

Abstract
U2 and U6 small nuclear (sn)RNAs are the only snRNAs directly implicated in catalyzing the splicing of pre-mRNA, but assembly and rearrangement steps prior to catalysis require numerous proteins. Previous studies have shown that the protein-free U2–U6 snRNA complex adopts two conformations in equilibrium, characterized by four and three helices surrounding a central junction. The four-helix conformer is strongly favored in the in vitro protein-free state, but the three-helix conformer predominates in spliceosomes. To analyze the role of the central junction in positioning elements forming the active site, we derived three-dimensional models of the two conformations from distances measured between fluorophores at selected locations in constructs representing the protein-free human U2–U6 snRNA complex by time-resolved fluorescence resonance energy transfer. Data describing four angles in the four-helix conformer suggest tetrahedral geometry; addition of Mg2+ results in shortening of the distances between neighboring helices, indicating compaction of the complex around the junction. In contrast, the three-helix conformer shows a closer approach between helices bearing critical elements, but the addition of Mg2+ widens the distance between them; thus in neither conformer are the critical helices positioned to favor the proposed triplex interaction. The presence of Mg2+ also enhances the fraction of the three-helix conformer, as does incubation with the Prp19-related protein RBM22, which has been implicated in the remodeling of the U2–U6 snRNA complex to render it catalytically active. These data suggest that although the central junction assumes a significant role in orienting helices, spliceosomal proteins and Mg2+ facilitate formation of the catalytically active conformer.
Keywords: spliceosome, U2–U6 snRNA complex, fluorescence, time-resolved FRET, junction
INTRODUCTION
The splicing of precursor messenger (pre-m)RNA is a fundamental biological process in which noncoding RNA segments (introns) are excised and flanking coding regions (exons) ligated to create a contiguous sequence of exons to be translated into protein. This process is catalyzed by the spliceosome, a huge and dynamic ribonucleoprotein complex located in a eukaryotic cell's nucleus involving five small nuclear (sn)RNAs (U1, U2, U4, U5, and U6) associated with specific proteins to form small nuclear ribonucleoprotein particles (snRNPs) and numerous non-snRNP splicing factors at different stages of assembly. The spliceosome promotes splicing through two transesterification steps (Will and Lührmann 2011): In the first step, the 2′-OH of a specific adenosine residue of the intron, called branch site, attacks the 5′ splice site, resulting in release of the first exon and an intron-3′-exon intermediate as a 2′–3′–5′ branched lariat intermediate. In the second step, the newly freed 3′-OH of the 5′ exon attacks the 3′ splice site, ligating the exons and releasing the lariat intron. These reactions were proposed to involve a two-metal ion center (Steitz and Steitz 1993), in which one Mg2+ ion activates the 2′OH branch site nucleophile and the other stabilizes the oxyanion leaving group (Gordon et al. 2000).
The protein-assisted assembly of a spliceosome on a pre-mRNA substrate involves a highly choreographed set of well-defined events in which snRNPs and protein splicing factors, some partially preassembled, are escorted to the assembling spliceosome or are released. Major conformational changes leading to discrete stages of assembly are mediated by eight helicases and numerous non-ATP-dependent remodeling proteins. Individual steps involve recognition of the 5′ splice site and branch site by the U1 and U2 snRNPs, respectively, followed by the introduction of remaining U5, U6, and U4 snRNP as a tri-snRNP (for review, see Will and Lührmann 2011). A major rearrangement catalyzed by a helicase leads to pairing between U2 and U6 snRNAs and positioning of the 5′ and 3′ splice sites by the U5 snRNP, accompanied by release of U1 and U4 snRNP to achieve an activated state (Bact). Subsequent helicase-mediated adjustments bring catalytic RNA components into proximity and proteins associated with the Prp19 complex (NTC) lead to local remodeling to promote the branching step (Chan et al. 2003; Makarova et al. 2004), followed by additional rearrangements, the second catalytic step, and finally dissociation of snRNPs. Thus, the ultimate goal of each of these protein-assisted stages is to usher new components to the assembly, catalyze RNA–RNA rearrangements, and to protect, expose, or juxtapose catalytic elements for catalysis at the appropriate time. Detailed images of spliceosomes trapped in defined stages produced by cryo-EM studies have contributed dramatically to our understanding of the landscape, interactions, and rearrangements of spliceosomal components throughout the cycle of assembly and activity in human (Bertram et al. 2017a,b) and yeast (S. pombe and S. cerevisiae; Yan et al. 2015, 2016, 2017; Wan et al. 2016; Bai et al. 2018; Zhan et al. 2018) spliceosomes.
The catalytic center of the spliceosome is a complex defined by the U2 and U6 snRNA (Fabrizio and Abelson 1990), the most highly conserved among the five snRNA sequences. Inter- and intramolecular base pairing between U2 and U6 snRNA lead to the formation of two intermolecular helices, Helix I and Helix II, and the U6 intramolecular stem–loop (ISL) surrounding a central junction. The ACAGAGA sequence within Helix I of U6 snRNA is involved in 5′ splice site selection and in promoting both steps of the splicing reaction (Luukkonen and Séraphin 1998; Mefford and Staley 2009); the U2 snRNA sequence opposing this ACAGAGA segment pairs with a region of the intron to position the branch site residue.
Multiple similarities between self-splicing Group II intron ribozyme and the spliceosome suggest shared ancestry, and crystal structures of the Group IIC intron of O. Iheyensis provided important information about common features about the tertiary interaction involving distant elements and metal ions to form the active site (Toor et al. 2008). Analogy between critical sequence elements and ion-binding features of group II introns and spliceosomes led to the hypothesis of an analogous motif in spliceosome (Keating et al. 2010) involving the ion binding site at the bulged U of the ISL, AGC triad, and the 3′ end of the ACAGAGA loop (Keating et al. 2010) to form catalytically essential metal-ion binding sites. Such a triple helix interaction was evidenced by cross-linking and genetic mutations in yeast spliceosomes in their cellular environment (Fica et al. 2014) and fully supported by cryo-EM based models (Anokhina et al. 2013; Yan et al. 2016) that established the absence of proteins in the immediate vicinity of the catalytic site.
Although a three-helix conformer has been identified in intact spliceosomes (Yan et al. 2016; Bertram et al. 2017b) and cellular systems (Anokhina et al. 2013), in vitro studies of the U2–U6 snRNA complex in its protein-free state have supported formation of two different secondary conformations characterized by three and four helices around the central junction (Sashital et al. 2004; Guo et al. 2009; Burke et al. 2012; Zhao et al. 2013; Karunatilaka and Rueda 2014) that are in dynamic equilibrium with each other (Zhao et al. 2013). The observation of alternative distributions is interesting because the conserved catalytic AGC triad is in a different pairing context in each conformer: It is paired with U6 snRNA to extend the base of U6 ISL in the four-helix variant and paired with U2 snRNA to form Helix 1b in the three-helix conformation; this difference suggests the possibility that the three-helix junction observed in the spliceosome facilitates formation of the active site (Sashital et al. 2004). However, genetic experiment found that the four-helix fold maintains a critical role at an unspecified stage in human cells (Wu and Manley 1989).
Interaction with multivalent metal ions is necessary and sufficient to facilitate folding of the Group II intron into a catalytic form in the absence of proteins. However, although in spliceosomes Mg2+ has an impact on the conformation of protein-free U2–U6 snRNA complex by nonspecific screening of RNA backbones near the central junction, it is not sufficient to create an active conformation to support the splicing reaction in yeast and human U2–U6 snRNA complexes because the stems bearing the catalytically critical components forming the active site are far apart in the protein-free state (Sashital et al. 2004; Guo et al. 2009; Burke et al. 2012; Zhao et al. 2013; Karunatilaka and Rueda 2014). However, evidence supports the need for spliceosomal proteins to facilitate formation and/or stabilization of an active conformation by the native U2–U6 snRNA complex (Hogg et al. 2010). Specifically, data implicate RBM22 to stabilize formation of the triple-helical active site (Rasche et al. 2012). Since only the three-helix conformer is observed in cryo-EM images of spliceosomes, and RBM22 is the only NTC-related protein to contact the U2–U6 snRNA complex directly, we speculate that RBM22 preferentially binds to the three-helix conformer, thereby favoring conformational redistribution into the three-helix conformer to facilitate formation of the active site.
To enhance our understanding of the role of the central junction and of divalent metal ions in facilitating formation of the active structure, we have investigated the orientation of the helical stems of the human U2–U6 snRNA complex in the absence of spliceosomal proteins. Toward this goal, we used time-resolved Förster resonance energy transfer (trFRET) to measure distances between stems of the wild-type and mutant complexes, and dependence on added Mg2+.
