

Abstract
Chronic obstructive pulmonary disease (COPD) is a progressive condition of chronic bronchitis, small airway obstruction, and emphysema, and represents a leading cause of death worldwide. While inflammation, fibrosis, mucus hypersecretion, and metaplastic epithelial lesions are hallmarks of this disease, their origins and dependent relationships remain unclear. Here we apply single-cell cloning technologies to lung tissue of patients with and without COPD. Unlike control lungs, which were dominated by normal distal airway progenitor cells, COPD lungs were inundated by three variant progenitors epigenetically committed to distinct metaplastic lesions. When transplanted to immunodeficient mice, these variant clones induced pathology akin to the mucous and squamous metaplasia, neutrophilic inflammation, and fibrosis seen in COPD. Remarkably, similar variants pre-exist as minor constituents of control and fetal lung and conceivably act in normal processes of immune surveillance. However, these same variants likely catalyze the pathologic and progressive features of COPD when expanded to high numbers.
Graphical Abstract
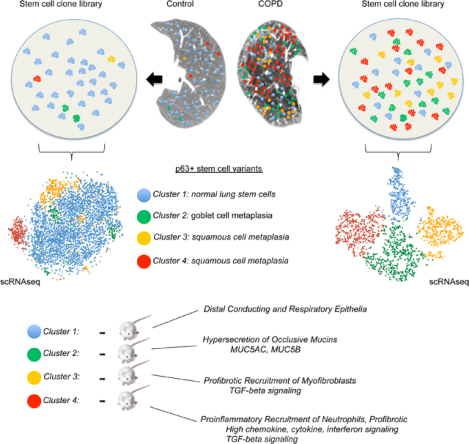
In Brief
The hallmark features of COPD (inflammation, fibrosis, and mucus hypersecretion) are driven by distinct pathogenic progenitors which pre-exist as minor populations in healthy lungs but dominate in the disease state relative to normal lung stem cells.
INTRODUCTION
Chronic obstructive pulmonary disease (COPD) is an inflammatory condition of the lung marked by chronic bronchitis, small airway occlusion, inflammation, fibrosis, and emphysematous destruction of alveoli (Barnes et al., 2015; Decramer et al., 2012; Hogg et al., 2004; McDonough et al., 2011). The global impact of COPD is enormous as evidenced by the 250 million patients with this condition and the 50%, 5-year survival of those with GOLD stage 3 and 4 disease (FEV1<50%) (Barnes et al., 2015; Decramer et al., 2012; Hogg et al., 2004; McDonough et al., 2011; Quaderi and Hurst, 2018; Singh et al., 2019). Risk factors include chronic exposure to cigarette smoke and environmental pollutants, early-life respiratory illnesses such as asthma, viral pneumonia, and perinatal bronchopulmonary dysplasia, as well as genetics (Busch et al., 2017; Fletcher and Peto, 1977; Martinez, 2016; Martinez et al., 2018; McGeachie et al., 2016). Given that 80% of COPD cases are associated with long-term chronic smoking, absolute abstinence will limit risk for this disease. However, the success of smoking cessation programs on patients with established COPD has been disappointing (Barnes, 2016; Fletcher and Peto, 1977; Gamble et al., 2007; Hogg and Timens, 2009; Miller et al., 2011; Scanlon et al., 2000; Wen et al., 2010), and most people with moderate COPD progress to severe disease (FEV1<30) in a 5- to 20-year interval despite tobacco cessation. The mechanistic basis for this dogged progression of COPD is unclear, but multiple studies have shown that lung inflammatory pathways remain activated years after smoking cessation (Barnes, 2016; Gamble et al., 2007; Miller et al., 2011; Scanlon et al., 2000; Wen et al., 2010).
The predisposing influences of early-life pulmonary disease, coupled with the general irreversibility of this condition in previous smokers, suggest that inertial processes may drive COPD. Consistent with this notion, squamous and goblet cell metaplasia typically observed in COPD likewise are not fully abolished by smoking cessation (Hogg and Timens, 2009; Lapperre et al., 2007; Raju et al., 2016; Zhou-Suckow et al., 2017). This persistence was surprising as metaplasia has been tied to acute effects of cigarette smoke and inflammatory cytokines on normal airway progenitors (Araya et al., 2007; Schamberger et al., 2015; Wills-Karp et al., 1998). Removal of these toxicants was expected to enable distal airway progenitors to resume their roles in restoring tissue homeostasis in small airways and terminal bronchioles (Kumar et al., 2011; Ray et al., 2016; Tanaka et al., 2018; Vaughan et al., 2015; Yang et al., 2018; Zuo et al., 2015). In this context, we speculated that some irreversible alterations in distal airway progenitors explain their inability to restore airway and lung homeostasis following smoking cessation.
In the present study, we leveraged robust technologies that enable the cloning of distal airway epithelial cells (Kumar et al., 2011; Zuo et al., 2015) to perform a comparative analysis of clonogenic cells in patients with and without COPD. The data revealed that the COPD distal airways are populated by a highly stereotyped triad of clonogenic cells in addition to the normal clonogenic cell type that predominates control lungs. We show here these variants are stably committed to metaplastic fates and autonomously drive pro-inflammatory and pro-fibrotic programs akin to those implicated in COPD pathology. Lastly, we show that a similar triad of clonogenic cells in COPD also exists in control adult and 13-, 14-, and 17-week fetal lung, albeit at low ratios to the predominant distal airway clone type seen in normal lung. Thus the variant clones predate any disease state and yet have features that could contribute to the emergence, pathology, or progression of COPD as a function of their numbers.
RESULTS
Heterogeneity of clonogenic cells in COPD
Libraries of clonogenic epithelial cells from resected lung tissue of 19 COPD and 11 control patients (Table S1) were generated by selective colony growth on lawns of irradiated, 3T3-J2 feeder cells (Kumar et al., 2011; Zuo et al., 2015; detailed in Methods). In brief, approximately 0.85+/−0.04% of all enzymatically digested lung cells yielded colonies from COPD patients and 0.15+/−0.02% from controls (Table S2). These libraries have a complexity of 2,000 to 20,000 independent colonies per cubic centimeter of lung tissue and consist exclusively of cells that co-express E-cadherin and p63 (Ecad+/p63+). Single cells from these patient-specific libraries have a clonogenicity of greater than 50% as judged by single cell-sorting to 384-well plates and give rise to discrete lines of highly immature, epithelial cells characterized by a long-term (>1yr) proliferative potential. Randomly sampled clones from libraries of control patients (SPN-12, −14) displayed whole genome expression profiles consistent with previously characterized clonogenic cells of distal airways (Kumar et al., 2011; Zuo et al., 2015) (Fig. 1B, C). In contrast, sampled clones from libraries derived from COPD patients (SPN-7, −13) yielded a more complex spectrum of gene expression profiles. In addition to the normal distal airway clones (hereafter “Cluster 1”), the majority of sampled clones showed whole genome expression profiles that diverged from Cluster 1 and readily segregated into three additional distinct clusters (Clusters 2, 3, and 4) (Fig. 1B,C). Despite this clonal heterogeneity in COPD, all clones from Clusters 1–4 shared the expression of established markers of distal airway progenitors (p63, KRT5), displayed a clonogenicity of 60+/−1.4%, and showed a long-term proliferative potential of at least 25 passages (>8mos) while maintaining a clonogenicity of 59.9+/−1.6% (Fig. 1D). Herein we leverage the regenerative properties of these patient-specific libraries of clonogenic cells and the single cell-derived clones from them to assess the properties of epithelial progenitors in COPD and control cases.
Figure 1. Epithelial clone heterogeneity in COPD.
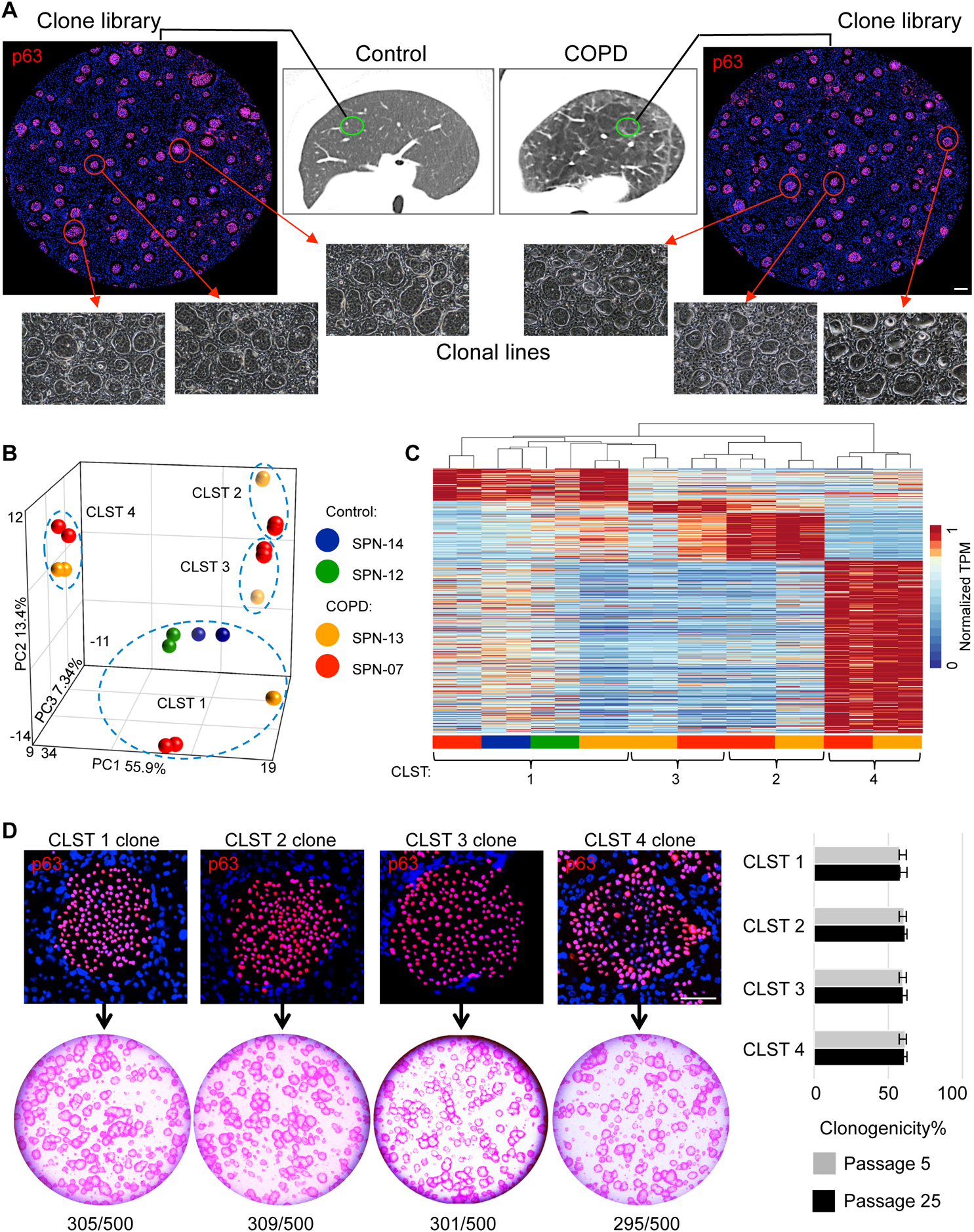
A. Schematic depicting workflow of generating p63+ epithelial colony pools from resected normal and COPD lung tissue. Individual colonies are captured and expanded individually to generate clonal cell lines. Scale bar, 500 μm. B. Principal component analysis of RNAseq differential expression genes (DEGs) (FDR<0.05) derived from multiple clones from control (SPN-12, −14) and COPD (SPN-13, −07) lungs. C. RNAseq expression heatmap of the clones depicted in the PCA plot is shown highlighting Cluster 1 (control) vs Clusters 1–4 in COPD (detailed in Table S4). D. Top, Individual colonies from cloned representatives of Clusters 1–4 stained with antibody to p63 showing uniform nuclear staining. Scale bar, 100 μm. Bottom, Rhodamine red-stained colonies arising on lawns of irradiated 3T3-J2 fibroblasts from 500 cells from each of cloned representatives of Clusters 1–4. Right, Histogram of clonogenicity based on percentage of plated cells that formed colonies using representative clones from Clusters 1–4 at passage 5 (P5) and 25 (P25). Data represented as mean ± SEM; See also Table S1 and Table S4.
Metaplastic fate commitment of COPD clones
In vitro differentiation of Cluster 1 clones from both COPD and control libraries yielded normal epithelia (NM) marked by p63+ basal cells and suprabasal, differentiated cells expressing SCGB1A1, SFTPB, and AQP4, similar to human epithelia of small airways and terminal bronchioles (Fig. 2A; Fig. S1A,B). In contrast, clones of Cluster 2 differentiated to a goblet cell metaplasia (GCM) marked by p63+ basal cells and differentiated goblet cells co-expressing SCGB1A1, MUC5AC, and MUC5B, whereas clones from Clusters 3 and 4 gave rise to squamous cell metaplasia (SCM) marked by immature p63 cells and differentiated, keratin 10 (Krt10)- and involucrin (IVL)-expressing cells (Fig. 2A; Fig. S1A).
Figure 2. Commitment of variant clones to metaplastic fates.
