

Age-related ventricular dilatation is considered a consequence of tissue atrophy, distinct from the ventriculomegaly seen in idiopathic normal pressure hydrocephalus. Aso et al. show that an age-related venous perfusion timing shift may be another factor driving ventricular enlargement in both normal ageing and pathology.
Keywords: brain ageing, normal pressure hydrocephalus, traumatic brain injury, venous drainage, ventricular enlargement
Abstract
Recently, age-related timing dissociation between the superficial and deep venous systems has been observed; this was particularly pronounced in patients with normal pressure hydrocephalus, suggesting a common mechanism of ventriculomegaly. Establishing the relationship between venous drainage and ventricular enlargement would be clinically relevant and could provide insight into the mechanisms underlying brain ageing. To investigate a possible link between venous drainage and ventriculomegaly in both normal ageing and pathological conditions, we compared 225 healthy subjects (137 males and 88 females) and 71 traumatic brain injury patients of varying ages (53 males and 18 females) using MRI-based volumetry and a novel perfusion-timing analysis. Volumetry, focusing on the CSF space, revealed that the sulcal space and ventricular size presented different lifespan profiles with age; the latter presented a quadratic, rather than linear, pattern of increase. The venous timing shift slightly preceded this change, supporting a role for venous drainage in ventriculomegaly. In traumatic brain injury, a small but significant disease effect, similar to idiopathic normal pressure hydrocephalus, was found in venous timing, but it tended to decrease with age at injury, suggesting an overlapping mechanism with normal ageing. Structural bias due to, or a direct causative role of ventriculomegaly was unlikely to play a dominant role, because of the low correlation between venous timing and ventricular size after adjustment for age in both patients and controls. Since post-traumatic hydrocephalus can be asymptomatic and occasionally overlooked, the observation suggested a link between venous drainage and CSF accumulation. Thus, hydrocephalus, involving venous insufficiency, may be a part of normal ageing, can be detected non-invasively, and is potentially treatable. Further investigation into the clinical application of this new marker of venous function is therefore warranted.
Introduction
A common consequence of senescence is shrinkage of brain tissue overall, which has led to the use of the term ‘age norm’ when diagnosing brain atrophy (Dubois et al., 2007). Considered to be a part of this phenomenon, age-related ventricular dilatation per se does not represent a disease condition (de Leon et al., 1987; Fjell et al., 2009). Nevertheless, prominent ventriculomegaly reflects pathological states, such as Alzheimer’s disease (Apostolova et al., 2012) and idiopathic normal pressure hydrocephalus (iNPH), both of which are prevalent among the elderly. Two major mechanisms have been postulated to underlie pathological ventricular dilatation: accelerated brain atrophy due to cell degeneration, and abnormal CSF accumulation. While brain atrophy is a common feature of neurodegenerative diseases, iNPH is thought to involve abnormal CSF absorption, although the exact mechanism is unknown. Purely age-dependent ventricular enlargement has been associated almost solely with parenchymal atrophy, due to neuronal loss (Apostolova et al., 2012), but the influence of CSF accumulation has not been well studied.
Recently, we reported that changes in cerebral venous drainage pattern commonly occur in normal ageing processes and iNPH (Satow et al., 2017), based on our findings with a novel blood-tracking technique that uses intrinsic deoxyhaemoglobin fluctuation as a virtual tracer. This technique allows broad measurements of perfusion timing, covering the venous aspect of the vasculature (Amemiya et al., 2013; Lv et al., 2013; Tong et al., 2017; Nishida et al., 2019). The analysis involves measuring the phase, or lag, in the spontaneous low-frequency oscillation (sLFO) (Tong et al., 2012, 2019) of the blood oxygenation level-dependent (BOLD) signal, which varies across brain parts. This sLFO originates from the systemic circulatory fluctuation and, therefore, primarily belongs to the noise component in functional MRI that is diminished by stringent data-cleaning procedures (Aso et al., 2017). We previously observed a relative perfusion timing shift with healthy ageing in the anterior periventricular area, a major territory of the deep venous system. This venous timing shift was further pronounced in iNPH and reversed by the diagnostic spinal tap test. These findings suggested a pathological change underlying iNPH, which represents exaggerated ageing, emerging as a venous timing dissociation between the superficial and deep venous systems. There are various explanations for iNPH pathology (Ringstad et al., 2017), and venous insufficiency has been considered as a candidate precipitating factor (Bateman, 2008; Beggs, 2013). However, the causality between the observed venous timing shift and iNPH development remains unclear. While venous insufficiency can affect CSF absorption, CSF drainage relieves ventriculomegaly in advanced iNPH, ameliorating impaired blood flow as well as symptoms (Virhammar et al., 2014). Further details of these age-associated phenomena may provide insight into the causal relationships and underlying physiology.
In this follow-up cross-sectional study with a larger cohort of healthy subjects (n = 225, age range 21–89 years), we investigated the relationship between venous perfusion timing and volumetric parameters. Along with ventricular size, the subarachnoid sulcal space was evaluated as an index of tissue atrophy. Both between- and within-age variabilities were compared. To explore the relationship of age-related effects with pathological ventriculomegaly, we further included patients with traumatic brain injury (TBI, n = 71, age range: 12–70 years), a condition presenting with both brain atrophy and ventricular enlargement, across a wide range of ages. Because TBI is an acquired condition that is highly independent of age and predisposition, its study may allow dissection of the multiple possible mechanisms of ventricular enlargement. The large control dataset was used to estimate and control for the effect of age, to explore the interactions between normal ageing and injury, and thereby provide insight into the general relationships between the venous timing shift and ventricular enlargement.
Materials and methods
Participants
The healthy control participants were paid volunteers without a history of neurological illness. After excluding seven subjects with excessive head motion (maximum framewise displacement >1.2 mm), 225 healthy control individuals were included in the study (n = 225, age range: 21–89 years, 137 males and 88 females).
Sixteen patients with TBI were excluded after data acquisition because of head motion. Table 1 shows the demographic and clinical information of the included TBI patients (n = 71). Severity was defined using the Glasgow Coma Scale or the Japan Coma Scale converted into the former (Namiki, 2007). Patients were recruited from those referred to the neuropsychology unit at the Department of Psychiatry and at the Department of Neurosurgery, Kyoto University Hospital. Inclusion criteria were as follows: (i) TBI sustained through significant trauma at least 3 months before the enrolment; and (ii) acute imaging findings of head trauma, including microhaemorrhage in the deep white matter or peri-brainstem haemorrhage. Exclusion criteria were a history of (i) another TBI; (ii) drug abuse; or (iii) neurological or psychiatric disorders before TBI onset. The diagnostic criterion for patients with diffuse axonal injury (DAI, n = 20) was absence of gross parenchymal lesions (e.g. lobar contusions, and haematoma/haemorrhage >2 mm diameter) on MRI. Fluid-attenuated inversion recovery (FLAIR) imaging was used to assess DAI in all patients. In 18 DAI patients who underwent susceptibility-weighted imaging, we classified the haemorrhagic lesions using the extended anatomical grading for DAI (Abu Hamdeh et al., 2017). Written informed consent was obtained from all participants, in accordance with the Declaration of Helsinki and the following protocols were approved by the Institutional Review Board of Kyoto University Hospital.
Table 1.