Our results show that the protein-free four-helix conformer that predominates in vitro adopts a roughly tetrahedral arrangement, and that the angles between helical stems bearing the catalytically integral elements including the AGC triad, ACAGAGA loop and U6 ISL around the junction decrease upon addition of Mg2+, resulting in a more compact structure. In contrast, the three-helix conformer observed in intact spliceosomes displays closer juxtaposition between the stems containing the catalytically essential elements in the absence of Mg2+, but these stems assume a markedly increased distance from each other upon addition of Mg2+. Although the data are consistent with the central junction maintaining a Mg2+-sensitive role in positioning of helices, in neither protein-free conformer at any concentration of Mg2+ are the regions bearing the catalytically essential elements close enough to interact with each other to form the triple helix defining the catalytic center. We also show here that RBM22 preferentially binds the three-helix conformer, thus favoring a shift in the fractional representation from the four-helix to three-helix conformer associated with catalytic function.
RESULTS
To analyze the role of the central junction in positioning the two helices critical to forming the active sites in both conformers, and to build a three-dimensional structural model of the orientation of the four helical stems of the majority form of the protein-free human U2–U6 snRNA complex, we used time-resolved (tr)FRET technique to measure distances between termini of complexes formed by paired fragments representing the U2 and U6 snRNA complex. RNA constructs are described in Materials and Methods and shown in Figure 1.
FIGURE 1.


Constructs representing the human U2–U6 snRNA complex used in these experiments. Modifications to helix length including truncation, deletion, or addition of hairpin loops to create “chimeric” strands, or nucleotide changes to favor base pairings, are described in Materials and Methods. Constructs are all depicted in the four-helix secondary structure; the AGC catalytic triad corresponds to nucleotides A53G54C55 of U6 snRNA. (A) Wild-type junction in which the ACAGAGA loop is “closed” by pairing with a complementary sequence (shown in red), with fluorescein as the Donor (D; Fl) and Cy3 as the Acceptor (A) on termini of helix III and U6 ISL, respectively (WTΔL). (B) Mutations in Stem I favor the four-helix conformer (individual nucleotide changed in red), also with a “closed” ACAGAGA loop and dyes on helix III and U6 ISL (4HJΔL1). (C) Four-helix mutant with “closed” ACAGAGA loop, and Fl and Cy3 dyes on termini of U6 ISL and Helix II, respectively (4HJΔL2). (D) Four-helix mutant with “closed” ACAGAGA loop with AF488 (D) and AF555 (A) dyes on termini of Helix III and Helix II, respectively (4HJΔL3). (E) Four-helix mutant with “closed” ACAGAGA loop with AF488 and AF555 dyes on Helix III and Stem Loop I, respectively (4HJΔL4). (F) Wild-type U2–U6 junction with wild-type ACAGAGA loop, and Fl and Cy3 on termini of Helix III and U6 ISL, respectively (WT). (G) Four-helix mutant with ACAGAGA loop, with Fl and Cy3 dyes on termini of Helix III and U6 ISL, respectively (4HJ). (H) The proposed three-helix junction conformer without an ACAGAGA loop (3HJ).
trFRET of a U2–U6 snRNA complex with the wild-type junction
Steady state measurement of FRET efficiency between D and A, calculated from the decrease in emission of D in the presence of A (corrected for emission of A alone when illuminated at the excitation wavelength of D, 495 nm) was measured to confirm FRET between donor and acceptor dyes attached to selected sites of the RNA complex (Supplemental Fig. S1). However, the resulting distance represents the mean distance in the four-helix junction and the three-helix junction of the wild-type, and therefore does not provide information to distinguish individual distances in a heterogeneous system. Thus, to estimate distances in each of the major conformers in this heterogeneous system, as well as the distribution of the populations, we utilized trFRET. The advantage of this approach is that the decay curve of a pulse-excited donor dye transferring energy to a suitable acceptor dye contains rate/distance information for all D-A distances represented in an ensemble weighted according to fractional representation in the ensemble; deconvolution of decay curves thus allows determination of the relative populations of signals from multiple conformers in a heterogeneous system.
We acquired interdye distance measurements between termini of Helix III and the U6 ISL of a U2–U6 snRNA construct, the two stems containing elements that interact to define the catalytic center. This construct (WTΔL; Fig. 1A) replaced the sequence of U2 snRNA opposing the ACAGAGA loop with a complementary duplex to create a continuous stem. Although nucleotides in the 5′ end of the U6 and the 3′ end of the U2 snRNA sequences are capable of forming nine Watson–Crick base pairs to form Helix III, this duplex is not observed in cellular (Anokhina et al. 2013) or spliceosomal (Yan et al. 2016; Bertram et al. 2017a) systems. However, this helix forms in the protein-free RNA complex and is useful in these experiments for stabilizing the position of the attached dye on the 5′ terminus of the U6 snRNA fragment. In this case, we measured FRET between a fluorescein Donor (D)-labeled 5′ end of the abbreviated U6 snRNA strand and the Cy3 Acceptor (A)-labeled U6 ISL.
We first measured the decay of emission of D alone (with an unlabeled U6–U2 chimeric strand), followed by D in the presence of A, and fitted decay curves with single exponential or the weighted sum of two exponential curves. The time-resolved decay curve of the D alone was fitted by a single exponential curve with a lifetime, τ, of 4.05 ± 0.008 ns and a CHISQ (χ2, a measure of fit to a model or curve, with an ideal fit approaching unity) value = 1.07. The decay curve for D in the presence of A (Supplemental Fig. S2A) was first fit with a single exponential curve with τDA = 3.69 ± 0.010 nsec and χ2 = 2.67, that is, a poor fit (Supplemental Fig. S2B). Fitting the curve with two decay components, however, with lifetimes of 3.87 ± 0.01 nsec (fractional amplitude of 91.4%) and 0.89 ± 0.06 nsec (fractional amplitude of 8.6%), for distances between the dyes of 93.0 and 45.3 Å, respectively, yielded a far better overall fit (χ2 = 1.15, Supplemental Fig. S2C). The next task was to match each set of distances with the conformer it represents independently without any assumptions.
trFRET of the four-helix conformer
To obtain distances related to individual conformers, we performed measurements using a construct with a modification of nucleotides near the junction that favored the four-helix structure (construct 4HΔL1; Fig. 1B) and compared results with those for the wild-type junction (WTΔL). Measurements of trFRET between the dyes in the construct 4HΔL1 labeled at the same sites as of construct WTΔL fit into a single exponential curve yielding a distance of 90.8 ± 0.6 Å (χ2 = 1.24), a value very similar to that of the deconvoluted four-helix conformer and very different from that of the minor conformer, confirming that the longer lifetime component in the decay curve deconvolution corresponds to the four-helix conformer, and the smaller component with the shorter lifetime therefore corresponds to a three-helix conformer.
Determination of Helix III-ISL distances in the three-helix conformer
We had previously designed a mutant that stabilizes formation of Helix Ib and disrupts the base pair formation in the lower region of U6 ISL in the three-helix conformation (Fig. 1H; Zhao et al. 2013). However, the appearance of a number of unexpected 19F NMR peaks suggests the presence of multiple subconformers induced by this mutation that are unrelated to native junction conformation. Therefore, we did not pursue this mutation further, but opted to calculate distances in the three-helix junction two independent ways: (i) deconvolution of the decay curve in the wild-type junction construct; and (ii) subtraction of the decay curve of the four-helix junction mutant from that of the wild-type construct. While each method includes a number of assumptions, the similarity in distance values achieved by the two methods increased confidence in the results.
Translation of distance information into junction angles
From the distance between termini of helical stems and estimated lengths of the stems (Helices III–I and ISL), we triangulated angles between stems. For construct 4HΔL1, the stem formed by Helices I and III is a continuous A-type helix of 24 nt (2.6 Å between centers of base pairs); with an extension of 8.5 Å for fluorescein and a 2 carbon amino linker and 5 Å due to addition of four more C–C bonds in six-carbon amino linker compared to two-carbon amino linker (see Materials and Methods), the total length is ∼(2.6 Å × 24) + 8.5 + 5 = 75.9 Å. The acceptor-Cy3 attached at U6 ISL stacks onto the RNA helix and therefore contributes to the length of the human U6 ISL as one additional nucleotide, for a total of 33.8 Å. The angle between the U6 ISL and Helix III/I in the construct 4HΔL1 from the directly measured distance reported above was 105.2 ± 1.2°. In comparison, from calculation of individual distances obtained from deconvolution of the WT decay curve, 93 and 45.3 Å for the four-helix and three-helix conformers, respectively, we obtained a similar value of ∼109.9° for the angle in the majority four-helix conformer (Fig. 3A) and 19° for the minor three-helix conformer (Fig. 3C). The smaller angle between stems in the three-helix junction indicates that even in the absence of all sequence elements necessary for formation of the catalytic triplex interaction (pairing of the ACAGAGA loop prevents any tertiary interaction), the position of ISL and Helix III/I stem is much closer than it is in the four-helix conformation.
FIGURE 3.

Predictive computational models of the two major conformers of the human U2–U6 snRNA complex obtained by simulation using SimRNA (Magnus et al. 2016), with distance restraints between dyes determined experimentally and visualized by NGLview (Nguyen et al. 2018). (A,B) Models of the four-helix junction conformer (Helix I/III in red; U6 ISL in blue; U2 Stem I folded behind the junction in green; Helix II in violet) (A) without Mg2+ and (B) with 40 mM Mg2+, showing a compaction of Helix I/III, ISL, and Helix II around the junction (Stem I is folded behind the junction in this view). (C,D) Models of calculated three-helix junction conformer with three helices (Helix I/III in red; U6 ISL in blue; Helix II in gray) (C) without Mg2+ and (D) with 40 mM Mg2+, showing a widened junction. The position of Helix II in the three-helix conformer was not experimentally determined but found to be in the lowest energy position by simulation.