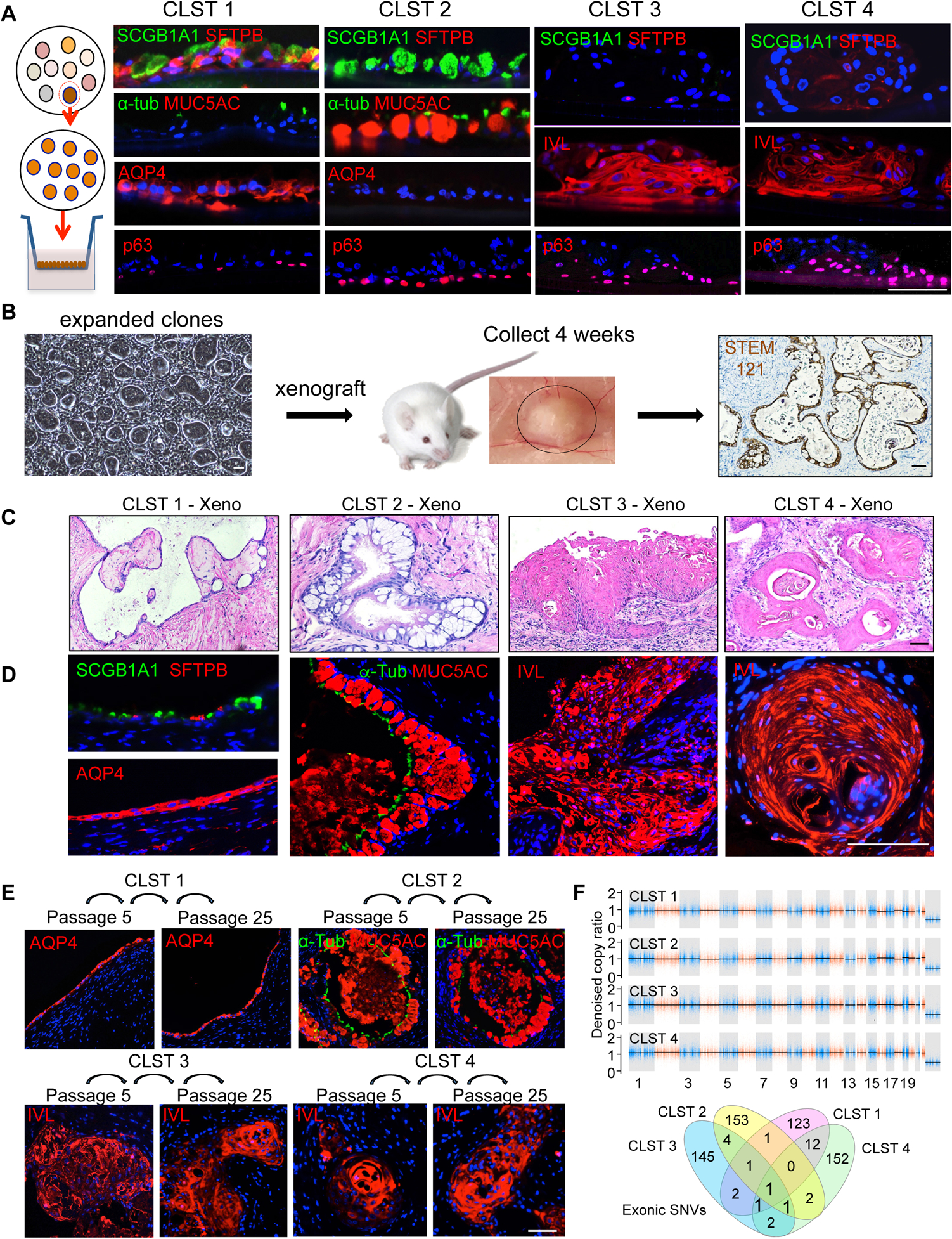
A. From left, Schematic of clonal expansion and differentiation in air-liquid interface (ALI) cultures assessed by immunofluorescence on histological sections. Cluster 1 clones differentiated to an epithelium characterized by expression of SCGB1A1, SFTPB, α-TUB and AQP4 and the absence of MUC5AC. Cluster 2 clones differentiated into an epithelium characterized by SCGB1A1+, MUC5AC+, AQP4− goblet cells. Cluster 3 and Cluster 4 clones differentiated into squamous epithelia that expressed involucrin (IVL) but not SCGB1A1, AQP4, or SFTPB. Scale bar, 100 μm. B. Schematic of subcutaneous transplants of immature cells from clones or library of clones into immunodeficient mice. Nodules formed at 4 weeks were processed for histology and immunostaining and showed polarized epithelia that reacted with antibodies to the human-specific marker STEM121. Scale bar, 100 μm. C., D. In vivo differentiation following subcutaneous transplantation of cloned representatives of Cluster 1 (SCGB1A1+, SFTPB+, AQP4+), Cluster 2 (MUC5AC+ GCM), and Clusters 3 and 4 (IVL+; SCM). Scale bar, 100 μm. E. Xenografts of Cluster 1–4 clones previously grown in vitro to passage 5 (P5) and passage 25 (P25) showing stability of fate differentiation and expression of Cluster-specific markers. Scale bar, 100um. F. Copy number and single nucleotide variation analysis derived from whole exome sequencing of representative clones of Clusters 1–4 from SPN-13 relative to patient blood. CNV events of larger than 10–20Kb were not detected in any of the clones. Venn diagram of all detected exonic SNVs (synonymous, non-synonymous, indels) in each clone relative to all others. See also Figure S1, Table S2 and Table S5.
We further tested the fates of cluster-specific clones in vivo by transplanting discrete clones into highly immunodeficient NSG (NODscidIL2ranull) (Shultz et al., 1995) mice. Briefly, 1 million cells of a designated clone were mixed with 50% Matrigel, injected subcutaneously, and the resulting xenograft nodule examined four weeks later (Fig. 2B). Significantly, normal control clones, as well as those from Cluster 1 of COPD patients, assembled into a polarized epithelium in vivo comprised of cells positive for p63, Krt5, SCGB1A1, AQP4, AQP5, SFTPB, and SFTPC (Fig. 2C,D) like that of normal human terminal bronchioles (Fig. S1B) or that produced by in vitro differentiation of Cluster 1 clones (Fig. 2A Fig. S1A). Also mirroring the in vitro ALI cultures, the xenografts of Cluster 2 clones from COPD libraries formed an epithelium dominated by large goblet cells (Fig. 2C, D; Fig. S1D) expressing MUC5AC and MUC5B (Fahy and Dickey, 2010; Kesimer et al., 2017; Okuda et al., 2019), and both Cluster 3 and 4 clone xenografts yielded squamous cell metaplasia expressing IVL and Krt10 (Fig. 2C,D; Fig S1E,F). Despite their commitment to histologically similar squamous cell metaplasia, Cluster 3 and 4 clones exhibit distinct gene expression profiles (Fig. 1B,C). Cluster 4 clones in particular constitutively express a broad array of genes related to inflammation and are denoted herein as ‘inflammatory SCM’ or ‘iSCM’ versus ‘SCM’ for Cluster 3 clones. Importantly, the respective differentiation fates of these clone types in xenografts proved to be remarkably stable to 250 days of continuous propagation in vitro, suggesting that these metaplasia are the consequences of highly stable, cell-autonomous fate programs (Fig. 2E). Lastly, whole exome DNA sequencing of Cluster 2, 3, and 4 clones derived from these COPD patients showed an absence of copy number variation events greater than 10Kb (Fig. 2F). Moreover, the 127–157 single nucleotide variation (synonymous, nonsynonymous, indels) events in these clones were consistent in number with other somatic cells (Lee-Six et al., 2018) and did not impact known tumor suppressor or oncogenes such as p16 or p53 (Fig. 2G; Fig. S1G; Table S5), arguing against the possibility these clones are related to neoplastic lesions.
Metaplastic lesions dominate end-stage COPD lung
Given the observation that the Clusters 1–4 clones expressed p63 and yet differentiated to cluster-specific metaplasia in vitro and in vivo, we asked if metaplasia in the distal airways of COPD lung exhibited a similar association with p63+ basal cells. To address this question, we morphometrically quantified the distribution of normal distal airway epithelia and metaplasia in histological sections of distal lung from five additional donors lacking lung or systemic disease as well as from an additional 10 lungs of Stage 4 COPD transplant recipients (Table S3). Whereas metaplastic lesions in the normal lungs were rare, the end-stage COPD distal airways were replete with goblet cell (MUC5AC+) and squamous cell (IVL+) metaplasia that, like normal distal airway epithelia, were subtended by basally-positioned p63+ cells (Fig. 3A). In quantitative terms, GCM occupied a mean of 32.7% of the distal airway epithelia (defined as that subtended by p63+ basal cells) of COPD lung versus 5.7% of the normal lungs, SCM occupied 26.8% of the COPD airways epithelia compared to less than 1% in normal lungs, and SCM associated with inflammation (iSCM) constituted approximately 21.6% of COPD airways and was not detected in normal lungs (Fig. 3B). We also examined the distribution of marker genes associated with the p63+ basal cells of normal (AQP5+), iSCM (CXCL8+), and SCM/GCM (TRPC6+) airway epithelia in the same normal and Stage 4 COPD lungs (Fig. 3C). Consistent with our morphometric analyses of lung metaplasia in COPD and control lung, CXCL8+ basal cells of iSCM comprised 19.3% of all p63+ distal airway basal cells of COPD lung while these were not observed in normal lungs. Fully 52% of the basal cells in COPD lung expressed TRPC6, a marker of both GCM and SCM, while fewer than 2% of basal cells showed TRPC6 expression in the normal lungs (Fig. 3D; Fig. S2). The close association of p63+ cells with the major forms of metaplasia in COPD is consistent with our finding that COPD lung possesses an array of p63+ clonogenic cells with absolute commitments to normal airway epithelia, goblet cell metaplasia, or squamous cell metaplasia. These findings are also in agreement with earlier studies that showed metaplasia in COPD and idiopathic pulmonary fibrosis were subtended by p63+ cells (Araya et al., 2007; Chilosi et al., 2002; Plantier et al., 2011; Seibold et al., 2013; Smirnova et al., 2016).
Figure 3. p63+ cells in COPD metaplasia and clone libraries.
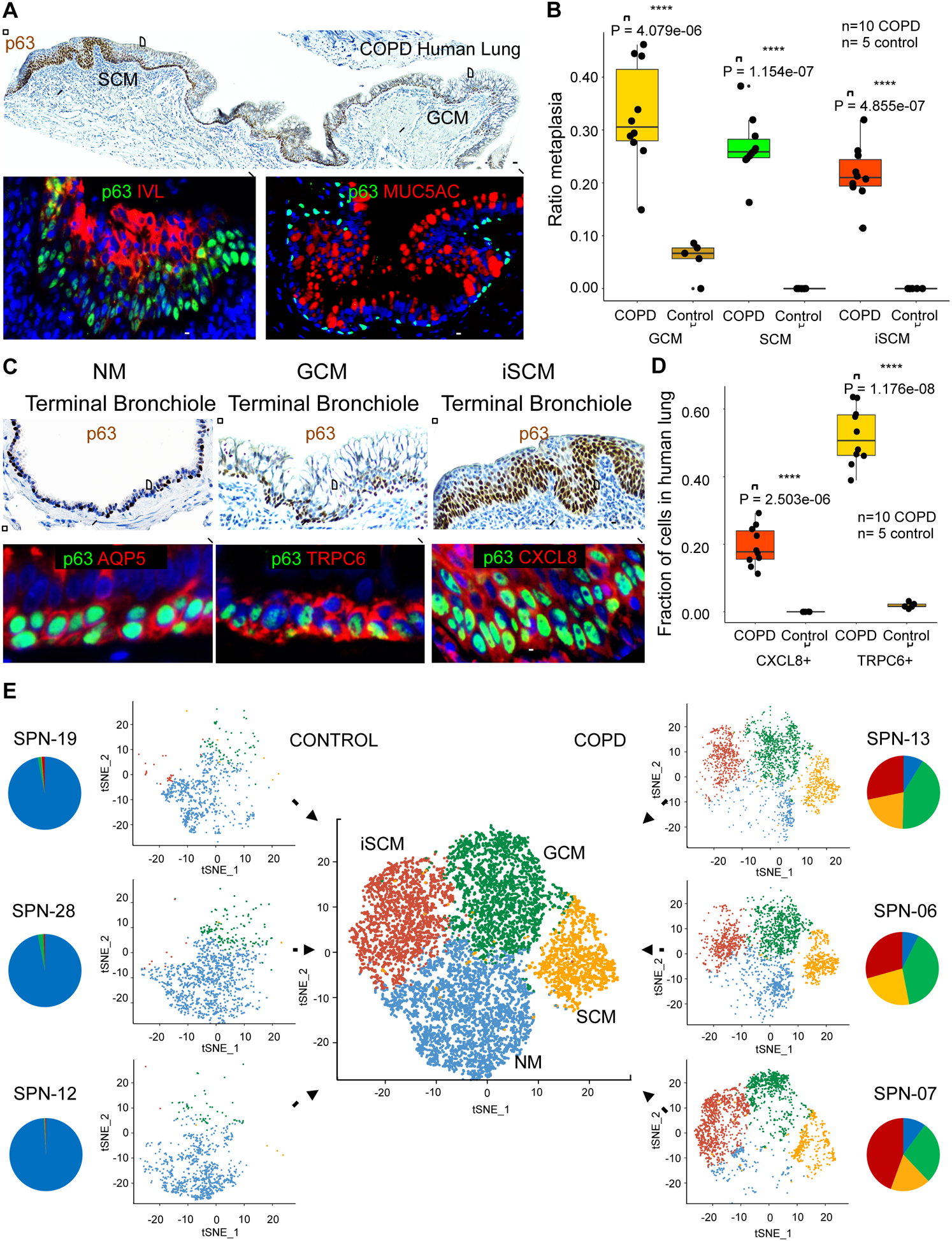
A. Top, p63 immunohistochemistry of distal lung of GOLD Stage 4 COPD showing contiguous regions of squamous and goblet cell metaplasia (SCM and GCM) subtended by p63+ basal cells (brown). Scale bar, 200 μm. Bottom, Immunofluorescence micrographs of expanded regions of SCM and GCM stained with antibodies to p63 (green), IVL (red) and MUC5AC (red). Scale bar, 100 μm. B. Box-Whisker plots for the linear occupancy of metaplastic lesions (GCM of P = 4.1e-06, SCM of P = 1.2e-07, and inflammatory SCM of P = 4.9e-07, Student’s t-test) across distal airways ten Stage 4 COPD lungs compared with five normal lungs without disease. C. Top, p63 immunohistochemistry micrographs of regions of COPD distal airways showing, from left, examples of terminal bronchiole, goblet cell metaplasia, and squamous cell metaplasia. Scale bar, 200 μm. Bottom, immunofluorescence micrographs of expanded regions of terminal bronchiole (p63+, Aqp5+), GCM (p63+, TRPC6+), and inflammatory SCM (p63+, CXCL8+). Scale bar, 100 μm. D. Box-Whisker plots of distribution of CXCL8+ (P = 2.5e-06, Student’s t-test) and TRPC6+ basal cells (P = 1.2e-08, Student’s t-test) in 10 cases of Stage 4 COPD distal lung compared with five normal lungs. E. Single and aggregate tSNE profiles of single cell RNAseq data of three COPD and three control clone libraries. Pie charts indicate the fractional contributions of clones of Clusters 1–4 to patient-specific clone libraries (NM, blue; GCM, green, SCM, orange, iSCM, red). See also Figure S2, Figure S3, Table S3, Table S6 and Table S7.
Clonal architecture of COPD and control libraries
We next asked if the widespread distribution of metaplastic lesions in the distal airways of end-stage COPD lung was reflected in the properties of the clonogenic cell libraries generated from our 19 COPD and 11 non-COPD cases. Single-cell RNA sequencing (scRNAseq) (Satija et al., 2015) analyses of three COPD and three control libraries revealed four major clusters with gene expression profiles similar to those identified by single colony sampling as Clusters 1–4 (Fig. 3E; Fig. S3). In particular, the three COPD lungs showed major contributions to the clonal architecture of these libraries by clones committed to GCM, SCM, and iSCM represented by Clusters 2, 3, and 4, respectively. The coherence between markers identified by scRNAseq and by RNAseq of representative clones of each cluster was high, suggesting that our sampling approach likely captured all major clone variants in the COPD patients (Fig. 3E; Fig. S3A). This analysis also indicated that while control libraries were dominated by Cluster 1 clones committed to distal airway epithelia, these libraries also harbored small percentages (6–12%) of metaplastic variants (Fig. 3E; Fig. S3B).
To develop a more simplified means of assessing the heterogeneity of clone libraries from COPD and Control patients, we exploited the consensus markers identified by scRNAseq and RNAseq of representative clones for quantitative flow cytometry (FACS) analysis. Antibodies to AQP5 were specific to Cluster 1 cells, CXCL8 for Cluster 4 cells, and TRPC6 antibodies for both Clusters 2 and 3 (Fig. 4A). This limited set of markers was used to quantify the relative distribution of clone variants across our 19 COPD and 11 Control patients (Fig. 4A, B; Table S1). Consistent with our analyses of scRNAseq profiles, the quantification of clone types by flow cytometric analyses of markers showed that variants (TRPC6+ for Clusters 2 and 3; CXCL8+ for Cluster 4) comprised 77.7+/−5% of all cells in the 19 COPD clone libraries, while in control specimens, 12.2+/−6% of the p63+ clonogenic cells in the 11 control libraries fell within Clusters 2–4 based on FACS analyses of markers TRPC6 and CXCL8 (P<2.2e-16; Fig.4B, Table S2; Fig. S4A).
Figure 4. Library composition and pro-inflammatory response in xenografts.
