Therefore, the miscoding rate results from the cumulative proofreading steps of (i) A-site accommodation of anticodon/codon duplex and (ii) translocation check point of the codon–anticodon mini helix prior movement from the A to the P site.
To gain further insights in decoding rules, we sought to identify all codon–anticodon combinations that are first efficiently accommodated in the A site and then further allowed to undergo translocation to the P site. We used the Internal Ribosome Entry Site (IRES) from Cricket Paralysis Virus (CrPV). This dicistrovirus contains in its viral RNA genome two open reading frames separated by an InterGenic Region (IGR) IRES (21,22). The IGR is able to promote translation initiation on any codon without the need of any translation initiation factor (eIF) or initiator tRNA (23–25). It folds into a sophisticated structure, which contains three pseudoknot structures PKI, PKII and PKIII (26). PK II and PKIII fold into compact domains that participate in ribosomal recruitment (27,28). The IRES is properly positioned in the decoding centre of the ribosome by two other RNA domains, SLIV and SLV that directly interact with the small ribosomal subunit 40S (24,29,30). Most importantly, PKI functionally mimics a codon–anticodon helix and is recognized by the ribosome in the same way as a cognate mRNA–tRNA codon–anticodon duplex is (Figure 1 and Supplementary Figure S1) (30–32). Recent cryo-EM studies have demonstrated that PKI enters the ribosome by interacting with the A site in an identical manner as an aminoacylated tRNA base-paired to its cognate codon (30,32). The IRES domains SLIV and SLV bind to the head of the ribosome, thereby restricting its flexibility, which allows the introduction of PKI in the A site (33). The same restriction of the ribosomal head movement is also observed during canonical translation (34,35). Indeed, PKI accurately mimics structurally and functionally a tRNA anticodon base paired with the three nucleotides of the codon (Supplementary Figure S1) (33). Moreover, biochemical experiments demonstrated that correct base pairing in PKI is a prerequisite for an active IGR IRES (36,37). Once PKI is loaded into the A site, the elongation factor eEF-2 promotes further translocation of PKI toward the P site like the codon–anticodon mini helix during canonical translation (33,38–40). When PKI is in the P site, contacts with SLIV and SLV are disrupted, leaving the A site free to accept the next aminoacylated tRNA and translation elongation of the native reporter protein can proceed (41). After translocation PKI dissociates in the E site and mimics the acceptor stem of an E-site tRNA (42).
Figure 1.

Microfluidic pipeline for high throughput screening of active codon-anticodon pairs. The Cricket Paralysis Virus IGR IRES was inserted in the 5′UTR of a reporter gene. PKI (shown in blue), which mimics a codon-anticodon duplex, was placed in frame with the coding sequence of Green Fluorescence Protein (GFP). Using this construct, we generated a cDNA library containing the IGR-GFP sequence with the full set of the possible 4096 (64 × 64) codon-anticodon combinations in frame with PKI. Each IGR-GFP cDNA variant was individualized into droplets (diluted to reach a 10% of occupancy) to limit droplet occupancy by more than one variant. Each variant was first PCR amplified prior to fusing each droplet with another one containing a coupled transcription/translation mixture made of T7 RNA polymerase and rabbit reticulocyte lysate. Upon an hour of incubation at 30°C, droplets were sorted based on their fluorescence. Fluorescent droplets containing active codon–anticodon combinations were recovered, pooled and their cDNA content analyzed by Next Generation Sequencing (NGS). The resulting sequences were compared to NGS sequencing of the starting library for normalization.
Altogether, these studies confirmed that PKI follows the same path than a codon–anticodon duplex namely (i) discrimination of codon/anticodon duplex in the A site and (ii) its subsequent translocation from the A to the P site. However, unlike tRNAs, PKI does not contain any modified nucleotides that are known to influence codon–anticodon interactions. We used PKI as a molecular scaffold to investigate the functional constraints imposed by the ribosome decoding centre and identify in a systematic fashion which codon/anticodon pairs of unmodified bases are able to support translation elongation. With an original approach that combines microfluidic technology with cell-free translation extracts, we could screen a library containing the 4096 codon/anticodon combinations (64 × 64).
MATERIALS AND METHODS
Microfluidic-assisted ultrahigh-throughput screening procedure
Gene library preparation
IRES gene library with randomized codon/anticodon mimicking regions was prepared by PCR amplifying the CrPV IGR coding template using a sense primer (5′GTCGTCTAATCCAGAGACCCCGGATCGGATATTAATACGACTCACTATAGGCAAAAATGTGATCTTGCTT3′) appending the T7 RNA polymerase promoter (underlined sequence) to the construct and an antisense primer (5′CGAAGTATCTTGAAATGTAGCNNNTAAATTTCTTAG-GTTTTTCGACTANNNAATCTGAAAAACCGCAGAGAGGGCTTCCTGG3′) with the codon/anticodon mimicking regions randomized (symbolized by N in antisense primer sequence) with a controlled ratio of 25/25/25/25 for A/C/G/T (Integrated DNA Technologies). A PCR mixture containing 0.02 ng/μl of CrPV IGR-containing template plasmid, 0.2 μM of each primer (Integrated DNA Technologies), 0.2 mM of each dNTP (Thermofisher), 0.04 U/μl of Phusion DNA polymerase (Thermofisher) and the corresponding buffer at the recommended concentration was subjected to an initial denaturation step of 2 min at 95°C followed by 25 cycles of: 30 s at 95°C, 30 s at 55°C, 1 min at 72°C, and terminated by a final extension of 10 min at 72°C. The PCR product was then purified on a 1% agarose TBE gel, the band containing the product of interest was excised, the DNA recovered using a ‘Wizard® SV Gel and PCR Clean-Up System’ kit (Promega) and quantified with a Nanodrop (Thermo Scientific). This PCR product was then used as primer in a second PCR reaction using as template the GFPMut2-containing plasmid (pGFP) we used in a previous study (43). This second PCR was performed in the same conditions as above but using 0.02 ng/μl of pGFP, 0.07 μM of the first PCR product (the region of the antisense primer complementary to GFP is italicized) and 0.2 μM of RevGFP primer (5′ GAAGCGGCCGCTCTAGATTAATTTAAATC3′). Finally, this second PCR product was purified on a 1% agarose gel and quantified as above. The proper randomization of the library was confirmed by NGS analysis (Supplementary Figure S2b). Importantly, even though some sequences were found slightly over-represented (dark blue combinations in the matrix), they did not introduce any bias in the selection process as they were not found in the enriched pools.
Droplet-based microfluidic screening
We used the same overall droplet-based microfluidics strategy we described before for the screening of ribozyme (44) or fluorogenic aptamer (45–47) gene libraries (Supplementary Figure S2a) with a few adaptations.
i. Digital droplet PCR
First, DNA molecules were individualized into 2.5 pl PCR mixture-containing droplets by diluting the DNA solution such that only 1 out 10 droplets initially contained a DNA molecule to limit multiple encapsulation events. To do so, a PCR mixture containing 0.13 pM of template DNA diluted into 200 ng/μl yeast total RNA (Ambion), 0.2 μM of FwdIRES-GFP (5′GTCGTCTAATCCAGAGACCCCGGATCGG3′) and RevGFP (5′ GAAGCGGCCGCTCTAGATTAATTTAAATC3′) primers, 0.2 mM of each dNTP (Thermofisher), 0.1% Pluronic F68 (Gibco), 0.7 mg/ml Dextran Texas-Red 70 000 MW (Thermofisher), Phire Hot Start II DNA polymerase (Thermofisher) and the corresponding buffer at recommended concentrations was dispersed into 2.5 pl droplets carried by a Novec7500 fluorinated oil (3M) supplemented with 3% of fluorosurfactant as described before (44). The emulsion was collected and thermocycled as above.
ii.In vitro gene expression
Upon thermal-cycling, amplified DNA-containing droplets were reinjected into a droplet fusion device where they were synchronized and fused with larger 17 pl on-chip produced droplets (generated as described in (45) containing an in vitro expression mixture made of 3 mM of the 20 amino acids, 1 U/μl RNasin® (Promega), 500 μM of the four NTP, 3 mM MgCl2, 80 mM KCl, 30 μl/ml Dextran Texas-Red 70 000 MW (Thermofisher), 50 μg/ml of T7 RNA polymerase purified in the lab and half of a volume of Rabbit Reticulocyte Lysate prepared as previously described (48). Pairwise droplets were fused, the emulsion was collected as described before (45) and incubated for 3 h at 30°C.
