

Abstract
The purpose of this study was to explore whether individual differences in glucocorticoid concentrations were associated with symptom improvement following exposure therapy for patients with social anxiety disorder. To do this, 60 participants with social anxiety disorder completed a randomized-controlled trial of exposure therapy, where participants were randomized to receive scopolamine-augmentation or placebo during their 7 exposure sessions. Scopolamine is an antimuscarinic which blocks the effects of acetylcholine and reduces autonomic arousal. During sessions 1, 4, 7, and during the post-treatment extinction assessment, participants provided up to 16 saliva samples (4 in each session). Pre-treatment, post-treatment, and at 1-month follow-up, participants completed the Liebowitz Social Anxiety Scale to monitor change in fear and avoidance symptoms. Elevated endogenous in-session cortisol during exposure sessions was associated with less symptom improvement from pre- to post-treatment and at 1-month follow-up. The association between elevated endogenous in-session cortisol and attenuated symptom change was not moderated by scopolamine treatment condition. Individuals with social anxiety disorder who have elevated neuroendocrine signaling may under-benefit from exposure therapy. This is the first study, to our knowledge, to examine whether endogenous in-session cortisol concentrations predict symptom changes following exposure therapy for the treatment of social anxiety disorder. More investigation of non-invasive and reliable biological markers that explain variability in responses to effective treatments are needed.
1.0. Introduction
Social anxiety disorder is characterized by intense and persistent fear of social or performance situations when exposed to unfamiliar people or potential scrutiny by others (American Psychiatric Association, 2013; World Health Organization, 1992). Social anxiety disorder is common; affecting approximately 13% of the population (Bandelow and Michaelis, 2015; Kessler et al., 2012; Merikangas et al., 2010). The prevalence of social anxiety disorder contributes significantly to the global burden of disease, such that 35% of men and 41% of women with the disorder report their symptoms to be “severely disabling” (Kessler et al., 2009). There are several efficacious treatments for social anxiety disorder. Among them, exposure therapy has emerged as a highly effective treatment, with large effect sizes relative to waitlist control as well as other established pharmacological and psychological treatments (Acarturk et al., 2009; Feske & Chambless, 1995; Mayo-Wilson et al., 2014; Stewart & Chambless, 2009). Yet, there remain individual differences in the efficacy of exposure therapy for social anxiety disorder, resulting in a clinically significant response rate of approximately 50% (Loerinc et al., 2015). In the present study, we explored whether endogenous glucocorticoid concentrations as measured by a common biomarker, salivary cortisol, identified individuals for whom exposure therapy was more or less effective.
Biomarkers may be critical for precision medicine, which seeks to identify which individuals benefit from which interventions (Insel, 2014). Salivary biomarkers are of particular interest due to their cost-benefit profile (Kuhlman and Mousavi, in press). Yet, only a few studies to date have explored functioning of the HPA axis via salivary cortisol as a predictor of response to exposure therapy for anxiety disorders, with none in social anxiety disorder. As of 2017, only six studies had explored whether individual differences in tonic or resting cortisol predict treatment responses for individuals with any anxiety disorder (Fischer and Cleare, 2017). When subjected to meta-analysis, this small number of studies did not support a reliable, aggregated association between tonic cortisol at pre-treatment or during treatment sessions and symptom reductions (Fischer and Cleare, 2017). Notably, the studies included in this meta-analysis were heterogeneous in both the clinical sample and the psychological treatment administered, and only one study even included participants with social anxiety disorder (Dierckx et al., 2012). In their pediatric sample, Dierckx and colleagues (2012) found that treatment responders and non-responders did not differ in diurnal cortisol indices prior to treatment. However, non-response was associated with an increase in total diurnal cortisol (AUCg) and a decrease in the cortisol awakening response from baseline to the 1-year follow-up assessment (Dierckx et al., 2012). Further, only four of the six studies used some form of exposure therapy as their intervention (Gaab et al., 2005; Lass-Hennemann & Michael, 2014; Meuret et al., 2015; Siegmund et al., 2011). Since this meta-analysis was published, Rauch and colleagues (2017) conducted a randomized-controlled trial (RCT) testing the effectiveness of prolonged exposure compared with present-centered therapy in 30 veterans with post-traumatic stress disorder. Using salivary cortisol collected three times across three treatment sessions, patients with a low response to treatment exhibited increases in cortisol reactivity across sessions compared with treatment responders (Rauch et al., 2017). Conceivably, individuals with social anxiety disorder with elevated cortisol or cortisol reactivity may also experience less symptom improvement following exposure therapy. Indeed, activation of the HPA axis via acute stress has been shown to impair extinction retrieval (Raio et al., 2014).
The purpose of this study was to examine whether glucocorticoid concentrations during exposure treatment, as measured by salivary cortisol, differentially predicted symptom reductions following exposure therapy among individuals with social anxiety disorder. We did this in a secondary analysis of salivary cortisol collected during a RCT for social anxiety disorder where participants were randomized to either receive exposure therapy alone or exposure therapy augmented by scopolamine. We hypothesized that individuals with elevated cortisol throughout their exposure treatments would report less symptom improvement relative to other participants.
There are complexities inherent to the interpretation of glucocorticoid concentrations during exposure therapy. Extinction learning is a purported key mechanism of exposure therapy, involving updating of excitatory conditioned stimulus (CS) – unconditioned stimulus (US) associations in memory (e.g., neutral facial expression – rejection) through the development of inhibitory CS-no US associations (neutral facial expression – no rejection), leading to extinction of the conditional fear response (Craske et al., 2019, 2014). The experience of fear involves a complex neurophysiological system that typically includes activation of the autonomic nervous system and the HPA axis (Charney & Deutch, 1996; Phelps & LeDoux, 2005; Shin & Liberzon, 2010). Activation of both of these systems increases circulation of catecholamines (e.g., norepinephrine) and glucocorticoids (e.g., cortisol in humans), respectively, which influence learning and memory (Schwabe et al., 2012). Indeed, both exogenous and endogenous glucocorticoids have been linked to reduced subjective fear during single phobic exposures (Soravia et al., 2006), norepinephrine and glucocorticoids have both been shown to enhance extinction at multiple levels of analysis (Singewald et al., 2015), and the effect of glucocorticoids on memory may depend upon the presence of norepinephrine (Roozendaal et al., 2006). Thus, the role of individual differences in endogenous cortisol concentrations during exposure therapy may be confounded by individual differences in autonomic nervous system activation occurring simultaneously.
Disentangling HPA from autonomic processes during exposure therapy can be accomplished pharmacologically. The drug scopolamine results in decreased autonomic nervous system activation (Liem-Moolenaar et al., 2011). This occurs because scopolamine antagonizes the excitatory effect of acetylcholine on norepinephrine neurons in the locus coeruleus (Engberg & Svensson, 1980). Data from the present RCT has shown that augmentation of exposure therapy with scopolamine causes reduced skin conductance (an index of sympathetic nervous system activation) during exposure sessions (Craske et al., 2019). Indeed, scopolamine has been proposed as a promising adjunct to exposure therapy because it can impede context-based learning (Luyten et al., 2017; Zelikowsky et al., 2013), thus reducing contextually-based return of fear and therefore relapse (Craske et al., 2019). Specifically, scopolamine blocks the effects of acetylcholine on muscarinic receptors within the hippocampus, which may impede contextual encoding of extinction learning (Zelikowsky et al., 2013), as well as inhibitory feedback of the HPA axis (Bhatnagar et al., 1997; Smythe et al., 1998). Comparing the association between in-session cortisol concentrations and symptom improvement when treatment was conducted with versus without scopolamine may elucidate the unique role of neuroendocrine signaling in exposure therapy through fear extinction. Thus, we also explored whether augmentation of exposure therapy with scopolamine would moderate the association between endogenous cortisol and symptom improvement.
