

Abstract
Recent studies have highlighted the key role of Casein kinase 1 δ (CK1δ) in the development of several neurodegenerative pathologies, such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS). So far, CK1δ inhibitors are noncovalent ATP competitive ligands and no drugs are currently available for this molecular target, hence the interest in developing new CK1δ inhibitors. The study aims to identify new inhibitors able to bind the enzyme; by a dual approach in silico/in vitro, the virtual screening has been performed on an in-house chemical library, which was previously designed and synthesized for other targets. The work can, therefore, be seen in the scaffold repurposing logic. The proposed strategy has led to the identification of two hits, having a novel scaffold in the landscape of CK1δ inhibitors and with an activity in the micromolar range.
Keywords: Casein kinase 1 δ, virtual screening, scaffold repurposing, consensus docking
The development of novel therapeutic approaches for the treatment of neurodegenerative diseases is still a great challenge. The discovery of the CK1 isoforms involvement in the development of neurodegenerative disorders has paved the way for the development of CK1 inhibitors. In particular, the physiopathological role of CK1 isoform δ in neurodegenerative diseases such as Alzheimer’s disease (AD), Parkinson’s disease (PD), and amyotrophic lateral sclerosis (ALS) has encouraged research for innovative therapeutic approaches.
The protein kinase CK1 isoform δ is encoded by the gene CSNK1D which is located on chromosome 17 (chromosomal localization 17q25). CK1δ human gene was characterized as a sequence of 1245 nucleotides which is transcribed into a 49 kDa protein consisting of 415 amino acids.
The poor substrate specificity of CK1 family members is supported by the fact that nearly 140 substrates are reported in the literature.1 CK1δ is an acidotropic protein kinase, which means it recognizes substrates containing acidic or phosphorylated amino acid residues. The canonical consensus sequence for CK1 is (P)S/T-X-X-S/T. Where (P)S/T indicates a phosphorylated serine or threonine residue. Nevertheless, CK1 can also phosphorylate the target if there is an N-terminal cluster of acidic residues or acidic amino acids in the N-3 position. This allows CK1 to play the role of priming kinase, activating the substrate for other kinases. Also, noncanonical sequences are recognized by CK1 such as the SLS motif.2
CK1 family members have several effectors able to modulate their expression and activity. X-ray studies demonstrate that the formation of homodimers could have a negative regulatory effect on CK1δ activity.3,4 Moreover, post-translational modifications as phosphorylation are involved in the regulation of CK1 activity. Ser318, Thr323, Ser328, Thr329, Ser331, and Thr337 are the main residues subjected to autophosphorylation. In addition to autophosphorylation, CK1δ is phosphorylated by other kinases including PKA, Akt, CLK2 (CDC-like kinase), PKC isoform α, and Chk1.2,5,6
Several studies have also underlined the importance of compartmentalization and subcellular localization in CK1 activity regulation. The subcellular localization of the kinases is mostly regulated by binding to intracellular structures or protein complexes.7,8
Dysregulations in expression or activity of CK1δ have been observed in cancer as well as in different neurodegenerative disorders such as AD, PD, and ALS.
CK1δ appears to be involved in different stages of AD development. The residues Ser202/Thr205 and Ser396/Thr404 have been identified as CK1δ phosphorylation sites on the Tau protein.9,10 Furthermore, the CK1 family is overexpressed in Alzheimer’s disease and CK1 isoforms colocalize with granulovacuolar degeneration bodies in the AD hippocampus.11
As concerns PD, it has been demonstrated that CK1 isoforms constitutively phosphorylate α-synuclein at Ser129. This suggests that CK1 mediated phosphorylation of the protein can play a key role in PD development.9
Moreover, recent studies have demonstrated that CK1δ phosphorylates many different sites of TAR (TransActive Response) DNA-binding protein 43 (TDP-43) in vitro.12 TDP-43 was identified as the major component of ALS protein aggregates, and it is responsible for the onset and progression of ALS. As a consequence, the identification of potent and selective inhibitors of CK1δ may provide an innovative therapeutic strategy for ALS.13
Initiating hit identification campaigns by using chemical scaffolds from an in-house library designed for other indications (scaffold repurposing) can speed up drug discovery in several therapeutic areas.13,14 Additionally, in silico approaches for the discovery of new kinase ligands is now mainly structure-driven, with the determination of the X-ray of several hundred structures of kinase-ligand complexes. Structure comparisons have been widely used to identify the most common and stabilizing interaction networks between ligands and their corresponding kinase binding sites. Regarding specifically CK1δ, nowadays 19 unique protein–inhibitor complexes are available from the Protein Data Bank (PDB). In parallel, docking-based virtual screening (DBVS) has extensively and successfully been used to identify potential hit compounds.14
Following this approach, in this work, we have performed a DBVS of an in-house chemical library composed by 431 compounds synthesized over more than 30 years of research in the field of oncology and directed to the inhibition of several molecular targets such as topoisomerase 1 and 2, aromatase, and tubulin. In particular, our computational pipeline was based on a combination of a canonical DBVS followed by a pharmacophore-driven filtering process of all obtained docking poses, as summarized in Figure 1.
