

Abstract
Adenium obesum (Forssk.) Roem. & Schult. is a promising medicinal plant belonging to the Apocynaceae family. It is a rich source of various phytochemicals such as cardiac glycosides, flavonoids, terpeniods, pregnanes etc. which have different pharmacological properties such as anticancer, antibacterial, acaricidal etc. While previous reports showed the anticancer activity of the aerial parts of the plant extract of A. obesum, the mechanisms of action of its chemical constituents are not known. The present study is aimed at elucidation of plausible mechanisms of anticancer activity of the plant by evaluating the binding interaction of its nine major selected compounds with macromolecular receptors implicated in the initiation and progression of cancer using various in silico approaches. Molecular docking results showed that the compound Δ16-3-Acetyldigitoxigenin (16-anhydro-3-acetylgitoxigenin) scored the best binding energy scores with the majority of the target proteins. The molecular binding of the compound was stabilized through hydrogen bonds as well as hydrophobic interactions, and also possesses favorable drug-like properties without significant toxicities.
Keywords: Molecular docking, Anticancer properties, Bioactive compounds, Desert rose, Adenium obesum, Apocynaceae, Macromolecular receptors, Binding affinity
1. Introduction
Every year, approximately 6.7 million deaths take place around the world due to various types of cancer (ACS, 2020). A wide array of cytotoxic agents and radiotherapy used in the cancer treatment have limitations in their usage e.g. side effects, and efficacy (Kim et al., 2007, Stopeck and Thompson, 2012); therefore, the development of better effective therapeutics for the cancer treatment from natural products remain continues because of its minimal side effects (Da Rocha et al., 2001, Vermani and Garg, 2002, Gurib-Fakim, 2006).
Although Adenium obesum (Forssk.) Roem. & Schult. (family Apocynaceae, commonly known as ‘Desert Rose’) is primarily an ornamental poisonous plant, the whole plant including latex has been used in traditional system of medicines for the treatment of various aliments e.g. skin lumps, wound, ear ache, rhinitis, gonorrhea and infectious diseases. It contains nearly 50 major chemicals constituents belonging to the class of cardenolides, flavonoids, pregnanes and triterpenes (Versiani et al., 2014). The recent reports (Almehdar et al., 2012, Hossain et al., 2017, Ali et al., 2019a, Ali et al., 2019b) established the anticancer activity of extract of aerial part of A. obesum but the modes of action of the chemical constituents have not been understood; therefore, the aim of the current study is to elucidate the plausible molecular mechanisms underlying the anticancer activity of A. obesum extract using in silico approaches.
2. Materials and methods
2.1. Preparation of ligand and receptor
The structures of nine major compounds of A. obesum (Table 1) were modeled using Chemsketch. The three-dimensional structures of selected macromolecular receptors e.g. (i) CDK-2 [PDB ID: 1DI8], (ii) CDK-6 [PDB ID: 1XO2], (iii-iv) Topoisomerases-I [PDB ID: 1T8I] and II [PDB ID: 1ZXM], (v) BCL-2 [PDB ID: 2O2F], (vi) VEGFR-2 [PDB ID: 2OH4], and (vii) Telomere: G-quadruplex [1L1H] were retrieved from Protein Data Bank (PDB). The 3-D structures of ligands optimized with MMFF94 force field (Halgren, 1996), and the receptors prepared following our previously described method (Gurung et al., 2016) were used to execute docking. The binding sites were defined by choosing grid boxes of suitable dimensions around the bound co-crystal ligands.
Table 1.
The major compounds derived from A. obesum selected for molecular docking.