2.0. Methods
2.1. Participants
Participants in this study were 60 individuals (58.3% female) with social anxiety disorder recruited for a clinical trial testing the efficacy of scopolamine-augmentation of exposure therapy. In order to be eligible for the trial, participants needed clinically severe symptoms (represented by diagnosis with a clinical severity rating > 3 on 0–8 point scale) and a score > 6 on a 0–8 scale of self-reported fear of public speaking. Participants were excluded from the study if they had bipolar disorder, symptoms of psychosis, currently smoked, had any medical conditions contraindicated by scopolamine, or tested positive for opiate or THC use in a urine drug screen.
2.2.1. Procedures
All study procedures were approved by the UCLA Institutional Review Board. This study was a double-blind, randomized controlled trial designed to assess the efficacy of scopolamine-augmentation of exposure therapy for social anxiety disorder (NCT01900301). Participants were recruited via public announcements (e.g., flyers at local colleges and Universities) and referrals to the UCLA Anxiety and Depression Research Center. All participants provided written, informed consent, and were assessed for social anxiety disorder via the Anxiety Disorders Interview Schedule for DSM-5 (ADIS-5) (Brown & Barlow, 2014). Eligible participants were then randomized to receive either .5 mg of scopolamine (n = 19), .6 mg of scopolamine (n = 20), or placebo (n = 21) intranasally at the start of their exposure sessions. Exposure therapy was administered via virtual reality, which demonstrates comparable effectiveness for social anxiety disorder relative to in-vivo exposure (Carl et al., 2019; Opriş et al., 2012; Powers & Emmelkamp, 2008). Therapy included 7 sessions, twice per week. During each exposure session, participants completed seven virtual reality speech tasks, each lasting 1-minute. Participants returned to the laboratory for an extinction test, as well as a test of context renewal, within 1-week of their final session. Participants also returned for a 1-month follow-up assessment which included symptom measures and a long-term extinction test. For more details on the sample and study procedures see Craske et al. (2019).
2.2.2. Measures
2.2.2.1. Endogenous cortisol
Cortisol concentrations were measured in saliva four times across exposure sessions 1, 4, 7, and during the post-treatment extinction assessment. Participants provided saliva samples 4 times throughout each session using absorbent cotton placed between their gum and cheek for 2 minutes and collected into a sterile salivette. Each sample was collected 30–45 minutes apart: immediately after initial arrival questionnaires and VR set-up, +30 after scopolamine/placebo administration/immediately before the 1st exposure trial, immediately after the 7th exposure trial, and +30 minutes after the end of their 7th exposure trial. Saliva samples during the post-treatment extinction test occurred upon arrival, 20 minutes after the first VR speech, 20 minutes after the second VR speech, and a final sample 20 minutes later. The first and second VR speech were extinction retest and context renewal counterbalanced across participants. Salivettes were stored at −20 degrees Celsius until assay for batch processing. Saliva samples were assayed for cortisol concentrations at the UCI Institute for Interdisciplinary Salivary Bioscience (https://iisbr.uci.edu/). Cortisol was assayed via ELISA using a commercially available enzyme immunoassay kit (Salimetrics, Inc.). The range of detection for the assay was 0.007 – 3.00 μg/dl. A subsample (15%) was assayed in duplicate and the inter-assay CV was 3.17%.
2.2.2.2. Social anxiety symptoms
Participants reported symptoms of social anxiety using the Liebowitz Social Anxiety Scale (LSAS) (Liebowitz, 1987) at pre-treatment, post-treatment, and at 1-month follow-up. The LSAS includes 24 situations such as “Going to a party” and “Speaking up at a meeting.” Participants were asked to indicate the degree to which they fear each situation according to a 4-point Likert scale where 0 = none and 3 = severe. Participants also responded with the frequency with which they avoid each situation where 0 = never and 3 = usually. Responses to each item were summed to create a total fear subscore and a total avoidance subscore which were used as our primary outcomes. Total fear and avoidance subscores can range from 0 to 72. Total scores greater than 30 were used to differentiate individuals with social anxiety disorder (Mennin et al., 2002; Rytwinski et al., 2009). The LSAS and its subscales demonstrate excellent internal reliability (Heimberg et al., 1999), and the internal reliably of the total score, total fear, and total avoidance scores were all excellent in this sample as well, αs ≥ .93.
2.3. Data analysis
All continuous variables were assessed for normality and heteroscedasticity and, with the exception of cortisol concentrations, were found to be sufficiently normally distributed to subject to multivariate analyses. Of a total 960 possible samples, 869 (90.5%) were collected and sent for assay. Of the 91 missing samples, 40 (44.0%) were missing because 10 participants missed their 7th session, 16 (17.6%) were missing due to protocol errors, and the remaining 35 (38.5%) were missing for other reasons. Among the assayed samples, 45 (5.2%) were flagged for quality control (e.g., low volume, discoloration, contamination) but did not result in any notably unusual values or influence our analyses, and 2 samples returned a value below the limit of detection for the assay and replaced with half of the detection limit, or 0.0035 μg/dl. Raw salivary cortisol concentrations were skewed and highly kurtotic, MCortisol = 0.17, SDCortisol = 0.32, skewness = 11.69, kurtosis = 175.69. Extreme values (n = 9) were winsorized to 3 SDs from the mean which improved the distribution of the variable but did not bring kurtosis within an acceptable range, MCortisol = 0.16, SDCortisol = 0.15, skewness = 3.30, kurtosis = 14.55. Cortisol concentrations were then transformed using the natural log (ln) transformation, MCortisol = 0.14, SDCortisol = 0.11, skewness = 2.50, kurtosis = 8.56. Endogenous in-session cortisol was computed by averaging all cortisol concentrations collected within a therapy session.
There were no significant differences between individuals in the scopolamine-augmentation and placebo conditions on any study variables or covariates, all ps > .35. There were no significant differences between male and female participants in social anxiety symptoms throughout the study, all ps > .24, nor were there sex differences in the number of saliva samples that contributed to the endogenous cortisol estimations, p = .37. Male and female participants did not differ in endogenous cortisol at session 1, p = .90, or session 4, p = .12, however female participants had non-significantly higher average endogenous cortisol during their final exposure therapy session (session 7), F(1,47) = 2.88, p = .097, and at the post-treatment extinction visit, F(1,57) = 3.82, p = .055. Therapy sessions took place throughout the business day and therefore saliva samples occurred between 8:04 am and 5:14 pm, MCollection Time = 12:24 pm (SDCollection Time=2:11). Higher cortisol concentrations were observed at sessions occurring earlier in the day, r = −.35, p < .001. As a result of these apparent and potential differences, sex, BMI, the number of saliva samples a participant provided during the trial, and the average collection time within each session were included as key covariates.
All hypotheses were tested separately for Total Avoidance and Total Fear subscales on the LSAS. We used linear mixed models with an unstructured covariance matrix, maximum likelihood estimation, and random effects for the intercept (symptoms at baseline) and slope (change in symptoms over time). These models consistent of two-levels, individual and session. Sessions (1, 4, 7, and post-treatment/1-month follow-up) were nested within participants, enabling symptoms to be modeled as a function of time (session), endogenous cortisol (session), treatment condition (scopolamine vs placebo; person), their interaction (cross-level), and our covariates. Scopolamine treatment conditions .5 mg and .6 mg were combined into one scopolamine condition because we had no hypotheses specific to dose. Coefficients with a p < .05 were considered significant.