iii. Droplet analysis and sort
Upon incubation, droplets were reinjected into a sorting device where they were spaced by a surfactant-free oil stream and their fluorescence was analyzed just before reaching the sorting junction (49). Droplets orange (Texas-red) fluorescence allowed discriminating in vitro expression droplets fused to a single PCR droplet from those unfused or fused with more than one PCR droplet as described in (44). Moreover, using the green fluorescence (GFP fluorescence) of these single-fused droplets allowed us to identify and sort those droplets displaying significant concentration of GFP, therefore containing variants able to support efficient translation initiation. The green fluorescence gates used for each experiment are summarized on Supplementary Figures S2d and S7a. Upon sorting, the recovered droplets were collected in a tube and broken by adding 50 μl of 1H,1H,2H,2H-perfluoro-1-octanol (Sigma-Aldrich) and 200 μl of 200 μg/ml yeast total RNA solution (Ambion). The selected genes were finally recovered by PCR-amplifying the DNA contained in a 2 μl aliquot of droplet lysate solution and introducing it in 100 μl of PCR reaction mixture containing 0.2 μM of FwdIRES-GFP (5′GTCGTCTAATCCAGAGACCCCGGATCGG3′) and RevGFP (5′ GAAGCGGCCGCTCTAGATTAATTTAAATC3′) primers, 0.2 mM of each dNTP (Thermofisher), 0.04 U/μl of Phusion DNA polymerase (Thermofisher) and the corresponding buffer at the recommended concentration. The mixture was then subjected to an initial denaturation step of 2 min at 95°C followed by 25 cycles of: 30 s at 95°C, 30 s at 55°C, 1 min at 72°C, and terminated by a final extension of 10 min at 72°C. The PCR product was then purified on a 1% agarose TBE gel, the band containing the product of interest was excised, the DNA recovered using a ‘Wizard® SV Gel and PCR Clean-Up System’ kit (Promega) and quantified with a Nanodrop (Thermo Scientific). The purified DNA was then either used to start a new round of screening or indexed and sequenced (see below).
The procedure described above was performed in two independent biological replicates using different batches of Rabbit reticulocytes extract. At each round, an average of 1.5 million of droplets were screened corresponding to a minimum of ∼37.5 times coverage (considering the 10% occupancy of PCR droplets).
Sequence analysis
Libraries indexing and sequencing
A 2 μl aliquot of droplet lysate was introduced in 100 μl of PCR reaction mixture containing 0.2 μM of Label-CrPV-Fwd (5′TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGGGCAAAAATGTGATCTTGCTTGTAAAT3′) and Label-CrPV-Rev (5′ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGGGACAACTCCAGTGAAAAGTTCTTC3′) primers, 0.2 mM of each dNTP (Thermofisher), 0.04 U/μl of Phusion DNA polymerase (Thermofisher) and the corresponding buffer at the recommended concentration. The mixture was then subjected to an initial denaturation step of 2 min at 95°C followed by 25 cycles of: 30 s at 95°C, 30 s at 55°C, 1 min at 72°C, and terminated by a final extension of 10 min at 72°C. PCR products were purified using a ‘Wizard® SV Gel and PCR Clean-Up System’ kit (Promega). The PCR product was then diluted down to 0.5 ng/μl into 25 μl of a PCR mixture containing 2.5 μl of each Nextera Index primer (Illumina), 0.2 mM of each dNTP (Thermofisher), 0.04 U/μl of Phusion DNA polymerase (Thermofisher) and the corresponding buffer at the recommended concentration. The mixture was subjected to an initial denaturation step of 3 min at 95°C followed by 20 cycles of: 30 s at 95°C, 30 s at 55°C, 30 s at 72°C and the run was concluded by 5 min of final extension at 72°C. PCR products were then purified on a 1% agarose gel as above. Indexed DNA libraries were then quantified as recommended by Illumina, loaded and analyzed on a MiSeq intrument using a MiSeq Reagent kit V2 300 cycles cartridge and a pair-end protocol.
Sequencing data analysis
Fastq data files were analyzed using a custom Python-written pipeline. Briefly, high quality reads (Q-score > 30) were recovered and only those sequences free of mutation outside the randomized region (i.e. codon/anticodon mimicking region) were conserved for the rest of the analysis. Next, we filtered out the experimental noise that corresponded mainly to sequences displaying mutations in the codon/anticodon region as a result of PCR and/or sequencing errors. These sequences were expected to have a significantly lower occurrence than the error-free DNA (a droplet was expected to contain ∼2.4 × 105 identical copies of the DNA). Therefore, monitoring the occurrence of the different sequences made possible identifying those underrepresented (Supplementary Figure S2c). Indeed, whereas in the case of evenly represented sequences (e.g. starting library, Supplementary Figure S2a and b) their occurrences linearly accumulate throughout the sequence population, the presence of a significantly underrepresented sub-population of sequences leads to a biphasic line whom the breakpoint can be used as a signal/noise threshold (Supplementary Figure S2b). Using this approach, only sequences with an occurrence frequency over the threshold (respectively set to 1.9 × 10−3 and 2.4 × 10−3 for replicates 1 and 2 of relaxed selection and set to 3.4 × 10−3 and 1.6 × 10−3 respectively for replicates 1 and 2 of stringent selection) were considered has ‘real’ signal (Supplementary Figure S2c). Furthermore, only sequences reliably found in both replicates were conserved for the establishment of codon/anticodon matrices (Supplementary Figures S2e and S7a). This led to 97 sequences in the relaxed selection conditions and 55 sequences in the stringent selection conditions. Finally, in the stringent selection conditions, only the 52 sequences shared with those reliably identified in the relaxed selection conditions were finally considered. The generally larger number of sequences found in replicate 2 correlates well with the apparently less resolutive sort (Supplementary Figure S2b and c).
tRNA transcript synthesis
tRNAs were in vitro transcribed by T7 RNA polymerase from templates generated by primer extension of overlapping DNA oligonucleotides (IDT) and purified on 10% denaturing PAGE. Transcripts were extracted by soaking gel slices two hours at 37°C in 50 mM KOAc and 200 mM KCl, pH 7, precipitated, and resuspended in H2O (50).
In vitro translation with rabbit reticulocyte lysates
Translation reactions were performed in self-made RRL extracts as previously described (51). Reactions were incubated at 30°C for 60 min and included 100 and 200 nM of each reporter mRNA transcript and 10.8 μCi [35S]Met. Aliquots of translation reactions were analyzed by 15% SDS-PAGE and Luciferase assays.
tRNA transfection in HeLa cells and metabolic assay
Adherent HeLa cells were maintained at 37°C, 5% CO2, in Dulbecco's modified Eagle's medium supplemented with fetal calf serum and antibiotics. Transfections were performed in 96-well plates at 90% confluency with 1.25 pmol (∼30 ng) or 2.5 pmol (∼60 ng) of control or impossible tRNAs using Lipofectamine 2000 (Invitrogen) according to manufacturer's instructions (52). Each transfection condition was independently replicated nine times.
Cell proliferation assays were performed 24 h after transfection by addition of 10 μl (1/10 of the culture volume) of WST-1 (G-biosciences). The formazan dye (yellow) produced by metabolically active cells was quantified using a multi-well spectrophotometer at 450 nm, 30 min after WST-1 addition (53). Unpaired t-tests (GraphPad software package) were performed to evaluate the statistical relevance of the differences in metabolic activities between control and impossible tRNAs.
RESULTS AND DISCUSSION
Ultrahigh-throughput screening of the complete anticodon/codon library
In order to profile exhaustively the different anticodon/codon pairs and identify those accepted and translocated by the ribosome, we designed a droplet-based microfluidic screening pipeline allowing the quantitative measurement of the expression of a green fluorescent protein-coding (GFP) reporter gene. We first prepared a construct in which the IGR IRES from CrPV was placed upstream the GFP coding sequence (Figure 1 and Supplementary Figure S1). The internal anticodon and codon mimicking sequences embedded in PKI pseudoknot recruit the ribosome and lead to GFP synthesis all the better the anticodon/codon pair is efficiently accepted by the ribosome, which makes possible to directly correlate anticodon/codon acceptancy with a fluorescent signal. Using this construct as a framework, we generated a library containing the 4096 (64 × 64) possible anticodon/codon combinations by randomizing both sequences (Figure 1 and Supplementary Figure S1). The library was then screened for variant able to support efficient translation (so anticodon/codon pairs readily accepted by the ribosome) using a droplet-based microfluidic workflow (Supplementary Figure S2a) similar to that previously used to analyze protein (54) and RNA gene libraries (44–47). Briefly, each DNA molecule of the library was individualized with an amplification mixture into picoliter-sized water-in-oil droplets serving as independent vessels. Upon PCR amplification, each droplet was fused one-to-one with a larger droplet containing an in vitro coupled transcription and translation mixture allowing genes to be transcribed and resulting mRNA to be translated (55), provided the anticodon/codon pair displayed by PKI is properly accommodated and used by the ribosome. Therefore, constructs in which an IRES-displayed anticodon/codon pair was accepted and validated by the ribosome were expected to support GFP synthesis, turning the corresponding droplets fluorescent and allowing for sorting them (49). Two replicates were performed (Supplementary Figure S2d) during which our capacity to generate and manipulate millions of such droplets in a single experiment, allowed us to screen the 4096 combinations contained in the starting library with a more than 30-time coverage. Next-generation sequencing was then used to assess the completeness of the starting library (Supplementary Figure S2b and c) and then to identify functional codon/anticodon pairs that were selected (Supplementary Figure S2e).
