

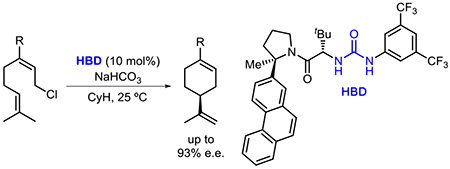
Abstract
Chiral urea derivatives are shown to catalyze enantioselective tail-to-head cyclization reactions of neryl chloride analogues. Experimental data are consistent with a mechanism in which π-participation by the nucleophilic olefin facilitates chloride ionization and thereby circumvents simple elimination pathways. Kinetic and computational studies support a cooperative mode of catalysis wherein two molecules of the urea catalyst engage the substrate and induce enantioselectivity through selective transition state stabilization.
Graphical Abstract
Carbocycles are ubiquitous motifs within natural and unnatural organic molecules, and their construction has been a primary research focus in synthetic organic chemistry since the inception of the field.1 Terpenes and terpenoids constitute one of the most important classes of carbocyclic natural products from both structural and functional perspectives.2 Their carbocyclic frameworks are constructed by terpene cyclase enzymes, which engage linear isoprenoid substrates of varying length.3 Cyclization of these polyolefins is initiated either through protonation of an olefin or epoxide in head-to-tail (HT) cyclizations, or through abstraction of an allylic pyrophosphate leaving group in tail-to-head (TH) cyclizations (Figure 1A).2–3 The reactivity of the resulting carbocationic intermediates is then modulated through a combination of substrate preorganization5 and non-covalent stabilizing interactions3,6 in the enzyme active site, resulting in selective rearrangements and carbon-carbon bond-forming reactions that ultimately give rise to an extraordinarily diverse array of natural products (Figure 1B).
Figure 1.
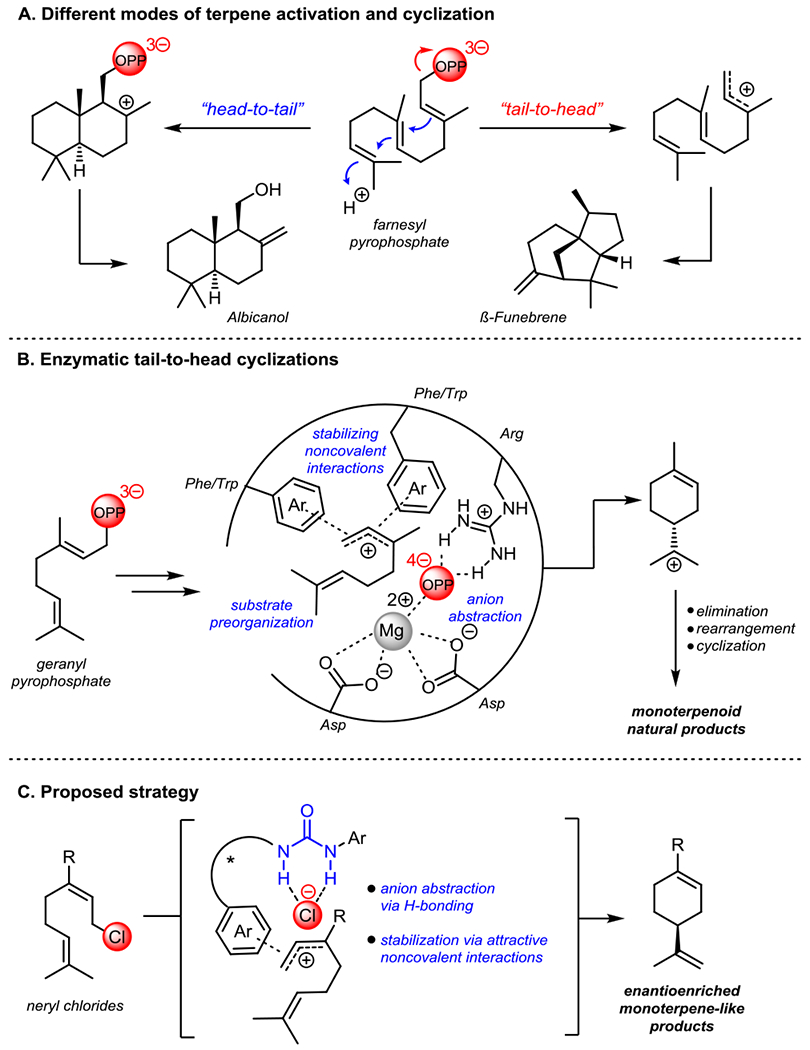
A) Head-to-tail and tail-to-head cyclization reactions. B) Schematic illustrating Nature’s strategy for controlled ionization-dependent cyclizations. C) Proposed strategy for enantioselective tail-to-head cyclizations catalyzed by chiral hydrogen-bond donors.
The remarkable ability of cyclase enzymes to generate carbocationic intermediates and channel their reactivity along specific pathways has long captured the imagination of chemists and motivated efforts to deploy analogous strategies in synthesis.7 However, the very features that make carbocations such powerful intermediates in biosynthesis also render their application outside of enzymatic chemistry quite challenging.4,8 Nonetheless, over the last 60 years organic chemists have made significant progress in mimicking the HT synthesis of steroidal ring systems by leveraging the propensity of these reactions to proceed through concerted, stereospecific mechanisms.9,10 In contrast, efforts to reproduce TH cyclizations using non-biological catalysts have generally resulted in unselective or thermodynamically controlled reactions.4,11 Pioneering studies from the laboratories of Shenvi4 and Tiefenbacher12 have revealed strategies for extending carbocation lifetime, unlocking the potential for non-enzymatic mimics of TH polycyclizations, but catalyst control over enantioselectivity has remained elusive. To our knowledge, the only reported enantioselective TH cyclizations13 employ a binol-derived leaving group as a chiral auxiliary.
