

Abstract
Current ovarian cancer maintenance therapy is limited by toxicity and no proven impact on overall survival. To study a maintenance strategy targeted at missense mutant p53, we hypothesized that the release of mutant p53 from mortalin inhibition by the SHetA2 drug combined with reactivation of mutant p53 with the PRIMA-1MET drug inhibits growth and tumor establishment synergistically in a mutant-p53 dependent manner. The Cancer Genome Atlas (TCGA) data and serous ovarian tumors were evaluated for TP53 and HSPA9/mortalin status. SHetA2 and PRIMA-1MET were tested in ovarian cancer cell lines and fallopian tube secretory epithelial cells using isobolograms, fluorescent cytometry, Western blots and ELISAs. Drugs were administered to mice after peritoneal injection of MESOV mutant p53 ovarian cancer cells and prior to tumor establishment, which was evaluated by logistic regression. Fifty-eight percent of TP53 mutations were missense and there were no mortalin mutations in TCGA high-grade serous ovarian cancers. Mortalin levels were sequentially increased in serous benign, borderline and carcinoma tumors. SHetA2 caused p53 nuclear and mitochondrial accumulation in cancer, but not in healthy, cells. Endogenous or exogenous mutant p53 increased SHetA2 resistance. PRIMA-1MET decreased this resistance and interacted synergistically with SHetA2 in mutant and wild type p53-expressing cell lines in association with elevated reactive oxygen species/ATP ratios. Tumor-free rates in animals were 0% (controls), 25% (PRIMA1MET), 42% (SHetA2) and 67% (combination). SHetA2 (p = 0.004) and PRIMA1MET (p = 0.048) functioned additively in preventing tumor development with no observed toxicity. These results justify the development of SHetA2 and PRIMA-1MET alone and in combination for ovarian cancer maintenance therapy.
Keywords: p53, mortalin, high-grade serous ovarian cancer, SHetA2, PRIMA-1
Introduction
Despite being cancer-free after primary cytoreductive surgery and platinum-based chemotherapy, approximately 70% of ovarian cancer patients will relapse after 3 years.1 Maintenance therapy using the FDA-approved PARP-1 inhibitor, olaparib, in BRCA mutation-positive patients, or the angiogenesis inhibitor, bevacizumab, in all other patients, was shown to prolong time to recurrence,2–4 however no improvement in overall survival was found in long term follow-up for bevacizumab or interim analysis of 21% of the data for olaparib.2,5 Both of these therapies have significant side effects that limit dosing and duration of treatment.2,5 Furthermore, olaparib caused an increased risk of developing secondary cancers.2
A rational approach for developing nontoxic maintenance therapy for ovarian cancer is to target p53 mutations. TP53 gene mutations occur with a 96–100% frequency in high-grade serous ovarian cancer (HGSOC), the most common and lethal type of ovarian cancer,6,7 which can arise from fallopian tube fimbriae.8 Mutation or loss of p53 protein can reduce cancer therapy response.9 Currently p53 is targeted by multiple anti-cancer drug strategies, including reactivation of the wild type p53 functions in missense mutant p53 (m-p53) and release of p53 from inhibitory proteins.10 PRIMA-1 is a p53 reactivator that modifies amino acids in the m-p53 core domain to restore proper protein conformation and function.11 Its methylated analog, PRIMA-1MET (APR-246), exhibited greater cancer cell line cytotoxicity and synergy with chemotherapy and is undergoing clinical investigation.12 A first-in-human clinical trial found PRIMA-1MET to have a favorable pharmacokinetic pro-file, to induce wild type p53 molecular and cellular activities in tumors and to be safe at plasma levels predicted to have therapeutic effects.13 Currently, clinicaltrials.gov reports multiple ongoing trials evaluating PRIMA-1MET in combination with chemotherapy for several malignancies, including recurrent ovarian cancer.
Another drug in development that can induce p53 is SHetA2 (NSC 726189), an orally bioavailable, small molecule drug that disrupts mortalin/p53 complexes in ovarian cancer cells.14 This drug is currently being developed for clinical trials based on its induction of apoptosis in cancer cells, while its effect on healthy cells is limited to G1 cell cycle arrest.15–19 SHetA2 also exhibited chemoprevention activity.20,21 Extensive preclinical studies conducted by the US National Cancer Institute (NCI) RAID and RAPID Programs, and others, demonstrated that SHetA2 did not cause mutagenicity,22 teratogenicity22 or toxicity,23 and exhibited a pharmacologic profile suitable for an oral chemoprevention agent.23–25 Peer-reviewed NCI R01 and PREVENT grants are supporting first-in-human clinical trials of SHetA2 capsules.
Our objective was to develop an m-p53-targeted strategy for maintenance therapy of HGSOC. We hypothesized that SHetA2 release of m-p53 from mortalin inhibition combined with reactivation of m-p53 by PRIMA-1 or PRIMA-1MET is synergistic in killing ovarian cancer cells that harbor p53 point mutations, and not in cancer cells with null p53 mutations, or in healthy cells.
