

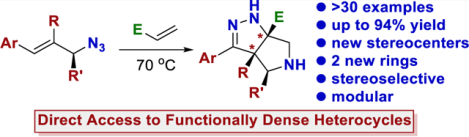
Abstract
Developing reactions to generate complex and modular building blocks in a concise and direct fashion remains a contemporary synthetic challenge. This work describes a stereo-selective cascade reaction between allylic azides and acrylates that directly generates tetrahydro-pyrrolo-pyrazole ring systems. These products contain up to four contiguous stereocenters, two of which may be tetrasubstituted carbon atoms attached to a nitrogen atom. Over 30 examples are provided with an average isolated yield of 71% (ranging from 40% to 94%). The reaction was easily scaled to use more than one gram of starting material, and the products can be readily diversified.
Graphical Abstract
INTRODUCTION
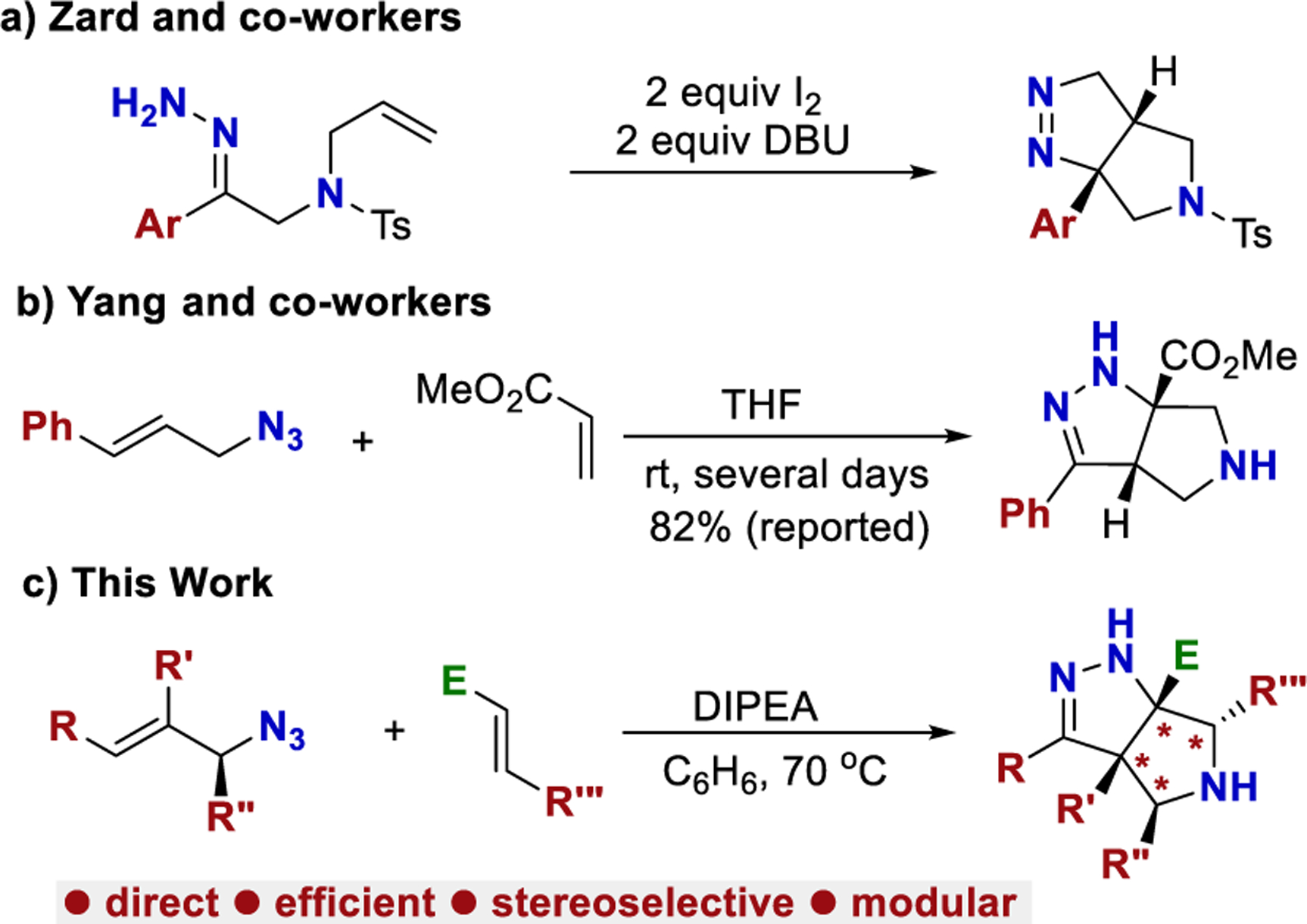
Nitrogen-containing heterocycles are ubiquitous in pharmaceuticals, agrochemicals, and natural products.1,2 Often, saturated heterocycles provide particularly advantageous properties.3−7 Specifically, substituted tetrahydro-pyrrolo-pyrazole heterocycles are known to be biologically active with a variety of potential applications (Figure 1).8−12 A direct and efficient synthesis of these functionally dense heterocycles from simple starting materials would be empowering.13,14 Classically, tetrahydro-pyrrolo-pyrazole heterocycles are generated through cycloaddition of a diazo species with activated maleimide derivatives.8,10 More recently, Zard demonstrated that hydrazones with pendant alkenes can be oxidized to the corresponding diazo species, which subsequently undergoes an intramolecular cycloaddition (Scheme 1a).15 Jahn demonstrated that a domino aza-Michael reaction can afford similar heterocycles using n-BuLi and NfN3.16,17
Figure 1.
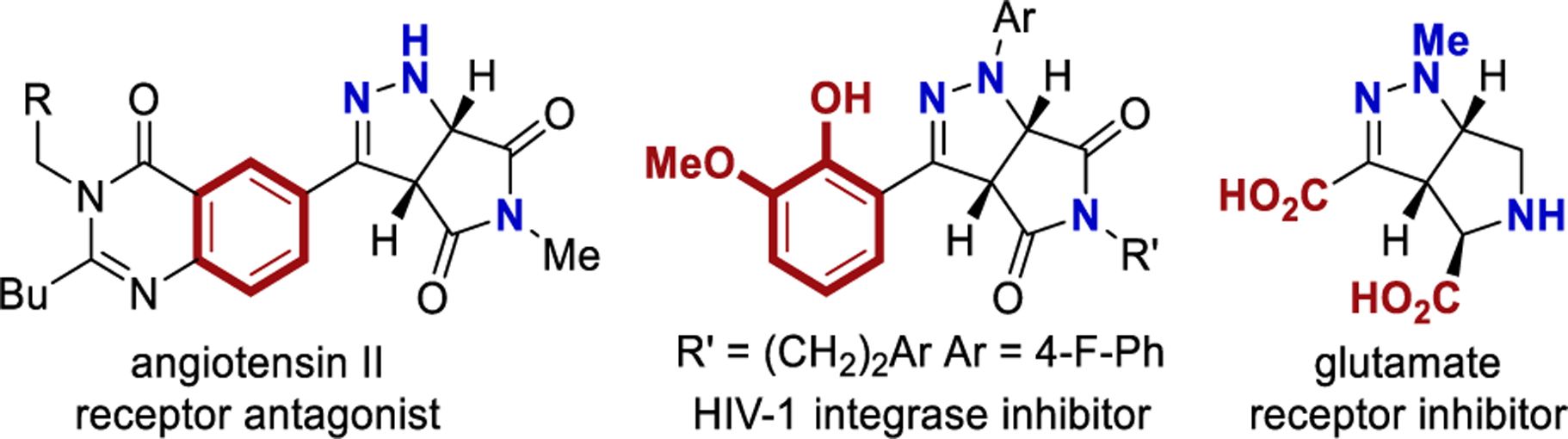
Biologically active fused heterocycles.
Scheme 1.
Synthesis of Tetrahydro-Pyrrolo-Pyrazoles
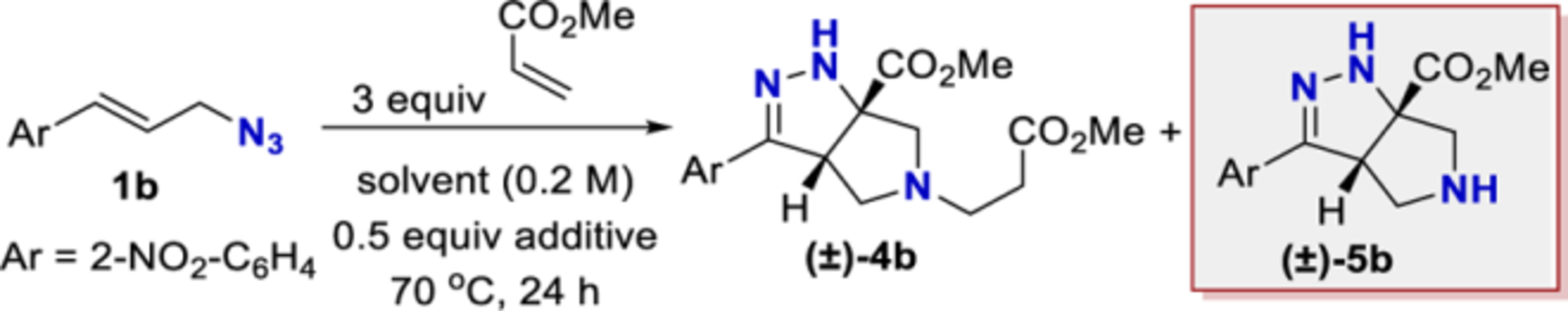
Recently, our lab has been developing synthetic methods that leverage the unique properties of allylic azides. We showed that allylic azides can react selectively at either the alkene18−20 or azide21 functional group. These results led us to ask if a reaction could synergistically utilize both functional groups. We were encouraged to find an example of such a reaction. Yang reported a cycloaddition cascade with cinnamyl azide and methyl acrylate to form tetrahydro-pyrrolo-pyrazole hetero-cycles (Scheme 1b).22,23 We were excited to build upon these reports. Unfortunately, neither report provided full experimental details. Our attempts to replicate the procedure using cinnamyl azide afforded a low yield (82% reported, less than 15% obtained). We speculated that this cascade could be a powerful tool if a more reproducible procedure could be developed and if the scope of this reaction was expanded.
This report provides a reoptimized and fully documented procedure that affords tetrahydro-pyrrolo-pyrazole hetero-cycles in high yields from simple cinnamyl azides. The substrate scope of Yang’s reaction is expanded to include (i) secondary and tertiary azides, (ii) cinnamyl azides substituted at the α or β carbon, (iii) the use of an enantioenriched azide, (iv) additional Michael acceptors, and (v) derivatization of the products (Scheme 1c).
RESULTS AND DISCUSSION
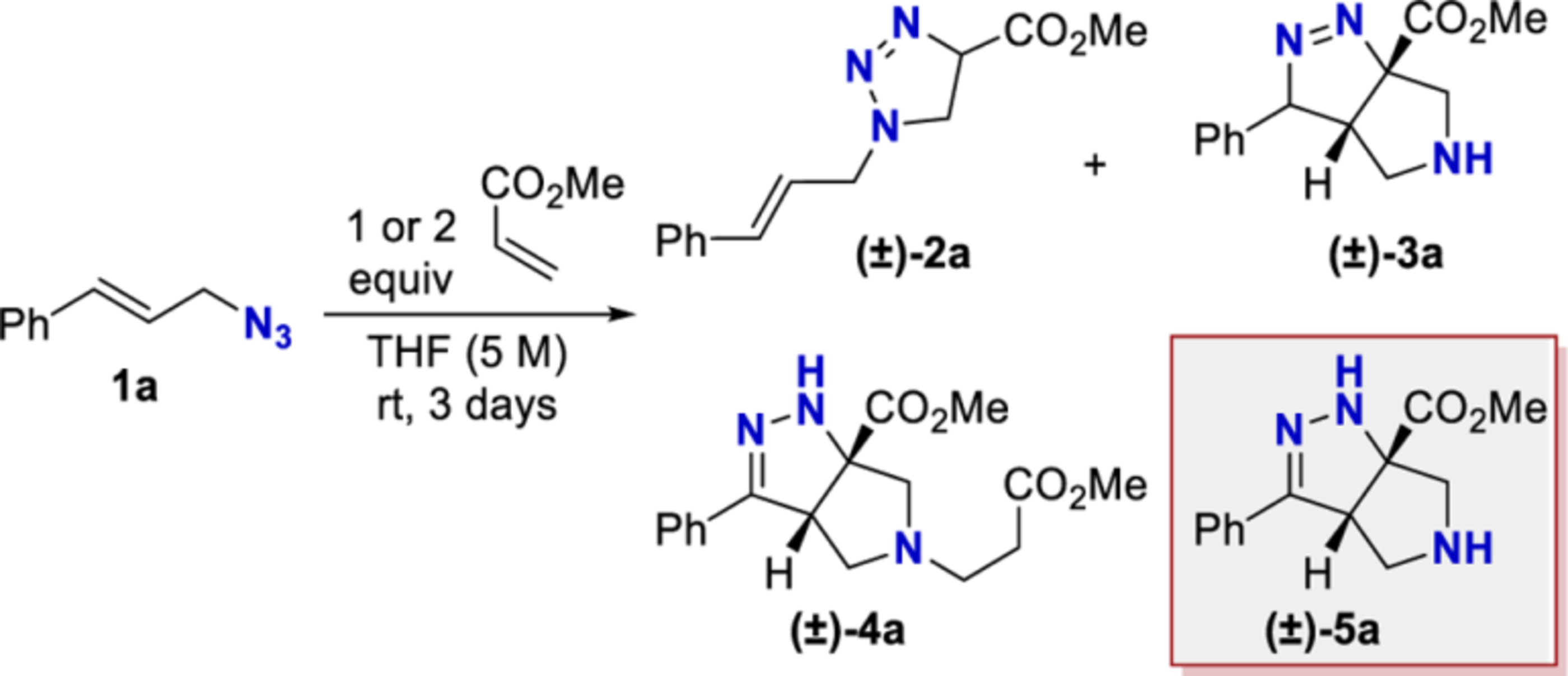
This study began by examining the thermally promoted cascade sequence between methyl acrylate and cinnamyl azide using the conditions originally reported by Yang (Table 1).22,23 Yang’s experimental procedure states that cinnamyl azide (5M in THF) was mixed with methyl acrylate (1 or 2 equiv) at room temperature for a time ranging between several hours to a few days.23 In our hands, the room-temperature reaction with 1 equiv of methyl acrylate did not fully consume the azide after 3 days (Table 1). Several intermediates and byproducts could be identified and quantified (2a–4a). A yield below 25% was observed, even if both observable tautomers of the desired product (3a and 5a) are combined. The reaction with cinnamyl azide 1a was extensively screened, and in no case did we observe a yield exceeding 70% (see Supporting Information, Tables S1−S3). These observations prompted a reoptimization of this cascade reaction.
Table 1.
Replicating Yang’s Reported Conditions
Reactions were conducted with azide 1a (90 μmol) and methyl acrylate (90 or 180 μmol) in THF (5 M) at rt. Conversion and yield were determined by 1H NMR. Reactions were run in duplicate, and the average value is reported. MA = methyl acrylate
Qualitatively, the cascade reaction appeared to proceed more efficiently with a cinnamyl azide featuring a polarizing aryl substituent. This was true regardless of if the substituent is commonly thought of as an electron-donating (e.g., OMe) or -withdrawing (e.g., NO2) group (vide infra). The substrate 2-nitrocinnamyl azide (1b) was selected for optimization along with methyl acrylate due to the availability of 2-nitrocinnamaldehyde (Table 2). Solvents with a range of polarities were examined (entries 1–5). Polar solvents promoted the conjugate addition that formed product 4b (entries 1 and 2), while less polar solvents suppressed the conjugate addition (entries 4 and 5). The observation that the formation of product 4b could be suppressed in less polar media is consistent with the need to form ionic intermediates during conjugate addition. When hexane was used, a significant amount of the conjugate addition was observed because the product was insoluble in hexane, resulting in a reaction that was pseudoneat in methyl acrylate (entry 3).
Table 2.
Cascade Optimization with Methyl Acrylate
 | |||||
|---|---|---|---|---|---|
| entry | solvent | additive | % 2ba | % 4ba | % 5ba |
| 1 | DMSO | nd | 14 | 77 | |
| 2 | MeOH | nd | 95 | n.d. | |
| 3 | hexane | nd | 54 | 35 | |
| 4 | THF | 34 | nd | 46 | |
| 5 | C6H6 | 37 | nd | 11 | |
| 6 | C6H6 | AcOH | nd | 67 | nd |
| 7 | C6H6 | HFIP | nd | 55 | 17 |
| 8 | C6H6 | DMAP | nd | 8 | 80 |
| 9 | C6H6 | pyridine | 5 | nd | 41 |
| 10 | C6H6 | TEA | nd | nd | 85 |
| 11 | C6H6 | DIPEA | nd | nd | 91 |
| 12 | PhMe | DIPEA | nd | nd | 83 |
| 13 | THF | TEA | nd | nd | 72 |
| 14 | THF | DIPEA | nd | nd | 78 |
Reactions were conducted with azide 1b (70 μmol), methyl acrylate (210 μmol), and an additive (35 μmol) in solvent (0.2 M) at 70 °C. Conversion and yield were determined by calibrated GC-FID analysis. Reactions were run in duplicate, and the average value is reported. Not detected = nd.
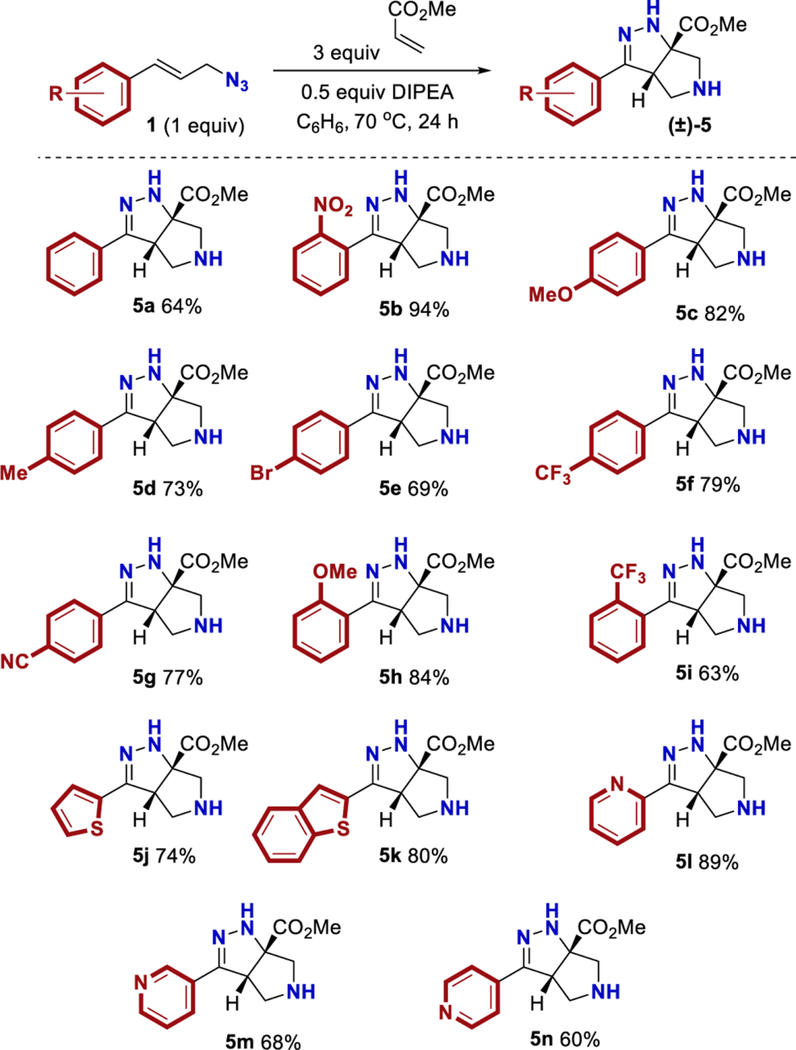
Having identified THF and benzene as the optimal solvents, several additives were examined. The addition of acetic acid or HFIP promoted the conjugate addition (entries 6 and 7). Prior work from L’abbe indicated that the triazoline to diazo-ester equilibrium could be facilitated by the addition of an amine base (vide infra).24,25 Thus, the addition of an amine base proved most effective at converting triazoline intermediate 2b to the desired product (entries 8–14). Given the complications associated with conjugate addition (e.g., polymerization of acrylate and over addition), it is perhaps unsurprising that DMAP and pyridine provided poor results and that less nucleophilic amine bases were preferred. Ultimately, conditions utilizing DIPEA in benzene (entry 11) were chosen as the optimal conditions. Toluene and THF were suitable alternative solvents (entries 12–14). It is worth noting that, at the end of the reaction, the desired product typically existed as a mixture of the two tautomers 3b and 5b. TFA was added during the workup to convert the mixture into the more stable conjugated tautomer 5b. In some instances, silica gel and DBU were also able to promote tautomerization (see Table 7 in Experimental Section). When compared to Yang’s original conditions, this reoptimization reflects a change in the (i) solvent, (ii) concentration, (iii) temperature, (iv) equivalents of acrylate, (v) time, and (vi) addition of DIPEA. The optimized reaction conditions work well with a variety of cinnamyl azides (Table 3). Compound 5b was isolated in 94% yield under the reoptimized conditions. The product from cinnamyl azide (5a) was crystalline, and diffraction analysis confirmed the structure (see Supporting Information). While the 64% yield of product 5a observed was a significant improvement over the data shown in Table 1, it was still lower than the yield originally reported by Yang.22,23 Other cinnamyl azides containing electron-donating groups (5c, 5d, and 5h) and electron-withdrawing groups (5f, 5g, and 5i) were tolerated. Substrates with ortho substitution were tolerated (5b, 5h, and 5i). Lastly, products with an electron-rich heteroarene (5j and 5k) or an electron-deficient heteroarene (5l−5n) were isolated in good yields. As previously noted, substrates with polarizing groups on the arene appeared to be better suited for this cascade, with some of the highest yields observed for products 5b and 5h as well as 5k and 5l. Recent attention has been focused on developing reactions that are effective with heterocycles,26,27 and the ability of this cascade to generate products 5j−5n under identical conditions should be considered an attribute.
