

Abstract
Myriad studies have linked type I interferon (IFN) to the pathogenesis of autoimmune diseases, including systemic lupus erythematosus (SLE). While increased levels of type I IFN are found in patients with SLE, and IFN blockade ameliorates disease in many mouse models of lupus, its precise roles in driving SLE pathogenesis remain largely unknown. Here, we dissected the effect of type I IFN sensing by CD4 T cells and B cells on the development of T follicular helper cells (TFH), germinal center (GC) B cells, plasmablasts, and anti-nuclear dsDNA IgG levels using the bm12 chronic graft versus host disease (cGVHD) model of SLE-like disease. Type I IFN sensing by B cells decreased their threshold for BCR signaling and increased their expression of MHCII, CD40, and Bcl-6, requirements for optimal GC B cell functions. In line with these data, ablation of type I IFN sensing in B cells significantly reduced the accumulation of GC B cells, plasmablasts, and autoantibodies. Ablation of type I IFN sensing in T cells significantly inhibited TFH expansion and subsequent B cell responses. In contrast to the effect in B cells, type I IFN did not promote proliferation in the T cells but protected them from NK cell-mediated killing. Consequently, ablation of either perforin or NK cells completely restored TFH expansion of IFNAR−/− TFH and, subsequently, restored the B cell responses. Together our data provide evidence for novel roles of type I IFN and immunoregulatory NK cells in the context of sterile inflammation and SLE-like disease.
Keywords: type I IFN, T follicular helper cells (TFH), Natural Killer cells (NK), B cells, Autoimmunity, Systemic Lupus Erythematosus (SLE)
Introduction
The overproduction of type I IFN is a prominent feature associated with the development of SLE and has been implicated in the pathology of other autoimmune diseases including Sjögren’s syndrome, systemic sclerosis, adult-onset rheumatoid arthritis, and type I diabetes mellitus (1, 2). A role for type I IFN in driving disease pathology has been demonstrated in several genetic and inducible murine models of SLE (3-7). Moreover, the therapeutic use of IFN to treat patients with hepatitis C virus and cancer has been associated with the induction of SLE (8-10), and the administration of IFN to lupus-prone mice induced earlier onset, lethal disease (11). Furthermore, a type I IFN gene signature correlates with disease severity in SLE patients (12, 13) and approaches blocking type I IFN or its receptor have shown some efficacy in clinical trials (14-17). While these studies strongly implicate type I IFN in promoting SLE pathogenesis, relatively little is known about the precise functions of IFN in SLE.
Type I IFN comprises 13 IFNα proteins, IFNβ, IFNκ, and IFNω, all of which signal through a common receptor, known as the interferon alpha and beta receptor, or IFNAR. IFNAR is expressed by essentially all nucleated cells and plays well described, key roles in antimicrobial defenses (18). In SLE, one prevailing hypothesis is that by some combination of genetic and environmental factors, an accumulation of dying cells leads to increased exposure to self-RNA and self-DNA, which induces high levels of IFN production by plasmacytoid dendritic cells (DCs) (19-21). Relatively few studies, however, have sought to characterize the mechanism of action of IFN specifically in SLE.
Studies of SLE peripheral blood-derived DCs by Blanco et al. indicated a likely role of IFN sensing by DCs in SLE pathogenesis (22). It is likely that type I IFN sensing by B cells and CD4 T cells also contributes to disease development, given that type I IFN is known to promote B cell activation, class switching, and support the generation of antibody-secreting plasma cells (23-25) and type I IFN sensing by CD4 T cells can dramatically enhance their priming in the context of certain viral infections and vaccines (25-27). Further dissection of the relative contribution of type I IFN sensing in different cell types in disease development and progression would provide valuable insights that could be exploited for therapeutic gain.
Here we investigated the potential role for type I IFN sensing by B cells and CD4 T cells in the bm12 chronic graft-versus-host disease model of SLE, a well-established murine model of SLE with clinical signs that correspond to those of SLE patients, including the development of ANA, lupus nephritis, and a recognized role for type I IFN (28, 29). Our data show that direct sensing of type I IFN by B cells is required for the maximal development of GC B cells, plasmablasts, and anti-dsDNA IgG through the upregulation of membrane-associated molecules that promote T:B interactions, induction of the GC-promoting transcription factor Bcl-6, and increasing the proliferative capacity to BCR stimuli. Direct sensing of type I IFN by CD4 T cells was critically important for TFH accumulation and subsequent disease development. However, in this scenario type I IFN sensing was required to protect the TFH cells from NK cell-mediated killing and elimination of NK cells or genetic ablation of perforin allowed for normal accumulation of IFNAR−/− TFH and disease associated sequelae.
Our data provide evidence for novel roles of type I IFN and immunoregulatory NK cells in SLE, providing rationale and potential new targets for the development of future combination immunotherapies.
Materials and Methods
Mice and cell lines
All experiments involving mice were conducted following protocols approved by Cincinnati Children’s Hospital Medical Center’s Institutional Animal Care and Use Committee (IACUC). Mice were maintained under specific pathogen-free conditions in accordance with guidelines by the Association for Assessment and Accreditation of Laboratory Animal Care International. C57BL/6J, B6.SJL-Ptpcra (CD45.1), B6.PL-Thy1a /CyJ (CD90.1), B6.129S2-Ighmtm1Cgn/J (μMT), C57BL/6-Prf1tm1Sdz/J (Prf1−/−), B6(C)-H2-Ab1bm12/KhEgJ (bm12), and B6.129(Cg)-Foxp3tm(DTR/GFP)Ayr/J (Foxp3-DTR) mice were originally purchased from The Jackson Laboratory (Bar Harbor, ME) and bred in-house. IFNAR1−/− mice were originally a gift from Dr. Jonathan Sprent and have since been bred and backcrossed to the C57Bl/6 background in our facility (30). Both male and female mice were used, but each experiment consisted of only one aged-matched gender. ISRE-L929 IFN reporter cells have been described previously (31). Mixed bone marrow BM) chimeric mice were generated using lethally irradiated mice of indicated strains on a C57BL/6 background. Mice received an initial dose of 700 RAD followed by an additional 475 RAD dose 3 hours later using a 137Cs irradiator (J L Shepherd & Associates, Inc., San Fernando, CA). BM from indicated strains (on different congenic backgrounds) were mixed at indicated ratios prior to reconstitution, and 6 million total BM cells were transferred via tail vein injection. All BM chimeric mice were given 12 weeks for hematopoietic reconstitution.
CD4 T cell isolation
For most in vivo and in vitro assays, CD4 T cells were sorted to >97% purity by negative selection using magnetic bead sorting technologies from Miltenyi Biotec (Bergisch Gladbach, Germany) and Biolegend (San Diego, Ca). Post sort purity was determined by flow staining for live cells, CD45, CD3 and CD4. In several experiments, cells were labeled with Cell Trace Violet (CTV) or CFSE as proliferation tracking dyes. For isolation of CD4bm12 T cells for the TFH in vivo killing assay, CD4bm12T cells were re-purified from spleens 2 weeks after their initial transfer by negative selection for CD4, followed by positive selection for the congenic marker using biotinylated Ab to CD45.1 (clone A20, Biolegend, CA) and anti-biotin labeled Miltenyi beads. Flow cytometric analysis showed >95% viability, >95% purity (CD45.1+CD4+) with >95% of the cells expressing TFH markers PD1 and CXCR5.
Bm12 model of cGVHD
The bm12 model experiments were performed as previously described(7, 28). Briefly, 7x106 naïve donor CD4bm12T cells from indicated strains were injected intraperitoneally into recipient mice of the specified strains. Fourteen days later, unless indicated otherwise, spleens were harvested, weighed, and processed into single cell suspensions for counting and flow staining. Serum was harvested by retro-orbital bleeding prior to sacrificing mice, processed using serum separator tubes (BD Biosciences, San Jose, CA) and stored at −80°C for analysis of cytokine and anti-dsDNA IgG levels.
In vivo depletion studies
NK cells were depleted via an intraperitoneal injection of 10ul anti-Asialo-GM1 rabbit antiserum (Wako, Richmond, VA) at −1,1, 5, and 10 days after CD4bm12T cell transfer. Parallel studies used NK1.1 (clone PK136, 25ug/mouse) on day −1 and 1. CD8 T cells were depleted by i.p. injection of 25ug anti-CD8β (clone YTS156.7.7, 20ug/mouse) one day prior to CD4bm12T cell transfer. Treg depletion in Foxp3-DTR mice was achieved by administration of DT (i.p. 20mg/kg) −1, 3, and 10 days after CD4bm12T cell transfer. Depletion efficacy was assessed at the end of the experiments by flow cytometry.