Demographic and clinical characteristics of participants
| Characteristic | TBI (n = 71) | TBI focal lesion (n = 51) | TBI DAI (n = 20) |
|---|---|---|---|
| Age | 40.9 (15.6) [12–70] | 40.7 (16.5) [12–70] | 41.7 (13.3) [21–63] |
| Male sex | 48 (68%) | 34 (67%) | 14 (70%) |
| Cause of injury | Traffic accident 63, fall 5, other 3 | Traffic accident 46, fall 3, other 2 | Traffic accident 17, fall 2, other 1 |
| Time from injury, months | 91.5 (91.7) [3–418] | 88.2 (88.8) [3–418] | 99.8 (100.61) [6–372] |
| Severity | Severe 38 (54%), moderate 11 (15%), mild 20 (28%), NA 2 (3%) | Severe 22 (43%), moderate 10 (20%), mild 17 (33%), NA 2 (4%) | Severe 16 (80%), moderate 1 (5%), mild 3 (15%) |
| Lesion location | – | Frontal 38, other 13 | – |
| Anatomical grading for DAI | – | – | Grade I: 3, Grade II:5 Grade III: 3, Grade IV: 3, Grade 0: 4, NA: 2 |
| WAIS-III | |||
| FIQ | 93.4 (18.6) | 96.1 (17.4) | 89.3 (20.9) |
| VIQ | 95.0 (18.3) | 97.3 (18.2) | 91.5 (19.1) |
| PIQ | 93.2 (16.3) | 95.3 (14.2) | 90.0 (19.6) |
| VC | 94.5 (17.9) | 97.6 (16.1) | 89.8 (20.3) |
| PO | 98.1 (14.1) | 99.4 (12.1) | 96.1 (17.4) |
| WM | 93.3 (17.1) | 96.9 (18.3) | 87.8 (14.4) |
| PS | 79.6 (20.5) | 86.0 (18.6) | 69.9 (20.4) |
Results are expressed as means (SD) [range], unless otherwise stated.
Anatomical grading for DAI: Grade I, hemispheric lesions; Grade II, hemispheric and additional corpus callosum lesions; Grade III, brainstem lesions; Grade IV, lesions in substantia nigra or mesencephalic tegmentum. Grade 0 indicates absence of haemorrhagic spots in chronic images but with evidence of microbleeds in acute X-ray computed tomography.
FIQ = full intelligence quotient; NA = not available; PIQ = performance intelligence quotient; PO = perceptual organization; PS = processing speed; SD = standard deviation; VC = verbal comprehension; VIQ = verbal intelligence quotient; WAIS-III = Wechsler Adult Intelligence Scale-Third Edition; WM = working memory.
Brain MRI
Images were acquired on a Siemens 3 T Trio Tim system with a 32-channel head-array coil. Resting state BOLD-MRI was acquired using standard functional MRI based on the gradient-echo, echo-planar imaging technique. The imaging parameters were as follows: repetition time = 2500 ms; slice thickness = 3.2 mm with a 25% gap; number of slices = 40; voxel resolution 3.3 × 3.3 × 3.2 mm3; flip angle = 80°. A 10-min scan (242 volumes) was acquired. Subjects were instructed to lie still with their eyes open. High-resolution T1-weighted images were acquired with the following parameters: repetition time = 2.3 s; echo time = 2.94 ms; flip angle = 9°; resolution = 256 × 256 × 160 mm3. A magnetic field map derived from the dual-echo gradient-echo dataset was also acquired to undistort the BOLD images using FUGUE/FSL.
The BOLD-MRI and T1 anatomical data were preprocessed using SPM12 (http://www.fil.ion.ucl.ac.uk/spm) and the FMRIB Software Library (www.fmrib.ox.ac.uk/fsl) with in-house MATLAB scripts for an automated pipeline. After slice-timing and motion corrections, B0 distortion correction was applied using FUGUE/FSL (Jenkinson et al., 2012). The head motion information obtained here was used to reject those participants with excessive motion by framewise displacement. The undistorted images were co-registered to the subject’s T1 image for normalization to the template space using the same deformation field. The normalized functional images were downsampled to 4 × 4 × 4 mm3 voxels to achieve a high temporal signal-to-noise ratio. At this stage, images were checked for motion-related signal deflection or dropout (Mazaika et al., 2009; Power et al., 2012). This procedure involved searching for time points satisfying two stringent criteria: (i) a global mean signal change between consecutive volumes exceeding 1%; and (ii) head displacement exceeding a Euclidian distance of ±1 mm per repetition time. The affected time points were replaced by linearly interpolated values. The data were then cleaned by multiple regression using head motion parameters, but the six rigid-body parameters were not directly included, because of the possible contamination of the global signal change when the subjects were stable (Freire and Mangin, 2001). The regression involved 24 parameters consisting of the first temporal derivatives of the motion parameters, their versions shifted by one repetition time, and squares of those 12 time series (Satterthwaite et al., 2013).
Each participants’ T1 anatomical image was first spatially normalized to the Montreal Neurological Institute (MNI) reference space by unified segmentation (Ashburner and Friston, 2005). The segmented images of all healthy control participants in the original space were fed into the Diffeomorphic Anatomical Registration using Exponentiated Lie algebra (DARTEL) pipeline for improved registration (Ashburner and Friston, 2005). The co-registered individual images were finally aligned to the MNI template again, using the deformation field for the average image. DARTEL was not used for patients with TBI because (i) no voxel-wise analyses were performed on the dataset; and (ii) the regions of interest for the venous clusters were valid in detecting the treatment effect in iNPH, despite the more pronounced ventriculomegaly (Satow et al., 2017).
Volumetric analysis
The 3D T1-weighted image was segmented into grey matter, white matter and CSF prior to the DARTEL spatial normalization in each individual. To measure the lateral ventricular volume in the native space of individual scans, a ventricular mask in the reference MNI space was transformed using the inverse deformation field for each subject. This ventricular mask template was created from the normalized CSF masks thresholded at 0.5 (in probability of a voxel belonging to CSF) and combined across subjects to cover the lateral ventricles in all subjects. For measurements of whole brain volumes, summed images of the grey and white matter masks were thresholded at 0.5 before counting the voxels. The sulcal space was defined by masking the CSF mask with a smoothed version of this parenchymal mask in order to exclude the subdural part of the CSF (see ‘Volumetric analyses and changes throughout the lifespan’ section). All volumes were obtained in cubic millimetres and their cubic root-represented size (equivalent to diameter) was indicated. Finally, relative sulcal space and ventricle and parenchymal sizes were calculated by dividing the values by the sum of these three metrics, which represented the total subarachnoid volume.
Additional analyses on grey and white matter volumes were performed with or without normalization by the total subarachnoid volume in order to rule out possible structural bias in venous timing measurement.
Perfusion lag-mapping
We used the most common approach for BOLD lag-mapping based on fixed seeds (Amemiya et al., 2013; Christen et al., 2015), but used an additional process for extracting the sLFO, or the lag = 0 signal, from the global mean signal (Aso et al., 2017; Satow et al., 2017). After image preprocessing, temporal band-pass filtering with a low passband (0.008–0.07 Hz) was applied to ensure that the phase was uniquely determined within the cross-correlation range of 14 s. The filtered time courses in each voxel were then resampled to a 0.5-s sampling interval. Figure 1A illustrates the steps in the lag-mapping procedure. The lag map assumed discrete values between −7 s and +7 s at a 0.5-s interval, in which positive values indicated phase advance, meaning that they represented the ‘arrival time’ to the global signal phase (Aso et al., 2017). There were voxels with no cross-correlogram peak or a negative peak correlation coefficient, suggesting ambiguity or tracking failure. These ‘holes’ were filled by 3D eroding and linear interpolation.
Figure 1.

Age-related changes in the perfusion lag map. (A) Steps in the BOLD perfusion lag-mapping procedure. Once the sLFO was determined by averaging the time series from voxels with lag = 0, it was used as the reference signal to measure the time shift for each brain voxel by the cross-correlogram peak. This final part was performed by creating a 3D time × space (voxel) × time-shift matrix and finding the maxima along the third dimension. (B) Average lag maps overlaid on the average anatomical T1 images (inverted contrast) for the young and the elderly group. Negative values (cool colours) indicate late phases relative to the global sLFO representing the centre of the vascular tree. Regions with age-related phase shifts are indicated by arrows. (C) Schematic of the present analytical model of the BOLD lag structure. Greyscale shadings in the vascular tree represent the intrinsic source of slow signal fluctuation. The right panel illustrates the relationship with conventional temporal metrics based on different reference points. (D) Regions presenting linear correlations with age. Positive shifts are indicated by warm colours. Overlaid are t-score maps from the voxel-wise multiple regression with a degree of freedom of 222 (n = 225; age and sex as independent variables; voxel-wise false discovery rate-corrected P < 0.05). These clusters were used as the deep/superficial venous regions-of-interest in the following analyses. Results with a more stringent threshold are presented in Supplementary Fig. 1.