Table 7.
Tautomerization with TFA and DBU
| additive (equiv) | time (h) | 9c′/9c |
|---|---|---|
| none | 0.5 | 55:45 |
| none | 18 | 55:45 |
| TFA (3) | 0.5 | 43:57 |
| TFA (3) | 18 | 0:100 |
| DBU (0.5) | 0.5 | 22:78 |
| DBU (0.5) | 18 | 0:100 |
Methyl (3aS,6aS)-5-Benzyl-3-(2-nitrophenyl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (9d).
Table 3.
Scope of the Cascade Reaction with Cinnamyl Azidesa
|
Yields are reported for isolated and purified products. Yield values reflect the average of duplicate trials.
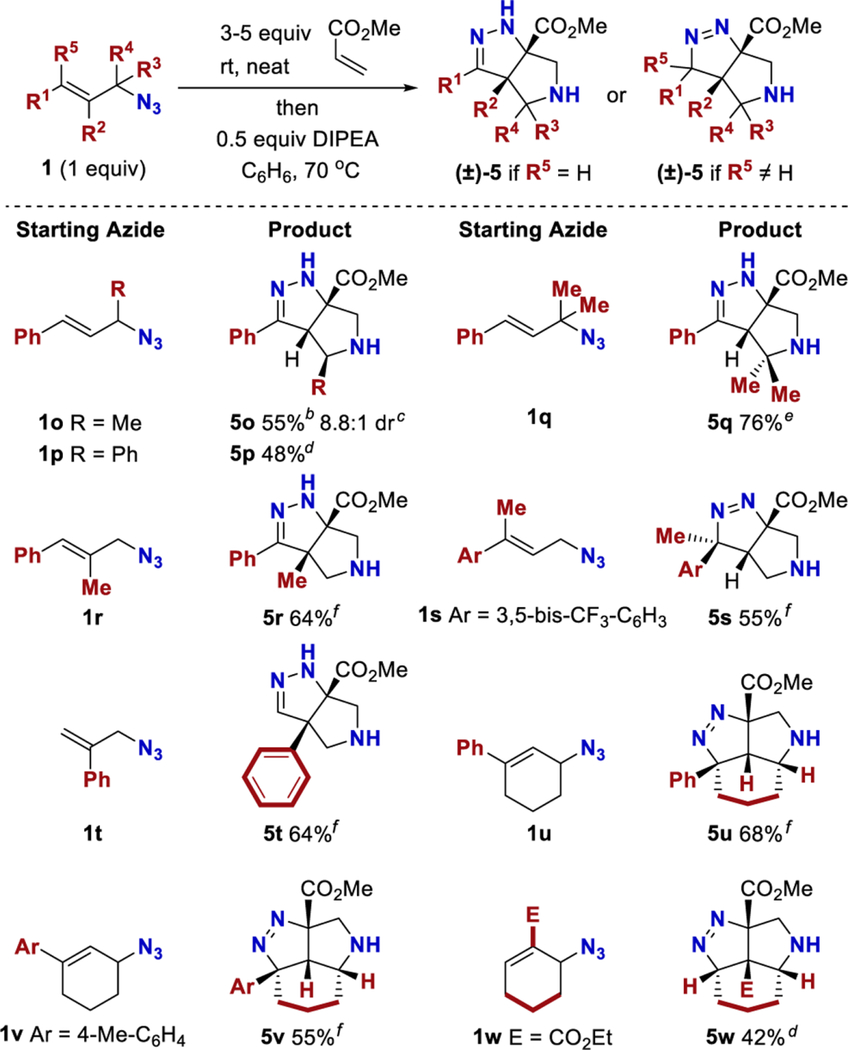
The substrate scope was further expanded (Table 4). A methyl or a phenyl group could be incorporated adjacent to the azide (5o and 5p). For product 5o, a minor diastereomer was observed that could arise from the Z-alkene isomer. An E/Z alkene isomerization could be occurring under the reaction conditions via the Winstein rearangement.28−30 The relative configuration of product 5p was assigned based on a crystal structure (see Supporting Information), and that of product 5o was assigned by analogy to product 5p. A minor diastereomer was not detected for compound 5p. Tertiary azide 1q afforded product 5q. A methyl group could be incorporated at the α or β position relative to the arene, affording compounds 5r and 5s. In both cases, a minor diastereomer was not detected. The relative configuration of compound 5s was assigned by 2D NMR and is consistent with the alkene geometry of the starting material as would be expected for a cycloaddition. The trans relationship between the methyl and hydrogen on starting azide 1s should be maintained in the product if the reaction proceeds through a concerted cycloaddition. A branched allylic azide was suitable and afforded product 5t, which demonstrated that a cinnamyl azide was not necessary. Product 5t is noteworthy because significant eclipsing interactions likely exist between the bridgehead substituents. Furthermore, a cyclic azide afforded tricyclic products (5u− 5w). A proximal arene was not required for the reaction to proceed, and product 5w was derived from unsaturated ester 1w.
Table 4.
Reaction Scope with Substituted Allylic Azidesa
|
Yields are reported for isolated and purified products. Yield values reflect the average of duplicate trials.
Initial 72 h incubation at rt with an additional 24 h at 70 °C.
Diastereomeric ratio determined by crude 1H NMR spectroscopy.
Reaction for 48 h at 70 °C.
Reaction for 72 h at 70 °C.
Initial 48 h incubation at rt with an additional 24 h at 70 °C.
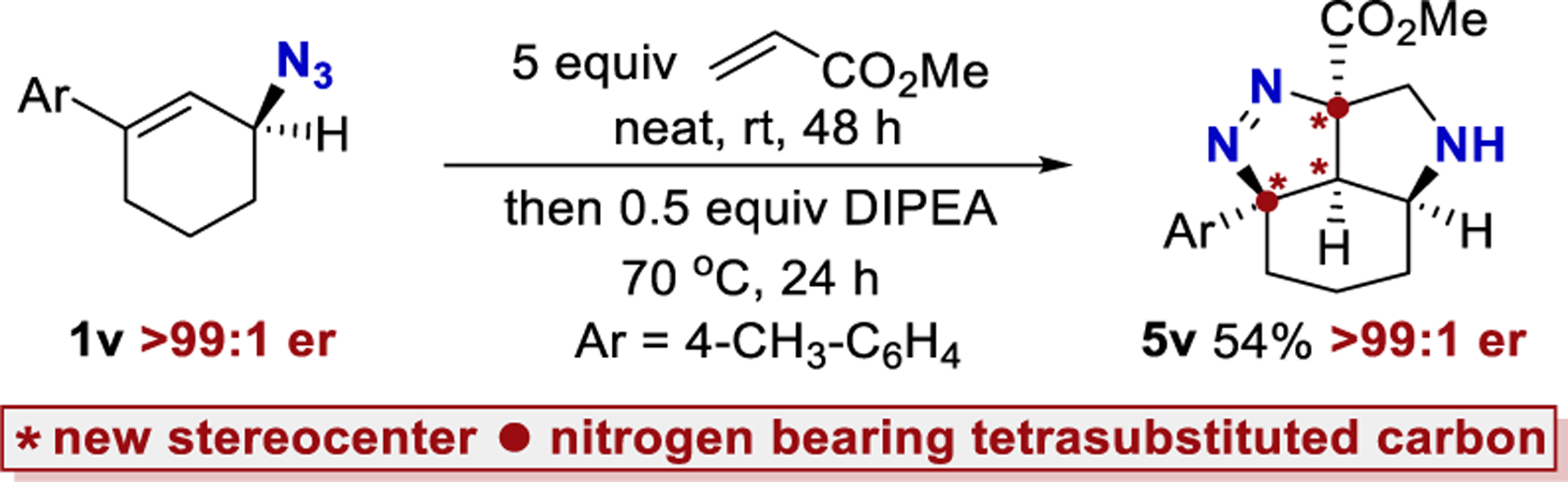
A single enantiomer of azide 1v (>99:1 er) was utilized (Scheme 2).30 Product 5v could be isolated as a single diastereomer with no loss in enantiomeric purity (>99:1 er). A crystal structure of (±)−5v·HCl was obtained to confirm its identity and relative stereochemistry (see Supporting Information). This represents a tremendous amplification of stereochemical complexity. Azide 1v contains only a single stereocenter. Product 5v contains four contiguous stereo-centers, three of which are generated from this cascade reaction. Two stereocenters are formed at tetrasubstituted carbon atoms that bear a nitrogen substituent, which are known to be particularly challenging stereocenters to establish.13,31
Scheme 2.
Stereospecific Cascade Reaction
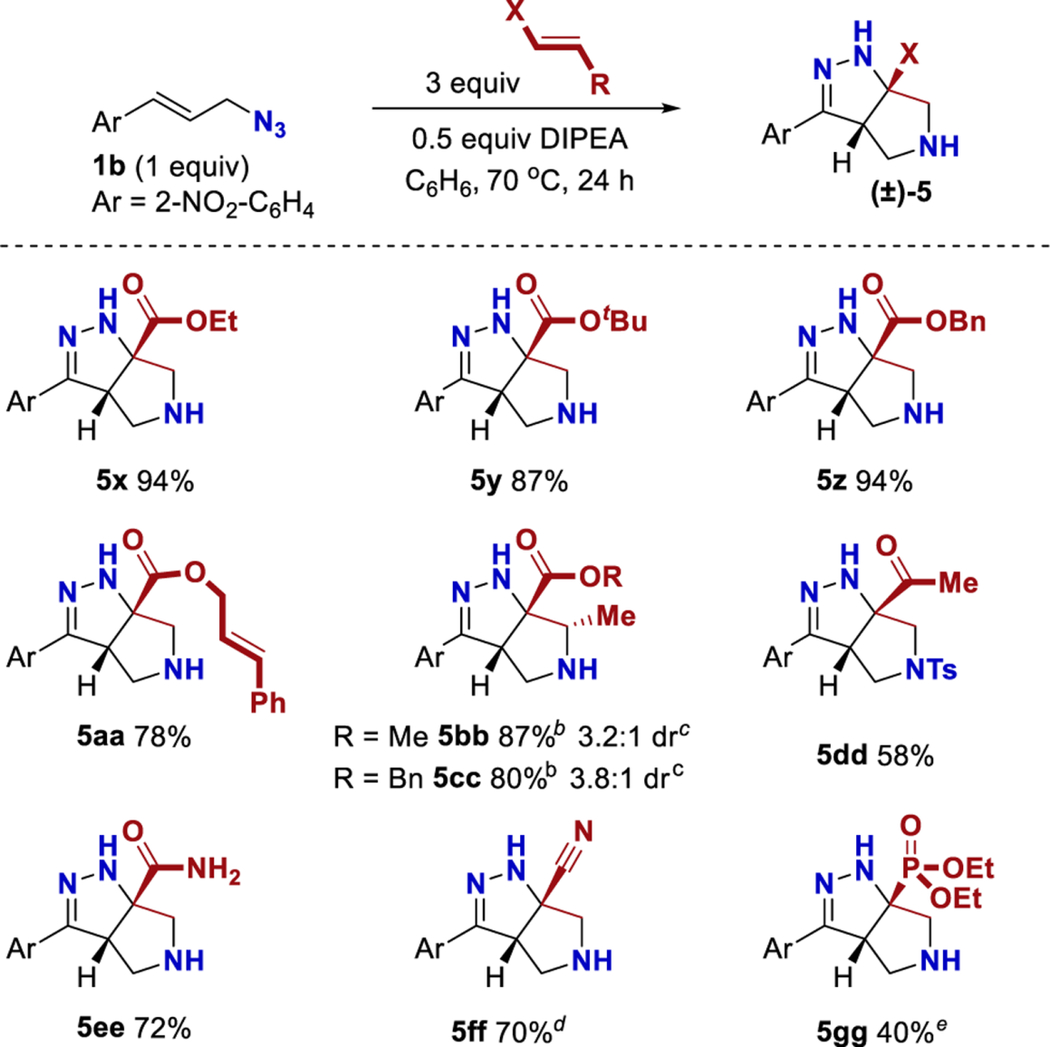
The reaction was investigated with other Michael acceptors (Table 5). Simple acrylates provided high yields, and ethyl (5x), tert-butyl (5y), and benzyl (5z) esters were isolated. The selective formation of cinnamate 5aa is interesting because the putative α-diazoester intermediate selectively engaged one of the two pendant alkenes. The use of methyl or benzyl crotonate afforded products 5bb and 5cc in high yields and modest dr’s. The relative configuration of compound 5bb was determined by X-ray diffraction analysis, and the relative configuration of product 5cc was assigned by analogy to product 5bb. The use of methyl vinyl ketone afforded exclusively the product of conjugate addition (4dd, see Experimental Section). To prevent this, TsCl and DMAP were added to trap the initial adduct prior to conjugate addition, which allowed the isolation of product 5dd. Other Michael acceptors including acrylamide (5ee), acrylonitrile (5ff), and diethyl vinyl phosphonate (5gg) were also effective as partners in this cascade reaction. This provides access to a diverse array of functional groups via this cascade.
Table 5.
Reaction Scope with Different Michael Acceptorsa
|
Yields are reported for isolated and purified products. Yield values reflect the average of duplicate trials.
Reaction for 5 days at 70 °C.
Diastereomeric ratio determined by crude 1H NMR spectroscopy.
Initial 24 h incubation neat at 40 °C. Then C6H6 and DIPEA were added for an additional 24 h at 40 °C.
The reaction was heated to 90 °C.
A plausible mechanism for this cascade reaction and side product formation is shown in Scheme 3. This proposal is based on the work of L’abbe24,25 and Yang.22,23 The cascade is thought to proceed via an initial Huisgen cycloaddition to afford triazoline 2.24,25,32−36 This cycloaddition generally proceeds with good selectivity, but the regioisomeric triazoline 2′ was frequently observed (~5%).24 When diethyl vinyl phosphonate was used as the Michael acceptor, triazoline 2gg′ was a significant byproduct (ca. 1:1). In some cases, the triazoline was stable to isomerization with a base and could be isolated. A crystal structure of one triazoline, derived from N-phenylmaleimide, was obtained to verify the structure (see Supporting Information).
Scheme 3.
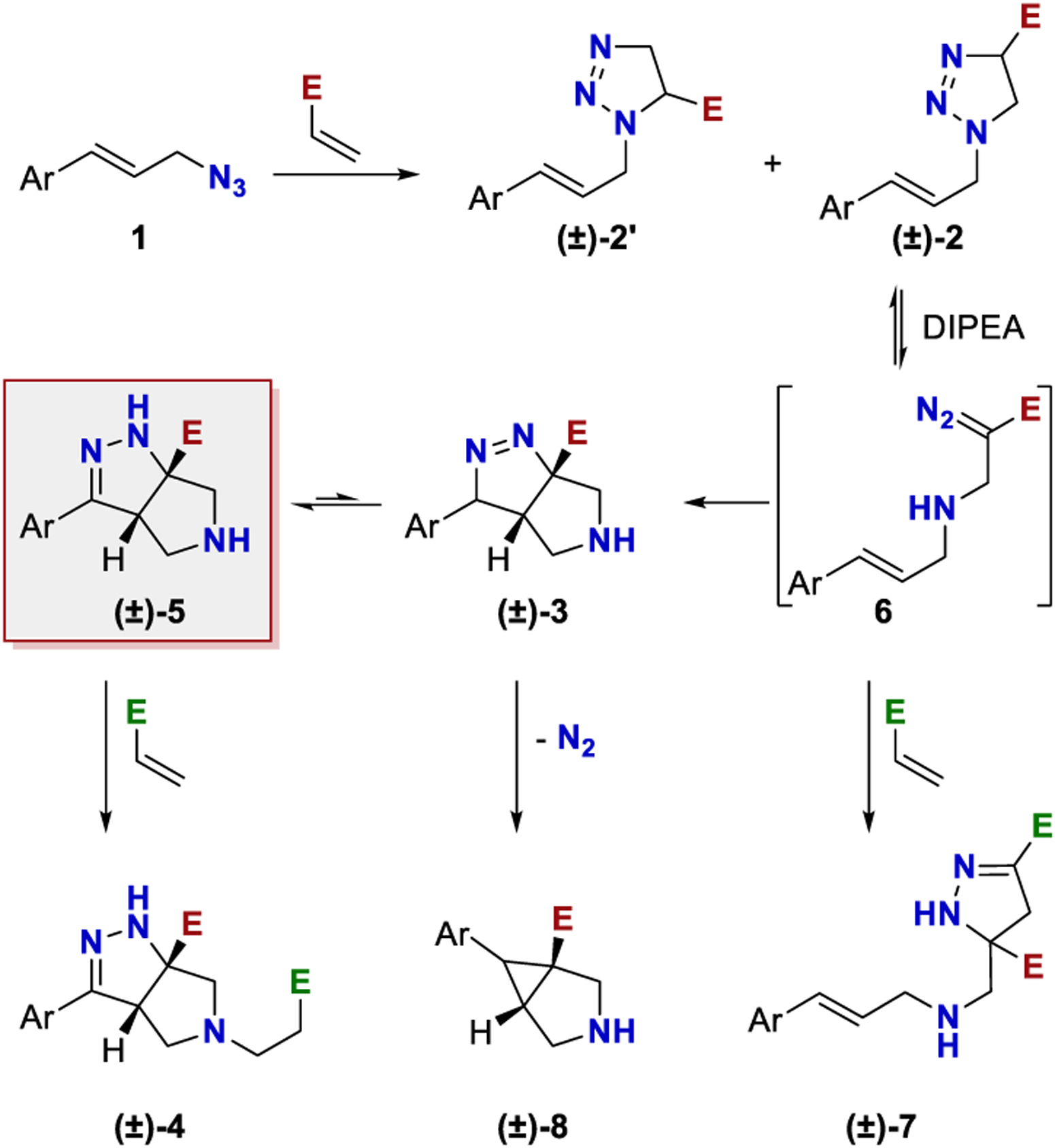
Mechanism of the Cascade Reaction
Triazoline 2 can equilibrate to achiral diazo species 6.24,37 Amine bases facilitate this equilibrium.24,25 The role of the amine is most likely to deprotonate at the α position of the ester. The resulting ammonium salt then can facilitate proton transfer to the distal nitrogen, leading to fragmentation and forming diazo compound 6. Intermediate 6 can undergo an intramolecular cycloaddition between the diazo and alkene to afford compound 3.22,23 Alternatively, in the presence of excess acrylate, diazo 6 can intercept a second equivalent of acrylate to form product 7.22,23,38,39 To mitigate this side product, a second procedure was developed for products where the intramolecular cyclization was slow (Table 4). The triazoline was preformed by mixing the azide and acrylate neat at room temperature. Excess acrylate was removed in vacuo. Then, the triazoline was heated in the presence of DIPEA in C6H6, which triggered the remainder of the cascade.
When compound 3 contains a hydrogen atom at the benzylic position, it can tautomerize to generate conjugated product 5.22,23 This tautomerization was readily accomplished with TFA or DBU during workup (see Experimental Section for details). When the benzylic position was tetrasubstituted, product 3 was isolated. Cyclopropane 8 was observed as a side product in a low yield for substrate 1j. This product likely arises from intermediate 3 via the loss of nitrogen.22,40 Lastly, products 3 and 5 are sufficiently nucleophilic to engage excess acrylate in a conjugate addition, affording compound 4.22,23 As shown in the optimization above (Table 2), less polar solvents generally minimized the conjugate addition by destabilizing ionic intermediates.
Conducting the cascade reaction in the presence of various electrophiles was a productive means to extend the cascade process (Table 6). The addition of Boc2O (9a), TsCl (9b), TrCl (9c), and CbzCl (9d) all afforded derivatized products directly in good yields. When TrCl was used, the product was initially isolated as a mixture of tautomers. Tautomerization of this mixture was slower than for other substrates, and it was promoted by exposure to 0.5 equiv of DBU, after which only the conjugated isomer was observed by 1H NMR.
Table 6.
Product Functionalization in Situ to Expand Cascadea
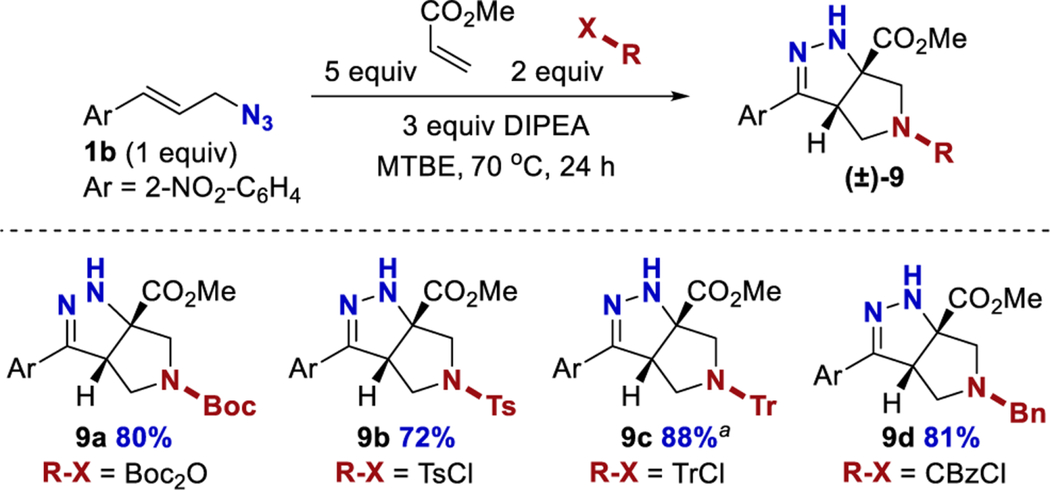
|
Yield reflects the sum of both tautomers. Yields are reported for isolated and purified products.