We also estimated the distance between U6 ISL and Helix I/III values for the minority three-helix conformer by subtraction of the measured decay curve for the fraction of four-helix fold (91.4%) from that of the (heterogeneous) WT. The decay curve of four-helix fold is well fitted to a single exponential decay curve with correlation coefficient, R2 = 0.9999, whereas the subtracted decay curve for the three-helix conformer is less perfectly fitted to a single exponential decay curve shown in Figure 2 (R2 = 0.97, calculated by SigmaPlot 13.0; Systat Software, Inc., San Jose California USA; www.systatsoftware.com), perhaps as the result of several subconformers; a similar conclusion was reached from marked 19F peak heterogeneity in NMR experiments (Zhao et al. 2013). The distance calculated between U6 ISL and Helix III for the remaining three-helix is 56.3 Å, corresponding to an angle of 43.3°. Although there is a noticeable difference in the angle obtained by deconvolution and subtraction (19° vs. 43.3° respectively), this disagreement may result from uncertainty associated with dye/linker mobility, heterogeneity of the three-helix conformer, the large difference in helix length, and/or the indirect nature of the calculation involved for analysis of this minority conformer. However, both values indicate an acute angle in the range consistent with the Y shape structure modeled by Butcher and coworkers shown previously for the yeast U2–U6 snRNA complex (Burke et al. 2012), implying that the junction has a role in positioning the two helices near to each other.
FIGURE 2.
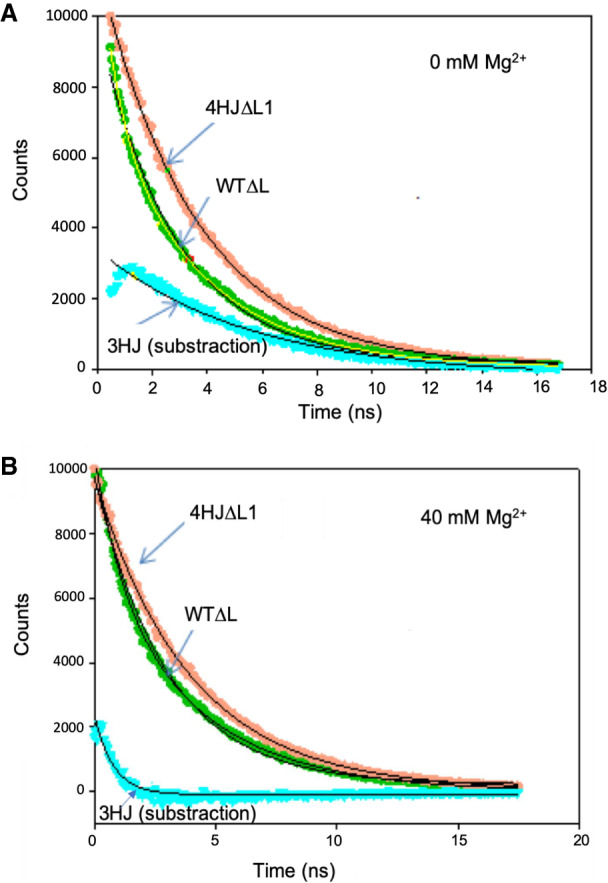
Calculation of a decay curve for the three-helix conformer (cyan) by subtraction of the experimentally obtained decay curves of fluorescein-labeled 4HJΔL1 (orange) from that of WTΔL (green). (A) Curves without added Mg2+ and (B) curves with added Mg2+ (40 mM). Concentration of dye-labeled RNA was 150 nM. trFRET measurements were recorded on a timescale of 20 nsec to a total of 10,000 counts in the peak. The decay curve of fluorescein on 4HJΔL1 and WTΔL were fit by a monoexponential and biexponential curve, respectively, with an excellent R2 (R2 > 0.999). The calculated decay curve for the three-helix conformer in each case was fitted with a monoexponential curve yielding a less perfect fit (R2 value ∼0.974 and 0.911 without and with Mg2+, respectively), suggesting a mix of several three-helix conformers (see Results for details).
Measurement of other distances in the four-helix conformer
To contribute to the three-dimensional topology models of the two conformers of the protein-free human U2–U6 snRNA complex, we also determined angles between dye pairs at other sites within the complex. In each case, we repeated trFRET measurements to determine interdye distances using constructs with dyes on different termini, and with either the wild-type or the helix mutated to form only the four-helix conformer. As in WTΔL and 4HJΔL, the ACAGAGA loop was eliminated by nucleotide substitution in the U2 snRNA strand to produce extensive WC pairing.
To measure the distance between the cleaved hairpin loop of U6 ISL and the terminus of Helix II we created a four-helix construct, 4HΔL2 (Fig. 1C), and labeled 5′ termini with fluorescein and Cy3, respectively. Results of trFRET measurements indicated a distance of 73.0 ± 1.0 Å, triangulated into an angle of 110.1 ± 2.4°. This angle calculation included an estimation that five U6 snRNA unpaired nucleotides (AAAUU) at the junction form a loop contributing ∼5 Å to the length of Helix II.
For measurements between Helices II and I/III, we labeled the 5′ termini of strands representing U2 snRNA (in Helix II) and U6 snRNA (in Helix III) (4HJΔL3; Fig. 1D). We used the AlexaFluor dye pair (AF488 and AF555) to take advantage of the longer R0 for the larger anticipated distance between the termini (Yuan et al. 2007). AF488 and AF555 have been shown by simulation to be relatively mobile in aqueous solution (Corry and Jayatilaka 2008); we therefore assumed they would behave like fluorescein in calculation of the length of the RNA duplex. The distance between probes at 5′ termini of U2 and U6 snRNA was 97.0 ± 1.0 Å, which triangulated to an angle of 116.7 ± 2.0°.
Finally, to measure the distance between Stem I of U2 snRNA and Helix III (4HJΔL4; Fig. 1E), we measured trFRET between AF488 labeled at 5′ of U6 snRNA and AF555 at internally labeled U16 of U2 Stem I. The distance from 5′ U6 snRNA to U2 stem I is 85.2 ± 0.8 Å, for an angle of 139.0 ± 4.0°. Using the distance measured here, we have simulated models for the orientation of stems for the measured four-helix conformer and for the calculated three-helix conformer of the human protein-free U2–U6 snRNA complex (Fig. 3).
Effect of Mg2+ on conformation of the human U2–U6 snRNA complex
Both spliceosomes and Group II introns are dependent upon interaction with Mg2+ for assembly, including RNA folding and tertiary interaction of key sequence elements (ACAGAGA loop, AGC triad and U74 of U6 ISL), as well as for catalytic activity. To investigate the impact of Mg2+ addition on the three-dimensional fold of the protein-free human U2–U6 snRNA complex, we repeated each of the measurements of interhelical distances in the presence of MgCl2 from concentrations of 5 mM (near cellular concentration; (Romani and Scarpa 1992) to 40 mM (the value used in previous in vitro studies (Guo et al. 2009). Addition of even a relatively low concentration of Mg2+, 5 mM, resulted in a 6.9% decrease in distance between termini of Helix I/III and U6 ISL in the four-helix conformation (4HJΔL1). At a maximal concentration of 40 mM Mg2+, the distance decreased by 13.8% from 90.8 ± 0.6Å to 78.3 ± 0.8 Å (Fig. 4), for a decrease in angle from 105.2 ± 1.2° to 81.3 ± 1.4°, that is, a closer proximity of Helix I/III and U6 ISL. The distance between Helix II and U6 ISL in construct 4HJΔL2 (Fig. 1C) decreased by 5.1 Å, for a decrease in angle from 110.1 ± 2.4° to 99.1 ± 2.0°. No significant change was observed for distance between Helix II and Helix I/III (in 4HJΔL3). The distance between Stem I and Helix I/III in construct 4HJΔL4 was lengthened slightly by 2.1 Å for an increase in angle from 139.0 ± 4.0° to 150.5 ± 5.6°, placing Stem I further “behind” the junction (Fig. 3B). Therefore, Mg2+ induced a decrease in each distance (or angle) in the four-helix conformer appear to be in the approach of helices toward the U6 ISL, originating from changes in stem orientation (Fig. 3B), resulting in a more compact folded structure. We had previously noted this compaction by increased migration measured by analytical ultracentrifugation in the presence of 40 mM Mg2+ (Zhao et al. 2013).
FIGURE 4.