3.0. Results
Table 1 displays descriptive statistics for all key study variables and bivariate correlations between them. Cortisol concentrations declined across sessions 1, 4, and post-treatment, session 1 F(3, 48) = 2.64, p = .06 and session 4 F(3, 59) = 6.88, p < .001 and post-treatment F(3, 53) = 3.72, p = .017, but did not change across session 7, F(3, 48) = 1.53, p = .22. On average, participants demonstrated a 51.6% increase in cortisol from arrival to the end of their 7th exposure trial in session 1, a 12.5% increase in session 4, a 5.5% decrease in session 7, and 22.3% increase at post-treatment. These within session increases in cortisol were driven by a minority of participants, such that within any given session ≤ 33.1% of participants exhibited a 20% increase in cortisol or greater from pre- to post-exposure trials. Importantly, individuals in the scopolamine-augmentation condition were more likely to exhibit at least a 20% increase in cortisol within the session, Session 1 d = .29, p = .08, Session 4 d = .32, p = .007, and Session 7 d = .22, p = .05. Controlling for the timing of the session, endogenous in-session cortisol did not change significantly across the trial, exposure only b = .016 (SE = .018), p = .37 and exposure with scopolamine-augmentation b = .013 (SE = .021), p = .55. See Figure 1 for raw cortisol concentrations across each exposure session.
Table 1.
Descriptive statistics and zero-order correlations between all study variables
| M (SD) | Correlations | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1. | 2. | 3. | 4. | 5. | 6. | 7. | 8. | 9. | 10. | 11. | ||
| In-session cortisol | ||||||||||||
| 1. Session 1 | 0.16 (0.17) | 1.00 | ||||||||||
| 2. Session 4 | 0.13 (0.08) | .45** | 1.00 | |||||||||
| 3. Session 7 | 0.22 (0.50) | .34* | .40** | 1.00 | ||||||||
| 4. Post-treatment extinction | 0.17 (0.19) | .44** | .44** | .56*** | 1.00 | |||||||
| Social anxiety symptoms (LSAS) | ||||||||||||
| Total Fear | ||||||||||||
| 5. Pre-treatment | 40.66 (13.69) | −.15 | −.07 | −.13 | .14 | 1.00 | ||||||
| 6. Post-treatment | 25.74 (12.00) | .09 | .14 | .29+ | .34* | .50*** | 1.00 | |||||
| 7. 1-month follow-up | 24.18 (12.41) | .28 | .18 | .41* | .19 | .40 | .86*** | 1.00 | ||||
| Total Avoidance | ||||||||||||
| 8. Pre-treatment | 39.43 (15.32) | −.07 | .07 | −.07 | .25 | .88*** | .45** | .38* | 1.00 | |||
| 9. Post-treatment | 24.28 (12.23) | .10 | .13 | .38* | .35* | .45** | .86*** | .71*** | .53*** | 1.00 | ||
| 10. 1-month follow-up | 22.30 (11.93) | .30+ | .18 | .49* | .32+ | .30 | .74*** | .87*** | .40* | .76*** | 1.00 | |
| Key covariates | ||||||||||||
| 11. BMI | 24.37 (4.82) | .12 | .13 | −.01 | .002 | .34* | .12 | .04 | .38* | .17 | .06 | 1.00 |
| 12. Number of samples contributed | 14.71 (1.47) | −.09 | −.02 | .11 | −.16 | −.02 | .09 | −.06 | −.01 | .04 | −.14 | .03 |
Note:
p < .10,
p < .05,
p < .01,
p <.001;
BMI = Body Mass Index; LSAS = Liebowitz Social Anxiety Scale. Note: Raw means are reported, however bivariate associations were conducted with winsorized and transformed variables as described in the data analysis section.
Figure 1.
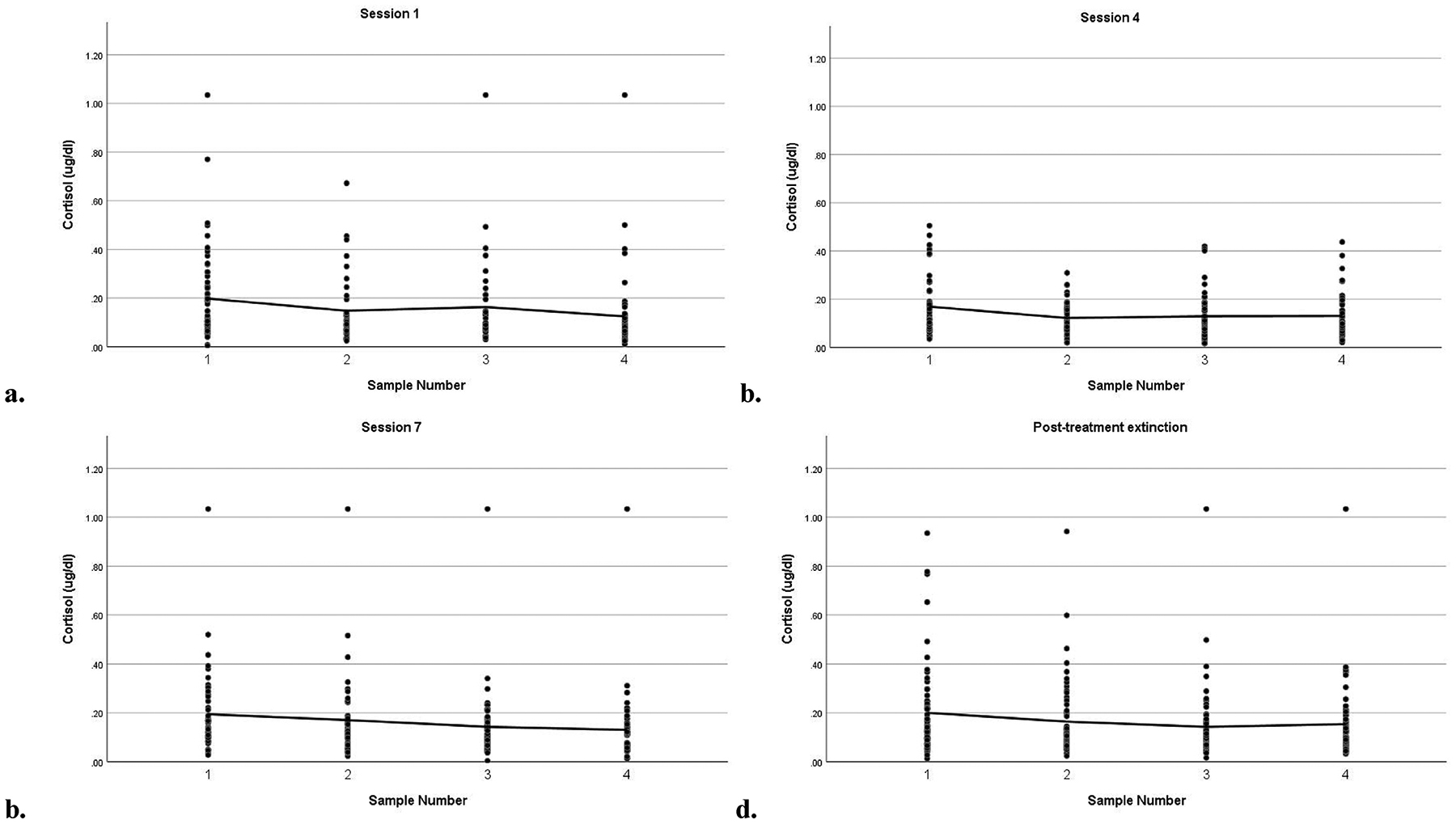
Endogenous in-session cortisol across exposure therapy sessions 1 (a), 4 (b), 7 (c), and at post-treatment (d)
Social anxiety symptoms decreased across the trial from pre- to post-treatment, Cohen’s d = 1.06, and from pre-treatment to 1-month follow-up, Cohen’s d = 1.09. Specifically, LSAS scores declined from 40.09 ± 14.18 at pre-treatment to 25.52 ± 13.21 at post-treatment and remained below the clinical cut-off (LSAS = 30) at the 1-month follow-up, 24.82 ± 13.93. In our multilevel model, time accounted for 50.9% of variance in total fear symptoms (AIC = 599.64 vs 1,220.43) and, on average, fear symptoms declined by two points on the total fear scale each session, b = −2.26, SE = .32, p < .001. Time accounted for 50.4% of variance in total avoidance symptoms (AIC = 614.05 vs 1,238.76) and, on average, avoidance symptoms declined by two points on the total avoidance scale each session, b = −2.32, SE = .33, p < .001.