The cascade cyclization could be conducted on a gram scale without any decrease in efficiency (Scheme 4). Each of the functional groups present in product 5b could be selectively derivatized. The nitro group could be selectively reduced to afford aniline 10. The ester present in product 5 could be reduced to afford alcohol 11 or hydrolyzed to acid 12. The pyrrolidine nitrogen could be alkylated, arylated, or acylated to afford products 9d, 13, and 14, respectively. Lastly, the hydrazine motif could be acylated, yielding compound 15. These results clearly highlight how the dense functionality present on the tetrahydro-pyrrolo-pyrazole core can be selectively elaborated and diversified.
Scheme 4.
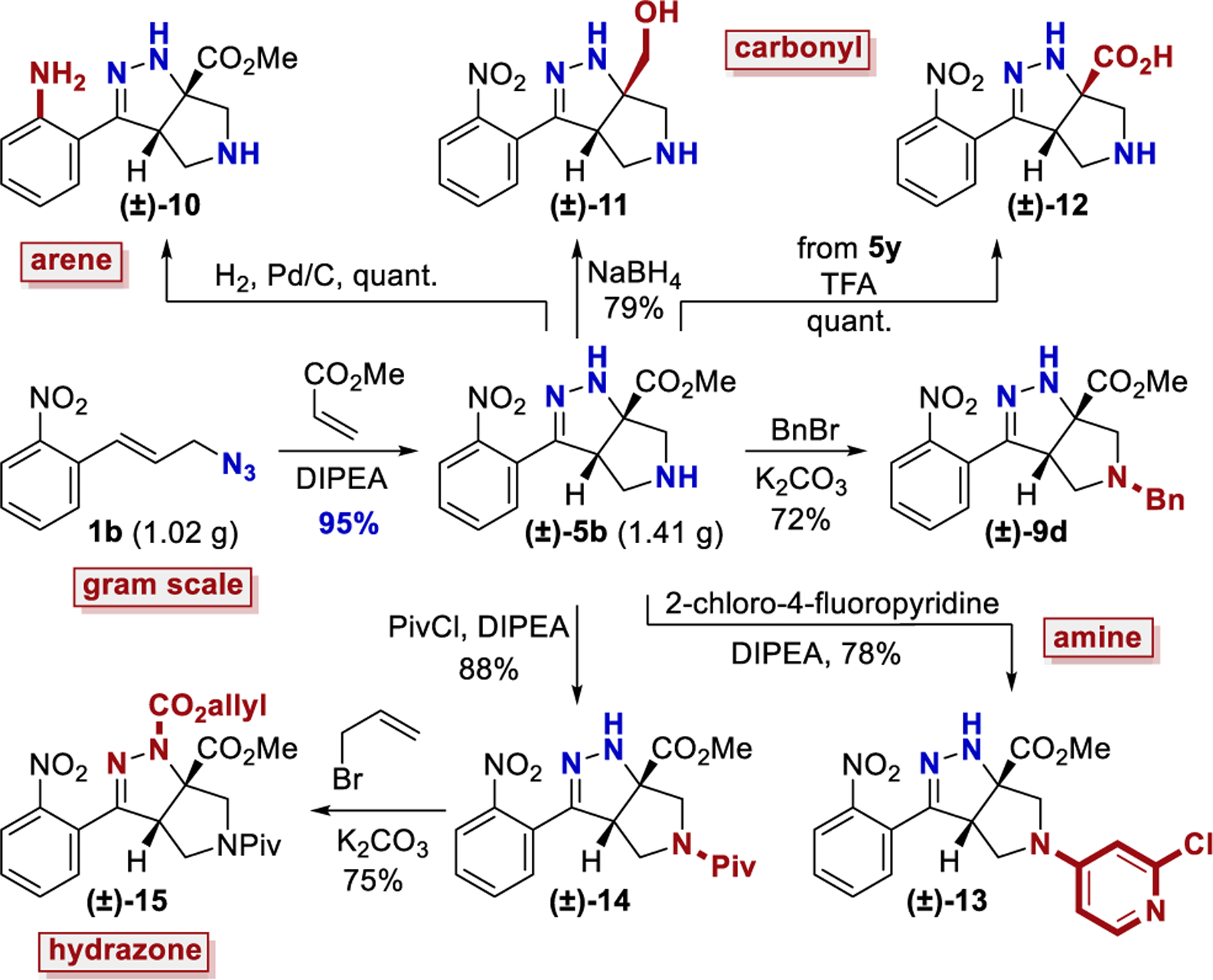
Gram-Scale Reaction and Product Derivatization
CONCLUSION
In conclusion, cinnamyl azides and Michael acceptors can engage in a modular cascade cyclization to directly generate fused tetrahydro-pyrrolo-pyrazole heterocycles. The reaction proceeds on an array of coupling partners. Furthermore, the products can be derivatized in situ. This reaction rapidly amplifies the complexity of two trivial starting materials and directly affords functionally dense bicyclic amine building blocks. This cascade is modular and amenable to diversification.
EXPERIMENTAL SECTION
Azide Safety.
Azides are known to be high-energy materials, and explosions have been reported when working with azides. In the course of this work, no issues were encountered. The majority of the azides synthesized in this report have C/N ratios equal to or above the recommended guideline of 3. Those with C/N ratios below 3 were prepaired on a small scale and extra precution was used when handeling them. Precautionary safety shields were used for all reactions using or producing more than 1 mmol of azide. Safety shields were used both in the fume hood and during rotary evaporation. All waste and aqueous solution that could be contaminated with azide anin were kept in individually labeled containers and were kept STRICTLY free of acid to avoid the accidental production on HN3: DO NOT use aqueous HCl during the workup in any of reaction that could result in HN3 formation. Further reading on azide safety is available.41−43
Methods and Reagents.
All reactions conducted at the elevated temperature used aluminum heating blocks with magnetic stirring bars (200 rpm). Reported temperatures were based on an external thermal couple. All commercially available chemicals were used without further purification. Dry tetrahydrofuran (THF), toluene, and methylene chloride were obtained from a commercial solvent system utilizing activated alumina columns under a positive pressure of argon. Thin-layer chromatography (TLC) was used for monitoring reaction progress. Visualization was conducted by using UV light, KMnO4, or PMA stains. Organic solutions were concentrated using a rotary evaporator under reduced pressure at or below 40 °C. Flash chromatography was performed on a Teledyne Isco CombiFlash Rf system utilizing normal phase precolumn load cartridges and gold high-performance columns. Structural assignments were made with additional information from COSY, HSQC, and HMBC experiments. All proton (1H) nuclear magnetic resonance spectra were recorded at 400 or 500 MHz. All carbon (13C) nuclear magnetic resonance spectra were recorded at 101 or 126 MHz. The fluorine (19F) nuclear magnetic resonance spectra were recorded at 376 MHz with proton decoupling. Chemical shifts are expressed in parts per million and are referenced to residual solvent (CDCl3: 7.26 ppm), to the central carbon in the NMR solvent (CDCl3: 77.2 ppm). Data is presented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, sept = septet, and m = multiplet), integration, and coupling constant in hertz (Hz). Infrared (IR) spectra were taken in a Nicolet Nexus 670 FT-IR with salt plates. IR spectra were reported in cm−1. Enantiomeric excesses were determined using a Shimadzu HPLC with a PDA detector and RegisPack 5 Micron column or Regis Reflect column (C-Amylose A; 3 μ, 250 mm × 4.6 mm).
Azide Synthesis.
The following cinnamyl azides were prepared through known procedures and have been previously characterized: 1a,44,45 1c–1d,44,45 1e–1f,44,46 1g,44,47 1h,18,44 1j–1k,44,47 1o,44,45 1p–1q,44 1t,46,48,49 1u–1v,30 1w,49−51 and 1hh.18,44
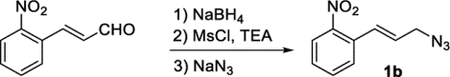
2-Nitrocinnamylazide (1b).
To a solution of 2-nitrocinnamalde-hyde (1.02 g, 5.77 mmol) in MeOH (13 mL) in an ice bath was added slowly NaBH4 (257 mg, 6.80 mmol). After 45 min, the reaction was quenched by the addition of H2O (25 mL), and the mixture was extracted with EtOAc (3 × 100 mL). The combined organic phases were washed with brine, dried (Na2SO4), and concentrated under reduced pressure. The crude material (1.20 g) was used without further purification. Characterization data for 2-nitrocinnamyl alcohol has been reported.52 The crude oil was reconstituted in DCM (20 mL) and cooled in an ice bath, and TEA (1.6 mL, 12 mmol) was added, followed by the dropwise addition of MsCl (0.67 mL, 8.7 mmol). After 1.5 h, the reaction was quenched by the addition of water and extracted with DCM (3 × 100 mL). The combined organic phases were dried (Na2SO4), filtered, and concentrated under reduced pressure. The crude oil was dissolved in acetone (5 mL) at rt, and an aqueous solution of NaN3 (5 mL, 1.9 M, 9.4 mmol) was added. After 18 h, the reaction was diluted with water and extracted with EtOAc (3 × 100 mL). The combined organic phases were washed with brine, dried (Na2SO4), filtered, and concentrated under reduced pressure. Final purification by column chromatography (EtOAc in hexanes, gradient elution 0–5%) afforded compound 1b (962 mg, 82% over three steps) as a yellow oil. 1H NMR (400 MHz, CDCl3): δ 8.00 (d, J = 8.4 Hz, 1H), 7.68–7.57 (m, 2H), 7.51–7.41 (m, 1H), 7.18 (d, J = 15.6 Hz, 1H), 6.22 (dt, J = 15.6, 6.4 Hz, 1H), 4.04 (d, J = 6.3 Hz, 2H). 13C{1H} NMR (101 MHz, CDCl3): δ 147.8, 133.3, 132.0, 129.9, 129.0, 128.7, 127.9, 124.7, 52.6. IR (NaCl, thin film, cm−1): 3072, 2927, 2850, 2094, 1524, 1344, 964. HRMS (CITOF): m/z [M + NH4]+ calcd for C9H12N5 O2 +, 222.0986; found, 222.0978.
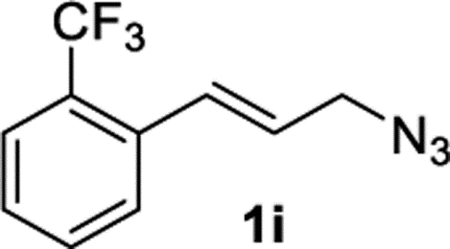
2-Trifluoromethylcinnamyl Azide (1i).
The preparation of compound 1i was adapted from a known procedure.44 Purification by column chromatography (gradient EtOAc/hexane from 0–40%) afforded compound 1i in 53% yield (603 mg) as a colorless oil. 1H NMR (400 MHz, CDCl3): δ 7.68 (d, J = 8.5 Hz, 1H), 7.66 (d, J = 8.8 Hz, 1H), 7.54 (t, J = 7.8 Hz, 1H), 7.40 (t, J = 7.7 Hz, 1H), 7.08 (d, J = 15.6 Hz, 1H), 6.24 (dt, J = 15.6, 6.5 Hz, 1H), 4.00 (d, J = 6.5 Hz, 2H). 13C{1H} NMR (101 MHz, CDCl3): δ 135.2 (q, JC–F = 1.8 Hz), 132.0, 130.4 (q, JC–F = 2.1 Hz), 127.8, 127.64, 127.63 (q, JC–F = 30.1 Hz), 126.9, 125.8 (q, JC–F = 5.7 Hz), 124.2 (q, JC–F = 274.9 Hz), 52.7. 19F{1H} NMR (376 MHz, CDCl3): δ −59.4. IR (NaCl, thin film, cm−1): 3063, 2198, 2104, 1314, 1160, 1125, 765. HRMS (CI-TOF): m/z [M + NH4]+ calcd for C10H12F3 N4 +, 245.1009; found, 245.1000.
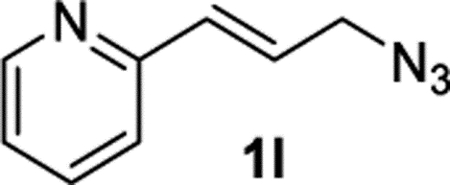
(E)-2-(3-Azidoprop-1-en-1-yl)pyridine (1l).
The preparation of compound 1l was adapted from a known procedure.44 Purification by column chromatography (gradient EtOAc/hexane from 0–50%) afforded compound 1l in 46% yield (369 mg) as a yellow oil. Note that cinnamyl azide 1l is not stable in storage at an ambient temperature and should be used immediately or stored in a freezer. 1H NMR (400 MHz, CDCl3): δ 8.59 (d, J = 4.8 Hz, 1H), 7.67 (td, J = 7.7, 1.8 Hz, 1H), 7.31 (d, J = 7.9 Hz, 1H), 7.19 (dd, J = 7.4, 4.9 Hz, 1H), 6.84–6.70 (m, 2H), 4.03 (d, J = 5.0 Hz, 1H). 13C{1H} NMR (101 MHz, CDCl3): δ 154.4, 149.7, 136.6, 133.4, 127.1, 122.7, 122.0, 52.5. IR (NaCl, thin film, cm−1): 3000, 2918, 2209, 1585, 1470, 1431, 975. HRMS (CI-TOF): m/z [M + H]+ calcd for C8H9N4+, 161.0822; found, 161.0821.
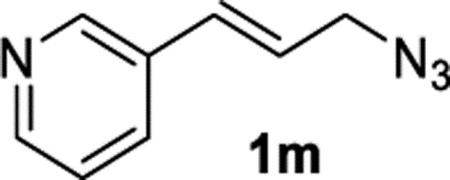
(E)-3-(3-Azidoprop-1-en-1-yl)pyridine (1m).
The preparation of compound 1m was adapted from a known procedure.44 Purification by column chromatography (gradient EtOAc/hexane from 0–70%) afforded compound 1m in 34% yield (272 mg) as a yellow oil. Note that cinnamyl azide 1m is not stable in storage at ambient temperature and should be used immediately or stored in a freezer. 1H NMR (400 MHz, CDCl3): δ 8.64 (s, 1H), 8.56–8.50 (m, 1H), 7.77–7.71 (m, 1H), 7.32–7.25 (m, 1H), 6.67 (d, J = 15.8 Hz, 1H), 6.33 (dtd J = 15.4, 6.3 Hz, 1H), 4.01 (d, J = 6.5 Hz, 2H). 13C{1H} NMR (101 MHz, CDCl3): δ 149.2, 148.5, 133.1, 131.7, 130.7, 125.0, 123.5, 52.8. IR (NaCl, thin film, cm−1): 3031, 2102, 1416, 1243, 971. HRMS (CI-TOF): m/z [M + H]+ calcd for C8H9N4+, 161.0822; found, 161.0821.
(E)-4-(3-Azidoprop-1-en-1-yl)pyridine (1n).
The preparation of compound 1n was adapted from a known procedure.44 Purification by column chromatography (DCM in hexanes, gradient elution 0–70%) afforded compound 1n in 62% yield (498 mg) as a yellow oil. Note that cinnamyl azide 1n is not stable in storage at ambient temperature and should be used immediately or stored in a freezer. 1H NMR (400 MHz, CDCl3): δ 8.59 (d, J = 6.2 Hz, 2H), 7.28 (d, J = 6.2 Hz, 2H), 6.63 (d, J = 15.9 Hz, 1H), 6.46 (dt, J = 15.9, 6.1 Hz, 1H), 4.03 (d, J = 6.1, 1.5 Hz, 2H). 13C{1H} NMR (101 MHz, CDCl3): δ 150.3, 143.3, 131.5, 127.6, 121.0, 52.5. IR (NaCl, thin film, cm−1): 3029, 2106, 1596, 1415, 974. HRMS (CI-TOF): m/z [M + H]+ calcd for C8H9N4+, 161.0822; found, 161.0821.
(3-Azido-2-methylprop-1-en-1-yl)benzene (1r).
The preparation of compound 1r was adapted from a known procedure.44 Purification by column chromatography (gradient EtOAc/hexane from 0–20%) afforded compound 1r in 60% yield (519 mg) as a colorless oil with an E/Z ratio of 85:15. The major product of the obtained material provided an identical 1H NMR spectrum.
2-(3,5-Bis(trifluoromethyl)phenyl)but-3-en-2-ol (1s′).
An oven-dried flask was equipped with a stir bar, fitted with a septum, sealed under argon, charged with THF (20 mL), and vinyl magnesium chloride (3.80 mL, 1.6 M in THF, 6.1 mmol). The solution was cooled in an acetone/dry ice bath. Dropwise addition of 3′,5′-bis(trifluoromethyl)acetophenone (0.90 mL, 5.0 mmol) to the flask was conducted over 5 min. The reaction was gradually warmed to an ambient temperature. After 16 h, the reaction was quenched by the addition of water (20 mL). The resulting solution was diluted with water (20 mL) and extracted with EtOAc (3 × 30 mL). The combined organic solution was washed with brine, dried (MgSO4), filtered, and concentrated under reduced pressure. Purification by column chromatography (EtOAc in hexanes, gradient elution 0– 40%) afforded target compound 1s′ as a colorless oil (360 mg, 25%). 1H NMR (400 MHz, CDCl3): δ 7.96 (s, 2H), 7.80 (s, 1H), 6.17 (dd, J = 17.3, 10.6 Hz, 1H), 5.79 (d, J = 17.3 Hz, 1H), 5.28 (d, J = 10.6 Hz, 1H), 2.15 (br, 1H), 1.72 (s, 3H). 13C{1H} NMR (101 MHz, CDCl3): δ 149.1, 143.3, 131.5 (q, JC–F = 33.4 Hz), 125.6 (m), 123.4 (q, JC–F = 273.9 Hz), 121.0 (sept, JC–F = 4.0 Hz), 114.3, 74.4, 29.5. 19F{1H} NMR (376 MHz, CDCl3): −62.8. IR (NaCl, thin film, cm−1): 3406, 2917, 1374, 1278, 1174, 1134. HRMS (ESI-TOF): m/z [M]+ calcd for C12H10F6O+, 284.0630; found, 284.0629.
(E)-1-(4-Chlorobut-2-en-2-yl)-3,5-bis(trifluoromethyl)benzene (1s″).
The synthesis of compound 1s″ was adapted from a known method with slight modification.53 An oven-dried flask was equipped with a stir bar, fitted with a septum, sealed under argon, and charged with compound 1s′ (281 mg, 1.00 mmol) and DCM (5 mL). The solution was cooled in an ice bath. The dropwise addition of thionyl chloride (0.29 mL, 4.0 mmol) to the flask was conducted over 5 min. The reaction was gradually warmed to an ambient temperature. After 16 h, the reaction was quenched by the addition of water (5 mL). The resulting solution was diluted with water (10 mL) and extracted with DCM (3 × 15 mL). The combined organic solution was washed with brine, dried (MgSO4), filtered, and concentrated under reduced pressure. Purification by column chromatography (hexanes as eluant) afforded target compound 1s″ as a colorless oil (107 mg, 37%). 1H NMR (400 MHz, CDCl3): δ 7.84 (s, 2H), 7.82 (s, 1H), 6.12 (tq, J = 7.9, 1.4 Hz, 1H), 4.30 (d, J = 7.9 Hz, 2H), 2.21 (s, 3H). 13C{1H} NMR (101 MHz, CDCl3): δ 144.3, 138.2, 131.8 (q, JC–F = 33.3 Hz), 126.2, 126.0 (m), 123.3 (q, JC–F = 273.7 Hz), 121.3 (sept, JC–F = 3.8 Hz), 40.1, 15.8. 19F{1H} NMR (376 MHz, CDCl3): −62.9. IR (NaCl, thin film, cm−1): 2950, 1380, 1280, 1182, 1132, 898. HRMS (EITOF): m/z [M − Cl]+ calcd for C12H9F6+, 267.0603; found, 267.0598.
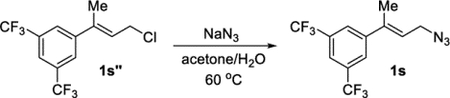
(E)-1-(4-Azidobut-2-en-2-yl)-3,5-bis(trifluoromethyl)benzene (1s).
A flask was charged with compound 1s″ (171 mg, 0.592 mmol) and acetone (1 mL) and equipped with a stir bar. An aqueous solution of sodium azide (1.0 mL, 0.96 M, 0.96 mmol) was added to the reaction at an ambient temperature. The flask was equipped with a condenser, and the reaction was heated to 60 °C. After 3 h, the reaction was cooled and diluted with water (5 mL). The resulting solution was extracted with EtOAc (3 × 15 mL). The combined organic solution was washed with brine, dried (MgSO4), filtered, and concentrated under reduced pressure. Purification by column chromatography (EtOAc in hexanes, gradient elution 0–40%) afforded target compound 1s as a colorless oil (148 mg, 85%). 1H NMR (400 MHz, CDCl3): δ 7.84 (s, 2H), 7.83 (s, 1H), 6.01 (tq, J = 7.2, 1.4 Hz, 1H), 4.01 (d, J = 7.1 Hz, 2H), 2.19 (s, 3H). 13C{1H} NMR (101 MHz, CDCl3): δ 144.4, 138.5, 131.8 (q, JC–F = 33.4 Hz), 126.0, 124.0, 123.3 (q, JC–F = 273.8 Hz), 121.3 (sept, JC–F = 3.8 Hz), 48.5, 16.3. 19F{1H} NMR (376 MHz, CDCl3): −62.9. IR (NaCl, thin film, cm−1): 2292, 2103, 1380, 1280, 1179, 1132. HRMS (CI-TOF): m/z [M − N2 + H]+ calcd for C12H10F6N+, 282.0712; found, 282.0705.
Formation of Triazoline 2b.
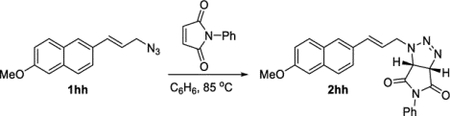
(3aS,6aR)-1-((E)-3-(6-Methoxynaphthalen-2-yl)allyl)-5-phenyl-3a,6a-dihydropyrrolo[3,4-d][1,2,3]triazole-4,6(1H,5H)-dione (2hh).