Selected cognate codon/anticodon combinations with Watson-Crick base pairs
Throughout this publication, we use the sign ‘/’ to indicate both orientations of base pairs (e.g. A/U for A–U or U–A) and the sign ‘o’ for standard wobble base between G and U (e.g. GoU for G–U). We found that only 97 combinations are efficiently accepted and translocated by the ribosome (Figure 2). Surprisingly, less than half of the combinations, 38 out of 97, are among the expected complementary Watson–Crick (W–C) pairs between anticodon and cognate codon (black squares on the diagonal of the matrix in Figure 2 and Supplementary Figure S3). Gaps in the diagonal correspond to the remaining 26 possible W–C combinations that were not selected (Figure 3 and Supplementary Figure S4). Among these combinations, a significant bias against A/U-content was observed and the higher the A/U content, the less the sequence was recovered (Figure 3A, B and Supplementary Figure S4). Indeed, only 1 instance out of eight A/U-free sequence pairs was missing, whereas 6 out of 12 (50%), 13 out of 24 (54%) and 6 out of 8 (75%) sequences were missing for sequence pairs containing respectively 1, 2 and 3 A/U base pairs. In 19 out of 26 instances, the missing combinations have a pyrimidine (Y) at the first anticodon position (numbered 34) and thus a purine (R) at position 3 of the codon (Supplementary Figure S4). These missing combinations include the AUG start codon and the three STOP codons. The only missing W–C combination without an A/U pair is the combination between anticodon CCC and codon GGG (corresponding to Gly) (Supplementary Figure S4).
Figure 2.
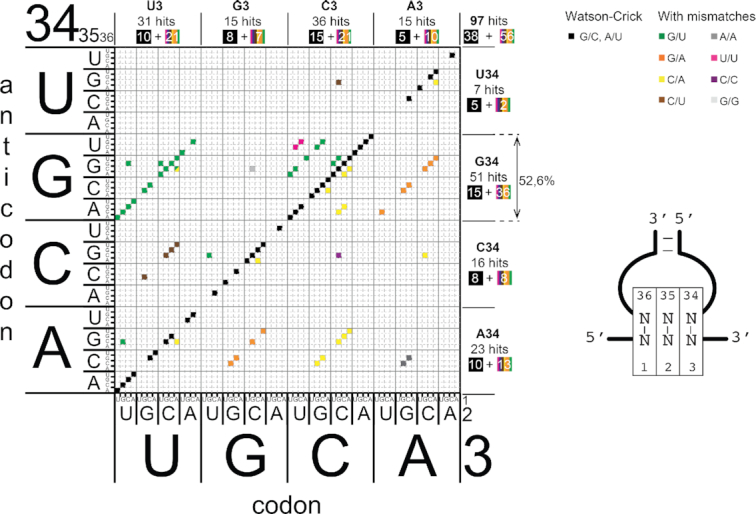
Matrix representing the codon–anticodon combinations selected by the microfluidic pipeline. The sequences of the active codon–anticodon pairs that are efficiently recognized by the ribosome are plotted on a matrix. The 64 codons are represented on the x-axis and 64 anticodons are represented on the y-axis. The nucleotides of the codons are numbered 1, 2 and 3 from 5′ to 3′. The nucleotides of the anticodons are numbered 34, 35, 36 from 5′ to 3′ according to their position in tRNAs. The active codon–anticodon combinations are represented on the matrix by black squares (Watson–Crick pairs are along the diagonal) and by coloured squares for combinations containing mismatches that are parallel to the diagonal. The total number of hits is indicated on the upper right part of the matrix. The number of selected codon–anticodon pairs containing A, C, G and U at position 34 (anticodon) are shown on the right of the matrix. The number of selected codon–anticodon pairs containing A, C, G and U at position 3 (codon) are shown above the matrix. In each case, the number of hits is decomposed in those along the main diagonal (in black squares) and those off-diagonal (in rainbow squares).
Figure 3.

Combinations of codon–anticodon containing Watson-Crick base pairs never isolated through the selection procedure. (A) Histogram showing the increase in the contribution of G/C base pairs in the selected W-C combinations (see Supplementary Figure S3 in supplementary material for more detailed distributions). The numbers are normalized to the total number of triplets containing 0 (8), 1 (24), 2 (24) or 3 (8) G/C pairs. (B) Histogram showing the increase in the contribution of A/U base pairs in the W–C combinations missing from the selection (see Supplementary Figure S4 in supplementary material for more detailed distributions). The numbers are normalized to the total number of triplets containing 0 (8), 1 (24), 2 (24) or 3 (8) A/U pairs. (C) Distribution of the missing Watson-Crick combination absent from the selection is plotted on the wheel of the genetic code (blue circles around the third codon base) (56). At the north of the wheel are shown the ‘strong’ or G/C-rich codon–anticodon triplets, at the south the ‘weak’ or A/U-rich codon–anticodon triplets and in the middle the ‘intermediate’ ones as measured by the Turner energy of the helical triplet.
The distribution of the missing Watson–Crick combinations absent from the selection is plotted on the wheel representation of the genetic code (56) in Figure 3C. At the north of the wheel are shown the ‘strong’ or G/C-rich codon–anticodon triplets, at the south the ‘weak’ or A/U-rich codon–anticodon triplets and in the middle the ‘intermediate’ ones as measured by the energy (57) of the helical triplet. The A/U-rich missing combinations confirm the contribution of nucleotide modifications in such codon/anticodon triplets to productive decoding in natural systems (58,59). In five of the codon boxes, all codons are selected (corresponding to the two 4-codon boxes Pro and Arg, and the three 2-codon boxes His, Gln and Ser). In the selected combinations, three codons were not represented, corresponding to Glu (GAR), Lys (AAR) and Met (AUG). This is also true for the 2-codon boxes corresponding to amino acids Leu (UUR) and Arg (AGR) (see Figure 3C and Supplementary Figure S5). Please note that we use the corresponding amino acid for commodity but, in the present experimental system, no amino acid is inserted.
Selected near-cognate codon/anticodon combinations
Besides W–C combinations, we also selected a significant number of combinations (59/97) with one or two mismatches at the three positions of the codon (Figure 2 and Supplementary Figure S5a). Overall, eight types of mismatches are accepted by the ribosome in the present experimental set-up (G/U, G/A, C/A, C/U, A/A, U/U, C/C and G/G). Most mismatches occur at the third position of the codon (Supplementary Figure S5b, c). Among the observed mismatches, the most frequent ones are G/U, G/A, C/A and C/U (Supplementary Figure S5c). Although mismatches are generally found in both orientations (Supplementary Figure S6a), there are two exceptions. First G/U mismatches observed at the third codon position are exclusively of the G34oU3 type and never of the U34oG3 type. Secondly, C/A mismatches at the second position of the codon are found only as C2/A35. Whereas G/U and C/A mismatches are found in all three positions of the triplets, G/A, C/U, A/A, C/C and G/G mismatches are only found in the third position (Supplementary Figure S5a and c). Overall, the most frequent mismatch is the G/U base pair, which was found 26 times in the 59 combinations. Likewise, in vivo the mis-incorporation frequency determined by mass spectrometry is ∼10−3–10−5, also confirming that G/U are the most frequent mismatches, responsible for 40% of mis-incorporated amino acids (17,60).
The distribution of mismatches at the first two positions leads to three interesting observations: (i) 7 out of 10 mismatches in the first position are C/A mismatches in both orientations; (ii) 8 out of 12 mismatches in the second position are G/U mismatches, 6 are between U2 and G35 and 2 between G2 and U35; (iii) only 2 C/A or U/U mismatches are in the second position (see Supplementary Figures S5c and S6a). Structural data have shown the presence of G/U pairs at both the first and second positions with a ‘Watson–Crick like’ geometry rather than a ‘wobble’ geometry (61). The present data provide further evidence that G/U base pairs can be accommodated not only at the A site of the ribosome decoding centre but can also be efficiently translocated from the A to the P site to proceed with elongation. This is further corroborated by other structural observations showing atypical Watson-Crick-like G/U base pair geometry in the P site of the tRNA (14). Interestingly, C/A mismatches at position 2 are exclusively observed in the C2–A35 orientation (Supplementary Figure S6a). Finally, G/A mismatches in both orientations are only observed in position 3 of the codon. G/A is a mismatch involving two large bases that can coexist only in position 3 but not in positions 1 and 2 due to tighter steric constraints (62). The same reasoning likely applies to G/G and A/A mismatches that are also observed only in position 3 of the codon (Supplementary Figure S5).
Interestingly, only 17 near-cognate events would lead to amino acid change (in normal ribosomal translation) and all those involve a G34-containing anticodon (see Supplementary Figure S8 and below).