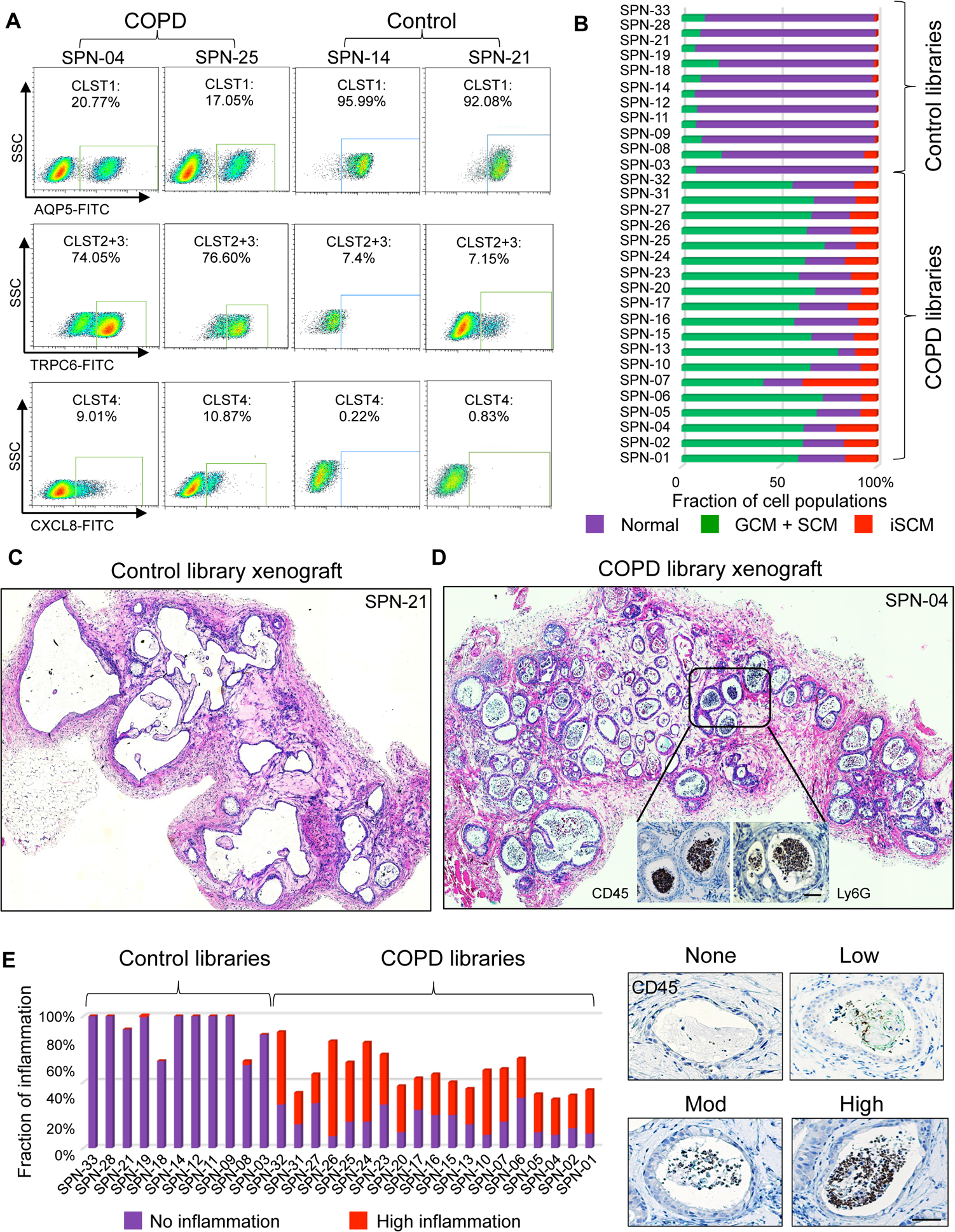
A. FACS profiles of COPD and control clone libraries using markers established from library scRNAseq and clonal RNAseq including anti-AQP5 (Cluster 1), anti-TRPC6 (Clusters 2+3), and anti-CXCL8 (Cluster 4). B. Histogram compiling FACS quantification data on the relative clone composition of each patient library. C. Histological sections of four-week xenograft of clone libraries from control (SPN-21) showing epithelial cysts devoid of luminal cells. D. Histological sections of four-week xenograft of clone libraries from COPD case (SPN-04) showing epithelial cysts marked by abundant infiltration of CD45+/Ly6G+ leukocytes (insets). E. Histogram depicting the quantification of leukocyte infiltration in xenografts of clone libraries from 11 control and 19 COPD patients based on degree of CD45+ cells in cysts (right). Scale bar, 100 μm. See also Figure S4.
Xenografts of COPD clone libraries drive neutrophilic inflammation
To assess the pathogenic potential of the COPD epithelial cells, we performed subcutaneous transplants of composite clone libraries of each of the 19 COPD patients and 11 control patients into immunodeficient mice. Over four weeks, clone libraries from control patients yielded epithelial cysts with appropriate basal-apical polarity around largely vacant spaces (Fig. 4C; Fig. S4B; Table S2). In contrast, clone libraries generated from COPD patients produced cysts occupied by dense arrays of epithelial and non-epithelial cells (Fig. 4D; Fig. S4C; Table S2). The majority of non-epithelial luminal cells were hematopoietic in origin as evidenced by anti-murine CD45 staining, and the majority of those were positive for antibodies to Ly6G, a marker of neutrophils (Fig. 4D). Similar patterns of leukocyte transepithelial migration (TEM) (Brazil and Parkos, 2016) are present in COPD lung (Barnes et al., 2015; Butler et al., 2018; Quint and Wedzicha, 2007). Quantification of the extent of neutrophil infiltration at the level of individual epithelial cysts revealed that 36.4+/−14% of these cysts scored “severe” (closest neutrophil packing) in the COPD clone library xenografts in contrast to only 0.3+/−0.9% of the cysts in control library xenografts (P=1.2e-09, Student’s t-test; Fig. 4E; Fig. S4D).
Given the robust inflammatory response triggered by xenografts of COPD clone libraries, we asked if this inflammatory activity was a function of one or more of the constituent clone types. Using pathway enrichment analysis algorithms for the gene expression profiles of the four clone types, the multiple inflammatory pathways highlighted in clones of Cluster 4 was striking including Antigen Presentation, Interferon Signaling, Graft-vs-Host responses, and Dendritic Cell Maturation among others, and their relative absence from clones of Clusters 1, 2, and 3 (Fig. 5A; Fig. S5A). We tested media conditioned by clones of Clusters 1–4 for their ability to trigger phase I and II activation of human airway microvasculature endothelial cells, an essential step in leukocyte recruitment (Filippi, 2016). Endothelial cell activation was assessed by the expression of Vcam1 (CD106), a vascular adhesion protein that binds VLA-1 (alpha4beta1 integrin) on monocytes and lymphocytes, and it was found that Cluster 1 clones from COPD patients showed no induction, whereas IL-13 challenge yielded a 30-fold induction of Vcam1 (Fig. S5B). In this same assay media conditioned by Cluster 2 (GCM) or Cluster 3 (SCM) clones, respectively, yielded 4- and 10-fold inductions of VCAM1, and while media from Cluster 4 (iSCM) clones induced Vcam1 by 60-fold (Fig. S5B). These data provide functional support for the notion that individual clone variants can promote, at least in vitro, an early step in inflammatory responses.
Figure 5. Cluster 4 COPD clones are constitutively hyperinflammatory.
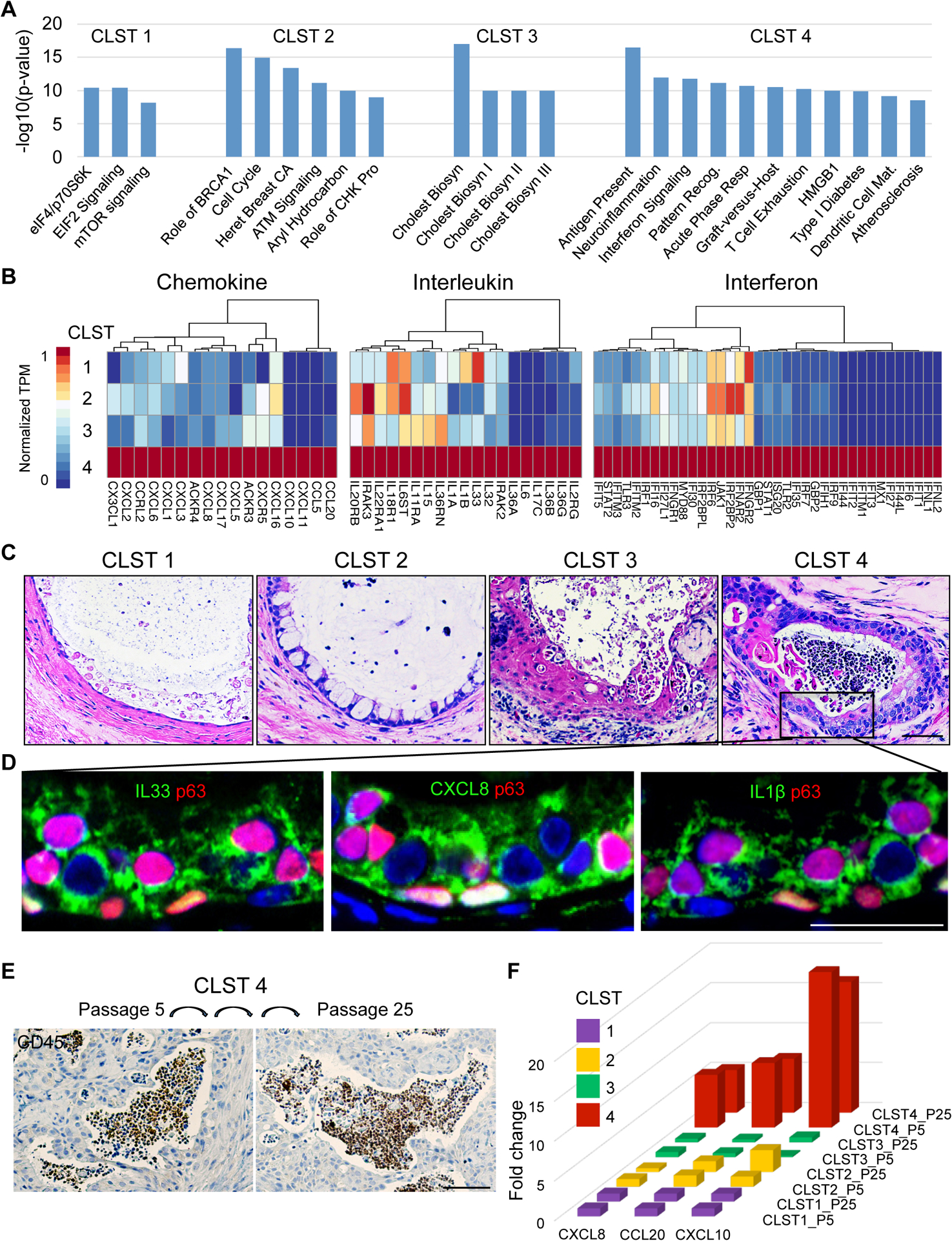
A. Histogram depicting most significant (P<1.0e-8) pathways determined by Ingenuity Pathway Analysis of RNAseq differentially expressed genes (FDR<0.05) of patient-matched clones representative of Clusters 1–4. B. Differential expression heatmaps of chemokine, interleukin, and interferon genes among RNAseq DEGs (FDR<0.05) of patient-matched clones representative of Clusters 1–4 (SPN-13). C. H&E on sections through four-week xenografts of patient-matched clones of Clusters 1–4 showing that only Cluster 4 clones are accompanied by abundant intraluminal leukocytes. Scale bar, 100 μm. D. Immunofluorescence micrographs of Cluster 4 xenografts revealing high expression in epithelia of inflammatory mediators including IL33, CXCL8, and IL1B. Scale bar, 50 μm. E. CD45 immunohistochemistry of xenografts derived from Cluster 4 clone grown in vitro to passage 5 and to passage 25. Scale bar, 200 μm. F. Histogram of CXCL8, CCL20, and CXCL10 gene expression in clonal representatives of Clusters 1–4 at in vitro passage 5 and passage 25. See also Figure S5.
A more detailed expression heatmap of the variant clones revealed that Cluster 4 clones in particular displayed a constitutive expression of a broad array of chemokines, interleukins, and interferon genes compared to clones from Cluster 1, 2, and 3 (Fig. 5B; Fig. S5C). Among the large array of Cluster 4-specific chemokines were key determinants of neutrophil responses including CXCL1, CXCL5, and CXCL8 (Barnes, 2016; Brazil and Parkos, 2016; Ponce-Gallegos et al., 2017; Traves et al., 2002), the lymphocyte and dendritic cell chemoattractant CCL20 (Demedts et al., 2007), and others such as CXCL10 and 11 implicated in Th1 responses (Tokunaga et al., 2018). In addition, Cluster 4 clones expressed a broad array of interleukin genes, including IL1α, IL1β, IL6, IL17C, and IL33, known to interact with multiple cellular mediators of the innate and adaptive immune response (Barnes, 2016; Byers et al., 2013; Holtzman et al., 2014). Lastly, this analysis highlighted Cluster 4 clones as a robust source of Type I and Type II interferon pathway genes typically induced by viral infection (Schneider et al., 2014). In fact Cluster 4 clones showed a constitutively high expression of genes implicated in the entire interferon-γ pathway, including those involved in recognition of pathogen-associated molecular signatures (PAMPs), such as TLR2, TLR3, and MYD88, transcription factors driving the interferon-response genes (IRF1, IRF6, IRF7, and IRF9), as well as genes in the JAK-STAT pathway that cooperate in driving the interferon response such as STAT1, STAT2, and JAK1 (Fig. 5B; Fig. S5C). Together with the patterns of chemokine and interleukin gene expression, these data suggest the potential of Cluster 4 clones to promote an inflammatory response involving diverse cells of the immune system.
To directly test the prediction that Cluster 4 clones were primarily responsible for host neutrophil responses in whole COPD library transplants (cf. Fig. 4D), we performed xenografts of the individual clone types in immunodeficient mice. Significantly, nodules formed by xenografts of Cluster 4 clones showed robust leukocyte infiltration (Fig. 5C). Consistent with their proinflammatory properties in xenografts, the epithelial cysts formed by Cluster 4 clones were unique among the four clone types by showing high protein expression of multiple inflammatory mediators such as IL33, CXCL8, and IL1β (Fig. 5D). Lastly, the leukocyte infiltration response to Cluster 4 clones proved stable for eight months of continuous propagation in vitro (Fig. 5E,F) suggesting that their proinflammatory activity is epigenetically maintained. Together, these data support the conclusion that Cluster 4 clones play a major and likely autonomous role in promoting the observed host leukocyte responses observed in the COPD clone library transplants.
Clonogenic variants drive fibrosis
Fibrosis of small airways and respiratory vasculature is an established feature of COPD (Araya and Nishimura, 2010; Aschner and Downey, 2016; Barnes, 2016; Hogg and Timens, 2009) and has been associated with TGFβ-induced activation of myofibroblasts (Black et al., 2019; Murphy-Ullrich and Suto, 2018). In this regard, we observed that many of the epithelial cysts in xenographs of clone libraries from all 19 COPD patients were surrounded by dense layers of fibroblast-like cells (Fig. 4d). Antibodies to alpha-smooth muscle actin (α-SMA), a key marker of activated myofibroblasts, labeled these submucosal assemblies along the majority (77.1+/−7.2%) of the perimeter of the epithelial cysts in COPD xenografts, whereas only relatively few of these cysts in control xenografts showed these myofibroblast arrays (12.9+/−8.1%; P=7.044e-15, Student’s t-test; Fig. 6A–C; Fig. S6A,B). Similar accumulations of submucosal myofibroblasts were observed in xenographs of single Cluster 3 or Cluster 4 clones, but not in xenografts of clones of Clusters 1 or 2 (Fig. 6D). These data link the two squamous cell metaplasia produced by Clusters 3 and 4 clones, but not goblet cell metaplasia nor normal airway epithelia, to the accumulation of myofibroblasts. Consistently, gene expression profiling of xenografts derived from clones of the respective Clusters revealed differential expression of multiple, pro-fibrotic genes in xenografts of clones of Cluster 3 and 4 that were not evident in xenografts of Cluster 1 (Fig. 6E). Among this large set of constitutively expressed genes implicated in the regulation of fibrosis were TGFβ3, GDF15, and TGFβR2, downstream genes including collagen isotypes (e.g. COL1A1), matrix metallopeptidases (MMP7, MMP13), and BACH1 (Dhakshinamoorthy et al., 2005), a transcriptional repressor of the Nrf2-activated anti-oxidant pathway known to suppress fibrosis in multiple tissues (Fig. 6F). Finally, like the proinflammatory effect of Cluster 4, the pro-fibrotic effects of clones of Clusters 3 and 4 were stable to long-term propagation in vitro (Fig. S6C).