Voxel-wise multiple regression for age-related changes in perfusion lag map
The lag maps were resliced to 2-mm isotropic voxels before statistical analysis. SPM12 was used for voxel-wise multiple linear regression including age (continuous) and sex (categorical) in 225 healthy subjects. To define region of interest for the subsequent analysis, the resulting t-score maps were thresholded at P = 0.05, with multiple comparison correction by the false discovery rate to map age-related changes towards up- and downstream. We used an attenuated threshold for region of interest definition, even though significant clusters were detected at a more stringent threshold of P = 0.05 (correcting for multiple comparisons using the family-wise error rate; see Supplementary Fig. 1), because we aimed to explore non-linear effects by allowing for deviations from first-order linear effects.
Arterial and venous transit time
For each individual lag map, a whole-brain voxel histogram was created to identify the peak phase, or lag, that should represent the centre of the vascular tree, which was close to zero in all cases. Using all voxels with lag values indicating upstream locations (i.e. larger phase advance) relative to the histogram peak, the arterial transit time was calculated by simply averaging the phase in seconds. Venous transit time was obtained in a similar manner using the downstream voxels, but converted to positive values to represent the temporal distance from the centre of the vascular tree. The sum of these two values was named global transit time. This is essentially different from the mean transit time based on PET or X-ray CT perfusion images and calculated for each voxel, but may carry information equivalent to the global average of those metrics, as a measure of blood flow velocity.
Statistical analysis
All individual metrics of interest from the volumetric and perfusion timing analyses were converted to corresponding W-scores, a covariate-adjusted Z-score relative to the normal control (Yau et al., 2015). Therefore, and to compare the lifespan profiles, healthy control datapoints were fitted to a quadratic model according to age, by using a linear least-squares algorithm in the MATLAB Curve Fitting Toolbox. The goodness-of-fit was expressed in R squares and corresponding P-values. Sex was not included as a factor because of the non-uniform distribution of male and female subjects and the sample size. To confirm the effect of sex in the healthy control dataset, ANCOVA was performed controlling for the full quadratic effect of age. Pearson’s correlation coefficient (r) was used to explore the relationships among the volumetric and perfusion markers as well as the effect of age on the venous timing change in TBI. We performed multiple linear regression including age, sex, DAI diagnosis, and time-after-injury on the W-score data of the TBI group. Deviations of the patients’ W scores from the age norm (W = 0) were tested using one-sample t-tests. Statistical significance was accepted at the 0.01 level (two-tailed).
Data availability
MATLAB codes for the perfusion lag-mapping can be downloaded at https://github.com/RIKEN-BCIL/BOLDLagMapping. Data supporting the findings of this study are available from the corresponding author upon request.
Results
Age-related changes in the perfusion lag map
Average lag maps created from two healthy control age groups, composed of either young (<50 years of age) or elderly subjects (50 years or older), showed a similar spatial distribution consistent with the gross vascular anatomy (Fig. 1B). The lag maps identified upstream positions in the vascular tree as areas with positive phase advance compared to the global sLFO phase (Fig. 1C). The lag, expressed in seconds, represents temporal distance, or travelling time to and from the centre of the vasculature. Importantly, this measurement cannot be directly compared with conventional techniques with different reference points, due to the variation in vascular path lengths (Jespersen and Østergaard, 2012; Tong and Frederick, 2012) (Fig. 1C, right). For example, the arrival time of contrast agents (or labelled blood in arterial spin labelling) from the artery is not directly inferred from the perfusion time lag. This flexibility of reference points in the vascular tree in turn allowed observation of the venous aspect of cerebral circulation (Tong et al., 2017).
The average maps displayed shrinkage of the anterior periventricular cluster in elderly subjects, based on an upstream phase shift. This perfusion timing shift may reflect prolongation of venous drainage time or shortening of path length, or both. The difference was evident in the anterior and inferior horns of the lateral ventricle and their nearby structures (arrows in Fig. 1B), but not in the dorsal and posterior parts of the ventricles that are outside of the deep venous system territory.
Age-related changes were formally located by voxel-wise linear regression (Fig. 1D). Sex was also included as a regressor, but no voxels presented significant effects at the same threshold. As expected, upstream age-related timing shifts were observed in adjacent structures of the deep venous system, whereas the opposite changes were predominantly located at the tentorial sinus, extending to the sinus confluence. The differential effects of age on the superficial and deep venous systems were thus reproduced in a larger sample (Satow et al., 2017). Region of interest analyses further revealed that the phase shifts followed quadratic patterns, despite the bias introduced by the choice of the linear regressor (Fig. 2A and B). In young participants, the deep cluster presented a delay (indicated by a negative phase advance) to the global mean, reflecting its position in the gross vasculature; thus diminished with age. An opposite trend was found in the superficial cluster. This venous drainage change was summarized by the phase difference between the two clusters, which fit better (Fig. 2C).
Figure 2.

Age-related changes in venous flow timing and volumetry. r2 indicates the R square of the quadratic fitting. (A and B) Individual phase advance (in seconds, positive values indicating upstream) from the clusters in Fig. 1C plotted against age. Shaded area: 95% confidence interval of the fitted curve. Dashed lines: 95% confidence interval estimated for the population. (C) Difference between the two clusters. (D–F) Cubic root of the sulcal space and the ventricular and parenchymal volumes. (G) Sum of these three measures representing the subarachnoid space. This value was used to calculate relative sulcal and ventricular sizes (H and I). (J) Temporal profiles from C, H, and I, normalized for comparison between 0 and 1. The first temporal derivatives are overlaid as dashed lines. (K and L) Temporal profiles of venous timing and ventricular size fitted separately for male and female subjects.
Volumetric analyses and changes throughout the lifespan
Volumetric measures from the 3D anatomical image confirmed significant age effects, replicating previous observations (Bigler et al., 2002; Fotenos et al., 2005) (Fig. 2D–F). The measurements were converted to the cubic-root equivalent of the volume to homogenize the variances for comparison. The increase in the sulcal space size showed a linear pattern, whereas the decrease in parenchymal size and the increase in ventricular size presented quadratic profiles (Fotenos et al., 2005). After correcting each parameter by dividing it by the total subarachnoid space (Fig. 2G), both CSF spaces presented an improved fit (Fig. 2H and I). For normalization, we used the total subarachnoid space, instead of the intracranial volume as a measure. This approach was motivated primarily by the possible confounding effect of an increase in subdural space with age (Fogelholm et al., 1975), which accompanies so-called subdural effusion (Liao and Xiao, 2017), in order to focus on the parenchymal shape changes. Additionally, some elderly cases demonstrated unstable image segmentation in the CSF-dura mater boundary, possibly reflecting structural changes within the dura (Haines et al., 1993).
Different temporal profiles of the venous flow timing shift and the two volumetric markers were evident in normalized time courses (Fig. 2J). The sulcal space change was compatible with earlier reports of tissue atrophy starting in the mid-20s (Pfefferbaum et al., 1994). In contrast, ventricular enlargement started slowly and exceeded the rate of sulcal enlargement at around the age of 50 years, slightly later than the venous timing shift. Figure 2K and L plot these two markers separately for male and female subjects. The effect of sex was not significant in either the venous shift [F(1,222) = 0.057, P = 0.81, ANCOVA with sex × quadratic age as a covariate; non-significant interaction P > 0.1] or the sex × ventricular size [F(1,222) = 1.6, P = 0.20; interaction P > 0.1]. Relatively poor R-square fitting was noted in males for each of the ageing markers, suggesting higher interindividual variability in males. Nevertheless, the slight temporal precedence of the venous timing change over the ventricular enlargement was preserved in both sexes.