In vivo NK cell killing assay
To assess the susceptibility of the TFH to NK cell-mediated killing, naïve WT and IFNAR−/− CD45.1 CD4bm12T cells were injected into Prf−/− mice. After 14 days, TFH were sorted and mixed in a 1:1 ratio with naïve CD4 T cells from CD90.1 mice. A total of 4 X106 cells were injected i.v. into intact and NK-depleted (anti-Asialo-GM1 treated) WT recipients. After 24 h, spleens were collected and the ratio of CD45.1 CD4bm12T cells to CD90.1 control CD4 T cells was determined by flow cytometry.
Serum cytokine and anti-dsDNA lgG analyses
Circulating cytokines were analyzed in the serum of mice by Luminex multiplex technology (Austin, TX). Serum type I IFN was measured using the highly sensitive ISRE-L929 bioassay. Serum levels of anti-dsDNA were determined using ELISA essentially as described previously (7, 28). Anti-dsDNA IgG levels are plotted as relative units towards a reference standard that was used in all experiments.
In vitro T and B cell assays
Spleens and lymph nodes were harvested from indicated strains and CD4 T cells or B cells were isolated by positive selection using magnetic bead sorting technology (CD4+ T cell and CD19+ B cell isolation kits, Miltenyi). Cells were labeled with a proliferation tracking dye (CTV or CFSE) per the manufacturer’s protocol and cultured in IMDM supplemented with 10% FBS, penicillin, streptomycin, and L-glutamine. CD4 T cells were stimulated with CD3 (1 μg/ml plate-bound, clone 17A2, Biolegend) and soluble CD28 (1 μg/ml, clone 37.51, Biolegend) in the absence or presence of 50-200 U/ml recombinant IFNα or IFNβ (PBL Assay Science, Piscataway, NJ). B cells were stimulated with goat-anti-mouse IgM F (ab’)2 fragment (10 μg/ml, unless otherwise noted, Jackson ImmunoResearch Laboratories, PA) and anti-mouse CD40 (10 μg/ml, unless otherwise noted, clone FGK4.5, BioXCell). Analysis was performed by flow cytometry at indicated timepoints.
Flow cytometry
Unless otherwise specified, cells were collected and immediately stained directly ex vivo. For the bm12 model, cells were stained essentially as described previously (7, 28) using combinations of a Fixable Viability dye and antibodies to CD3, CD4, PD1, CXCR5, ICOS, CD45.1/CD45.2, CD90.1/CD90.2 and CD25. Intracellular staining for Bcl-6, Foxp3, Bcl-2, Bcl-X, Bim, Bcl2A1, Ki67, and pH2AX was performed using the FoxP3 Staining Buffer Set (eBioscience, San Diego, CA). NK cells were defined as NK1.1+NKp46+ and were evaluated further using combinations of antibodies toward CD27, CD11b, NKG2A, NKG2D, Ly49A, C, D, F, G2, H, and I, CD244 and CD122. Staining for NK cell ligands included CD48, ICAM1, ICAM2, H2-Kb/Db, Rae1, Mult1, and NCR1-Fc followed by a secondary Ab. B cells were stained for viability, B220, CD19, GL7, Fas, and CD138. In vitro assessment of B cells included MHCII, CD40, and Bcl-6. For in vivo depletion studies, efficacy was assessed by staining for NK cells by NK1.1 and NKp46 and CD8 T cells by CD8α, CD3, and CD4. All Abs and dyes were purchased from Biolegend or eBioscience, (except for NCR1-Fc which purchased from R&D Systems). Samples were collected on a Fortessa flow cytometer with FACSDiva software (BD Biosciences), and data were analyzed with FlowJo software (Tree Star, Ashland, OR).
Statistical analysis
Data were analyzed using Prism software (GraphPad Software, Inc). Unless stated otherwise, the data are represented as means ± SEM and group sizes for each experiment are provided in the legends. Data were evaluated using a Student’s t test unless otherwise specified. A p-value of ≤0.05 was considered statistically significant.
Results
Type I IFN sensing by the hematopoietic compartment is required for disease development in the bm12 cGVHD model of SLE
Analysis of serum cytokines showed that type I IFN levels increased over time, concomitant with an increase in the serum levels of the type I IFN-inducible chemokines CXCL9 and CXCL10 (Fig. 1A). Additional inflammatory cytokines including TNFα, IL-12, and IL-6 were also induced, though these were present in relatively low levels and at generally delayed kinetics compared to the type I IFNs and the IFN-inducible chemokines (Fig. 1A).
Figure 1. Type I IFN sensing by hematopoietic cells is critical for disease development.
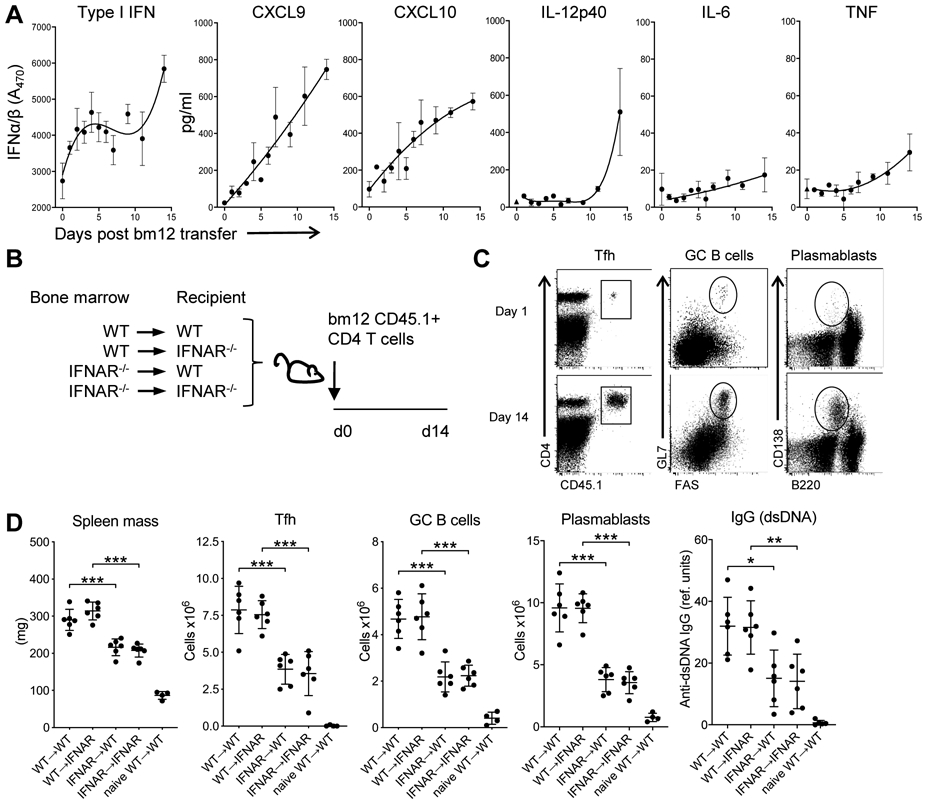
A. C57BL/6 mice were injected with 7 x 106 purified WT CD45.1 CD4bm12 T cells. At the indicated days after injection, serum type I IFN was analyzed by bioassay and additional cytokines and chemokines were analyzed by multiplex technology. The dotted line indicates the limit of detection for each analyte. Data are shown as mean± SEM (n=2-3/timepoint). B. Schema of bone-marrow chimeric mice experiments. All bone-marrow donors and recipients were on a CD45.2 background. BM-chimeric mice were injected with purified CD45.1+ WTbm12 CD4 T cells (n=6-7/group) and mice were sacrificed after 14 days. C. Representative flow cytometry plots depict the gating strategy for analysis of splenic T cell (in total live splenocytes), GC B cells (in live CD19+ cells) and plasmablasts (in total live splenocytes). D. Splenic mass, and absolute splenic number of TFH (live, CD4+CD45.1+PD1+CXCR5+), GC B cells, plasma B cells, and anti-dsDNA IgG levels 14 days after CD45.1+ WTbm12 CD4 T cell transfer. Naïve WT→WT represents WT mice reconstituted with WT bone marrow that did not receive CD45.1+ WTbm12 CD4 T cells. Data from one representative experiment (out of 2 experiments with n=6-7/group) is shown as mean ± SEM, where *p≤0.05.
To assess the relative contribution of type IFN sensing in the hematopoietic and non-hematopoietic compartments to disease development, we generated bone marrow chimeric mice where WT and IFNAR−/− mice were reconstituted with either WT or IFNAR−/− BM. After 12 weeks, CD45.1 CD4bm12T cells were transferred and splenic mass, development of TFH (PD1+CXCR5+ within live CD4+CD45.1+ cells), GC B cells (live, CD19+Fas+GL7+), plasmablasts (live, B220med/low CD138+), and anti-dsDNA IgG levels were assessed 2 weeks later (Fig. 1B,C) (28).