We hypothesized that it might be possible to achieve enantioselectivity in TH cyclizations with a small-molecule catalyst by mimicking nature’s strategy of controlled generation and selective stabilization of key high-energy cationic intermediates and transition states. In particular, we sought to draw on advances in dual-hydrogen-bond-donor (HBD) catalysis, which have revealed that chiral urea and thioureas are capable of inducing enantioselectivity in reactions involving cationic intermediates generated by anion abstraction.14 Moreover, specifically tailored HBD catalysts have been shown to induce enantioselectivity through non-covalent stabilizing interactions similar to those present in the active sites of cyclase enzymes.10e,15 Herein we report the development of a urea-catalyzed enantioselective cyclization of neryl chloride derivatives (Figure 1C). Mechanistic analysis has provided key insights into the basis of reactivity and stereoinduction, including the revelation that π-participation by the nucleophilic olefin during ionization is critical to the success of the enantioselective transformation.
In preliminary studies, geranyl chloride and neryl chloride (1a) were found to display dramatic differences in reactivity in the presence of the achiral bis-aryl urea 6 and a stoichiometric base (Figure 2). Geranyl chloride underwent a very slow reaction at room temperature, with significant formation of uncyclized elimination products. In contrast, the reaction of neryl chloride (1a) proceeded to high conversion under the same conditions, leading predominantly to the formation of cyclic products 2a-4a. While enantioselective variants of the cyclization of 1a could be promoted with chiral dual HBD catalysts, only very modest levels of enantioselectivity (up to 34% e.e.) were attained in the formation of limonene (2a) despite the evaluation of a wide assortment of chiral hydrogen-bond-donor catalysts and reaction conditions (see SI for details).
Figure 2.
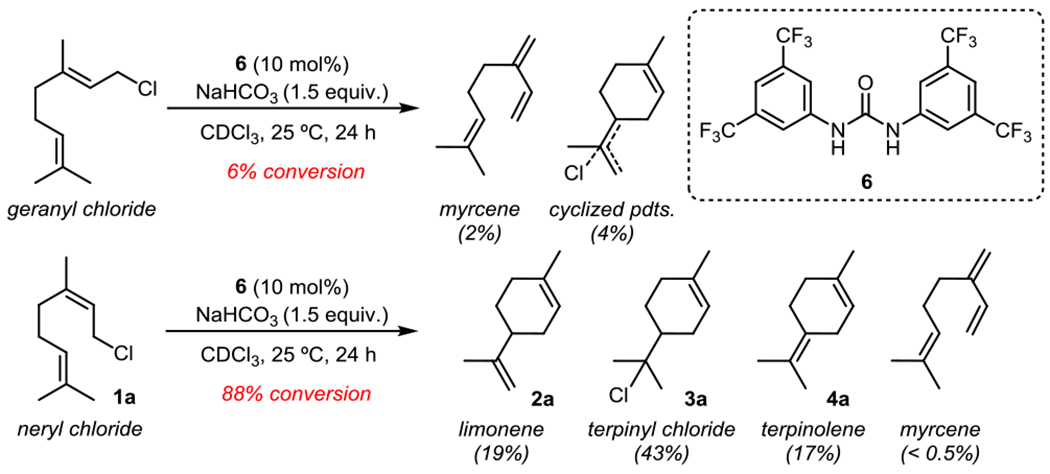
Differing reactivity observed in the urea-catalyzed cyclization of E and Z isomers. Conversions and yields were assessed from crude reaction mixtures using 1H NMR with mesitylene as an internal quantitative standard.
Recognizing that 1a might be a particularly challenging substrate for asymmetric induction due to its limited structural features, we explored variations to the structure of the reactants. Introduction of a phenyl substituent as a potential catalyst-recognition element in place of the C3 methyl group (1b) led to significant improvements in enantioselectivity. Urea 7a was identified as the optimal catalyst for this substrate, promoting cyclization to 2b in 63% NMR yield and 87% e.e. at room temperature (Figure 3). In addition to 2b, alkyl chloride 3b was formed in 20% yield with similar e.e. (86%), consistent with both products arising from a common intermediate; 3b could be converted to 2b and 4b in 83% combined yield (2b:4b = 10:1) via collidine-promoted elimination.13b The remainder of the mass balance consisted of two achiral cyclization products: 12% yield of tetrasubstituted olefin 4b and 5% yield of conjugated diene 5b, which we propose forms via a [1,2] hydride shift followed by elimination.16 Consistent with our prior observations using geranyl chloride, the Z isomer of 1b was found to undergo very slow reaction promoted by 7a (5% conversion after 24 h), with 2b generated in only 50% e.e. (see SI for details).
Figure 3.