To further corroborate the selection matrix, we chose several representative selected combinations, namely purely W–C combinations, combinations containing mismatches with a few combinations that were not selected taken as negative controls. We inserted them in a reporter mRNA containing the IGR upstream of the Renilla luciferase coding region in order to measure the corresponding translation efficiency. Using this experimental set-up, we determined that the present microfluidic pipeline allowed us to select combinations that promote translation efficiency >20% compared to the Wt IGR (Supplementary Figure S6b). Likewise, we confirmed the preferential orientation of G34/U3 mismatch at position 3 of the codon as deduced from the selection results (Supplementary Figure S6c).
The types of selected combinations indicate that the IRES-based microfluidic pipeline recapitulates faithfully cellular decoding rules
A/U rich combinations require modification in the anticodon
In cells, tRNAs contain critical nucleotide modifications in the ASL. Although every anticodon nucleotide can be modified, positions 32, 34, 37 and 38 (Supplementary Figure S1) play critical roles in translation fidelity (63,64). In the present in vitro transcribed PKI and its associated IGR-GFP reporter, these modified nucleotides are absent. The observation that nearly all of the missing W–C combinations contain between one and three A/U base pairs strongly suggests that A/U base pairs are weak interactions not stable enough on the ribosome to allow efficient translocation without modifications in the tRNA ASL (63). Especially, the lack of modifications also correlates with the absence in the selected pools of combinations with A or U at the 1st and 3rd positions of the codon. Both require modifications at either position 37 (63) or at position 34 (16,65) of the anticodon, respectively (see below) (Supplementary Figure S1). There is particularly a poor representation of combinations with a U or a C at the first position 34 of the anticodon (19 out of 26 cases), indeed U34 and C34 are nucleotides that are frequently modified in tRNAs (16,65) (Figure 3B).
As expected, the absence of modified nucleotides in the PKI of the reporter IGR-GFP does not allow the selection of the A/U-rich codon–anticodon combinations since they are not stable enough on the ribosome and cannot promote GFP translation. Thus, the gaps in the W–C diagonal mainly correspond to codons that require post-transcriptional modifications in the tRNA ASL to be efficiently decoded by the eukaryotic ribosome.
Position 3 of the codon is the most permissive
Among the three positions of the codon, it is expected that position 3 of the codon is the most permissive one (66). As expected, most of the mismatches are observed on position 3 of the codon (Supplementary Figure S5b, c). This is consistent with fundamental decoding rules in both prokaryotic and eukaryotic ribosomes (66).
Most of the G34/U3 wobble combinations are efficiently selected
Remarkably, 12 of the 16 possible combinations with a G34oU3 pair were selected; the four missing ones contain A/U base pairs at positions 1 and/or 2 in addition to the G34oU3, which suggests that the binding affinity of the corresponding complexes is too low to support translational activity in the modification-free PKI. Standard wobble base pairs between G and U are widely used in cells for decoding synonymous codons; the observation that the present system selected almost all of them confirms that PKI accurately mimics the structure and the function of a genuine codon/anticodon mini helix on the ribosome. Additional evidence comes from the asymmetry observed in the G/U pairs at the third position where only G34oU3 and not the inverted U34oG3 base pair was observed. This observation is in agreement with crystallographic data showing the expected G34oU3 wobble pair while the U34oG3 pair was observed only in presence of U34 modification and then only in a tautomeric Watson–Crick-like geometry (67) or in a novel type of pair in which the U instead of moving in the major groove as in standard wobble pair moves in the minor groove (68). These alternative geometries are imposed by the ribosome decoding center grip together with the presence of modified nucleotides shaping the ASL and the codon/anticodon helix (16,56). Using a Renilla luciferase reporter assay, we confirmed that such a preferential orientation is also observed in the context of the IGR PKI, which again confirms that PKI is a biologically relevant mimic of the codon–anticodon helix (Supplementary Figure S6c).
The most frequently accepted mismatches are G/U, G/A and C/A
In order to assess the efficiency of the selected codon–anticodon combinations to promote translation, we re-screened the enriched libraries from both replicates and performed another screen with a higher sorting cut-off (only the 50% most fluorescent droplets displaying a signal above the background were recovered) and (this new selection was called ‘stringent selection’) (Supplementary Figure S7a, b). Under these ‘stringent’ conditions, only 52 combinations are present in both replicates ((Supplementary Figure S7b). However, the distribution of mismatches is similar to the first selection, again most of the mismatches are observed on position 3 of the codon. The combinations containing the G34oU3 wobble mismatch are still largely represented (10/16 possible combinations). Moreover, the combinations contain only three types of mismatches G/U, G/A and C/A indicating that they are better accepted by the ribosome. Interestingly, triplets involving G/U, G/A and C/U mismatches are responsible for the most prevalent amino acid substitutions observed in vivo (60). In contrast, A/A, U/U, C/C and G/G are not present in the ‘stringent’ selection, suggesting that they are less efficiently tolerated by the ribosome, which is in agreement with in vivo observations.
We realize that the PKI-based reporter system does not fully mimic a tRNA anticodon–codon duplex. First, unlike tRNA molecules, the IRES is delivered to the ribosome in the absence of elongation factors, therefore the system eliminates initial selection of cognate tRNA molecules and competition between tRNAs and factors. Secondly, the IRES forms extensive contacts with the ribosome compared to tRNAs and in the IRES structure, the ‘anticodon-like’ triplet is covalently linked to the ‘codon-like’ triplet. Such factors could affect tRNA selection and proofreading in the A site. Nevertheless, the results of the selections strongly indicate that the PKI-based microfluidic-assisted selection using an IGR-GFP reporter system does recapitulate faithfully most fundamental aspects of both bacterial and eukaryotic decoding. Altogether, the present data suggest that eukaryotic ribosomes (rabbit) follow the same basic geometrical rules as the bacterial decoding center (68) and that decoding centeres across the different domains of life likely share a similar if not identical structural grip.
More than half of the combinations contain G34 in the anticodon
Unexpectedly, the majority of the selected combinations (51/97, 52.6%) contained G34 in the anticodon (Figure 2). These comprise 15 W–C combinations G34–C3 (15 of the 16 possible), 12 wobble combinations G34–U3 (12 of the 16 possible) suggesting that combinations with a G34 are well accepted by the ribosome. Surprisingly, G34 is also present in 24 combinations containing mismatches (Figure 2), suggesting the sole presence of G34 allows mismatches at the three positions of the codon. In terms of decoding, the consequence of such mismatches would be detrimental for translation fidelity. Indeed, many of these combinations would promote miscoding issues because they enable base pairing of tRNA anticodons with near-cognate codons (Supplementary Figure S8). The resulting potential miscoding errors are almost exclusively observed in the decoding of the 4-codon boxes (equivalent to Leu (CUN), Ala, Val, Thr, Ser (UCN), Pro, Arg (CGN) and Gly). The high occurrence of mismatches in combinations with G34 prompted us to examine the distribution of G34 in tRNA genes decoding the 4-codon boxes from several eukaryotic species. As previously shown (65), G34-containing tRNAs are very rarely found in eukaryotic tRNAs that decode 4-codon boxes (Figure 4a and Supplementary Figure S9). Indeed G34-containing tRNAs are virtually absent from all eukaryotic genomes in the tRNAs decoding 4-codon boxes; A34 tRNAs (normally modified into I34, see for example (69)) are used instead. Gly is the only exception for which, in all three kingdoms, a G34-containing tRNA decodes the C3- and U3-ending codons (and A34 is never observed (65)). In contrast to bacterial tRNAs for Gly, the eukaryotic tRNAGly do not exhibit the potential for 32–38 WC pair (mainly C/C or C/A in all eukaryotes tested on the GtRNAdb database (70)). Explanations have been suggested for the presence of eukaryotic G34-containing tRNAGly: structural incompatibility of A34 in the anticodon loop (71) or frame maintenance (72). We note also the conserved presence of GU pairs in the alignments of anticodon stem of eukaryotic tRNAGly (70).
Figure 4.