Three biomarkers in patients with TBI
The 71 TBI patients, some of whom had been diagnosed with DAI (n = 21), presented deviations from the healthy controls in all markers, including the venous timing shift (Fig. 3A). All non-DAI patients with TBI (n = 51) had visible focal parenchymal lesions mainly in the frontal and temporal lobes (Supplementary Fig. 2A). To isolate the disease effects, data were age-adjusted using the control dataset (age-adjusted Z, or W-score). As depicted in Fig. 3B and Supplementary Fig. 2B–D, all markers showed significant injury effects, in line with earlier reports of accelerated ageing in TBI (Cole et al., 2015; Irimia et al., 2018). Moreover, there was a significant effect of age at injury on the venous timing shift, resulting in a diminishing deviation with age (Fig. 3B, Pearson’s r = −0.427; P = 0.0008; n = 71, multiple linear regression including age, sex, DAI diagnosis, and time after injury). This finding was replicated after dividing the patients into a DAI and a focal TBI group, despite the tissue defect in the latter (Table 2). No significant effect of DAI diagnosis was found for the other three volumetric markers (multiple linear regression including age, sex, DAI diagnosis, and time after injury in 71 patients), except for marginally larger ventricles in the DAI group [F(1,64) = 3.3, P = 0.073]. The linear trend in TBI’s venous state was also independent of clinical severity according to the subgroup analysis (Pearson’s r = −0.454 and P = 0.004 in severe TBI only; n = 38). Such an interaction with age was not detected in the markers for proportional brain atrophy (Supplementary Fig. 2). However, there was another age-at-injury effect in the absolute total subarachnoid space that was used to normalize the CSF space measures (Fig. 3C, r = 0.515; P = 0.000004), which originated from absolute parenchymal loss (Supplementary Fig. 3G; r = 0.504; P = 0.000008), in which a young age was correlated with increased atrophy. This finding was preserved within the severe TBI subgroup, again indicating the absence of bias from severity (Pearson’s r = −0.550 and P = 0.001; n = 38). Because the time after injury was <9 years in our patient group (median and maximum, 54 and 418 weeks) and correlated poorly with age (Pearson’s r = 0.050; variance inflation factor = 1.047 from the multiple linear regression), these interactions are likely related to the age at injury per se.
Figure 3.

Age-adjusted values of the venous timing shift and volumetry. Healthy controls (HC) = dots; TBI = open circles; DAI = filled circles. (A) Perfusion timing difference between the deep and superficial clusters from the patients with TBI (n = 71). The healthy control values presented in Fig. 2C are underlaid for comparison (n = 225). Inset: Average anatomical image of the TBI group after spatial normalization presented in the same slice locations used in Fig. 1A, indicating sufficient alignment for the region of interest analysis. Note that the volumetric measurements were made in the native subject space. (B) Values after age adjustment revealed a significant effect of injury relative to the healthy controls at baseline (P = 0.00022, n = 71, one-sample t-test). In addition, there was an effect of age at injury indicated by the regression line (red, n = 71). Pearson’s correlation coefficient is shown at the top with the corresponding P-value. (C) Interaction between disease effect and age was also found in the absolute total subarachnoid volume, suggesting another overlapping mechanism between disease and normal ageing. (D) Relationship between sulcal space and ventricular size. Correlation coefficients for the TBI and healthy controls groups are displayed (top and bottom, respectively) with a corresponding regression line for the TBI group. (E–J) Relationship with venous timing shift is shown for absolute total subarachnoid volume (E), sulcal space size (F), ventricular size (G), parenchymal size (H), absolute grey matter (I) and white matter (J) volumes after age adjustment. Correlation coefficients for the TBI (top, pink) and healthy controls (bottom, white) groups are displayed. *Statistically significant at P < 0.001, uncorrected for multiple comparisons.
Table 2.
Correlations with age at injury
| All TBI (n = 71) |
DAI (n = 20) |
TBI focal (n = 51) |
||||
|---|---|---|---|---|---|---|
| r | P | r | P | r | P | |
| Deep superficial lag | −0.427 | 0.0002 | −0.498 | 0.025 | −0.417 | 0.0023 |
| Sulcal space size | −0.143 | 0.234 | −0.036 | 0.881 | −0.175 | 0.220 |
| Ventricular size | 0.111 | 0.355 | −0.070 | 0.768 | 0.159 | 0.264 |
| Parenchymal size | −0.041 | 0.734 | −0.233 | 0.322 | 0.010 | 0.942 |
Perfusion timing differences between the deep and superficial venous regions and the three volumetric measures are compared. Adjustments were made for present age using healthy control data, with corresponding P-values from the univariate Pearson’s correlation. Volumes are converted to the cubic root equivalent and corrected for total subarachnoid volume. Patients with TBI who did not meet the criteria for DAI were classified TBI focal. There was a correlation between the venous timing abnormality and age at injury. Values in bold indicate significant correlations.
Ventricular size and sulcal space were significantly correlated even after age adjustment, possibly reflecting the shared effect of tissue atrophy (Fig. 3C). The pronounced correlation in TBI compared to healthy controls further supports this interpretation. In contrast, there were only weak, non-significant correlations between the venous timing shift and any of the volume metrics, including the total subarachnoid, and grey and white matter volumes in TBI (P > 0.01, Pearson’s correlation, Fig. 3D–J). We conducted another multiple regression to examine how the age-corrected ventricular size in TBI is accounted for by a combination of atrophy (i.e. sulcal space size) and venous state (Supplementary Fig. 2E). This analysis was expected to reveal the relative contributions of these two on ventriculomegaly. The effect of atrophy on ventricular size was significant, while the effect of venous timing shift was weak (P = 0.072). In the healthy control group, the correlation was also weak, but significant, with ventricular size possibly reflecting a causal link between venous drainage and ventriculomegaly (Fig. 3G).
To compare the present findings with previous reports, grey and white matter were analysed (Supplementary Fig. 3). Supplementary Fig. 3A and D show the normal lifespan profiles of these structures, which were similar to a previous report (Fotenos et al., 2005). There was significant white matter reduction in TBI patients (P < 10−10, Supplementary Fig. 3E), again confirming previous reports, while the grey matter volume was within the normal range (P = 0.36, Supplementary Fig. 3B) (Hukkelhoven et al., 2003). However, there was an unexpected trend in grey matter of TBI that young and elderly patients display opposite changes, indicating another interaction, but in the absence of disease effect. Bias from clinical severity was not evident, again, because the trend was still found after excluding the mild/moderate cases (Pearson’s r = −0.662 and P = 0.00001; n = 38). Neither of these two metrics were correlated with venous timing shift (Supplementary Fig. 3C and F). Sum of grey and white matter, or parenchymal volume, also presented a trend but with significant disease effect (P = 0.000008), which was similar to the findings for the total subarachnoid space (Fig. 3C). As our main analysis of CSF spaces involved correction by the total subarachnoid space, corrected grey and white matter values were also investigated (Supplementary Fig. 3I–L). The white matter reduction had a significant effect on the total volume, making it difficult to judge the implication of the proportionally increased grey matter volume (P < 10−12, Supplementary Fig. 3I and J). Nonetheless, we failed to find any additional age-at-injury effect in TBI.
Blood transit times estimated from the lag map
The arterial and venous transit times were calculated from individual lag maps, as temporal distance averaged over voxels from/to the centre of the vascular tree (Fig. 4). The sum of these two parameters, that is, the global transit time, is a measure of voxel histogram width and is different from the mean transit time defined for each voxel based on other modalities. It nevertheless yielded a similar range of values, with an age-related prolongation consistent with earlier studies (Fujishima and Omae, 1980; Mihara et al., 2003). The highest correlation coefficient was found for venous transit time, indicating an age-related prolongation of venous drainage time.
Figure 4.