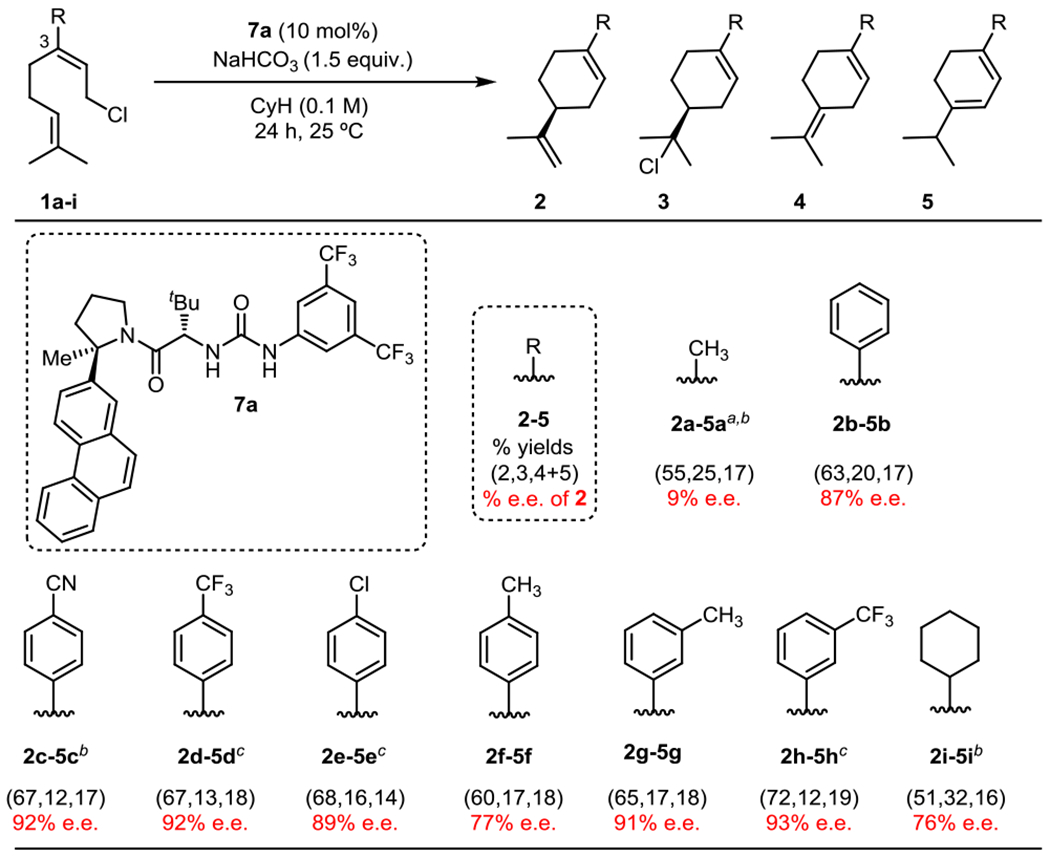
Substrate scope. All reactions were performed on 0.15 mmol scale and proceeded to complete conversion. E.e. values are for products 2a-i. Alkyl chlorides 3b, 3h, and 3i were generated in 86% e.e., 91% e.e., and 70% e.e., respectively. Conversions and yields were assessed from crude reaction mixtures using 1H NMR with mesitylene as an internal standard. aReaction run in C6D12; b72 hr. reaction time; c48 hr. reaction time.
Variation of the electronic and steric properties of the C3 aryl substituent in 1 was explored in cyclization reactions catalyzed by 7a (Figure 3). Electronic perturbation of the C3 aryl group of 1 revealed that the highest levels of e.e. were attained with electron-deficient substrates. Improved enantioselectivity was also observed upon substitution of the meta position with either electron-donating or withdrawing groups. While urea 7a catalyzed the formation of limonene 2a (R = Me) with low (< 10%) enantioselectivity, the cyclohexyl-substituted analog 2i was formed in 76% e.e. It is therefore apparent both steric and electronic properties of the substrate play important roles in enantioinduction.
The dramatic differences in reactivity and enantioselectivity observed in the 7a-catalyzed cyclizations of the E and Z isomers of 1b (vide supra) indicated that both the rate- and enantiodetermining steps differed for the two isomers, suggesting that they might react through fundamentally different mechanisms. While the Z isomer of 1b must undergo rearrangement prior to cyclization,17 the nucleophilic olefin of 1b can interact with the allyl electrophile in a preorganized structure, potentially facilitating chloride ionization through anchimeric assistance.18
The role of the nucleophilic olefin in the rate-determining step of the cyclization of 1b was assessed in a kinetic isotope effect (KIE) experiment.19 Starting material recovered from one-pot competition experiments between 1b and 1b-d1 revealed enrichment in the protio isotopologue corresponding to kH/kD = 0.944(3) (Figure 4A). This small, secondary inverse KIE is consistent with direct involvement of the distal olefin in the rate-determining step, with partial rehybridization of the vinylic carbon from sp2 to sp3 and a small degree of C–C bond formation in the transition state.20,21
Figure 4.
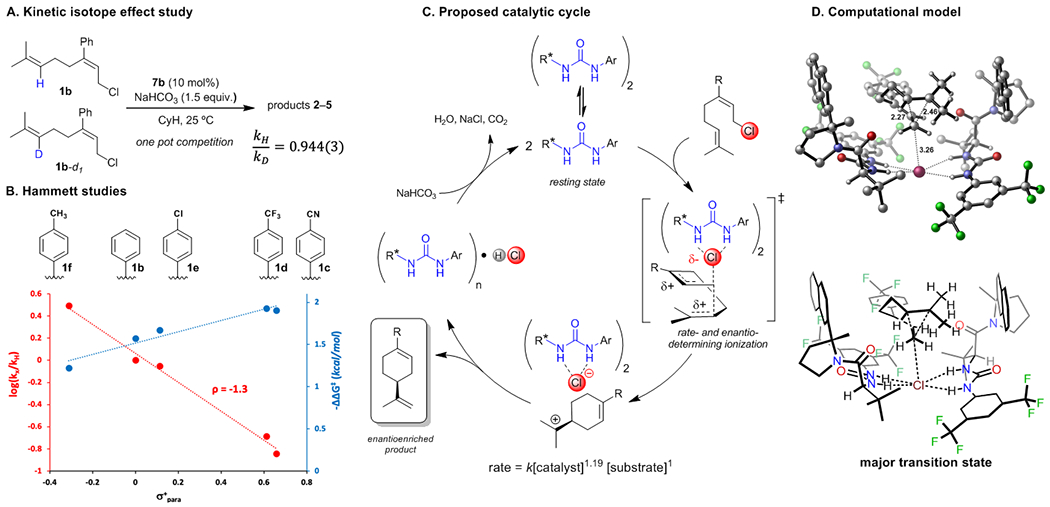
Mechanistic studies. A) One-pot competition secondary H/D KIE experiment. B) Hammett studies. In red: Relative rates of cyclization of 1b-1f promoted by catalyst 7b. In blue: Enantioselectivities (expressed as –ΔΔG‡= RTln(enantiomer ratio), T = 25 °C) in the formation of 2b-2f promoted by 7a. C) Proposed catalytic cycle based on the KIE data and the experimentally determined rate law. D) Transition state model for the pathway leading to the major enantiomeric product in the cyclization of 1d. Key bond lengths are reported in Angstroms. Calculations were carried out at PCM (CyH) – B3LYP-D3(BJ)/6-311+G(d,p) // B3LYP/6-31G(d).
