

Abstract
A precisely designed chiral squaramide derivative is shown to promote the highly enantioselective addition of trimethylsilyl bromide (TMSBr) to a broad variety of 3-substituted and 3,3-disubstituted oxetanes. The reaction provides direct and general access to synthetically valuable 1,3-bromohydrin building blocks from easily accessed achiral precursors. The products are readily elaborated both by nucleophilic substitution and through transition-metal-catalyzed cross-coupling reactions. The enantioselective catalytic oxetane ring opening was employed as part of a 3-step, gram-scale synthesis of pretomanid, a recently-approved medication for the treatment of multi-drug-resistant tuberculosis. Heavy-atom kinetic isotope effect (KIE) studies are consistent with enantiodetermining delivery of bromide from the H-bond-donor (HBD) catalyst to the activated oxetane. While the nucleophilicity of the bromide ion is expected to be attenuated by association to the HBD, overall rate acceleration is achieved by enhancement of Lewis acidity of the TMSBr reagent through anion-abstraction.
Graphical abstract
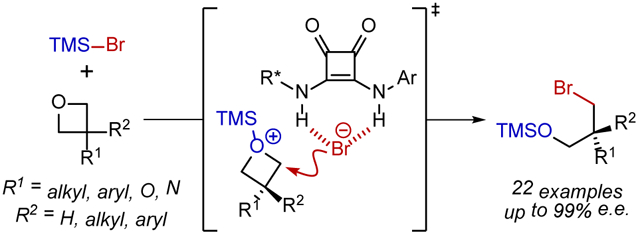
Chiral anion-binding catalysis has emerged as a powerful strategy for enantioselective additions to cationic intermediates through their non-covalent association to catalyst-bound spectator anions.1,2 In most applications identified to date, the chiral catalyst-anion complex mediates stereoinduction in the addition of an external nucleophile (Figure 1A). An interesting variation to the anion-binding catalysis concept arises when the catalyst-bound anion also acts as the nucleophile in the enantiodetermining bond construction.3,4 At least in principle, such an approach can provide more precise control over stereoselectivity through specific association of both nucleophile and electrophile to the chiral catalyst. However, H-bonding from the catalyst would also be expected to attenuate the reactivity of the nucleophile relative to an uncatalyzed, racemic pathway.5 This fundamental reactivity challenge can be circumvented if the catalyst also promotes the generation of the reactive ion-pair, as demonstrated elegantly by Gouverneur3f,g in the specific context of phase-transfer reactions of alkali metal fluorides (Figure 1B). Following the recent discovery that H-bond donors such as chiral squaramides can activate silyl triflates via anion binding to promote enantioselective transformations,6 we were drawn to an alternative and possibly general approach to catalysis of nucleophile delivery by applying the anion-binding principle to activation of Lewis acids bearing nucleophilic counterions. Anion abstraction from the promoter should result in enhanced Lewis acidity, providing a general platform to access highly-reactive, cationic, electrophilic intermediates ion paired with a catalyst-bound nucleophilic anion (Figure 1C).
Figure 1.

(A) Conventional approach to anion-binding catalysis in which the catalyst binds a spectator anion. (B) Alternative reactive mode in anion-binding catalysis involving delivery of the bound anionic nucleophile to a cationic electrophile. Hydrogen bonding attenuates the reactivity of the nucleophile, but rate acceleration has been achieved via phase-transfer catalysis. (C) Catalyst-promoted ionization of an anionic nucleophile from a neutral Lewis acid-nucleophile complex allows for H-bond donor catalyzed anion delivery.
We chose to explore the anion-binding effect on nucleophile-bearing Lewis acids in the context of additions of TMSBr to prochiral oxetanes (Fig. 2A). Enantioselective ring-opening of 3-substituted oxetanes provides a route to valuable 3-carbon chiral building blocks from simple, synthetically-accessible precursors.7 Several examples of enantioselective openings of 3-susbtituted oxetanes with intramolecular nucleophiles have been identified.8,9 However, more generally applicable highly enantioselective reactions involving intermolecular nucleophilic addition are limited to two pioneering examples from Sun and coworkers involving chiral phosphoric acid-catalyzed addition of mercaptobenzothiazole and HCl.10 Here we report the successful application of a chiral squaramide catalyst to promote the ring-opening addition of TMSBr to prochiral oxetane substrates with unprecedented substrate scope.