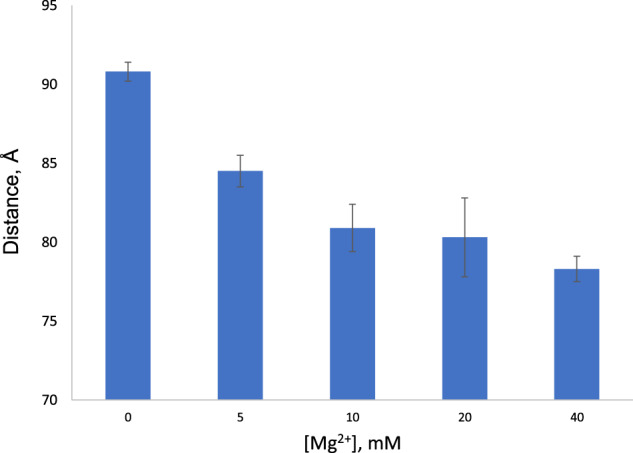
Decrease in distance between dyes attached to Helix I/III and U6 ISL in construct 4HJΔL1 with respect to increasing Mg2+ concentration from 0 to 40 mM. Error bars indicate ± standard deviation. Experimental details in Materials and Methods.
We then investigated changes in the distribution of four- and three-helix conformers upon addition of 40 mM Mg2+. The distance between Helix I/III and U6 ISL in heterogeneous WTΔL shortened by 3.5 Å with added Mg2+, a value much smaller than the change in four-helix construct (12.5 Å). However, deconvolution of the decay curves of WTΔL upon addition of Mg2+ revealed approximately twice the fraction of the three-helix conformer relative to its fraction in the absence of Mg2+ (increase from 8.6% to 17% of the total), but also induced marked changes in stem orientation. By deconvolution, the distance between Helix I/III and U6 ISL in the three-helix junction conformer in 40 mM Mg2+ increased to 67.7 Å (from 45.3 Å), triangulated to an angle of 63.1° (from 19°); calculation by subtraction (Fig. 3B) yielded a change in distance to 68.9 Å (from 56.3 Å) triangulated to an angle of 65.2° (from 43.3°). By either approach, this change agrees with data from single molecule FRET studies illustrating that addition of Mg2+ widens the distance between these stems in the three-helix conformer (Guo et al. 2009). Thus, the effect of Mg2+ on the three-helix conformer (Fig. 3D) is opposite to what we observed in four-helix conformer (Fig. 3B).
Role of the ACAGAGA loop in helix orientation
To examine the effect of the ACAGAGA loop (i.e., an open loop in the presence of the naturally occurring U2 snRNA sequence) to the folding of the complex, we performed the same measurements for the distance between Helix I/III and U6 ISL with the wild-type complex (construct WT; Fig. 1F) and the four-helix mutant (construct 4HJ; Fig. 1G), both with a native (open) ACAGAGA loop. In both cases, results of trFRET measurements show a small increase of interdye distances, from 80.0 ± 2.2 Å (construct WTΔL) to 84.0 ± 1.5 Å (construct WT); and from 84.0 ± 1.2 Å (4HJ) to 90.8 ± 0.6 Å (4HJΔL1), suggesting that the ACAGAGA loop contributes a small kink to Helix I/III in the protein-free U2–U6 snRNA complex. There was also no observable change in measurements repeated on constructs in which the region of U2 snRNA was paired with a short intron fragment representing the branch site (pairing confirmed by change in electrophoretic mobility of components using nondenaturing PAGE; data not shown).
We had noted previously that the distribution between junction conformations was not altered significantly in 19F-NMR spectra in response to the presence or absence of the ACAGAGA loop (Zhao et al. 2013). In none of these cases did inclusion of the native ACAGAGA loop, with or without pairing to an intron segment, facilitate increased proximity or interaction between Helix I/III and the U6 ISL, even in the presence of 40 mM Mg2+.
Binding of NTC-related protein RBM22 to U2–U6 snRNA complexes
Although the folded state of the U2–U6 snRNA complex starts to form in the Bact stage, we anticipate that binding of RBM22 (or Cwc2 in yeast), in association with NTC proteins, in addition to anchoring distant regions of the RNA complex, acts as a scaffold for the central junction and precise positioning of the catalytic Mg2+. Therefore, we investigated the relative affinity of recombinant RBM22 for the wild-type junction (construct WTΔL) and four-helix junction mutant (construct 4HJΔL1) by EMSA gel described in Materials and Methods. Results from EMSA measurements (Supplemental Fig. S3) exhibit a greater affinity of RBM22 for the wild-type complex, with a Kd of 8.3 ± 1.2 µM, compared with a Kd of 37.1 ± 10.5 µM for the four-helix junction mutant (Fig. 5).
FIGURE 5.
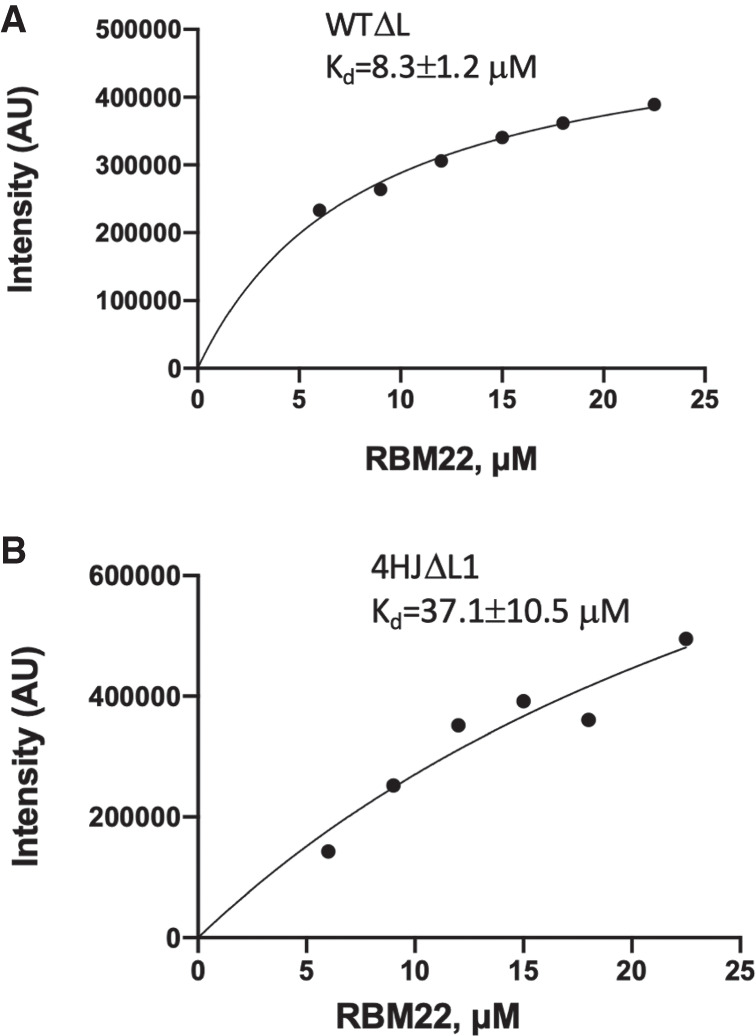
Analysis of binding affinity of contruct WTΔL (A) and contruct 4HJΔL1 (B) to RBM22. Concentration of RNA was constant at 15 µM and RBM22 concentration varied from 6 µM to 22.5 µM, corresponding to the RNA:RBM22 ratio from 1:0.4 to 1:1.5. Binding curves were fitted by a one-site binding (hyperbola) model using Prism. Goodness of fit was evaluated by least squared fit R2 with 95% confidence intervals.
DISCUSSION
Our goal was to analyze the contribution of the central junction of the spliceosomal U2–U6 snRNA complex in facilitating splicing activity by positioning of the helices associated with formation of the catalytic center. Using protein-free human snRNA complexes labeled at termini or internally with fluorescent dyes, results acquired from trFRET measurements enabled characterization of orientation of helical stems about the central junction of the two major folds of the RNA complex characterized by four and three stems emanating from the central junction, and changes in conformation and distribution of populations upon incubation with Mg2+. We also show that the NTC-related protein RBM22 preferentially binds to the three-helix conformer of the snRNA complex, suggesting that this junction provides a recognition/binding site for the protein.
Ensemble and single-molecule (sm)FRET techniques have been used to analyze orientations of stems around Holliday junctions in DNA (Clegg et al. 1994), junctions in hairpin and other ribozymes (Walter et al. 1998; Tan et al. 2003; Liu et al. 2007), Group II introns (Steiner et al. 2008), and spliceosomal junctions (Yuan et al. 2007; Guo et al. 2009; Karunatilaka and Rueda 2014) and time-resolved luminescence resonance energy transfer was used to estimate locations of multiple ion-binding sites in a protein-free U2–U6 snRNA complex (Yuan et al. 2007). In this report we use trFRET techniques to probe three-dimensional orientations of stems around three- and four-helix junction conformations of the human U2–U6 snRNA complex, and Mg2+-dependent changes in the distribution and conformations of those junctions, in such a system.