Hammett analysis conducted using catalyst 7b established that reaction rate correlates linearly with σ+para in the reactions of 1b-1f (Figure 4B), consistent with the buildup of positive charge on the C3 carbon during the rate-determining step. Enantioselectivity values for the same substrates also correlate directly with σ+para. The increased levels of asymmetric induction in electron-deficient substrates may be a consequence of differential extents of olefin participation during chloride displacement. For electron-deficient substrates, a higher degree of anchimeric assistance from the distal olefin would be expected on the basis of a diminished ability to support positive charge at C3. A greater degree of C–C bond formation would be expected to result in a more highly ordered enantiodetermining transition state.18g
Kinetic analysis of the reaction catalyzed by urea 7b revealed a first-order dependence of rate on substrate 1b, 0th order dependence on base, and a kinetic order in catalyst of 1.19 (see SI for details). Aryl pyrrolidine urea and thiourea hydrogen-bond donors such as 7 are prone to dimerization both in the solid state and in nonpolar organic solvents,22 so a mixed resting state of monomeric and dimeric 7b could account for the observed non-integer order in catalyst. This possibility was supported through isothermal titration calorimetric studies, which revealed the presence of a roughly 70:30 equilibrium mixture of dimeric and monomeric 7b in cyclohexane at [7b]total = 0.01 M (see SI for details). Thus, the observed kinetic order in [7b] can be ascribed to a mixed dimer-monomer resting state and a rate-determining transition state containing two molecules of catalyst. Based on the results of the kinetic analyses, Hammett studies, and the KIE experiment, we propose the catalytic cycle depicted in Figure 4C, where concerted rate- and enantioselectivity-determining chloride ionization and carbon-carbon bond formation is promoted through the cooperative action of two molecules of the urea catalyst.14d,23
Having established the stoichiometry and general features of the key selectivity-determining transition state, we sought to explore the factors responsible for enantioinduction through the use of computational modeling (see SI for computational details). Density functional theory (DFT) calculations identified energy-minimized transition state structures for the major and minor enantiomeric cyclization pathways of 1d promoted by two molecules of 7b.24 Consistent with experimental observations, chloride ionization was characterized by olefinic participation (Figure 4D, forming C–C bond: 2.27 Å, breaking C–Cl bond: 3.26 Å). The lowest energy computed cyclization transition state is partially encapsulated within the dimeric catalyst assembly, with catalyst naphthyl groups positioned in close proximity to developing positive charge. The mode by which the aryl substituents on the catalyst influence enantioselectivity was assessed experimentally. Kinetic analysis conducted on the cyclization of 1b using catalysts 7a-7d revealed a positive correlation between reaction rate and enantioselectivity (Figure 5).25 Decomposition of the observed rate into contributions from the major and minor enantiomeric pathways15c reveals that the effect is far more pronounced for the major pathway; the catalyst aryl pyrrolidine stabilizes the transition state leading to the minor enantiomer to a lesser extent. Thus, it can be concluded that stabilizing aromatic interactions are at least partially responsible for enantioinduction.26
Figure 5.
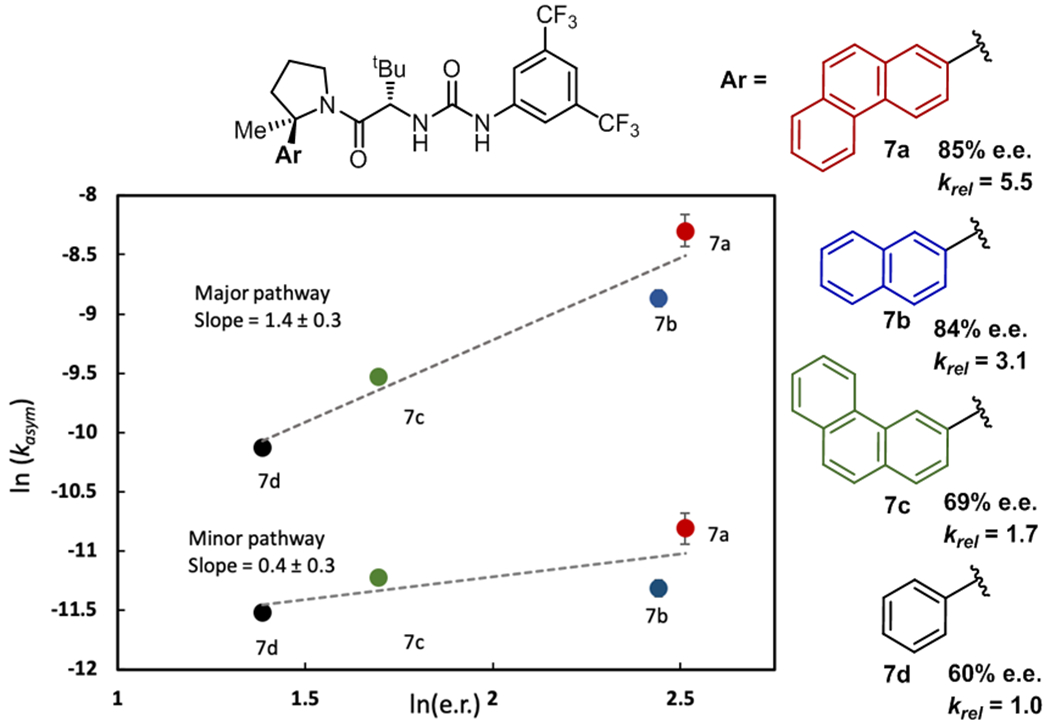
Effect of catalyst aryl substituents on reaction rate and enantioselectivity.