Figure 2.

A) Model reaction: enantioselective opening of oxetanes with TMSBr. B) Catalyst screening data for a series of arylpyrrolidino squaramides.
The addition of TMSBr to 3-phenyloxetane (1a) was selected as a model reaction.11 Squaramide hydrogen-bond-donor catalysts6,12 bearing a 2-arylpyrrolidino amide were identified as particularly effective, with the the aryl substituent having a marked effect on enantioselectivity and reproducibility (Fig. 2B). Systematic reaction and catalyst development (Fig. S1-S5) led to the identification of 9-phenanthryl squaramide 3a as the optimal catalyst for the synthesis of silylated bromohydrin 2a, catalyzing its formation in quantitative yields and with 96-98% e.e. over >20 runs.13
Squaramide 3a was found to catalyze the opening of a broad range of 3-subsituted and 3,3-disubstituted oxetanes in high levels of e.e. (Figure 3). With 3-aryl oxetanes, both electron donating and withdrawing substituents could be introduced, with only ortho substitution impacting enantioselectivity adversely (1a–h). Weakly Lewis basic functional groups such as nitriles (1f) and esters (1g) had no deleterious effect on the reaction, and aryl ether spectator groups remained intact (1h). Oxetanes bearing protected alcohol and amine functionality (1i–m) as well as simple saturated alkyl groups (1n-p) all underwent reaction with high enantioselectivity. The reaction could even be extended successfully to certain 3,3'-disubstituted oxetane substrates, which underwent stereoselective ring opening to provide products bearing fully substituted stereocenters (1q–v) with moderate-to-high enantioselectivity.
Figure 3.

Isolated yield and enantiomeric enrichments measured for the asymmetric oxetane opening at 0.4 mmol scale. See SI for details on methods for e.e. determination, reproducibility studies, and the assignment of absolute configuration. a Isolated as a 12.5 : 1 ratio of ROTMS to ROH product. b 48-hr reaction time. c −25 °C. d 72-hr reaction time. e 7.5 mol% 3a. f −65 °C.
Alkyl bromide 2a was examined as a model substrate for potential product derivatizations (Fig. 4A) and was found to undergo facile substitution with azide, cyanide, and thiophenolate nucleophiles. Recent advances in transition-metal-catalyzed cross-coupling chemistry14 provide further opportunities for product elaborations; for example, we found that 2a engaged effectively in cobalt-catalyzed arylations.15 Moreover, the polarity of the electrophilic alkyl bromide could be inverted either by copper-catalyzed borylation,16 or through metal-halogen exchange, allowing 2a to function as the nucleophilic partner in a C(sp2)–C(sp3) cross coupling.17 Overall, the diverse range of products that can be accessed directly from 2a illustrates the synthetic versatility of these chiral bromohydrin building blocks.
Figure 4.

A) Product elaborations: all reported yields are for the entire sequence of reactions starting from 1a a) standard reaction conditions with 0.4 mmol 1a; b) NaCN; c) NaN3; d) NaSPh; e) p-TolMgBr, Co(acac)3, TMEDA; f) B2Pin2, CuCl, Xantphos, t-BuOK; g) NaI in MeCN then solvent swap to Et2O, t-BuLi, ZnCl2, Pd(dppf)Cl2, R-I. See SI for detailed procedures. B) Glycidol- and oxetane-based strategies to C3 chiral derivatives. C) Gram-scale synthesis of pretomanid: Ar = 4-(trifluoromethoxy)phenyl h) 4-(trifluormethoxy)benzyl bromide (1.2 equiv.), NaH (1.2 equiv.), 2-Me-THF (1.0 M), 60 °C, 12 hr; i) 3a (2 mol%), TMSBr (1.1 equiv.), t-BuOMe (0.25 M), −80 °C, 24 hr; j) 2-chloro-4-nitroimidazole (2.0 equiv.), Et3N (2.1 equiv.), NaI (1.0 equiv.) DMF (0.25 M), 115 °C, 24 hr, then cool to 23 °C and add MeOH (1.0 M) and NaOH (5.0 equiv.), 30 min.