WT→WT and WT→IFNAR−/− recipients showed comparable increases in splenic mass, frequencies and absolute numbers of TFH, GC B cells, plasmablasts and anti-dsDNA IgG levels, indicating that type I IFN sensing by the non-hematopoietic compartment was not required for disease development (Fig. 1D). In contrast, IFNAR−/−→WT mice showed significantly diminished disease compared to mice reconstituted with WT BM. There was no added effect of loss of IFNAR on the non-hematopoietic compartment (IFNAR−/−→IFNAR−/− compared to IFNAR−/−→WT) (Fig. 1D). Together these data identify type I IFN sensing by the hematopoietic compartment as the critical component in disease development.
Type I IFN sensing by B cells is required for optimal GC and plasmablast formation
As it is likely that multiple components in the hematopoietic compartment require type I IFN sensing to confer disease development, we next assessed the contribution of type I IFN sensing on the development of GC B cells, plasmablasts and anti-dsDNA IgG levels.
To generate mice that lacked IFNAR only on the B cell compartment, WT mice were reconstituted with a mix of bone marrow from B cell deficient μMT mice together with either WT or IFNAR−/− BM in a 10:1 ratio, effectively generating mice where the B cells are either WT or IFNAR−/− (Fig. 2Ai). Transfer of CD45.1 CD4bm12 T cells into the WT/μMT mice resulted the expansion of TFH and induction of GC B cells, plasmablasts and anti-dsDNA IgG. However, in the IFNAR−/−/μMT recipients, GC B cells, plasmablasts and anti-dsDNA IgG levels were significantly reduced, suggesting a direct effect of type I IFN sensing on B cells (Fig. 2B). However, IFNAR−/−/μMT recipient mice also had lower levels of TFH than WT/μMT recipient mice. TFH and GC B cell are reciprocally supportive of each other: the frequency of TFH cells is associated with the magnitude of the B cell response, and GC B cells have been shown to promote TFH expansion (32-34). Therefore, the observed reduction in B cell responses in the IFNAR−/−/μMT mice could result from changes in reciprocal TFH:B cell interactions, rather than a sole effect on B cells.
Figure 2. B cell IFN sensing augments plasmablast and GC B cell development.
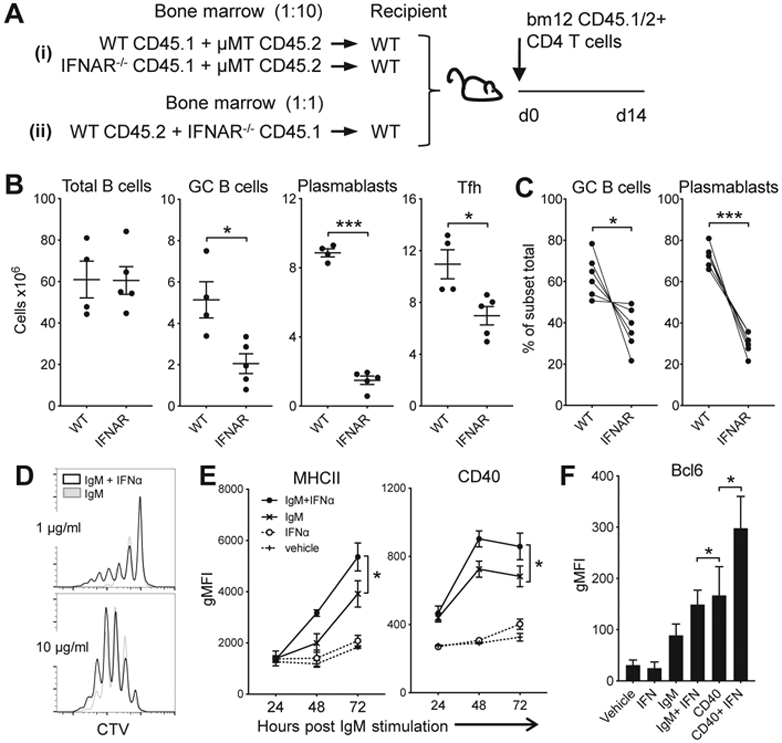
Ai and Aii. Schemas used for the mixed bone marrow chimeras in B and C, respectively. B. Bone marrow from WT or IFNAR−/− CD45.1 mice was combined with bone marrow of B cell-deficient CD45.2 μMT mice at a 1:10 ratio and transplanted into lethally irradiated C57BL/6 mice. After 12 weeks, mice received CD45.1/2 WTbm12 CD4 T cells. B cell and donor TFH response 2 weeks after T cell transfer presented as absolute number of cells per spleen. C. Bone marrow from WT (CD45.2) and IFNAR−/− (CD45.1) mice was combined at a 1:1 ratio and transplanted into lethally irradiated C57BL/6 mice. Twelve weeks later, mice received CD45.1/2 WTbm12 CD4 T cells and composition of the B cell compartments in each part of the graft was assessed. Depicted are the percentages of WT vs. IFNAR−/− cells within GC or plasmablast B cell subsets normalized to the percentage of each genotype within the total B cell graft. For B and C, data from one representative experiment (out of 2 experiments with n=4-6/group) is shown as mean ± SEM, where *p≤0.05 and ***p<0.001. D. CTV-labeled purified B cells were cultured with 1 or 10 μg/ml anti-IgM and anti-CD40, with or without 100 U/ml IFNα and proliferation was assessed 96 h later. E/F. Purified B cells were cultured under the indicated conditions with or without 50 U/ml IFNα. Expression of CD40 and MHCII was assessed over time and Bcl-6 expression was assessed after 3 days by flow cytometry. Data are shown as mean ± SEM (n=3-4/group for each timepoint), where *p≤0.05 (one of 2 experiments is shown).
To circumvent this problem, we generated chimeric mice with WT (CD45.2) and IFNAR−/− (CD45.1) BM mixed in a 1:1 ratio (Fig. 2Aii). In this setting the WT B cells facilitate the development of TFH, allowing the assessment of the direct effect of type I IFN sensing on B cell responses during disease by the comparison of WT and IFNAR−/− B cells in the same animal. Upon transfer of CD45.1/2 CD4bm12 T cells, all mice developed high frequencies of TFH and anti-dsDNA IgG levels (data not shown). However, compared to the WT B cell compartment, the development of both GC B cells and plasmablasts was significantly reduced in the IFNAR−/− B cell compartment (Fig. 2C). Together these data demonstrate a critical role for type I IFN signaling in the B cell response that cannot be overcome by other mediators involved in the disease development.
Type I IFN sensing by B cells has been shown to promote B cell survival and lower the threshold for BCR signaling, enhancing the proliferation of naïve B cells when stimulated with anti-IgM ((35) and Fig. 2D). Indeed, B cells exposed to IgM and type I IFN also increase expression of MHCII and CD40, molecules involved with cognate T:B interaction and requirements for optimal GC formation (Fig. 2E). Moreover, type I IFN sensing in IgM- and CD40-stimulated B cells increased the expression of Bcl-6, the transcription factor that is associated with GC B cell formation (Fig. 2F).
Together these data indicate that type I IFN has a direct effect on B cells and promotes B cell responses on the transcriptional level as well as stimulatory capacity towards T cells.
TFH cells require type I IFN for their accumulation
Genetic ablation of IFNAR can completely prevent disease development in various experimental models of SLE (4-6). While our data show that lack of IFN sensing by the hematopoietic compartment was critically important for disease development, the transfer of IFNAR-sufficient CD45.1 CD4bm12T cells into IFNAR recipients still resulted in partial disease (Fig. 1D and (7)). As these transferred cells are the drivers of disease, we next tested the contribution of IFN sensing by the donor CD4bm12T cells on disease development.