| Compounds | Name | Structure | Class | Parts | References |
|---|---|---|---|---|---|
| 1 | Δ16-3-Acetyldigitoxigenin (16-anhydro-3-acetylgitoxigenin) | 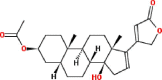 |
Cardiac glycosides | Stem | Vethaviyasar and John (1982) |
| 2 | 12β-Hydroxypregna-4,6,16-triene-3,20-dione (neridienoneA) | 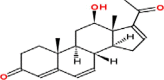 |
Pregnanes | Stem, roots, leaves | Yamauchi and Abe (1990) Nakamura et al. (2000) |
| 3 | 12β-Hydroxypregna-4,6-diene-3,20-dione (16,17-dihydroneridienone A) |
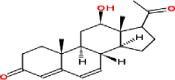 |
Pregnanes | Stem, roots, leaves | Yamauchi and Abe (1990) Nakamura et al. (2000) |
| 4 | 12β-Hydroxpregna-4,16-diene-3,20-dione | 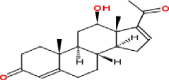 |
Pregnanes | Leaves | Nakamura et al. (2000) |
| 5 | 12β-Hydroxypregn-4-ene-3,20-dione | 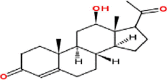 |
Pregnanes | Leaves | Nakamura et al. (2000) |
| 6 | Dihydroifflaionic acid | 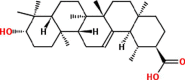 |
Triterpenoids | Aerial | Hoffmann and Cole (1977) |
| 7 | Lup-20(29)-ene-3,28-diol (betulin) | 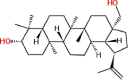 |
Triterpenoids | Stem bark | Tijjani et al. (2012) |
| 8 | Quercetin 3,3′-dimethyl ether | 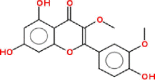 |
Flavonoids | Aerial | Hoffmann and Cole (1977) |
| 9 | Kaempferol 3-methyl ether | 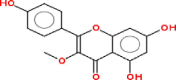 |
Flavonoids | Aerial | Hoffmann and Cole (1977) |
2.2. Molecular docking
The Lamarckian Genetic Algorithm was used for performing molecular docking using AutoDock4.2 (Morris et al., 2009) considering the docking parameters from our previously described method (Gurung et al., 2016). A total number of 50 independent docking runs were performed for each ligand. The conformations were grouped under clusters by considering a difference of less than 2.0 Å of root mean square deviation (RMSD). The lowest free energy of binding (ΔG) and the lowest inhibition constant (Ki) were considered for choosing the most favorable binding pose. The molecular interactions between the compounds and receptors were studied using LigPlot + v 1.4.5 (Laskowski and Swindells, 2011).
2.3. Validation of docking method
In order to check the suitability of molecular docking parameters and algorithm to reproduce the native binding poses, redocking experiment was performed using the co-crystal ligands.
2.4. Determination of physicochemical properties of the compounds
DataWarrior program version 4.6.1 was used for the determination of various physicochemical properties of the selected compounds such as drug likeness and toxicity (Sander et al., 2015).
3. Results and discussion
The three dimensional structure of nine major compounds [Δ16-3-Acetyldigitoxigenin (16-anhydro-3-acetylgitoxigenin), 12β-Hydroxypregna-4,6,16-triene-3,20-dione (neridienoneA), 12β-Hydroxypregna-4,6-diene-3,20-dione (16,17-dihydroneridienone A), 12β-Hydroxpregna-4,16-diene-3,20-dione, 12β-Hydroxypregn-4-ene-3,20-dione, Dihydroifflaionic acid, Lup-20(29)-ene-3,28-diol (betulin), Quercetin 3,3′-dimethyl ether, Kaempferol 3-methyl ether] from A. obesum were modeled and optimized. The optimized structures were used further for molecular docking studies (Table 1). Before performing molecular docking studies, we validated the docking protocol and algorithm through redocking experiment. In all the cases, the root mean square deviation (RMSD) between the docked and native co-crystal position were found to be less than 2 Å. This indicates that the docking protocols, and parameters employed in the present study can reliably predict the native conformations of the compounds (Januar et al., 2012).
The results of molecular docking are shown in Table 2. It is evident that compound 1 [(Δ16-3-Acetyldigitoxigenin (16-anhydro-3-acetylgitoxigenin)] was best docked to CDK-2 with ΔG of −11.39 kcal/mol and Ki of 4.50 nM, which is significantly lower than the co-crystal ligand having ΔG of −8.04 kcal/mol and Ki of 1270 nM. LigPlot + results as shown in Fig. 1 indicates that the compound 1 was able to establish four hydrogen bonds through Lys33, Leu83 and Lys89. Further, this binding was strengthened by hydrophobic interactions with Ile10, Val18, Ala31, Val64, Phe80, Glu81, Phe82, His84, Gln85, Asp86, Leu134, Ala144, Asp145 and Leu148.
Table 2.