The oxetane-opening methodology presents an interesting alternative to well-established synthetic strategies for accessing three-carbon chiral building blocks based on glycidol or epichlorohydrin derivatives (Figure 4B).18 In particular, the identity of the C2 group can be set in the prochiral oxetane substrate, thereby avoiding potentially multi-step late-stage functional-group manipulations required in routes involving epoxide ring-opening. This advantage is illustrated in the synthesis of the recently approved tuberculosis drug pretomanid19 (Figure 4C), which was prepared previously by Reider, Sorensen and coworkers in an elegant 5-step route from enantioenriched (R)-3-chloro-1,2-propanediol.20a Readily accessible oxetane 1w underwent highly enantioselective ring-opening to yield TMS-protected bromohydrin 2w in 98% e.e.21 Gratifyingly, 2-chloro-4-nitroimadzole, which was identified in the Reider and Sorensen synthesis as a non-explosive alternative to 2,4-dinitroimidazole,20a underwent alkylation by 2w with complete regioselectivity followed by SNAr annulation to yield the desired product. Both intermediates were formed in sufficient purity to be carried forward without purification, and only a recrystallization of the final product was required to access analytically-pure pretomanid (10) in >99% e.e. The synthetic route avoids protecting-group manipulation steps and provides access to pretomanid in just 3 steps.
As an initial step toward establishing the basis for exquisite enantiocontrol in oxetane ring-opening reactions with squaramide 3a, we endeavored to determine whether bromide delivery was indeed the enantiodetermining step as proposed at the outset of reaction development. To address this question, the 12C / 13C KIE at the site of bromide attack was determined through analysis of starting material recovered at partial conversion from a one-pot competition between doubly labeled oxetane 13C2-1a and unlabeled isotopologue 1a (Fig. 5).22 If bromide-promoted ring opening were substrate-committing, and thus, enantiodetermining, a primary KIE consistent with C─O bond cleavage would be expected. In contrast, if a step preceding ring opening such as oxetane silylation were irreversible then only a small, secondary KIE would be anticipated. Irreversible silylation would not necessarily preclude enantiodetermining bromide delivery, but it would allow for the possibility that silylation of 1a was enantiodetermining.23 Subjection of a mixture of 1a and 13C2-1a to the catalytic reaction conditions led to the observation of a large, primary KIE (k12/k13 = 1.126(9)), fully consistent with reversible silylation and enantioselectivity-determining bromide delivery (see SI for full details of the KIE studies).
Figure 5.

One-pot competition KIE between 1a and 13C2-1a. A primary KIE of 1.126(9) was measured, indicating that oxetane silylation must be reversible, supporting enantiodetermining bromide delivery.
In conclusion, the chiral squaramide-catalyzed addition of TMSBr to 3-aryl, 3-alkyl, and 3-heteroatom substituted oxetanes as well as certain 3,3-disubstituted oxetanes provides a general enantioselective synthesis of protected 1,3-bromohydrin derivatives. The products of these reactions can be elaborated through a variety of nucleophilic substitution reactions, and the utility of the method is illustrated in the 3-step, gram-scale synthesis of the TB drug pretomanid. Heavy-atom KIE studies are consistent with enantiodetermining bromide delivery by the catalyst to an activated oxetane. This strategy overcomes the intrinsic deactivation of nucleophiles that accompanies association with an H-bond donor and holds promise as a broadly applicable approach to asymmetric catalysis of addition reactions.
Supplementary Material
ACKNOWLEDGMENT
Financial support for this work was provided by the NIH through GM043214 and a postdoctoral fellowship to Z.K.W. We thank Dr. E. E. Kwan for discussions regarding the KIE studies, and Dr. S.-L. Zheng for X-ray data collection and structure determination.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: https://pubs.acs.org/doi/10.1021/jacs.0c03991.