Genetic studies by Manley identified the importance of formation of the four-helix conformer in human cells (Wu and Manley 1989), although the stage in which this conformer was critical was not determined. In contrast, genetic studies in yeast identified a three-helix conformer as the critical conformer (Madhani and Guthrie 1992). Further conformational analysis was performed for protein-free U2–U6 snRNA complexes in vitro, supporting the predominance of either the four-helix conformer (Sashital et al. 2004), the three-helix conformer (Sashital et al. 2004), or a combination of the two in yeast (Guo et al. 2009) or human (Zhao et al. 2013) complexes. These studies suggest the likelihood that the two alternative conformations with a small energy difference and distribution between the two may depend on experimental conditions. In particular, we confirmed coexistence of the predominant four-helix conformer and three-helix conformer and calculated a very small difference in ΔG between the two conformations from NMR peak volumes (4.7 kJ/mol), and verified that the two are in dynamic exchange (Zhao et al. 2014). In contrast with evidence for an equilibrium distribution between conformers in vitro, recent results from biochemical structure probing in human cells (Anokhina et al. 2013) and by cryo-EM studies of human (Bertram et al. 2017b) and yeast (Yan et al. 2015; Rauhut et al. 2016; Plaschka et al. 2017; Wan et al. 2019) spliceosomes illustrate that the more open three-helix junction is generally observed in the intact spliceosomes suggesting that this conformation is associated with catalysis.
The four-helix junction model, measured either by deconvolution of the compound curve of the heterogeneous wild-type junction or by direct analysis of the mutant favoring the four-helix fold, indicated a roughly tetrahedral arrangement of helical stems in the absence of Mg2+. Upon addition of Mg2+, the angle between Helix I/III and the ISL decreases with increasing concentration of Mg2+, accompanied by a decrease in the angle between the ISL and Helix II. Approach of these two intermolecular stems to the intramolecular (U6) ISL suggests overall compaction of the complex. This finding agrees with a decrease in the Stokes radius and axial ratio (a/b) measured in the presence of Mg2+ measured by analytical ultracentrifugation (Zhao et al. 2013). There is no direct equivalence between the two measurements (i.e., “compaction” measured by a change in sedimentation velocity relates only to a change in Stokes radius and does not reveal specific changes in specific angles); however, we note that the trend of the two Mg2+-induced changes is consistent. This compaction/decrease in angle between ISL and Helix I of the four-helix junction conformer suggests that Mg2+ assists in bringing the catalytically essential elements into proximity. However, this decrease in distance remains insufficient to induce formation of the triplex by itself.
However, cryo-EM data suggest that the three-helix conformer is associated with catalytic activity—so we also investigated the impact of Mg2+ on the three-helix conformer by deconvolution of the compound curve of the wild-type and, independent, by subtraction of the measured four-helix curve of that from the wild-type.
In contrast with the Mg2+-induced approach of critical stems in the four-helix conformer, distances between the same stems in the minority fraction characterized by the more open three-helix junction, exhibited a close position of the stems in the Mg2+ free state, but responded to Mg2+ by a greatly increased separation between the catalytically essential elements. Results were qualitatively similar for both methods. Moreover, addition of Mg2+ to complexes containing the heterogeneous wild-type junction displayed a partial conversion from the four- to the three-helix conformer (∼8.6% to ∼17% of the total). Therefore, the presence of Mg2+ induces changes not only in the equilibrium distribution of junction conformers but also the positioning of helical stems around the junction. This Mg2+ induced opening/separation is quite surprising and contrary to what one would expect for metalloribozyme. However, in agreement with our finding, Guo et al. (2009) identified the same behavior in yeast in the presence of Mg2+.
Our previously published 19F-NMR study similarly indicated a shift in the distribution of conformers to increase the relative population of the three-helix conformer from ∼13% of the total to ∼17%, corroborating the fitting of the decay curve obtained by trFRET and further supporting the conclusion that interaction with, or screening by, Mg2+ shifts the conformation of the junction toward the AGC presentation that facilitates catalytic site formation. Single molecule FRET studies by Rueda and coworkers on the protein-free yeast U2–U6 snRNA complex showed the coexistence of at least three conformations: four-helix structure, intermediate and three-helix structure (Guo et al. 2009). It is possible that such an intermediate, in equilibrium with other three-helix subconformers, contributed to the broad and heterogeneous peak observed in our 19F spectra (and would not have been directly detected by either our NMR or ensemble trFRET approaches). If so, the previous value of ∼13% for the population of three-helix subconformers from 19F NMR is actually an overestimate of the population of several three-helix folds or intermediate(s) and is likely to explain the difference between measurements of distribution by NMR (Zhao et al. 2013) and trFRET (this work). Our NMR acquisition includes a maximum of 5 mM Mg2+ and ∼0.35 mM RNA (for a ratio of ∼14:1), considerably less than the current ratio of 40 mM Mg2+ to 150 nM RNA (267,000:1), so this difference in conditions may contribute to some variation in observed behavior. Not surprising with such a small difference in energy between two conformers, we found that the three- and four-helix conformers of the junction exchange on a subsecond time scale (Zhao et al. 2014)
Rueda and coworkers also observed a significant Mg2+ dependence in the smFRET data of yeast U2–U6 snRNA complexes labeled at the 5′ terminus of U6 and the U6 ISL loop, noting that an increase in the concentration of Mg2+ from 0 to 40 mM induced a decrease in the population of complexes displaying high FRET efficiency (assigned to the four-helix conformer) from 64% to 19% (Guo et al. 2009). In a subsequent study of the human U2–U6 snRNA complex (Karunatilaka and Rueda 2014), the authors reported that an increase in the concentration of Mg2+ from 10 to 40 mM resulted in a decrease in the relative population of the four-helix conformer from 13% to 5% and in an increase of the three-helix conformer from 44% to 69%. In both studies, they attributed this significant change entirely to conversion from four-helix to three-helix junction and did not address the possibility of ion-induced change in stem orientation in either junction conformer that contributes to such a significant change in FRET efficiency. That strict consideration might explain the difference in the relative population of the two conformers compared to our findings, which were based on direct analysis of the four-helix junction mutant confirmed by 19F NMR (Zhao et al. 2014).
In these experiments, we monitored changes in multiple distances for both the four-helix mutant and wild-type (mix of four- and three-helix junctions) U2–U6 snRNA complexes with increasing concentrations of Mg2+, from which we calculated change in fraction and orientations of the junction. By either analysis, results of this experiment provided clear evidence that Mg2+, in addition to increasing the fraction of the three-helix conformer, induces a marked increase in the angle between Helix I/III and the ISL (as well as a more moderate decrease in the angle between these stems in the four-helix conformer). The marked increase in separation between Helix I/III and the ISL upon interaction with Mg2+ would decrease the likelihood that the two stems will interact to promote catalysis. These findings provide definitive evidence that both the junction conformer and helix orientation undergo conformational change in the presence of Mg2+.
These data suggest that although the junction acts as a Mg2+-sensitive pivot in positioning the stems, it alone does not drive the proximity of the stems needed for tertiary interaction. Mg2+ clearly brings the catalytically integral elements in the protein-free four-helix junction conformer closer but it is not sufficient to form a catalytically active conformation, and the three-helix conformer identified in cryo-EM images in the activated spliceosome is actually driven further apart from the active conformation for catalysis. It has been proposed that the four-helix junction that predominates in the protein-free state (Zhao et al. 2013) exists only to protect the premature formation of active site (Sashital et al. 2004), or is simply just a lower energy conformer in vitro.
While these studies focused on behavior of junction alone, parallel experiments on complexes in which the ACAGAGA loop was restored, with or without pairing of the U2 snRNA side of the ACAGAGA loop with a short fragment to form a branch site helix, found no detectible difference in the distance between the termini of Helix III and the ISL, even in the presence of high concentrations of Mg2+ (data not shown). Inclusion of the region of the ACAGAGA sequence that participates in the long-range interaction, in the protein-free system, is insufficient to promote folding. This observation suggests that although all elements required for catalytic activity are present in the RNA, the inability to form or stabilize the active site by the U2–U6 snRNA complex alone is mirrored by the exceptionally slow (and low-yield) rate of catalysis by the RNA alone, and even then only a modified reaction by sequences with multiple mutations to stabilize interaction between the U2–U6 snRNA complex and an intron strand (Valadkhan and Manley 2001; Valadkhan et al. 2007, 2009).
These long-range interactions are structurally equivalent to their counterparts in the Group II intron, in which a triple helix is defined by interaction of the Domain 5 bulge and J2/3 with the major groove edge of the catalytic AGC triad (Toor et al. 2008). Crystal structures of a Group IIC self-splicing intron identified the importance of an elaborate network of long-range interactions to stabilize the catalytic DV and other components (Toor et al. 2010); this situation is not paralleled in the U2–U6 snRNA complex in spliceosomes, where regions of snRNAs not directly involved in forming the catalytic site are dispersed in a protein-rich environment (Zhang et al. 2019 and references therein). The role of RNA–RNA tertiary interactions involving multiple domains to stabilize the catalytic center in the Group II intron is in stark contrast with the reliance on protein–RNA interactions to stabilize the catalytic core of the spliceosome, and points to a vital difference between the two splicing systems.