Compared to transfer of WT CD4bm12 T cells, transfer of IFNAR−/−CD45.1 CD4bm12 T cells (into WT recipients) resulted in a significantly reduced disease, illustrated by reduced TFH, GC B cells, plasmablasts and anti-dsDNA IgG levels (Fig. 3A,C). Importantly, transfer of IFNAR−/− CD45.1 CD4bm12 T cells into IFNAR−/− recipients completely abrogated disease development. Since TFH and GC B cell responses are reciprocally supportive of one another, we considered the possibility that type I IFN may have acted in trans to promote TFH expansion. We reasoned that type I IFN sensing by CD4bm12 T cells may increase their expression of costimulatory molecules such as IL-21 that support a more robust germinal center B cell response. An augmented GC B cell response may subsequently drive greater Tfh expansion. To test this hypothesis, we co-transferred congenically marked WT CD4bm12 T cells (CD45.2) and IFNAR−/−CD4bm12 T cells (CD45.1) into CD45.1/2 WT recipients (Fig. 3B). In this model the WT CD4bm12 T cells drive comparable GC B cell and plasmablast responses to those found in mice that received WT CD4 bm12 T cells only. Direct comparison between the WT and IFNAR−/−CD4bm12 T cells in the same animals still showed a significant reduction in TFH development in the IFNAR−/−CD4bm12 T cells (Fig. 3C), indicating a critical role of type I IFN sensing by the T cells that could not be overcome by other inflammatory mediators.
Figure 3. T cells require type I IFN sensing for TFH accumulation.
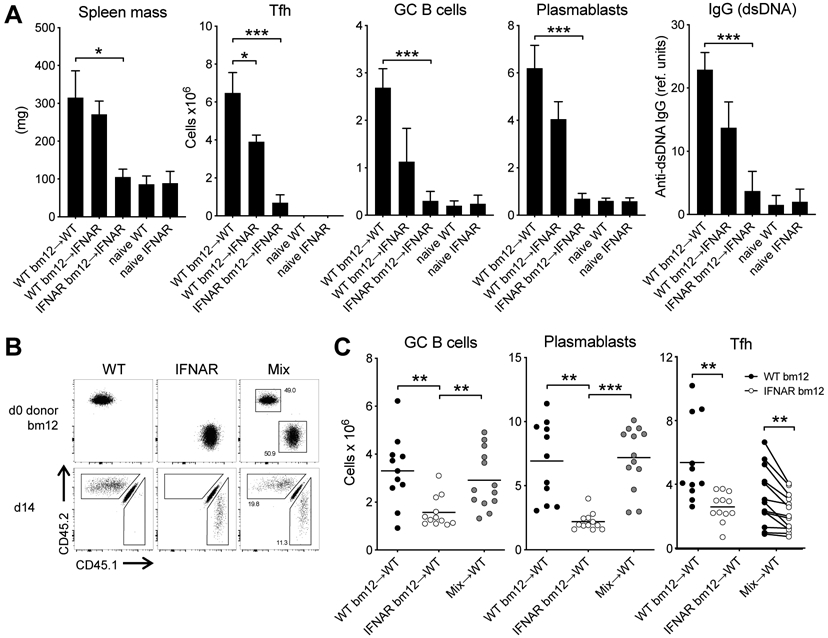
WT and IFNAR−/− CD45.1+CD4bm12 T cells were transferred into WT or IFNAR−/− recipients and responses were assessed 2 weeks later. Data from naive WT and naïve IFNAR mice are included as reference. A. Splenic mass, and absolute number of splenic donor TFH, GC B cells, plasmablasts and anti-dsDNA IgG levels are plotted. Data from one representative experiment (out of 2 experiments with n=4-5/group) is shown as mean ± SEM, where *p≤0.05, and ***p<0.001. B. WT (CD45.2) and IFNAR−/− (CD45.1) CD4bm12 T cells were mixed in a 1:1 ratio and transferred into WT CD45.1/2 recipients. As control, a parallel group of mice received only WT or only IFNAR−/− CD4bm12 T cells. All mice received a total of 7x106 CD4bm12 T cells. Flow data shows cells before injection and 2 weeks after injection (gated on live CD4+ cells). C. Absolute number of splenic GC B cells, plasmablasts and donor TFH in mice that received either WT, IFNAR−/− CD4bm12 T cells or the mixture. Data from one representative experiment (out of 3 experiments with n=11-12/group), where *p≤0.05, **p<0.01, and ***p<0.001.
Type I IFN is not required for proliferation or TFH development
To identify the mechanisms that drive the reduced IFNAR−/− TFH accumulation, we first assessed when TFH accumulation of WT and IFNAR−/− CD45.1 CD4bm12 T started to diverge. A time-course indicated that small differences were apparent within 3 days of transfer and became significant within 5 days after transfer (Fig. 4A). Proliferation dye indicated that only a proportion of transferred WT and IFNAR−/− CD4bm12 T cells proliferated and that the WT and IFNAR−/−CD4bm12 T cells that did proliferate showed a similar loss of the dye, indicating similar proliferation rates (Fig. 4B). The absolute number of non-proliferating cells remained similar between WT and IFNAR−/− CD4bm12 T cells while the absolute number of proliferating cells was significantly reduced for the IFNAR−/− CD4bm12 T cells (Fig. 4B). Analysis of Ki67 expression was low/absent in undivided cells and high in divided cells, resulting in a trend towards reduced Ki67 levels in the bulk IFNAR−/−CD4bm12 T cell population (Fig. 4C). A comparable percentage of WT and IFNAR CD4bm12 T cells also expressed phospho-gamma H2AX (pγH2AX), a well-characterized marker for the DNA damage response (DDR), shown to be highly elevated in in vivo activated and proliferating T cells (36).
Figure 4. Normal development but lack of accumulation of IFNAR−/− TFH.
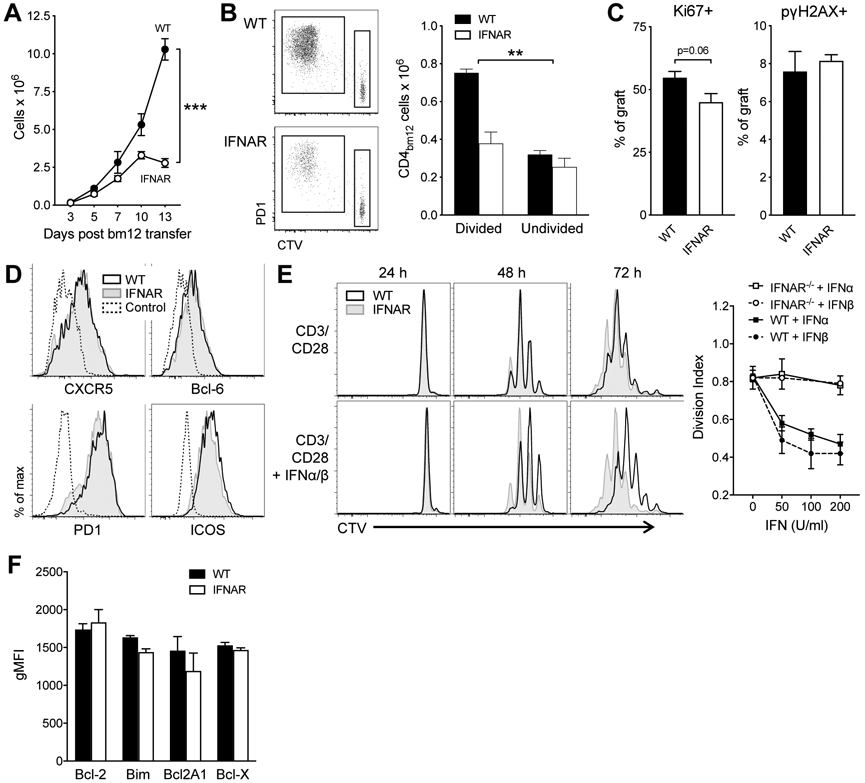
A. CTV-labeled WT and IFNAR−/− CD45.1 CD4bm12 T cells were transferred into CD45.2 WT recipients and the absolute number of splenic donor cells was assessed over time. Data are shown as mean ± SEM, with n=5-6/group, and exhibit significant interaction by a 2-way ANOVA (p<0.001). B-D. CTV-labeled WT or IFNAR−/− CD45.1+ CD4+ bm12 T cell cells, or control CTV-labeled CD45.1+ CD4+ H-2Kb T cells were transferred into B6 mice. Spleens were harvested 5 days later. B. Representative CTV-dilution plots (gated on live CD4+ CD45.1+) and absolute numbers of divided and undivided donor CD4bm12 T cells 5 days after transfer. Data are shown as mean ± SEM, with n=4-5/group, and exhibit significant interaction by a 2-way ANOVA (p<0.01). C. Proportion of Ki67+ and pγH2AX+ cells within the proliferating cells 5 days after transfer. D. Comparable expression of TFH-associated molecules in proliferating donor WT and IFNAR−/− CD45.1+ CD4bm12 T cells 5 days after transfer. Data are gated on live CD45.1+ CD4+ bm12 CTV-diluted cells, or CD45.1+ CD4+ undiluted control cells. E. Inhibition of proliferation by type I IFN. Purified naïve WT (CD45.2) and IFNAR−/− (CD45.1) CD4bm12 T cells were mixed (1:1 ratio), CTV labeled, and co-cultured with CD3/CD28 in the absence or presence of type I IFN. Proliferation was assessed 24, 48, and 72 h later. Representative CTV dilution plots are shown from 1 of 3 independent experiments. Day 3 proliferation data for varying concentrations of IFNα and IFNβ are also plotted using the Divisional Index calculated by the FlowJow proliferation modeling tool. F. Expression of pro- and anti-apoptotic markers in proliferating WT (black bar) and IFNAR−/− (white bar) CD4bm12 T cells 3 days after transfer (gating on live CD45.1+ CD4+ CTV diluted cells). Data are shown as mean ± SEM, with n=3-4/group.