Figure 1.
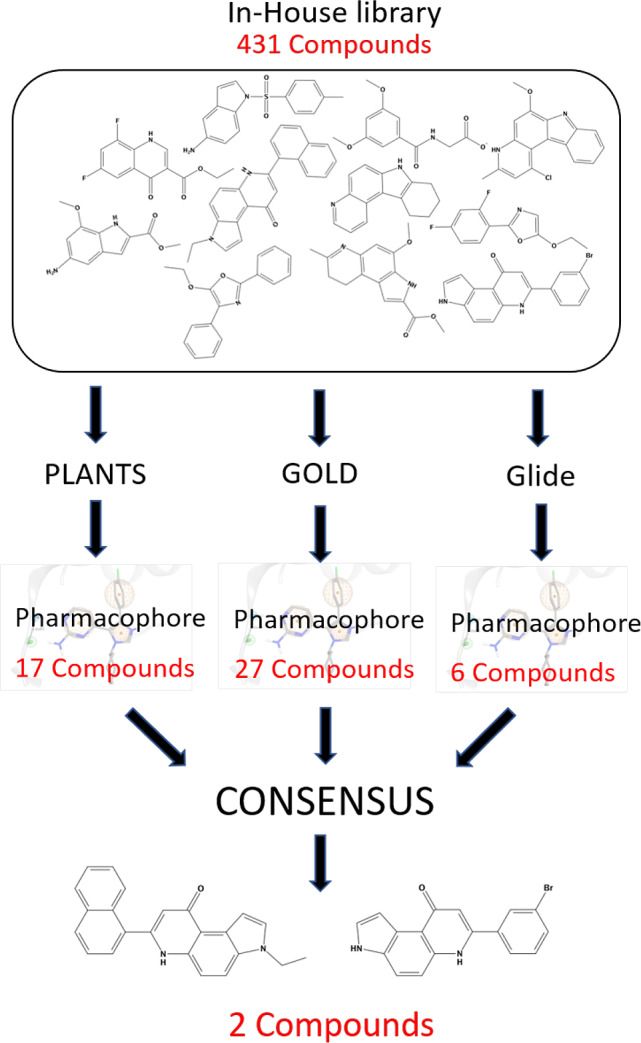
Workflow for hit compound identification.
The primary goal of this study is to verify if in our in-house library there were some ligands characterized by a scaffold not yet used among the already known inhibitors of CK1 and that was therefore susceptible to a later phase of optimization. After the preliminary in silico screening, the most promising candidates have undergone in vitro tests to confirm whether they have shown a detectable inhibition of CK1δ activity. Interestingly, we have identified two hit compounds, that share the pyrrolo[3,2-f]quinolinone moiety as key-scaffold, that are able to inhibit CK1δ activity in the micromolar range. This repurposed scaffold is now subject to further study for the construction of focused libraries for the necessary phase of optimization of its pharmacodynamic and pharmacokinetic properties.
Materials and Methods
Preparation of the Virtual Database for the Docking Protocol Calculation
The preparation of the in-house chemical library for the DBVS consisted in the enumeration of the tautomeric state and selection of the most stable one (when more than one tautomeric state is possible), the generation of the three-dimensional coordinates, the assignation of the correct ionization state for a given pH, and the calculation of the atomic partial charges.