The binding energies and inhibition constants of selected compounds derived from A. obesum docked against molecular targets.
| Compounds | Drug targets (PDB Entries) |
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CDK-2 (1DI8) |
CDK-6 (1XO2) |
Topoisomerase-II (1ZXM) |
BCL-2 (2O2F) |
VEGFR-2 (2OH4) |
Telomere: G-quadruplex (1L1H) |
Topoisomerase-I (1T8I) |
||||||||
| BE (kcal/mol) | Ki(nM) | BE (kcal/mol) | Ki(nM) | BE (kcal/mol) | Ki(nM) | BE (kcal/mol) | Ki(nM) | BE (kcal/mol) | Ki(nM) | BE (kcal/mol) | Ki(nM) | BE (kcal/mol) | Ki(nM) | |
| 1 | −11.39 | 4.50 | −8.93 | 282.69 | −11.45 | 4.03 | −8.47 | 618.23 | −10.17 | 35.14 | −5.61 | 77,660 | −10.94 | 9.49 |
| 2 | −10.10 | 39.54 | −10.11 | 39.13 | −9.05 | 231.69 | −8.23 | 923.49 | −9.74 | 73.07 | −7.55 | 2940 | −9.13 | 201.63 |
| 3 | −10.01 | 46.25 | −9.99 | 47.72 | −8.96 | 268.59 | −8.23 | 933.55 | −9.21 | 177.53 | −7.30 | 4470 | −9.34 | 143.28 |
| 4 | −10.06 | 42.53 | −9.96 | 49.96 | −8.98 | 262.49 | −8.24 | 905.38 | −8.73 | 401.36 | −7.38 | 3920 | −9.81 | 64.68 |
| 5 | −9.98 | 48.73 | −10.03 | 44.34 | −8.84 | 333.04 | −8.37 | 736.14 | −9.29 | 154.49 | −7.43 | 3570 | −9.69 | 78.71 |
| 6 | −11.20 | 6.20 | −6.02 | 38,380 | −7.95 | 1490 | −8.55 | 540.37 | −8.41 | 679.17 | −6.26 | 25,950 | −9.82 | 63.21 |
| 7 | −10.30 | 28.15 | −6.43 | 19,240 | −9.40 | 129.67 | −8.73 | 400.37 | −7.97 | 1430 | −7.35 | 4100 | −9.65 | 85.11 |
| 8 | −8.30 | 819.79 | −9.20 | 181.34 | −7.42 | 3640 | −5.95 | 43.29 | −9.11 | 211.74 | −8.92 | 287.67 | −9.03 | 240.57 |
| 9 | −7.74 | 2110 | −9.05 | 232.35 | −7.20 | 5280 | −6.05 | 36,840 | −8.69 | 426.31 | −8.46 | 628.78 | −8.61 | 487.63 |
| Co-crystal ligand | −8.04 | 1270 | −8.26 | 882.71 | −11.11 | 7.24 | −11.01 | 8.56 | −12.46 | 0.738 | −11.97 | 1.68 | −10.75 | 13.23 |
Fig. 1.

The binding modes and LigPlot + results for receptor-ligand interactions. The molecular interaction between: CDK-2 and Δ16-3-Acetyldigitoxigenin (16-anhydro-3-acetylgitoxigenin) (A); CDK-6 and 12β-Hydroxypregna-4,6,16-triene-3,20-dione (neridienoneA) (B); Topoisomerase-II and Δ16-3-Acetyldigitoxigenin (16-anhydro-3-acetylgitoxigenin) (C); BCL-2 and Lup-20(29)-ene-3,28-diol (betulin) (D); VEGFR-2 and Δ16-3-Acetyldigitoxigenin (16-anhydro-3-acetylgitoxigenin) (E); Telomere: G-quadruplex and Quercetin 3,3′-dimethyl ether (F); Topoisomerase-I and Δ16-3-Acetyldigitoxigenin (16-anhydro-3-acetylgitoxigenin) (G). The hydrogen bonds are represented by green dashed lines with the bond distance. The residues contributing to the hydrophobic interactions are indicated with red arcs with spikes.