Figure 6. Cluster 3 and 4 clones drive host myofibroblast activation.
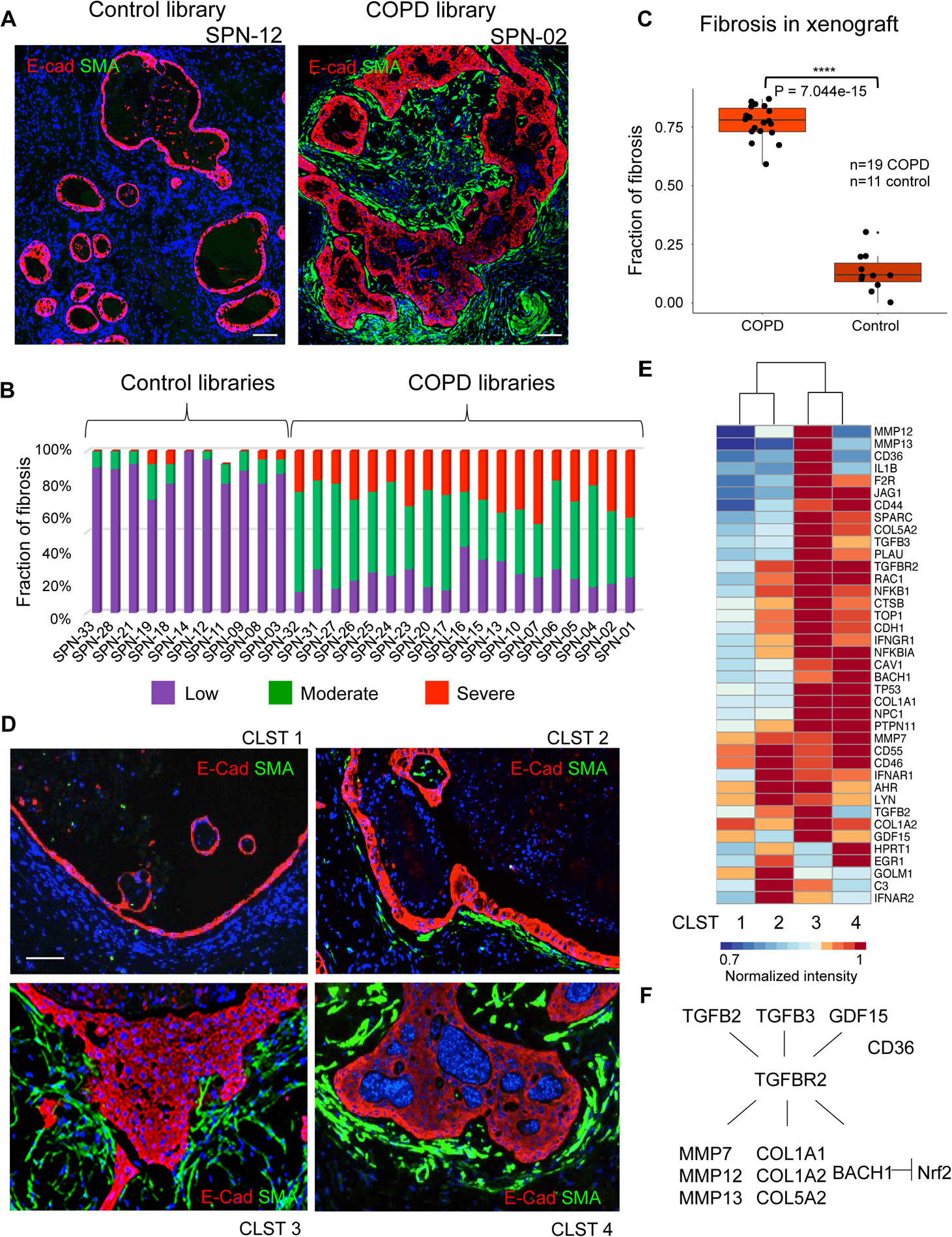
A. Immunofluorescence micrographs of xenografts derived from control case (SPN-12; left) and COPD case (SPN-02; right) clone libraries stained with antibodies to the myofibroblast marker alpha-smooth muscle actin (SMA). Scale bar, 200 μm. B. Quantification of myofibroblast submucosal accumulation in xenografts based on general scale applied to cysts within 11 control and 19 COPD clone library transplants. C. Box-Whisker plot representation of fibrosis accumulation about cysts in each of 19 COPD and 11 control library xenografts (P = 7.0e-15, Student’s t-test). D. Immunofluorescence micrographs of xenografts derived from patient-matched clones of Clusters 1–4 using antibodies to E-cadherin (ECAD, red) and alpha-smooth muscle actin (SMA, green). Scale bar, 100 μm. E. Differential expression heatmap of fibrosis-related genes (1.5-fold, p<0.05) of xenografts derived from patient-matched clones representative of Clusters 1–4. F. Schematic TGF-β pathway including genes differentially expressed in clones of Clusters 3 and 4. See also Figure S6.
Pre-existence of variant clone types in normal and fetal lungs
If the variant clones that dominate COPD lung indeed contribute to the disease process, their origin becomes a central question. These variant clones, all of which express p63, conceivably arise from an epigenetic ‘reprogramming’ of normal, p63+ distal airway clones. Alternatively, these variant cells might already exist as distinct lineages in the normal distal airway epithelia, albeit at low ratios to the distal airway progenitor. In support of the pre-existence model, both single cell RNA sequencing and FACS profiles of control lungs indicated an aggregate representation of variant clones of approximately 10% (Fig. 3E, 4B), and direct cloning from control libraries showed that these variants possessed corresponding metaplastic fate commitment and pathological activity in xenografts (Fig. S7A). Consistently, expression data of Cluster 1–4 clones from control case SPN-14 showed significant overlap with those of COPD cases (Fig. S7B; Table S8; Fig. S3A). However, we could not rule out the possibility that at least some of the control cases had ‘sub-clinical” disease reflected by these variant clones. To more definitively address this question, we examined the clonal architecture of libraries generated from 13- and 17-week fetal lungs (Fig. 7A, Fig. S7C). Remarkably, scRNAseq profiles of the clone libraries of these pseudoglandular fetal lungs (Schittny, 2017) revealed minor populations of clones similar to those of Cluster 2, 3, and 4 variants (in aggregate, 17–34%) among a majority of normal Cluster 1 clones (Fig. 7B; Fig. S7D,E). The similarity of these four fetal clone types with those of Clusters 1–4 from COPD cases was confirmed by their respective differentiation fate in vitro and upon xenografting, at the level of gene expression, and finally by their corresponding potential to drive mucus hypersecretion (GCM), fibrosis (SCM and iSCM), and neutrophilic inflammation (iSCM) (Fig. 7C,D). The pre-existence of these variant clone types in both normal adult lung and in developing fetal lung favors some unknown, normal function of these variants in the lung as well as the notion that the evolution of COPD involves a pathological and irreversible expansion of these variant populations that, in turn, drives the disease process.
Figure 7. Identification of variant clone types in normal and fetal lung.
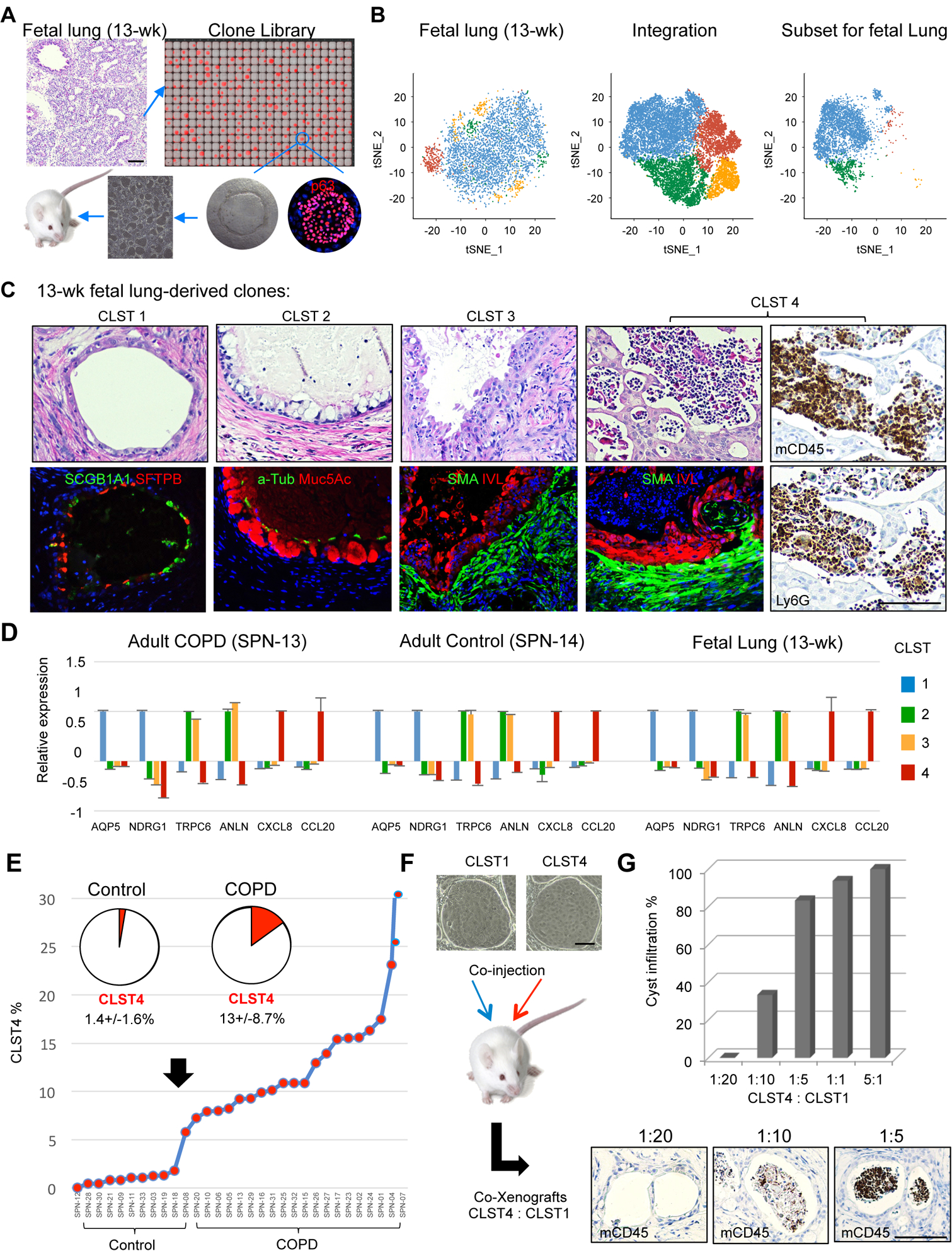
A. Schematic of clone library generation from pseudoglandular fetal lung and analysis by scRNAseq and xenografting. B. Single cell RNA sequencing of clone library from 13-wk fetal lung yielding tSNE profile (left), integration with three adult control and three COPD libraries (middle), and the fetal subset profile based on the integrated profile (right). C. Histology and indicated marker analysis of xenografts of 13-wk fetal clones corresponding to Clusters 1–4. Cluster 4 xenografts are further assessed by immunohistochemistry with antibodies to CD34 and Ly6G. D. Ratio of qPCR-determined marker expression across Cluster 1–4 clones from COPD, Control, and 13-wk fetal lung. Data represented as mean ± SEM. Cluster 1 markers, AQP5 and NDRG1; Clusters 2 and 3, TRPC6 and ANLN; Cluster 4, CXCL8 and CCL20. E. Percentage composition of Cluster 4 clones across 11 Control and 19 COPD clone libraries. F. Schematic for generating xenografts from defined ratios of Cluster 4 and Cluster 1 cells. G. Histogram of quantification of host inflammatory response to co-xenografts of Cluster 4 and Cluster 1 clones based on CD45 and Ly6G monitoring of cystic infiltration by leukocytes. See also Figure S7 and Table S8.
DISCUSSION
The abnormal clonogenic cell types in COPD lungs offer new insights into the natural history of COPD lung disease. The fates and biological properties of the three major clone types (GCM, SCM, and iSCM) in the COPD lung appear to be autonomously maintained and, in aggregate, dominate the epithelia of moderate and end-stage COPD. There are two implications of this work. One is that these clonogenic variants are stem cells respectively committed to metaplastic lesions that, in turn, promote the fibrotic and inflammatory processes observed in COPD. The second is that these variant cells or the factors they secrete may represent important therapeutic targets in this disease.
The clonal analysis described here was instrumental in defining the heterogeneity of clonogenic epithelial cells in COPD. Unlike the normal Cluster 1 clones committed to distal bronchiolar fates, the three variant types exhibited distinct pathologic phenotypes, with Cluster 2 clones producing a GCM marked by the excess mucin secretion implicated in airway occlusion (Fahy and Dickey, 2010; Kesimer et al., 2017; Wedzicha, 2017), while SCM (Cluster 3) clones induced an activation of airway endothelial cells in vitro and, in immunodeficient mice, a host myofibroblast response linked to fibrosis. Cluster 4 clones not only activated endothelial cells in vitro but triggered both submucosal myofibroblast recruitment and leukocyte infiltration upon xenografting to immunodeficient mice. Given that the unparsed clone libraries from all 19 COPD patients induced leukocyte infiltration whereas only 1 of the 11 controls did so, as well as the major role of inflammation in this disease (Barnes, 2016; Brazil and Parkos, 2016; Butler et al., 2018), we predict that Cluster 4 clones will figure prominently in the evolution of COPD.
It should be emphasized that the inflammatory genes expressed in proliferating clones isolated from COPD lung are constitutively activated for months even in a sterile, in vitro environment (Fig. 5). The inflammatory pathways manifest in these cells are linked to the activation of a broad array of the innate and adaptive immune cell populations associated with COPD, including neutrophils, monocyte-derived macrophages and dendritic cells, as well as a host of T-helper subsets, natural killer cells, and other lineages such as innate lymphoid cells. Many of these immune cell types impact the development of fibrosis (Kasembeli et al., 2018; Lee and Kalluri, 2010). Notably, despite the broad range of the inflammatory pathways expressed in Cluster 4 clones, only neutrophil responses were apparent in xenografts of these cells. At least some of this restricted host response can be attributed to strain of immunodeficient mice (NOD/scid/IL2rγnull) used in these studies, which lacks mature B and T cells, natural killer cells, and shows defects in monocyte-derived macrophages and dendritic cells (Ito et al., 2012; Shultz et al., 2005; Shultz et al., 1995). Therefore, we were unable to assess whether other hematopoietic lineages associated with COPD (Barnes, 2016; Suzuki et al., 2017) might be impacted as well by the variant clones. Xenografts of these clones in immunodeficient strains having a greater complement of hematopoietic lineages may provide a more complete assessment of interactions between these clones and the immune system, as would syngeneic studies of of homologous variant clones in genetically tractable model organisms.