The Tautomers application, which is included in the OpenEye toolkit QUACPAC, enumerates the most reasonable tautomeric forms of the molecule. Subsequently, the FixpKa program (also included in the Openeye toolkit QUACPAC) can be used to assign the most probable molecule ionization state for pH 7.4. The 3D conformations were generated by Corina Classic.15,16 To determine the partial charges of each compound, the Molcharge application (also included in the Openeye toolkit QUACPAC) in accordance with the MMFF94 force field was used. The library structures are reported in the SI.
Selection of the Best Protocols through DockBench and Virtual Screening
All 19 Holo Crystal Structures of CK1δ were retrieved from the Protein Data Bank (PDB). These structures were prepared with the MOE Structure Preparation tool.17 If more than one chain is reported in the crystal data file, the best-solved chain was selected. The highest occupancy alternative for each residue with alternate locations was selected. The system was protonated with the Protonate3D tool (which assigns the most probable protonation state at selected pH) using the AMBER 10 force field. The partial charges of the system (protein and ligand) were calculated, and the hydrogen atoms were minimized. The cocrystallized ligands were saved in a separate database for the following analyses while the protein structures were saved after removing ions, solvent, or other molecules used to obtain the crystal formation. This procedure is sped up by the use of a platform called DockBench.18 The software is based on a self-docking analysis. Briefly, each cocrystallized ligand is docked using the docking protocols, and the ability of each protocol in reproducing the pose of the crystallographic complex is evaluated. For each structure-docking protocol pair, minimum (RMSDmin) and average RMSD (RMSDave) values with respect to the X-ray binding mode were calculated. Twenty poses for each molecule were generated and analyzed. The VS was performed using GOLD (Scoring Function: Goldscore), PLANTS (Scoring Function: chemplp), and Glide (standard precision). The results were evaluated using a consensus strategy.
Interaction Energy Fingerprint (IEF)
The per residue analysis was performed using the software MOE (Molecular Operating Environment)17 and the SVL programming language.
The electrostatic interaction energies were measured through the Coulombic function, and they were expressed in kcal/mol, while the hydrophobic contribution resulted from the contact surfaces analysis performed by MOE and is associated with a dimensionless score. To rationalize the binding mode of each compound, the interaction energy values can be translated into heat maps called Interaction Energy Fingerprint (IEF)
Generation of the Pharmacophore Model
The conformation originated from docking was further filtered by a pharmacophore model. The alignment and the superimposition of CK1δ crystal structures have allowed a comparison between different ligands and the detection of common interaction features. The identification of the main features to build the pharmacophore model for CK1δ ligands has required a visual investigation of the protein–ligand crystallographic complexes in addition to information from the previous IEF analysis.
The pharmacophoric query design and the consequential search were performed using the MOE pharmacophore modeling tools.17
CK1δ Activity Assay
Compounds were evaluated toward CK1δ (full length, ThermoFisher) with the KinaseGlo luminescence assay (Promega) slightly modifying a procedure reported in the literature.13 In detail, luminescent assays were performed in black 96-well plates, using the following buffer: 50 mM HEPES (pH 7.5),1 mM EDTA, 1 mM EGTA, and 15 mM magnesium acetate. Compound PF-670462 (IC50 = 7.7 nM) was used as positive control for CK1δ19 while DMSO/buffer solution was used as negative control. In a typical assay, 10 μL of inhibitor solution (dissolved in DMSO at 10 mM concentration and diluted in assay buffer to the desired concentration) and 10 μL (26 nM) of enzyme solution were added to the well, followed by 20 μL of assay buffer containing 0.1% casein substrate and 4 μM ATP. The final DMSO concentration in the reaction mixture did not exceed 1–2%. After 10 min of incubation at 30 °C the enzymatic reactions were stopped with 40 μL of KinaseGlo reagent (Promega).
Luminescence signal (relative light unit, RLU) was recorded after 10 min at 30 °C using Tecan Infinite M100. For IC50 determination, ten different inhibitor concentrations ranging from 100 and 0.026 μM were used. IC50 values are reported as means ± standard errors of three independent experiments. Data were analyzed using GraphPad Prism software (version 8.0).