Upon transfer, both WT and IFNAR−/−CD4bm12 T cells rapidly adopted a TFH phenotype and both genotypes expressed comparable levels of PD1, CXCR5, ICOS, and Bcl-6 (Fig. 4D). Together these data suggest that differences in proliferation rate, either directly driven by type I IFN or as a consequence of different phenotype development, were not responsible for the differences in TFH accumulation.
In vivo proliferation data can be skewed by silent clearance of dying cells or migration away from target organs. Analysis of a wide variety of lymphoid and non-lymphoid tissues eliminated the differences in migratory capacity as a critical contributor to the phenotype as all lymphoid tissues and the peritoneal cavity showed similar reduction in IFNAR−/− CD4bm12 T cells and the transferred cells (WT and IFNAR−/−) were not detectable in non-lymphoid tissues at this timepoint (not shown).
To assess the effect of IFN sensing on CD4 T cell proliferation and survival in vitro, purified CD4bm12 T cells from WT (CD45.2) and IFNAR−/− (CD45.1) donors were combined at a 1:1 ratio, labeled with a proliferation tracking dye, and stimulated with anti-CD3/CD28 in the absence or presence of type I IFN. Type I IFN inhibited the proliferation of activated WT, but not IFNAR−/− CD4 T cells, demonstrating that the anti-proliferative effect of IFN involved direct signaling in the CD4bm12 T cells (Fig. 4E). As these data were seemingly at odds with the phenomenon observed in vivo, where IFN sensing allowed for the increased accumulation of WT CD4bm12 T cells, we therefore shifted our focus toward the potential effect of type I IFN on the survival of these cells. However, analysis of pro- and anti-apoptotic molecules did not show a difference between WT and IFNAR−/−CD4bm12 T cells 5 days after transfer and IFNAR−/− CD45.1 CD4bm12 T cell had comparable levels of pγH2AX+ cells (Fig. 4C,F). In line with this finding, both WT and IFNAR−/− CD45.1 CD4bm12 T cells showed similar rates of cell death when cultured ex vivo in medium or in the presence of type I IFN (data not shown).
Type I IFN sensing protects TFH from perforin-dependent cell death
Since type I IFN imparted no obvious cell-intrinsic survival advantage, we hypothesized that IFN may be protecting TFH from cell-extrinsic pressure, either control by regulatory T cells or killing by NK cells or CD8 T cells.
The frequency and total number of CD4+Foxp3+CD25+ cells in the spleens did increase slightly between day 3 and day 7 after transfer, but no difference between WT and IFNAR−/− recipient groups was seen. Similarly, 7 days after transfer, WT and IFNAR−/− CD45.1 CD4bm12 T cells contained comparable proportions of Tregs and elimination of recipient Tregs before and during the T cell transfer did not restore the IFNAR−/− CD4bm12 TFH accumulation (Fig S1.). These observations argue against a specific role for Treg-mediated suppression of the IFNAR−/− TFH.
To assess susceptibility to killing, WT and IFNAR−/−CD4bm12 T cells were transferred into WT or perforin-deficient (Prf1−/−) recipients. Transfer of IFNAR−/−CD4bm12 T cells into Prf1−/− recipients completely restored accumulation of IFNAR−/−CD4bm12 TFH cells concomitant with increases in plasmablasts, GC, and anti-dsDNA IgG levels (Fig. 5).
Figure 5. Type I IFN protects TFH from perforin-mediated destruction.
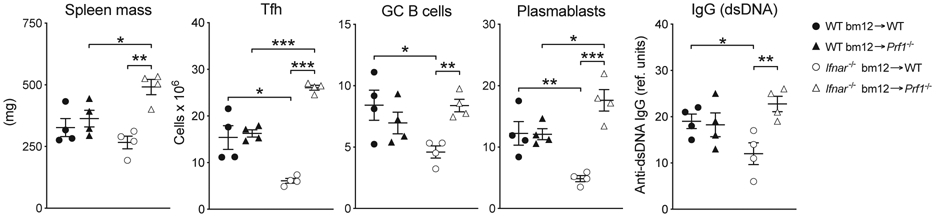
WT (black bar) or IFNAR−/− (white bar) CD4bm12 T cells were transferred into C57BL/6 (+) or Perforin-deficient (−) animals. Two weeks after transfer, splenic weight, and the total number of splenocytes, donor TFH, GC B cells, and plasmablasts was determined. Total anti-dsDNA IgG levels in the serum were determined by ELISA. A representative experiment (of 4) is shown as mean ± SEM, with n=4/group, where *p≤0.05, **p<0.01, and ***p<0.001.
Type I IFN protects TFH cells from NK cell-mediated killing
Based on the kinetic accumulation data, both CD8 T cells and NK cells could be involved in the elimination of the IFNAR−/− TFH. Depletion of CD8 T cells before IFNAR−/−CD4bm12 T cell transfer did not restore TFH accumulation, nor the number of GC B cells, plasmablasts and anti-dsDNA IgG levels (Fig. 6A).
Figure 6. Type I IFN protects TFH cells from NK cell-mediated killing.
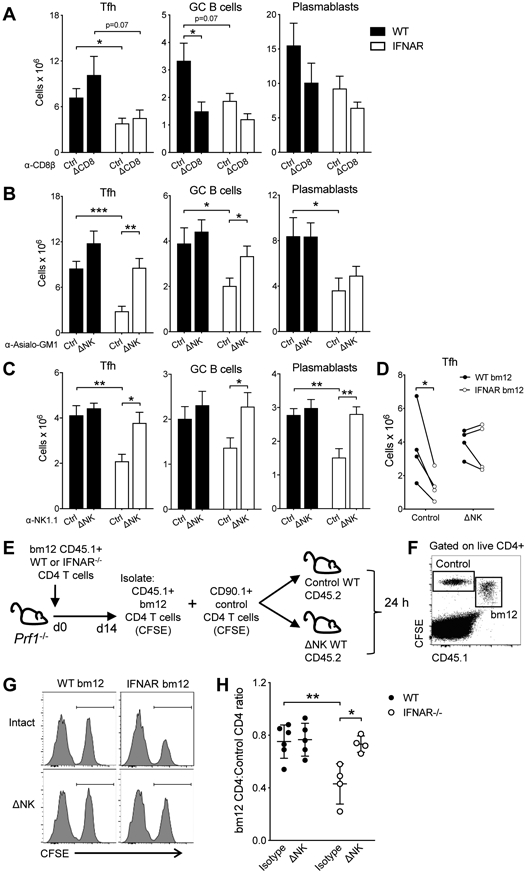
A-C. WT (black bar) or IFNAR−/− (white bar) CD45.1 CD4bm12 T cells were transferred into intact or antibody-depleted CD45.2 mice. Two weeks after transfer, mice were assessed for the absolute splenic number of donor TFH, GC B cells and plasmablasts in each graft. Mice were kept intact or treated with anti-CD8β (A), anti-Asialo-GM1 (B) or anti-NK1.1 (C) antibodies. D. WT (CD45.2, black circles) and IFNAR −/− (CD45.1, white circles) CD4bm12 T cells were mixed in a 1:1 ratio and transferred into intact or anti-Asialo-GM1 treated (ΔNK) CD45.1/2 recipients. The absolute splenic number of donor TFH in each graft was determined after 2 weeks. A representative experiment for each depletion approach is shown (out of 2-5 individual experiments/depletion approach with each experiment n=4-6/group) as mean ± SEM, where *p≤0.05, **p<0.01, and ***p<0.001. E-H. WT and IFNAR−/− CD645.1+ CD90.2+ CD4bm12 T cells were transferred into Prf−/− CD45.2+ CD90.2+ recipients. After 2 weeks, CD45.1 CD4 bm12 T cells were sorted, CFSE-labeled and mixed in a 1:1 ratio with CFSE-labeled control CD90.1+ CD4 T cells. A total of 4x106 T cells was transferred into intact or NK-depleted (anti-Asialo-GM1) CD45.2+ CD90.2+ WT mice. The ratio of CD45.1+ CD4 bm12 T cells to control cells was assessed 24 h later by flow cytometry. E. Schema of transfer approach. F. Example of flow cytometric data upon gating on live and CD4+ cells. G. Examples of histograms of CD90.1 control CD4 T cells and WT and IFNAR−/− CD45.1 CD4 bm12 T cells in different recipients (gated on live CD4+ CFSE+ cells). H. Ratio of CD45.1 CD4 bm12 T cells/CD90.1 CD4 T cells in each mouse. Black circles, WT CD4bm12 T cells; white circles, IFNAR−/− CD4bm12 T cells. Data are expressed as mean ± SEM with n=4-6/group, where *p≤0.05 and **p<0.01.