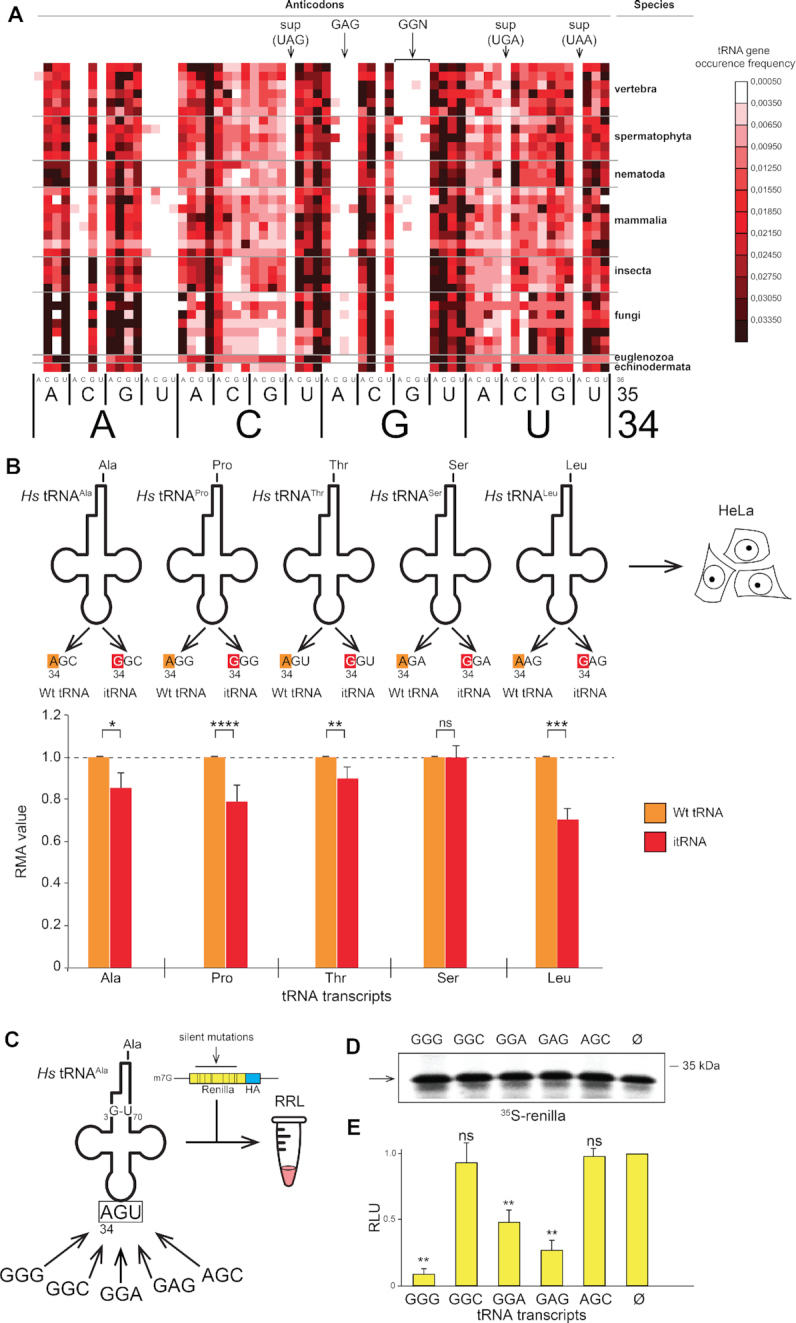
G at position 34 of the anticodon is prone to miscoding in eukaryotes. (A) The heat map represents, in various eukaryotic genomes, the ratio between the number of genes encoding any of the 64 anticodon combinations and the total number of tRNA genes. The colour code of the heat map is shown on the right, from black for highly represented genes to white for tRNA genes that are absent in genomes. Arrows indicate the suppressor tRNA genes containing anticodons corresponding to the stop codons and tRNA genes containing the anticodons 34GGN36 and 34GAG36. (B) Human tRNA transcripts tRNAAla,Pro,Thr,Ser,Leu containing 34ANN36 anticodons and their impossible tRNA (itRNA) counterparts, which contains respectively a single substitution of A at position 34 of the anticodons to G, are introduced into HeLa cells. The histogram represents the Relative Metabolic Activity (RMA) average value measured by WST1 assay for each itRNA transcript (in red) normalized to the corresponding Wt transcript (in orange) (n = 9, Statistical Student Test, ns: P-value > 0.05, *: P ≤ 0.05, **: P ≤ 0.01, ***: P ≤ 0.001, ****: P ≤ 0.0001). The error bars represent the standard deviation for each value. (C) Human tRNAAla transcripts containing the major determinant 3G-U70 for Alanyl-tRNA synthetase with variable anticodon sequences are introduced in Rabbit Reticulocyte Lysates together with a synthetic HA-tagged Renilla reporter mRNA that contains silent mutations at Phenylalanine, Proline, Threonine, Valine, Leucine and Serine codons in order to enrich the proportions of following codons Phenylalanine (UUC), Proline (CCC), Threonine (ACU, ACC), Valine (GUC), Leucine (CUC), Serine (UCC, UCU) according to Supplementary Figure S9. (D) The yield of Renilla proteins synthesized in the presence of tRNAAla transcripts of without transcript (Ø) is evaluated by SDS-PAGE of 35S-methionine-labeled proteins. (E) Histogram representing the relative luciferase activities of the synthesized Renilla proteins normalized to the luciferase activity obtained in absence of tRNA transcript (Ø) (n = 3, ns: P-value>0.05, **: P ≤ 0.01).
tRNAs with G34 are toxic miscoders in eukaryotes
The microfluidic-assisted screenings hinted that a G34 in the anticodon of tRNA decoding 4-box codons is potentially detrimental for translation fidelity in eukaryotes due to an increased miscoding capability. Since these tRNAs are virtually absent from eukaryotic genomes, we named such tRNAs ‘impossible tRNAs’ or itRNAs. In order to investigate the origin of the counter-selection against itRNAs during eukaryotic evolution, we assembled those species artificially. For that purpose, we synthesized control/wild type human tRNAAla,Pro,Thr,Ser,Leu containing A34 and their corresponding itRNA counterparts in which only the nucleotide A34, was substituted by G34. We then transfected them into human HeLa cells and assessed cellular fitness through a standard WST-1 metabolic assay (Figure 4B). Strictly speaking WST-1 assays directly measure the metabolization of formazan by mitochondria. Mitochondrial fitness is a widely accepted proxy for the overall cellular fitness. Although the rate of protein synthesis very often correlates with cellular health, WST-1 assays should not be used to directly assess the dynamic of translation. We would like to emphasize that we did not employ WST-1 assay to draw conclusions on the effects of our tRNA constructs on translation. As anticipated from the microfluidic screenings, when G34 is present, the cell general metabolism is significantly affected indicating that itRNAAla,Pro,Thr,Leu are indeed toxic to a certain degree, itRNASer with a G34 being the only non-toxic itRNA. To demonstrate that the toxicity is actually due to miscoding induced by itRNAs, we used human tRNAAla as a scaffold to test the impact of G34-containing anticodons. We assembled different chimeras tRNAAla displaying ambiguous G34-anticodons (Figure 4C). The alanine system is best suited to anticodon manipulation because human alanyl-tRNA synthetase (AlaRS) discriminates tRNAAla solely by recognizing the unique feature G3-U70 in the acceptor stem of the tRNA (73,74). Therefore, it is possible to change the whole anticodon of tRNAAla without affecting its ability to be alanylated by AlaRS. We generated human tRNAAla transcripts containing the following G34-anticodons (G34GG, G34GC, G34GA, G34AG) and AGC the alanine anticodon as a negative control. Miscoding capacity was measured with a reporter C-terminally HA-tagged Renilla luciferase gene in which we introduced silent mutations to enrich the luciferase coding sequence in codons prone to be potentially misdecoded by itRNAs according to the combinations that were identified from the microfluidic screenings (Supplementary Figure S8). Reporter mRNAs were then translated in vitro using rabbit reticulocyte lysates and the different tRNAAla chimeras. None of the tRNAAla constructs affected the overall translation yield of Renilla reporter protein in a significant way (Figure 4D). However, the luciferase activity was significantly affected in the presence of hybrid tRNA transcripts containing GGG, GGA and GAG anticodons suggesting that these tRNAs promote alanine insertion at unexpected positions in the Renilla luciferase coding sequence thereby affecting the luciferase activity (Figure 4C). To demonstrate the miscoding capability of these chimeric tRNAAla transcripts, we purified the produced Renilla luciferase protein using a C-terminal HA-tag. Mass spectrometry analysis of in vitro synthesized and purified luciferase revealed the presence of peptides containing alanine residues at mutated codons. These residues were inserted by the chimeric tRNAs at non-alanine codons indicating that the transcripts are efficiently aminoacylated by AlaRS and functional in the ribosome (Supplementary Figure S10a). We also found another peptide containing an alanine residue that was inserted by tRNAAlaGGA at a CCC codon. Therefore, we show that this hybrid tRNAAlaGGA promotes miscoding of a CCC codons, demonstrating without ambiguity that G at position 34 allows the decoding of non-cognate CCC codons displaying a C/A mismatch at codon position 1 (C1/A36) (Supplementary Figure S10b). Remarkably, this unexpected 1CCC3/34GGA36 combination is one of those identified in the microfluidic-based screening (marked by an asterisk in Supplementary Figure S8). Altogether, these experiments confirm that in a eukaryotic system the sole presence of G34 in tRNAs decoding 4-codon boxes induces miscoding induced by the formation of mismatches at positions 1 and 2 of the codon.