The compound 2 [12β-Hydroxypregna-4,6,16-triene-3,20-dione (neridienoneA)] was best docked to CDK-6 with a binding energy of −10.11 kcal/mol and Ki of 39.13 nM, which is much lower than the co-crystal ligand having binding energy −8.26 kcal/mol and Ki of 882.71 nM. It formed three hydrogen bonds with Lys43, His100, Val101 and residues involved in hydrophobic interactions include Ile19, Asp102, Gln103, Asp104, Gln149, Leu152, Ala162 and Asp163. Again, compound 1 [(Δ16-3-Acetyldigitoxigenin (16-anhydro-3-acetylgitoxigenin)] was found to be best docked to Topoisomerase-II with a binding energy of −11.45 and Ki of 4.03 nM which is slightly lower than the co-crystal ligand having a binding energy of −11.11 kcal/mol and Ki of 7.24 nM. It formed five hydrogen bonds with Arg98, Asn120, Asn163, Gly164 and Lys378 and hydrophobic interaction with residues Glu87, Asn91, Asn95, Lys123, Ile125, Ile141, Phe142, Ser148, Asn150, Gly161, Arg162, Tyr165, Gly166, Ala167, Thr215 and Gln376.
The best docked compound for BCL-2 was found to be compound 7 [Lup-20(29)-ene-3,28-diol (betulin)] with a binding energy of −8.73 kcal/mol and Ki of 400.37 nM which was found to higher than the co-crystal ligand with a binding energy of −11.01 kcal/mol and Ki of 8.56 nM. It was able to establish only one hydrogen bond with Tyr105 and hydrophobic interactions with residues Phe101, Asp108, Phe109, Met112, Leu134, Gly142, Arg143 and Ala146. Compound 1 [(Δ16-3-Acetyldigitoxigenin (16-anhydro-3-acetylgitoxigenin)] was also best docked to VEGFR-2 with a binding energy of −10.17 kcal/mol and Ki of 35.14 nM which was found to be higher than the co-crystal ligand with binding energy of −12.46 kcal/mol and Ki of 0.738 nM. It showed good interaction with VEGFR-2 through one hydrogen bond with residue Glu915 and hydrophobic interaction via residues Leu838, Ala864, Glu883, Val897, Val914, Phe916, Cys917, Gly920, Leu1033, Cys1043, Asp1044 and Phe1045.
The best docked ligand for Telomere:G-quadruplex was compound 8 [Quercetin 3,3′-dimethyl ether] with a binding energy of −8.92 kcal/mol and Ki of 287.67 nM which was significantly higher than the co-crystal ligand with a binding energy of −11.97 kcal/mol and Ki of 1.68 nM. Compound 8 [Quercetin 3,3′-dimethyl ether] formed two hydrogen bonds with bases DT1006 and DG1009 and hydrophobic interactions via bases Dt1007, Dt1008, Dg2001 and Dg2012. Compound 1 [(Δ16-3-Acetyldigitoxigenin (16-anhydro-3-acetylgitoxigenin)] is also best docked to Topoisomerase-I with a binding energy of −10.94 kcal/mol and Ki of 9.49 nM which is slightly lower than the co-crystal ligand with a binding energy of −10.75 kcal/mol and Ki of 13.23 nM. It formed three hydrogen bonds with Asn419, Ile420, Met428 and hydrophobic interactions via residues Asn352, Glu356, Lys374, Arg375, Trp416, Glu418, Lys425, Tyr426 and Ile427.
The physicochemical properties of the docked compounds are tabulated in Table 3. The majority of the compounds obeyed the Lipinski’s rule of five (ROF) (Lipinski, 2004) except for compounds 6 [Dihydroifflaionic acid] and 7 [Lup-20(29)-ene-3,28-diol (betulin)] which showed one violation as their cLogP values were higher than the permissible limits. Compounds 1 [Δ16-3-Acetyldigitoxigenin (16-anhydro-3-acetylgitoxigenin)], 6 [Dihydroifflaionic acid], 7 [Lup-20(29)-ene-3,28-diol (betulin)], 8 [Quercetin 3,3′-dimethyl ether] and 9 [Kaempferol 3-methyl ether] were found to be non-mutagenic, non-tumorigenic, non-irritant and without any adverse effects on reproductive health. Compounds 2 [12β-Hydroxypregna-4,6,16-triene-3,20-dione (neridienoneA)], 3 [12β-Hydroxypregna-4,6-diene-3,20-dione (16,17-dihydroneridienone A)], 4 [12β-Hydroxpregna-4,16-diene-3,20-dione] and 5 [12β-Hydroxypregn-4-ene-3,20-dione] also showed similar results except for their possible toxicity on reproductive health. The majority of the compounds showed a good drug likeness score except for compounds 1 [Δ16-3-Acetyldigitoxigenin (16-anhydro-3-acetylgitoxigenin)], 6 [Dihydroifflaionic acid] and 7 [Lup-20(29)-ene-3,28-diol (betulin)] which exhibited negative drug likeness score. The other physicochemical properties such as topological surface area (TPSA) and number of rotatable bonds (RB) were also found to be within permissible limits (TPSA ≤ 140 Å2 and RB ≤ 10).