This study raises important questions on the origin of the variant clones detected in COPD, how they become hegemonic, and what they mean to the etiology and progression of the disease. Taking just the Cluster 4 clone (iSCM) that promotes host neutrophilic and fibrotic responses as an example, quantitative FACS profiling showed that it represents 13+/−8% of all clonogenic cells across 19 COPD lungs whereas only 1.4+/−1.6% of those in Control lungs (Fig. 7E). These numbers raise the possibility that the COPD phenotype becomes apparent at some percentage threshold of these clones. We tested this hypothesis in a tangential manner by asking what ratios of Cluster 4 to Cluster 1 clones in co-xenografts would elicit a host neutrophil response (Fig. 7F). The resulting xenografts induced a minimal inflammatory response when Cluster 4 clones comprised less than 5% of the total clones, whereas ratios between 10 and 20% of Cluster 4 clones triggered a robust neutrophil response (Fig. 7G). These findings support the notion that a threshold phenomenon, tied to the relative ratios of these variant cells, could be relevant to both the onset of clinical symptoms and the vectored progression of COPD. We anticipate that additional modeling of these variant clones singly or in combination will contribute to understanding their roles in COPD.
Given that similar variant stem cells preexist in control adult lungs as well as in lungs at multiple stages of fetal development, it is likely that these variants play some role in the normal lung perhaps as sentinels for pathogen incursions. Understanding the responses of these variants in normal lung may provide insights into their intrinsic functions, and how these cells might be suborned to disease. In this regard, key questions remain for how these minor variants become major variants in COPD. How do early-life pulmonary events or chronic smoking set in motion their numerical expansion, do genetic variations impact these expansion dynamics, does the disease process itself further alter these variants during the expansion process, and can this expansion be therapeutically preempted or reversed?
The overall findings here set the stage for more detailed studies of correlations between the abundance of such clones and COPD stage as well as their correlation with the known regional, intrapulmonary variations in COPD pathology (Nambu et al., 2016). We should note here that multiple studies have highlighted the possibility that alterations in epithelial cells in idiopathic pulmonary fibrosis (IPF), chronic rhinosinusitis, and COPD may contribute to the pathology of these conditions (Byers et al., 2013; Habiel et al., 2018; Ordovas-Montanes et al., 2018; Vieira Braga et al., 2019; Xu et al., 2016). While those studies did not examine clonogenic cells per se, their findings support the speculation that many if not all chronic lung conditions involve pathogenic clone variants of the sort described here.
If indeed these clones contribute to COPD, multiple opportunities become available to limit their impact on disease progression. These include neutralizing one or more of the pathogenically relevant chemokines or cytokines secreted by individual variants, targeting particular variants with cell surface-directed antibodies, or through the discovery of small molecules that selectively affect one or more of these clone types. This latter strategy could be predicated on the observation that COPD patients retain normal clones that would potentially compensate for the loss of their pathogenic counterparts.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Wa Xian (wxian@uh.edu).
Materials Availability
The unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
The microarray, single-cell RNA-seq, RNA-seq, and exome sequencing datasets generated during this study are available at GEO: GSE118950, SRA: PRJNA514053, and SRA: PRJNA492749. A list of software used in this study can be found in the Key Resources Table.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Human Subjects
The lung tissues used in this study were from resected lobes, lung transplants, or fetal demise cases that were obtained under informed consent as de-identified material under approved institutional review board protocols at the University of Connecticut Health Sciences (IRB# 08-310-1), the University of Iowa Carver College of Medicine (IRB# 199507432), the University of Texas Health Sciences Center, Houston (HSC-MS-08–0354/HSC-MS-15–1049), and the Brigham and Women’s Hospital, Boston, MA (2009P002281), respectively. Relevant patient data is listed in Table S2.
Primary Cell Culture
The primary cells used in this study were derived from human lung tissues. All cells were cultured in StemECHO™PU culture medium (Nüwa Medical Systems, Inc., Houston, USA). The cells were tested as mycoplasma-free. Single Nucleotide Polymorphisms (SNPs) detected in RNA-seq data were used as genotyping marker to ensure the authentication of each cell line.
Animals
All animal experiments have been approved by the Animal Care Committee at the University of Houston (IACUC 16–002). Human lung cell variants were subcutaneously transplanted to NSG (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ) mice. Animal studies were conducted under maximum containment in an animal biosafety level 2 facility. NSG mice were immune-compromised, drug or test naive, and housed in a sterile condition. Both male and female mice (6–8 weeks old) were utilized in these studies. At 3 weeks post-transplant, mice were humanely euthanized according to IACUC-approved criteria.
Cell Lines
Human Lung Microvascular Endothelial Cells (HMVEC-L, human [Homo sapiens]) were obtained from Lonza (https://bioscience.lonza.com/lonza_bs/CH/en/). HMVEC-L cells were cultured in EGMTM −2 MV medium purchased from Lonza (Cat# CC-3202). All cell lines were incubated at 37°C with 7.5% CO2.
METHOD DETAILS
In vitro culture of clonogenic cells from 19 COPD and 11 control lungs
Minced lung tissue was digested in 2 mg/ml collagenase type IV (Gibco, USA) at 37°C for 30–60 min with agitation. Dissociated cells were passed through a 70 μm Nylon mesh (Falcon, USA) to remove masses and then were washed four times in cold F12 media, and seeded onto a feeder layer of lethally irradiated 3T3-J2 cells as described (Kumar et al., 2011; Zuo et al., 2015) and grown in StemECHO™PU culture medium (Nüwa Medical Systems, Inc., Houston, USA). In each case, approximately 2 to 4 × 106 cells were obtained per cubic centimeter of lung tissue after erythrocyte lysis of which 200–400 thousand expressed E-cadherin. 1 million of these dissociated lung cells were seeded onto lawns of irradiated 3T3-J2 feeder cells. In the control cases, 1,500 to 2,300 colonies were observed at P0 after seeding, indicating that 0.15% to 0.23% of all cells were clonogenic. For COPD cases, we obtained 4,500 to 8,000 colonies at P0, indicating that 0.45% to 0.8% of all lung cells were clonogenic. The culture media was changed every two days. Colonies were digested by 0.25 % trypsin-EDTA solution (Gibco, USA) for 5–8 min and passaged every 7 to 10 days. Colonies were trypsinized by TrypLE Express solution (Gibco, USA) for 8–15 min at 37°C and cell suspensions were passed through 30 μm filters (Miltenyi Biotec, Germany). Approximately 20,000 epithelial cells were seeded to each well of 6-well plate. Cloning cylinder (Pyrex, USA) and high vacuum grease (Dow Corning, USA) were used to select single colonies for pedigrees. Gene expression analyses were performed on cells derived from passage 4–10 (P4-P10) cultures.
Histology validation set of five control and 10 Stage-4 COPD lungs
To validate the localization of clone-specific metaplasia in situ, we performed histopathology and immunolocalization studies on serial sections from five control lungs from patients without lung or systemic disease (Okuda et al., 2019) and from 10 patients with Stage 4 COPD. These histological sections were obtained under IRB approval from the University of North Carolina (03–1396), the University of Iowa Carver College of Medicine (199507432), and the University of Texas Health Sciences Center at Houston (HSC-MS-08–0354/HSC-MS-15–1049). Data relevant to the patients of this validation set is presented on Table S3. Student’s t-test was used to determine the statistical difference between groups. P < 0.05 was considered statistically significant.
Histology and immunostaining
Histology, hematoxylin and eosin (H&E) staining, immunohistochemistry, and immunofluorescence were performed using standard techniques. For immunofluorescence and immunohistochemistry, 4% paraformaldehyde-fixed, paraffin embedded tissue slides were subjected to antigen retrieval in citrate buffer (pH 6.0, Sigma-Aldrich, USA) at 120 °C for 20 min, and a blocking procedure was performed with 5% bovine serum albumin (BSA, Sigma-Aldrich, USA) and 0.05 % Triton X-100 (Sigma-Aldrich, USA) in DPBS(−) (Gibco, USA) at room temperature for 1 hr. The sources of primary antibodies used in this study include: Rabbit monoclonal cytokeratin 5 antibody (RM-2106-S; Thermo Fisher, USA); mouse monoclonal human cytokeratin 5 antibody (NCL-L-CK5; Leica Biosystems, Germany); mouse monoclonal CC10 antibody (sc-365992; Santa Cruz Biotechnology, USA); rabbit polyclonal AQP4 antibody (HPA014784; Sigma-Aldrich, USA); rabbit polyclonal SFTPB antibody (HPA034820; Sigma-Aldrich, USA); mouse monoclonal acetylated tubulin T7451 (Sigma-Aldrich, USA); rat monoclonal mouse CD45 antibody 14-0451-85 (Thermo Fisher, USA); mouse monoclonal human CD45 MAB1430-SP (R&D systems, USA); rat monoclonal mouse Ly6G MAB1037 (R&D systems, USA); mouse monoclonal alpha smooth muscle actin antibody (ab7817; Abcam, UK); rabbit polyclonal Involucrin antibody (HPA055211; Sigma-Aldrich, USA); mouse monoclonal Cytokeratin 10 antibody (904301; BioLegend, USA); rabbit polyclonal Muc5B antibody (ab87376; Abcam, UK); rabbit polyclonal Muc5AC antibody (ab78660; Abcam, UK); goat polyclonal E-Cadherin antibody (AF648; R&D Systems, USA). Secondary antibodies used here are Alexa Fluor-488 or Alexa Fluor-594 Donkey anti-goat/mouse/rabbit IgG antibody (Thermo Fisher, USA). All images were captured by using the Inverted Eclipse Ti-Series (Nikon, Japan) microscope with Lumencor SOLA light engine and Andor Technology Clara Interline CCD camera and NIS-Elements Advanced Research v.4.13 software (Nikon, Japan) or LSM 780 confocal microscope (Carl Zeiss, Germany) with LSM software. Bright field cell culture images were obtained on an Eclipse TS100 microscope (Nikon, Japan) with Digital Sight DSFi1camera (Nikon, Japan) and NIS-Elements F3.0 software (Nikon, Japan).
In vitro differentiation
Air-liquid interface (ALI) culture of epithelial clones was performed as described (Kumar et al., 2011; Zuo et al., 2015). Briefly, Transwell inserts (Corning, USA) were coated with 20% Matrigel (BD biosciences, USA) and incubated at 37 °C for 30 min to polymerize. 200,000 irradiated 3T3-J2 cells were seeded to each Transwell insert and incubated at 37 °C, 7.5 % CO2 incubator overnight. QuadroMACS Starting Kit (LS) (Miltenyi Biotec, Germany) was used to purify epithelial by removal of feeder cells. 200,000–300,000 cells were seeded into each Transwell insert and cultured in StemECHO™PU media. At confluency (3–7 days), the apical media on the inserts was removed through careful pipetting and the cultures were continued in differentiation media (PneumaCult-ALI Media, STEMCELL Technologies, Vancouver) for an additional 14–21 days prior to harvesting. The differentiation media was changed every one or two days.
Xenografts in immunodeficient mice
One million epithelial cells were harvested by trypsinization, mixed with 50% Matrigel (Becton Dickinson, Palo Alto) to a volume of 100 ul and injected subcutaneously in NSG (NODscid IL2ranull) (Shultz et al., 1995) mice (Jackson Laboratories, Bar Harbor) and harvested two or four weeks later.
RNA sample preparation
For cell colonies, RNA was isolated using PicoPure RNA Isolation Kit (Life Technologies, USA). For ALI and Xenografts structure, RNA was isolated using Trizol RNA Isolation Kit (Life Technologies, USA). RNA quality (RNA integrity number, RIN) was measured by analysis Agilent 2100 Bioanalyzer and Agilent RNA 6000 Nano Kit (Agilent Technologies, USA). RNAs having a RIN > 8 were used for microarray analysis.
Flow cytometry analysis
Clonogenic cell libraries from patients with or without COPD were trypsinized and harvested as single cell suspension. Feeders were removed as mentioned above and approximately 300,000 epithelial cells were fixed and permeabilized by using Fixation/Permeabilization Solution Kit (BD biosciences, USA, cat. 554714). After a blocking procedure with Permeabilization solution at 4 °C for 30 min, cells were incubated with primary and Alexa Fluor 488 Secondary antibodies (Thermo Fisher, USA) for 1hr at 4 °C, with five washing events between each step. Primary antibodies used in these experiments include: rabbit monoclonal anti-AQP5 antibody ab92320 (Abcam, UK), rabbit polyclonal TRPC6 antibody (18236–1-AP; Proteintech, USA), mouse monoclonal anti-human IL-8/CXCL8 antibody (MAB208; R&D, USA). Samples were collected and analyzed with on a Sony SH800S Cell Sorter (Sony Biotechnology, USA).
Transcriptomic sequencing data analysis
All RNA-seq libraries were sequenced on Illumina NovaSeq 6000 with 150 bp pair-end reads. Raw reads were trimmed to remove low quality bases (phred score < 20) and sequencing adapter leftovers using Trim Galore (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/) (Martin, 2011). Potential mouse genomic DNA contaminant reads were removed for further analysis using Xenome (Conway et al., 2012). Trimmed RNAseq reads were mapped to the human genome (UCSC hg19) using Salmon (version 0.9.1) with default settings (Patro et al., 2017). Alignment results were then input to DEseq2 (Love et al., 2014) for differential expression analysis with default settings and FDR less than 0.05. Batch effects were estimated and corrected using RUVSeq (Risso et al., 2014). The heatmaps with hierarchical clustering analysis of the global gene expression pattern in different samples were performed using pheatmap package (https://cran.rproject.org/web/packages/pheatmap/index.html) (Li et al., 2018) in R (version 3.5.1). The pathway enrichment analysis was performed using Enrichr (Chen et al., 2013) and Ingenuity Pathway Analysis (IPA) tools (Kramer et al., 2014).
Sequence alignment of single cell RNA sequencing
The single cell mRNA sequencing (scRNA-seq) libraries were established using the 10X Genomics Chromium system (Single Cell 3’ Reagent Kit v2). The scRNA-seq libraries were sequenced on the Illumina HiSeq × Ten with 10K cells for three COPD cases and fetal lung case. For three normal cases, the scRNA-seq libraries were sequenced on the Illumina NextSeq 500 with 2K cells. Demultiplexing, alignment and UMI-collapsing were performed using the Cellranger toolkit (version 2.1.0, 10X Genomics) (Ferguson and Chen, 2020). The raw paired-end reads were trimmed to 26 bps for Read1 and 98 bps for Read2. The trimmed reads were mapped to both the human genome (hg19) and the mouse genome (mm10). The reads uniquely mapped to the human genome were used for downstream analysis.
Single cell RNA sequencing
The scRNA-seq data analyses were performed using the Seurat package (version 2.3.4) (Satija et al., 2015). We kept the genes with expression in at least three cells, and excluded cells expressing less than 200 genes. We also excluded the cells with high mitochondrial percentage or with an outlier level of UMI content. The normalization was performed using the global-scaling normalization method, which normalizes the gene expression measurements for each cell by the total expression, and then multiplies by 10,000, and finally log-transforms the result. The variable genes were identified using a function to calculate average expression and dispersion for each gene, divides these genes into bins, and then calculates a z-score for dispersion within each bin (“x.low.cutoff = 0.0125”, “x.high.cutoff = 3”, and “y.cutoff = 0.5”). We scaled the data to regress out the variation of mitochondrial gene expression.
We performed PCA based on the scaled data to identify significant principal components (PCs). We selected the PCs with p-values less than 0.01 as input to perform clustering analysis and visualization by t-SNE. We detected the marker genes in each cell subpopulation using two methods of Wilcoxon rank sum test and DESeq2. For Wilcoxon rank sum test, we used the default parameter. For DEseq2, we kept the marker genes with average log-fold change above 0.1 and adjust p-value fewer than 0.05.