Nucleotides 32 and 38 correlate with R34 in 4-codon boxes
Very specific covariations at nucleotides 32 and 38 (as well as 31 and 39) were shown to participate in the anticodon loop conformation (58,75,76) and in the modulation of the codon/anticodon interactions (42). Early experiments showed the role of nucleotide 32 for discriminating the Gly codons (77,78). Later, experiments on E.coli tRNAAla (G34CC) revealed that the nucleotides A32 and U38 in this tRNA were critical for accurate decoding (79,80). Indeed, when 32–38 could not form a potential Watson-Crick pair (e.g. A32 and C38), the mutated tRNAAla promoted miscoding and lethal toxicity in bacteria. This prompted us to examine the distribution of nucleotides 32 and 38 in Homo sapiens. In contrast to prokaryotes, nucleotides 32 and 38 are not complementary in any 4-codon box tRNA (Figure 5). By analogy to the prokaryotic tRNAs, we then speculated that the toxicity induced by G34 in itRNAs was actually anticorrelated with the potential for nucleotides 32 and 38 to form a standard Watson-Crick pair in these tRNAs. In order to demonstrate this, we introduced in the toxic tRNAAla,Pro,Thr,Leu-G34, a point mutation that enables Watson-Crick base pairing between nucleotides 32 and 38 (Figure 5). As predicted, tRNAAla,Pro,Leu, for which the itRNA counterparts are the most toxic, are not toxic anymore when 32–38 can form a Watson-Crick base pair. Unexpectedly, this is not the case for tRNAThr-G34, which is more toxic with a 32–38 bp. In contrast to the three others, the wt tRNAThr contains a m3C32 that could be critical for other enzymatic reactions such as aminoacylation or tRNA modification. m3C32 is positively charged and the effects of the introduction of such a modification are unknown. Overall, these results confirm that G34 is toxic in such tRNAs only when nucleotides 32–38 cannot form a standard Watson–Crick pair.
Figure 5.
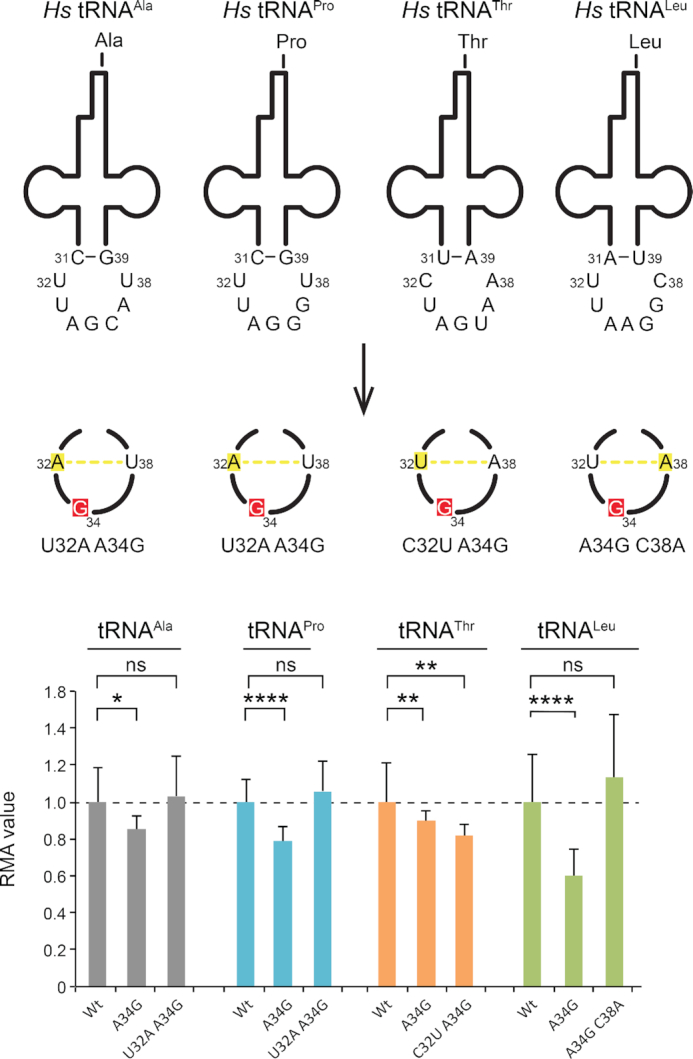
Anticodons with a G34 are prone to miscoding but not when 32–38 can form a Watson-Crick base pair. Human wild type tRNA transcripts tRNAAla,Pro,Thr,Leu containing 34ANN36 anticodons and their impossible tRNA (itRNA) counterparts that contain a single substitution of A at position 34 of the anticodons to G (in red) and double mutants containing an additional single substitution that enables base pairing between 32 and 38 (in green). The itRNAAla,Pro,Thr,Leu G34 were used as toxicity controls. The three types of tRNA transcripts were introduced into HeLa cells. The histogram represents the Relative Metabolic Activity (RMA) average value measured by WST1 assay for each tRNA transcript normalized to the corresponding Wt transcript (n = 6, Statistical Student's t-test, ns: P-value > 0.05, *: P ≤ 0.05, **: P ≤ 0.01, ***: P ≤ 0.001, ****: P ≤ 0.0001). The error bars represent the standard deviation for each value.
Nucleotides 32 and 38 are respectively at the beginning and end of the anticodon loop just following the anticodon stem. The presence of complementary bases at those two positions can induce the formation of an additional base pair in the anticodon stem. This introduces an additional rotation to the stem and re-orients the anticodon triplet in an unfavorable position for base pairing with the codon triplet. In other words, the conformation of the anticodon is no longer properly pre-organized for productive base pairing with the codon (76,78–80). Such a terminal 32–38 bp is naturally dynamic and only some fraction of the tRNA population will at any moment contain the base pair. By mass action, this automatically leads to a reduction in the free energy of tRNA–mRNA binding (42). This mechanism applies particularly to tRNAs forming G/C-rich anticodon/codon triplets where U32/A38 occurs instead of the more common C32/A38. In the early experiments mentioned above (77,78), replacing U32 by C32 led to an undiscriminating tRNA. The replacement of G34 by A34 (modified in I34) leads to base pairings with less energy content. Thus, strong G34-containing tRNAs would favor nucleotide combinations at 32 and 38 that are able to form a Watson–Crick pair, while weak A34(I34)-containing tRNAs will not need this additional tRNA constraints.
In conclusion, the combined use of a microfluidic-based analysis pipeline and of cell-free translation extracts faithfully recapitulates the main structural trends of molecular recognition in eukaryotic translation. The data show that, in the absence of competitor tRNAs, release factors and protein sequence effects, productive translation relies entirely on the stability of the codon/anticodon triplet. In addition, in the absence of tRNA modifications in the anticodon loop, tRNA binding to the ribosomal decoding centre is either unproductive or leads to extensive miscoding, especially with G34-containing tRNAs. Finally, the nature of the base at R34-containing tRNAs correlates with nucleotide conservations at positions 32 and 38 for smooth decoding: G34-tRNAs favor combinations of 32 and 38 that have the potential to form a Watson-Crick pair while A34-tRNAs do not. Bacteria evolved to maintain G34 and the constraint on 32 and 38, while eukaryotes selected A34(I34) instead.
DATA AVAILABILITY
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Supplementary Material
ACKNOWLEDGEMENTS
We are grateful to Friedrich Preusser, Fatima Alghoul and Lauriane Gross for technical assistance and we also warmly thank Sandrine Koechler and Abdelmalek Alioua from the IBMP Gene Expression Analysis facility for technical assistance with high-throughput sequencing. The facility is supported and funded as part of the LabEx NetRNA and MitoCross. The mass spectrometry instrumentation was granted from Université de Strasbourg (IdEx 2015 Equipement mi-lourd).
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Agence Nationale pour la Recherche [Ribofluidix, ANR-17-CE12-0025-01/02 and RFS, ANR-15-CE11-0021-01]; University of Strasbourg [PEPS 2015-IDEX]; Centre National de la Recherche Scientifique; LabEx [ANR-10-LABX-0036_NETRNA]; French National Research Agency. Funding for open access charge: CNRS.
Conflict of interest statement. Eric Westhof is Executive Editor of NAR.