Higher concentrations of cortisol during exposure sessions were associated with less symptom improvement. Individuals with below average in-session cortisol in our sample showed an average decrease in total fear symptoms of 17.33 (SD = 1.13) and an average decrease in total avoidance symptoms of 15.52 (SD=1.50) from Session 1 to post-treatment. Individuals with above average in-session cortisol in our sample showed an average decrease in total fear symptoms of 14.04 (SD = 0.8) and an average decrease in total avoidance symptoms of 13.55 (SD = 0.46). This corresponds to large effect sizes for in-session cortisol on change in fear, d = 3.27 95%CI[2.29, 4.25], and change in avoidance, d = 1.66 95%CI[0.92, 2.41]. With the current sample, the study had greater than 95% power to detect this association at 95% reliability.
When cortisol was added to the multi-level model of total fear symptoms, higher endogenous cortisol was not associated with any differences in symptoms at treatment onset, b = −2.46, SE = 2.27, p = .28, but was associated with less symptom decline over time, b = 1.38, SE = .42, p = .002. Similarly, when cortisol was added to the model of total avoidance symptoms, higher endogenous cortisol was not associated with any differences in symptoms at treatment onset, b = −1.53, SE = 2.84, p = .59, but was associated with less symptom decline over time, b = 1.40, SE = .53, p = .011. These patterns did not change when adjusting for key covariates, such as female sex, BMI, the number of samples a participant contributed to their cortisol estimations, and sample collection times. Table 2 provides model fit parameters and coefficient estimates for social anxiety symptoms across treatment and follow-up as a function of endogenous cortisol and adjusted for key covariates. Figure 2 illustrates changes in social anxiety symptoms over time as a function of endogenous in-session cortisol.
Table 2.
Coefficient estimates of social anxiety symptoms as a function of endogenous in-session cortisol during exposure therapy
| Total Fear | Total Avoidance | Performance - Fear | Performance - Avoid | |
|---|---|---|---|---|
| AIC | 602.84 | 607.23 | 505.70 | 482.21 |
| Predictor | b (SE) | b (SE) | b (SE) | b (SE) |
| Intercept | 38.43 (2.69)*** | 37.01 (2.95)*** | 20.49 (1.36)*** | 18.86 (1.46)*** |
| Session | −1.81 (0.57)** | −1.36 (0.65)* | −0.91 (0.34)** | −0.67 (0.31)* |
| Cortisol (ln) | −3.24 (3.22) | −2.57 (2.82) | −1.62 (1.28) | −0.70 (1.30) |
| Session × cortisol (ln) | 1.54 (0.54)** | 1.68 (0.61)** | 0.86 (0.31)** | 0.87 (0.29)** |
| Covariates | ||||
| BMI | 0.81 (0.42)+ | 0.94 (0.46)* | 0.50 (0.21)* | 0.64 (0.23)** |
| Session × BMI | −0.04 (0.07) | −0.04 (.08) | −0.03 (0.04) | −0.05 (0.03) |
| Female | 2.91 (4.52) | 5.06 (5.03) | −0.08 (2.31) | 1.28 (2.45) |
| Session × female | −0.61 (0.78) | −1.16 (0.91) | 0.03 (0.45) | −0.32 (0.42) |
| Number of samples | 0.77 (0.81) | 0.53 (0.89) | 0.23 (0.41) | 0.13 (0.44) |
| Session × number of Samples | −0.16 (0.23) | −0.31 (0.25) | −0.13 (0.14) | −0.22 (0.12)+ |
| Collection time | 1.09E-5 (2.89E-4) | −0.0001 (0.0003) | 3.06E-5 (0.0002) | −3.36E-5 (0.0002) |
| Session × collection time | 1.01E-5 (5.05E-5) | −3.49E-5(5.89E-5) | −1.02 (2.87E-5) | −2.33 (2.71E-5) |
Note:
p < .10,
p < .05,
p < .01,
p <.001;
BMI = Body mass index
Figure 2.
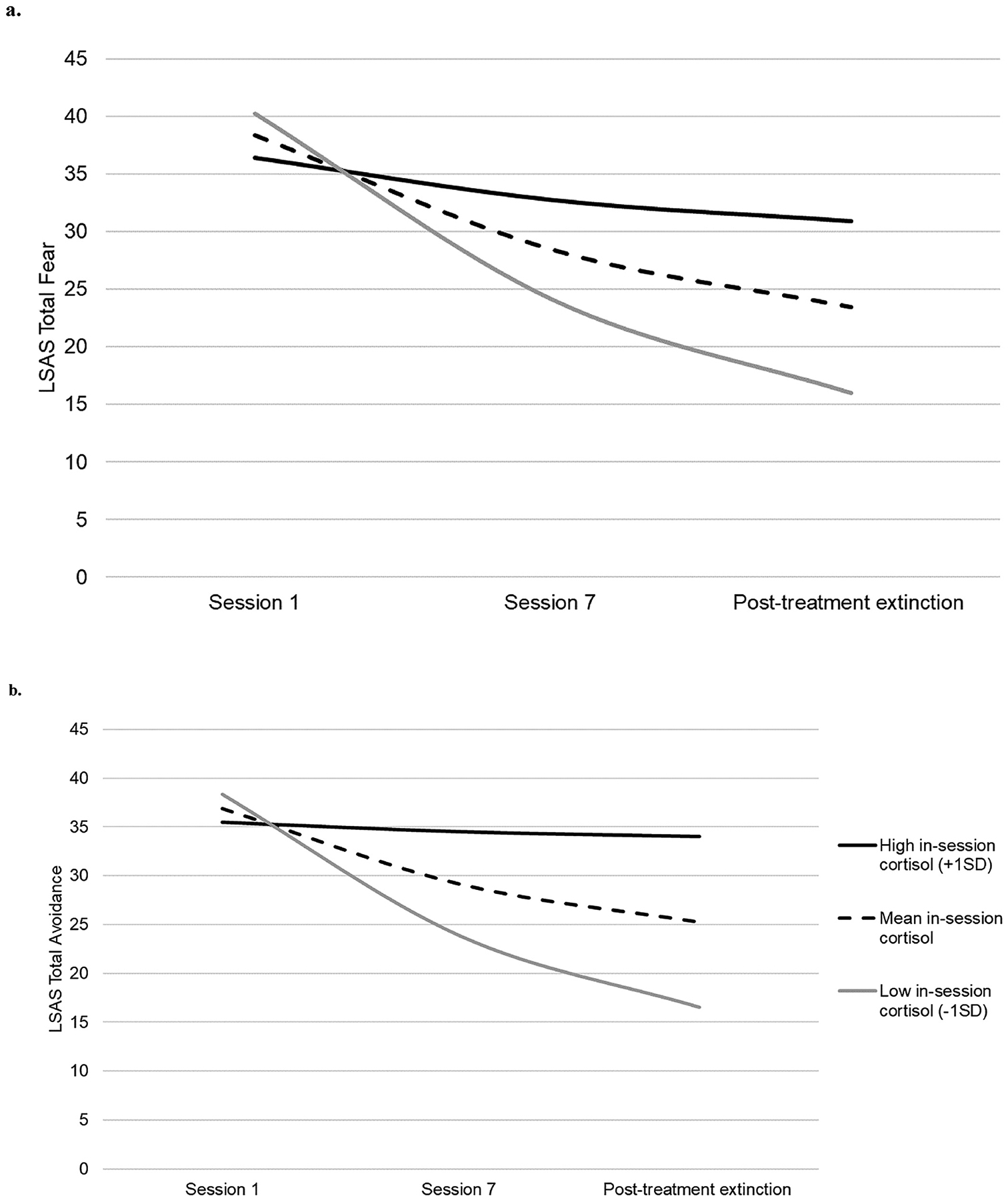
Estimated social anxiety symptoms of a) fear and b) avoidance during and after exposure therapy as a function of endogenous in-session cortisol during treatment sessions
The association between endogenous cortisol during exposure sessions and symptom changes across the trial was not moderated by treatment condition. For total fear symptoms, there was no significant interaction between endogenous in-session cortisol and scopolamine condition at treatment onset, b = 7.06, SE = 4.63, p = .14, nor on symptom change over time, b = −1.46, SE = 1.13, p = .20. The results were similar for total avoidance symptoms, such that there was no significant interaction between endogenous in-session cortisol and scopolamine-augmentation of exposure treatment at treatment onset, b = 7.90, SE = 4.78, p = .10, nor on symptom change over time, b = −1.06, SE = 1.03, p = .31.