Experimental and characterization data of catalyst and substrate syntheses, procedures and analytical data for enantioselective reactions, procedure and analytical data for product elaborations, details of KIE experiment (PDF)
Crystallographic data for 26a (derivative of 2a) (CIF)
Crystallographic data for 26o (derivative of 2o) (CIF)
Crystallographic data for 2t (CIF)
Crystallographic data for 26u (derivative of 2u) (CIF)
The authors declare no competing financial interest.
REFERENCES
- (1) (a).Doyle AG; Jacobsen EN Small-molecule H-bond donors in asymmetric catalysis. Chem. Rev 2007, 107, 5713–5743. [DOI] [PubMed] [Google Scholar]; (b) Brak K; Jacobsen EN Asymmetric ion-pairing catalysis. Angew. Chem., Int. Ed 2013, 52, 534–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Chiral Brønsted acids have also been employed effectively to generate analogous chiral ion-pair intermediates through association of protonated species to the conjugate base of the chiral acid catalyst. For a recent reviews see ref. 1b and: Parmar D; Sugiono E; Raja S; Rueping M Complete field guide to asymmetric BINOL-phosphate derived Bronsted acid and metal catalysis: history and classification by mode of activation; Bronsted acidity, hydrogen bonding, ion pairing, and metal phosphates. Chem. Rev 2014, 114, 9047–9153. [DOI] [PubMed] [Google Scholar]
- (3) (a).Zuend SJ; Coughlin MP; Lalonde MP; Jacobsen EN Scaleable catalytic asymmetric Strecker syntheses of unnatural α-amino acids. Nature 2009, 461, 968–970. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Zuend SJ; Jacobsen EN; Mechanism of amido-thiourea catalyzed enantioselective imine hydrocyanation: transition state stabilization via multiple non-covalent interactions. J. Am. Chem. Soc 2009, 131, 15358–15374. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Birrell JA; Desrosiers J-N; Jacobsen EN Enantioselective acylation of silyl ketene acetals through fluoride anion-binding catalysis. J. Am. Chem. Soc 2011, 133, 13872–13875. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) De CK; Mittal N; Seidel D A dual-catalysis approach to the asymmetric Steglich rearrangement and catalytic enantioselective addition of O-acylated azalactones to isoquinolines. J. Am. Chem. Soc 2011, 133, 16802–16805. [DOI] [PubMed] [Google Scholar]; (e) Jarvis CL; Hirschi JS; Vetticatt MJ; Seidel D Catalytic enantioselective synthesis of lactams through formal [4+2] cycloaddition of imines with homophthalic anhydride. Angew. Chem., Int. Ed 2017, 56, 2670–2674. [DOI] [PubMed] [Google Scholar]; (f) Pupo G; Ibba F; Ascough DMH; Vicini AC; Ricci P; Christensen KE; Pfeifer L; Morphy JR; Brown JM; Paton RS; Gouverneur V Asymmetric nucleophilic fluorination under hydrogen bonding phase-transfer catalysis. Science 2018, 360, 638–642. [DOI] [PubMed] [Google Scholar]; (g) Pupo G; Vicini AC; Ascough DMH; Ibba F; Christensen KE; Thompson AL; Brown JM; Paton RS; Gouverneur V Hydrogen bonding phase-transfer catalysis with potassium fluoride: enantioselective synthesis of β-fluoroamines. J. Am. Chem. Soc 2019, 141, 2878–2883. [DOI] [PubMed] [Google Scholar]
- (4).Nucleophilic attack by catalyst-bound anions on neutral electrophiles is frequently proposed in reactions catalyzed by bifunctional hydrogen-bond donors. See ref. 4a and 4b for representative mechanistic studies, and ref. 4c for an example of halide delivery in a hydrochlorinative aziridine opening. (a) Hamza A; Schubert G; Soos T; Papai I Theoretical studies on the bifunctionality of chiral thiourea-based organocatalysts: competing routes to C-C bond formation. J. Am. Chem. Soc 2006, 128, 13151–13160. [DOI] [PubMed] [Google Scholar]; (b) Izzo JA; Myshchuk Y; Hirschi JS; Vetticatt MJ Transition state analysis of an enantioselective Michael addition by a bifunctional thiourea organocatalyst. Org. & Biomol. Chem 2019, 17, 3934–3939. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mita T; Jacobsen EN Bifunctional asymmetric catalysis with hydrogen chloride: enantioselective ring opening of aziridines catalyzed by a phosphinothiourea. Synlett 2009, 10, 1680–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Anslyn EV; Dougherty DA Modern Physical Organic Chemistry; University Science Books: Sausalito, CA, 2006: pp 643–646. [Google Scholar]
- (6) (a).Banik SM; Levina A; Hyde AM; Jacobsen EN Lewis acid enhancement by hydrogen-bond donors for asymmetric catalysis. Science 2017, 10, 761–764. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wendlandt AE; Vangal P; Jacobsen EN Quaternary stereocenters via an enantioconvergent catalytic SN1 reaction. Nature 2018, 556, 447–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7) (a).Burkhard JA; Wuitschik G; Rogers-Evans M; Muller K; Carreira EM Oxetanes as versatile elements in drug discovery and synthesis. Angew. Chem., Int. Ed 2010, 49, 9052–9067. [DOI] [PubMed] [Google Scholar]; (b) Wuitschik G; Carreira EM; Wagner B; Fischer H; Parilla I; Schuler F; Rogers-Evans M; Müller K Oxetanes in drug discovery: structural and synthetic insights. J. Med. Chem 2010, 53, 3227–3246. [DOI] [PubMed] [Google Scholar]; (c) Ahmad S; Yousaf M; Mansha A; Rasool N; Zahoor AF; Hafeez F; Rizvi SMA Ring-opening reactions of oxetanes: a review of methodology development and synthetic applications. Synth. Comm 2016, 46, 1397–1416. [Google Scholar]; (d) Bull JA; Croft RA; Davis OA; Doran R; Morgan KF Oxetanes: recent advances in synthesis, reactivity, and medicinal chemistry. Chem. Rev 2016, 116, 12150–12233. [DOI] [PubMed] [Google Scholar]
- (8) (a).Loy RN; Jacobsen EN Enantioselective intramolecular openings of oxetanes catalyzed by (salen)Co(III) complexes: access to enantioenriched tetrahydrofurans. J. Am. Chem. Soc 2009, 131, 2786–2787. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen Z; Wang B; Wang Z; Zhu G; Sun J Complex bioactive alkaloid-type polycycles through efficient catalytic asymmetric multicomponent aza-Diels-Alder reactions of indoles with oxetane as directing group. Angew. Chem., Int. Ed 2013, 52, 2027–2031. [DOI] [PubMed] [Google Scholar]; (c) Chen Z; Wang Z; Sun J Catalytic enantioselective synthesis of tetrahydroisoquinolines and their analogues bearing a C4 stereocenter: formal synthesis of (+)-(8S,13R)-cyclocelabenzine. Chem. Eur. J 2013, 19, 8426–8430. [DOI] [PubMed] [Google Scholar]; (d) Wang Z; Chen Z; Sun J Catalytic asymmetric nucleophilic openings of 3-substituted oxetanes. Org. & Biomol. Chem 2014, 12, 6028–6032. [DOI] [PubMed] [Google Scholar]; (e) Yang W; Sun J Organocatalytic enantioselective synthesis of 1,4-dioxanes and other oxa-heterocycles by oxetane desymmetrization. Angew. Chem., Int. Ed 2016, 55, 1868–1871. [DOI] [PubMed] [Google Scholar]; (f) Zhang R; Guo W; Duan M; Houk KN; Sun J Asymmetric desymmetrization of oxetanes for the synthesis of chiral tetrahydrothiophenes and tetrahydroselenophenes. Angew. Chem., Int. Ed 2019, 58, 18055–18060. An enantioselective ring expansion of 3-substituted oxetanes has also been developed: [DOI] [PubMed] [Google Scholar]; (g) Yin Q; You S-L Asymmetric chlorination/ring expansion for the synthesis of α-quaternary cycloalkanones. Org. Lett 2014, 14, 1810–1813. [DOI] [PubMed] [Google Scholar]