These findings imply that the context of the U2–U6 snRNA complex in the intact spliceosome contributes to shifting toward the three-helix conformer. Protein components clearly fulfill an essential role in facilitating the fold of the U2–U6 snRNA complex into a catalytically active fold, which starts to form in the Bact stage, although images (Yan et al. 2016) imply that the catalytic Mg2+ ions are not in place until the B* stage. The NTC-related protein RBM22 in the human spliceosome (related to Cwc2 in S. cerevisiae) is implicated in the final folding activity (McGrail et al. 2009; Rasche et al. 2012; Schmitzová et al. 2012). The essential role of Cwc2 in yeast spliceosomes was demonstrated by effects of its deletion from yeast spliceosomes, that is, complete inhibition of the first step of splicing; subsequent supplementation of exogenous Cwc2 rescued the catalytic activity. Rasche et al. (2012) have demonstrated that these NTC-related proteins make contact with U6 snRNA in the 5′ terminus and the upper part of the ISL—although it was shown that residues of the protein residing in its RRM, finger, and region connecting the Torus and RRM domains may be involved in this binding, it is not clear precisely which region of the protein recognizes which region of the RNA.
RBM22 has been shown to have interaction with U2–U6 snRNA shown by cross-links between the protein and the RNA in the ISL loop and the 5′ single stranded region of U6, ACAGAGA (Rasche et al. 2012). However, other work in our laboratory (J Ciavarella, W Perea, NL Greenbaum, unpubl.) have shown that these regions only account for a fraction of binding affinity, suggesting that additional contacts that do not participate in cross-linking are important for interaction, and perhaps for their role in RNA folding. One possibility is the unpaired region of the central junction, especially the more open junction observed in the three-helix conformer.
We proposed that a role of RBM22, as well as a structural role for Mg2+, is to overcome the small energy barrier to favor the three-helix junction conformer favored in vivo. We showed that RBM22 binds more tightly to the wild-type complex than it does to the four-helix junction mutant, suggesting that RBM22 preferentially binds the more open junction in the three-helix conformer in addition to other sites identified by cross-linking (Rasche et al. 2012). This preference for the three-helix conformer shifts equilibrium values of the two junction conformers to favor the three-helix form observed in active spliceosomes, thereby explaining the prevalence of this conformation in the spliceosome, where it would permit formation of the catalytically active form.
MATERIALS AND METHODS
Design and synthesis of RNA constructs
To determine three-dimensional conformations of stems surrounding the central junction of the protein-free U2–U6 snRNA complex, we first designed, prepared, and paired RNA oligomers labeled with fluorescent dyes at designated sites.
To create a pairing representing the human U2–U6 snRNA complex with the native (WT) sequence in the region of the central junction that would be amenable to measurement of distances between helix termini by FRET, we designed two strands: (i) a 32 nt truncated U6 strand started from the 5′ terminal end of U6 snRNA (i.e., starting at G33, 8 nt upstream of the ACAGAGA sequence), and terminated just before the hairpin pentaloop of the ISL, synthesized by IDT; and (ii) a U6–U2 “chimeric” oligomer starting immediately 3′ of the ISL hairpin loop and connecting with the 5′ stem of U2 snRNA at the end of Helix II via a GCAA tetraloop to the U2 snRNA strand through U46 of the native sequence. The native sequence at the 5′ terminus of the human U6 snRNA oligomer includes nine nucleotides capable of forming Watson–Crick base pairs with complementary nucleotides at the 3′ end of the U2 snRNA oligomer. Although we observe these base pairs in vitro (Zhao et al. 2013), this pairing has been shown not to occur in vivo (Anokhina et al. 2013) but since the duplex formation observed in the absence of other spliceosomal components is useful for measurement of FRET-based distances between termini of stems, and thus of angles, between Helix I and the U6 ISL, we have maintained such a duplex in each of our constructs.
To create a construct that focused solely on the native junction without the added flexibility in the Helix I/III stem from the large open ACAGAGA region, we also modified the sequence of U2 snRNA opposing the ACAGAGAA loop of U6 snRNA to form a complementary Helix I/III stem (WTΔL; Fig. 1A). All nucleotides within the region forming the central junction are present in the wild-type sequences; modifications include truncation of sequence regions beyond paired stems and, in some cases, removal of the ISL pentaloop; a change in the 3′ terminal nucleotide of the truncated U6 strand (U64) and the 5′ nt of the chimeric U6/U2 strand (A70) to create a C–G pair (maintaining base pairing adjacent to the deleted hairpin loop of U6 ISL) and facilitate transcription of the chimeric strand. The chimeric strand was prepared by in vitro transcription techniques using double-stranded DNA templates (IDT) and T7 polymerase overexpressed and purified in the laboratory, followed by gel purification and electroelution of the final product.
We next used a construct, based on WTΔL, that we had previously designed (Zhao et al. 2013) to limit junction conformation to the four-helix conformer by mutating several U2 Stem I nucleotides: mutating G12–C21 to C12–G21, as well as U17U18 to C17G18 (construct 4HJΔL1). These modifications disrupt base pairing of the AGA triad in U6 snRNA to U2 snRNA (disfavoring the three-helix fold) and stabilize Stem I (to favor the four-helix fold), respectively. All other modifications described in WTΔL, including the closing of the ACAGAGA loop, were maintained (Fig. 1B).
To measure the distance between the U6 ISL and Helix II while maintaining the same junction features as in 4HJΔL1, we made changes to the design of 4HJΔL1 to enable attachment of linkers and dyes to the 5′ terminus of U2 snRNA (4HJΔL2; Fig. 1C). In design of this construct, the connecting tetraloop GCAA was “moved” from the end of Helix II to the end of Helix I/III, resulting in a chimeric strand starting with the 5′ terminus of U2 snRNA, and connecting to the 5′ end of U6 snRNA by the tetraloop. We also removed five base pairs from Helix III (this helix was not dye-labeled in this experiment) to compensate for addition of the loop and added two base pairs to U6 ISL to increase dye-labeling yield. The 5′ 31 nt fragment representing the 5′ side of the ISL and U6 strand of Helix II as synthesized by Dharmacon (synthesized with a 5′ phosphate to facilitate linker attachment); the longer chimeric strand representing U2 snRNA and the 5′ segment of U6 snRNA was generated by in vitro transcription as described above.
To measure the distance between 5′ of U2 snRNA and 5′ of U6 snRNA, we designed a construct favoring four-helix junction fold used in construct 4HJΔL3 (Fig. 1D). The mismatch (U6)U90–(U2)U10 in Helix II was modified to form a Watson–Crick pair by changing (U2)U10 to (U2)A10; Helix II and Helix III have the same number of base pairs as 4HJΔL1.
To measure the distance between 5′ of U6 snRNA and internal U16 of Stem I, we designed construct 4HJΔL4 (Fig. 1E). The U6 strand, which represents the U6 snRNA sequences between Helix III and Helix II, including the ISL pentaloop, was transcribed and purified as above. The U2 strand containing a linker-labeled internal U16 was synthesized chemically (IDT); U16 was incorporated as the amino-modified phosphoramidite iUniAmM. The six-carbon linker is attached covalently through the C5 of uridine, providing a free primary amine that attaches to a fluorophore by the same reaction as attachment at the terminus of an oligomer.
Finally, to assess the impact of the ACAGAGA loop on angles between Helix I/III and the ISL as a result of flexibility or long-range interactions with elements in the ISL, we reintroduced the native U2 snRNA sequence opposing the U6 ACAGAGA sequence in two constructs, WTΔL and 4HJΔL1 to create WT and 4HJ, respectively (Fig. 1F,G).
All transcribed RNAs were purified by ethanol precipitation and electrophoresis on a 20% polyacrylamide gel. Desired RNA bands were eluted with an Elutrap device at 4°C, 100 V overnight. The concentration of RNA was determined by absorbance at 260 nm.
Pairing of strands
The base pairing of all RNA constructs was performed in nondenaturing 15% PAGE gel. For each RNA complex, 40 pmol of each RNA strand were mixed and annealed by heating at 85°C for 3 min and cooling at room temperature for 10 min prior to loading on the gel in a buffer of Tris (30 mM)-HEPES (60 mM), pH 7.6, 30 mM NaCl, 1 mM EDTA. As controls, individual strands were subjected to the same process. Samples were then applied to nondenaturing 15% PAGE at 4°C, 120 V, for 4 h in a buffer of Tris-HEPES, pH 7.6. Gels were stained with ethidium bromide and visualized under UV light at 302 nm (and/or visualized by fluorescence of attached dyes). Complete pairing was confirmed by appearance of a single band with slower electrophoretic mobility than either of the individual strands (Supplemental Fig. S5).
Linker and dye labeling
The donors (fluorescein NHS ester, AF488 NHS ester) and the acceptors (Cy3 NHS ester, AF555 NHS ester) were attached covalently to linker-modified 5′ phosphate termini or a linker-modified internal uridine residue (e.g., construct 4HJΔL4) via a two-carbon (in the transcribed RNAs) or six-carbon (in the chemically synthesized RNAs) primary diamine linker. The two-carbon primary diamine linker was added to 5′ phosphate termini using protocol Tech tip #30 provided by Thermo Scientific (ThermoFisher.com). A 10 times excess of fluorophore (250 µg dissolved in 14 µL DMSO) was added to 10 nmol of amine-modified RNA in 0.1 M sodium tetraborate buffer, pH 8.5, to a total volume of 100 µL in a darkened room. The reaction mixtures were mixed occasionally during the first 2 h and then left to incubate overnight at room temperature. The dye labeled RNAs were purified by ethanol precipitation followed by 20% polyacrylamide gel electrophoresis and eluted by a “crush and soak” method. The dye-labeling yield was determined by absorbance of RNA and the fluorophore at 260 nm and the excitation wavelength of each dye, respectively. The base pairing of labeled RNA strands was tested as described above to confirm that labeling did not diminish pairing efficiency.