Results and Discussion
The first step of our work was the identification of a suitable docking protocol on which to base the DBVS of our in-house library. To this purpose, we performed a benchmark of the 17 docking protocols applied to 19 ligand– CK1δ complexes. This procedure was sped up by the use of a platform for a self-docking comparison called DockBench.18 The results of the DockBench Analysis are visualized through the use of heatmap plots. In each plot, the vertical axis shows the docking protocols while the horizontal axis represents the protein–ligand complexes. A color code, from blue to red, displays the RMSD value. The plot in Figure 2 summarizes the minimum value of RMSD (RMSDMIN) calculated for each docking protocol on each protein–ligand complex; blue spots represent low RMSD values while red ones indicate higher values. The average RMSD value (RMSDAVE) of poses generated by each docking protocol for each protein–ligand complex was also considered (Figure 2, right plot) reporting a similar profile to RMSDMIN. According to these metrics, the crystal structure selected for the subsequent molecular docking analyses was 3UZP since it has resulted in one of the protein structures for which molecular docking better reproduces the crystal structure pose with different protocols.
Figure 2.

Heatmaps summarizing the performances of molecular docking benchmark in the self-docking procedure. (Panel A) RMSD lower value obtained by each docking protocol (y-axis) for each protein–ligand complex (x-axis). (Panel B) RMSD average value obtained by each docking protocol (y-axis) for each protein–ligand complex (x-axis).
The comparison of the different docking protocols on the complex 3UZP revealed that several different algorithms were able to nicely reproduce the experimental geometries showing RMSDMIN below 0.55 Å (Table 1). Encouraged by these performances, we decided to maximize the conformational sampling by using three different docking protocols in the Virtual Screening: GOLD20 coupled to Goldscore Scoring Function, PLANTS21,22 coupled to Chemplp Scoring Function,23 and Glide-sp.24 This strategy, usually named consensus docking,25 is a method to improve the reliability of docking results; it consists in the parallel use of several docking protocols based on different search algorithms and in the interpolation of the results of these.
Table 1. Lowest and Average RMSD Values of the Three Docking Protocols Selected from the Benchmark.
| RMSDMIN | RMSDAVE | |
|---|---|---|
| GOLD - Goldscore | 0.29 Å | 0.54 Å |
| PLANTS - Chemplp | 0.35 Å | 1.73 Å |
| GLIDE - SP | 0.55 Å | 5.33 Å |
In this view, the selection of the protocols not only satisfies the benchmark results but also respects the fundamental requirement to have an orthogonal search algorithm. Indeed, PLANTS relies on an ant colony optimization algorithm for the search algorithm, GOLD on a genetic algorithm, and Glide on a systematic search. Ten poses for each molecule of the chemical library were hence calculated generating a total of 12930 ligand conformations.
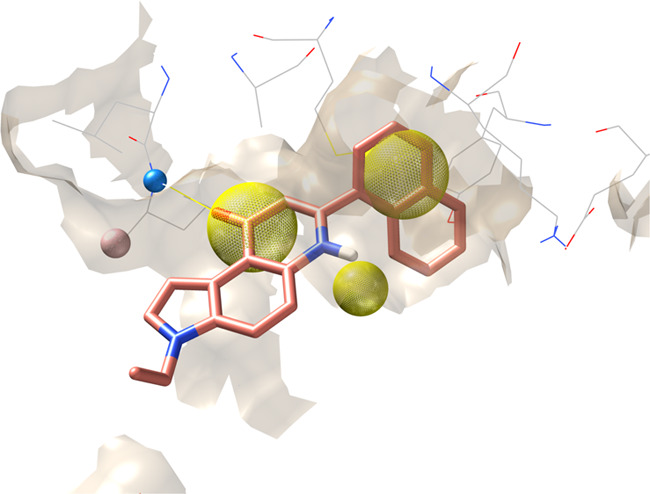
To analyze the VS output instead of using a classical scoring function we adopted a geometrical based method based on a structure-based pharmacophore developed on the same data set of CK1δ holo-complexes used in the previous benchmark. The alignment and the superimposition of CK1δ crystal structures have allowed a comparison between different ligands and the detection of common interaction features to build the pharmacophore model. In addition, a qualitative analysis of the molecular interaction was carried out by considering the interaction energy fingerprints (IEF) of the 19 ligands in our data set (Figure 3). By coupling the geometrical alignment and IEFs it was confirmed the relevance of interactions with the hinge region of the kinase. In particular, Leu 85 plays a key role in establishing two hydrogen bonds with most of the cocrystallized ligands. The hypothesis of the Leu 85 key role is strongly supported by studies reported in the literature.26−28 For this reason, the H-bond interaction with the backbone of this residue has been included in the pharmacophore model. In addition, the superimposition of the compounds revealed the presence of aromatic moieties for most structures; their presence guarantees a strong hydrophobic contribution as confirmed in the hydrophobic fingerprint (Figure 3). All these analyses were summarized in a pharmacophore having five features: two hydrogen bonds (one acceptor and one donor) and three hydrophobic ones (Figure 4).