Contaminating 3T3-J2 fibroblast cells were identified by murine reads. In addition, the cells in S stage of cell cycle were identified based on the marker gene of SLBP (Nestorowa et al., 2016). The cells in G2 or M stage of cell cycle were identified based on the marker genes of UBE2C, AURKA, CENPA, CDC20, HMGB2, CKS2, and CKS1B. The cells in G0 stage of cell cycle were identified based on the marker genes of G0S2. In addition, the ambiguous cells with few marker genes were also removed, which could possibly correspond to sequencing low quality cells. Finally, we integrated the clean data of six cases to perform clustering analysis and visualization by t-SNE.
Expression microarray and bioinformatics
Total RNAs obtained from immature colonies and ALI-differentiated epithelia were used for microarray preparation with WT Pico RNA Amplification System V2 for amplification of DNA and Encore Biotin Module for fragmentation and biotin labeling (NuGEN Technologies, USA). All samples were prepared according to manufacturer’s instructions and hybridized onto GeneChip Human Exon 1.0 ST array (Affymetrix, USA). GeneChip operating software was used to process Cel files and calculate probe intensity values. To validate sample quality, quality checks were conducted using Affymetrix Expression Console software. The intensity values were log2-transformed and imported into the Partek Genomics Suite 6.6 (Partek Incorporated, USA) (Wodehouse et al., 2019). Exons were summarized to genes and a 1-way ANOVA was performed to identify differentially expressed genes. Unsupervised clustering and heatmap generation were performed with sorted datasets by Euclidean distance based on average linkage by Partek Genomics Suite 6.6.
Whole genome exome sequencing data analysis
For exome capture and high-throughput sequencing, genomic DNA was isolated from single cell-derived pedigrees and 1000ng of DNA per sample were sheared, end-repaired, A-tailed and adaptor-ligated. Exome capture was conducted using the Agilent SureSelect Human All Exon V6 Kit following the manufacturer’s standard protocols. The libraries were sequenced on an Illumina HiSeq × ten platform (150 bp paired-end model). The raw sequencing data were quality controlled by Trimmomatic (Bolger et al., 2014) version 0.36 to trim low-quality bases, filtered by Xenome (Conway et al., 2012) version 1.0.1 to remove mouse sequences, aligned to reference genome (hg19) via BWA-mem (Li and Durbin, 2010) version 0.7.15, realigned through GATK (McKenna et al., 2010) version 3.8.0 in regions near indels (Mills_and_1000G_gold_standard indels), and recalibrated with default settings following the best practices pipeline of GATK (Van der Auwera et al., 2013). Somatic SNVs and Indels were called by Manta (Chen et al., 2016) version 1.3.2 and Strelka (Kim et al., 2018) version 2.9.2 and annotated with ANNOVAR (Yang and Wang, 2015). We require that somatic mutations situated at the targeted capturing regions, pass the Strelka default filters, and have only two genotypes present, a minimum of 5% mutant reads fraction, a minimum 5 mutant reads coverage, and minimum 15 total reads coverage. The corresponding matched normal sample should be homozygous wild type at the mutation sites. Somatic mutations were further filtered to remove possible germline mutations based on a panel of 27 normal samples. Somatic mutations with allele frequencies larger than 0.01 in 1000 Genome database (Sudmant et al., 2015) or gnomAD database v2 (Konrad J. Karczewski, 2019) were discarded as well. The somatic allelic copy number variants were called by the best practices pipeline of GATK version 4.0.4 (DePristo et al., 2011; Van der Auwera et al., 2013). The modeled copy number segments with less than 7.5 Kbp in length or less than 15 common variants were filtered out. We used default settings in all software. The oncogenes and tumor suppressors were fetched from the OncoKB database (Chakravarty et al., 2017).
QUANTIFICATION AND STATISTICAL ANALYSIS
Unpaired two-tailed student’s t-test was used to determine the statistical significance between two groups. Statistical analyses were performed using R (version 3.5.1). The “n” numbers for each experiment are provided in the text and figures. P < 0.05 was considered statistically significant. Asterisks denote corresponding statistical significance *p < 0.05; **p < 0.01; ***p < 0.001 and ****p < 0.0001.
Supplementary Material
Figure S1. In vitro and in vivo differentiation of Cluster 1–4 clones; Related to Figure 2. A. H&E staining of histological sections of air-liquid interface (ALI)-differentiated Cluster 1–4 clones. Scale bar, 100 μm. B. Left, Immunohistochemistry of p63 antibody staining (brown) of the distal airway of normal human lung. Scale bar, 500 μm. Right from top, Magnification of indicated terminal bronchiole epithelium region of p63 immunohistochemistry; immunofluorescence micrograph of terminal bronchiolar epithelium stained with DAPI, anti-p63, anti-SCGB1A1, and anti-SFTPB, and immunofluorescence micrograph of terminal bronchiolar epithelium showing anti-p63 and anti-AQP4 staining. Alveo, alveolar epithelia. Scale bar, 100 μm. C-F. Immunofluorescence micrographs of xenograft sections derived from transplants of clones of Clusters 1–4 probed with the indicated antibodies. Scale bar, 100 μm. G. Venn diagrams showing overlap between nonsynonymous SNV events detected from whole genome exome sequencing of clones of Clusters 1–4 from COPD (SNP-13) lung and 202 oncogenes and 231 tumor suppressor genes (detailed in Table S5).
Figure S2. Distribution of metaplasia in Stage 4 COPD lung; Related to Figure 3. A. Immunohistochemistry micrograph of section of distal lung of normal donor showing distribution of p63 staining (brown) in bronchioles and terminal bronchioles. Scale bar, 1,000 μm. Boxed region indicates region presented as inset right top) and corresponding region of serial section examined by p63 and AQP5 immunofluorescence (lower right). Scale bar of insets, 100 μm. B. Immunohistochemistry micrograph of Stage 4 COPD distal lung stained with p63 antibodies (brown). (*) denote regions of submucosal inflammation. Scale bar, 1,000 μm. Boxed regions corresponding to inflammatory SCM, GCM, and SCM are shown at higher magnification below along with immunofluorescence micrographs of similar regions from serial sections stained with antibodies to p63/CXLC8 (iSCM), p63/TRPC6 (GCM), and p63/TRPC6 (SCM). Inset region scale bars, 100 μm.
Figure S3. Clone variant analysis by single cell RNA-seq; Related to Figure 3. A. Differential gene expression determined by scRNAseq of three COPD and three control clone libraries and the corresponding differential gene expression determined by RNAseq of discrete clones of Clusters 1–4 of SPN-13 (detailed in Tables S6, S7). B. tSNE profiles of single cell RNAseq data derived from clone libraries from the individual COPD and control cases. C. Mapping of cluster-specific gene expression markers onto an integrated tSNE profile assembled from three Control and three COPD cases.
Figure S4. Leukocyte transepithelial migration in xenografts; Related to Figure 4. A. Box-Whisker plot representation of Cluster 2–4 FACS proportions in each of 19 COPD and 11 control libraries (P<2.2e-16, Student’s t-test). B.,C. Histological sections through nodules formed four weeks after transplantation of clonogenic cell pools of six representative non-COPD cases (upper) and COPD cases (lower) to immunodeficient mice. Scale bar, 200 μm. D. Box-Whisker plot representation of severe inflammation fraction in each of 19 COPD and 11 control library xenografts (P= 1.2e-09, Student’s t-test).
Figure S5. Inflammatory gene expression by clones of Clusters 1 to 4; Related to Figure 5. A. Top 10 pathways of differentially expressed genes (FDR<0.05) of patient-matched clones representative of Clusters 1–4 using Ingenuity Pathway Analysis (IPA) software. B. Immunofluorescence of Vcam1 (red) in human lung endothelial cells following exposure to media conditioned by clones of Clusters 1–4 compared to IL-13 challenge. Scale bar, 100 μm. C. Expression heatmaps (FDR<0.05) depicting pair-wise gene expression analysis of Clusters 1–4 focused on chemokine, interleukin, and interferon genes.
Figure S6. Fibrosis in xenografts; Related to Figure 6. A., B. Immunofluorescence micrographs of sections of nodules formed four weeks after transplantation of clone libraries from six representative control cases (upper) and COPD cases (lower). Green, E-cad, E-cadherin; red, SMA, anti-alpha smooth muscle actin. Scale bar, 200 μm. C. Immunofluorescence micrographs of E-cadherin (Ecad) and alpha smooth muscle actin (SMA) expression in xenografts of clones of Cluster 3 (left) and Cluster 4 (right) from in vitro passage 5 and passage 25. Scale bar, 100 μm.
Figure S7. Variant clones in normal adult and fetal lung; Related to Figure 7. A. Histology and marker analysis of xenografts of cloned representatives of Clusters 1–4 from adult Control lung (SPN-14). B. Left, Principal component analysis of RNAseq profiles of Clusters 1–4 clones from control lung (SPN-14) and those of Clusters 1–4 of two COPD cases (SPN-7 and SPN-13). Right, Expression heatmap comparing RNAseq profiles of Clusters 1–4 clones of control lung (SPN-14) with those of clones of COPD cases SPN-7 and SPN-13 (detailed in Table S8). C. Histology of pseudoglandular epithelia of 13-wk fetal lung and marker immunofluorescence including antibodies to p63, AQP5, AQP4, α-TUB, and SFTPB. D. Left, Distribution of cluster-specific expression markers on scRNAseq tSNE profile of clone library from 13-wk fetal lung. Right, Pie chart showing distribution of variants in 13-wk fetal library. E. Dot plots of expression for cluster-specific marker genes in the integrated tSNE profiles of lung clones from three adult Controls, three COPD cases, and the 13-wk fetal lung. The size of the dot corresponds to the percentage of cells expressing the feature in each cluster. The color represents the average expression level.
Table S1. Patient-derived clone library information related to Figure 1
Footnotes:
NA: not available
INF: inflammation (leukocytes)
SCM: squamous metaplasia
GCM: goblet cell metaplasia
ABN: abnormality
ns inter pneu: non-specific interstitial pneumonia
sarc: sarcoidosis
CA: cancer
DIPNECH: diffuse idiopathic pulmonary neuroendocrine cell hyperplasia
DLCO: carbon monoxide diffusing capacity
Leukocyte TEM: transepithelial migration
UCONN Health: University of Connecticut Health Center
UTHSC: University of Texas Health Science Center
UIHC: University of Iowa Hospitals & Clinics
Table S2. Clone library and xenograft information related to Figure 2
Footnotes:
SPN: Study Patient Number
NA: not available
INF: inflammation (leukocytes)
SCM: squamous metaplasia
GCM: goblet cell metaplasia
Leukocyte TEM: transepithelial migration
Table S3. Histology validation set information related to Figure 3
Footnotes:
NA: not available
INF: inflammation (leukocytes)
SCM: squamous metaplasia
GCM: goblet cell metaplasia
ABN: abnormalities
*Sections from patients reported in Okuda et al., 2019.
UNC: University of North Carolina at Chapel Hill
UTHSC: The University of Texas Health Science Center at Houston
UIHC: University of Iowa Hospitals & Clinics
Table S5. Exome-seq related to Figure S1G
Table S6. scRNAseq related to Figure S3A
Table S7. RNAseq related to Figure S3A
Table S8. Differential gene expression between Clusters 1–4 clones of control case and COPD cases related to Fig. S7B
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit monoclonal Cytokeratin 5 | Thermo Fisher | RRID: AB_2538529 |
| Mouse monoclonal human Cytokeratin 5 | Leica Biosystems | RRID: AB_442073 |
| Mouse monoclonal CC10 | Santa Cruz | RRID: AB_2183388 |
| Rabbit polyclonal AQP4 | Sigma-Aldrich | RRID: AB_1844967 |
| Rabbit polyclonal SFTPB | Sigma-Aldrich | RRID: AB_2674347 |
| Mouse monoclonal acetylated tubulin | Sigma-Aldrich | RRID: AB_609894 |
| Rat monoclonal mouse CD45 | Thermo Fisher | RRID: AB_657749 |
| mouse monoclonal human CD45 | R&D systems | RRID: AB_2174120 |
| Rat monoclonal mouse Ly6G | R&D systems | RRID: AB_2232806 |
| mouse monoclonal alpha smooth muscle Actin | Abcam | RRID: AB_262054 |
| Rabbit polyclonal Involucrin | Sigma-Aldrich | RRID: AB_2682739 |
| Mouse monoclonal Cytokeratin 10 | BioLegend | RRID: AB_2565038 |
| Rabbit polyclonal Muc5B | Abcam | RRID: AB_10712492 |
| E-Cadherin Antibody | R&D Systems | RRID: AB_355504 |
| Human IL-8/CXCL8 Antibody | R&D Systems | RRID: AB_2249110 |
| Aquaporin 5 antibody [EPR3747] | Abcam | RRID: AB_2049171 |
| Anti-TRPC6 Rabbit Polyclonal Antibody | Proteintech | RRID: AB_10859822 |
| Anti-p63 antibody (4a4) | Abcam | RRID: AB_305870 |
| Bacterial and Virus Strains | ||
| N/A | N/A | N/A |
| Biological Samples | ||
| Human Lung samples from COPD and non-COPD patients | UCONN Health; UTHSC; UIHC | See Table S1 for a list of Patients included in this study |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Trizol Reagent | Thermo Fisher | Cat#15596018 |
| Feeder removal beads | Miltenyi Biotec | Cat#130–095-531 |
| Human IL-13 Recombinant Protein | eBioscience™ | Cat# BMS351 |
| Power SYBR Green PCR master mix | Thermo Fisher | Cat#4368706 |
| TrypLE™ Express Enzyme (1X), phenol red | Thermo Fisher | Cat#12605036 |
| Corning™ Matrigel™ GFR Membrane Matrix | Fisher Scienitific | Cat# CB-40230 |
| COLLAGENASE TYPE IV | Life Technologies | Cat# 17104–019 |
| StemECHO™PU Ground-State Stem Cell Culture System | Nüwa Medical Systems, Inc. | http://www.stemecho.com |
| Critical Commercial Assays | ||
| Fixation/Permeabilization Solution Kit (Cytofix/Cytoperm) | BD Biosciences | Cat#554714 |
| Maxima First Strand cDNA Synthesis Kit for RT-qPCR, with dsDNase | Thermo Fisher | Cat# K1671 |
| DNeasy Blood & Tissue Kit (250) | Qiagen | Cat#69506 |
| GeneChip™ Human Exon 1.0 ST Array | Affymetrix | Cat#900650 |
| Deposited Data | ||
| Expression microarray raw data | This paper | GSE118950 |
| Whole genome exome sequencing data | This paper | PRJNA492749 |
| RNAseq data | This paper | PRJNA492749 |
| scRNAseq data | This paper | PRJNA514053 |
| Statistical analysis raw data | This paper | https://data.mendeley.com/drafts/tf8kyp2ghx |
| Experimental Models: Cell Lines | ||
| HMVEC-L–Human Lung Microvascular Endothelial Cells | Lonza | Cat# CC-2527 |
| Experimental Models: Organisms/Strains | ||
| Mouse: NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ | The Jackson Laboratory | Stock No:005557 | NSG |
| Oligonucleotides | ||
| N/A | N/A | N/A |
| Recombinant DNA | ||
| N/A | N/A | N/A |
| Software and Algorithms | ||
| SH800S Cell Sorter Software version 2.1 | SH800S Cell Sorter | https://www.sonybiotechnology.com/us/instruments/sh800s-cell-sorter/software/ |
| Trim Galore | Martin, 2011 | https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| Xenome | Conway et al., 2012 | http://www.nicta.com.au/bioinformatics |
| Salmon | Patro et al., 2017 | https://salmon.readthedocs.io/en/latest/salmon.html |
| DEseq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| RUVSeq | Risso et al., 2014 | https://bioconductor.org/packages/release/bioc/html/RUVSeq.html |
| Pheatmap | Li et al., 2018 | https://cran.rproject.org/web/packages/pheatmap/index.html |
| Enrichr | Chen et al., 2013 | https://amp.pharm.mssm.edu/Enrichr/ |
| Ingenuity Pathway Analysis | Kramer et al., 2014 | https://www.qiagenbioinformatics.com/products/features/?gclid=Cj0KCQiAl5zwBRCTARIsAIrukdM9LVWhBGtDPLVPTgsVLN1_N-jK9bcU5ZniWNXs-UihFNPyaxcOcK8aAl2tEALw_wcB |
| Cellranger toolkit | Ferguson and Chen, 2020 | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| Seurat | Satija et al., 2015 | https://satijalab.org/seurat/ |
| Partek Genomics Suite 6.6 | Wodehouse et al., 2019 | https://www.partek.com/partek-genomics-suite/ |
| Trimmomatic | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| BWA-mem | Li and Durbin, 2010 | http://bio-bwa.sourceforge.net/ |
| GATK | McKenna et al., 2010; Van der Auwera et al., 2013 | https://software.broadinstitute.org/gatk/ |
| Manta | Chen et al., 2016 | https://github.com/Illumina/manta |
| Strelka | Kim et al., 2018 | https://github.com/target/strelka |
| ANNOVAR | Yang and Wang, 2015 | http://annovar.openbioinformatics.org/en/latest/ |
| GnomAD database v2 | Karczewski et al., 2019 | https://gnomad.broadinstitute.org/ |
| 1000 Genome database | Sudmant et al., 2015 | https://www.internationalgenome.org/ |
| OncoKB database | Chakravarty et al., 2017 | https://www.oncokb.org/ |
Highlights.