A 4 mL vial was charged with azide 1hh (119.4 mg, 0.500 mmol), N-phenylmaleimide (86.6 mg, 0.500 mmol), and benzene (0.5 mL). The vial was sealed and heated to 85°C. After 24 h, the reaction was cooled. The crude solid was collected using a Hirsch funnel and washed with hexanes (3 × 2 mL). This afforded compound 2hh as a white solid (179.5 mg, 81%). 1H NMR (400 MHz, CDCl3): δ 7.78–7.67 (m, 3H), 7.59 (dd, J = 8.6, 1.8 Hz, 1H), 7.53–7.41 (m, 3H), 7.32–7.26 (m, 2H), 7.21–7.12 (m, 2H), 6.91 (d, J = 15.8 Hz, 1H), 6.31 (ddd, J = 15.7, 8.6, 5.4 Hz, 1H), 5.69 (d, J = 10.9 Hz, 1H), 5.02 (dd, J = 15.1, 5.4 Hz, 1H), 4.56 (d, J = 11.0 Hz, 1H), 4.52 (dd, J = 15.2, 8.6 Hz, 1H), 3.95 (s, 3H). 13C{1H} NMR (126 MHz, CD3CN): δ 171.9, 170.7, 158.0, 134.5, 134.3, 131.82, 131.79, 129.5, 129.2, 129.0, 128.9, 127.2, 126.9, 126.4, 124.1, 122.7, 119.0, 106.0, 82.4, 58.2, 55.0, 51.2. IR (NaCl, thin film, cm−1): 2935, 1723, 1600, 1477, 1378, 1178. HRMS (ESI-TOF): m/z [M − N2 + H]+ calcd for C24H20N4NaO3+, 435.1428; found, 435.1433.
Reaction Optimization.
Procedure for Table 1.
A stock solution of azide 1a (87.8 mg, 0.552 mmol) and naphthalene (18.8 mg, 0.147 mmol, internal standard) was prepared in THF (0.10 mL). Individual 4 mL vials were charged with 30 μL portions of this solution and methyl acrylate (10 or 20 μL). The vials were sealed under air at rt. After 72 h, the reactions were concentrated under reduced pressure and analyzed by 1H NMR. All of the reactions were run in duplicate, and the average values are reported.
Procedure for Table 2 (Solvent Screening Procedure).
A stock solution of azide 1b (197 mg, 0.964 mmol) and naphthalene (54.9 mg, 0.429 mmol, internal standard) was prepared in DCM (455 μL). Individual 4 mL vials were charged with 50 μL portions of this solution, and the DCM was evaporated. The respective solvent (370 μL) was added to each vial, followed by methyl acrylate (20 μL, 0.22 mmol). The vials were sealed under air and heated to 70 °C. After 24 h, the reactions were cooled to ambient temperature, and TFA (28 μL) was added. After 10 min, each reaction was diluted with K2CO3 (sat. aq 1 mL) and EtOAc (1 mL). The phases were separated, and the organic layer was analyzed by GC-FID to determine the yield. All of the reactions were run in duplicate, and the average values are reported.
Procedure for Table 2 (Additive Screening Procedure).
A stock solution of azide 1b (263 mg, 1.29 mmol), methyl acrylate (0.35 mL, 3.9 mmol), and naphthalene (166 mg, 1.30 mmol, internal standard) was prepared in benzene (6.5 mL). Individual 4 mL vials were charged with 380 μL portions of this solution. One additive (0.5 equiv) was added individually to each vial. The vials were sealed under air and heated to 70 °C. After 24 h, the reactions were cooled to an ambient temperature, and TFA (28 μL) was added. After 10 min, each reaction was diluted with a saturated K2CO3 aqueous solution (1 mL) and EtOAc (1 mL). The phases were separated, and the organic layer was analyzed by GC-FID to determine percent yields. All of the reactions were run in duplicate, and the average values are reported.
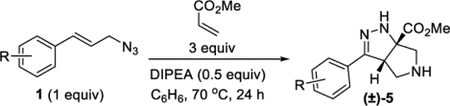
General Procedure I.
Procedure for the single-step synthesis of tetrahydro-pyrrolo-pyrazoles, example given for R = 2-NO2. A 20 mL vial was sequentially charged with azide 1b (81.7 mg, 0.400 mmol, 1 equiv), benzene (2.0 mL), methyl acrylate (0.11 mL, 1.2 mmol, 3 equiv), and DIPEA (35 μL, 0.20 mmol, 0.5 equiv). The vial was sealed with a Teflon-lined cap and heated to 70 °C. After 24 h, the reaction was cooled to an ambient temperature, and the solvent was removed under reduced pressure. The reaction was reconstituted in DCM (2 mL), and TFA (0.15 mL, 2.0 mmol, 5 equiv) was added. (Note that the treatment with TFA helped promote the tautomerization to the hydrazone. This step was not necessary; however, skipping it resulted in a mixture of tautomers.) After 5 min, the solvent was removed under reduced pressure. Final purification by column chromatography (IPA in DCM with 1% TEA, gradient elution 0–10%) afforded compound 5b (109 mg, 93%) as an orange oil.
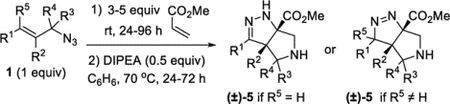
General Procedure II.
Procedure for the two operation synthesis of tetrahydro-pyrrolo-pyrazoles, example given for R1 = Ph, R2 = Me, R3 = R4 = R5 = H. A 4 mL vial was charged with azide 1r (86.8 mg, 0.501 mmol, 1 equiv) and methyl acrylate (0.23 mL, 2.5 mmol, 5 equiv). The vial was sealed with a Teflon-lined cap, and the reaction was stirred at an ambient temperature. After 48 h, the reaction was transferred to a 20 mL vial and then concentrated under reduced pressure at an ambient temperature. The residual methyl acrylate was removed under a high vacuum for 15 min. The crude triazoline was reconstituted in benzene (2.0 mL), and DIPEA (44 μL, 0.25 mmol, 0.5 equiv) was added. The vial was sealed with a Teflon-lined cap and heated to 70 °C. After 24 h, the reaction was cooled, and the solvent was removed under reduced pressure. Final purification by column chromatography (IPA in DCM with 1% TEA, gradient elution 0– 10%) afforded compound 5r (91.0 mg, 70%) as a colorless oil.
General Procedure III.
Procedure for the single-step synthesis of the N-protected tetrahydro-pyrrolo-pyrazoles, example given for R–X = Boc2O and R = Boc. A 20 mL vial was charged with azide 1b (61.0 mg, 0.30 mmol, 1 equiv) and MTBE (1.5 mL). Di-tert-butyl dicarbonate (0.14 mL, 0.61 mmol, 2 equiv) was added to the reaction, followed by methyl acrylate (0.14 mL, 1.5 mmol, 5 equiv) and DIPEA (0.16 mL, 0.92 mmol, 3 equiv). The vial was sealed with a Teflon-lined cap, and the reaction was heated to 70 °C. After 24 h, the reaction was cooled to an ambient temperature, and the mixture was diluted with water (5 mL). The resulting solution was extracted with DCM (3 × 20 mL). The combined organic phase was washed with brine, dried (MgSO4), filtered, and concentrated under reduced pressure. Purification by column chromatography (EtOAc in hexanes, gradient elution 0–100%) afforded target compound 9a as a yellow solid (92.9 mg, 80%).
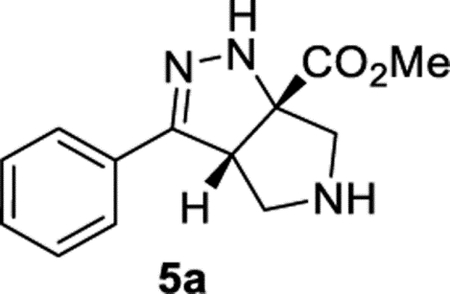
Methyl (3aS,6aS)-3-Phenyl-3a,4,5,6-tetrahydropyrrolo[3,4-c]-pyrazole-6a(1H)-carboxylate (5a).
General Procedure I was used, and the product was isolated in 66% (83.3 mg) and 61% (77.9 mg) yields in duplicate trials. An average of 64% is reported. The material provided similar 1H NMR as previously reported.23 1H NMR (400 MHz, CDCl3): δ 7.63–7.58 (m, 2H), 7.40–7.30 (m, 3H), 6.57 (br, 1H), 4.20 (dd, J = 7.0, 3.6 Hz, 1H), 3.80 (s, 3H), 3.34–3.26 (m, 3H), 3.17 (d, J = 12.1 Hz, 1H), 2.50 (br, 1H). 13C{1H} NMR (126 MHz, CDCl3): δ 174.2, 153.5, 131.5, 129.2, 128.9, 126.3, 81.5, 61.6, 58.6, 54.8, 53.1. IR (NaCl, thin film, cm−1): 3317, 2951, 1732, 1679, 1446, 1272, 1219. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C13H15N3NaO2+, 268.1056; found, 268.1059.
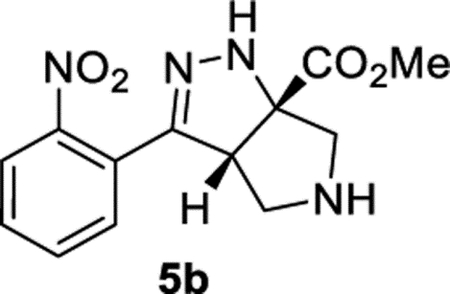
Methyl (3aS,6aS)-3-(2-Nitrophenyl)-3a,4,5,6-tetrahydropyrrolo-[3,4-c]pyrazole-6a(1H)-carboxylate (5b).
General Procedure I was used, and the product was isolated in 93% (109 mg) and 94% (108 mg) yields in duplicate trials. The average of 94% is reported. 1H NMR (400 MHz, CD2Cl2): δ 7.81–7.72 (m, 1H), 7.69–7.60 (m, 1H), 7.57–7.47 (m, 2H), 6.78 (br, 1H), 4.21 (dd, J = 7.9, 2.1 Hz, 1H), 3.85 (s, 3H), 3.29 (d, J = 12.1 Hz, 1H), 3.24 (dt, J = 12.5, 1.9 Hz, 1H), 3.18–3.10 (m, 2H), 2.25 (br, 1H). 13C{1H} NMR (101 MHz, CD2Cl2): δ 173.5, 149.1, 149.0, 132.2, 129.7, 129.4, 126.2, 124.1, 81.2, 61.4, 60.0, 53.9, 52.9. IR (NaCl, thin film, cm−1): 3338, 2950, 1736, 1729, 1528, 1273. HRMS (ESI-TOF): m/z [M + H]+ calcd for C13H15N4O4+, 291.1088; found, 291.1089.
Gram-Scale Synthesis of 5b.
A 100 mL round-bottom flask was equipped with a stir bar and charged with azide 1b (1.02 g, 5.00 mmol), methyl acrylate (1.36 mL, 15.0 mmol, 3 equiv), and benzene (25 mL). DIPEA (0.44 mL, 2.5 mmol, 0.5 equiv) was added to the flask, and the flask was sealed with a glass stopper. The reaction was heated to 70 °C. After 24 h, the reaction was cooled, and TFA (1.9 mL, 25 mmol) was added to the flask. After 5 min, the solvent was removed under reduced pressure. Final purification by column chromatography (IPA in DCM with 1% TEA, gradient elution 0– 10%) afforded compound 5b (1.41 g, 97%) as an orange oil. A duplicate trial afforded compound 5b (1.06 g, 92%). The average of 95% is reported.
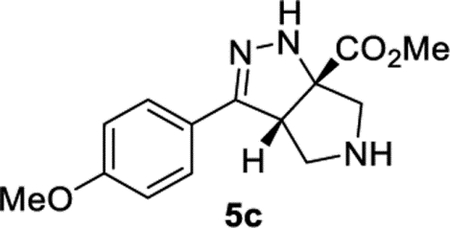
Methyl (3aS, 6aS)-3-(4-Methoxyphenyl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (5c).
General Procedure I was used, and the product was isolated in 79% (112 mg) and 84% (118 mg) yields in duplicate trials. The average of 82% is reported. 1H NMR (500 MHz, C6D6): δ 7.58 (d, J = 8.8 Hz, 2H), 6.72 (d, J = 8.7 Hz, 2H), 6.55 (br, 1H), 3.97 (apparent t, J = 5.3 Hz, 1H), 3.26 (s, 3H), 3.18 (s, 3H), 3.07 (br, 2H), 3.04–3.00 (m, 2H), 1.74 (br, 1H). 13C{1H} NMR (126 MHz, C6D6): δ 173.8, 160.2, 152.2, 128.0, 125.0, 114.0, 81.5, 61.5, 58.7, 54.6, 54.5, 51.8. IR (NaCl, thin film, cm−1): 3323, 2953, 2840, 1731, 1672, 1608, 1516, 1253, 1220, 1176, 1037. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C14H17N3NaO3+, 298.1162; found, 298.1161.
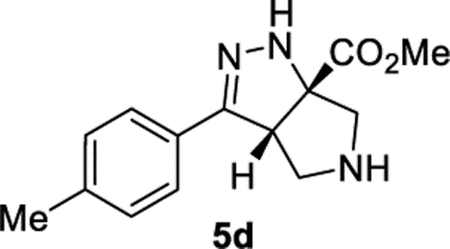
Methyl (3aS,6aS)-3-(p-Tolyl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]-pyrazole-6a(1H)-carboxylate (5d).
General Procedure I was used, and the product was isolated in 72% (103 mg) and 74% (116 mg) yields in duplicate trials. The average of 73% is reported. 1H NMR (500 MHz, C6D6): δ 7.57 (d, J = 8.0 Hz, 2H), 6.95 (d, J = 7.9 Hz, 2H), 6.59 (br, 1H), 3.98 (apparent t, J = 5.3 Hz, 1H), 3.18 (s, 3H), 3.05 (br, 2H), 3.00 (d, J = 5.3 Hz, 2H), 2.07 (s, 3H), 1.67 (br, 1H). 13C{1H} NMR (126 MHz, CDCl3): δ 174.1, 153.6, 139.2, 129.4, 128.6, 126.2, 81.2, 61.4, 58.5, 54.6, 53.0, 21.4. IR (NaCl, thin film, cm−1): 3324, 2952, 2873, 1731, 1436, 1272, 1220, 1039. HRMS (ESITOF): m/z [M + H]+ calcd for C14H18N3O2+, 260.1394; found, 260.1397.
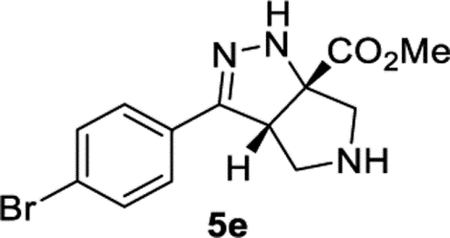
Methyl (3aS,6aS)-3-(4-Bromophenyl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (5e).
General Procedure I was used, and the product was isolated in 67% (86.9 mg) and 71% (90.4 mg) yields in duplicate trials. The average of 69% is reported. 1H NMR (500 MHz, C6D6): δ 7.25–7.19 (m, 4H), 6.58 (br, 1H), 3.79 (dd, J = 8.1, 2.4 Hz, 1H), 3.19 (s, 3H), 3.02 (br, 2H), 2.90 (dd, J = 12.3, 8.0 Hz, 1H), 2.82 (dd, J = 12.4, 2.5 Hz, 1H), 2.09 (br, 1H). 13C{1H} NMR (126 MHz, C6D6): δ 173.4, 151.0, 131.6 (tentatively assigned as 2C), 131.0, 122.5, 81.6, 61.3, 57.9, 54.3, 51.8. IR (NaCl, thin film, cm−1): 3324, 2951, 1730, 1491, 1399, 1271, 1220, 1070. HRMS (ESI-TOF): m/z [M + H]+ calcd for C13H15BrN3O2+, 324.0342; found, 324.0342.
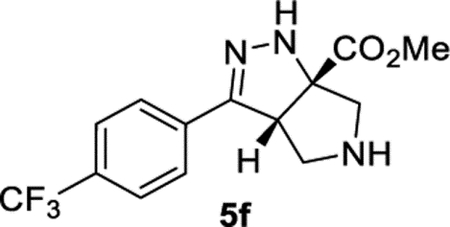
Methyl (3aS,6aS)-3-(4-(Trifluoromethyl)phenyl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (5f).
General Procedure I was used, and the product was isolated in 78% (103 mg) and 80% (98.6 mg) yields in duplicate trials. The average of 79% is reported. 1H NMR (400 MHz, C6D6): δ 7.39 (d, J = 8.2 Hz, 2H), 7.30 (d, J = 8.6 Hz, 2H), 6.63 (br, 1H), 3.81 (dd, J = 8.0, 2.4 Hz, 1H), 3.21 (s, 3H), 3.01 (br, 2H), 2.92 (dd, J = 12.3, 8.0 Hz, 1H), 2.81 (dd, J = 12.3, 2.5 Hz, 1H), 1.70 (br, 1H). 13C{1H} NMR (101 MHz, C6D6): δ 173.3, 150.4, 135.5, 131.2 (q, JC–F = 271.8 Hz), 129.7 (q, JC–F = 32.5 Hz), 126.0, 125.3 (q, JC–F = 3.8 Hz), 81.9, 61.4, 57.8, 54.3, 51.9. 19F{1H} NMR (471 MHz, CDCl3): δ −62.72. IR (NaCl, thin film, cm−1): 3305, 2955, 1732, 1327, 1274, 1220, 1165, 1120, 1068. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C14H14F3N3NaO2+,336.0930; found, 336.0927.
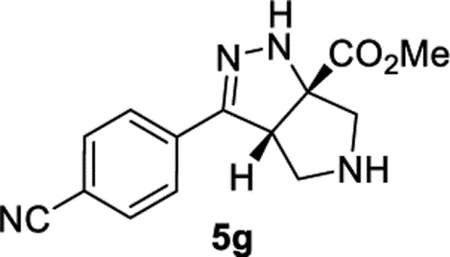
Methyl (3aS,6aS)-3-(4-Cyanophenyl)-3a,4,5,6-tetrahydropyrrolo-[3,4-c]pyrazole-6a(1H)-carboxylate (5g).
General Procedure I was used, and the product was isolated in 76% (104 mg) and 77% (103 mg) yields in duplicate trials. The average of 77% is reported. 1H NMR (400 MHz, CD2Cl2): δ 7.75–7.69 (m, 2H), 7.69–7.65 (m, 2H), 6.82 (br, 1H), 4.20 (dd, J = 8.0, 2.5 Hz, 1H), 3.82 (s, 3H), 3.38–3.22 (m, 3H), 3.20 (d, J = 12.1 Hz, 1H), 2.19 (br, 1H). 13C{1H} NMR (101 MHz, CD2Cl2): δ 173.6, 150.8, 136.1, 132.4, 126.2, 118.7, 111.6, 82.0, 61.3, 57.8 54.4, 52.8. IR (NaCl, thin film, cm−1): 3331, 3050, 2954, 2226, 1728, 1606, 1579, 1435, 1274, 1220. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C14H14N4NaO2+, 293.1009; found, 293.1010.
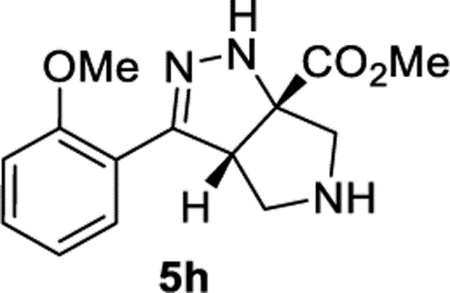
Methyl (3aS,6aS)-3-(2-Methoxyphenyl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (5h).
General Procedure I was used, and the product was isolated in 88% (121 mg) and 79% (109 mg) yields in duplicate trials. The average of 84% is reported. 1H NMR (500 MHz, C6D6): δ 8.26 (dd, J = 7.9, 1.7 Hz, 1H), 7.02 (td, J = 7.8, 1.7 Hz, 1H), 6.82 (t, J = 7.5 Hz, 1H), 6.72 (br, 1H), 6.44 (d, J = 8.3 Hz, 1H), 4.50 (dd, J = 8.5, 2.6 Hz, 1H), 3.93 (br, 1H), 3.30–3.19 (m, 3H), 3.23 (s, 3H), 3.18–3.09 (m, 1H), 3.14 (s, 3H). 13C{1H} NMR (126 MHz, C6D6): δ 173.7, 156.6, 151.1, 129.9, 129.3, 121.0, 120.9, 111.4, 80.3, 60.5, 60.0, 54.5, 54.4, 51.9. IR (NaCl, thin film, cm−1): 3318, 2950, 1737, 1688, 1680, 1599, 1245, 1200, 1130. HRMS (ESI-TOF): m/z [M + H]+ calcd for C14H18N3O3+, 276.1343; found, 276.1340.
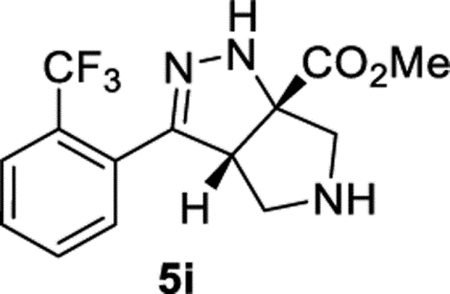
Methyl (3aS,6aS)-3-(2-(Trifluoromethyl)phenyl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (5i).