Figure 3.

Interaction energy fingerprint (IEF). Per residue electrostatic (upper plot) and the hydrophobic contribution (lower plot) interaction for each crystallographic ligand (reported on the y-axis) of CK1δ. For electrostatic interaction the colorimetric scale is blue to red while for the hydrophobic contribution is white to green.
Figure 4.
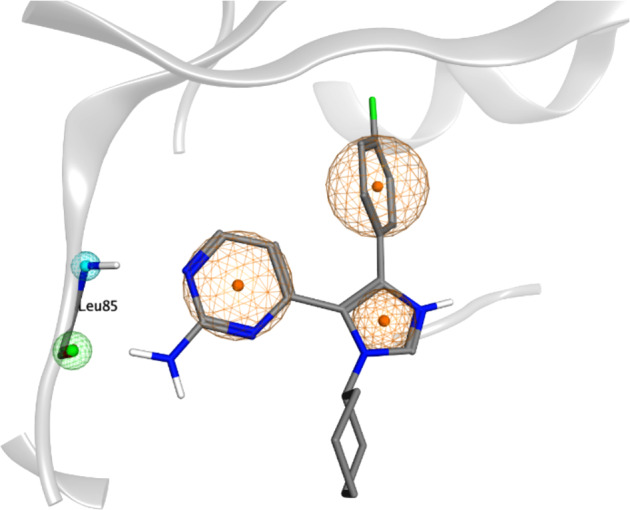
Pharmacophoric model superposed to the crystallographic complex ligand 0CK-CK1δ (PDB ID: 3UZP). The orange sphere represents an aromatic feature, while the blue and the green ones indicate respectively the presence of a hydrogen bond donor (HBD) and a hydrogen bond acceptor (HBA) mediating the interaction with Leu 85.
To filter out the conformations obtained from the DBVS the following criterion was used: only the poses that satisfy at least three features of the pharmacophore model were retained including the mandatory presence of at least one donor/acceptor feature. The pharmacophoric filter was applied to each docking protocol separately in order to obtain three independent lists. Only the molecules that satisfy the pharmacophore model in each docking protocol were retained. In this way, we were able to select two molecules: compounds 1 and 2 (Figure 5). For compound 2, the bromophenyl group fills the hydrophobic pocket formed between the side chains of Lys 38, Met 80, and the gatekeeper residue Met 82, while the pyrrolo-quinolinone scaffold occupies the outer portion of the binding site. The carbonyl oxygen of the pyrrolo-quinolinone portion maintains the recurrent interaction with the hinge region, especially with the backbone NH of Leu 85. In addition, π–CH interactions occur between the pyridone and pyrrole moieties and nonpolar amino acids as Ile 15 and Ile 23.
Figure 5.

Resulting pose for compounds 1 (panel A) and 2 (panel B). CK1δ binding site is reported using the ribbon representation (light gray). The key residue Leu 85 in the hinge region is explicated by stick representation.
For compound 1, the hydrogen bond with Leu 85 is conserved as well as the CH−π interaction with Ile 23. The hydrophobic pocket is widely occupied by the naphthyl group while the ethyl-substituted pyrrole faces outward.
In Figure 6 are reported the IEF of the two compounds while in the Supporting Information are reported two comparisons between the electrostatic interactions of compounds 1 and 2 and the crystallographic ligand 0CK.
Figure 6.