Table 3.
The physicochemical properties of the compounds derived from A. obesum [- (none), h (high)].
| Compounds | Mol weight | cLogP | cLogS | HBA | HBD | TPSA | Drug likeness | Mutagenic | Tumorigenic | Reproductive Effective | Irritancy | Rotatable Bonds |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 414.54 | 3.3025 | −4.414 | 5 | 1 | 72.83 | −0.27262 | – | – | – | – | 3 |
| 2 | 326.434 | 2.787 | −3.651 | 3 | 1 | 54.37 | 1.9744 | – | – | h | – | 1 |
| 3 | 328.45 | 2.8915 | −3.915 | 3 | 1 | 54.37 | 1.9131 | – | – | h | – | 1 |
| 4 | 328.45 | 3.0624 | −3.879 | 3 | 1 | 54.37 | 1.9189 | – | h | h | – | 1 |
| 5 | 330.466 | 3.1669 | −4.143 | 3 | 1 | 54.37 | 1.8595 | – | – | h | – | 1 |
| 6 | 456.708 | 6.0021 | −6.111 | 3 | 2 | 57.53 | −2.3517 | – | – | – | – | 1 |
| 7 | 442.725 | 6.7202 | −6.296 | 2 | 2 | 40.46 | −23.933 | – | – | – | – | 2 |
| 8 | 331.299 | 0.8411 | −2.164 | 7 | 3 | 113.29 | 1.5726 | – | – | – | – | 3 |
| 9 | 301.273 | 0.9111 | −2.146 | 6 | 3 | 104.06 | 1.5726 | – | – | – | – | 2 |
Thus, the present molecular docking studies revealed structural insights into possible binding modes of major active compounds of A. obesum, and identified the best docked compound for each target. The compound 1 (16-anhydro-3-acetylgitoxigenin) was found to be best docked (showed a high binding affinity, good number of hydrogen bonds and hydrophobic interactions with their respective molecular targets which play a key role in the pathogenesis of cancer) to four targets CDK-2, Topoisomerase-II, VEGFR-2 and Topoisomerase-I whereas Compound 2 (12β-Hydroxypregna-4,6,16-triene-3,20-dione), Compound 7 (Lup-20(29)-ene-3,28-diol) and Compound 8 (Quercetin 3,3′-dimethyl ether) were found to be best docked to CDK-6, BCL-2 and Telomere:G-quadruplex respectively with favorable drug-like properties; and thus, these compounds can be promising leads for the design of specific target inhibitors which would help with management of the disease.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
The authors would like to extend their sincere appreciation to the Deanship of Scientific Research at King Saud University for its funding of the research through the research group project #RG-1438-015. J. Lee thanks to Chungnam National University, Daejeon, Republic of Korea for the funding support. The authors thank the Deanship of Scientific Research and RSSU at King Saud University for their technical support.
Footnotes
Peer review under responsibility of King Saud University.
Contributor Information
Arun Bahadur Gurung, Email: arunbgurung@gmail.com.
Joongku Lee, Email: joongku@cnu.ac.kr.