- (9).Several enantioselective reactions have also been developed that employ 2-substituted oxetanes as substrates for ring expansion: (a) Nozaki H; Moriuti S; Takaya H; Noyori R Asymmetric induction in carbenoid reactions by means of a dissymmetric copper chelate. Tet. Lett 1966, 7, 5239–5244. [Google Scholar]; (b) Nozaki H; Takaya H; Moritui S; Noyori R Homogeneous catalysis in the decomposition of diazo compounds by copper chelates: asymmetric carbenoid reactions. Tetrahedron 1968, 24, 3655–3669. [Google Scholar]; (c) Ito K; Katsuki T Asymmetric carbene C-O insertion reaction using optically active bipyridine-copper complex as a catalyst. Ring expansion of oxetanes to tetrahydrofurans. Chem. Lett 1994, 23, 1857–1860. [Google Scholar]; (d) Ito K; Yoshitake M; Katsuki T Enantioselective synthesis of trans-whisky lactone by using newly developed asymmetric ring expansion reaction of oxetane as a key step. Chem. Lett 1995, 24, 1027–1028. [Google Scholar]; (e) Ito K; Yoshitake M; Katsuki T Enantiospecific ring expansion of oxetanes: stereoselective synthesis of tetrahydrofurans. Heterocycles 1996, 42, 305–317. [Google Scholar]; (f) Ito K; Fukuda T; Katsuki T A new methodology for efficient construction of 2,7-dioxabicyclo[3.3.0]octane derivatives. Synlett 1997, 4, 387–389. [Google Scholar]; (g) Ito K; Fukuda T; Katsuki T A new enantiospecific approach to the bislactone structure: formal syntheses of (+)-avenaciolide and (−)-isoavenaciolide. Heterocycles 1997, 46, 401–411. [Google Scholar]; (h) Lo MM-C; Fu GC Applications of planar-chiral heterocycles in enantioselective catalysis: Cu(I)/bisazaferrocene-catalyzed asymmetric expansion of oxetanes to tetrahydrofurans. Tetrahedron 2001, 57, 2621–2634. [Google Scholar]; (i) Guo B; Schwarzwalder G; Njardarson JT Catalytic ring expansion of vinyl oxetanes: asymmetric synthesis of dihydropyrans using chiral counterion catalysis. Angew. Chem., Int. Ed 2012, 51, 5675–5678. [DOI] [PubMed] [Google Scholar]
- (10) (a).Wang Z; Chen Z; Sun J Catalytic enantioselective intermolecular desymmetrization of 3-substituted oxetanes. Angew. Chem., Int. Ed 2013, 52, 6685–6688. [DOI] [PubMed] [Google Scholar]; (b) Yang W; Wang Z; Sun J Enantioselective oxetane ring opening with chloride: unusual use of wet molecular sieves for the controlled release of HCl. Angew. Chem., Int. Ed 2016, 55, 6954–6958. For pioneering examples of moderately enantioselective intermolecular oxetane openings with organolithium reagents, see: [DOI] [PubMed] [Google Scholar]; (c) Mizuno M; Kanai M; Iida A; Tomioka K Chiral ligand controlled enantioselective opening of oxirane and oxetane. Tet. Asymm 1996, 7, 2483–2484. [Google Scholar]; (d) Mizuno M; Kanai M; Iida A; Tomioka K An external chiral ligand controlled enantioselective opening of oxirane and oxetane by organolithiums. Tetrahedron 1997, 53, 10699–10708. [Google Scholar]
- (11).Kricheldorf HR; Morber G; Regel W Syntheses of alkyl bromides from ethers and bromotrimethylsilane. Synthesis 1981, 5, 383–384. [Google Scholar]
- (12).Malerich JP; Hagihara K; Rawal VH Chiral squaramide derivatives are excellent hydrogen bond donor catalysts. J. Am. Chem. Soc 2008, 130, 14416–14417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Variability in the e.e. was observed to be catalyst-dependent and traced to the effect of adventious water. The addition of up to 4 mol% of H2O or other protic additives had little effect on the enantioselectivity of ring opening of 1a catalyzed by 3a (Fig. S6 entries 1 and 2, Fig. S7 entry 1), but higher loadings of protic additives led to decreases in enantioselectivity ranging from moderate (Fig. S6 entries 3 and 4, Fig. S7 entry 2) to significant (Fig. S7 entry 3). The effect of catalytic amounts of HBr and the unqiue features that allow 3a to catalyze the transformation with consistently high levels of e.e. are the subject of ongoing study, and will be discussed in a separate report.