In summary, we have developed a highly enantioselective cyclization reaction of neryl chloride analogues catalyzed by chiral ureas. Reactions proceed through a concerted pathway in which π-participation by the nucleophilic olefin facilitates ionization of the leaving group, thereby avoiding direct elimination products. A network of attractive non-covalent interactions involving two molecules of the urea serves to stabilize the cyclization transition state and induce enantiocontrol. Concerted mechanisms have been proposed to play key roles in enzymatic3b,27 and synthetic reactions9,28 involving formal cationic intermediates, and they likely underlie the attainment of high chemo- and enantioselectivity in the present system. Future studies will be aimed at leveraging the principles uncovered here toward more complex transformations such as polycyclization reactions.
Supplementary Material
ACKNOWLEDGMENT
Financial support for this work was provided by the NIH through GM043214. We thank Dr. Andreas Rötheli for helpful discussions. We thank Mr. Ethan Magno for assistance in preparative-HPLC purifications and Dr. Shao-Liang Zheng for X-ray data collection and structure determination.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: https://pubs.acs.org/doi/10.1021/jacs.0c02665
Experimental procedures and characterization data of catalyst and substrate syntheses, procedures and analytical data for enantioselective reactions, details of mechanistic studies, and computational studies (PDF)
Crystallographic data for 2d (CIF)
The authors declare no competing financial interest.
REFERENCES
- (1).Some of the most conspicuous examples:; (a) Dieckmann W Zur Kenntniss der Ringbildung aus Kohlenstoffketten. Chem. Ber 1894, 27, 102–103. [Google Scholar]; (b) Robinson R LXIII . A Synthesis of Tropinone. J.Chem.Soc 1917,111,762–768. [Google Scholar]; (c) Diels O; Alder K Synthesen in der hydroaromatischen Reihe, I. Justus Liebigs Ann. Chem 1928, 460, 98–122; [Google Scholar]; (d) Rapson WS; Robinson R Experiments on the synthesis of substances related to the sterols. Part II. A new general method for the synthesis of substituted cyclohexenones J. Chem. Soc 1935, 1285–1288. [Google Scholar]; (e) Woodward RB; Sondheimer F; Taub D; Heusler K; McLamore WM The Total Synthesis of Steroids. J. Am. Chem. Soc 1952, 74, 4223–4251. [Google Scholar]; (f) Fu GC; Grubbs RH The Application of Catalytic Ring-Closing Olefin Metathesis to the Synthesis of Unsaturated Oxygen Heterocycles. J. Am. Chem. Soc 1992, 114, 5426–5427. [Google Scholar]
- (2).(a) Breitmaier E Terpenes: Flavors, Fragrances, Pharmaca, Pheromones; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, 2006. [Google Scholar]; (b) Terpenes and Terpenoids; Perveen S; Al-Taweel A, Eds. IntechOpen, 2018. [Google Scholar]
- (3).(a) Wendt KU; Schulz GE Isoprenoid Biosynthesis: Manifold Chemistry Catalyzed by Similar Enzymes. Structure 1998, 6, 127–133. [DOI] [PubMed] [Google Scholar]; (b) Davis EM; Croteau R Cyclization Enzymes in the Biosynthesis of Monoterpenes, Sesquiterpenes and Diterpenes. Top. Curr. Chem 2000, 209, 53–95. [Google Scholar]; (c) Wendt KU; Schulz GE; Corey EJ; Liu DR Enzyme Mechanisms for Polycyclic Triterpene Formation. Angew. Chem. Int. Ed 2000, 39, 2812–2833. [PubMed] [Google Scholar]; (d) Christianson DW Structural Biology and Chemistry of the Terpenoid Cyclases. Chem. Rev 2006, 106, 3412–3442. [DOI] [PubMed] [Google Scholar]; (e) Christianson DW Structural and Chemical Biology of Terpenoid Cyclases. Chem. Rev 2017, 117, 11570–11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Pronin SV; Shenvi RA Synthesis of highly strained terpenes by non-stop tail-to-head polycyclization. Nat. Chem 2012, 4, 915–920. [DOI] [PubMed] [Google Scholar]
- (5).(a) Sato H; Mitsuhashi T; Yamazaki M; Abe I; Uchiyama M Inherent Atomic Mobility Changes in Carbocation Intermediates During the Sesterterpene Cyclization Cascade. Beilstein J. Org. Chem 2019, 15, 1890–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sato H; Mitsuhashi T; Yamazaki M; Abe I; Uchiyama M Computational Studies on Biosynthetic Carbocation Rearrangements Leading to Quiannulatene: Initial Conformation Regulates Biosynthetic Route, Stereochemistry, and Skeleton Type. Angew. Chem. Int. Ed 2018, 57, 14752–14757. [DOI] [PubMed] [Google Scholar]; (c) Hong YJ; Tantillo DJ Consequences of Conformational Preorganization in Sesquiterpene Biosynthesis: Theoretical Studies on the Formation of the Bisabolene, Curcumene, Acoradiene, Zizaene, Cedrene, Duprezianene, and Sesquithuriferol Sesquiterpenes. J. Am. Chem. Soc 2009, 131, 7999–8015. [DOI] [PubMed] [Google Scholar]