4.0. Discussion
To our knowledge, this was the first study to examine endogenous cortisol as a predictor of symptom improvement among individuals undergoing exposure therapy for social anxiety disorder. Elevated endogenous cortisol during exposure sessions was associated with less symptom improvement relative to participants with average or low in-session cortisol. Further, the association between cortisol during treatment and symptom improvement was independent of scopolamine-augmentation. Endogenous in-session cortisol during exposure therapy may be a useful biomarker for identifying individuals for whom exposure therapy is likely to be effective.
Participants with elevated endogenous in-session cortisol exhibited less symptom improvement at post-treatment and 1-month follow-up. This was partially consistent with a previous trial of prolonged exposure for veterans with posttraumatic stress disorder for whom a pattern of increasing in-session cortisol reactivity across treatment predicted treatment non-response (Rauch et al., 2017), as well as a study of youth for whom elevated diurnal cortisol was linked to persistence of anxiety symptoms following treatment (Dierckx et al., 2012). There are several ways in which elevated in-session cortisol may interfere with exposure therapy. For example, individuals with a propensity for elevated endogenous cortisol may form more intractable fear-related memories. Indeed, youth with an elevated cortisol awakening response are at greater risk for developing social anxiety disorder (Adam et al., 2014). Fear memories formed under high concentrations of glucocorticoids are also resistant to subsequent extinction (Chakraborty & Chattarji, 2019) possibly due to alterations in synaptic plasticity in the amygdala and hippocampus (Goldwater et al., 2009; Vyas et al., 2002). Elevated endogenous cortisol during exposure sessions may also interfere with extinction learning. Increases in glucocorticoids (either through exogenous administration or as a result of acute stress) can impair the retrieval of emotional memories (de Quervain et al., 2019). Extinction learning depends on a discrepancy between the expected and actual outcome (Rescorla & Wagner, 1972). In social anxiety disorder, extinction learning results from a discrepancy between the expectation of rejection when engaging in social interaction and the non-occurrence of the aversive outcome. Glucocorticoid-impaired retrieval of emotional memories may lead to less expectation of the unconditioned stimulus (rejection) and impair the acquisition of extinction learning. Elevated cortisol may also impair extinction retrieval once acquired. Indeed, acute stress-related activation of the HPA axis is associated with impaired retrieval of extinction (fear recovery) (Raio et al., 2014). That being said, future studies are needed to address the possibility that HPA axis activity during exposure sessions was increasing as a result of poor responses to treatment. The timing of our symptom and cortisol measures did not enable us to test the potential bidirectional nature of these observations.
The present findings add to a small and inconsistent literature linking neuroendocrine functioning to treatment outcomes. Much of this equivocal evidence can be clarified by carefully examining the neuroendocrine index used in each study. Functioning of the HPA axis can be indexed in a number of ways, such as through the cortisol awakening response (Clow et al., 2010; Wüst et al., 2000), diurnal decline in cortisol across the day (Adam et al., 2017), acute reactivity to an exogenous challenge (Dickerson and Kemeny, 2004), and trait cortisol concentrations (Doane et al., 2015). Each of these indices represents unique underlying neurophysiology and has been linked to differentiated psychosocial experiences and health outcomes (cf. Kuhlman et al., 2016, 2015; Vrshek-Schallhorn et al., 2013). Specifically, the cortisol awakening response has been linked to the capacity for the HPA axis to respond to the environment, in part because this index increases with greater daily demands and stressors (Adam et al., 2006; Clow et al., 2010; Wüst et al., 2000). The capacity for the HPA axis to respond to the environment has been linked to better treatment outcomes for exposure therapy. For example, individuals undergoing exposure therapy for panic disorder exhibit higher cortisol awakening responses on exposure days compared to mornings on days without exposure, and larger cortisol awakening responses on exposure days predicted better responses to treatment (Meuret et al., 2015). Further, exposure therapy for spider phobia was found to be more effective in the morning than the evening because endogenous cortisol concentrations are higher earlier in the day (Lass-Hennemann & Michael, 2014). In both studies, the indices of higher cortisol concentrations may indicate the capacity for the HPA axis to respond to the environment, rather than persistently elevated concentrations.
In contrast, the measure of cortisol in this study is unlikely to represent reactivity of the HPA axis to exposures. While there was heterogeneity in HPA axis function during exposure sessions, less than one third of our sample exhibited at least a 20% increase in cortisol in any given exposure session, and cortisol values declined on average throughout the sessions, if they showed any change at all. Further, subjective reports of fear via SUDS ratings were not related to in-session cortisol concentrations, and autonomic inhibition via scopolamine-augmentation did not moderate our findings. This last observation is particularly important given that in-session HPA reactivity disproportionately occurred in the scopolamine-augmentation condition. Scopolamine is known to impair inhibitory feedback of the HPA axis (Bhatnagar et al., 1997; Smythe et al., 1998), thus leading to higher glucocorticoid exposure throughout the session independent of acute activation. Thus, for the vast majority of our participants, endogenous in-session cortisol likely represents tonic HPA axis regulation.
At first glance, our finding of an inverse association between cortisol and symptom improvement is contrary to a dominant theory in the field. Indeed, there is strong experimental evidence that glucocorticoids actually enhance extinction learning (Bentz et al., 2010; Singewald et al., 2015), thus leading many to hypothesize that elevated cortisol during exposure therapy may predict better treatment responses. However, acute versus sustained elevations in glucocorticoids likely exert different effects on cognition and its underlying neurocircuitry (Hermans et al., 2014). Specifically, acute increases in catecholamines and glucocorticoids exert short-term and non-genomic influences on cognitive systems via the salience network, while sustained elevations in glucocorticoids lead to genomic effects, particularly within the executive control system, and predominantly occur after the catecholamine response has terminated (Hermans et al., 2014). Taken together, more attention to the underlying neurobiology represented by each HPA axis index is needed to better characterize neuroendocrine predictors of treatment response to exposure therapy. Indices that represent the capacity for the HPA axis to respond to the environment may be more likely to predict better responses to exposure therapy while cortisol indices that represent chronically elevated cortisol with little variation over time may predict poorer responses. Post hoc analyses showed that individuals in our sample who exhibited at least a 20% increase in cortisol during any of our sessions did not differ in their change in symptoms from pre- to post-treatment. However, given the low rate of observable cortisol reactivity to exposure sessions in our sample and other study design characteristics (e.g., lack of a no treatment control), this question warrants further investigation in a study designed for this specific purpose.