General Procedure I was used, and the product was isolated in 76% (95 mg), 56% (70 mg), and 56% (74 mg) yields in triplicate trials. The average of 63% is reported. 1H NMR (400 MHz, CD2Cl2): δ 7.77 (d, J = 7.2 Hz, 1H), 7.63 (t, J = 7.1 Hz, 1H), 7.57–7.50 (m, 2H), 6.74 (br, 1H), 4.25 (dd, J = 7.6, 2.6 Hz, 1H), 3.86 (s, 3H), 3.29 (d, J = 12.0 Hz, 1H), 3.20–3.05 (m, 3H), 2.21 (br, 1H). 13C{1H} NMR (101 MHz, CD2Cl2): δ 173.8, 151.4, 131.9, 131.6 (q, JC–F = 2.1 Hz), 130.5, 128.8, 128.1 (q, JC–F = 30.8 Hz), 126.8 (q, JC–F = 5.6 Hz), 124.1 (q, JC–F = 274.4 Hz), 81.0, 62.0, 61.4, 53.6, 52.8. 19F{1H} NMR (376 MHz, CD2Cl2): δ −58.7. IR (NaCl, thin film, cm−1): 3329, 2956, 2877, 1729, 1601, 1436, 1312, 1274, 1217, 1170, 1127, 1035, 766. HRMS (ESI-TOF): m/z [M + H]+ calcd for C14H15F3N3O2+, 314.1111; found, 314.1112.
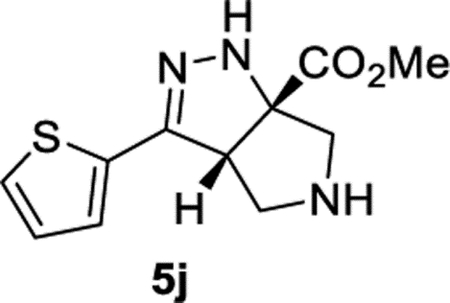
Methyl (3aS,6aS)-3-(Thiophen-2-yl)-3a,4,5,6-tetrahydropyrrolo-[3,4-c]pyrazole-6a(1H)-carboxylate (5j).
General Procedure I was used, and the product was isolated in 70% (88.7 mg) and 77% (95.8 mg) yields in duplicate trials. The average of 74% is reported. 1H NMR (400 MHz, CD3CN): δ 7.38 (d, J = 5.1 Hz, 1H), 7.14 (d, J = 3.5 Hz, 1H), 7.04 (dd, J = 4.9, 3.8 Hz, 1H), 6.61 (br, 1H), 4.17 (dd, J = 7.5, 3.0 Hz, 1H), 3.76 (s, 3H), 3.30–3.20 (m, 2H), 3.18 (d, J = 11.9 Hz, 1H), 3.13 (d, J = 12.1 Hz, 1H).1.30 (br, 1H). 13C{1H} NMR (101 MHz, CD3CN): δ 173.4, 148.3, 136.0, 127.6, 126.6, 126.5, 80.9, 60.3, 58.9, 53.6, 52.4. IR (NaCl, thin film, cm−1): 3277, 2952, 1731, 1659, 1433, 1413, 1271, 1220. HRMS (ESI-TOF): m/z [M + H]+ calcd for C11H14N3O2S+, 252.0801; found, 252.0805.
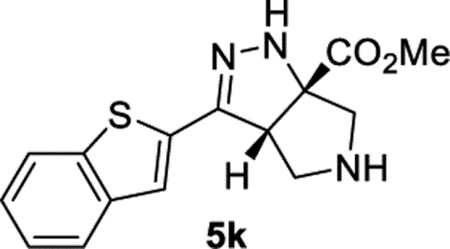
Methyl (3aS,6aS)-3-(Benzo[b]thiophen-2-yl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (5k).
General Procedure I was used, and the product was isolated in 74% (89.4 mg) and 85% (103 mg) yields in duplicate trials. The average of 80% is reported. 1H NMR (500 MHz, CD2Cl2): δ 7.86–7.72 (m, 2H), 7.44–7.32 (m, 2H), 7.28 (s, 1H), 6.67 (br, 1H), 4.26 (dd, J = 8.0, 2.2 Hz, 1H), 3.84 (s, 3H), 3.46 (d, J = 12.2 Hz, 1H), 3.41–3.28 (m, 2H), 3.21 (d, J = 12.1 Hz, 1H), 2.28 (br, 1H). 13C{1H} NMR (126 MHz, CD2Cl2): δ 173.6, 149.2, 139.8, 139.7, 136.0, 125.3, 124.6, 123.8, 122.8, 122.2, 82.0, 61.4, 59.0, 54.6, 52.8. IR (NaCl, thin film, cm−1): 3304, 2936, 1725, 1414, 1266, 1210, 836. HRMS (ESI-TOF): m/z [M + H]+ calcd for C15H16N3O2S+, 302.0958; found, 302.0956.
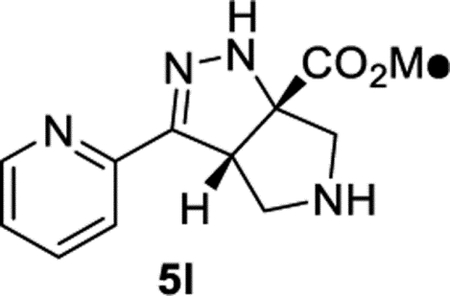
Methyl (3aS,6aS)-3-(Pyridin-2-yl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (5l).
General Procedure I was used, and the product was isolated in 93% (115 mg) and 84% (104 mg) yields in duplicate trials. The average of 89% is reported. 1H NMR (500 MHz, C6D6): δ 8.34 (d, J = 4.8 Hz, 1H), 8.07 (d, J = 8.0 Hz, 1H), 7.02 (td, J = 7.8, 1.7 Hz, 1H), 6.77 (br, 1H), 6.60–6.52 (m, 1H), 4.52 (dd, J = 7.9, 2.6 Hz, 1H), 3.44–3.31 (m, 2H), 3.14 (s, 3H), 3.09 (d, J = 12.0 Hz, 1H), 3.05 (d, J = 12.0 Hz, 1H), 1.98 (br, 1H). 13C{1H} NMR (126 MHz, C6D6): δ 173.7, 153.6, 151.9, 148.8, 135.4, 122.3, 120.6, 81.0, 61.3, 58.8, 552, 51.7. IR (NaCl, thin film, cm−1): 3272, 2950, 1743, 1680, 1437, 1284, 1202, 1133. HRMS (ESI-TOF): m/z [M + H]+ calcd for C12H15N4O2+, 247.1190; found, 247.1197.
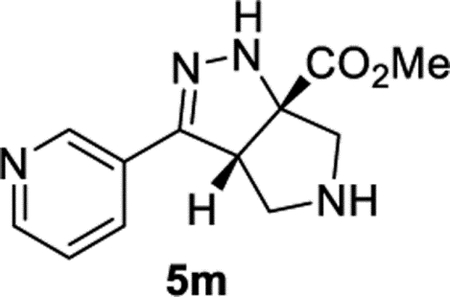
Methyl (3aS,6aS)-3-(Pyridin-3-yl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (5m).
General Procedure I was used, and the product was isolated in 72% (88.0 mg) and 63% (76.4 mg) yields in duplicate trials. The average of 68% is reported. 1H NMR (400 MHz, CD2Cl2): δ 8.80 (dd, J = 2.3, 0.9 Hz, 1H), 8.54 (dd, J = 4.8, 1.7 Hz, 1H), 7.96 (ddd, J = 8.0, 2.3, 1.7 Hz, 1H), 7.31 (ddd, J = 8.0, 4.8, 0.9 Hz, 1H), 6.78 (br, 1H), 4.22 (dd, J = 7.9, 2.6 Hz, 1H), 3.81 (s, 3H), 3.38–3.23 (m, 3H), 3.19 (d, J = 12.2 Hz, 1H), 2.42 (br, 1H). 13C{1H} NMR (101 MHz, CD2Cl2): δ 173.8, 150.1, 149.5, 147.3, 132.9, 127.7, 123.4, 81.5, 61.2, 58.0, 54.3, 52.8. IR (NaCl, thin film, cm−1): 3308, 2953, 1730, 1591, 1434, 1274, 1221. HRMS (ESITOF): m/z [M + H]+ calcd for C12H15N4O2+, 247.1190; found, 247.1189.
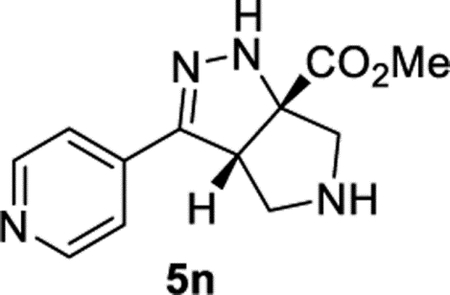
Methyl (3aS,6aS)-3-(Pyridin-4-yl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (5n).
General Procedure I was used, and the product was isolated in 70% (86.7 mg), 49% (61.1 mg), and 61% (74.7 mg) yields in triplicate trials. The average of 60% is reported. 1H NMR (400 MHz, CD2Cl2): δ 8.59 (d, J = 6.2 Hz, 2H), 7.46 (d, J = 6.2 Hz, 2H), 6.88 (br, 1H), 4.19 (dd, J = 7.9, 2.6 Hz, 1H), 3.82 (s, 3H), 3.37–3.23 (m, 3H), 3.20 (d, J = 12.1 Hz, 1H), 2.52 (br, 1H). 13C{1H} NMR (101 MHz, CD2Cl2): δ 173.5, 150.2, 150.1, 138.9, 119.9, 81.9, 61.3, 57.6, 54.3, 52.8. IR (NaCl, thin film, cm−1): 3295, 2954, 2923, 1732, 1599, 1434, 1415, 1275, 1219. HRMS (ESITOF): m/z [M + H]+ calcd for C12H15N4O2+, 247.1190; found, 247.1189.
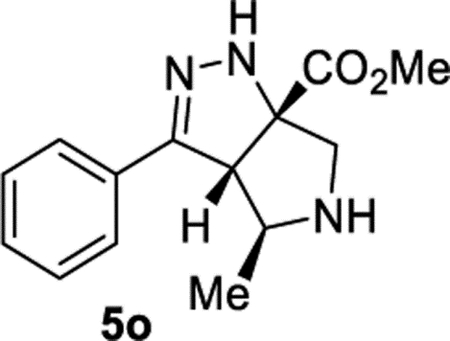
Methyl (3aS,4S,6aS)-4-Methyl-3-phenyl-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (5o).
General Procedure II was used, and the product was isolated in 63% and 46% yields. The average of 55% is reported. The dr was determined by crude 1H NMR to be 8.81:1 and 8.71:1 in duplicate trials. The average dr of 8.8:1 was reported. Final purification by column chromatography (gradient IPA/acetone from 0 to 40%) afforded the major diastereomer. 1H NMR (400 MHz, CDCl3): δ 7.66–7.60 (m, 2H), 7.43–7.33 (m, 3H), 6.58 (br, 1H), 3.86 (d, J = 2.6 Hz, 1H), 3.84 (s, 3H), 3.57 (qd, J = 6.9, 2.6 Hz, 1H), 3.46 (d, J = 12.3 Hz, 1H), 3.26 (d, J = 12.3 Hz, 1H), 2.45 (br, 1H), 1.42 (d, J = 6.9 Hz, 3H). 13C{1H} NMR (101 MHz, CDCl3): δ 174.1, 153.1, 131.6, 129.1, 128.7, 126.1, 82.3, 65.0, 61.7, 58.6, 53.1, 21.1. IR (NaCl, thin film, cm−1): 3319, 2961, 1731, 1445, 1271, 1227. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C14H17N3NaO2+, 282.1213; found, 282.1202.
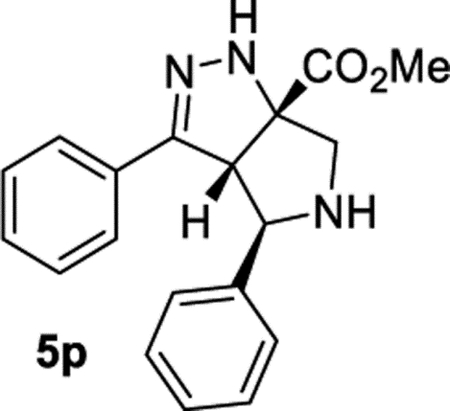
Methyl (3aS,4R,6aS)-3,4-Diphenyl-3a,4,5,6-tetrahydropyrrolo-[3,4-c]pyrazole-6a(1H)-carboxylate (5p).
General Procedure I was used, and the reaction time was 48 h. Final purification by column chromatography (IPA in hexanes with 1% TEA, gradient elution 10– 40%) afforded product 5p in 48% (59.2 mg) and 48% (58.5 mg) yields in duplicate trials. The average of 48% is reported. 1H NMR (500 MHz, CDCl3): δ 7.54–7.49 (m, 4H), 7.42 (apparent t, J = 7.5 Hz, 2H), 7.39–7.34 (m, 1H), 7.33–7.26 (m, 3H), 6.66 (br, 1H), 4.52 (d, J = 4.0 Hz, 1H), 4.40 (d, J = 4.0 Hz, 1H), 3.87 (s, 3H), 3.59 (d, J = 12.1 Hz, 1H), 3.31 (d, J = 12.1 Hz, 1H) 2.77 (br, 1H). 13C{1H} NMR (126 MHz, CDCl3): δ 174.0, 153.0, 142.2, 131.4, 129.1, 128.9, 128.6, 127.7, 127.3, 126.5, 82.5, 69.7, 64.2, 59.5, 53.1. IR (NaCl, thin film, cm−1): 3333, 3031, 2953, 1732, 1446, 1274, 1225. HRMS (ESITOF): m/z [M + Na]+ calcd for C19H19N3NaO2+, 344.1369; found, 344.1364.
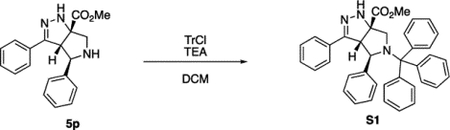
Methyl (3aS,4R,6aS)-3,4-Diphenyl-5-trityl-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (S1).
To a solution of compound 5p (160 mg, 0.498 mmol) in DCM (3 mL) was added TEA (0.14 mL, 1.0 mmol) and TrCl (168 mg, 0.602 mmol) at rt. After 6 h, the reaction was quenched by the addition of H2O and extracted with DCM. The combined organic phases were dried (Na2SO4), filtered, and concentrated under reduced pressure. Final purification by recrystallization in EtOAc/hexanes (ca. 1:5) afforded compound S1 (194 mg, 69%) as a white solid. 1H NMR (500 MHz, C6D6) δ 7.71 (d, J = 7.5 Hz, 2H), 7.39 (d, J = 7.8 Hz, 6H), 7.04–6.98 (m, 4H), 6.98–6.94 (m, 3H), 6.94–6.90 (m, 7H), 6.88–6.83 (m, 3H), 5.31 (s, 1H), 4.46 (s, 1H), 3.62 (d, J = 10.5 Hz, 1H), 3.46 (d, J = 10.6 Hz, 1H), 3.07 (s, 3H). 13C{1H} NMR (126 MHz, C6D6) δ 172.7, 151.9, 144.0, 143.6, 132.3, 129.8, 129.2, 128.5, 128.4, 128.3, 127.3, 127.1, 126.5, 125.8, 77.1, 73.9, 67.3, 64.1, 58.4, 51.9. IR (NaCl, thin film, cm−1): 3058, 3031, 2952, 1727, 1690, 1597, 1491, 1448, 1265, 1221. HRMS (ESI-TOF): m/z [M − CH3]+ calcd for C37H30N3O2+, 548.2333; found, 548.2297.
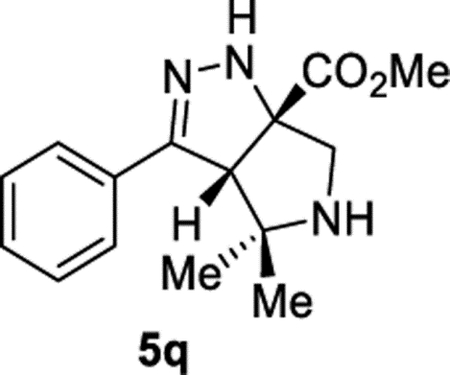
Methyl (3aS,6aS)-4,4-Dimethyl-3-phenyl-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (5q).
General Procedure I was used and the reaction time was 72 h. The product was isolated in 82% (113 mg) and 70% (95.4 mg) yields in duplicate trials. The average of 76% is reported. 1H NMR (400 MHz, CD3CN): δ 7.69–7.61 (m, 2H), 7.44–7.32 (m, 3H), 6.79 (br, 1H), 3.91 (s, 1H), 3.76 (s, 3H), 3.37 (d, J = 12.5 Hz, 1H), 3.12 (d, J = 12.5 Hz, 1H), 2.33 (br, 1H), 1.35 (s, 3H), 0.94 (s, 3H). 13C{1H} NMR (101 MHz, CD3CN): δ 174.2, 151.9, 133.4, 128.5, 128.4, 126.1, 84.9, 66.1, 66.0, 56.0, 52.4, 28.3, 25.2. IR (NaCl, thin film, cm−1): 3331, 2962, 1730, 1446, 1270, 1241. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C15H19N3NaO +2, 296.1369; found, 296.1366.
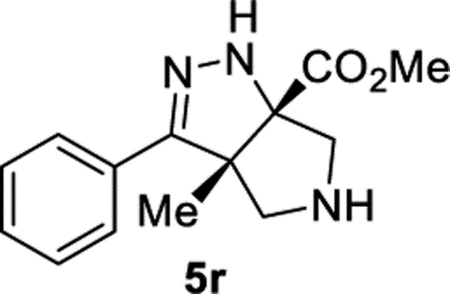
Methyl (3aS,6aS)-3a-Methyl-3-phenyl-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (5r).
General Procedure II was used, and the product was isolated in 70% (91.0 mg) and 57% (73.1 mg) yields in duplicate trials. The average of 64% is reported. 1H NMR (400 MHz, CD2Cl2): δ 7.75–7.61 (m, 2H), 7.49–7.29 (m, 3H), 6.35 (br, 1H), 3.81 (s, 3H), 3.74 (d, J = 12.3 Hz, 1H), 3.50 (d, J = 12.3 Hz, 1H), 3.15 (d, J = 12.3 Hz, 1H), 2.98 (d, J = 12.3 Hz, 1H), 2.50 (br, 1H), 1.41 (s, 3H). 13C{1H} NMR (101 MHz, CD2Cl2): δ 172.7, 154.5, 131.5, 128.5, 128.4, 126.6, 84.7, 66.8, 61.5, 60.0, 52.5, 19.6. IR (NaCl, thin film, cm−1): 3322, 2951, 1727, 1444, 1263. HRMS (ESI-TOF): m/z [M + H]+ calcd for C14H18N3O2+, 260.1394; found, 260.1399.
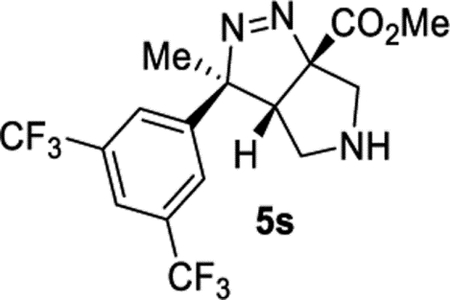
Methyl (3S,3aR,6aS)-3-(3,5-Bis(trifluoromethyl)phenyl)-3-methyl-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(3H)-carboxylate (5s).
General Procedure II was used, and the product was isolated in 64% (72.3 mg) and 45% (51.0 mg) yields in duplicate trials. The average of 55% is reported. 1H NMR (400 MHz, CDCl3): δ 7.81 (s, 1H), 7.66 (s, 2H), 3.83 (d, J = 12.1 Hz, 1H), 3.67 (s, 3H), 3.41 (d, J = 12.1 Hz, 1H), 3.28 (d, J = 12.0 Hz, 1H), 2.99 (dd, J = 12.0, 7.1 Hz, 1H), 2.64 (d, J = 7.0 Hz, 1H), 1.88 (s, 3H), 1.58 (br, 1H). 13C{1H} NMR (101 MHz, CDCl3): δ 169.2, 147.0, 131.9 (q, JC–F = 33.5 Hz), 125.5 (m), 123.1 (q, JC–F = 273.9 Hz), 121.4 (sept, JC–F = 3.9 Hz), 109.3, 97.1, 55.1, 52.9, 51.2, 50.2, 22.2. 19F{1H} NMR (376 MHz, CDCl3): −62.9. IR (NaCl, thin film, cm−1): 3323, 2957, 2848, 1739, 1375, 1279, 1182, 1135. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C16H15F6N3NaO2+, 418.0961; found, 418.0949.
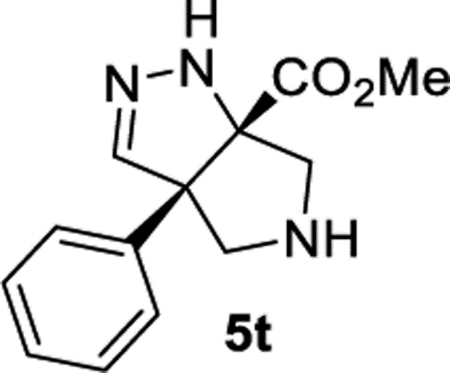
Methyl (3aS,6aS)-3a-Phenyl-3a,4,5,6-tetrahydropyrrolo[3,4-c]-pyrazole-6a(1H)-carboxylate (5t).
General Procedure II was used, and the product was isolated in 67% (80.6 mg) and 60% (79.7 mg) yields in duplicate trials. The average of 64% is reported. 1H NMR (500 MHz, CD2Cl2): δ 7.37 (t, J = 7.5 Hz, 2H), 7.30 (t, J = 7.3 Hz, 1H), 7.23 (d, J = 7.9 Hz, 2H), 6.57 (s, 1H), 6.35 (br, 1H), 3.70 (d, J = 12.6 Hz, 1H), 3.55 (d, J = 12.2 Hz, 1H), 3.45 (d, J = 12.6 Hz, 1H), 3.25 (d, J = 12.2 Hz, 1H), 3.17 (s, 3H), 2.32 (br, 1H). 13C{1H} NMR (126 MHz, CD2Cl2): δ 171.8, 147.2, 137.2, 128.3, 127.6, 127.3, 83.9, 75.8, 61.5, 57.3, 51.9. IR (NaCl, thin film, cm−1): 3315, 2948, 1728, 1446, 1434, 1259, 1069. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C13H15N3NaO2+, 268.1056; found, 268.1063.