FRET measurements
In these studies, we used FRET measurements to determine distances <100 Å from a donor D to an acceptor A attached to two sites within a paired complex. The rate of energy transferred from D to A is measured by the decrease of Donor emission or by decrease in the lifetime of the D at the wavelength where only D emits when A is presented. Samples for FRET experiments were made using the same solution conditions used for pairing experiments, with the one difference that [RNA] in FRET samples was 150 nM.
Steady state measurements were performed on Fluorolog-3 from Horiba Scientific. We excited fluorescein at 495 nm (5 nm bandwidth) and measured Cy3 emission intensity at 518 nm. RNA strands labeled with D or A only were used as control samples. For time-resolved measurements (also performed on the Horiba instrument), fluorescence decay curves of D in RNA solutions (150 nM) were recorded on a timescale of 20 nsec to a total of 10,000 counts in the peak (5 nm bandwidth). To measure the lifetime of D only, we used control samples in which the dye labeled RNA was paired with the unlabeled RNA (same RNA with the acceptor used for FRET measurements). In FRET samples, D labeled RNA and A labeled RNA were paired with each other. Instrument response function was measured by Ludox scattering solution in water (internal calibration by instrument). For experiments testing the effect of titration of Mg2+, each aliquot of Mg2+ was assayed on a separate sample.
Donor decay lifetimes in the absence and presence of acceptor
All measurements were performed at room temperature in Tri-HEPES buffer, pH 7.6. The fluorescence lifetime of the D in the presence or absence of the acceptor should be measured at wavelength where no emission of the acceptor is observed. We excited samples at 495 nm and recorded the signal at 518 nm with fluorescein; 490 and 525 nm with AF488 dye. The decay curves were fitted with two-exponential equation giving αi (fractional amplitude associated with each lifetime) and τi (lifetime components of D):
where is normalized to unity. The results were used to calculate average lifetime of two decay time components, which was used to calculate FRET efficiencies. The average lifetime of D was calculated by τavg = . For curves best fit by a biexponential decay, the fitted curve is deconvoluted into two individual lifetimes, τ1 and τ2, each with its fractional contribution to the total: τ1 + σ1 with relative amplitude of α1 and τ2 + σ2 with relative amplitude of α2 in which σi represents the standard deviation for lifetime i.
The quality of fit was judged by reduced chi-squared value (reduced χ2), which is expected to be near unity for a good fit. The FRET efficiency was determined by a decrease in the lifetime of D in the presence of A (E = 1 − τDA/τD) and results are given in Table 1.
TABLE 1.
Distances between dyes labeled at termini of U2–U6 snRNA constructs used in this study in solution as described in Materials and Methods, without and with 40 mM added Mg2+

Calculation of donor-acceptor distance
The distance between D and A, R, is:
where R0 is the Förster distance between D and A at which E (FRET efficiency) is 50%; R0 = 56 Å (Norman et al. 2000) for the fluorescein-Cy3 pair and 70 Å for AF488–AF555 pair.
Calculation of angles between stems
Distances between two termini were translated into estimated angles between stems X and Y. Calculation of stem length assumed A-type helical stacking parameters and included the approximation that the Cy3 dye + linker stacked onto RNA helices, contributing ∼2.6 Å, equivalent to an additional base pair, to the length of a helix. Fluorescein is mobile in solution due to its positive charge (Norman et al. 2000). In order to calculate the length of helices, we estimated the overall contribution of fluorescein to the length of RNA helices by performing trFRET on dye-labeled B-type DNA duplexes of 8 and 18 bp. Resulting data indicated that fluorescein with a two-carbon primary diamine linker contributes ∼8.5 Å to the length of a duplex.
Simulation and visualization of hU2–U6 snRNA models
The predictive computational model of two major conformations of hU2–U6 snRNA complex was obtained by simulation using SimRNA (Magnus et al. 2016), an automated and online interface for modeling three-dimensional structure of RNAs. SimRNA uses a coarse-grained representation of RNA molecule; the Monte-Carlo method to sample the conformational space and statistical potential to introduce the interaction in the RNA folding process. We used RNA sequences, secondary structure restrains (base pairs), and distance restrains obtained from tr-FRET as parameters of the RNA folding simulation. The modeled RNA structures were then visualized by NGLview (Nguyen et al. 2018).
Electrophoretic mobility shift assay (EMSA) for U2U6–RBM22 interaction analysis
RBM22 was expressed and purified as described in the Supplemental Information. The concentration of RNA (wild-type junction and four-helix junction mutant) was maintained constant at 10 µM while we varied concentration of RBM22 from 5 µM to 15 µM. The RNA-RBM22 mixtures were prepared in NaPi buffer pH 6.5 and incubated at room temperature for 1 h, then loaded on an EMSA gel. The gel was run in 30 mM MOPs, 25 mM Histidine buffer pH 6.5 at 4°C for 90 min and then stained with Commasie Blue overnight. Only RBM22 and RNA-RBM22 complex bands were exposed after staining with Coommasie Blue. The RNA–protein band was visualized at 302 nm.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Professor David Lilley for his helpful advice testing dye mobility, and Wendy Li and Kenel Zhao for assistance with sample preparation. N.L.G. acknowledges financial support from the City University of New York PSC-CUNY award program (TRADB-48-370) and from the National Science Foundation (#MCB-0929394).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.073379.119.
REFERENCES
- Anokhina M, Bessonov S, Miao Z, Westhof E, Hartmuth K, Lührmann R. 2013. RNA structure analysis of human spliceosomes reveals a compact 3D arrangement of snRNAs at the catalytic core. EMBO J 32: 2804–2818. 10.1038/emboj.2013.198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai R, Wan R, Yan C, Lei J, Shi Y. 2018. Structures of the fully assembled Saccharomyces cerevisiae spliceosome before activation. Science 360: 1423–1429. 10.1126/science.aau0325 [DOI] [PubMed] [Google Scholar]
- Bertram K, Agafonov DE, Dybkov O, Haselbach D, Leelaram MN, Will CL, Urlaub H, Kastner B, Lührmann R, Stark H. 2017a. Cryo-EM structure of a pre-catalytic human spliceosome primed for activation. Cell 170: 701–713 e711. 10.1016/j.cell.2017.07.011 [DOI] [PubMed] [Google Scholar]
- Bertram K, Agafonov DE, Liu WT, Dybkov O, Will CL, Hartmuth K, Urlaub H, Kastner B, Stark H, Lührmann R. 2017b. Cryo-EM structure of a human spliceosome activated for step 2 of splicing. Nature 542: 318–323. 10.1038/nature21079 [DOI] [PubMed] [Google Scholar]
- Burke JE, Sashital DG, Zuo X, Wang YX, Butcher SE. 2012. Structure of the yeast U2/U6 snRNA complex. RNA 18: 673–683. 10.1261/rna.031138.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan SP, Kao DI, Tsai WY, Cheng SC. 2003. The Prp19p-associated complex in spliceosome activation. Science 302: 279–282. 10.1126/science.1086602 [DOI] [PubMed] [Google Scholar]
- Clegg RM, Murchie AI, Lilley DM. 1994. The solution structure of the four-way DNA junction at low-salt conditions: a fluorescence resonance energy transfer analysis. Biophys J 66: 99–109. 10.1016/S0006-3495(94)80765-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corry B, Jayatilaka D. 2008. Simulation of structure, orientation, and energy transfer between AlexaFluor molecules attached to MscL. Biophys J 95: 2711–2721. 10.1529/biophysj.107.126243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio P, Abelson J. 1990. Two domains of yeast U6 small nuclear RNA required for both steps of nuclear precursor messenger RNA splicing. Science 250: 404–409. 10.1126/science.2145630 [DOI] [PubMed] [Google Scholar]