The results of this study should be considered in the context of several limitations. First, the observations in this study cannot yet be used for precision medicine. In order for an index of HPA axis functioning to have utility in precision medicine, it would need to be assessed prior to treatment selection and delivery. However, in this study persistent elevations in cortisol across treatment sessions predicted less symptom improvement. Ongoing efforts to characterize the underlying neurobiology of different cortisol indices (e.g., Abelson et al., 2019; Deuschle et al., 1998b, 1998a), such as elevated in-session cortisol, will be helpful in identifying pre-treatment assessments that will serve the goals of precision medicine. Second, our results can only conclude that individuals with high endogenous in-session cortisol benefitted less from exposure therapy relative to individuals with average or below average in-session cortisol. The present trial was designed to test the effectiveness of scopolamine-augmentation for exposure therapy and therefore all participants received treatment. Assessment of both symptoms and endogenous cortisol in a no-treatment control group would help to clarify whether individuals with high endogenous in-session cortisol benefited from exposure relative to those who did not receive any treatment. Similarly, future studies of this nature would benefit from having endogenous cortisol concentrations from participants outside of the exposure therapy context. Third, our participants varied in the number of saliva samples provided during their exposure treatment. As a result, AUC could only have been computed for a small subset of participants (n = 27). We chose to maximize the data available for these analyses by using average cortisol concentrations during each session as the individual neuroendocrine signaling index. All analyses were adjusted for the number of samples each participant contributed to the study, however estimates of endogenous in-session cortisol for individuals with missing data are still likely to be less reliable than those computed from complete data.
There are several highly effective treatments for social anxiety disorder. Biomarkers such as salivary cortisol may be useful, cost-effective, and informative measures of individual differences in complex neurophysiology that can be used to optimize mental health services. Functioning of the HPA axis has long been implicated in the pathophysiology of anxiety disorders (Charney & Deutch, 1996; Owens & Nemeroff, 1993; Pine, 1999; Shin & Liberzon, 2010), including social anxiety disorder (Dieleman et al., 2015), and has also been identified as a mechanism through which exposure therapy is effective (Bentz et al., 2010). This study adds to a growing field of salivary predictors of treatment responses. Elevated in-session cortisol concentrations during exposure therapy predicted poorer symptom improvement in individuals with social anxiety disorder. The results of this study could be extended by identifying treatments for social anxiety disorder which disproportionately benefit individuals with elevated in-session cortisol concentrations.
Highlights.
Exposure therapy was less effective for individuals with high in-session cortisol
Inhibition of autonomic activation during exposure sessions did not alter the link between cortisol and symptom change
A more sensitive, pre-treatment measure of individual differences in glucocorticoid regulation is needed
Acknowledgements and Role of the Funding Source
Data collection for this study was made possible by the NIMH (R34MH101359; PI: Craske). The authors thank research coordinators, including Natalie Arbid, Abigail Branch, and Richard Kim, as well as the participants in the study. The composition of this manuscript was made possible by the National Institute of Mental Health through a career development award (K08MH112773; PI: Kuhlman). The funding organizations for this study were not involved in writing of the manuscript or the decision to submit the article for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors report no conflicts of interest.
References
- Abelson JL, Kirschbaum C, Herman J, 2019. Cortisol measures in saliva and hair: What is their biological meaning and scientific utility? Psychoneuroendocrinology. [Google Scholar]
- Acarturk C, Cuijpers P, Straten A. van, Graaf R. de, 2009. Psychological treatment of social anxiety disorder: a meta-analysis. Psychol. Med 39, 241–254. 10.1017/S0033291708003590 [DOI] [PubMed] [Google Scholar]
- Adam EK, Hawkley LC, Kudielka BM, Cacioppo JT, 2006. Day-to-day dynamics of experience–cortisol associations in a population-based sample of older adults. Proc. Natl. Acad. Sci 103, 17058–17063. 10.1073/pnas.0605053103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam EK, Quinn ME, Tavernier R, McQuillan MT, Dahlke KA, Gilbert KE, 2017. Diurnal cortisol slopes and mental and physical health outcomes: A systematic review and meta-analysis. Psychoneuroendocrinology 83, 25–41. 10.1016/j.psyneuen.2017.05.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam EK, Vrshek-Schallhorn S, Kendall AD, Mineka S, Zinbarg RE, Craske MG, 2014. Prospective associations between the cortisol awakening response and first onsets of anxiety disorders over a six-year follow-up – 2013 Curt Richter Award Winner. Psychoneuroendocrinology 44, 47–59. 10.1016/j.psyneuen.2014.02.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Psychiatric Association, 2013. Diagnostic and statistical manual of mental disorders (DSM-5®). American Psychiatric Pub. [Google Scholar]
- Bandelow B, Michaelis S, 2015. Epidemiology of anxiety disorders in the 21st century. Dialogues Clin. Neurosci 17, 327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentz D, Michael T, de Quervain DJ, Wilhelm FH, 2010. Enhancing exposure therapy for anxiety disorders with glucocorticoids: from basic mechanisms of emotional learning to clinical applications. J. Anxiety Disord 24, 223. [DOI] [PubMed] [Google Scholar]
- Bhatnagar S, Costall B, Smythe JW, 1997. Hippocampal cholinergic blockade enhances hypothalamic-pituitary-adrenal responses to stress. Brain Res. 766, 244. [DOI] [PubMed] [Google Scholar]
- Brown TA, Barlow DH, 2014. Anxiety and related disorders interview schedule for DSM-5. N (ADIS-5)—adult and lifetime version New York, NY: Oxford University Press. [Google Scholar]
- Carl E, Stein AT, Levihn-Coon A, Pogue JR, Rothbaum B, Emmelkamp P, Asmundson GJG, Carlbring P, Powers MB, 2019. Virtual reality exposure therapy for anxiety and related disorders: A meta-analysis of randomized controlled trials. J. Anxiety Disord., Virtual reality applications for the anxiety disorders 61, 27–36. 10.1016/j.janxdis.2018.08.003 [DOI] [PubMed] [Google Scholar]
- Chakraborty P, Chattarji S, 2019. Timing is everything: differential effects of chronic stress on fear extinction. Psychopharmacology (Berl.) 236, 73. [DOI] [PubMed] [Google Scholar]