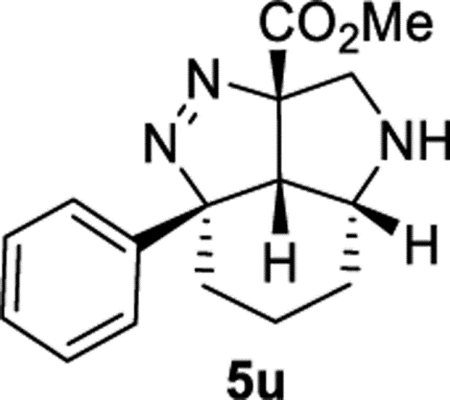
Methyl (2aS,2a1R,5aR,7aS)-2a-Phenyl-2a,2a1,3,4,5,5a,6,7-octa-hydro-7aH-pyrrolo[4,3,2-cd]indazole-7a-carboxylate (5u).
General Procedure II was used, and the product was isolated in 67% (76.2 mg) and 69% (78.3 mg) yields in duplicate trials. The average of 68% is reported. 1H NMR (400 MHz, CD2Cl2): δ 7.36–7.28 (m, 2H), 7.28–7.19 (m, 3H), 4.00 (d, J = 11.8 Hz, 1H), 3.49–3.41 (m, 1H), 3.44 (s, 3H), 3.29 (d, J = 11.8 Hz, 1H), 3.18–3.09 (m, 1H), 2.86 (d, J = 8.2 Hz, 1H), 1.97 (ddd, J = 14.6, 13.6, 3.6 Hz, 1H), 1.91–1.83 (m, 1H), 1.71–1.56 (m, 2H), 1.49–1.40 (m, 1H), 1.35–1.21 (m, 1H). 3C{1H} NMR (101 MHz, CD2Cl2): δ 169.0, 145.3, 128.2, 126.8, 125.0, 108.8, 96.2, 57.1, 53.6, 52.1, 47.1, 36.8, 26.1, 16.0. IR (NaCl, thin film, cm−1): 3336, 2934, 2861, 1735, 1445, 1433, 1286, 1212. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C16H19N3NaO2+, 308.1369; found, 308.1370.
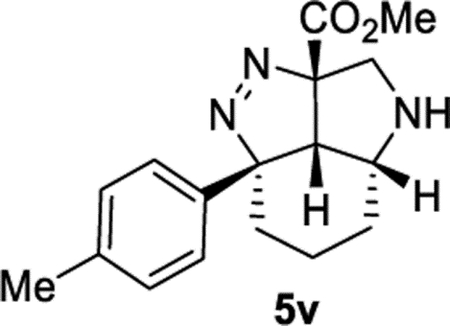
Methyl (2aS,2a1R,5aR,7aS)-2a-(p-Tolyl)-2a,2a1,3,4,5,5a,6,7-octahydro-7aH-pyrrolo[4,3,2-cd]indazole-7a-carboxylate (5v).
General Procedure II was used, and the product was isolated in 62% (74.3 mg) and 47% (58.1 mg) yields in duplicate trials. The average of 55% is reported. 1H NMR (400 MHz, CD3CN): δ 7.13 (br, 4H), 3.90 (d, J = 11.4 Hz, 1H), 3.44 (s, 3H), 3.44–3.36 (m, 1H), 3.21 (d, J = 11.5 Hz, 1H), 3.05–2.96 (m, 1H), 2.85 (d, J = 8.2 Hz, 1H), 2.30 (s, 3H), 2.17 (br, 1H), 2.01–1.89 (m, 1H), 1.81–1.70 (m, 1H), 1.63 (tt, J = 14.0, 4.0 Hz, 1H), 1.58–1.47 (m, 1H), 1.38–1.25 (m, 1H). 13C{1H} NMR (101 MHz, CD3CN): δ 169.1, 142.5, 136.5, 128.8, 125.0, 108.4, 95.9, 56.8, 53.3, 51.8, 46.4, 36.5, 25.9, 20.0, 15.9. IR (NaCl, thin film, cm−1): 2931, 2861, 1736, 1433, 1287, 1212. HRMS (ESITOF): m/z [M + Na]+ calcd for C17H21N3NaO +2, 322.1526; found, 322.1528.
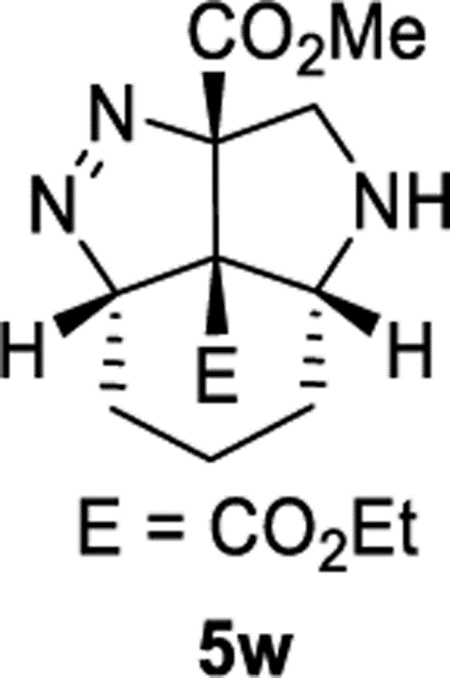
2a1-Ethyl 7a-Methyl (2aS,2a1S,5aR,7aS)-3,4,5,5a,6,7-Hexahydro-2a1H-pyrrolo[4,3,2-cd]indazole-2a1,7a(2aH)-dicarboxylate (5w).
General Procedure I was used with a minor modifications. The reaction time was 48 h and column chromatography was performed with IPA in hexanes with 1% TEA, gradient elution 10–60%. The product was isolated in 45% (60.2 mg) and 39% (51.9 mg) yields in duplicate trials. The average of 42% is reported. 1H NMR (500 MHz, CDCl3): δ 4.91 (dd, J = 6.2, 2.1 Hz, 1H), 4.15–4.08 (m, 2H), 3.90 (d, J = 12.7 Hz, 1H), 3.69 (s, 3H), 3.52 (d, J = 12.7 Hz, 1H), 3.46 (d, J = 4.1 Hz, 1H), 3.00 (d, J = 14.5 Hz, 1H), 1.93 (tdd, J = 14.3, 6.2, 3.7 Hz, 1H), 1.86 (d, J = 14.1 Hz, 1H), 1.61–1.49 (m, 2H), 1.22 (t, J = 7.2 Hz, 3H), 1.19–1.07 (m, 1H). 13C{1H} NMR (126 MHz, CDCl3): δ 171.6, 166.4, 111.7, 89.5, 62.3, 61.9, 57.7, 52.8, 52.4, 25.4, 25.1, 14.8, 14.1. IR (NaCl, thin film, cm−1): 2939, 2854, 1733, 1446, 1269, 1245, 1106, 1050. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C13H19N3NaO4+, 304.1268, found, 304.1273.
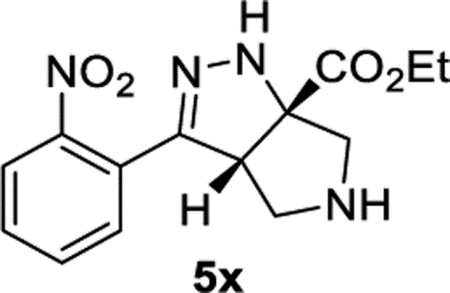
Ethyl (3aS,6aS)-3-(2-Nitrophenyl)-3a,4,5,6-tetrahydropyrrolo-[3,4-c]pyrazole-6a(1H)-carboxylate (5x).
General Procedure I was used with a minor modifications. The reaction was concentrated and purified directly (no TFA equilibration was necessary). The product was isolated in 93% (98.0 mg) and 95% (109 mg) yields in duplicate trials. The average of 94% is reported. 1H NMR (500 MHz, CDCl3): δ 7.80 (d, J = 8.1 Hz, 1H), 7.60 (apparent t, J = 7.6 Hz, 1H), 7.53– 7.45 (m, 2H), 6.72 (br, 1H), 4.31 (q, J = 7.1 Hz, 2H), 4.16 (dd, J = 8.1, 2.2 Hz, 1H), 3.31 (d, J = 12.1 Hz, 1H), 3.23 (d, J = 12.5 Hz, 1H), 3.16–3.08 (m, 2H), 2.21 (br, 1H), 1.34 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ 173.1, 149.8, 149.0, 132.5, 130.0, 129.6, 126.8, 124.4, 81.6, 62.3, 61.5, 60.1, 54.1, 14.3. IR (NaCl, thin film, cm−1): 3329, 2982, 2873, 1726, 1529, 1349, 1267, 1214. HRMS (ESITOF): m/z [M + Na]+ calcd for C14H16N4NaO4+, 327.1064; found, 327.1071.
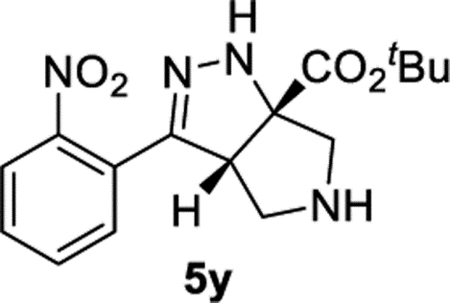
tert- Butyl (3aS,6aS)-3-(2-Nitrophenyl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (5y).
General Procedure I was used with a minor modifications. The reaction was concentrated and purified directly (no TFA equilibration was necessary). The product was isolated in 85% (90.6 mg) and 89% (95.2 mg) yields in duplicate trials. The average of 87% is reported. 1H NMR (500 MHz, CDCl3): δ 7.82 (d, J = 8.0 Hz, 1H), 7.59 (apparent t, J = 7.6 Hz, 1H), 7.51–7.45 (m, 2H), 6.66 (br, 1H), 4.08 (dd, J = 8.1, 2.2 Hz, 1H), 3.27 (d, J = 12.0 Hz, 1H), 3.18 (d, J = 12.4 Hz, 1H), 3.12–3.05 (m, 2H), 2.16 (br, 1H), 1.52 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3): δ 172.2, 150.1, 148.9, 132.6, 130.2, 129.6, 127.1, 124.5, 83.3, 82.2, 61.4, 60.1, 54.1, 28.1. IR (NaCl, thin film, cm−1): 3329, 2979, 2934, 2873, 1722, 1530, 1369, 1347, 1284, 1259, 1159. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C16H20N4NaO4+, 355.1377; found, 355.1372.
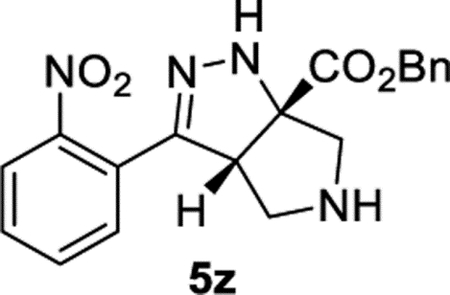
Benzyl (3aS,6aS)-3-(2-Nitrophenyl)-3a,4,5,6-tetrahydropyrrolo-[3,4-c]pyrazole-6a(1H)-carboxylate (5z).
General Procedure I was used with a minor modifications. The reaction was concentrated and purified directly (no TFA equilibration was necessary). The product was isolated in 91% (123 mg) and 96% (124 mg) yields in duplicate trials. The average of 94% is reported. 1H NMR (500 MHz, CDCl3): δ 7.80 (d, J = 8.0 Hz, 1H), 7.60 (apparent t, J = 7.6 Hz, 1H), 7.49 (apparent t, J = 8.7 Hz, 2H), 7.43–7.34 (m, 5H), 6.75 (br, 1H), 5.28 (s, 2H), 4.20 (dd, J = 8.1, 2.1 Hz, 1H), 3.32 (d, J = 12.1 Hz, 1H), 3.23 (d, J = 12.5 Hz, 1H), 3.17 (d, J = 12.1 Hz, 1H), 3.12 (dd, J = 12.5, 8.0 Hz, 1H), 2.24 (br, 1H). 13C{1H} NMR (126 MHz, CDCl3): δ 173.0, 149.6, 149.0, 135.1, 132.4, 130.0, 129.6, 128.84, 128.75, 128.4, 126.7, 124.4, 81.5, 67.9, 61.5, 60.2, 54.1. IR (NaCl, thin film, cm−1): 3329, 2936, 2874, 1729, 1530, 1347, 1267, 1213. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C19H18N4NaO4+, 389.1220; found, 389.1228.
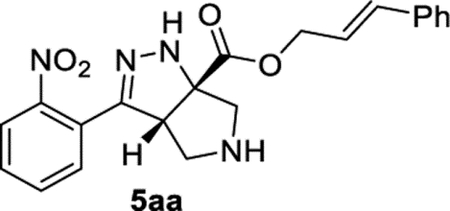
Cinnamyl (3aS,6aS)-3-(2-Nitrophenyl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (5aa).
General Procedure I was used with a minor modifications. The reaction was concentrated and purified directly (no TFA equilibration was necessary). The product was isolated in 83% (75.3 mg) and 72% (59.6 mg) yields in duplicate trials. The average of 78% is reported. 1H NMR (500 MHz, CDCl3): δ 7.82 (d, J = 8.0 Hz, 1H), 7.61 (apparent t, J = 7.6 Hz, 1H), 7.54–7.48 (m, 2H), 7.44 (d, J = 7.7 Hz, 2H), 7.36 (apparent t, J = 7.5 Hz, 2H), 7.32–7.27 (m, 1H), 6.76– 6.70 (m, 2H), 6.34 (dt, J = 15.9, 6.6 Hz, 1H), 4.92 (d, J = 6.6 Hz, 2H), 4.22 (d, J = 7.6 Hz, 1H), 3.36 (d, J = 12.1 Hz, 1H), 3.26 (d, J = 12.5 Hz, 1H), 3.23–3.12 (m, 2H), 2.23 (br, 1H). 13C{1H} NMR (101 MHz, CDCl3): δ 172.9, 149.7, 148.9, 135.9, 135.5, 132.4, 129.9, 129.5, 128.7, 128.4, 126.8, 126.7, 124.4, 122.1, 81.5, 66.8, 61.5, 60.1, 54.0. IR (NaCl, thin film, cm−1): 3339, 3027, 2937, 2874, 1728, 1529, 1346, 1264, 1209. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C21H20N4NaO4+, 415.1377; found, 415.1380.
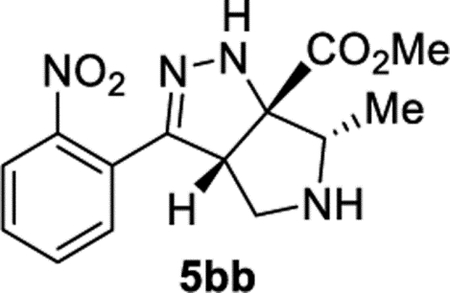
Methyl (3aS,6S,6aS)-6-Methyl-3-(2-nitrophenyl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (5bb).
General Procedure I was used with a minor modifications. The reaction time was 5 days, and the reaction was concentrated and purified directly (no TFA equilibration was necessary). The product was isolated in 83% (53.1 mg) and 90% (79.9 mg) yields in duplicate trials with a 3.2:1 dr. The average of 87% is reported. 1H NMR (500 MHz, CDCl3): δ 7.82 (d, J = 8.3 Hz, 1H), 7.61 (apparent t, J = 7.6 Hz, 1H), 7.54–7.48 (m, 2H), 6.47 (br, 1H), 4.21 (t, J = 5.1 Hz, 1H), 3.87 (s, 3H), 3.40 (q, J = 6.5 Hz, 1H), 3.18 (d, J = 5.1 Hz, 2H), 2.02 (br, 1H), 1.23 (d, J = 6.4 Hz, 3H). 1H NMR (500 MHz, CDCl3) minor diastereomer: δ 7.80 (d, J = 8.0 Hz, 1H), 7.61 (apparent t, J = 7.6 Hz, 1H), 7.55–7.48 (m, 2H), 6.67 (br, 1H), 4.32 (dd, J = 8.5, 4.1 Hz, 1H), 3.86 (s, 3H), 3.47–3.38 (m, 2H), 3.05 (dd, J = 12.5, 4.2 Hz, 1H), 1.16 (d, J = 6.9 Hz, 3H). 13C{1H} NMR (126 MHz, CDCl3): δ173.7, 150.3, 148.8, 132.3, 129.9, 129.5, 126.5, 124.3, 83.4, 65.8, 60.4, 53.1, 52.7, 13.3. IR (NaCl, thin film, cm−1): 3348, 2955, 2882, 1731, 1531, 1435, 1378, 1355, 1274, 1208. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C14H16N4NaO +4, 327.1064; found, 327.1068.
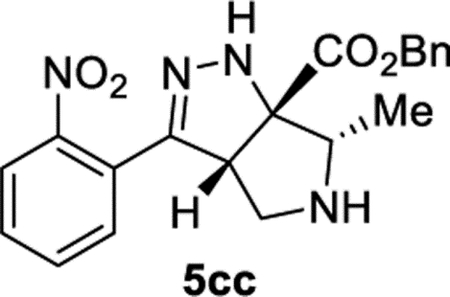
Benzyl (3aS,6S,6aS)-6-Methyl-3-(2-nitrophenyl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (5cc).
General Procedure I was used with a minor modifications. The reaction time was 5 days, and the reaction was concentrated and purified directly (no TFA equilibration was necessary). The product was isolated in 83% (70.6 mg) and 76% (66.6 mg) yields in duplicate trials with a 3.8:1 dr. The average of 80% is reported. 1H NMR (500 MHz, CDCl3): δ 7.82 (d, J = 8.0 Hz, 1H), 7.61 (apparent t, J = 7.6 Hz, 1H), 7.53–7.47 (m, 2H), 7.45–7.35 (m, 5H), 6.49 (br, 1H), 5.29 (s, 2H), 4.23 (t, J = 5.1 Hz, 1H), 3.41 (q, J = 6.4 Hz, 1H), 3.17 (d, J = 5.1 Hz, 2H), 2.06 (br, 1H), 1.20 (d, J = 6.8 Hz, 3H). 1H{1H} NMR (400 MHz, CDCl3) minor diastereomer: δ 7.79 (d, J = 7.9 Hz, 1H), 7.64–7.56 (m, 1H), 7.54–7.48 (m, 2H), 7.43–7.36 (m, 5H), 6.69 (br, 1H), 5.27 (d, J = 12.0 Hz, 1H), 5.26 (d, J = 12.0 Hz, 1H), 4.33 (dd, J = 8.4, 4.1 Hz, 1H), 3.47–3.35 (m, 2H), 3.03 (dd, J = 12.5, 4.1 Hz, 1H), 1.08 (d, J = 7.0 Hz, 3H). 13C NMR (126 MHz, CDCl3): δ 173.1, 150.3, 148.8, 135.1, 132.4, 129.9, 129.5, 128.8, 128.7, 128.3, 126.5, 124.4, 83.4, 67.8, 65.8, 60.4, 52.6, 13.3. IR (NaCl, thin film, cm−1): 3342, 2970, 2878, 1727, 1533, 1377, 1353, 1266, 1203. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C20H20N4NaO +4, 403.1377; found, 403.1375.
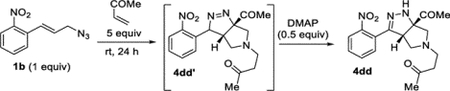
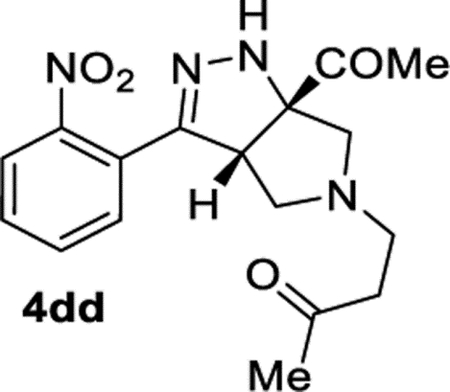
4-((3aS,6aS)-6a-Acetyl-3-(2-nitrophenyl)-3a,4,6,6atetrahydropyrrolo[3,4-c]pyrazol-5(1H)-yl)butan-2-one (4dd).
A 4 mL vial was charged with azide 1b (61.3 mg, 0.300 mmol) and methyl vinyl ketone (0.13 mL, 1.6 mmol). The vial was sealed with a Teflon-lined cap, and the reaction was stirred at an ambient temperature. After 24 h, excess methyl vinyl ketone was removed under reduced pressure (<15 Torr). The resulting oil was directly purified by column chromatography (EtOAc in hexanes, gradient elution 0–80%), which afforded compound 4dd′ as a yellow oil. Compound 4dd′ was isolated in 95% (98.4 mg) and 79% (81.7 mg) in duplicate trials. The average of 87% is reported. Compound 4dd′ tends to gradually tautomerize to compound 4dd at an ambient temperature. Pure 4dd can be synthesized using the procedure above with minor modifications. The crude 4dd′ was reconstituted in a 20 mL vial with DCM (2 mL), and DMAP (18 mg, 0.15 mmol) was added. The vial was sealed with a Teflon-lined cap, and the reaction was stirred at an ambient temperature. After 16 h, the solvent was removed under reduced pressure. Purification by column chromatography (IPA in DCM with 1% TEA, gradient elution 0–10%) afforded 4dd as a yellow oil (37.7 mg, 38%), along with recovered unreacted 4dd′.
1H NMR (400 MHz, CDCl3): δ 7.93 (d, J = 8.2 Hz, 1H), 7.65 (t, J = 7.6 Hz, 1H), 7.58–7.50 (m, 2H), 6.50 (br, 1H), 4.02 (dd, J = 7.5, 3.7 Hz, 1H), 3.02 (d, J = 10.0 Hz, 1H), 2.91 (d, J = 10.0 Hz, 1H), 2.86–2.70 (m, 3H), 2.70–2.64 (m, 1H), 2.60 (t, J = 7.1 Hz, 2H), 2.37 (s, 3H), 2.16 (s, 3H). 13C{1H} NMR (101 MHz, CDCl3): δ 207.3, 206.2, 149.6, 148.1, 133.0, 131.2, 129.7, 127.2, 124.5, 82.2, 64.3, 57.6, 56.3, 49.0, 42.7, 29.9, 25.3. IR (NaCl, thin film, cm−1): 3328, 2913, 2807, 1710, 1529, 1354. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C17H20N4NaO4+, 367.1377; found, 367.1374.