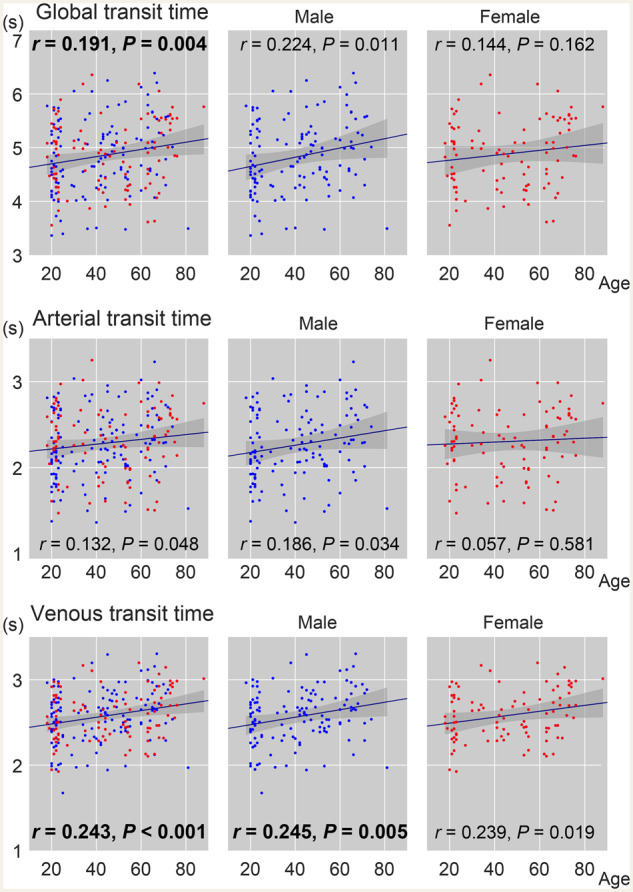
Age-related changes in transit time measures from the lag map. The global transit time is the sum of its arterial and venous components. Red dots indicate females (n = 88/225 in total). Pearson’s correlation coefficient with age is inserted with the corresponding P-value (P < 0.01 in bold).
Discussion
The present data supported the view that there are at least two mechanisms underlying ventricular enlargement in general, one accompanying a change in the venous drainage pattern, and the other most probably related to primary neuronal loss. Independent contributions of these two mechanisms are suggested by the weak correlations between the venous timing shift and morphological changes, in addition to the different lifespan profiles. Such a dissociation in temporal profiles has been reported in grey and white matter volume changes with age, presumably due to the ventricular enlargement that selectively affects the latter (Fotenos et al., 2005). For the timing shift between the superficial and deep venous systems, an interaction between the TBI effect and age at injury suggests a shared mechanism with the pure effect of age. This observation is generally compatible with the long-standing concept that TBI accelerates ageing (Corkin et al., 1989; Lou et al., 2018); however, venous drainage change has never been considered to be a factor generally promoting ventriculomegaly, and thus merits further investigation. It is noteworthy that we failed to find an effect of DAI in TBI patients in our main analyses, possibly because 92% of the TBI with focal lesions were caused by traffic accidents in which DAI pathology might coexist. The sample size may also have been too small to capture a reduced effect. Focal structural changes, including callosal atrophy, may have affected the region of interest analysis (Supplementary Fig. 2). However, we also failed to find a strong correlation suggesting structural bias in the venous timing shift. Accordingly, we discuss TBI patients as one group below.
Age-related changes found in the venous timing shift and in the ventricular size showed similar quadratic patterns, with the former presenting a slight temporal precedence. However, the difference between the fitted curves was too small to support a direction of causality. It is possible that the latter confounded voxel-wise analysis, thereby creating a spurious correlation; however, this account is unlikely, for the following reasons. First, as previously argued (Satow et al., 2017), age-related atrophy should exist in other brain–CSF boundaries, such as cortical surfaces, but these did not present similar changes in this study. Moreover, our finding was confined to the vicinity of the deep venous structures, instead of randomly occurring around the ventricles. Second, ventricular enlargement should primarily result in expansion, rather than shrinkage, of the periventricular structure. Finally, after removing the age effect, there was only a weak correlation between any of the volumetric parameters, particularly ventricular size, and the venous timing shift. This was more evident for TBI patients with prominent ventriculomegaly, further ruling out such a methodological bias. Importantly, the lack of a quantitative correlation also suggested that any physiological link between these two phenomena is not based on a temporally immediate effect.
The perfusion lag-mapping technique was initially proposed as a non-invasive alternative for evaluating arterial diseases (Lv et al., 2013; Christen et al., 2015; Nishida et al., 2019). Theoretically, it uses the slow variation in deoxyhaemoglobin concentration as a virtual contrast agent (Aso et al., 2019). Arterial spin labelling, another non-invasive method, has a limited tracking range, determined by the longitudinal relaxation time of the blood (<2 s), and is hence incapable of tracking the venous flow under normal conditions (Le et al., 2012). On the other hand, lag mapping has been confirmed to address this aspect of the vasculature (Tong et al., 2012; Satow et al., 2017), although a standardized interpretation is yet to be established because of the lack of validation techniques for postcapillary flow timing (Santini et al., 2008). The lack of clear vascular territories in venous drainage further complicates the interpretation. Because the cerebral veins have no valves, flow patterns are flexible (Andeweg, 1996). We indeed observed such dynamic changes within a few days in patients with iNPH, before and after the spinal tap test (Satow et al., 2017), consistent with the known immediate treatment effect, which is possibly related to cerebral blood flow recovery (Keong et al., 2016). However, there are at least two possible explanations for the change in the lag map commonly found in TBI, iNPH, and normal ageing. A phase shift towards zero in the perfusion lag map can logically represent either (i) an upward shift in the vascular tree; or (ii) shortening of the path length with unchanged drainage time. Strictly, path length also belongs to the time domain and is therefore influenced by flow velocity.
The former scenario involves earlier blood arrival, but extended drainage time, hence indicating deep venous insufficiency. This scenario may seem compatible with theories on venous collagenosis playing a role in age-related white matter pathology (Moody et al., 1995). However, the observed changes were distributed along the ventricular walls that should normally be downstream to the medullary veins and, hence, small vessel disease would not explain the change. Downstream to the current cluster is the vein of Galen, a single vessel draining the whole deep system. Outflow from this vein depends on its distinct and variable structure (Dagain et al., 2008) such as its junction with the straight sinus. This structure has been associated with disease conditions (Bateman, 2004; Beggs, 2013). However, this is in contradiction with a series of studies on NPH reporting normal straight sinus flow (Bateman, 2008). Age-related changes were not observed either, although the small sample size of earlier studies might not allow a comparison with the current study or generalized conclusions (Bateman et al., 2008). The second scenario does not face these discrepancies, because it assumes constant drainage time, but requires further reduction of the blood arrival time with age. Although this might seem counter-intuitive, it is possible that the perfusion timing shift that we observed is merely a relative phenomenon, forming part of broader changes. A body of evidence indicates decreased internal jugular vein flow in both iNPH (Kuriyama et al., 2008) and TBI (Pomschar et al., 2013) as well as in normal ageing (Chung et al., 2010). This internal jugular vein insufficiency is thought to be compensated by the venous plexuses that are drained by the cervical and external jugular veins (De Vis et al., 2018). If there is increased resistance or pressure in the dominating superficial system (Portnoy et al., 1994; Bateman, 2003; Kuriyama et al., 2011), there may be a downward shift of the whole lag structure, leaving the deep system at a relatively early phase. The deep venous system reportedly contributes only around 20% of the internal jugular vein flow and is therefore unlikely to cause such a widespread change (De Vis et al., 2018). Our observation of prolonged venous transit time with age is consistent with this view. In the case of iNPH, the deep venous system is spared and the spinal tap test may ameliorate the superficial venous congestion. Indeed, tight high convexity has been reported to predict the response to treatment (Narita et al., 2016), implying a mechanical effect on sagittal sinus flow (Kuriyama et al., 2011). Importantly, the treatment effect does not necessarily indicate a primary role for CSF accumulation; disease development may involve a vicious circle of events (Keong et al., 2016; Bräutigam et al., 2019). Figure 5 illustrates the possible causal links based on the above discussion. The mechanism underlying age-related venous drainage change remains to be clarified; although a causal relationship between the venous timing shift and the ventricular enlargement would be of major clinical relevance.
Figure 5.