In contrast, NK cell depletion by either NK1.1 or anti-Asialo-GM1 prior to IFNAR−/−CD4bm12 T cell transfer restored TFH accumulation and subsequent GC B cell, plasmablast and anti-dsDNA IgG level development (Fig. 6B,C). To eliminate the effect of the changes on the GC B cell response on the accumulation of the TFH, WT (CD45.2) and IFNAR−/− (CD45.1) CD4bm12 T cells were transferred together into CD45.1/2 NK-cell depleted mice (as in fig 3). Again, NK cell depletion restored IFNAR−/− TFH accumulation, further confirming that direct type I IFN sensing by TFH protected them from NK cell-mediated killing (Fig. 6D).
To further confirm that IFNAR−/−CD4bm12 TFH cells are target cells for NK cells, Prf−/− mice were injected with WT or IFNAR−/−CD4bm12 T cells and TFH were harvested 2 weeks later. Cells were added in a 1:1 ratio with CD90.1 CD4 control T cells and transferred into intact or NK-depleted WT mice. The ratio of CD4bm12 T cells:CD90.1 control cells was assessed 24 h later (Fig. 6E,F). As expected, the ratio of WT CD4bm12 T cells/control T cells in intact and NK-depleted recipients did not change significantly. Transfer of IFNAR−/−CD4bm12 TFH into intact WT recipients resulted in a significant distortion of the TFH/control T cell ratio. In support of our in vivo studies, the IFNAR−/−CD4bm12 TFH /control CD4 T cell ratio remained unaltered and comparable to the WT TFH/control T cell ratio when cells were transferred into NK-depleted WT mice (Fig. 6G,H).
Importantly, the protective effect of type I IFN sensing by the TFH was not shared by the B cells. In mixed BM-chimeric mice from Fig. 2 (WT/IFNAR 1:1), NK-depletion did not restore GC B cell and plasmablast numbers in the IFNAR−/− B cell compartment (Fig. S2). These data indicate that although both the T cell and the B cell response required type I IFN sensing for their accumulation, the underlying mechanisms are distinct.
Discussion
A connection between the overproduction of type I IFN and the development of SLE has been well established, yet the mechanisms by which IFN promotes disease pathology have not been fully elucidated. Here we showed that type I IFN enhances development of pathogenic T and B cell responses in the bm12 cGVHD model of SLE-like disease through two different mechanisms: directly promoting B cell proliferation and GC differentiation and protecting TFH from NK cell-mediated killing.
Our observations that type I IFN enhances B cell responses fits well with existing data on the direct and indirect roles of type I IFN and induction of humoral responses (23-25, 35, 37). Type I IFNs have been shown to promote B cell survival and isotype switching through the induction of BLyS and APRIL in DC and macrophages (24, 38). However, our mixed bone marrow chimera models indicate that lack of type I IFN sensing by B cells cannot be overcome by other inflammatory mediators, implicating a more important contribution of direct type I IFN sensing in the B cell response. Intrinsic direct effects of type I IFN sensing encompass reducing BCR signaling threshold, increasing expression of molecules involved in cognate and reciprocal T:B cell interactions, cytokine production, migration, survival, as well as plasmablast development (25, 35, 37, 39-41). While our bone marrow chimera experiment would indicate a limited role for increased cytokine production, it is likely that combinations of several above-mentioned mechanisms are involved in the type I IFN-associated boosting of the B cell response in SLE. IFNAR−/− B cells were still able to mount a partial response, pointing to the involvement of other stimulatory mechanisms in the auto-antibody response. Further dissection of the relative contribution of these mechanisms might point to additional pathways that can be therapeutically targeted in disease, either with or without type I IFN interfering therapy.
Our studies show that type I FN sensing by TFH cells protects them from NK cell-mediated killing which is crucial for their accumulation and promotion of the B cell response. The potential for immunoregulation of T cell activity by NK cells has been uncovered in recent years. Mounting evidence has established a role for NK cells in limiting T cell responses toward lymphocytic choriomeningitis virus (LCMV) by direct killing of virus-specific T cells (42-45). Recently, NK cytotoxicity toward LCMV-specific T cells was shown to be inhibited by type I IFN (44, 46). These reports were consistent with data published over four decades earlier, in which Hansson, Welsh, and colleagues showed that either LCMV infection, the induction of type I IFN by poly I:C, or pre-treatment with exogenous IFN reduced the sensitivity of normal mouse thymocytes to NK cells in vitro (47, 48). Here, we present the novel finding that type I IFN also inhibits in vivo regulation of activated CD4 T cells by NK cells in a sterile model of systemic autoimmune disease. Taken together, these data suggest the intriguing possibility that peripheral tolerance may be maintained in part by the default NK killing of highly active T cells, and that activated T cells can be licensed by type I IFN during certain conditions. Aberrant overproduction of type I IFN in a healthy individual could contribute to the inadvertent licensing of self-reactive T cells and might thus be capable of initiating autoimmune disease. This may be one mechanism behind the induction of autoimmunity, including SLE, which occurs in some patients after the administration of type I IFN to treat conditions such as chronic hepatitis C viral infection (8-10).
Several studies in SLE patients and experimental mouse models of SLE-like disease indicate that NK cells are dysregulated and decreased in number and cytolytic capacity (49-55). In the bm12 cGVHD model of SLE, NK cells are reduced and display a more immature phenotype (Fig. S3A-C). In addition to the relative paucity of intermediate maturity (CD11b+CD27+) NK cells, the NK cells remaining showed reductions in a number of activating and inhibitory NK cell receptors (Fig. S3D). However, the remaining NK cells were still able to kill IFNAR−/− TFH, indicating that at least part of their cytolytic machinery was functional. As this remaining cytolytic capacity was not sufficient to affect WT TFH accumulation and WT TFH numbers were maintained in the in vivo NK cell killing assay (Fig. 6), this raises the possibility that WT TFH are more resistant to NK cell-mediated killing through the differential expression of activating and inhibitory NK cell ligands. Comparing expression of various known NK cell activating and inhibiting ligands on WT and IFNAR−/− TFH in intact mice showed notable shifts in expression of activating (Rae1, Mult-1, CD48, ICAM-1/2) and inhibitory (H-2Db/Kb) NK ligands. We consistently observed increases in the expression of several NK cell receptor activating ligands in CD4bm12 TFH cells compared to CD4 T cells from naïve mice, however these changes were not unique to IFNAR−/− TFH cells from NK-depleted mice, suggesting the primary mechanism of protection from NK killing mediated by type I IFN was not through decreasing these particular signals (Fig. S4). Class I MHC (H2-Db and H2-Kb) was increased in WT TFH compared to IFNAR−/− TFH and naïve CD4 T cells, indicating one possible mechanism for type I IFN-induced protection from NK cell killing, though these shifts were relatively small and were not completely restored in the IFNAR−/− TFH upon NK cell depletion. One of the recent studies on NK immunoregulation in the context of LCMV showed that type I IFN protected SMARTA and P14 T cells by upregulating an–as of yet–unidentified Ncr1 ligand or ligands (46). However, we were unable to detect differences in Ncr1 ligands on our polyclonal endogenous WT and IFNAR−/− TFH upon staining with an Ncr1-hIgG fusion protein. These data suggest that the lack of WT TFH sensitivity to NK cells could involve a complex combination of signals, a critical effector ligand or ligands that is/are still unknown, a TFH-intrinsic intracellular compensatory mechanism for the cytolytic assaults, or a combination of these.
With the rise of therapeutics that target the type I IFN pathway it will be important to evaluate the relative effect of the treatment on the different immune cells. While one can envision that simply eliminating or reducing type I IFN would render the TFH susceptible to NK cell-mediated killing, it does not necessarily reinvigorate the disease-affected NK cells. The capacity of the host to properly kill the TFH is critically important as our in vitro and in vivo data indicates that type I IFN has an anti-proliferative effect. In vitro stimulation of human and mouse T cells in the presence of type I IFN significantly inhibits cell division (Fig. 4 and (56, 57)). Moreover, in the absence of perforin, IFNAR−/− TFH showed significantly more accumulation than WT TFH (Fig. 5), illustrating the in vivo proliferative potential of TFH in the absence of type I IFN sensing. This raises the interesting possibility that bolstering NK cell numbers or their functionality might be an appropriate therapeutic approach when combined with type I IFN-interfering drugs.