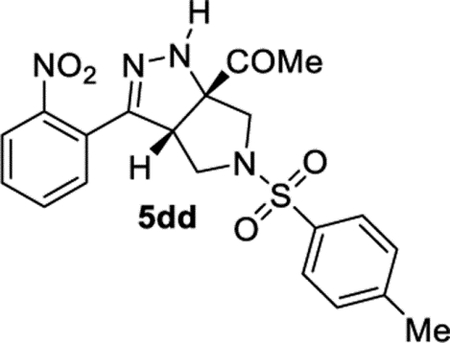
1-((3aS,6aS)-3-(2-Nitrophenyl)-5-tosyl-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazol-6a(1H)-yl)ethan-1-one (5dd).
General Procedure III was used with a minor modification. DMAP was used to replace DIPEA. The product was isolated in 49% (62.0 mg) and 66% (90.4 mg) yields in duplicate trials. The average of 58% is reported. 1H NMR (400 MHz, CDCl3): δ 8.04 (d, J = 8.2 Hz, 1H), 7.71 (t, J = 7.6 Hz, 1H), 7.67 (d, J = 8.2 Hz, 2H), 7.63–7.55 (m, 2H), 7.36 (d, J = 8.0 Hz, 2H), 6.41 (br, 1H), 4.06 (dd, J = 7.8, 3.3 Hz, 1H), 3.62 (d, J = 10.6 Hz, 1H), 3.50 (d, J = 10.7 Hz, 1H), 3.32 (dd, J = 10.1, 3,4 Hz, 1H), 3.10 (dd, J = 10.1, 7.7 Hz, 1H), 2.46 (s, 3H), 2.40 (s, 3H). 13C{1H} NMR (101 MHz, CDCl3): δ 205.0, 148.4, 147.8, 144.5, 133.7, 131.8, 131.5, 130.3, 130.0, 128.0, 126.7, 124.8, 81.7, 57.3, 56.7, 51.3, 25.5, 21.6. IR (NaCl, thin film, cm−1): 3334, 2292, 2868, 1712, 1529, 1345, 1163. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C20H20N4NaO5S+, 451.1047; found, 451.1036.
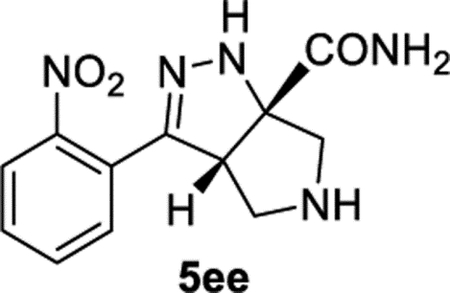
(3aS,6aS)-3-(2-Nitrophenyl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]-pyrazole-6a(1H)-carboxamide (5ee).
General Procedure I was used with a minor modifications. During the reaction, the product precipitated as a yellow solid. The solid was purified by recrystallization in MeOH/DCM/hexanes (1:2:10), and the product was isolated in 75% (82.5 mg) and 68% (69.6 mg) in duplicate trials. The average of 72% is reported. 1H NMR (500 MHz, DMSO): δ 7.77 (d, J = 8.0 Hz, 1H), 7.68–7.65 (m, 2H), 7.62 (br, 1H), 7.57–7.51 (m, 1H), 7.41 (br, 2H), 4.03 (d, J = 7.4 Hz, 1H), 3.12 (d, J = 11.9 Hz, 1H), 3.07–2.98 (m, 2H), 2.88 (d, J = 11.9 Hz, 1H), 2.28 (br, 1H). 13C{1H} NMR (126 MHz, DMSO): δ 175.4, 149.1, 145.8, 132.4, 129.9, 129.4, 125.5, 124.1, 79.9, 61.2, 59.8, 54.2. IR (NaCl, thin film, cm−1): 3289, 3189, 1630, 1526, 1420, 1356. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C12H13N5NaO3+, 298.0911; found, 298.0921.
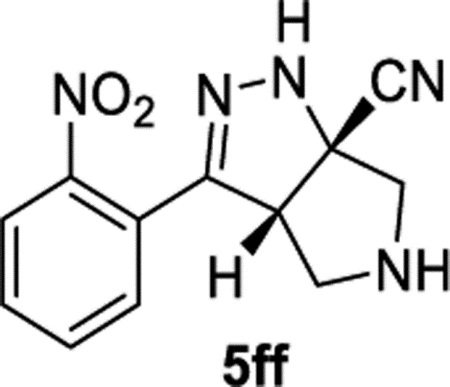
(3aS,6aS)-3-(2-Nitrophenyl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]-pyrazole-6a(1H)-carbonitrile (5ff).
General Procedure II was used with a minor modifications. Both parts of the reaction were heated at 40 °C for 24 h. The product was purified by recrystallization (EtOAc/hexanes). The product was isolated in 68% (66.3 mg) and 72% (64.4 mg) yields in duplicate trials. The average of 70% is reported. 1H NMR (500 MHz, DMSO-d6): δ 8.36 (br, 1H), 7.85 (d, J = 8.0 Hz, 1H), 7.75–7.66 (m, 2H), 7.62–7.56 (m, 1H), 4.48 (dd, J = 7.9, 2.5 Hz, 1H), 3.30 (d, J = 12.2 Hz, 1H), 3.18–3.07 (m, 2H), 2.96 (d, J = 12.1 Hz, 1H), 2.68 (br, 1H). 13C{1H} NMR (126 MHz, DMSO-d6): δ 148.9, 146.5, 132.7, 130.3, 130.1, 124.7, 124.3, 120.8, 67.7, 61.7, 61.4, 53.5. IR (NaCl, thin film, cm−1): 3320, 2926, 2237, 1529, 1345. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C12H11N5NaO2+, 280.0805; found, 280.0816.
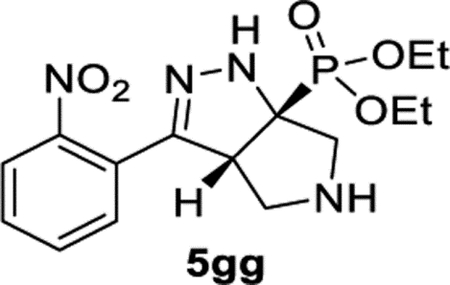
Diethyl ((3aR,6aR)-3-(2-Nitrophenyl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazol-6a(1H)-yl)phosphonate (5gg).
General Procedure I was used, and the product was isolated in 38% (56.4 mg) and 42% (62.0 mg) yields in duplicate trials. The average of 40% is reported. 1H NMR (500 MHz, CDCl3): δ 7.78 (d, J = 8.1 Hz, 1H), 7.61 (apparent t, J = 7.6 Hz, 1H), 7.53–7.48 (m, 2H), 6.18 (d, J = 7.3 Hz, 1H), 4.34–4.17 (m, 5H), 3.31 (dd, J = 12.7, 6.0 Hz, 1H), 3.24 (dd, J = 12.6, 3.4 Hz, 1H), 3.16 (d, J = 12.4 Hz, 1H), 3.03 (dd, J = 12.5, 7.6 Hz, 1H), 2.09 (br, 1H), 1.36 (t, J = 7.1 Hz, 6H). 13C{1H} NMR (126 MHz, CDCl3): δ 149.1, 148.2 (d, JC−P = 7.3 Hz), 132.2, 129.8, 129.4, 126.3, 124.3, 74.6 (d, JC−P = 159.7 Hz), 63.4 (d, JC−P = 7.0 Hz), 62.9 (d, JC−P = 7.3 Hz), 59.4 (d, JC−P = 13.8 Hz), 58.2, 53.7 (d, JC−P = 13.6 Hz), 16.6 (d, JC−P = 5.5 Hz), 16.5 (d, JC−P = 5.6 Hz). 31P{1H} NMR (162 MHz, CDCl3): δ 23.3. IR (NaCl, thin film, cm−1): 3245, 2979, 2928, 2873, 1532, 1236, 1048. HRMS (ESITOF): m/z [M + Na]+ calcd for C15H21N4NaO5P+, 391.1142; found, 391.1141.
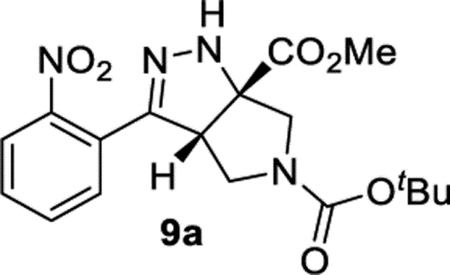
5-(tert-Butyl) 6a-Methyl (3aS,6aS)-3-(2-nitrophenyl)-3a,4-dihydropyrrolo[3,4-c]pyrazole-5,6a(1H,6H)-dicarboxylate (9a).
General Procedure III was used, and the product was isolated in 80% (92.9 mg) as a single tautomer. 1H NMR (400 MHz, CD3CN): 7.80 (d, J = 8.0 Hz, 1H), 7.69 (t, J = 7.6 Hz, 1H), 7.62–7.54 (m, 2H), 7.05 (br, 1H), 4.28 (dd, J = 8.9, 4.4 Hz, 1H), 3.88–3.78 (m, 2H), 3.80 (s, 3H), 3.72–3.65 (m, 1H), 3.60–3.52 (m, 1H), 1.43 (s, 9H). 13C{1H} NMR (101 MHz, CD3CN): 171.8, 153.6, 148.8, 147.9, 132.4, 130.0, 129.7, 125.2, 124.0, 79.5, 55.9 (br, tentatively assigned as 2C), 52.7, 51.4, 49.3, 27.5. IR (NaCl, thin film, cm−1): 3345, 2979, 2870, 1737, 1693, 1530, 1400, 1367. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C18H22N4NaO6+, 413.1432; found, 413.1424.
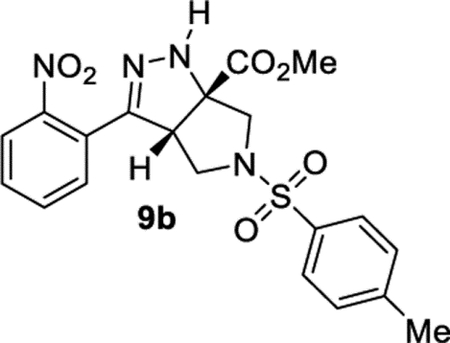
Methyl (3aS,6aS)-3-(2-Nitrophenyl)-5-tosyl-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (9b).
General Procedure III was used with R–X = 4-toluenesulfonyl chloride (TsCl). Product 9b was isolated in 74% (126.7 mg) and 69% (91.6 mg) yields in duplicate trials. An average of 72% is reported. 1H NMR (400 MHz, CDCl3): δ 7.93 (d, J = 8.1 Hz, 1H), 7.63 (d, J = 8.3 Hz, 2H), 7.65 (t, J = 7.6 Hz, 1H), 7.55 (t, J = 7.8 Hz, 1H), 7.49 (d, J = 7.7 Hz, 1H), 7.34 (d, J = 8.0 Hz, 2H), 6.64 (br, 1H), 4.21 (dd, J = 8.3, 3.9 Hz, 1H), 3.82 (s, 3H), 3.74 (d, J = 10.2 Hz, 1H), 3.46 (d, J = 10.3 Hz, 1H), 3.37 (dd, J = 10.0, 3.9 Hz, 1H), 3.29 (dd, J = 10.1, 8.3 Hz, 1H), 2.45 (s, 3H). 13C{1H} NMR (101 MHz, CDCl3): δ 171.2, 148.8, 147.9, 144.3, 133.2, 132.2, 131.2, 130.1, 129.9, 127.9, 126.3, 124.6, 77.8, 58.7, 56.0, 53.6, 51.3, 21.6. IR (NaCl, thin film, cm−1): 3332, 2956, 2857, 1738, 1530, 1346, 1164. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C20H20N4NaO6S+, 467.0996; found, 467.1000.
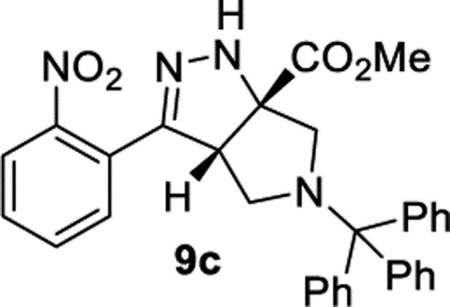
Methyl (3aS,6aS)-3-(2-Nitrophenyl)-5-trityl-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (9c).
General Procedure III was used and with R–X = triphenylmethyl chloride (TrCl). Product 9c was isolated in 88% (140.7 mg) as a tautomeric mixture 9c′/9c = 1.5:1. 1H NMR (500 MHz, CDCl3): δ 7.84 (d, J = 8.1 Hz, 1H), 7.76 (d, J = 7.7 Hz, 1H), 7.66 (t, J = 7.6 Hz, 1H), 7.55– 7.46 (m, 7H), 7.28–7.21 (m, 6H), 7.19–7.14 (m, 3H), 6.76 (br, 1H), 4.02 (d, J = 7.2 Hz, 1H), 3.75 (s, 3H), 3.44 (d, J = 9.4 Hz, 1H), 3.05 (d, J = 9.6 Hz, 1H), 2.09–2.00 (m, 2H). 13C{1H} NMR (126 MHz, CDCl3): δ 172.6, 149.3, 148.5, 132.3, 130.6, 129.3, 129.1, 127.9, 127.6, 126.8, 126.3, 124.3, 75.9, 73.4, 58.4, 54.2, 53.0, 50.4. IR (NaCl, thin film, cm−1): 3349, 2057, 2952, 2831, 1736, 1595, 1530, 1448, 1264, 1071, 738. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C32H28N4NaO4+, 555.2003; found, 555.2018.
Note.

The trityl-protected product was isolated as a mixture of the unconjugated (9c′) and conjugated (9c) isomers. To determine how to convert the mixture into a single isomer, a stock solution of the 9c′/9c mixture (45.2 mg, 84.6 μmol) and 1,3,5-trimethoxybenzene (6.4 mg, 38 μmol) in CDCl3 (2.1 mL) was divided into three portions. TFA (6.5 μL, 38 μmol) was added to one portion, DBU (2.1 μL, 6.4 μmol) was added to the second portion, and the third was left with no additive. The three samples were analyzed by 1H NMR after 0.5 and 18 h. After 18 h, the samples containing TFA or DBU had fully converted to the conjugated tautomer (6d).
Methyl (3aS,6aS)-5-Benzyl-3-(2-nitrophenyl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (9d).
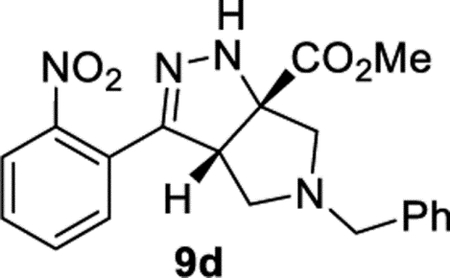
General Procedure III was used with R–X = benzyl chloroformate (CbzCl). Product 9d was isolated in 81% (93.4 mg) as a single tautomer. 1H NMR (400 MHz, CDCl3): δ 7.86 (d, J = 8.1 Hz, 1H), 7.62–7.54 (m, 1H), 7.53–7.45 (m, 2H), 7.40–7.31 (m, 3H), 7.29 (d, J = 6.8 Hz, 1H), 7.27–7.21 (m, 1H), 6.61 (br, 1H), 4.22 (dd, J = 7.6, 2.5 Hz, 1H), 3.85 (s, 3H), 3.77 (d, J = 13.2 Hz, 1H), 3.59 (d, J = 13.2 Hz, 1H), 3.21 (d, J = 9.5 Hz, 1H), 2.91–2.81 (m, 2H), 2.62 (d, J = 9.5, 7.7 Hz, 1H). 13C{1H} NMR (101 MHz, CDCl3): δ 172.9, 150.0, 148.2, 138.2, 132.6, 130.9, 129.5, 128.5, 128.3, 127.2, 127.0, 124.3, 77.1, 65.9, 58.6, 57.0, 56.6, 53.1. IR (NaCl, thin film, cm−1): 3345, 2956, 2804, 1734, 1532, 1347, 1275, 1227. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C20H20N4NaO4+, 403.1377; found, 403.1373.
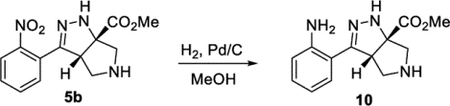
Methyl (3aS,6aS)-3-(2-Aminophenyl-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (10).
A vial was sequentially charged with compound 5b (61 mg, 0.21 mmol), MeOH (1 mL), and Pd/C (6 mg, 10 wt % Pd). The vial was sealed with a septum and purged with hydrogen gas using a hydrogen balloon without stirring. After 10 min, the vial was equipped with 2 hydrogen balloons under ambient pressure. After 4 h, the reaction was filtered through a short plug of silica gel and rinsed with MeOH (5 mL). The solvent was removed under reduced pressure. This procedure afforded the product 10 in 100% (54 mg) as a yellow oil. 1H NMR (500 MHz, CD2Cl2): δ 7.17–7.06 (m, 2H), 6.76–6.66 (m, 2H), 5.89 (br, 1H), 4.29 (d, J = 8.3 Hz, 1H), 3.81 (s, 3H), 3.43– 3.30 (m, 2H), 3.29 (d, J = 12.0 Hz, 1H), 3.22 (d, J = 12.0 Hz, 1H). 13C{1H} NMR (126 MHz, CD2Cl2): δ 174.2, 155.8, 147.3, 129.4, 128.8, 116.1, 115.6, 113.2, 79.6, 61.0, 59.8, 55.0, 52.8. IR (NaCl, thin film, cm−1): 3449, 3314, 2948, 1730, 1613, 1495, 1453, 1267, 1124. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C13H16N4NaO +2, 283.1165; found, 283.1149.
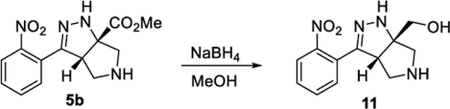
((3aS,6aS)-3-(2-Nitrophenyl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]-pyrazol-6a(1H)-yl)methanol (11).
To a solution of compound 5b (48.6 mg, 0.168 mmol) in MeOH (2 mL) was added NaBH4 (28.4 mg, 0.751 mmol). The reaction was sealed under air and heated to 60 °C. After 1 h, additional NaBH4 (20.6 mg, 0.545 mmol) was added. After 1 h, the reaction was quenched by the addition of H2O and brine. The reaction mixture was extracted with EtOAc (6 × 20 mL). The combined organic phases were dried (Na2SO4) and concentrated under reduced pressure to afford compound 11 (34.5 mg, 79%). 1H NMR (500 MHz, CDCl3): δ 7.80 (d, J = 8.1 Hz, 1H), 7.61 (apparent t, J = 7.6 Hz, 1H), 7.54–7.47 (m, 2H), 5.99 (br, 1H), 3.80 (s, 2H), 3.74 (d, J = 7.7 Hz, 1H), 3.19 (d, J = 12.3 Hz, 1H), 3.13 (d, J = 12.2 Hz, 1H), 3.04 (dd, J = 12.4, 7.7 Hz, 1H), 2.89 (d, J = 12.3 Hz, 1H), 2.57 (br, 1H). 13C{1H} NMR (126 MHz, MeOD): δ 149.0, 148.8, 132.1, 129.8, 129.1, 126.8, 123.9, 79.4, 63.5, 57.0, 55.6, 51.6. IR (NaCl, thin film, cm−1): 3290, 2921, 2868, 1528, 1347. HRMS (ESITOF): m/z [M + Na]+ calcd for C12H14N4NaO3+, 285.0958; found, 285.0956.
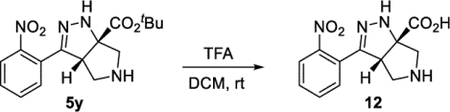
(3aS,6aS)-3-(2-Nitrophenyl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]-pyrazole-6a(1H)-carboxylic acid (12).
To a solution of bicyclic amine 5y (38.7 mg, 0.117 mmol) in DCM (0.5 mL) was added TFA (0.5 mL) at an ambient temperature. After 4 h, the reaction mixture was concentrated under reduced pressure to afford the bis-TFA salt (59.7 mg) in a quantitative yield. 1H NMR (500 MHz, MeOD:TFA 50:1): δ 7.96 (d, J = 8.0 Hz, 1H), 7.75 (apparent t, J = 7.6 Hz, 1H), 7.70–7.63 (m, 2H), 4.51 (dd, J = 9.8, 3.4 Hz, 1H), 3.88 (d, J = 12.0 Hz, 1H), 3.76 (d, J = 12.0 Hz, 1H), 3.71 (dd, J = 12.5, 9.6 Hz, 1H), 3.52 (dd, J = 12.4, 3.6 Hz, 1H). 13C{1H} NMR (126 MHz, MeOD/TFA 50:1): δ 170.9, 148.5, 147.4, 132.7, 130.4, 130.0, 125.2, 124.2, 77.9, 54.8, 54.6, 49.4. IR (NaCl, thin film, cm−1): 3323, 2917, 1779, 1721, 1659, 1519, 1344, 1204, 1160. HRMS (ESI-TOF): m/z [M + H]+ calcd for C12H13N4O4+, 277.0931; found, 277.0922.
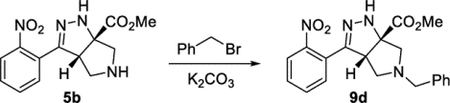
Methyl (3aS,6aS)-5-Benzyl-3-(2-nitrophenyl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (9d).
A 20 mL vial was charged with compound 5b (112 mg, 0.384 mmol) and acetonitrile (9 mL). Potassium carbonate (160 mg, 1.16 mmol) and benzyl bromide (55 μL, 0.46 mmol) were added consecutively. The vial was sealed with a Teflon-lined cap, and the reaction was heated to 55°C. After 16 h, the reaction was diluted with water (5 mL). The resulting solution was extracted with EtOAc (3 × 20 mL). The combined organic phases were dried (Na2SO4) and concentrated under reduced pressure. Purification by column chromatography (EtOAc in hexanes, gradient elution 0–70%) afforded compound 9d as a yellow oil (104 mg, 72%). The material obtained provided an identical 1H NMR spectra above.