- Charney DS, Deutch A, 1996. A Functional Neuroanatomy of Anxiety and Fear: Implications for the Pathophysiology and Treatment of Anxiety Disorders. Crit. Rev. Neurobiol 10 10.1615/CritRevNeurobiol.v10.i3-4.70 [DOI] [PubMed] [Google Scholar]
- Clow A, Hucklebridge F, Stalder T, Evans P, Thorn L, 2010. The cortisol awakening response: More than a measure of HPA axis function. Neurosci. Biobehav. Rev 35, 97–103. 10.1016/j.neubiorev.2009.12.011 [DOI] [PubMed] [Google Scholar]
- Craske MG, Fanselow M, Treanor M, Bystritksy A, 2019. Cholinergic Modulation of Exposure Disrupts Hippocampal Processes and Augments Extinction: Proof-of-Concept Study With Social Anxiety Disorder. Biol. Psychiatry [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craske MG, Treanor M, Conway CC, Zbozinek T, Vervliet B, 2014. Maximizing exposure therapy: An inhibitory learning approach. Behav. Res. Ther 58, 10–23. 10.1016/j.brat.2014.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Quervain D, Wolf OT, Roozendaal B, 2019. Glucocorticoid-induced enhancement of extinction-from animal models to clinical trials. Psychopharmacology (Berl.) 236, 183–199. 10.1007/s00213-018-5116-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuschle M, Schweiger U, Gotthardt U, Weber B, Körner A, Schmider J, Standhardt H, Lammers C-H, Krumm B, Heuser I, 1998a. The combined dexamethasone/corticotropin-releasing hormone stimulation test is more closely associated with features of diurnal activity of the hypothalamo—pituitary—adrenocortical system than the dexamethasone suppression test. Biol. Psychiatry 43, 762–766. 10.1016/S0006-3223(97)00276-X [DOI] [PubMed] [Google Scholar]
- Deuschle M, Weber B, Colla M, Müller M, Kniest A, Heuser I, 1998b. Mineralocorticoid Receptor also Modulates Basal Activity of Hypothalamus-Pituitary-Adrenocortical System in Humans. Neuroendocrinology 68, 355–360. 10.1159/000054384 [DOI] [PubMed] [Google Scholar]
- Dickerson SS, Kemeny ME, 2004. Acute stressors and cortisol responses: A theoretical integration and synthesis of laboratory research. Psychol. Bull 130, 355–391. 10.1037/0033-2909.130.3.355 [DOI] [PubMed] [Google Scholar]
- Dieleman GC, Huizink AC, Tulen JHM, Utens EMWJ, Creemers HE, van der Ende J, Verhulst FC, 2015. Alterations in HPA-axis and autonomic nervous system functioning in childhood anxiety disorders point to a chronic stress hypothesis. Psychoneuroendocrinology, This issue includes a Special Section on Biomarkers in the Military - New Findings from Prospective Studies 51, 135–150. 10.1016/j.psyneuen.2014.09.002 [DOI] [PubMed] [Google Scholar]
- Dierckx B, Dieleman G, Tulen JH, Treffers PD, Utens EM, Verhulst FC, Tiemeier H, 2012. Persistence of anxiety disorders and concomitant changes in cortisol. J. Anxiety Disord 26, 635. [DOI] [PubMed] [Google Scholar]
- Doane LD, Chen FR, Sladek MR, Van Lenten SA, Granger DA, 2015. Latent trait cortisol (LTC) levels: Reliability, validity, and stability. Psychoneuroendocrinology 55, 21–35. 10.1016/j.psyneuen.2015.01.017 [DOI] [PubMed] [Google Scholar]
- Engberg G, Svensson TH, 1980. Pharmacological analysis of a cholinergic receptor mediated regulation of brain norepinephrine neurons. J. Neural Transm 49, 137–150. 10.1007/BF01245220 [DOI] [PubMed] [Google Scholar]
- Feske U, Chambless DL, 1995. Cognitive behavioral versus exposure only treatment for social phobia: A meta-analysis. Behav. Ther 26, 695–720. 10.1016/S0005-7894(05)80040-1 [DOI] [Google Scholar]
- Fischer S, Cleare AJ, 2017. Cortisol as a predictor of psychological therapy response in anxiety disorders-Systematic review and meta-analysis. J. Anxiety Disord 47, 60–68. 10.1016/j.janxdis.2017.02.007 [DOI] [PubMed] [Google Scholar]
- Gaab J, Jucker P, Staub F, Ehlert U, 2005. Mind over matter: Psychobiological effects of exposure therapy in arachnophobia. Z. Klin. Psychol. Psychother 34, 121–132. 10.1026/1616-3443.34.2.121 [DOI] [Google Scholar]
- Goldwater DS, Pavlides C, Hunter RG, Bloss EB, Hof PR, McEwen BS, Morrison JH, 2009. Structural and functional alterations to rat medial prefrontal cortex following chronic restraint stress and recovery. Neuroscience 164, 798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heimberg RG, Horner KJ, Juster HR, Safren SA, Brown EJ, Schneier FR, Liebowitz MR, 1999. Psychometric properties of the Liebowitz Social Anxiety Scale. Psychol. Med 29, 199. [DOI] [PubMed] [Google Scholar]
- Hermans EJ, Henckens MJAG, Joëls M, Fernández G, 2014. Dynamic adaptation of large-scale brain networks in response to acute stressors. Trends Neurosci. 37, 304–314. 10.1016/j.tins.2014.03.006 [DOI] [PubMed] [Google Scholar]
- Insel TR, 2014. The NIMH Research Domain Criteria (RDoC) Project: Precision Medicine for Psychiatry. Am. J. Psychiatry 171, 395–397. 10.1176/appi.ajp.2014.14020138 [DOI] [PubMed] [Google Scholar]
- Kessler RC, Aguilar-Gaxiola S, Alonso J, Chatterji S, Lee S, Ormel J, Üstün TB, Wang PS, 2009. The global burden of mental disorders: An update from the WHO World Mental Health (WMH) Surveys*. Epidemiol. Psychiatr. Sci 18, 23–33. 10.1017/S1121189X00001421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Petukhova M, Sampson NA, Zaslavsky AM, Wittchen H-U, 2012. Twelve-month and lifetime prevalence and lifetime morbid risk of anxiety and mood disorders in the United States. Int. J. Methods Psychiatr. Res 21, 169–184. 10.1002/mpr.1359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman KR, Geiss EG, Vargas I, Lopez-Duran NL, 2015. Differential associations between childhood trauma subtypes and adolescent HPA-axis functioning. Psychoneuroendocrinology 54, 103–114. 10.1016/j.psyneuen.2015.01.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhlman KR, Mousavi Z, in press Applications of Salivary Bioscience to Precision Medicine, in: Salivary Bioscience: Foundations of Interdisciplinary Saliva Research and Applications. Springer Nature. [Google Scholar]
- Kuhlman KR, Repetti RL, Reynolds BM, Robles TF, 2016. Change in parent-child conflict and the HPA-axis: Where should we be looking and for how long? Psychoneuroendocrinology 68, 74–81. 10.1016/j.psyneuen.2016.02.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lass-Hennemann J, Michael T, 2014. Endogenous cortisol levels influence exposure therapy in spider phobia. Behav. Res. Ther 60, 39–45. 10.1016/j.brat.2014.06.009 [DOI] [PubMed] [Google Scholar]
- Liebowitz MR, 1987. Social phobia. Mod. Probl. Pharmacopsychiatry 22, 141–173. [DOI] [PubMed] [Google Scholar]
- Liem-Moolenaar M, de Boer P, Timmers M, Schoemaker RC, van Hasselt JGC, Schmidt S, van Gerven JMA, 2011. Pharmacokinetic–pharmacodynamic relationships of central nervous system effects of scopolamine in healthy subjects. Br. J. Clin. Pharmacol 71, 886–898. 10.1111/j.1365-2125.2011.03936.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loerinc AG, Meuret AE, Twohig MP, Rosenfield D, Bluett EJ, Craske MG, 2015. Response rates for CBT for anxiety disorders: Need for standardized criteria. Clin. Psychol. Rev 42, 72–82. 10.1016/j.cpr.2015.08.004 [DOI] [PubMed] [Google Scholar]