References
- Ali A.Q., Abou-Tarboush F.M., Al-Anazi K.M., Ali M.A., Al-Hemaid F.M., Hailan W.A.Q., Mahmoud A.H., Abul Farah M. Cytogenotoxic effects of Adenium obesum seeds extract on breast cancer cells. Saudi J. Biol. Sci. 2019;26:547–553. doi: 10.1016/j.sjbs.2018.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Cancer Society . American Cancer Society; Atlanta: 2020. Cancer Facts & Figures 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A.Q., Farah M.A., Abou-Tarboush F.M., Al-Anazi K.M., Ali M.A., Lee J., Hailan W.A.Q., Mahmoud A.H. Cytogenotoxic effects of Adenium obesum seeds extracts on breast cancer cells. Saudi J. Biol. Sci. 2019;26:547–553. doi: 10.1016/j.sjbs.2018.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almehdar H., Abdallah H.M., Osman A.-M.M., Abdel-Sattar E.A. In vitro cytotoxic screening of selected Saudi medicinal plants. J. Nat. Med. 2012;66:406–412. doi: 10.1007/s11418-011-0589-8. [DOI] [PubMed] [Google Scholar]
- Da Rocha A.B., Lopes R.M., Schwartsmann G. Natural products in anticancer therapy. Curr. Opin. Pharmacol. 2001;1:364–369. doi: 10.1016/s1471-4892(01)00063-7. [DOI] [PubMed] [Google Scholar]
- Gurib-Fakim A. Medicinal plants: traditions of yesterday and drugs of tomorrow. Mol. Aspects Med. 2006;27:1–93. doi: 10.1016/j.mam.2005.07.008. [DOI] [PubMed] [Google Scholar]
- Gurung A.B., Ali M.A., Bhattacharjee A., AbulFarah M., Al-Hemaid F., Abou-Tarboush F.M., Al-Anazi K.M., Al-Anazi F.S.M., Lee J. Molecular docking of the anticancer bioactive compound proceraside with macromolecules involved in the cell cycle and DNA replication. Genet. Mol. Res. 2016;15 doi: 10.4238/gmr.15027829. [DOI] [PubMed] [Google Scholar]
- Halgren T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996;17:490–519. doi: 10.1002/(SICI)1096-987X(199604)17:5/6<490::AID-JCC1>3.0.CO;2-P. [DOI] [Google Scholar]
- Hossain M.A., Akhtar M.S., Said S., Al-Abri T.H.A. Two new flavonoids from Adenium obesum grown in Oman. J. King Saud Univ. 2017;29:62–69. [Google Scholar]
- Januar H.I., Dewi A.S., Marraskuranto E., Wikanta T. In silico study of fucoxanthin as a tumor cytotoxic agent. J. Pharm. Bioallied Sci. 2012;4:56. doi: 10.4103/0975-7406.92733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.-W., Hong G.-H., Lee H.-H., Choi S.-H., Chun B.-G., Won C.-K., Hwang I.K., Won M.H. Effect of colloidal silver against the cytotoxicity of hydrogen peroxide and naphthazarin on primary cultured cortical astrocytes. Int. J. Neurosci. 2007;117:387–400. doi: 10.1080/00207450600592016. [DOI] [PubMed] [Google Scholar]
- Laskowski R.A., Swindells M.B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. J. Chem. Inf. Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- Lipinski C.A. Lead- and drug-like compounds: the rule-of-five revolution. Drug Discov. Today. Technol. 2004;1:337–341. doi: 10.1016/j.ddtec.2004.11.007. [DOI] [PubMed] [Google Scholar]
- Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander T., Freyss J., von Korff M., Rufener C. DataWarrior: an open-source program for chemistry aware data visualization and analysis. J. Chem. Inf. Model. 2015;55:460–473. doi: 10.1021/ci500588j. [DOI] [PubMed] [Google Scholar]
- Stopeck, A.T., Thompson, P.A., 2012. Breast cancer treatment and anagement. WebMD Heal. Prof. Network) Retrieved Sept. 29, 2012.
- Vermani K., Garg S. Herbal medicines for sexually transmitted diseases and AIDS. J. Ethnopharmacol. 2002;80:49–66. doi: 10.1016/s0378-8741(02)00009-0. [DOI] [PubMed] [Google Scholar]
- Versiani M.A., Ahmed S.K., Ikram A., Ali S.T., Yasmeen K., Faizi S. Chemical constituents and biological activities of Adenium obesum (Forsk.) Roem. et Schult. Chem. Biodivers. 2014;11:171. doi: 10.1002/cbdv.201200254. [DOI] [PubMed] [Google Scholar]
Further Reading
- Pullaiah T. Daya Books; 2006. Encyclopaedia of World Medicinal Plants. [Google Scholar]