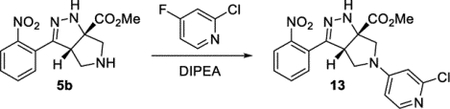
Methyl (3aS,6aS)-5-(2-Chloropyridin-4-yl)-3-(2-nitrophenyl)-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (13).
To a solution of compound 5b (61.0 mg, 0.210 mmol) in DMF (0.5 mL) were added DIPEA (75 μL, 0.42 mmol) and 2-chloro-4-fluoropyridine (76.8 mg, 0.586 mmol). The reaction was sealed under air and heated to 80 °C. After 16 h, the reaction was cooled to rt and diluted with H2O. The reaction mixture was extracted with Et2O. The combined organic phases were washed with H2O, brine, dried (Na2SO4), and concentrated under reduced pressure. Final purification by column chromatography (EtOAc in hexanes with 1% TEA, gradient elution 10–65%) afforded compound 13 (65.9 mg, 78%). 1H NMR (500 MHz, CDCl3): δ 7.99 (d, J = 5.9 Hz, 1H), 7.96 (d, J = 8.1 Hz, 1H), 7.68 (apparent t, J = 7.5 Hz, 1H), 7.61–7.55 (m, 2H), 6.94 (s, 1H), 6.38 (s, 1H), 6.30 (d, J = 5.9 Hz, 1H), 4.39 (dd, J = 9.5, 5.7 Hz, 1H), 3.92 (s, 3H), 3.86 (s, 2H), 3.71 (t, J = 9.7 Hz, 1H), 3.47 (dd, J = 10.1, 5.7 Hz, 1H). 13C{1H} NMR (126 MHz, CDCl3): δ 171.8, 152.9, 152.2, 149.9, 149.3, 148.2, 133.2, 130.7, 130.3, 126.3, 124.7, 107.2, 106.9, 78.8, 57.4, 54.7, 53.7, 51.2. IR (NaCl, thin film, cm−1): 3337, 2954, 2868, 1736, 1594, 1529, 1498. HRMS (ESITOF): m/z [M + Na]+ calcd for C18H16ClN5NaO4+, 424.0783; found, 424.0796.
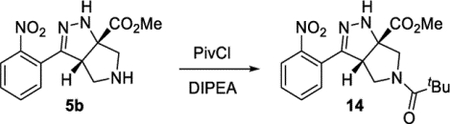
Methyl (3aS,6aS)-3-(2-Nitrophenyl)-5-pivaloyl-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-6a(1H)-carboxylate (14).
A 20 mL vial was charged with compound 5b (67.1 mg, 0.231 mmol) and DCM (1 mL). Pivaloyl chloride (57 μL, 0.46 mmol) was added, followed by DIPEA (76 μL, 0.46 mmol). The vial was sealed with a Teflon-lined cap, and the reaction was stirred at an ambient temperature. After 16 h, the reaction was quenched by the addition of water (5 mL). The resulting solution was extracted with DCM (3 × 10 mL). The combined organic solution was washed with brine, dried (MgSO4), filtered, and concentrated under reduced pressure. Purification by column chromatography (EtOAc in hexanes, gradient elution 0–100%) afforded compound 14 as a yellow solid (75.7 mg, 88%). 1H NMR (400 MHz, CDCl3): δ 7.94 (d, J = 8.2 Hz, 1H), 7.63 (apparent t, J = 7.6 Hz, 1H), 7.59–7.50 (m, 2H), 6.75 (br, 1H), 4.21–4.15 (m, 1H), 4.19 (d, J = 11.7 Hz, 1H), 3.97 (d, J = 11.8 Hz, 1H), 3.89 (s, 3H), 3.88–3.74 (m, 2H), 1.23 (s, 9H). 13C{1H} NMR (101 MHz, CDCl3): 176.4, 171.9, 150.0 (br), 148.1, 133.1, 130.9, 130.0, 126.5, 124.6, 78.5 (br), 57.2, 54.6, 53.5, 50.6, 38.8, 27.5. IR (NaCl, thin film, cm−1): 3331, 2529, 2959, 2868, 1738, 1624, 1533, 1141, 1365, 1348, 1276, 1233. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C18H22N4NaO5+, 397.1482; found, 397.1464.
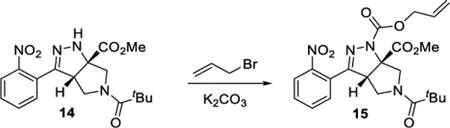
1-Allyl 6a-Methyl (3aS,6aS)-3-(2-Nitrophenyl)-5-pivaloyl-3a,4,5,6-tetrahydropyrrolo[3,4-c]pyrazole-1,6a-dicarboxylate (15).
To a solution of compound 14 (52.1 mg, 0.139 mmol) in DMF (0.5 mL) were added K2CO3 (83.2 mg, 0.602 mmol) and allyl bromide (0.10 mL, 1.6 mmol). The reaction was sealed under air and heated to 60 °C. After 13 h, the reaction was cooled to rt and diluted with water. The reaction mixture was extracted with Et2O. The combined organic phases were washed with water, brine, dried (Na2SO4), and concentrated under reduced pressure. Final purification by column chromatography (IPA in hexanes, gradient elution 0–15%) afforded compound 15 (44.5 mg, 70%). 1H NMR (500 MHz, CDCl3): δ 8.18 (d, J = 8.2 Hz, 1H), 7.75 (apparent t, J = 7.5 Hz, 1H), 7.66 (apparent t, J = 7.9 Hz, 1H), 7.60 (d, J = 7.7 Hz, 1H), 6.01–5.91 (m, 1H), 5.38 (d, J = 17.2 Hz, 1H), 5.27 (d, J = 10.5 Hz, 1H), 4.81 (dd, J = 13.3, 5.8 Hz, 1H), 4.77–4.67 (m, 2H), 4.37 (d, J = 12.7 Hz, 1H), 4.20 (d, J = 12.2 Hz, 1H), 4.13 (d, J = 8.5 Hz, 1H), 3.91 (s, 3H), 3.52–3.40 (m, 1H), 1.26 (s, 9H). 13C{1H} NMR (126 MHz, CDCl3): δ 177.0, 168.8, 153.3, 152.2, 147.6, 134.2, 132.6, 131.8, 131.1, 125.9, 125.2, 119.1, 67.5, 57.2, 55.1, 54.2, 53.6, 49.4, 38.9, 27.6. IR (NaCl, thin film, cm−1): 2974, 1747, 1704, 1633, 1531, 1435, 1415, 1346, 1277. HRMS (ESI-TOF): m/z [M + Na]+ calcd for C22H26N4NaO7+, 481.1694; found, 481.1710.
Supplementary Material
ACKNOWLEDGMENTS
Dr. Victor Young Jr. and Ryan Daley are thanked for their assistance with X-ray crystallography. Duke Ho is thanked for assistance in preparing several known cinnamyl azides. This research was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award no. R35GM124718. We also acknowledge NIH Shared Instrumentation Grant no. S10OD011952. Both A.S.C. and E.C.L. gratefully acknowledge support from the Wayland E. Noland Fellowship.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.0c00535.
Crystallographic data for compound 2hh (CIF)
Crystallographic data for compound 5a (CIF)
Crystallographic data for compound 5bb (CIF)
Crystallographic data for compound 5v·HCl (CIF)
Optimization procedures and tables; NMR spectra of all new compounds; HPLC traces; X-ray data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- (2).Wang Y-Y; Bode JW Olefin Amine (OLA) Reagents for the Synthesis of Bridged Bicyclic and Spirocyclic Saturated N-Hetero-cycles by Catalytic Hydrogen Atom Transfer (HAT) Reactions. J. Am. Chem. Soc 2019, 141, 9739–9745. [DOI] [PubMed] [Google Scholar]
- (3).Lovering F; Bikker J; Humblet C Escape from Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem 2009, 52, 6752–6756. [DOI] [PubMed] [Google Scholar]
- (4).Lovering F Escape from Flatland 2: Complexity and Promiscuity. MedChemComm 2013, 4, 515–519. [Google Scholar]
- (5).Meanwell NA Improving Drug Design: An Update on Recent Applications of Efficiency Metrics, Strategies for Replacing Problematic Elements, and Compounds in Nontraditional Drug Space. Chem. Res. Toxicol 2016, 29, 564–616. [DOI] [PubMed] [Google Scholar]
- (6).Orr STM; Ripp SL; Ballard TE; Henderson JL; Scott DO; Obach RS; Sun H; Kalgutkar AS Mechanism-Based Inactivation (MBI) of Cytochrome P450 Enzymes: Structure– Activity Relationships and Discovery Strategies To Mitigate Drug– Drug Interaction Risks. J. Med. Chem 2012, 55, 4896–4933. [DOI] [PubMed] [Google Scholar]
- (7).Ritchie TJ; MacDonald SJF; Young RJ; Pickett SD The Impact of Aromatic Ring Count on Compound Developability: Further Insights by Examining Carbo- and Hetero-Aromatic and -Aliphatic Ring Types. Drug Discovery Today 2011, 16, 164–171. [DOI] [PubMed] [Google Scholar]
- (8).Levin JI; Venkatesan AM; Chan PS; Bailey TK; Vice G; Coupet J 6-Substituted Quinazolinone Angiotensin II Receptor Antagonists. Bioorg. Med. Chem. Lett 1994, 4, 1819–1824. [Google Scholar]
- (9).Møllerud S; Pinto A; Marconi L; Frydenvang K; Thorsen TS; Laulumaa S; Venskutonyte R; Winther S; Moral AMC; Tamborini L; Conti P; Pickering DS; Kastrup JS Structure and Affinity of Two Bicyclic Glutamate Analogues at AMPA and Kainate Receptors. ACS Chem. Neurosci 2017, 8, 2056–2064. [DOI] [PubMed] [Google Scholar]
- (10).Liu G-N; Luo R-H; Zhou Y; Zhang X-J; Li J; Yang L-M; Zheng Y-T; Liu H Synthesis and Anti-HIV-1 Activity Evaluation for Novel 3a,6a-Dihydro-1H-Pyrrolo[3,4-c]Pyrazole-4,6-Dione Derivatives. Molecules 2016, 21, 1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Chen P; Feng D; Qian X; Apgar J; Wilkening R; Kuethe JT; Gao YD; Scapin G; Cox J; Doss G; Eiermann G; He H; Li X; Lyons KA; Metzger J; Petrov A; Wu JK; Xu S; Weber AE; Yan Y; Roy RS; Biftu T Structure-Activity-Relationship of Amide and Sulfonamide Analogs of Omarigliptin. Bioorg. Med. Chem. Lett 2015, 25, 5767–5771. [DOI] [PubMed] [Google Scholar]
- (12).Deaton DN; Haffner CD; Henke BR; Jeune MR; Shearer BG; Stewart EL; Stuart JD; Ulrich JC 2,4-Diamino-8-Quinazoline Carboxamides as Novel, Potent Inhibitors of the NAD Hydrolyzing Enzyme CD38: Exploration of the 2-Position Structure-Activity Relationships. Bioorg. Med. Chem 2018, 26, 2107–2150. [DOI] [PubMed] [Google Scholar]
- (13).Campos KR; Coleman PJ; Alvarez JC; Dreher SD; Garbaccio RM; Terrett NK; Tillyer RD; Truppo MD; Parmee ER The Importance of Synthetic Chemistry in the Pharmaceutical Industry. Science 2019, 363, 244–252. [DOI] [PubMed] [Google Scholar]
- (14).Blakemore DC; Castro L; Churcher I; Rees DC; Thomas AW; Wilson DM; Wood A Organic Synthesis Provides Opportunities to Transform Drug Discovery. Nat. Chem 2018, 10, 383–394. [DOI] [PubMed] [Google Scholar]
- (15).Quiclet-Sire B; Zard SZ Observations on the Reaction of Hydrazones with Iodine: Interception of the Diazo Intermediates. Chem. Commun 2006, 1831–1832. [DOI] [PubMed] [Google Scholar]
- (16).Kapras V; Pohl R; Císařová I; Jahn U Asymmetric Domino Aza-Michael Addition/[3 + 2] Cycloaddition Reactions as a Versatile Approach to α,β,γ-Triamino Acid Derivatives. Org. Lett 2014, 16, 1088–1091. [DOI] [PubMed] [Google Scholar]
- (17).Just D; Hernandez-Guerra D; Kritsch S; Pohl R; Císařová I; Jones PG; Mackman R; Bahador G; Jahn U Lithium Chloride Catalyzed Asymmetric Domino Aza-Michael Addition/[3 + 2] Cycloaddition Reactions for the Synthesis of Spiro- and Bicyclic α,β,γ-Triamino Acid Derivatives. Eur. J. Org. Chem 2018, 2018, 5213–5221. [Google Scholar]
- (18).Carlson AS; Calcanas C; Brunner RM; Topczewski JJ Regiocontrolled Wacker Oxidation of Cinnamyl Azides. Org. Lett 2018, 20, 1604–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Ott AA; Goshey CS; Topczewski JJ Dynamic Kinetic Resolution of Allylic Azides via Asymmetric Dihydroxylation. J. Am. Chem. Soc 2017, 139, 7737–7740. [DOI] [PubMed] [Google Scholar]
- (20).Porter MR; Shaker RM; Calcanas C; Topczewski JJ Stereoselective Dynamic Cyclization of Allylic Azides: Synthesis of Tetralins, Chromanes, and Tetrahydroquinolines. J. Am. Chem. Soc 2018, 140, 1211–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Liu E-C; Topczewski JJ Enantioselective Copper Catalyzed Alkyne–Azide Cycloaddition by Dynamic Kinetic Resolution. J. Am. Chem. Soc 2019, 141, 5135–5138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Yang C; Shen H One Pot Multiple-Steps Reactions of Allyl Azide and Alkenes Carrying Electron-Withdrawing Groups. Tetrahedron Lett. 1993, 34, 4051–4054. [Google Scholar]
- (23).Yang C-H; Sherf H-J; Wang R-H; Wang J-C 2,3,7-Triazabicyclo[3.3.0]Octenes Prepared by Tandem Cascade Reaction of Allyl Azides and Olefinic Dipolarophiles. J. Chin. Chem. Soc 2002, 49, 95–102. [Google Scholar]
- (24).Broeckx W; Overbergh N; Samyn C; Smets G; L’abbé G Cycloaddition Reactions of Azides with Electron-Poor Olefins. Tetrahedron 1971, 27, 3527–3534. [Google Scholar]
- (25).L’abbé G Decomposition and Addition Reactions of Organic Azides. Chem. Rev 1969, 69, 345–363. [Google Scholar]
- (26).Nadin A; Hattotuwagama C; Churcher I Lead-Oriented Synthesis: A New Opportunity for Synthetic Chemistry. Angew. Chem., Int. Ed 2012, 51, 1114–1122. [DOI] [PubMed] [Google Scholar]
- (27).Kutchukian PS; Dropinski JF; Dykstra KD; Li B; Dirocco DA; Streckfuss EC; Campeau L-CC; Cernak T; Vachal P; Davies IW; Krska SW; Dreher SD Chemistry Informer Libraries: A Chemoinformatics Enabled Approach to Evaluate and Advance Synthetic Methods. Chem. Sci 2016, 7, 2604–2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Gagneux A; Winstein S; Young WG Rearrangement of Allyl Azides. J. Am. Chem. Soc 1960, 82, 5956–5957. [Google Scholar]
- (29).Carlson AS; Topczewski JJ Allylic Azides: Synthesis, Reactivity, and the Winstein Rearrangement. Org. Biomol. Chem 2019, 17, 4406–4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ott AA; Packard MH; Ortuño MA; Johnson A; Suding VP; Cramer CJ; Topczewski JJ Evidence for a Sigmatropic and an Ionic Pathway in the Winstein Rearrangement. J. Org. Chem 2018, 83, 8214–8224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Nugent TC Chiral Amine Synthesis: Methods, Developments and Applications, 1st ed.; Wiley, 2010. [Google Scholar]
- (32).Huisgen R Proceedings of the Chemical Society. October 1961. Proc. Chem. Soc 1961, 357–396. [Google Scholar]
- (33).Huisgen R; Szeimies G; Möbius L 1.3-Dipolar Cyclo-additions. XXXII. Kinetics of the Addition of Organic Azides to Carbon-Carbon Multiple Bonds. Chem. Ber 1967, 100, 2494–2507. [Google Scholar]
- (34).Liddon JTR; Lindsay-Scott PJ; Robertson J Secondary Products from Intramolecular Cycloadditions of Azidoalkyl Enol Ethers and Azidoalkyl Vinyl Bromides: 1-Azadienes, Their Reactions with Diphenylketene, and Radical Cyclizations To Form Bi- and Tricyclic Lactams. J. Org. Chem 2019, 84, 13780–13793. [DOI] [PubMed] [Google Scholar]
- (35).Xie S; Lopez SA; Ramström O; Yan M; Houk KN 1,3-Dipolar Cycloaddition Reactivities of Perfluorinated Aryl Azides with Enamines and Strained Dipolarophiles. J. Am. Chem. Soc 2015, 137, 2958–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Krivopalov VP; Shkurko OP 1,2,3-Triazole and Its Derivatives. Development of Methods for the Formation of the Triazole Ring. Russ. Chem. Rev 2005, 74, 339–379. [Google Scholar]
- (37).Herdeis C; Schiffer T Synthesis of Nonracemic 2,3,6-Trisubstituted Piperidine Derivatives from Sugar Lactones via Tandem Wittig [2 + 3] Cycloaddition Reaction. A Novel Entry to Prosopis and Cassia Alkaloids. Tetrahedron 1999, 55, 1043–1056. [Google Scholar]
- (38).Yang CH; Lee LT; Yang JH; Wang Y; Lee GH Spiropyrazolines from Tandem Reaction of Azides and Alkyl Vinyl Ketones. Tetrahedron 1994, 50, 12133–12142. [Google Scholar]
- (39).Teng J-T; Yang C-H A Stereoselective Preparation of 1,2,7-Triazabicyclo[3.3.0]Oct-2-Enes. J. Chin. Chem. Soc 1998, 45, 375–380. [Google Scholar]
- (40).Louhichi N; Msaddek M Synthesis and Photolysis of Hexahydropyrrolo [3,4-c] Pyrazole Derivatives. J. Chem. Res 2007, 10, 569–571. [Google Scholar]
- (41).Kolb HC; Finn MG; Sharpless KB Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem., Int. Ed 2001, 40, 2004–2021. [DOI] [PubMed] [Google Scholar]
- (42).Bräse S; Gil C; Knepper K; Zimmermann V Organic Azides: An Exploding Diversity of a Unique Class of Compounds. Angew. Chem., Int. Ed 2005, 44, 5188–5240. [DOI] [PubMed] [Google Scholar]
- (43).Bräse S; Banert K Organic Azides Syntheses and Applications; John Wiley & Sons, Ltd: Chichester, 2010. [Google Scholar]
- (44).Goswami PP; Suding VP; Carlson AS; Topczewski JJ Direct Conversion of Aldehydes and Ketones into Azides by Sequential Nucleophilic Addition and Substitution. Eur. J. Org. Chem 2016, 2016, 4805–4809. [Google Scholar]
- (45).Rueping M; Vila C; Uria U Direct Catalytic Azidation of Allylic Alcohols. Org. Lett 2012, 14, 768–771. [DOI] [PubMed] [Google Scholar]
- (46).Ojo OS; Miranda O; Baumgardner KC; Bugarin A Practical Regio- and Stereoselective Azidation and Amination of Terminal Alkenes. Org. Biomol. Chem 2018, 16, 9354–9358. [DOI] [PubMed] [Google Scholar]
- (47).Chen H; Yang W; Wu W; Jiang H Palladium-Catalyzed Regioselective Azidation of Allylic C–H Bonds under Atmospheric Pressure of Dioxygen. Org. Biomol. Chem 2014, 12, 3340–3343. [DOI] [PubMed] [Google Scholar]
- (48).Smith SM; Hoang GL; Pal R; Khaled MOB; Pelter LSW; Zeng XC; Takacs JM γ-Selective Directed Catalytic Asymmetric Hydroboration of 1,1-Disubstituted Alkenes. Chem. Commun 2012, 48, 12180–12182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Hattori K; Okitsu O; Tabuchi S; Taniguchi K; Nishio M; Koyama S; Seki J; Sakane K Discovery of New Diphenyloxazole Derivatives Containing a Pyrrolidine Ring: Orally Active Prostacyclin Mimetics. Bioorg. Med. Chem. Lett 2005, 15, 3279–3283. [DOI] [PubMed] [Google Scholar]
- (50).Villieras J; Rambaud M; Graff M The Wittig-Horner Reaction in Heterogenous Media VIII1. Cyclisation During the Aldolisation Step from Aqueous Glutaraldehyde. Synth. Commun 1986, 16, 149–156. [Google Scholar]
- (51).Yamada M; Ichikawa T; Ii M; Itoh K; Tamura N; Kitazaki T Novel Cyclohexene Derivatives as Anti-Sepsis Agents: Synthetic Studies and Inhibition of NO and Cytokine Production. Bioorg. Med. Chem 2008, 16, 3941–3958. [DOI] [PubMed] [Google Scholar]
- (52).Chiou W-H; Kao C-L; Tsai J-C; Chang Y-M Domino Rh-Catalyzed Hydroformylation–Double Cyclization of o-Amino Cinnamyl Derivatives: Applications to the Formal Total Syntheses of Physostigmine and Physovenine. Chem. Commun 2013, 49, 8232–8234. [DOI] [PubMed] [Google Scholar]
- (53).Ardolino MJ; Morken JP Congested C–C Bonds by Pd-Catalyzed Enantioselective Allyl–Allyl Cross-Coupling, a Mechanism-Guided Solution. J. Am. Chem. Soc 2014, 136, 7092–7100. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.