Interaction energy fingerprint (IEF) for compounds 1 and 2. Per residue electrostatic (upper plot) and the hydrophobic contribution (lower plot). For electrostatic interaction the colorimetric scale is blue to red while for the hydrophobic contibution it is white.
To verify the accuracy of the results obtained by our computational pipeline, the two selected candidates were tested using a conventional in vitro kinase activity inhibitory assay.
The IC50 values against CK1δ were of 15.22 ± 2.71 μM for compound 1 and 12.95 ± 3.21 μM for compound 2, respectively (Figure 3 and 4 on SI). Despite the fact that the two selected molecules showed an inhibitory effect of CK1δ activity in the micromolar range, it is worth underlining that they were initially designed for completely different targets and, consequently, the repurposing aim of a novel scaffold can be considered as achieved. In fact, pyrrolo[3,2-f]quinolinone represents a novel scaffold for designing new CK1δ inhibitors. It is interesting to note how the strategy of in-house chemical library repurposing can be now particularly useful to cherry-pick from the library the closest analogs to our hit for developing a very preliminary structure–activity-relationship useful to quickly investigate the role of certain molecular decoration. However, as already anticipated, this repurposed scaffold is now subject to further study for the construction of focused libraries for the necessary phase of optimization of its pharmacodynamic and pharmacokinetic properties. Interestingly, during the writing of this work a new ck1d crystal has been released (PDB code: 6RCH) cocrystallized with a ligand having a naphthyl substituent positioned like the one suggested by us.
Concluding, the preliminary results here described supporting the fact that the suggested computational pipeline could represent an alternative valuable strategy to efficiently analyze the unexplored chemical space.
Acknowledgments
MMS lab is very grateful to Chemical Computing Group, OpenEye, and Acellera for the scientific and technical partnership. MMS lab gratefully acknowledges the support of NVIDIA Corporation with the donation of the Titan V GPU, used for this research.
Glossary
Abbreviations
- CK1δ
Casein kinase 1 δ
- CK1
Casein kinase isoform 1
- AD
Alzheimer’s disease
- PD
Parkinson’s disease
- ALS
amyotrophic lateral sclerosis
- LBVS
ligand-based virtual screening
- SBVS
structure-based virtual screening
- PDB
Protein Data Bank
- RMSD
root mean square deviation
- IEF
interaction energy fingerprint
Supporting Information Available
. The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00028.
Figure 1. Per residue electrostatic comparison among Compound 1 and OCK (3UZP ligand). Figure 2. Per residue electrostatic comparison among Compound 2 and OCK (3UZP ligand). Figure 3. Concentration–inhibition curve of Compound 1 at human CK 1d. Data were collected from three independent experiments performed in duplicate. Figure 4. Concentration–inhibition curve of Compound 2 at human CK 1d. Data were collected from three independent experiments performed in duplicate. (PDF)
Library structures (PDF)
Casein kinase 1 delta molecules retained after pharmacophore filtering for GLIDE docking protocol (MP4)
Casein kinase 1 delta molecules retained after pharmacophore filtering for GOLD docking protocol (MP4)
Casein kinase 1 delta molecules retained after pharmacophore filtering for PLANTS docking protocol (MP4)
Author Contributions
⊥ E.C. and G.B. contributed equally. All authors have given approval to the final version of the manuscript.
This scientific work has been financially supported by MIUR (PRIN2017, n. 2017MT3993).
The authors declare no competing financial interest.