- (6).(a) Pandit J; Danley DE; Schulte GK; Mazzalupo S; Pauly TA; Hayward CM; Hamanaka ES; Thompson JF; Harwood HJ Crystal Structure of Human Squalene Synthase. J. Biol. Chem 2000, 275, 30610–30617. [DOI] [PubMed] [Google Scholar]; b) Whittington DA; Wise WL; Urbansky M; Coates RM; Croteau RB; Christianson DW Bornyl diphosphate synthase: Structure and strategy for carbocation manipulation by a terpenoid cyclase. PNAS 2002, 99, 15375–15380. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Aaron JA; Lin X; Cane DE; Christianson DW Structure of Epi-Isozizaene Synthase from Streptomyces coelicolor A3(2), a Platform for New Terpenoid Cyclization Templates. Biochemistry 2010, 49, 1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Baer P; Rabe P; Citron CA; de Oliveira Mann CC; Kaufmann N; Groll M; Dickschat JS Hedycaryol Synthase in Complex with Nerolidol Reveals Terpene Cyclase Mechanism. ChemBioChem. 2014, 15, 213–216. [DOI] [PubMed] [Google Scholar]; (e) Morehouse BR; Kumar RP; Matos JO; Olsen SN; Entova S; Oprian DD Functional and Structural Characterization of a (+)-Limonene Synthase from Citrus sinensis. Biochemistry 2017, 56, 1706–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).For reviews, see:; (a) Yoder RA; Johnston JN A Case Study in Biomimetic Total Synthesis: Polyolefin Carbocyclizations to Terpenes and Steroids. Chem. Rev 2005, 105, 4730–4756. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Maimone TJ; Baran PS Modern Synthetic Efforts Toward Biologically Active Terpenes. Nat. Chem. Biol 2007, 3, 396–407. [DOI] [PubMed] [Google Scholar]; (c) Ungarean CN; Southgate EH; Sarlah D Enantioselective polyene cyclizations. Org. Biomol. Chem 2016, 14, 5454–5467. [DOI] [PubMed] [Google Scholar]
- (8).Wendlandt AE; Vangal P; Jacobsen EN Quaternary stereocentres via an enantioconvergent catalytic SN1 reaction. Nature 2018, 556, 447–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Stork G; Burgstahler AW The Stereochemistry of Polyene Cyclization. J. Am. Chem. Soc 1955, 77, 5068–5077. [Google Scholar]; (b) Eschenmoser A; Ruzicka L; Jeger O; Arigoni D Eine stereochemische Interpretation der biogenetischen Isoprenregel bei den Triterpenen. Helv. Chim. Acta 1955, 38, 1890–1904. [Google Scholar]; (c) Johnson WS Nonenzymic Biogenetic-like Olefinic Cyclizations. Acc. Chem. Res 1968, 1, 1–8. [DOI] [PubMed] [Google Scholar]; (d) van Tamelen EE Bioorganic chemistry: sterols and acrylic terpene terminal epoxides. Acc. Chem. Res 1968, 1, 111–120. [Google Scholar]; (e) Johnson WS Biomimetic Poylene Cyclizations. Angew. Chem. Int. Ed. 1976, 15, 9–17. [DOI] [PubMed] [Google Scholar]; (f) van Tamelen EE; Hwu JR Direct Total Synthesis of Traditional Sterols by Tricyclization of Polyunsaturated Cyclohexene Oxides. J. Am. Chem. Soc 1983, 105, 2490–2491. [Google Scholar]; (g) Kronja O; Orlović M; Humksi K; Borčić S Lack of a Secondary β-Deuterium Kinetic Isotope Effect in the Solvolysis of 2-Chloro-3-hydrosqualene. A Case of Extended π-Participation and Indication of Concerted Biomimetic Polycyclization. J. Am. Chem. Soc 1991, 113, 2306–2308. [Google Scholar]; (h) Malnar I; Humski K; Kronja O Hammett ρ+ Values as Kinetic Evidence for the Concerted Biomimetic Bicyclization Mechanism. J. Org. Chem 1998, 63, 3041–3044. [Google Scholar]; (i) Huang AX; Xiong Z; Corey EJ An Exceptionally Short and Simple Enantioselective Total Synthesis of Pentacyclic Triterpenes of the β-Amyrin Family. J. Am. Chem. Soc 1999, 121, 9999–10003. [Google Scholar]; (j) Bogensätter M; Limberg A; Overman LE; Tomasi AL Enantioselective Total Synthesis of Kinesin Motor Protein Inhibitor Adociasulfate 1. J. Am. Chem. Soc 1999, 121, 12206–12207. [Google Scholar]; (k) Eschenmoser A; Arigoni D Revisited after 50 Years: The ‘Stereochemical Interpretation of the Biogenetic Isoprene Rule for the Triterpenes’. Helv. Chim. Acta 2005, 88, 3011–3050. [Google Scholar]
- (10).For examples of enantioselective variants, see:; (a) Ishihara K; Nakamura S; Yamamoto H The First Enantioselective Biomimetic Cyclization of Polyprenoids. J. Am. Chem. Soc 1999, 121, 4906–4907. [Google Scholar]; (b) Kumazawa K; Ishihara K; Yamamoto H Tin(IV) Chloride-Chiral Pyrogallol Derivatives as New Lewis Acid-Assisted Chiral Brønsted Acids for Enantioselective Polyene Cyclization. Org. Lett 2004, 6, 2551–2554. [DOI] [PubMed] [Google Scholar]; (c) Sakakura A; Ukai A; Ishihara K Enantioselective halocyclization of polyprenoids induced by nucleophilic phosphoramidites. Nature 2007, 445, 900–903. [DOI] [PubMed] [Google Scholar]; (d) Rendler S; MacMillan DWC Enantioselective Polyene Cyclization via Organo-SOMO Catalysis. J. Am. Chem. Soc 2010, 132, 5027–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Knowles RR; Lin S; Jacobsen EN Enantioselective Thiourea-Catalyzed Cationic Polycyclizations. J. Am. Chem. Soc 2010, 132, 5030–5032. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Sethofer SG; Mayer T; Toste DF Gold(I)-Catalyzed Enantioselective Polycyclization Reactions. J. Am. Chem. Soc 2010, 132, 8276–8277. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Snyder SA; Treitler DS; Brucks AP Simple Reagents for Direct Halonium-Induced Poylene Cyclizations. J. Am. Chem. Soc 2010, 132, 14303–14314. [DOI] [PubMed] [Google Scholar]; (h) Surendra K; Corey EJ Highly Enantioselective Proton-Initiated Polycyclization of Polyenes. J. Am. Chem. Soc 2012, 134, 11992–11994. [DOI] [PubMed] [Google Scholar]; (i) Felix RJ; Munro-Leighton C; Gagné MR Electrophilic Pt(II) Complexes: Precision Instruments for the Initiation of Transformations Mediated by the Cation-Olefin Reaction. Acc. Chem. Res 2014, 47, 2319–2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).(a) Gutsche CD; Maycock JR; Chang CT; Acid-Catalyzed Cyclization of Farnesol and Nerolidol. Tetrahedron 1968, 24, 859–876. [Google Scholar]; (b) Andersen NH; Syrdal DD Chemical Simulation of the Biosynthesis of Cedrene. Tetrahedron Lett. 1972, 24, 2455–2458. [Google Scholar]; (c) Ohta Y; Hirose Y Electrophile induced cyclization of farnesol. Chem. Lett 1972, 1, 263–266. [Google Scholar]
- (12).(a) Zhang Q; Tiefenbacher K Terpene Cyclization Catalysed inside a Self-Assembled Cavity. Nat. Chem 2015, 7, 197–202. [DOI] [PubMed] [Google Scholar]; (b) Zhang Q; Catti L; Pleiss J; Tiefenbacher K Terpene Cyclizations inside a Supramolecular Catalyst: Leaving-Group-Controlled Product Selectivity and Mechanistic Studies. J. Am. Chem. Soc 2017, 139, 11482–11492. [DOI] [PubMed] [Google Scholar]; (c) Zhang Q; Rinkel J; Goldfuss B; Dickschat JS; Tiefenbacher K Sesquiterpene Cyclizations Catalysed inside the Resorcinarene Capsule and Application in the Short Synthesis of Isolongifolene and Isolongifolenone. Nat. Catal 2018, 1, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Pahima E; Zhang Q; Tiefenbacher K; Major DT Discovering Monoterpene Catalysis Inside Nanocapsules with Multiscale Modeling and Experiments. J. Am. Chem. Soc 2019, 141, 6234–6246. [DOI] [PubMed] [Google Scholar]; (e) Zhang Q; Tiefenbacher K Sesquiterpene Cyclizations inside the Hexameric Resorcinarene Capsule: Total Synthesis of δ-Selinene and Mechanistic Studies. Angew. Chem. Int. Ed 2019, 58, 12688–12695. [DOI] [PubMed] [Google Scholar]; (f) Merget S; Catti L; Piccini GM; Tiefenbacher K Requirements for Terpene Cyclizations inside the Supramolecular Resorcinarene Capsule: Bound Water and Its Protonation Determine Catalytic Activity. J. Am. Chem. Soc 2020, 142, 4400–4410. [DOI] [PubMed] [Google Scholar]
- (13).(a) Sakane S; Fujiwara J; Maruoka K; Yamamoto H Chiral leaving group. Biogenetic-type asymmetric synthesis of limonene and bisabolenes. J. Am. Chem. Soc 1983, 105, 6154–6155. [Google Scholar]; (b) Nakamura H; Ishihara K; Yamamoto H Lewis Acid-Activated Chiral Leaving Group: Enantioselective Electrophilic Addition to Prochiral Olefins. J. Org. Chem 2002, 67, 5124–5137. [DOI] [PubMed] [Google Scholar]
- (14).(a) Raheem IT; Thiara PS; Peterson EA; Jacobsen EN Enantioselective Pictet-Spengler-Type Cyclizations of Hydroxylactams: H-Bond Donor Catalysis by Anion Binding. J. Am. Chem. Soc 2007, 129, 13404–13405. [DOI] [PubMed] [Google Scholar]; (b) Reisman SE; Doyle AG; Jacobsen EN Enantioselective Thiourea-Catalyzed Additions to Oxocarbenium ions. J. Am. Chem. Soc 2008, 130, 7198–7199. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Brown AR; Kuo W-H.; Jacobsen EN, Enantioselective Catalytic α-Alkylation of Aldehydes via an SN1 Pathway. J. Am. Chem. Soc 2010, 132, 9286–9288. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ford DD; Lehnherr D; Kennedy CR; Jacobsen EN Anion-Abstraction Catalysis: The Cooperative Mechanism of α-Chloroether Activation by Dual Hydrogen-Bond Donors. ACS Catal. 2016, 6, 4616–4620. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) for a review, see:Brak K; Jacobsen EN Asymmetric Ion-Pairing Catalysis. Angew. Chem. Int. Ed 2013, 52, 534–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).(a) Knowles RR; Jacobsen EN Attractive noncovalent interactions in asymmetric catalysis: Links between enzymes and small molecule catalysts. PNAS 2010, 107, 20678–20685. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kennedy R; Lin S; Jacobsen E; The Cation-π Interaction in Small-Molecule Catalysis, Angew. Chem. Int. Ed 2016, 55, 12596–12624. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lin S; Jacobsen EN Thiourea-catalyzed ring opening of episulfonium ions with indole derivatives by means of stabilizing non-covalent interactions. Nat. Chem 2012, 4, 817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lafever R; Croteau R Hydride Shifts in the Biosynthesis of the p-Menthane Monoterpenes α-Terpinene, γ-Terpinene, and β-Phellandrene. Arch. Biochem. Biophys 1993, 301, 361–366. [DOI] [PubMed] [Google Scholar]
- (17).A 1,3 transposition of the chloride to the benzylic position would be required followed either by SN1(2)’ attack of the nucleophilic olefin or further 1,3 rearrangement to form 1b. Alternatively, Z-1b could undergo sequential ionization, allyl cation isomerization, and attack of the nucleophilic olefin. For a discussion on the plausibility of allyl cation isomerization, see ref. 12a.