While the current data set justify targeting type I IFN in SLE like disease, additional studies are needed to more precisely appreciate the many functions of type I IFN in order to enable the rational design of more targeted therapies for SLE and other autoimmune diseases with pathological type I IFN production.
Supplementary Material
Acknowledgements
We are grateful to Dr. Stephen Waggoner for providing reagents and discussion, to Jeff Bailey and Victoria Sumney in the Comprehensive Mouse and Cancer Core for help with bone marrow chimera experiments, to Alyssa Sproles for technical support with the Luminex assays, and to Monica DeLay and the CCHMC Flow Core for flow cytometry and cell sorting support.
This work was supported in part by the Lupus Research Institute, NCI grant CA138617, NIDDK grant DK090978, Charlotte Schmidlapp Award (to EMJ), and the Albert J. Ryan Fellowship and NIAID training grant T32-AI074491-09 (to JK).
Footnotes
Disclosures
The authors have no financial conflicts of interest to disclose.
References
- 1.Banchereau J, and Pascual V. 2006. Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity 25: 383–392. [DOI] [PubMed] [Google Scholar]
- 2.Pascual V, Farkas L, and Banchereau J. 2006. Systemic lupus erythematosus: all roads lead to type I interferons. Curr Opin Immunol 18: 676–682. [DOI] [PubMed] [Google Scholar]
- 3.Baccala R, Gonzalez-Quintial R, Schreiber RD, Lawson BR, Kono DH, and Theofilopoulos AN. 2012. Anti-IFN-alpha/beta receptor antibody treatment ameliorates disease in lupus-predisposed mice. J Immunol 189: 5976–5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jorgensen TN, Roper E, Thurman JM, Marrack P, and Kotzin BL. 2007. Type I interferon signaling is involved in the spontaneous development of lupus-like disease in B6.Nba2 and (B6.Nba2 x NZW)F(1) mice. Genes Immun 8: 653–662. [DOI] [PubMed] [Google Scholar]
- 5.Nacionales DC, Kelly-Scumpia KM, Lee PY, Weinstein JS, Lyons R, Sobel E, Satoh M, and Reeves WH. 2007. Deficiency of the type I interferon receptor protects mice from experimental lupus. Arthritis Rheum 56: 3770–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Santiago-Raber ML, Baccala R, Haraldsson KM, Choubey D, Stewart TA, Kono DH, and Theofilopoulos AN. 2003. Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J Exp Med 197: 777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klarquist J, Hennies CM, Lehn MA, Reboulet RA, Feau S, and Janssen EM. 2014. STING-mediated DNA sensing promotes antitumor and autoimmune responses to dying cells. J Immunol 193: 6124–6134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ahmed MM, Berney SM, Wolf RE, Hearth-Holmes M, Hayat S, Mubashir E, Vanderheyde H, Chang WL, and King JW. 2006. Prevalence of active hepatitis C virus infection in patients with systemic lupus erythematosus. Am J Med Sci 331: 252–256. [DOI] [PubMed] [Google Scholar]
- 9.Ramos-Casals M, Font J, Garcia-Carrasco M, Cervera R, Jimenez S, Trejo O, de la Red G, Sanchez-Tapias JM, and Ingelmo M. 2000. Hepatitis C virus infection mimicking systemic lupus erythematosus: study of hepatitis C virus infection in a series of 134 Spanish patients with systemic lupus erythematosus. Arthritis Rheum 43: 2801–2806. [DOI] [PubMed] [Google Scholar]
- 10.Wilson LE, Widman D, Dikman SH, and Gorevic PD. 2002. Autoimmune disease complicating antiviral therapy for hepatitis C virus infection. Semin Arthritis Rheum 32: 163–173. [DOI] [PubMed] [Google Scholar]
- 11.Mathian A, Weinberg A, Gallegos M, Banchereau J, and Koutouzov S. 2005. IFN-alpha induces early lethal lupus in preautoimmune (New Zealand Black x New Zealand White) F1 but not in BALB/c mice. J Immunol 174: 2499–2506. [DOI] [PubMed] [Google Scholar]
- 12.Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, and Pascual V. 2003. Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 197: 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kennedy WP, Maciuca R, Wolslegel K, Tew W, Abbas AR, Chaivorapol C, Morimoto A, McBride JM, Brunetta P, Richardson BC, Davis JC Jr., Behrens TW, and Townsend MJ. 2015. Association of the interferon signature metric with serological disease manifestations but not global activity scores in multiple cohorts of patients with SLE. Lupus Sci Med 2: e000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Narain S, and Furie R. 2016. Update on clinical trials in systemic lupus erythematosus. Curr Opin Rheumatol 28: 477–487. [DOI] [PubMed] [Google Scholar]
- 15.Mok CC 2017. Biological and targeted therapies of systemic lupus erythematosus: evidence and the state of the art. Expert Rev Clin Immunol 13: 677–692. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka Y, Takeuchi T, Okada M, Ishii T, Nakajima H, Kawai S, Nagashima T, Hayashi N, Wang L, and Tummala R. 2019. Safety and tolerability of anifrolumab, a monoclonal antibody targeting type I interferon receptor, in Japanese patients with systemic lupus erythematosus: a multicenter, phase 2, open-label study. Mod Rheumatol: 1–18. [DOI] [PubMed] [Google Scholar]
- 17.Yao Y, Richman L, Higgs BW, Morehouse CA, de los Reyes M, Brohawn P, Zhang J, White B, Coyle AJ, Kiener PA, and Jallal B. 2009. Neutralization of interferon-alpha/beta-inducible genes and downstream effect in a phase I trial of an anti-interferon-alpha monoclonal antibody in systemic lupus erythematosus. Arthritis Rheum 60: 1785–1796. [DOI] [PubMed] [Google Scholar]
- 18.Trinchieri G 2010. Type I interferon: friend or foe? J Exp Med 207: 2053–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baccala R, Hoebe K, Kono DH, Beutler B, and Theofilopoulos AN. 2007. TLR-dependent and TLR-independent pathways of type I interferon induction in systemic autoimmunity. Nat Med 13: 543–551. [DOI] [PubMed] [Google Scholar]
- 20.Biermann MH, Veissi S, Maueroder C, Chaurio R, Berens C, Herrmann M, and Munoz LE. 2014. The role of dead cell clearance in the etiology and pathogenesis of systemic lupus erythematosus: dendritic cells as potential targets. Expert Rev Clin Immunol 10: 1151–1164. [DOI] [PubMed] [Google Scholar]
- 21.Gaipl US, Munoz LE, Grossmayer G, Lauber K, Franz S, Sarter K, Voll RE, Winkler T, Kuhn A, Kalden J, Kern P, and Herrmann M. 2007. Clearance deficiency and systemic lupus erythematosus (SLE). J Autoimmun 28: 114–121. [DOI] [PubMed] [Google Scholar]
- 22.Blanco P, Palucka AK, Gill M, Pascual V, and Banchereau J. 2001. Induction of dendritic cell differentiation by IFN-alpha in systemic lupus erythematosus. Science 294: 1540–1543. [DOI] [PubMed] [Google Scholar]
- 23.Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, and Banchereau J. 2003. Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity 19: 225–234. [DOI] [PubMed] [Google Scholar]
- 24.Le Bon A, Schiavoni G, D'Agostino G, Gresser I, Belardelli F, and Tough DF. 2001. Type i interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 14: 461–470. [DOI] [PubMed] [Google Scholar]
- 25.Le Bon A, Thompson C, Kamphuis E, Durand V, Rossmann C, Kalinke U, and Tough DF. 2006. Cutting edge: enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. J Immunol 176: 2074–2078. [DOI] [PubMed] [Google Scholar]
- 26.Havenar-Daughton C, Kolumam GA, and Murali-Krishna K. 2006. Cutting Edge: The direct action of type I IFN on CD4 T cells is critical for sustaining clonal expansion in response to a viral but not a bacterial infection. J Immunol 176: 3315–3319. [DOI] [PubMed] [Google Scholar]
- 27.Le Bon A, Durand V, Kamphuis E, Thompson C, Bulfone-Paus S, Rossmann C, Kalinke U, and Tough DF. 2006. Direct stimulation of T cells by type I IFN enhances the CD8+ T cell response during cross-priming. J Immunol 176: 4682–4689. [DOI] [PubMed] [Google Scholar]