COPD lung is dominated by three pathogenic stem cell variants
These variants are epigenetically committed to metaplastic lesions found in COPD
Variants drive key COPD features of inflammation, fibrosis, and airway obstruction
Similar variants in fetal and normal lung likely serve sentinel functions
ACKNOWLEDGEMENTS
This work was supported by grants from the Cancer Prevention Research Institute of Texas (CPRIT; RR150104 to WX and RR1550088 to FM), the National Institutes of Health (1R01DK115445-01A1 to WX, 1R01CA241600-01 and U24CA228550 to FM, R01 DK047967 to JFE, 5R01 HL138510 to HK-Q, P01 HL108808 and R01 HL136961 to RCB, NIH, NHLBI, R01 HL129795 to BFD, 1R01 HL136370-01A1 to K.R.P.), the US Dept. of Defense (W81XWH-17-1-0123 to WX), and the American Gastroenterology Association Research and Development Pilot Award in Technology (to WX), and the Cystic Fibrosis Foundation (BOUCHE15R0 to RCB) (DICKEY18G0 to BFD). WX and FDM are CPRIT Scholars in Cancer Research. We thank all the members in the Xian-McKeon laboratory for helpful discussions and support. We thank H. Green for advice and support. Dedicated to Hazel and Richard T. and the valiant patients who have made this study possible.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
W.X., F.D.M., W.R., S.W., J.X., M.D., and M.V. have filed patents related to technologies used in the present work. M.V., F.D.M., and W.X. have financial interests in Nüwa Medical Systems, Inc., Houston, TX, USA and Tract pharmaceuticals, Inc., Houston, TX, USA.
REFERENCES
- Araya J, Cambier S, Markovics JA, Wolters P, Jablons D, Hill A, Finkbeiner W, Jones K, Broaddus VC, Sheppard D, et al. (2007). Squamous metaplasia amplifies pathologic epithelial-mesenchymal interactions in COPD patients. J Clin Invest 117, 3551–3562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya J, and Nishimura SL (2010). Fibrogenic reactions in lung disease. Annu Rev Pathol 5, 77–98. [DOI] [PubMed] [Google Scholar]
- Aschner Y, and Downey GP (2016). Transforming Growth Factor-beta: Master Regulator of the Respiratory System in Health and Disease. Am J Respir Cell Mol Biol 54, 647–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes PJ (2016). Inflammatory mechanisms in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol 138, 16–27. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Burney PG, Silverman EK, Celli BR, Vestbo J, Wedzicha JA, and Wouters EF (2015). Chronic obstructive pulmonary disease. Nat Rev Dis Primers 1, 15076. [DOI] [PubMed] [Google Scholar]
- Black LM, Lever JM, and Agarwal A (2019). Renal Inflammation and Fibrosis: A Double-edged Sword. J Histochem Cytochem 67, 663–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brazil JC, and Parkos CA (2016). Pathobiology of neutrophil-epithelial interactions. Immunol Rev 273, 94–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch R, Hobbs BD, Zhou J, Castaldi PJ, McGeachie MJ, Hardin ME, Hawrylkiewicz I, Sliwinski P, Yim JJ, Kim WJ, et al. (2017). Genetic Association and Risk Scores in a Chronic Obstructive Pulmonary Disease Meta-analysis of 16,707 Subjects. Am J Respir Cell Mol Biol 57, 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler A, Walton GM, and Sapey E (2018). Neutrophilic Inflammation in the Pathogenesis of Chronic Obstructive Pulmonary Disease. COPD 15, 392–404. [DOI] [PubMed] [Google Scholar]
- Byers DE, Alexander-Brett J, Patel AC, Agapov E, Dang-Vu G, Jin X, Wu K, You Y, Alevy Y, Girard JP, et al. (2013). Long-term IL-33-producing epithelial progenitor cells in chronic obstructive lung disease. J Clin Invest 123, 3967–3982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty D, Gao J, Phillips SM, Kundra R, Zhang H, Wang J, Rudolph JE, Yaeger R, Soumerai T, Nissan MH, et al. (2017). OncoKB: A Precision Oncology Knowledge Base. JCO Precis Oncol 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Schulz-Trieglaff O, Shaw R, Barnes B, Schlesinger F, Kallberg M, Cox AJ, Kruglyak S, and Saunders CT (2016). Manta: rapid detection of structural variants and indels for germline and cancer sequencing applications. Bioinformatics 32, 1220–1222. [DOI] [PubMed] [Google Scholar]
- Chilosi M, Poletti V, Murer B, Lestani M, Cancellieri A, Montagna L, Piccoli P, Cangi G, Semenzato G, and Doglioni C (2002). Abnormal re-epithelialization and lung remodeling in idiopathic pulmonary fibrosis: the role of deltaN-p63. Lab Invest 82, 1335–1345. [DOI] [PubMed] [Google Scholar]
- Conway T, Wazny J, Bromage A, Tymms M, Sooraj D, Williams ED, and Beresford-Smith B (2012). Xenome--a tool for classifying reads from xenograft samples. Bioinformatics 28, i172–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decramer M, Janssens W, and Miravitlles M (2012). Chronic obstructive pulmonary disease. Lancet 379, 1341–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demedts IK, Bracke KR, Van Pottelberge G, Testelmans D, Verleden GM, Vermassen FE, Joos GF, and Brusselle GG (2007). Accumulation of dendritic cells and increased CCL20 levels in the airways of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 175, 998–1005. [DOI] [PubMed] [Google Scholar]
- DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M, et al. (2011). A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhakshinamoorthy S, Jain AK, Bloom DA, and Jaiswal AK (2005). Bach1 competes with Nrf2 leading to negative regulation of the antioxidant response element (ARE)-mediated NAD(P)H:quinone oxidoreductase 1 gene expression and induction in response to antioxidants. J Biol Chem 280, 16891–16900. [DOI] [PubMed] [Google Scholar]
- Fahy JV, and Dickey BF (2010). Airway mucus function and dysfunction. N Engl J Med 363, 2233–2247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson A, and Chen K (2020). Analysis of Transcriptional Profiling of Immune Cells at the Single-Cell Level. Methods Mol Biol 2111, 47–57. [DOI] [PubMed] [Google Scholar]
- Filippi MD (2016). Mechanism of Diapedesis: Importance of the Transcellular Route. Adv Immunol 129, 25–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher C, and Peto R (1977). The natural history of chronic airflow obstruction. Br Med J 1, 1645–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamble E, Grootendorst DC, Hattotuwa K, O’Shaughnessy T, Ram FS, Qiu Y, Zhu J, Vignola AM, Kroegel C, Morell F, et al. (2007). Airway mucosal inflammation in COPD is similar in smokers and ex-smokers: a pooled analysis. Eur Respir J 30, 467–471. [DOI] [PubMed] [Google Scholar]
- Habiel DM, Espindola MS, Jones IC, Coelho AL, Stripp B, and Hogaboam CM (2018). CCR10+ epithelial cells from idiopathic pulmonary fibrosis lungs drive remodeling. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, Cherniack RM, Rogers RM, Sciurba FC, Coxson HO, et al. (2004). The nature of small-airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 350, 2645–2653. [DOI] [PubMed] [Google Scholar]
- Hogg JC, and Timens W (2009). The pathology of chronic obstructive pulmonary disease. Annu Rev Pathol 4, 435–459. [DOI] [PubMed] [Google Scholar]
- Holtzman MJ, Byers DE, Brett JA, Patel AC, Agapov E, Jin X, and Wu K (2014). Linking acute infection to chronic lung disease. The role of IL-33-expressing epithelial progenitor cells. Ann Am Thorac Soc 11 Suppl 5, S287–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito R, Katano I, Ida-Tanaka M, Kamisako T, Kawai K, Suemizu H, Aiso S, and Ito M (2012). Efficient xenoengraftment in severe immunodeficient NOD/Shi-scid IL2rgammanull mice is attributed to a lack of CD11c+B220+CD122+ cells. J Immunol 189, 4313–4320. [DOI] [PubMed] [Google Scholar]
- Kasembeli MM, Bharadwaj U, Robinson P, and Tweardy DJ (2018). Contribution of STAT3 to Inflammatory and Fibrotic Diseases and Prospects for its Targeting for Treatment. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kesimer M, Ford AA, Ceppe A, Radicioni G, Cao R, Davis CW, Doerschuk CM, Alexis NE, Anderson WH, Henderson AG, et al. (2017). Airway Mucin Concentration as a Marker of Chronic Bronchitis. N Engl J Med 377, 911–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Scheffler K, Halpern AL, Bekritsky MA, Noh E, Kallberg M, Chen X, Kim Y, Beyter D, Krusche P, et al. (2018). Strelka2: fast and accurate calling of germline and somatic variants. Nat Methods 15, 591–594. [DOI] [PubMed] [Google Scholar]
- Karczewski Konrad J., L.C.F., Tiao Grace, Cummings Beryl B., Jessica Alföldi Qingbo Wang, Collins Ryan L., Laricchia Kristen M., Ganna Andrea, Birnbaum Daniel P., Gauthier Laura D., Brand Harrison, Solomonson Matthew, Watts Nicholas A., Rhodes Daniel, Moriel Singer-Berk Eleina M. England, Seaby Eleanor G., Kosmicki Jack A., Walters Raymond K., Tashman Katherine, Farjoun Yossi, Banks Eric, Poterba Timothy, Wang Arcturus, Seed Cotton, Whiffin Nicola, Chong Jessica X., Samocha Kaitlin E., Emma Pierce-Hoffman Zachary Zappala, O’Donnell-Luria Anne H., Minikel Eric Vallabh, Weisburd Ben, Lek Monkol, Ware James S., Vittal Christopher, Armean Irina M., Bergelson Louis, Cibulskis Kristian, Connolly Kristen M., Covarrubias Miguel, Donnelly Stacey, Ferriera Steven, Gabriel Stacey, Gentry Jeff, Gupta Namrata, Jeandet Thibault, Kaplan Diane, Llanwarne Christopher, Munshi Ruchi, Novod Sam, Petrillo Nikelle, Roazen David, Valentin Ruano-Rubio Andrea Saltzman, Schleicher Molly, Soto Jose, Tibbetts Kathleen, Tolonen Charlotte, Wade Gordon, Talkowski Michael E., The Genome Aggregation Database Consortium, Neale Benjamin M., Daly Mark J., MacArthur Daniel G.(2019). Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv. [Google Scholar]
- Kramer A, Green J, Pollard J Jr., and Tugendreich S (2014). Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar PA, Hu Y, Yamamoto Y, Hoe NB, Wei TS, Mu D, Sun Y, Joo LS, Dagher R, Zielonka EM, et al. (2011). Distal airway stem cells yield alveoli in vitro and during lung regeneration following H1N1 influenza infection. Cell 147, 525–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapperre TS, Sont JK, van Schadewijk A, Gosman MM, Postma DS, Bajema IM, Timens W, Mauad T, Hiemstra PS, and Group GS (2007). Smoking cessation and bronchial epithelial remodelling in COPD: a cross-sectional study. Respir Res 8, 85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SB, and Kalluri R (2010). Mechanistic connection between inflammation and fibrosis. Kidney Int Suppl, S22–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee-Six H, Obro NF, Shepherd MS, Grossmann S, Dawson K, Belmonte M, Osborne RJ, Huntly BJP, Martincorena I, Anderson E, et al. (2018). Population dynamics of normal human blood inferred from somatic mutations. Nature 561, 473–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GM, Zhang CL, Rui RP, Sun B, and Guo W (2018). Bioinformatics analysis of common differential genes of coronary artery disease and ischemic cardiomyopathy. Eur Rev Med Pharmacol Sci 22, 3553–3569. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J 17, 10–12. [Google Scholar]
- Martinez FD (2016). Early-Life Origins of Chronic Obstructive Pulmonary Disease. N Engl J Med 375, 871–878. [DOI] [PubMed] [Google Scholar]
- Martinez FJ, Han MK, Allinson JP, Barr RG, Boucher RC, Calverley PMA, Celli BR, Christenson SA, Crystal RG, Fageras M, et al. (2018). At the Root: Defining and Halting Progression of Early Chronic Obstructive Pulmonary Disease. Am J Respir Crit Care Med 197, 1540–1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonough JE, Yuan R, Suzuki M, Seyednejad N, Elliott WM, Sanchez PG, Wright AC, Gefter WB, Litzky L, Coxson HO, et al. (2011). Small-airway obstruction and emphysema in chronic obstructive pulmonary disease. N Engl J Med 365, 1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeachie MJ, Yates KP, Zhou X, Guo F, Sternberg AL, Van Natta ML, Wise RA, Szefler SJ, Sharma S, Kho AT, et al. (2016). Patterns of Growth and Decline in Lung Function in Persistent Childhood Asthma. N Engl J Med 374, 1842–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. (2010). The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller M, Cho JY, Pham A, Friedman PJ, Ramsdell J, and Broide DH (2011). Persistent airway inflammation and emphysema progression on CT scan in ex-smokers observed for 4 years. Chest 139, 1380–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy-Ullrich JE, and Suto MJ (2018). Thrombospondin-1 regulation of latent TGF-beta activation: A therapeutic target for fibrotic disease. Matrix Biol 68–69, 28–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambu A, Zach J, Schroeder J, Jin G, Kim SS, Kim YI, Schnell C, Bowler R, and Lynch DA (2016). Quantitative computed tomography measurements to evaluate airway disease in chronic obstructive pulmonary disease: Relationship to physiological measurements, clinical index and visual assessment of airway disease. Eur J Radiol 85, 2144–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestorowa S, Hamey FK, Pijuan Sala B, Diamanti E, Shepherd M, Laurenti E, Wilson NK, Kent DG, and Gottgens B (2016). A single-cell resolution map of mouse hematopoietic stem and progenitor cell differentiation. Blood 128, e20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda K, Chen G, Subramani DB, Wolf M, Gilmore RC, Kato T, Radicioni G, Kesimer M, Chua M, Dang H, et al. (2019). Localization of Secretory Mucins MUC5AC and MUC5B in Normal/Healthy Human Airways. Am J Respir Crit Care Med 199, 715–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordovas-Montanes J, Dwyer DF, Nyquist SK, Buchheit KM, Vukovic M, Deb C, Wadsworth MH, 2nd, Hughes TK, Kazer SW, Yoshimoto E, et al. (2018). Allergic inflammatory memory in human respiratory epithelial progenitor cells. Nature 560, 649–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patro R, Duggal G, Love MI, Irizarry RA, and Kingsford C (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods 14, 417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plantier L, Crestani B, Wert SE, Dehoux M, Zweytick B, Guenther A, and Whitsett JA (2011). Ectopic respiratory epithelial cell differentiation in bronchiolised distal airspaces in idiopathic pulmonary fibrosis. Thorax 66, 651–657. [DOI] [PubMed] [Google Scholar]
- Ponce-Gallegos MA, Ramirez-Venegas A, and Falfan-Valencia R (2017). Th17 profile in COPD exacerbations. Int J Chron Obstruct Pulmon Dis 12, 1857–1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quaderi SA, and Hurst JR (2018). The unmet global burden of COPD. Glob Health Epidemiol Genom 3, e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quint JK, and Wedzicha JA (2007). The neutrophil in chronic obstructive pulmonary disease. J Allergy Clin Immunol 119, 1065–1071. [DOI] [PubMed] [Google Scholar]