Schematic illustration of the causal links suggested by the present study’s data. Insufficiency of the superficial system of the cerebral vein is assumed to be the primary event. CSF accumulation and reduced internal jugular outflow (purple background) have been reported, while blue background indicates interpretation of the results of the present study.
Our previous study suggested a shared mechanism that makes iNPH a disease of the elderly (Satow et al., 2017). In fact, there is no established histological finding for post-mortem diagnosis of iNPH, supporting this view (Relkin et al., 2005). The present observations further indicate a unified model that applies to both normal ageing and communicating hydrocephalus. One plausible mechanism would be a slowly accumulating effect of venous dysfunction on ventriculomegaly, which is in accordance with the low correlation between the present venous state and ventricular size. An opposite causal direction is unlikely; as observed in the response to iNPH treatment, the ventricular enlargement itself is highly irreversible (Virhammar et al., 2014). Nevertheless, the lag map was markedly changed by the spinal tap test, indicating that the venous flow is independent of the absolute ventricular size (Satow et al., 2017). In iNPH, since the classic model posited the unique anatomy of the deep venous system as a factor in CSF accumulation, venous involvement has been a popular concept (Andeweg, 1991; Portnoy et al., 1994). Many models have been proposed, involving abnormal CSF absorption, tissue compliance, and the recent glymphatic hypotheses (Bateman, 2003; Bateman and Siddique, 2014; Ringstad et al., 2017), but most of these are not mutually exclusive and may share factors. Although confirmation by longitudinal studies is necessary, the perfusion lag map may not only represent a disease biomarker, but may also help to elucidate the underlying mechanism.
Because none of the patients with TBI had been diagnosed with secondary NPH, the venous timing shift itself in the patient group was an unexpected finding. Even in the case of iNPH, the discrepancy between clinical and radiological manifestations is prominent (Iseki et al., 2014). Accordingly, parenchymal atrophy in TBI may further confound the diagnosis of secondary NPH. This has already led to the suggestion that post-traumatic hydrocephalus is underdiagnosed (Kammersgaard et al., 2013), as was supported by the observation that ventricular size does not immediately reflect CSF dynamics. It is hence undeniable that some patients with TBI have (or had) subclinical NPH pathology, which could potentially benefit from early detection and treatment. Moreover, we found an age-related diminution of the perfusion shift in TBI, suggesting a shared underlying process between TBI and normal ageing. However, this does not account for the similar interaction found in total subarachnoid and the parenchymal volumes, suggesting another overlapping mechanism, namely cell degeneration (although cautious interpretation is needed; as discussed below). Lack of clear correlation between venous timing shift and volume losses suggests that these interactions are separable. The mechanism of ventricular enlargement involving venous insufficiency is thus likely to be independent of neuronal loss.
Measurement of the age-related venous timing dissociation is non-invasive and hence repeatable, and may thus be useful if proven as an ageing/hydrocephalus marker. It shares the same limitation as functional MRI, with image distortion or signal dropout due to susceptibility effects of haemosiderin, metal implants, and air/tissue interfaces that typically affects the orbitofrontal cortices and brain base. This type of signal loss was indeed present in our TBI patients, but the effect was spatially limited and was not as large as the tissue loss caused by focal lesions (Supplementary Fig. 2). Moreover, the effect of the signal loss is constant over time and is therefore not expected to affect the lag-mapping procedure that yields temporal information. The most problematic factor was head motion during scanning; for this reason, we used a strict inclusion criterion based on framewise displacement.
While our main CSF volumetric analysis involved normalization by total subarachnoid space, which was supported by a slight improvement of the fitting, grey and white matter volumes were not easy to interpret because they directly affect the total volume. In contrast to grey matter, white matter atrophy is less evident in ageing and is barely accelerated by Alzheimer’s disease (Thompson et al., 2003; Fotenos et al., 2005). One explanation could be that the tissue loss can occur in the form of histological changes, such as perivascular space enlargement (Roher et al., 2003), which can escape detection by volumetry. As these structures are tightly attached to each other, they cannot change shape independently. In this respect, sulcal space could be an independent marker of total tissue loss. Another unresolved issue regarding grey matter/white matter parcellation is the interaction between age at injury and disease effect in absolute grey matter volume. In our study, the increased volume in the elderly TBI patients, as compared to healthy control participants, was completely unexpected. It is more likely that there was a pre-existing bias mediated by survival rate; indeed brain atrophy has been linked to a higher incidence of subdural bleeding (Adhiyaman et al., 2002). Nevertheless, this unresolved issue may have affected the parenchymal and total subarachnoid volume measurements, requiring a cautious interpretation. It is also important to note that there was no significant correlation with venous timing dissociation, supporting an independent origin.
Among non-perfusion-based markers, one candidate phenomenon connecting the communicating hydrocephalus with ageing is iron deposition. As has been stressed in the TBI studies, haemorrhage, whether intraventricular or not, is a major predictive factor of hydrocephalus. Iron deposition is not only a mediating factor of post-haemorrhagic hydrocephalus, but also a recent focus of brain ageing research (Ashraf et al., 2018). Another change in the brain commonly found in these conditions is decreased tissue stiffness, a relatively new biomarker that stems from magnetic resonance elastography (Sack et al., 2009). This phenomenon, consistently found in the periventricular regions in ageing, TBI, and iNPH, is particularly interesting in relation to the lag map change in the present data. Taking advantage of the non-invasive nature of BOLD-based lag-mapping, prospective and longitudinal assessments in a large cohort may provide insight into this phenomenon. In defining the disease entity of NPH, Adams and others (1965) described it as ‘symptomatic occult hydrocephalus’. Current efforts to prevent secondary hydrocephalus may benefit from the lag-mapping technique, and the definition of a broader spectrum of ‘occult’ hydrocephalic processes might allow detection and treatment in future.
Supplementary Material
Acknowledgement
We would like to thank Editage (www.editage.jp) for English language editing.
Funding
This study was supported by Grants-in-Aid for Scientific Research on Innovative Areas (JP15H05875, JP16H06395, and JP16H06397) and a Grant-in-Aid for Scientific Research C (19K09480) from the Japan Society for the Promotion of Science (JSPS) and the Takeda Science Foundation awarded to T.A. This work was also supported by an Industrial Disease Clinical Research Grant (150502-02) awarded to T.M. and a Health Labor Sciences Research Grant, a Grant-in-Aid for C (17K10327) from the JSPS, and a research grant from the General Insurance Association of Japan awarded to K.U. and Brain/MINDS-beyond from the Japan Agency for Medical Research and Development (AMED) (Grant Number JP19dm0307003, JP19dm0307004, JP19dm0307006, JP19dm0307102, and JP19dm0107089).
Competing interests
The authors report no competing interests.