- Luyten L, Nuyts S, Beckers T, 2017. Low-dose systemic scopolamine disrupts context conditioning in rats. J. Psychopharmacol. Oxf. Engl 31, 667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayo-Wilson E, Dias S, Mavranezouli I, Kew K, Clark DM, Ades AE, Pilling S, 2014. Psychological and pharmacological interventions for social anxiety disorder in adults: a systematic review and network meta-analysis. Lancet Psychiatry 1, 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennin DS, Fresco DM, Heimberg RG, Schneier FR, Davies SO, Liebowitz MR, 2002. Screening for social anxiety disorder in the clinical setting: using the Liebowitz Social Anxiety Scale. J. Anxiety Disord 16, 661–673. 10.1016/S0887-6185(02)00134-2 [DOI] [PubMed] [Google Scholar]
- Merikangas KR, He J, Burstein M, Swanson SA, Avenevoli S, Cui L, Benjet C, Georgiades K, Swendsen J, 2010. Lifetime prevalence of mental disorders in U.S. adolescents: Results from the National Comorbidity Survey Replication-Adolescent Supplement (NCS-A). J. Am. Acad. Child Adolesc. Psychiatry 49, 980–989. 10.1016/j.jaac.2010.05.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meuret AE, Trueba AF, Abelson JL, Liberzon I, Auchus R, Bhaskara L, Ritz T, Rosenfield D, 2015. High cortisol awakening response and cortisol levels moderate exposure-based psychotherapy success. Psychoneuroendocrinology 51, 331–340. 10.1016/j.psyneuen.2014.10.008 [DOI] [PubMed] [Google Scholar]
- Opriş D, Pintea S, García- Palacios A, Botella C, Szamosközi Ş, David D, 2012. Virtual reality exposure therapy in anxiety disorders: a quantitative meta-analysis. Depress. Anxiety 29, 85–93. 10.1002/da.20910 [DOI] [PubMed] [Google Scholar]
- Owens MJ, Nemeroff CB, 1993. The role of corticotropin-releasing factor in the pathophysiology of affective and anxiety disorders: laboratory and clinical studies., in: Ciba Foundation Symposium; p. 296. [DOI] [PubMed] [Google Scholar]
- Phelps EA, LeDoux JE, 2005. Contributions of the Amygdala to Emotion Processing: From Animal Models to Human Behavior. Neuron 48, 175–187. 10.1016/j.neuron.2005.09.025 [DOI] [PubMed] [Google Scholar]
- Pine DS, 1999. Pathophysiology of childhood anxiety disorders. Biol. Psychiatry 46, 1555–1566. 10.1016/S0006-3223(99)00115-8 [DOI] [PubMed] [Google Scholar]
- Powers MB, Emmelkamp PMG, 2008. Virtual reality exposure therapy for anxiety disorders: A meta-analysis. J. Anxiety Disord 22, 561–569. 10.1016/j.janxdis.2007.04.006 [DOI] [PubMed] [Google Scholar]
- Raio CM, Brignoni-Perez E, Goldman R, Phelps EA, 2014. Acute stress impairs the retrieval of extinction memory in humans. Neurobiol. Learn. Mem., Stress and the regulation of memory: From basic mechanisms to clinical implications 112, 212–221. 10.1016/j.nlm.2014.01.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch S. a. M., King AP, Liberzon I, Sripada RK, 2017. Changes in Salivary Cortisol During Psychotherapy for Posttraumatic Stress Disorder: A Pilot Study in 30 Veterans. J. Clin. Psychiatry 78, 599–603. 10.4088/JCP.15m10596 [DOI] [PubMed] [Google Scholar]
- Rescorla RA, Wagner AR, 1972. A theory of Pavlovian conditioning: Variations in the effectiveness of reinforcement and nonreinforcement, in: Black AH, Prokasy WF (Eds.), Classical Conditioning II: Current Research and Theory. Appleton-Century-Crofts, New York, pp. 64–99. [Google Scholar]
- Roozendaal B, Okuda S, de Quervain DJ-F, McGaugh JL, 2006. Glucocorticoids interact with emotion-induced noradrenergic activation in influencing different memory functions. Neuroscience, Neuroactive Steroids: Old Players in a New Game 138, 901–910. 10.1016/j.neuroscience.2005.07.049 [DOI] [PubMed] [Google Scholar]
- Rytwinski NK, Fresco DM, Heimberg RG, Coles ME, Liebowitz MR, Cissell S, Stein MB, Hofmann SG, 2009. Screening for social anxiety disorder with the self-report version of the Liebowitz Social Anxiety Scale. Depress. Anxiety 26, 34–38. 10.1002/da.20503 [DOI] [PubMed] [Google Scholar]
- Schwabe L, Joëls M, Roozendaal B, Wolf OT, Oitzl MS, 2012. Stress effects on memory: an update and integration. Neurosci. Biobehav. Rev 36, 1740. [DOI] [PubMed] [Google Scholar]
- Shin LM, Liberzon I, 2010. The Neurocircuitry of Fear, Stress, and Anxiety Disorders. Neuropsychopharmacology 35, 169–191. 10.1038/npp.2009.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegmund A, Köster L, Meves AM, Plag J, Stoy M, Ströhle A, 2011. Stress hormones during flooding therapy and their relationship to therapy outcome in patients with panic disorder and agoraphobia. J. Psychiatr. Res 45, 339. [DOI] [PubMed] [Google Scholar]
- Singewald N, Schmuckermair C, Whittle N, Holmes A, Ressler KJ, 2015. Pharmacology of cognitive enhancers for exposure-based therapy of fear, anxiety and trauma-related disorders. Pharmacol. Ther 149, 150–190. 10.1016/j.pharmthera.2014.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smythe JW, Bhatnagar S, Murphy D, Timothy C, Costall B, 1998. The effects of intrahippocampal scopolamine infusions on anxiety in rats as measured by the black-white box test. Brain Res. Bull 45, 89. [DOI] [PubMed] [Google Scholar]
- Soravia LM, Heinrichs M, Aerni A, Maroni C, Schelling G, Ehlert U, Roozendaal B, de Quervain DJ-F, 2006. Glucocorticoids reduce phobic fear in humans. Proc. Natl. Acad. Sci. U. S. A 103, 5585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart RE, Chambless DL, 2009. Cognitive-behavioral therapy for adult anxiety disorders in clinical practice: a meta-analysis of effectiveness studies. J. Consult. Clin. Psychol 77, 595–606. 10.1037/a0016032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vrshek-Schallhorn S, Doane LD, Mineka S, Zinbarg RE, Craske MG, Adam EK, 2013. The cortisol awakening response predicts major depression: Predictive stability over a 4-year follow-up and effect of depression history. Psychol. Med 43, 483–493. 10.1017/S0033291712001213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyas A, Mitra R, Shankaranarayana RB, Chattarji S, 2002. Chronic stress induces contrasting patterns of dendritic remodeling in hippocampal and amygdaloid neurons. J. Neurosci. Off. J. Soc. Neurosci 22, 6810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- World Health Organization, 1992. The ICD-10 classification of mental and behavioural disorders: clinical descriptions and diagnostic guidelines. Geneva: World Health Organization. [Google Scholar]
- Wüst S, Wolf J, Hellhammer DH, Federenko I, Schommer N, Kirschbaum C, 2000. The cortisol awakening response-normal values and confounds. Noise Health 2, 79. [PubMed] [Google Scholar]
- Zelikowsky M, Hast TA, Bennett RZ, Merjanian M, Nocera NA, Ponnusamy R, Fanselow MS, 2013. Cholinergic blockade frees fear extinction from its contextual dependency. Biol. Psychiatry 73, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]