- (14) (a).Luh T-Y; Leung M-K; Wong K-T Transition metal-catalyzed activation of aliphatic C-X bonds in carbon-carbon bond formation. Chem. Rev 2000, 100, 3187–3204. [DOI] [PubMed] [Google Scholar]; (b) Netherton MR; Fu GC Nickel-catalyzed cross-couplings of unactivated alkyl halides and pseudohalides with organometallic compounds. Adv. Synth. Catal 2004, 346, 1525–1532. [Google Scholar]; (c) Frisch AC; Beller M Catalysts for cross-coupling reactions with non-activated alkyl halides. Angew. Chem., Int. Ed 2004, 44, 674–688. [DOI] [PubMed] [Google Scholar]; (d) Terao J; Kambe N Transition metal-catalyzed C-C bond formation reactions using alkyl halides. Bull. Chem. Soc. Jpn 2006, 79, 663–672. [Google Scholar]; (e) Cahiez G; Moyeux A Cobalt-catalyzed cross-coupling reactions. Chem. Rev 2010, 110, 1435–1462. [DOI] [PubMed] [Google Scholar]
- (15).Cahiez G; Chaboche C; Duplais C; Moyeux A A new efficient catalytic system for the chemoselective cobalt-catalyzed cross-coupling of aryl Grignard reagents with primary and secondary alkyl bromides. Org. Lett 2009, 11, 277–280. [DOI] [PubMed] [Google Scholar]
- (16).Ito H; Kubota K Copper(I)-catalyzed boryl substitution of unactivated alkyl halides. Org. Lett 2012, 14, 890–893. [DOI] [PubMed] [Google Scholar]
- (17).Jana R; Pathak TP; Sigman MS Advances in transition metal (Pd,Ni,Fe)-catalyzed cross-coupling reactions using alkyl-organometallics as reaction partners. Chem. Rev 2011, 111, 1417–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18) (a).Gao Y; Hanson RM; Klunder JM; Ko SY; Masamune H; Sharpless KB Catalytic Asymmetric Epoxidation and Kinetic Resolution: Modified Procedures Including in Situ Derivatization. J. Am. Chem. Soc 1987, 109, 5765–5780. [Google Scholar]; (b) Hanson RM The Synthetic Methodology of Nonracemic Glycidol and Related 2,3-Epoxy Alcohols. Chem. Rev 1991, 91, 437–475. [Google Scholar]; (c) Tokunaga M; Larrow JF; Kakiuchi F; Jacobsen EN Asymmetric Catalysis with Water: Efficient Kinetic Resolution of Terminal Epoxides by Means of Catalytic Hydrolysis. Science 1997, 277, 936–938. [DOI] [PubMed] [Google Scholar]; (d) Furrow ME; Schaus SE; Jacobsen EN Practical Access to Highly Enantioenriched C-3 Building Blocks via Hydrolytic Kinetic Resolution. J. Org. Chem 1998, 63, 6776–6777. [DOI] [PubMed] [Google Scholar]; (e) Kasai N; Suzuki T; Furukawa Y Chiral C3 epoxides and halohydrins: Their preparation and synthetic application. J. Mol. Cat. B: Enzymatic 1998, 4, 237–252. [Google Scholar]; (f) Schaus SE; Brandes BD; Larrow JF; Tokunaga M; Hansen KB; Gould AE; Furrow ME; Jacobsen EN Highly Selective Hydrolytic Kinetic Resolution of Terminal Epoxides Catalyzed by Chiral (salen)CoIII Complexes. Practical Synthesis of Enantioenriched Terminal Epoxides and 1,2-Diols. J. Am. Chem. Soc 2002, 124, 1307–1315. [DOI] [PubMed] [Google Scholar]; (g) Larrow JF; Hemberger KE; Jasmin S; Kabir H; Morel P Commercialization of the hydrolytic kinetic resolution of racemic epoxides: toward the economical