- Fica SM, Mefford MA, Piccirilli JA, Staley JP. 2014. Evidence for a group II intron-like catalytic triplex in the spliceosome. Nat Struct Mol Biol 21: 464–471. 10.1038/nsmb.2815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon PM, Sontheimer EJ, Piccirilli JA. 2000. Metal ion catalysis during the exon-ligation step of nuclear pre-mRNA splicing: extending the parallels between the spliceosome and group II introns. RNA 6: 199–205. 10.1017/S1355838200992069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Karunatilaka KS, Rueda D. 2009. Single-molecule analysis of protein-free U2–U6 snRNAs. Nat Struct Mol Biol 16: 1154–1159. 10.1038/nsmb.1672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg R, McGrail JC, O'Keefe RT. 2010. The function of the NineTeen Complex (NTC) in regulating spliceosome conformations and fidelity during pre-mRNA splicing. Biochem Soc Trans 38: 1110–1115. 10.1042/BST0381110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karunatilaka KS, Rueda D. 2014. Post-transcriptional modifications modulate conformational dynamics in human U2-U6 snRNA complex. RNA 20: 16–23. 10.1261/rna.041806.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keating KS, Toor N, Perlman PS, Pyle AM. 2010. A structural analysis of the group II intron active site and implications for the spliceosome. RNA 16: 1–9. 10.1261/rna.1791310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Bokinsky G, Walter NG, Zhuang X. 2007. Dissecting the multistep reaction pathway of an RNA enzyme by single-molecule kinetic “fingerprinting”. Proc Natl Acad Sci 104: 12634–12639. 10.1073/pnas.0610597104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luukkonen BG, Séraphin B. 1998. A role for U2/U6 helix Ib in 5′ splice site selection. RNA 4: 915–927. 10.1017/S1355838298980591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhani HD, Guthrie C. 1994. Randomization-selection analysis of snRNAs in vivo: evidence for a tertiary interaction in the spliceosome. Genes Dev 8: 1071–1086. 10.1101/gad.8.9.1071 [DOI] [PubMed] [Google Scholar]
- Magnus M, Boniecki MJ, Dawson W, Bujnicki JM. 2016. SimRNAweb: a web server for RNA 3D structure modeling with optional restraints. Nucleic Acids Res 44: W315–W319. 10.1093/nar/gkw279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova OV, Makarov EM, Urlaub H, Will CL, Gentzel M, Wilm M, Lührmann R. 2004. A subset of human 35S U5 proteins, including Prp19, function prior to catalytic step 1 of splicing. EMBO J 23: 2381–2391. 10.1038/sj.emboj.7600241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGrail JC, Krause A, O'Keefe RT. 2009. The RNA binding protein Cwc2 interacts directly with the U6 snRNA to link the nineteen complex to the spliceosome during pre-mRNA splicing. Nucleic Acids Res 37: 4205–4217. 10.1093/nar/gkp341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford MA, Staley JP. 2009. Evidence that U2/U6 helix I promotes both catalytic steps of pre-mRNA splicing and rearranges in between these steps. RNA 15: 1386–1397. 10.1261/rna.1582609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen H, Case DA, Rose AS. 2018. NGLview—interactive molecular graphics for Jupyter notebooks. Bioinformatics 34: 1241–1242. 10.1093/bioinformatics/btx789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman DG, Grainger RJ, Uhrín D, Lilley DM. 2000. Location of cyanine-3 on double-stranded DNA: importance for fluorescence resonance energy transfer studies. Biochemistry 39: 6317–6324. 10.1021/bi992944a [DOI] [PubMed] [Google Scholar]
- Plaschka C, Lin PC, Nagai K. 2017. Structure of a pre-catalytic spliceosome. Nature 546: 617–621. 10.1038/nature22799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasche N, Dybkov O, Schmitzová J, Akyildiz B, Fabrizio P, Lührmann R. 2012. Cwc2 and its human homologue RBM22 promote an active conformation of the spliceosome catalytic centre. EMBO J 31: 1591–1604. 10.1038/emboj.2011.502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauhut R, Fabrizio P, Dybkov O, Hartmuth K, Pena V, Chari A, Kumar V, Lee CT, Urlaub H, Kastner B, et al. 2016. Molecular architecture of the Saccharomyces cerevisiae activated spliceosome. Science 353: 1399–1405. 10.1126/science.aag1906 [DOI] [PubMed] [Google Scholar]
- Romani A, Scarpa A. 1992. Regulation of cell magnesium. Arch Biochem Biophys 298: 1–12. 10.1016/0003-9861(92)90086-C [DOI] [PubMed] [Google Scholar]
- Sashital DG, Cornilescu G, McManus CJ, Brow DA, Butcher SE. 2004. U2–U6 RNA folding reveals a group II intron-like domain and a four-helix junction. Nat Struct Mol Biol 11: 1237–1242. 10.1038/nsmb863 [DOI] [PubMed] [Google Scholar]
- Schmitzová J, Rasche N, Dybkov O, Kramer K, Fabrizio P, Urlaub H, Lührmann R, Pena V. 2012. Crystal structure of Cwc2 reveals a novel architecture of a multipartite RNA-binding protein. EMBO J 31: 2222–2234. 10.1038/emboj.2012.58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner M, Karunatilaka KS, Sigel RK, Rueda D. 2008. Single-molecule studies of group II intron ribozymes. Proc Natl Acad Sci 105: 13853–13858. 10.1073/pnas.0804034105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steitz TA, Steitz JA. 1993. A general two-metal-ion mechanism for catalytic RNA. Proc Natl Acad Sci 90: 6498–6502. 10.1073/pnas.90.14.6498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan E, Wilson TJ, Nahas MK, Clegg RM, Lilley DM, Ha T. 2003. A four-way junction accelerates hairpin ribozyme folding via a discrete intermediate. Proc Natl Acad Sci 100: 9308–9313. 10.1073/pnas.1233536100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toor N, Keating KS, Taylor SD, Pyle AM. 2008. Crystal structure of a self-spliced group II intron. Science 320: 77–82. 10.1126/science.1153803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toor N, Keating KS, Fedorova O, Rajashankar K, Wang J, Pyle AM. 2010. Tertiary architecture of the Oceanobacillus iheyensis group II intron. RNA 16: 57–69. 10.1261/rna.1844010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valadkhan S, Manley JL. 2001. Splicing-related catalysis by protein-free snRNAs. Nature 413: 701–707. 10.1038/35099500 [DOI] [PubMed] [Google Scholar]
- Valadkhan S, Mohammadi A, Wachtel C, Manley JL. 2007. Protein-free spliceosomal snRNAs catalyze a reaction that resembles the first step of splicing. RNA 13: 2300–2311. 10.1261/rna.626207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valadkhan S, Mohammadi A, Jaladat Y, Geisler S. 2009. Protein-free small nuclear RNAs catalyze a two-step splicing reaction. Proc Natl Acad Sci 106: 11901–11906. 10.1073/pnas.0902020106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter F, Murchie AI, Lilley DM. 1998. Folding of the four-way RNA junction of the hairpin ribozyme. Biochemistry 37: 17629–17636. 10.1021/bi9821115 [DOI] [PubMed] [Google Scholar]
- Wan R, Yan C, Bai R, Huang G, Shi Y. 2016. Structure of a yeast catalytic step I spliceosome at 3.4 Å resolution. Science 353: 895–904. 10.1126/science.aag2235 [DOI] [PubMed] [Google Scholar]
- Wan R, Bai R, Yan C, Lei J, Shi Y. 2019. Structures of the catalytically activated yeast spliceosome reveal the mechanism of branching. Cell 177: 339–351 e313. 10.1016/j.cell.2019.02.006 [DOI] [PubMed] [Google Scholar]
- Will CL, Lührmann R. 2011. Spliceosome structure and function. Cold Spring Harb Perspect Biol 3: a003707 10.1101/cshperspect.a003707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Manley JL. 1989. Mammalian pre-mRNA branch site selection by U2 snRNP involves base pairing. Genes Dev 3: 1553–1561. 10.1101/gad.3.10.1553 [DOI] [PubMed] [Google Scholar]
- Yan C, Hang J, Wan R, Huang M, Wong CC, Shi Y. 2015. Structure of a yeast spliceosome at 3.6 Å resolution. Science 349: 1182–1191. 10.1126/science.aac7629 [DOI] [PubMed] [Google Scholar]
- Yan C, Wan R, Bai R, Huang G, Shi Y. 2016. Structure of a yeast activated spliceosome at 3.5 Å resolution. Science 353: 904–911. 10.1126/science.aag0291 [DOI] [PubMed] [Google Scholar]
- Yan C, Wan R, Bai R, Huang G, Shi Y. 2017. Structure of a yeast step II catalytically activated spliceosome. Science 355: 149–155. 10.1126/science.aak9979 [DOI] [PubMed] [Google Scholar]
- Yuan F, Griffin L, Phelps L, Buschmann V, Weston K, Greenbaum NL. 2007. Use of a novel Förster resonance energy transfer method to identify locations of site-bound metal ions in the U2-U6 snRNA complex. Nucleic Acids Res 35: 2833–2845. 10.1093/nar/gkm134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Yan C, Zhang X, Lei J, Shi Y. 2018. Structure of a human catalytic step I spliceosome. Science 359: 537–545. 10.1126/science.aar6401 [DOI] [PubMed] [Google Scholar]
- Zhang L, Vielle A, Espinosa S, Zhao R. 2019. RNAs in the spliceosome: insight from cryoEM structures. Wiley Interdiscip Rev RNA 10: e1523 10.1002/wrna.1523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Bachu R, Popovic M, Devany M, Brenowitz M, Schlatterer JC, Greenbaum NL. 2013. Conformational heterogeneity of the protein-free human spliceosomal U2–U6 snRNA complex. RNA 19: 561–573. 10.1261/rna.038265.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Anklin C, Greenbaum NL. 2014. Use of 19F NMR methods to probe conformational heterogeneity and dynamics of exchange in functional RNA molecules. Methods Enzymol 549: 267–285. 10.1016/B978-0-12-801122-5.00012-X [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.