REFERENCES
- 1. Drummond D.A., Wilke C.O.. Mistranslation-Induced protein misfolding as a dominant constraint on Coding-Sequence evolution. Cell. 2008; 134:341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ruan B., Palioura S., Sabina J., Marvin-Guy L., Kochhar S., LaRossa R.A., Soll D.. Quality control despite mistranslation caused by an ambiguous genetic code. Proc. Natl. Acad. Sci. U.S.A. 2008; 105:16502–16507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kohanski M.A., Dwyer D.J., Wierzbowski J., Cottarel G., Collins J.J.. Mistranslation of membrane proteins and two-component system activation trigger antibiotic-mediated cell death. Cell. 2008; 135:679–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mohler K., Mann R., Ibba M.. Isoacceptor specific characterization of tRNA aminoacylation and misacylation in vivo. Methods. 2017; 113:127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Francklyn C.S. DNA polymerases and aminoacyl-tRNA synthetases: shared mechanisms for ensuring the fidelity of gene expression. Biochemistry. 2008; 47:11695–11703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Soll D. The accuracy of aminoacylation–ensuring the fidelity of the genetic code. Experientia. 1990; 46:1089–1096. [DOI] [PubMed] [Google Scholar]
- 7. Ribas de Pouplana L., Santos M.A.S., Zhu J.H., Farabaugh P.J., Javid B.. Protein mistranslation: friend or foe. Trends Biochem. Sci. 2014; 39:355–362. [DOI] [PubMed] [Google Scholar]
- 8. Jørgensen F., Kurland C.G.. Processivity errors of gene expression in Escherichia coli. J. Mol. Biol. 1990; 215:511–521. [DOI] [PubMed] [Google Scholar]
- 9. Wohlgemuth I., Pohl C., Mittelstaet J., Konevega A.L., Rodnina M. V. Evolutionary optimization of speed and accuracy of decoding on the ribosome. Philos. Trans. R. Soc. B Biol. Sci. 2011; 366:2979–2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Manickam N., Nag N., Abbasi A., Patel K., Farabaugh P.J.. Studies of translational misreading in vivo show that the ribosome very efficiently discriminates against most potential errors. RNA. 2014; 20:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Parker J. Errors and alternatives in reading the universal genetic code. Microbiol. Rev. 1989; 53:273–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kramer E.B., Farabaugh P.J.. The frequency of translational misreading errors in E. coli is largely determined by tRNA competition. RNA-A Publ. RNA Soc. 2007; 13:87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kramer E.B., Vallabhaneni H., Mayer L.M., Farabaugh P.J.. A comprehensive analysis of translational missense errors in the yeast Saccharomyces cerevisiae. RNA. 2010; 16:1797–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Demeshkina N., Jenner L., Westhof E., Yusupov M., Yusupova G.. A new understanding of the decoding principle on the ribosome. Nature. 2012; 484:256–259. [DOI] [PubMed] [Google Scholar]
- 15. Rozov A., Demeshkina N., Westhof E., Yusupov M., Yusupova G.. Structural insights into the translational infidelity mechanism. Nat. Commun. 2015; 6:7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rozov A., Demeshkina N., Khusainov I., Westhof E., Yusupov M., Yusupova G.. Novel base-pairing interactions at the tRNA wobble position crucial for accurate reading of the genetic code. Nat. Commun. 2016; 7:10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rozov A., Wolff P., Grosjean H., Yusupov M., Yusupova G., Westhof E.. Tautomeric G•U pairs within the molecular ribosomal grip and fidelity of decoding in bacteria. Nucleic Acids Res. 2018; 46:7425–7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rodnina M.V., Wintermeyer W.. The ribosome as a molecular machine: the mechanism of tRNA-mRNA movement in translocation. Biochem. Soc. Trans. 2011; 39:658–662. [DOI] [PubMed] [Google Scholar]
- 19. Voorhees R.M., Ramakrishnan V.. Structural basis of the translational elongation cycle. TL - 82. Annu. Rev. Biochem. 2013; 82:203–236. [DOI] [PubMed] [Google Scholar]
- 20. Demeshkina N., Jenner L., Yusupova G., Yusupov M.. Interactions of the ribosome with mRNA and tRNA. Curr. Opin. Struct. Biol. 2010; 20:325–332. [DOI] [PubMed] [Google Scholar]
- 21. Wilson J.E., Powell M.J., Hoover S.E., Sarnow P.. Naturally occurring dicistronic cricket paralysis virus RNA is regulated by two internal ribosome entry sites. Mol. Cell Biol. 2000; 20:4990–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sasaki J., Nakashima N.. Translation initiation at the CUU codon is mediated by the internal ribosome entry site of an insect picorna-like virus in vitro. J. Virol. 1999; 73:1219–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pestova T.V., Lomakin I.B., Hellen C.U.T.. Position of the CrPV IRES on the 40S subunit and factor dependence of IRES/80S ribosome assembly. EMBO Rep. 2004; 5:906–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schuler M., Connell S.R., Lescoute A., Giesebrecht J., Dabrowski M., Schroeer B., Mielke T., Penczek P.A., Westhof E., Spahn C.M.. Structure of the ribosome-bound cricket paralysis virus IRES RNA. Nat. Struct. Mol. Biol. 2006; 13:1092–1096. [DOI] [PubMed] [Google Scholar]
- 25. Pestova T.V., Hellen C.U.T.. Translation elongation after assembly of ribosomes on the Cricket paralysis virus internal ribosomal entry site without initiation factors or initiator tRNA. Genes Dev. 2003; 17:181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Costantino D., Kieft J.S.. A preformed compact ribosome-binding domain in the cricket paralysis-like virus IRES RNAs. RNA. 2005; 11:332–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Filbin M.E., Kieft J.S.. Toward a structural understanding of IRES RNA function. Curr. Opin. Struct. Biol. 2009; 19:267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Au H.H., Cornilescu G., Mouzakis K.D., Ren Q., Burke J.E., Lee S., Butcher S.E., Jan E.. Global shape mimicry of tRNA within a viral internal ribosome entry site mediates translational reading frame selection. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:E6446–E6455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Spahn C.M.T., Jan E., Mulder A., Grassucci R.A., Sarnow P., Frank J.. Cryo-EM visualization of a viral internal ribosome entry site bound to human ribosomes: The IRES functions as an RNA-based translation factor. Cell. 2004; 118:465–475. [DOI] [PubMed] [Google Scholar]
- 30. Fernàndez I.S., Bai X.C., Murshudov G., Scheres S.H.W., Ramakrishnan V.. Initiation of translation by cricket paralysis virus IRES requires its translocation in the ribosome. Cell. 2014; 157:823–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Costantino D.A., Pfingsten J.S., Rambo R.P., Kieft J.S.. tRNA-mRNA mimicry drives translation initiation from a viral IRES. Nat. Struct. Mol. Biol. 2008; 15:57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Koh C.S., Brilot A.F., Grigorieff N., Korostelev A.A.. Taura syndrome virus IRES initiates translation by binding its tRNA-mRNA-like structural element in the ribosomal decoding center. Proc. Natl. Acad. Sci. USA. 2014; 111:9139–9144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Murray J., Savva C.G., Shin B.-S., Dever T.E., Ramakrishnan V., Fernández I.S.. Structural characterization of ribosome recruitment and translocation by type IV IRES. Elife. 2016; 5:e13567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hussain T., Llácer J.L., Fernández I.S., Munoz A., Martin-Marcos P., Savva C.G., Lorsch J.R., Hinnebusch A.G., Ramakrishnan V.. Structural changes enable start codon recognition by the eukaryotic translation initiation complex. Cell. 2014; 159:597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Erzberger J.P., Stengel F., Pellarin R., Zhang S., Schaefer T., Aylett C.H.S., Cimermančič P., Boehringer D., Sali A., Aebersold R. et al.. Molecular architecture of the 40S⋅eIF1⋅eIF3 translation initiation complex. Cell. 2014; 158:1123–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kanamori Y., Nakashima N.. A tertiary structure model of the internal ribosome entry site (IRES) for methionine-independent initiation of translation. RNA. 2001; 7:266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jan E., Sarnow P.. Factorless ribosome assembly on the internal ribosome entry site of cricket paralysis virus. J. Mol. Biol. 2002; 324:889–902. [DOI] [PubMed] [Google Scholar]
- 38. Yamamoto H., Nakashima N., Ikeda Y., Uchiumi T.. Binding mode of the first aminoacyl-tRNA in translation initiation mediated by Plautia stali intestine virus internal ribosome entry site. J. Biol. Chem. 2007; 282:7770–7776. [DOI] [PubMed] [Google Scholar]