- Raju SV, Kim H, Byzek SA, Tang LP, Trombley JE, Jackson P, Rasmussen L, Wells JM, Libby EF, Dohm E, et al. (2016). A ferret model of COPD-related chronic bronchitis. JCI Insight 1, e87536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray S, Chiba N, Yao C, Guan X, McConnell AM, Brockway B, Que L, McQualter JL, and Stripp BR (2016). Rare SOX2(+) Airway Progenitor Cells Generate KRT5(+) Cells that Repopulate Damaged Alveolar Parenchyma following Influenza Virus Infection. Stem Cell Reports 7, 817–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso D, Ngai J, Speed TP, and Dudoit S (2014). Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol 32, 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satija R, Farrell JA, Gennert D, Schier AF, and Regev A (2015). Spatial reconstruction of single-cell gene expression data. Nat Biotechnol 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlon PD, Connett JE, Waller LA, Altose MD, Bailey WC, Buist AS, Tashkin DP, and Lung Health Study Research, G. (2000). Smoking cessation and lung function in mild-to-moderate chronic obstructive pulmonary disease. The Lung Health Study. Am J Respir Crit Care Med 161, 381–390. [DOI] [PubMed] [Google Scholar]
- Schamberger AC, Staab-Weijnitz CA, Mise-Racek N, and Eickelberg O (2015). Cigarette smoke alters primary human bronchial epithelial cell differentiation at the air-liquid interface. Sci Rep 5, 8163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schittny JC (2017). Development of the lung. Cell Tissue Res 367, 427–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider WM, Chevillotte MD, and Rice CM (2014). Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 32, 513–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seibold MA, Smith RW, Urbanek C, Groshong SD, Cosgrove GP, Brown KK, Schwarz MI, Schwartz DA, and Reynolds SD (2013). The idiopathic pulmonary fibrosis honeycomb cyst contains a mucocilary pseudostratified epithelium. PLoS One 8, e58658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shultz LD, Lyons BL, Burzenski LM, Gott B, Chen X, Chaleff S, Kotb M, Gillies SD, King M, Mangada J, et al. (2005). Human lymphoid and myeloid cell development in NOD/LtSz-scid IL2R gamma null mice engrafted with mobilized human hemopoietic stem cells. J Immunol 174, 6477–6489. [DOI] [PubMed] [Google Scholar]
- Shultz LD, Schweitzer PA, Christianson SW, Gott B, Schweitzer IB, Tennent B, McKenna S, Mobraaten L, Rajan TV, Greiner DL, et al. (1995). Multiple defects in innate and adaptive immunologic function in NOD/LtSz-scid mice. J Immunol 154, 180–191. [PubMed] [Google Scholar]
- Singh D, Agusti A, Anzueto A, Barnes PJ, Bourbeau J, Celli BR, Criner GJ, Frith P, Halpin DMG, Han M, et al. (2019). Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease: the GOLD science committee report 2019. Eur Respir J 53. [DOI] [PubMed] [Google Scholar]
- Smirnova NF, Schamberger AC, Nayakanti S, Hatz R, Behr J, and Eickelberg O (2016). Detection and quantification of epithelial progenitor cell populations in human healthy and IPF lungs. Respir Res 17, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Fritz MH, et al. (2015). An integrated map of structural variation in 2,504 human genomes. Nature 526, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Sze MA, Campbell JD, Brothers JF 2nd, Lenburg ME, McDonough JE, Elliott WM, Cooper JD, Spira A, and Hogg JC (2017). The cellular and molecular determinants of emphysematous destruction in COPD. Sci Rep 7, 9562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka Y, Yamaguchi M, Hirai S, Sumi T, Tada M, Saito A, Chiba H, Kojima T, Watanabe A, Takahashi H, et al. (2018). Characterization of distal airway stem-like cells expressing N-terminally truncated p63 and thyroid transcription factor-1 in the human lung. Exp Cell Res 372, 141–149. [DOI] [PubMed] [Google Scholar]
- Tokunaga R, Zhang W, Naseem M, Puccini A, Berger MD, Soni S, McSkane M, Baba H, and Lenz HJ (2018). CXCL9, CXCL10, CXCL11/CXCR3 axis for immune activation - A target for novel cancer therapy. Cancer Treat Rev 63, 40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traves SL, Culpitt SV, Russell RE, Barnes PJ, and Donnelly LE (2002). Increased levels of the chemokines GROalpha and MCP-1 in sputum samples from patients with COPD. Thorax 57, 590–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. (2013). From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43, 11 10 11–11 10 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan AE, Brumwell AN, Xi Y, Gotts JE, Brownfield DG, Treutlein B, Tan K, Tan V, Liu FC, Looney MR, et al. (2015). Lineage-negative progenitors mobilize to regenerate lung epithelium after major injury. Nature 517, 621–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira Braga FA, Kar G, Berg M, Carpaij OA, Polanski K, Simon LM, Brouwer S, Gomes T, Hesse L, Jiang J, et al. (2019). A cellular census of human lungs identifies novel cell states in health and in asthma. Nat Med 25, 1153–1163. [DOI] [PubMed] [Google Scholar]
- Wedzicha JA (2017). Airway Mucins in Chronic Obstructive Pulmonary Disease. N Engl J Med 377, 986–987. [DOI] [PubMed] [Google Scholar]
- Wen Y, Reid DW, Zhang D, Ward C, Wood-Baker R, and Walters EH (2010). Assessment of airway inflammation using sputum, BAL, and endobronchial biopsies in current and ex-smokers with established COPD. Int J Chron Obstruct Pulmon Dis 5, 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, and Donaldson DD (1998). Interleukin-13: central mediator of allergic asthma. Science 282, 2258–2261. [DOI] [PubMed] [Google Scholar]
- Wodehouse T, Demopoulos M, Petty R, Miraki-Moud F, Belhaj A, Husband M, Fulton L, Randive N, Oksche A, Mehta V, et al. (2019). A randomized pilot study to investigate the effect of opioids on immunomarkers using gene expression profiling during surgery. Pain 160, 2691–2698. [DOI] [PubMed] [Google Scholar]
- Xu Y, Mizuno T, Sridharan A, Du Y, Guo M, Tang J, Wikenheiser-Brokamp KA, Perl AT, Funari VA, Gokey JJ, et al. (2016). Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight 1, e90558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, and Wang K (2015). Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat Protoc 10, 1556–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Riccio P, Schotsaert M, Mori M, Lu J, Lee DK, Garcia-Sastre A, Xu J, and Cardoso WV (2018). Spatial-Temporal Lineage Restrictions of Embryonic p63(+) Progenitors Establish Distinct Stem Cell Pools in Adult Airways. Dev Cell 44, 752–761 e754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou-Suckow Z, Duerr J, Hagner M, Agrawal R, and Mall MA (2017). Airway mucus, inflammation and remodeling: emerging links in the pathogenesis of chronic lung diseases. Cell Tissue Res 367, 537–550. [DOI] [PubMed] [Google Scholar]
- Zuo W, Zhang T, Wu DZ, Guan SP, Liew AA, Yamamoto Y, Wang X, Lim SJ, Vincent M, Lessard M, et al. (2015). p63(+)Krt5(+) distal airway stem cells are essential for lung regeneration. Nature 517, 616–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. In vitro and in vivo differentiation of Cluster 1–4 clones; Related to Figure 2. A. H&E staining of histological sections of air-liquid interface (ALI)-differentiated Cluster 1–4 clones. Scale bar, 100 μm. B. Left, Immunohistochemistry of p63 antibody staining (brown) of the distal airway of normal human lung. Scale bar, 500 μm. Right from top, Magnification of indicated terminal bronchiole epithelium region of p63 immunohistochemistry; immunofluorescence micrograph of terminal bronchiolar epithelium stained with DAPI, anti-p63, anti-SCGB1A1, and anti-SFTPB, and immunofluorescence micrograph of terminal bronchiolar epithelium showing anti-p63 and anti-AQP4 staining. Alveo, alveolar epithelia. Scale bar, 100 μm. C-F. Immunofluorescence micrographs of xenograft sections derived from transplants of clones of Clusters 1–4 probed with the indicated antibodies. Scale bar, 100 μm. G. Venn diagrams showing overlap between nonsynonymous SNV events detected from whole genome exome sequencing of clones of Clusters 1–4 from COPD (SNP-13) lung and 202 oncogenes and 231 tumor suppressor genes (detailed in Table S5).
Figure S2. Distribution of metaplasia in Stage 4 COPD lung; Related to Figure 3. A. Immunohistochemistry micrograph of section of distal lung of normal donor showing distribution of p63 staining (brown) in bronchioles and terminal bronchioles. Scale bar, 1,000 μm. Boxed region indicates region presented as inset right top) and corresponding region of serial section examined by p63 and AQP5 immunofluorescence (lower right). Scale bar of insets, 100 μm. B. Immunohistochemistry micrograph of Stage 4 COPD distal lung stained with p63 antibodies (brown). (*) denote regions of submucosal inflammation. Scale bar, 1,000 μm. Boxed regions corresponding to inflammatory SCM, GCM, and SCM are shown at higher magnification below along with immunofluorescence micrographs of similar regions from serial sections stained with antibodies to p63/CXLC8 (iSCM), p63/TRPC6 (GCM), and p63/TRPC6 (SCM). Inset region scale bars, 100 μm.
Figure S3. Clone variant analysis by single cell RNA-seq; Related to Figure 3. A. Differential gene expression determined by scRNAseq of three COPD and three control clone libraries and the corresponding differential gene expression determined by RNAseq of discrete clones of Clusters 1–4 of SPN-13 (detailed in Tables S6, S7). B. tSNE profiles of single cell RNAseq data derived from clone libraries from the individual COPD and control cases. C. Mapping of cluster-specific gene expression markers onto an integrated tSNE profile assembled from three Control and three COPD cases.
Figure S4. Leukocyte transepithelial migration in xenografts; Related to Figure 4. A. Box-Whisker plot representation of Cluster 2–4 FACS proportions in each of 19 COPD and 11 control libraries (P<2.2e-16, Student’s t-test). B.,C. Histological sections through nodules formed four weeks after transplantation of clonogenic cell pools of six representative non-COPD cases (upper) and COPD cases (lower) to immunodeficient mice. Scale bar, 200 μm. D. Box-Whisker plot representation of severe inflammation fraction in each of 19 COPD and 11 control library xenografts (P= 1.2e-09, Student’s t-test).
Figure S5. Inflammatory gene expression by clones of Clusters 1 to 4; Related to Figure 5. A. Top 10 pathways of differentially expressed genes (FDR<0.05) of patient-matched clones representative of Clusters 1–4 using Ingenuity Pathway Analysis (IPA) software. B. Immunofluorescence of Vcam1 (red) in human lung endothelial cells following exposure to media conditioned by clones of Clusters 1–4 compared to IL-13 challenge. Scale bar, 100 μm. C. Expression heatmaps (FDR<0.05) depicting pair-wise gene expression analysis of Clusters 1–4 focused on chemokine, interleukin, and interferon genes.
Figure S6. Fibrosis in xenografts; Related to Figure 6. A., B. Immunofluorescence micrographs of sections of nodules formed four weeks after transplantation of clone libraries from six representative control cases (upper) and COPD cases (lower). Green, E-cad, E-cadherin; red, SMA, anti-alpha smooth muscle actin. Scale bar, 200 μm. C. Immunofluorescence micrographs of E-cadherin (Ecad) and alpha smooth muscle actin (SMA) expression in xenografts of clones of Cluster 3 (left) and Cluster 4 (right) from in vitro passage 5 and passage 25. Scale bar, 100 μm.
Figure S7. Variant clones in normal adult and fetal lung; Related to Figure 7. A. Histology and marker analysis of xenografts of cloned representatives of Clusters 1–4 from adult Control lung (SPN-14). B. Left, Principal component analysis of RNAseq profiles of Clusters 1–4 clones from control lung (SPN-14) and those of Clusters 1–4 of two COPD cases (SPN-7 and SPN-13). Right, Expression heatmap comparing RNAseq profiles of Clusters 1–4 clones of control lung (SPN-14) with those of clones of COPD cases SPN-7 and SPN-13 (detailed in Table S8). C. Histology of pseudoglandular epithelia of 13-wk fetal lung and marker immunofluorescence including antibodies to p63, AQP5, AQP4, α-TUB, and SFTPB. D. Left, Distribution of cluster-specific expression markers on scRNAseq tSNE profile of clone library from 13-wk fetal lung. Right, Pie chart showing distribution of variants in 13-wk fetal library. E. Dot plots of expression for cluster-specific marker genes in the integrated tSNE profiles of lung clones from three adult Controls, three COPD cases, and the 13-wk fetal lung. The size of the dot corresponds to the percentage of cells expressing the feature in each cluster. The color represents the average expression level.
Table S1. Patient-derived clone library information related to Figure 1
Footnotes:
NA: not available
INF: inflammation (leukocytes)
SCM: squamous metaplasia
GCM: goblet cell metaplasia
ABN: abnormality
ns inter pneu: non-specific interstitial pneumonia
sarc: sarcoidosis
CA: cancer
DIPNECH: diffuse idiopathic pulmonary neuroendocrine cell hyperplasia
DLCO: carbon monoxide diffusing capacity
Leukocyte TEM: transepithelial migration
UCONN Health: University of Connecticut Health Center
UTHSC: University of Texas Health Science Center
UIHC: University of Iowa Hospitals & Clinics
Table S2. Clone library and xenograft information related to Figure 2
Footnotes:
SPN: Study Patient Number
NA: not available
INF: inflammation (leukocytes)
SCM: squamous metaplasia
GCM: goblet cell metaplasia
Leukocyte TEM: transepithelial migration
Table S3. Histology validation set information related to Figure 3
Footnotes:
NA: not available
INF: inflammation (leukocytes)
SCM: squamous metaplasia
GCM: goblet cell metaplasia
ABN: abnormalities
*Sections from patients reported in Okuda et al., 2019.
UNC: University of North Carolina at Chapel Hill
UTHSC: The University of Texas Health Science Center at Houston
UIHC: University of Iowa Hospitals & Clinics
Table S5. Exome-seq related to Figure S1G
Table S6. scRNAseq related to Figure S3A
Table S7. RNAseq related to Figure S3A
Table S8. Differential gene expression between Clusters 1–4 clones of control case and COPD cases related to Fig. S7B
Data Availability Statement
The microarray, single-cell RNA-seq, RNA-seq, and exome sequencing datasets generated during this study are available at GEO: GSE118950, SRA: PRJNA514053, and SRA: PRJNA492749. A list of software used in this study can be found in the Key Resources Table.