Supplementary Material
References
- Knippschild U.; Gocht A.; Wolff S.; Huber N.; Löhler J.; Stöter M. The Casein Kinase 1 Family: Participation in Multiple Cellular Processes in Eukaryotes. Cell. Signalling 2005, 17 (6), 675–689. 10.1016/j.cellsig.2004.12.011. [DOI] [PubMed] [Google Scholar]
- Knippschild U.; Krüger M.; Richter J.; Xu P.; García-Reyes B.; Peifer C.; Halekotte J.; Bakulev V.; Bischof J. The CK1 Family: Contribution to Cellular Stress Response and Its Role in Carcinogenesis. Front. Oncol. 2014, 4 (May), 1–33. 10.3389/fonc.2014.00096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longenecker K. L.; Roach P. J.; Hurley T. D. Crystallographic Studies of Casein Kinasc I δ: Toward a Structural Understanding of Auto-Inhibition. Acta Crystallogr., Sect. D: Biol. Crystallogr. 1998, 54, 473. 10.1107/S0907444997011724. [DOI] [PubMed] [Google Scholar]
- Hirner H.; Günes C.; Bischof J.; Wolff S.; Grothey A.; Kühl M.; Oswald F.; Wegwitz F.; Bösl M. R.; Trauzold A.; et al. Impaired CK1 Delta Activity Attenuates SV40-Induced Cellular Transformation in Vitro and Mouse Mammary Carcinogenesis in Vivo. PLoS One 2012, 7, e29709. 10.1371/journal.pone.0029709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bischof J.; Randoll S. J.; Süßner N.; Henne-Bruns D.; Pinna L. A.; Knippschild U. CK1δ Kinase Activity Is Modulated by Chk1-Mediated Phosphorylation. PLoS One 2013, 8, e68803. 10.1371/journal.pone.0068803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graves P. R.; Roach P. J. Role of COOH-Terminal Phosphorylation in the Regulation of Casein Kinase Iδ. J. Biol. Chem. 1995, 270, 21689. 10.1074/jbc.270.37.21689. [DOI] [PubMed] [Google Scholar]
- Milne D. M.; Looby P.; Meek D. W. Catalytic Activity of Protein Kinase CK1δ (Casein Kinase 1 δ) Is Essential for Its Normal Subcellular Localization. Exp. Cell Res. 2001, 263, 43. 10.1006/excr.2000.5100. [DOI] [PubMed] [Google Scholar]
- Xu P.; Ianes C.; Gärtner F.; Liu C.; Burster T.; Bakulev V.; Rachidi N.; Knippschild U.; Bischof J.. Structure, Regulation, and (Patho-)Physiological Functions of the Stress-Induced Protein Kinase CK1 Delta (CSNK1D); Elsevier B.V, Gene 2019, 715, 144005. 10.1016/j.gene.2019.144005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez D. I.; Gil C.; Martinez A. Protein Kinases CK1 and CK2 as New Targets for Neurodegenerative Diseases. Med. Res. Rev. 2011, 31, 924. 10.1002/med.20207. [DOI] [PubMed] [Google Scholar]
- Li G.; Yin H.; Kuret J. Casein Kinase 1 Delta Phosphorylates Tau and Disrupts Its Binding to Microtubules. J. Biol. Chem. 2004, 279, 15938. 10.1074/jbc.M314116200. [DOI] [PubMed] [Google Scholar]
- Schwab C.; Demaggio A. J.; Ghoshal N.; Binder L. I.; Kuret J.; McGeer P. L. Casein Kinase 1 Delta Is Associated with Pathological Accumulation of Tau in Several Neurodegenerative Diseases. Neurobiol. Aging 2000, 21, 503. 10.1016/S0197-4580(00)00110-X. [DOI] [PubMed] [Google Scholar]
- Nonaka T.; Suzuki G.; Tanaka Y.; Kametani F.; Hirai S.; Okado H.; Miyashita T.; Saitoe M.; Akiyama H.; Masai H.; et al. Phosphorylation of TAR DNA-Binding Protein of 43 KDa (TDP-43) by Truncated Casein Kinase 1δ Triggers Mislocalization and Accumulation of TDP-43. J. Biol. Chem. 2016, 291, 5473. 10.1074/jbc.M115.695379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salado I. G.; Redondo M.; Bello M. L.; Perez C.; Liachko N. F.; Kraemer B. C.; Miguel L.; Lecourtois M.; Gil C.; Martinez A.; et al. Protein Kinase CK-1 Inhibitors as New Potential Drugs for Amyotrophic Lateral Sclerosis. J. Med. Chem. 2014, 57, 2755. 10.1021/jm500065f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozza G.; Gianoncelli A.; Montopoli M.; Caparrotta L.; Venerando A.; Meggio F.; Pinna L. A.; Zagotto G.; Moro S. Identification of Novel Protein Kinase CK1 Delta (CK1δ) Inhibitors through Structure-Based Virtual Screening. Bioorg. Med. Chem. Lett. 2008, 18, 5672. 10.1016/j.bmcl.2008.08.072. [DOI] [PubMed] [Google Scholar]
- 3D Structure Generator CORINA Classic, Molecular Networks GmbH, Nuremberg, Germany, www.mn-am.com. Date Accessed 2020-04-24.