Glossary
- BOLD =
blood-oxygenation level-dependent
- DAI =
diffuse axonal injury
- iNPH =
idiopathic normal pressure hydrocephalus
- sLFO =
spontaneous low-frequency oscillation
- TBI =
traumatic brain injury
References
- Abu Hamdeh S, Marklund N, Lannsjö M, Howells T, Raininko R, Wikström J, et al. Extended anatomical grading in diffuse axonal injury using MRI: hemorrhagic lesions in the substantia nigra and mesencephalic tegmentum indicate poor long-term outcome. J Neurotrauma 2017; 34: 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams RD, Fisher CM, Hakim S, Ojemann RG, Sweet WH.. Symptomatic occult hydrocephalus with normal cerebrospinal-fluid pressure. N Engl J Med 1965; 273: 117–126. [DOI] [PubMed] [Google Scholar]
- Adhiyaman V, Asghar M, Ganeshram KN, Bhowmick BK.. Chronic subdural haematoma in the elderly. Postgrad Med J 2002; 78: 71–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amemiya S, Kunimatsu A, Saito N, Ohtomo K.. Cerebral hemodynamic impairment: assessment with resting-state functional MR imaging. Radiology 2013; 270: 1–8. [DOI] [PubMed] [Google Scholar]
- Andeweg J. Concepts of cerebral venous drainage and the aetiology of hydrocephalus. J Neurol Neurosurg Psychiatry 1991; 54: 830–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andeweg J. The anatomy of collateral venous flow from the brain and its value in aetiological interpretation of intracranial pathology. Neuroradiology 1996; 38: 621–628. [DOI] [PubMed] [Google Scholar]
- Apostolova LG, Green AE, Babakchanian S, Hwang KS, Chou Y-Y, Toga AW, et al. Hippocampal atrophy and ventricular enlargement in normal aging, mild cognitive impairment and Alzheimer’s disease. Alzheimer Dis Assoc Disord 2012; 26: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner J, Friston KJ.. Unified segmentation. Neuroimage 2005; 26: 839–51. [DOI] [PubMed] [Google Scholar]
- Ashraf A, Clark M, So P-W. The Aging of Iron Man. Front Aging Neurosci 2018; 10: 65. [DOI] [PMC free article] [PubMed]
- Aso T, Jiang G, Urayama S, Fukuyama H.. A resilient, non-neuronal source of the spatiotemporal lag structure detected by bold signal-based blood flow tracking. Front Neurosci 2017; 11: 256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aso T, Urayama S, Fukuyama H, Murai T.. Axial variation of deoxyhemoglobin density as a source of the low-frequency time lag structure in blood oxygenation level-dependent signals. PLoS One 2019; 14: e0222787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman GA. The reversibility of reduced cortical vein compliance in normal-pressure hydrocephalus following shunt insertion. Neuroradiology 2003; 45: 65–70. [DOI] [PubMed] [Google Scholar]
- Bateman GA. Pulse wave encephalopathy: a spectrum hypothesis incorporating Alzheimer’s disease, vascular dementia and normal pressure hydrocephalus. Med Hypotheses 2004; 62: 182–187. [DOI] [PubMed] [Google Scholar]
- Bateman GA. The pathophysiology of idiopathic normal pressure hydrocephalus: cerebral ischemia or altered venous hemodynamics? Am J Neuroradiol 2008; 29: 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman GA, Levi CR, Schofield P, Wang Y, Lovett EC.. The venous manifestations of pulse wave encephalopathy: Windkessel dysfunction in normal aging and senile dementia. Neuroradiology 2008; 50: 491–497. [DOI] [PubMed] [Google Scholar]
- Bateman GA, Siddique SH.. Cerebrospinal fluid absorption block at the vertex in chronic hydrocephalus: obstructed arachnoid granulations or elevated venous pressure? Fluids Barriers CNS 2014; 11: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beggs CB. Venous hemodynamics in neurological disorders: an analytical review with hydrodynamic analysis. BMC Med 2013; 11:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigler ED.Anderson CV, Blatter DD, Andersob CV. Temporal lobe morphology in normal aging and traumatic brain injury. AJNR Am J Neuroradiol 2002; 23: 255–66. [PMC free article] [PubMed] [Google Scholar]
- Bräutigam K, Vakis A, Tsitsipanis C.. Pathogenesis of idiopathic normal pressure hydrocephalus: a review of knowledge. J Clin Neurosci 2019; 61: 10–13. [DOI] [PubMed] [Google Scholar]
- Christen T, Jahanian H, Ni WW, Qiu D, Moseley ME, Zaharchuk G.. Noncontrast mapping of arterial delay and functional connectivity using resting-state functional MRI: A study in Moyamoya patients. J Magn Reson Imaging 2015; 41: 424–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung C-P, Lin Y-J, Chao A-C, Lin S-J, Chen Y-Y, Wang Y-J, et al. Jugular venous hemodynamic changes with aging. Ultrasound Med Biol 2010; 36: 1776–1782. [DOI] [PubMed] [Google Scholar]
- Cole JH, Leech R, Sharp DJ, for the Alzheimer’s disease neuroimaging initiative. prediction of brain age suggests accelerated atrophy after traumatic brain injury: brain age prediction after TBI. Ann Neurol 2015; 77: 571–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corkin S, Rosen TJ, Sullivan EV, Clegg RA.. Penetrating head injury in young adulthood exacerbates cognitive decline in later years. J Neurosci 1989; 9: 3876–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Leon MJ, George AE, Tomanelli J, Christman D, Kluger A, Miller J, et al. Positron emission tomography studies of normal aging: a replication of PET III and 18-FDG using PET VI and 11-CDG. Neurobiol Aging 1987; 8: 319–323. [DOI] [PubMed] [Google Scholar]
- De Vis JB, Lu H, Ravi H, Hendrikse J, Liu P.. Spatial distribution of flow and oxygenation in the cerebral venous drainage system. J Magn Reson Imaging 2018; 47: 1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagain A, Vignes JR, Dulou R, Dutertre G, Delmas JM, Guerin J, et al. Junction between the great cerebral vein and the straight sinus: An anatomical, immunohistochemical, and ultrastructural study on 25 human brain cadaveric dissections. Clin Anat 2008; 21: 389–397. [DOI] [PubMed] [Google Scholar]
- Dubois B, Feldman HH, Jacova C, DeKosky ST, Barberger-Gateau P, Cummings J, et al. Research criteria for the diagnosis of Alzheimer’s disease: revising the NINCDS–ADRDA criteria. Lancet Neurol 2007; 6: 734–746. [DOI] [PubMed] [Google Scholar]
- Fjell AM, Walhovd KB, Fennema-Notestine C, McEvoy LK, Hagler DJ, Holland D, et al. One year brain atrophy evident in healthy aging. J Neurosci 2009; 29: 15223–15231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogelholm R, Heiskanen O, Waltimo O.. Chronic subdural hematoma in adults. J Neurosurg 1975; 42: 43–46. [DOI] [PubMed] [Google Scholar]
- Fotenos AF, Snyder AZ, Girton LE, Morris JC, Buckner RL.. Normative estimates of cross-sectional and longitudinal brain volume decline in aging and AD. Neurology 2005; 64: 1032–1039. [DOI] [PubMed] [Google Scholar]
- Freire L, Mangin JF.. Motion correction algorithms may create spurious brain activations in the absence of subject motion. Neuroimage 2001; 14: 709–722. [DOI] [PubMed] [Google Scholar]
- Fujishima M, Omae T.. Brain blood flow and mean transit time as related to aging. Gerontology 1980; 26: 104–107. [DOI] [PubMed] [Google Scholar]
- Haines DE, Harkey HL, Al-Mefty O.. The “subdural” space: a new look at an outdated concept. Neurosurgery 1993; 32: 111–120. [DOI] [PubMed] [Google Scholar]
- Hukkelhoven CWPM, Steyerberg EW, Rampen AJJ, Farace E, Habbema JDF, Marshall LF, et al. Patient age and outcome following severe traumatic brain injury: an analysis of 5600 patients. J Neurosurg 2003; 99: 666–673. [DOI] [PubMed] [Google Scholar]
- Irimia A, Van Horn JD, Vespa PM.. Cerebral microhemorrhages due to traumatic brain injury and their effects on the aging human brain. Neurobiol Aging 2018; 66: 158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iseki C, Takahashi Y, Wada M, Kawanami T, Adachi M, Kato T.. Incidence of idiopathic normal pressure hydrocephalus (iNPH): a 10-year follow-up study of a rural community in Japan. J Neurol Sci 2014; 339: 108–112. [DOI] [PubMed] [Google Scholar]
- Jenkinson M, Beckmann CF, Behrens TEJ, Woolrich MW, Smith SM.. FSL. Neuroimage 2012; 62: 782–790. [DOI] [PubMed] [Google Scholar]