- 28.Klarquist J, and Janssen EM. 2015. The bm12 Inducible Model of Systemic Lupus Erythematosus (SLE) in C57BL/6 Mice. J Vis Exp: e53319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morris SC, Cohen PL, and Eisenberg RA. 1990. Experimental induction of systemic lupus erythematosus by recognition of foreign Ia. Clin Immunol Immunopathol 57: 263–273. [DOI] [PubMed] [Google Scholar]
- 30.Kolumam GA, Thomas S, Thompson LJ, Sprent J, and Murali-Krishna K. 2005. Type I interferons act directly on CD8 T cells to allow clonal expansion and memory formation in response to viral infection. J Exp Med 202: 637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang Z, Georgel P, Du X, Shamel L, Sovath S, Mudd S, Huber M, Kalis C, Keck S, Galanos C, Freudenberg M, and Beutler B. 2005. CD14 is required for MyD88-independent LPS signaling. Nat Immunol 6: 565–570. [DOI] [PubMed] [Google Scholar]
- 32.Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, and Sallusto F. 2013. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity 38: 596–605. [DOI] [PubMed] [Google Scholar]
- 33.Stebegg M, Kumar SD, Silva-Cayetano A, Fonseca VR, Linterman MA, and Graca L. 2018. Regulation of the Germinal Center Response. Front Immunol 9: 2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kerfoot SM, Yaari G, Patel JR, Johnson KL, Gonzalez DG, Kleinstein SH, and Haberman AM. 2011. Germinal center B cell and T follicular helper cell development initiates in the interfollicular zone. Immunity 34: 947–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Braun D, Caramalho I, and Demengeot J. 2002. IFN-alpha/beta enhances BCR-dependent B cell responses. Int Immunol 14: 411–419. [DOI] [PubMed] [Google Scholar]
- 36.McNally JP, Millen SH, Chaturvedi V, Lakes N, Terrell CE, Elfers EE, Carroll KR, Hogan SP, Andreassen PR, Kanter J, Allen CE, Henry MM, Greenberg JN, Ladisch S, Hermiston ML, Joyce M, Hildeman DA, Katz JD, and Jordan MB. 2017. Manipulating DNA damage-response signaling for the treatment of immune-mediated diseases. Proc Natl Acad Sci U S A 114: E4782–E4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kiefer K, Oropallo MA, Cancro MP, and Marshak-Rothstein A. 2012. Role of type I interferons in the activation of autoreactive B cells. Immunol Cell Biol 90: 498–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wolf AI, Mozdzanowska K, Quinn WJ 3rd, Metzgar M, Williams KL, Caton AJ, Meffre E, Bram RJ, Erickson LD, Allman D, Cancro MP, and Erikson J. 2011. Protective antiviral antibody responses in a mouse model of influenza virus infection require TACI. J Clin Invest 121: 3954–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shiow LR, Rosen DB, Brdickova N, Xu Y, An J, Lanier LL, Cyster JG, and Matloubian M. 2006. CD69 acts downstream of interferon-alpha/beta to inhibit S1P1 and lymphocyte egress from lymphoid organs. Nature 440: 540–544. [DOI] [PubMed] [Google Scholar]
- 40.Badr G, Saad H, Waly H, Hassan K, Abdel-Tawab H, Alhazza IM, and Ahmed EA. 2010. Type I interferon (IFN-alpha/beta) rescues B-lymphocytes from apoptosis via PI3Kdelta/Akt, Rho-A, NFkappaB and Bcl-2/Bcl(XL). Cell Immunol 263: 31–40. [DOI] [PubMed] [Google Scholar]
- 41.de Goer de Herve MG, Durali D, Dembele B, Giuliani M, Tran TA, Azzarone B, Eid P, Tardieu M, Delfraissy JF, and Taoufik Y. 2011. Interferon-alpha triggers B cell effector 1 (Be1) commitment. PLoS One 6: e19366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Welsh RM, and Waggoner SN. 2013. NK cells controlling virus-specific T cells: Rheostats for acute vs. persistent infections. Virology 435: 37–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Waggoner SN, Cornberg M, Selin LK, and Welsh RM. 2011. Natural killer cells act as rheostats modulating antiviral T cells. Nature 481: 394–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu HC, Grusdat M, Pandyra AA, Polz R, Huang J, Sharma P, Deenen R, Kohrer K, Rahbar R, Diefenbach A, Gibbert K, Lohning M, Hocker L, Waibler Z, Haussinger D, Mak TW, Ohashi PS, Lang KS, and Lang PA. 2014. Type I interferon protects antiviral CD8+ T cells from NK cell cytotoxicity. Immunity 40: 949–960. [DOI] [PubMed] [Google Scholar]
- 45.Lang PA, Lang KS, Xu HC, Grusdat M, Parish IA, Recher M, Elford AR, Dhanji S, Shaabani N, Tran CW, Dissanayake D, Rahbar R, Ghazarian M, Brustle A, Fine J, Chen P, Weaver CT, Klose C, Diefenbach A, Haussinger D, Carlyle JR, Kaech SM, Mak TW, and Ohashi PS. 2012. Natural killer cell activation enhances immune pathology and promotes chronic infection by limiting CD8+ T-cell immunity. Proc Natl Acad Sci U S A 109: 1210–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Crouse J, Bedenikovic G, Wiesel M, Ibberson M, Xenarios I, Von Laer D, Kalinke U, Vivier E, Jonjic S, and Oxenius A. 2014. Type I interferons protect T cells against NK cell attack mediated by the activating receptor NCR1. Immunity 40: 961–973. [DOI] [PubMed] [Google Scholar]
- 47.Hansson M, Kiessling R, Andersson B, and Welsh RM. 1980. Effect of interferon and interferon inducers on the NK sensitivity of normal mouse thymocytes. J Immunol 125: 2225–2231. [PubMed] [Google Scholar]
- 48.Welsh RM, Karre K, Hansson M, Kunkel LA, and Kiessling RW. 1981. Interferon-mediated protection of normal and tumor target cells against lysis by mouse natural killer cells. J Immunol 126: 219–225. [PubMed] [Google Scholar]
- 49.Voynova EN, Skinner J, and Bolland S. 2015. Expansion of an atypical NK cell subset in mouse models of systemic lupus erythematosus. J Immunol 194: 1503–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Henriques A, Teixeira L, Ines L, Carvalheiro T, Goncalves A, Martinho A, Pais ML, da Silva JA, and Paiva A. 2013. NK cells dysfunction in systemic lupus erythematosus: relation to disease activity. Clin Rheumatol 32: 805–813. [DOI] [PubMed] [Google Scholar]
- 51.Pan LZ, Dauphinee MJ, Ansar Ahmed S, and Talal N. 1986. Altered natural killer and natural cytotoxic cellular activities in lpr mice. Scand J Immunol 23: 415–423. [DOI] [PubMed] [Google Scholar]
- 52.Ye Z, Ma N, Zhao L, Jiang ZY, and Jiang YF. 2016. Differential expression of natural killer activating and inhibitory receptors in patients with newly diagnosed systemic lupus erythematosus. Int J Rheum Dis 19: 613–621. [DOI] [PubMed] [Google Scholar]
- 53.Hervier B, Beziat V, Haroche J, Mathian A, Lebon P, Ghillani-Dalbin P, Musset L, Debre P, Amoura Z, and Vieillard V. 2011. Phenotype and function of natural killer cells in systemic lupus erythematosus: excess interferon-gamma production in patients with active disease. Arthritis Rheum 63: 1698–1706. [DOI] [PubMed] [Google Scholar]
- 54.Erkeller-Yuksel FM, Lydyard PM, and Isenberg DA. 1997. Lack of NK cells in lupus patients with renal involvement. Lupus 6: 708–712. [DOI] [PubMed] [Google Scholar]
- 55.Park YW, Kee SJ, Cho YN, Lee EH, Lee HY, Kim EM, Shin MH, Park JJ, Kim TJ, Lee SS, Yoo DH, and Kang HS. 2009. Impaired differentiation and cytotoxicity of natural killer cells in systemic lupus erythematosus. Arthritis Rheum 60: 1753–1763. [DOI] [PubMed] [Google Scholar]
- 56.Bekisz J, Baron S, Balinsky C, Morrow A, and Zoon KC. 2010. Antiproliferative Properties of Type I and Type II Interferon. Pharmaceuticals (Basel) 3: 994–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tanabe Y, Nishibori T, Su L, Arduini RM, Baker DP, and David M. 2005. Cutting edge: role of STAT1, STAT3, and STAT5 in IFN-alpha beta responses in T lymphocytes. J Immunol 174: 609–613. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.