- (18).Nucleophilic assistance has previously been proposed for the cyclization of nerol and linalool derivatives under solvolysis conditions. See:; (a) Rittersdorf W; Cramer F Cyclization of Nerol and Linalool on Solvolysis of Their Phosphate Esters. Tetrahedron, 1968, 24, 43–52. [Google Scholar]; (b) Bunton CA; Leresche JP; Hachey D Deuterium Isotope Effects in Cyclization of Monoterpenoids. Tet. Lett 1972, 24, 2431–2434. [Google Scholar]; (c) Winstein S; Valkanas G; Wilcox CF Jr. The Solvolysis of Linalyl p-Nitrobenzoate and the Stereochemical Aspects of the Resulting 1-3 and 1-5 Rearrangements. J. Am. Chem. Soc 1972, 94, 2286–2290. [Google Scholar]; (d) Astin KB; Whiting MC Allylic Carbonium Ions. Part II. Solvolysis and Cyclisation of Some Monoterpene 2,4-Dinitrophenyl Ethers. J. Chem. Soc. Perkin. Trans. II 1976, 10, 1160–1165. [Google Scholar]; (e) Cori O; Chayet L; Perez LM; Bunton CA; Hachey D Rearrangement of Linalool, Geraniol, and Nerol and Their Derivatives. J. Org. Chem 1986, 51, 1310–1316. [Google Scholar]; However, other studies have found evidence against alkene nucleophilic assistance:; (f) Brody EP; Gutsche CD The Mechanism of the Acid-Catalyzed Decomposition of the Farnesyl Phosphates. Tetrahedron 1977, 33, 723–729. [Google Scholar]; (g) Poulter CD; King C-HR Model Studies of Terpene Biosynthesis. A Stepwise Mechanism for Cyclization of Nerol to α-Terpineol. J. Am. Chem. Soc 1982, 104, 1422–1424. [Google Scholar]; The importance of nucleophilic assistance most likely varies depending on the precise details of the system under investigation.
- (19).Catalyst 7b was used and was found to promote cyclization of 1b to 2-5b in quantitative yield and similar e.e. of 2b (86%) as compared to the reaction catalyzed by 7a. See SI for details.
- (20).A similar kH/kD KIE of 0.925 was reported by Bunton and co-workers in solvolysis studies of neryl chloride. See reference 18b.
- (21).Rapid, reversible ionization followed by rate-limiting olefin addition to an allyl cation intermediate cannot be ruled out a priori but would be expected to involve a significant degree of rehybridization at the nucleophilic carbon and therefore display a much larger inverse secondary KIE.
- (22).Ford DD; Lehnherr D; Kennedy CR; Jacobsen EN On- and Off-Cycle Catalyst Cooperativity in Anion-Binding Catalysis. J. Am. Chem. Soc 2016, 138, 7860–7863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Enantioselectivities were observed to be invariant as a function of [7b] at 5, 10, and 15 mol% loadings, rendering highly unlikely any participation of a competitive monomeric pathway.
- (24).On the basis of prior mechanistic studies from our group (see ref. 22), we assumed a 4-H binding geometry to chloride. Transition states characterized by 2-H binding to chloride were not modeled.
- (25).There is no detectable background reaction of 1 under the reaction conditions in the absence of catalysts 7.
- (26).For detailed investigations on how cationic C–H···π interactions can modify potential energy surfaces in terpene cyclase chemistry see:; (a) Hong YJ; Tantillo DJ C–H···π interactions as modulators of carbocation structure – implications for terpene biosynthesis. Chem. Sci 2013, 4, 2512–2518. [Google Scholar]; (b) Hong YJ; Tantillo DJ Tension between Internal and External Modes of Stabilization in Carbocations Relevant to Terpene Biosynthesis: Modulate Minima Depth via C–H···π Interactions. Org. Lett 2015, 17, 5388–5391. [DOI] [PubMed] [Google Scholar]
- (27).(a) Tantillo DJ Recent excursions to the borderlands between the realms of concerted and stepwise: carbocation cascades in natural product biosynthesis. J. Phys. Org. Chem 2008, 21, 561–570. [Google Scholar]; (b) Tantillo DJ The carbocation continuum in terpene biosynthesis—where are the secondary cations? Chem. Soc. Rev 2010, 39, 2847–2854. [DOI] [PubMed] [Google Scholar]; (c) Tantillo DJ Biosynthesis via carbocations: Theoretical studies on terpene formation. Nat. Prod. Rep 2011, 28, 1035–1053. [DOI] [PubMed] [Google Scholar]
- (28).(a) Khomutnyk YY; Arguelles AJ; Winschel GA; Sun Z; Zimmerman PM; Nagorny P Studies of the Mechanism and Origins of Enantioselectivity for the Chiral Phosphoric Acid-Catalyzed Stereoselective Spiroketalization Reactions. J. Am. Chem. Soc 2016, 138, 444–456. [DOI] [PubMed] [Google Scholar]; (b) Tsuji N; Kennemur JL; Buyck T; Lee S; Prevost S; Kaib PSJ; Bykov D; Fares C; List B Activation of olefins via asymmetric Brønsted acid catalysis. Science 2018, 359, 1501–1505. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.