- Sadowski J.; Gasteiger J.; Klebe G. Comparison of Automatic Three-Dimensional Model Builders Using 639 X-Ray Structures. J. Chem. Inf. Model. 1994, 34, 1000. 10.1021/ci00020a039. [DOI] [Google Scholar]
- Chemical Computing Group ULC, Molecular Operating Environment (MOE), 2019.01. 1010 Sherbrooke St. West, Suite #910, Montreal, QC, Canada, H3A 2R7, 2019.
- Cuzzolin A.; Sturlese M.; Malvacio I.; Ciancetta A.; Moro S. DockBench: An Integrated Informatic Platform Bridging the Gap between the Robust Validation of Docking Protocols and Virtual Screening Simulations. Molecules 2015, 20, 9977. 10.3390/molecules20069977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettayeb K.; Oumata N.; Echalier A.; Ferandin Y.; Endicott J. A.; Galons H.; Meijer L. CR8, a Potent and Selective, Roscovitine-Derived Inhibitor of Cyclin-Dependent Kinases. Oncogene 2008, 27 (44), 5797–5807. 10.1038/onc.2008.191. [DOI] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and Validation of a Genetic Algorithm for Flexible Docking. J. Mol. Biol. 1997, 267, 727. 10.1006/jmbi.1996.0897. [DOI] [PubMed] [Google Scholar]
- Korb O.; Stützle T.; Exner T. E. An Ant Colony Optimization Approach to Flexible Protein–Ligand Docking. Swarm Intell 2007, 1, 115. 10.1007/s11721-007-0006-9. [DOI] [Google Scholar]
- Korb O.; Stützle T.; Exner T. E.. PLANTS: Application of Ant Colony Optimization to Structure-Based Drug Design. In Lecture Notes in Computer Science (including subseries Lecture Notes in Artificial Intelligence and Lecture Notes in Bioinformatics ); 2006. 10.1007/11839088_22. [DOI] [Google Scholar]
- Korb O.; Stützle T.; Exner T. E. Empirical Scoring Functions for Advanced Protein-Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84. 10.1021/ci800298z. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: A New Approach for Rapid, Accurate Docking and Scoring. 2. Enrichment Factors in Database Screening. J. Med. Chem. 2004, 47, 1750. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Houston D. R.; Walkinshaw M. D. Consensus Docking: Improving the Reliability of Docking in a Virtual Screening Context. J. Chem. Inf. Model. 2013, 53, 384. 10.1021/ci300399w. [DOI] [PubMed] [Google Scholar]
- Mente S.; Arnold E.; Butler T.; Chakrapani S.; Chandrasekaran R.; Cherry K.; Dirico K.; Doran A.; Fisher K.; Galatsis P.; et al. Ligand-Protein Interactions of Selective Casein Kinase 1δ Inhibitors. J. Med. Chem. 2013, 56 (17), 6819–6828. 10.1021/jm4006324. [DOI] [PubMed] [Google Scholar]
- Wager T. T.; Galatsis P.; Chandrasekaran R. Y.; Butler T. W.; Li J.; Zhang L.; Mente S.; Subramanyam C.; Liu S.; Doran A. C.; et al. Identification and Profiling of a Selective and Brain Penetrant Radioligand for in Vivo Target Occupancy Measurement of Casein Kinase 1 (CK1) Inhibitors. ACS Chem. Neurosci. 2017, 8 (9), 1995–2004. 10.1021/acschemneuro.7b00155. [DOI] [PubMed] [Google Scholar]
- García-Reyes B.; Witt L.; Jansen B.; Karasu E.; Gehring T.; Leban J.; Henne-Bruns D.; Pichlo C.; Brunstein E.; Baumann U.; et al. Discovery of Inhibitor of Wnt Production 2 (IWP-2) and Related Compounds As Selective ATP-Competitive Inhibitors of Casein Kinase 1 (CK1) δ/ϵ. J. Med. Chem. 2018, 61 (9), 4087–4102. 10.1021/acs.jmedchem.8b00095. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.