- Jespersen SN, Østergaard L.. The roles of cerebral blood flow, capillary transit time heterogeneity, and oxygen tension in brain oxygenation and metabolism. J Cereb Blood Flow Metab 2012; 32: 264–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammersgaard LP, Linnemann M, Tibæk M.. Hydrocephalus following severe traumatic brain injury in adults. Incidence, timing, and clinical predictors during rehabilitation. NeuroRehabilitation 2013; 33: 473–480. [DOI] [PubMed] [Google Scholar]
- Keong NCH, Pena A, Price SJ, Czosnyka M, Czosnyka Z, Pickard JD.. Imaging normal pressure hydrocephalus: theories, techniques, and challenges. Neurosurg Focus 2016; 41: E11. [DOI] [PubMed] [Google Scholar]
- Kuriyama N, Tokuda T, Miyamoto J, Takayasu N, Kondo M, Nakagawa M.. Retrograde jugular flow associated with idiopathic normal pressure hydrocephalus. Ann Neurol 2008; 64: 217–221. [DOI] [PubMed] [Google Scholar]
- Kuriyama N, Tokuda T, Yamada K, Akazawa K, Hosoda M, Sakai K, et al. Flow velocity of the superior sagittal sinus is reduced in patients with idiopathic normal pressure hydrocephalus. J Neuroimaging 2011; 21: 365–369. [DOI] [PubMed] [Google Scholar]
- Le TT, Fischbein NJ, André JB, Wijman C, Rosenberg J, Zaharchuk G.. Identification of venous signal on arterial spin labeling improves diagnosis of dural arteriovenous fistulas and small arteriovenous malformations. Am J Neuroradiol 2012; 33: 61–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao C-C, Xiao F.. Subdural effusion protects the aging brain from harmful ventriculomegaly. Med Hypotheses 2017; 108: 108–114. [DOI] [PubMed] [Google Scholar]
- Lou D, Du Y, Huang D, Cai F, Zhang Y, Li T, et al. Traumatic brain injury alters the metabolism and facilitates Alzheimer’s disease in a murine model. Mol Neurobiol 2018; 55: 4928–4939. [DOI] [PubMed] [Google Scholar]
- Lv Y, Margulies DS, Cameron Craddock R, Long X, Winter B, Gierhake D, et al. Identifying the perfusion deficit in acute stroke with resting-state functional magnetic resonance imaging. Ann Neurol 2013; 73: 136–140. [DOI] [PubMed] [Google Scholar]
- Mazaika PK, Hoeft F, Glover GH, Reiss AL.. Methods and software for fMRI analysis of clinical subjects. Neuroimage 2009; 47: S58. [Google Scholar]
- Mihara F, Kuwabara Y, Tanaka A, Yoshiura T, Sasaki M, Yoshida T, et al. Reliability of mean transit time obtained using perfusion-weighted MR imaging; comparison with positron emission tomography. Magn Reson Imaging 2003; 21: 33–39. [DOI] [PubMed] [Google Scholar]
- Moody DM, Brown WR, Challa VR, Anderson RL.. Periventricular venous collagenosis: association with leukoaraiosis. Radiology 1995; 194: 469–476. [DOI] [PubMed] [Google Scholar]
- Namiki J. Difficulty and inaccuracy of assessment of the consciousness level by the Glasgow Coma Scale: comparison with the Japan Coma Scale. J Jpn Soc Emerg Med 2007; 10: 20–25. [Google Scholar]
- Narita W, Nishio Y, Baba T, Iizuka O, Ishihara T, Matsuda M, et al. High-convexity tightness predicts the shunt response in idiopathic normal pressure hydrocephalus. Am J Neuroradiol 2016; 37: 1831–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida S, Aso T, Takaya S, Takahashi Y, Kikuchi T, Funaki T, et al. Resting-state functional magnetic resonance imaging identifies cerebrovascular reactivity impairment in patients with arterial occlusive diseases: a pilot study. Neurosurgery 2019; 85: 680–8. [DOI] [PubMed] [Google Scholar]
- Pfefferbaum A, Mathalon DH, Sullivan EV, Rawles JM, Zipursky RB, Lim KO.. A quantitative magnetic resonance imaging study of changes in brain morphology from infancy to late adulthood. Arch Neurol 1994; 51: 874–887. [DOI] [PubMed] [Google Scholar]
- Pomschar A, Koerte I, Lee S, Laubender RP, Straube A, Heinen F, et al. MRI evidence for altered venous drainage and intracranial compliance in mild traumatic brain injury. PLoS One 2013; 8: e55447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portnoy HD, Branch C, Castro ME, Branch C, Hospital HF, Portnoy HD, et al. The relationship of intracranial venous pressure to hydrocephalus. Childs Nerv Syst 1994; 10: 29–35. [DOI] [PubMed] [Google Scholar]
- Power JD, Barnes KA, Snyder AZ, Schlaggar BL, Petersen SE.. Spurious but systematic correlations in functional connectivity MRI networks arise from subject motion. Neuroimage 2012; 59: 2142–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Relkin N, Marmarou A, Klinge P, Bergsneider M, Black PMcL. Diagnosing idiopathic normal-pressure hydrocephalus. Neurosurgery 2005; 57: S2–4-S2-16. [DOI] [PubMed] [Google Scholar]
- Ringstad G, Vatnehol SAS, Eide PK.. Glymphatic MRI in idiopathic normal pressure hydrocephalus. Brain 2017; 140: 2691–2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roher AE, Kuo Y-M, Esh C, Knebel C, Weiss N, Kalback W, et al. Cortical and leptomeningeal cerebrovascular amyloid and white matter pathology in Alzheimer’s disease. Mol Med 2003; 9: 112–122. [PMC free article] [PubMed] [Google Scholar]
- Sack I, Beierbach B, Wuerfel J, Klatt D, Hamhaber U, Papazoglou S, et al. The impact of aging and gender on brain viscoelasticity. Neuroimage 2009; 46: 652–657. [DOI] [PubMed] [Google Scholar]
- Santini F, Patil S, Meckel S, Scheffler K, Wetzel SG.. Double-reference cross-correlation algorithm for separation of the arteries and veins from 3D MRA time series. J Magn Reson Imaging 2008; 28: 646–654. [DOI] [PubMed] [Google Scholar]
- Satow T, Aso T, Nishida S, Komuro T, Ueno T, Oishi N, et al. Alteration of venous drainage route in idiopathic normal pressure hydrocephalus and normal aging. Front Aging Neurosci 2017; 9: 387. [DOI] [PMC free article] [PubMed]
- Satterthwaite TD, Elliott MA, Gerraty RT, Ruparel K, Loughead J, Calkins ME, et al. An improved framework for confound regression and filtering for control of motion artifact in the preprocessing of resting-state functional connectivity data. Neuroimage 2013; 64: 240–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PM, Hayashi KM, Zubicaray Gd, Janke AL, Rose SE, Semple J, et al. Dynamics of gray matter loss in Alzheimer’s disease. J Neurosci 2003; 23: 994–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Frederick B deB.. Concurrent fNIRS and fMRI processing allows independent visualization of the propagation of pressure waves and bulk blood flow in the cerebral vasculature. Neuroimage 2012; 61: 1419–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Hocke LM, Frederick BB.. Low frequency systemic hemodynamic “noise” in resting state BOLD fMRI: characteristics, causes, implications, mitigation strategies, and applications. Front Neurosci 2019; 13: 787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Hocke LM, Licata SC, deB. Frederick B.. Low-frequency oscillations measured in the periphery with near-infrared spectroscopy are strongly correlated with blood oxygen level-dependent functional magnetic resonance imaging signals. J Biomed Opt 2012; 17: 1060041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Lindsey KP, Hocke LM, Vitaliano G, Mintzopoulos D, Frederick BdB.. Perfusion information extracted from resting state functional magnetic resonance imaging. J Cereb Blood Flow Metab 2017; 37: 564–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virhammar J, Laurell K, Ahlgren A, Cesarini KG, Larsson E-M.. Idiopathic normal pressure hydrocephalus: cerebral perfusion measured with pCASL before and repeatedly after CSF removal. J Cereb Blood Flow Metab 2014; 34: 1771–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yau W-YW, Tudorascu DL, McDade EM, Ikonomovic S, James JA, Minhas D, et al. Longitudinal assessment of neuroimaging and clinical markers in autosomal dominant Alzheimer’s disease: a prospective cohort study. Lancet Neurol 2015; 14: 804–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
MATLAB codes for the perfusion lag-mapping can be downloaded at https://github.com/RIKEN-BCIL/BOLDLagMapping. Data supporting the findings of this study are available from the corresponding author upon request.