large-scale production of enantiopure epichlorohydrin. Tet. Asymm 2003, 14, 3589–3592. [Google Scholar]; (h) Larrow JF; Quigley PF Industrial Applications of the Jacobsen Hydrolytic Kinetic Resolution Technology In Comprehensive Chirality; Carreira EM, Yamamoto H, Eds.; Elsevier: Amsterdam, 2012; Vol. 9, pp 129–146. [Google Scholar]; (i) Singh GS; Mollet K; D’hooghe M; De Kimpe N Epihalohydrins in Organic Synthesis. Chem. Rev 2013, 113, 1441–1498. [DOI] [PubMed] [Google Scholar]
- (19).TB Alliance: News: FDA Approves New Treatment for Highly Drug-Resistant Forms of Tuberculosis (August 14, 2019). https://www.tballiance.org/news/fda-approves-new-treatment-highly-drug-resistant-forms-tuberculosis (accessed April 11, 2020).
- (20) (a).Marsini MA; Reider PJ; Sorensen EJ A Concise and Convergent Synthesis of PA-824. J. Org. Chem 2010, 75, 7479–7482. For other syntheses of pretomanid see: [DOI] [PubMed] [Google Scholar]; (b) Baker WR; Shaopei C; Keeler EL Nitro-[2,1-b]imidazopyran Compounds and Antibacterial Uses Thereof. U.S. Patent 6087358, 2000.; (c) Orita A; Miwa K; Otera J Integration of Solventless Reaction in a Multi-Step Process: Application to an Efficient Synthesis of PA-824. Adv. Synth. Catal 2007, 349, 2136–2144. [Google Scholar]; (d) Thompson AM; Blaser A; Anderson RF; Shinde SS; Franzblau SG; Ma Z; Denny WA; Palmer BD Synthesis, Reduction Potentials, and Antitubercular Activity of Ring A/B Analogues of the Bioreductive Drug (6S)-2-Nitro-6-{[4-(trifluoromethoxy)benzyl]oxy}-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (PA-824). J. Med. Chem 2009, 52, 637–645. [DOI] [PubMed] [Google Scholar]; (e) Thompson AM; O’Connor PD; Marshall AJ; Blaser A; Yardley V; Maes L; Gupta S; Launay D; Braillard S; Chatelain E; Wan B; Franzblau SG; Ma Z; Cooper CB; Denny WA Development of (6R)-2-Nitro-6-[4-(trifluoromethoxy)phenoxy]-6,7-dihydro-5H-imidazo[2,1-b][1,3]oxazine (DNDI-8219): A New Lead for Visceral Leishmaniasis. J. Med. Chem 2018, 61, 2329–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).The catalytic reaction was found to slow down on larger scale, but this could be compensated for by increasing the concentration to 0.25 M. The basis for the dependence of rate on reaction scale is related to the effect of adventitious water noted in ref. 13 and will be discussed fully in a separate report.
- (22).The one-pot intermolecular competition experiment is the only option that allows accurate and diagnostic determination of the KIE in this system. For a lucid discussion of the applications of intramolecular, one-pot, and two-pot competition KIE experiments see: Simmons EM; Hartwig JF On the Interpretation of Deuterium Kinetic Isotope Effects in C—H Bond Functionalizations by Transition Metal Complexes. Angew. Chem., Int. Ed 2012, 51, 3066–3072. [DOI] [PubMed] [Google Scholar]
- (23).For an example where substrate activation rather than anion delivery is proposed to be enantiodetermining, see ref. 3b.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.