- 39. Zhu J., Korostelev A., Costantino D.A., Donohue J.P., Noller H.F., Kieft J.S.. Crystal structures of complexes containing domains from two viral internal ribosome entry site (IRES) RNAs bound to the 70S ribosome. Proc. Natl. Acad. Sci. U.S.A. 2011; 108:1839–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Petrov A., Grosely R., Chen J., O’Leary S.E., Puglisi J.D.. Multiple parallel pathways of translation initiation on the CrPV IRES. Mol. Cell. 2016; 62:92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Muhs M., Hilal T., Mielke T., Skabkin M.A., Sanbonmatsu K.Y., Pestova T.V., Spahn C.M.T.. Cryo-EM of ribosomal 80s complexes with termination factors reveals the translocated cricket paralysis virus IRES. Mol. Cell. 2015; 57:422–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pisareva V.P., Pisarev A.V., Fernández I.S.. Dual tRNA mimicry in the cricket paralysis virus IRES uncovers an unexpected similarity with the hepatitis C virus IRES. Elife. 2018; 7:e34062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Woronoff G., Ryckelynck M., Wessel J., Schicke O., Griffiths A.D., Soumillion P.. Activity-Fed Translation (AFT) assay: a new high-throughput screening strategy for enzymes in droplets. Chembiochem. 2015; 16:1343–1349. [DOI] [PubMed] [Google Scholar]
- 44. Ryckelynck M., Baudrey S., Rick C., Marin A., Coldren F., Westhof E., Griffiths A.D.. Using droplet-based microfluidics to improve the catalytic properties of RNA under multiple-turnover conditions. RNA. 2015; 21:458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Autour A., Westhof E., Ryckelynck M.. ISpinach: a fluorogenic RNA aptamer optimized for in vitro applications. Nucleic Acids Res. 2016; 44:2491–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Autour A., Jeng S.C.Y., Cawte A.D., Abdolahzadeh A., Galli A., Panchapakesan S.S.S., Rueda D., Ryckelynck M., Unrau P.J.. Fluorogenic RNA Mango aptamers for imaging small non-coding RNAs in mammalian cells. Nat. Commun. 2018; 9:656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bouhedda F., Fam K.T., Collot M., Autour A., Marzi S., Klymchenko A., Ryckelynck M.. A dimerization-based fluorogenic dye-aptamer module for RNA imaging in live cells. Nat. Chem. Biol. 2019; 16:69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Pelham H.R.B., Jackson R.J.. An efficient mRNA-Dependent translation system from reticulocyte lysates. Eur. J. Biochem. 1976; 67:247–256. [DOI] [PubMed] [Google Scholar]
- 49. Baret J.-C., Miller O.J., Taly V., Ryckelynck M., El-Harrak A., Frenz L., Rick C., Samuels M.L., Hutchison J.B., Agresti J.J. et al.. Fluorescence-activated droplet sorting (FADS): efficient microfluidic cell sorting based on enzymatic activity. Lab Chip. 2009; 9:1850. [DOI] [PubMed] [Google Scholar]
- 50. Eriani G., Karam J., Jacinto J., Richard E.M., Geslain R.. MIST, a novel approach to reveal hidden substrate specificity in aminoacyl-tRNA synthetases. PLoS One. 2015; 10:e0130042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Martin F., Barends S., Jaeger S., Schaeffer L., Prongidi-Fix L., Eriani G.. Cap-assisted internal initiation of translation of histone H4. Mol. Cell. 2011; 41:197–209. [DOI] [PubMed] [Google Scholar]
- 52. Geslain R., Pan T.. Functional ZtRNA isodecoders. J. Mol. Biol. 2010; 396:821–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Geslain R., Cubells L., Bori-Sanz T., Álvarez-Medina R., Rossell D., Martí E., de Pouplana L.R.. Chimeric tRNAs as tools to induce proteome damage and identify components of stress responses. Nucleic Acids Res. 2010; 38:e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fallah-Araghi A., Baret J.-C., Ryckelynck M., Griffiths A.D.. A completely in vitro ultrahigh-throughput droplet-based microfluidic screening system for protein engineering and directed evolution. Lab Chip. 2012; 12:882. [DOI] [PubMed] [Google Scholar]
- 55. Mazutis L., Araghi A.F., Miller O.J., Baret J.C., Frenz L., Janoshazi A., Taly V., Miller B.J., Hutchison J.B., Link D. et al.. Droplet-based microfluidic systems for high-throughput single DNA molecule isothermal amplification and analysis. Anal. Chem. 2009; 81:4813–4821. [DOI] [PubMed] [Google Scholar]
- 56. Grosjean H., Westhof E.. An integrated, structure- and energy-based view of the genetic code. Nucleic Acids Res. 2016; 44:8020–8040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Chen J.L., Dishler A.L., Kennedy S.D., Yildirim I., Liu B., Turner D.H., Serra M.J.. Testing the nearest neighbor model for canonical RNA base pairs: Revision of GU parameters. Biochemistry. 2012; 51:3508–3522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Olejniczak M., Dale T., Fahlman R.P., Uhlenbeck O.C.. Idiosyncratic tuning of tRNAs to achieve uniform ribosome binding. Nat. Struct. Mol. Biol. 2005; 12:788–793. [DOI] [PubMed] [Google Scholar]
- 59. Fahlman R.P., Dale T., Uhlenbeck O.C.. Uniform binding of aminoacylated transfer RNAs to the ribosomal A and P sites. Mol. Cell. 2004; 16:799–805. [DOI] [PubMed] [Google Scholar]
- 60. Zhang Z., Shah B., Bondarenko P. V. G/U and certain wobble position mismatches as possible main causes of amino acid misincorporations. Biochemistry. 2013; 52:8165–8176. [DOI] [PubMed] [Google Scholar]
- 61. Rozov A., Westhof E., Yusupov M., Yusupova G.. The ribosome prohibits the G•U wobble geometry at the first position of the codon-anticodon helix. Nucleic Acids Res. 2016; 44:6434–6441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Murphy F.V., Ramakrishnan V.. Structure of a purine-purine wobble base pair in the decoding center of the ribosome. Nat. Struct. Mol. Biol. 2004; 11:1251–1252. [DOI] [PubMed] [Google Scholar]
- 63. Machnicka M.A., Milanowska K., Oglou O.O., Purta E., Kurkowska M., Olchowik A., Januszewski W., Kalinowski S., Dunin-Horkawicz S., Rother K.M. et al.. MODOMICS: a database of RNA modification pathways - 2013 update. Nucleic Acids Res. 2013; 41:D262–D267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Björk G.R., Hagervall T.G.. Transfer RNA modification: presence, synthesis, and function. EcoSal Plus. 2014; 6:doi:10.1128/ecosalplus.ESP-0007-2013. [DOI] [PubMed] [Google Scholar]
- 65. Grosjean H., de Crécy-Lagard V., Marck C.. Deciphering synonymous codons in the three domains of life: Co-evolution with specific tRNA modification enzymes. FEBS Lett. 2010; 584:252–264. [DOI] [PubMed] [Google Scholar]
- 66. Crick F.H.C. Codon—anticodon pairing: The wobble hypothesis. J. Mol. Biol. 1966; 19:548–555. [DOI] [PubMed] [Google Scholar]
- 67. Weixlbaumer A., Murphy F.V., Dziergowska A., Malkiewicz A., Vendeix F.A.P., Agris P.F., Ramakrishnan V.. Mechanism for expanding the decoding capacity of transfer RNAs by modification of uridines. Nat. Struct. Mol. Biol. 2007; 14:498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Rozov A., Demeshkina N., Westhof E., Yusupov M., Yusupova G.. New structural insights into translational miscoding. Trends Biochem. Sci. 2016; 41:798–814. [DOI] [PubMed] [Google Scholar]
- 69. Auxilien S., Crain P.F., Trewyn R.W., Grosjean H.. Mechanism, specificity and general properties of the yeast enzyme catalysing the formation of inosine 34 in the anticodon of transfer RNA. J. Mol. Biol. 1996; 262:437–458. [DOI] [PubMed] [Google Scholar]
- 70. Chan P.P., Lowe T.M.. GtRNAdb 2.0: An expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016; 44:D184–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Saint-Léger A., Bello C., Dans P.D., Torres A.G., Novoa E.M., Camacho N., Orozco M., Kondrashov F.A., De Pouplana L.R.. Saturation of recognition elements blocks evolution of new tRNA identities. Sci. Adv. 2016; 2:e1501860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Janvier A., Despons L., Schaeffer L., Tidu A., Martin F., Eriani G.. A tRNA-mimic strategy to explore the role of G34 of tRNAGly in translation and codon frameshifting. Int. J. Mol. Sci. 2019; 20:3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. McClain W.H., Chen Y.M., Foss K., Schneider J.. Association of transfer RNA acceptor identity with a helical irregularity. Science. 1988; 242:1681–1684. [DOI] [PubMed] [Google Scholar]
- 74. Hou Y.M., Schimmel P.. A simple structural feature is a major determinant of the identity of a transfer RNA. Nature. 1988; 333:140–145. [DOI] [PubMed] [Google Scholar]
- 75. Auffinger P., Westhof E.. An extended structural signature for the tRNA anticodon loop. RNA. 2001; 7:334–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Olejniczak M., Uhlenbeck O.C.. tRNA residues that have coevolved with their anticodon to ensure uniform and accurate codon recognition. Biochimie. 2006; 88:943–950. [DOI] [PubMed] [Google Scholar]
- 77. Lustig F., Borén T., Claesson C., Simonsson C., Barciszewska M., Lagerkvist U.. The nucleotide in position 32 of the tRNA anticodon loop determines ability of anticodon UCC to discriminate among glycine codons. Proc. Natl. Acad. Sci. U.S.A. 1993; 90:3343–3347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Claesson C., Lustig F., Borén T., Simonsson C., Barciszewska M., Lagerkvist U.. Glycine codon discrimination and the nucleotide in position 32 of the anticodon loop. J. Mol. Biol. 1995; 247:191–196. [DOI] [PubMed] [Google Scholar]
- 79. Murakami H., Ohta A., Suga H.. Bases in the anticodon loop of tRNAGGCAla prevent misreading. Nat. Struct. Mol. Biol. 2009; 16:353–358. [DOI] [PubMed] [Google Scholar]
- 80. Ledoux S., Olejniczak M., Uhlenbeck O.C.. A sequence element that tunes Escherichia coli tRNAGGCAla to ensure accurate decoding. Nat. Struct. Mol. Biol. 2009; 16:359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.