Materials and Methods
Cell lines, plasmids and drugs
Ovarian cancer cells lines A2780 (RRID:CVCL_0134), OV-90 (RRID:CVCL_3768), SKOV3 (RRID:CVCL_0532), COV362 (RRID:CVCL_2420), OVSAHO (RRID:CVCL_3114), Caov3 (RRID:CVCL_0201) and OVCAR4 (RRID:CVCL_1627) (all from American Type Culture Collection/ATCC Manassas, VA) were cultured in RPMI1640 medium containing 10% FBS and 1× antibiotic/antimycotic. The MESOV/GFP-luc (RRID: CVCL_CZ92) cell line (gift from Dr Francois Moisan, Stanford University) (MESOV) was cultured in DMEM-high glucose medium with 10% FBS. SKOV3 cells stably transfected with mutant p53 (R248W, R273H) or parent vector were provided by Jeremy Chien, PhD (University of Kansas Medical Center). Human fallopian tube secretory epithelial cells (hFTSECs) were isolated from fallopian tube fimbriae surgical specimens under an IRB approved protocol as previously described.26 All cell lines were authenticated by autosomal short tandem repeat (STR) profiles determination within 3 years and comparison with reference databases by the University of Arizona Genetics Core and IDEXX BioAnalytics (Columbia, MO). Experiments were performed with cell lines free of mycoplasma contamination. Cultures were transfected with plasmids HSPA9 pLX304 or pLKO-p53-shRNA-427, PG13-Luc (Addgene, Watertown, MA) and treated with SHetA2 and PRIMA-1 (National Cancer Institute PREVENT Program) and PRIMA-1MET (Cayman Chemicals, Ann Arbor, MI) dissolved in dimethyl sulfoxide (DMSO). Caspase 3 inhibitor Z-DEVD-FMK and N-acetyl-L-Cysteine (Cayman Chemicals) were used to study the mechanism of drug action. PRIMA-1MET was synthesized for the animal studies by the Center for Cancer Prevention and Drug Development (Stephenson Cancer Center) according to published methods.27
TP53 mutation identification
Exome sequencing data from 429 TCGA ovarian cancer cases was analyzed for genome-wide mutations as previously described7 and illustrated (Supporting Information Fig. S3). The genomic coordinates of these mutations were converted from human genome build hg18 to hg19 using the UCSC Genome Browser liftOver tool (http://genome.ucsc.edu/cgi-bin/hgLiftOver). A total of 405 high-confidence mutations were located within the transcript locus of p53. Mutations were assigned to exons of ENSEMBL p53 transcripts based on their location and predicted mutation effect.
Mortalin expression in ovarian tissue
The Gynecologic Oncology Group (GOG) Disease Progression Tissue Microarray (TMA) was stained for mortalin using Abcam rabbit polyclonal antibody ab53098. Prior to staining the TMA, antibody dilutions and other immunohistochemical staining conditions were optimized on HGSOC sections using no primary antibody as negative controls. Heat-induced epitope retrieval was performed on formalin-fixed, paraffin-embedded TMA sections using a pressurized decloaking chamber (Biocare Medical, Pacheco, CA) in citrate buffer (pH 6.0) at 99°C for 18 min and then in 3% hydrogen peroxide at room temperature for 20 min. After incubation with primary antibody, positive signal was developed with a rabbit polymer–horseradish peroxidase secondary detection kit (Biocare Medical, Pacheco, CA) and diaminobenzidine (Sigma, St. Louis, MO). Digitized images of immunohistochemically-stained slides were generated and evaluated for mortalin positive staining intensities using the Positive Pixel Count algorithm software (Aperio Technologies, Inc., Vista, CA). Positivity scores (total number of positive pixels divided by total number of pixels) of the mortalin staining were derived separately for epithelial and stromal cells by tuning a Genie algorithm to differentiate between these two cell types on mortalin-stained ovarian tissues. Marked-up images of the staining intensity measurements generated by the Aperio imaging software were reviewed manually and individual cores that exhibited artifacts, lack of tumor on the section or inaccurate categorization of stromal versus epithelial cells were eliminated from the analysis. Each patient was represented on the slide by one to three sections. Individual cores that exhibited >10-fold difference from the two other cores from the same patient were eliminated from the averages.
Subcellular fraction enrichment
Cytoplasmic and nuclear protein fractions were isolated from cultures using NE-PER Nuclear and Cytoplasmic Extraction Reagents (ThermoFisher, Waltham, MA) and the Mitochondria/Cytosol Fractionation Kit (ThermoFisher) and protein concentrations determined with BCA reagent (ThermoFisher).
Western blots
Proteins were extracted from cell cultures using M-PER mammalian protein extraction reagent (ThermoFisher) and volumes corresponding to 30 μg determined with BCA reagent (ThermoFisher) were electrophoresed onto a SDS-PAGE gel and transferred to PVDF membranes. Membranes were blocked (5% dry skim milk powder in TBST buffer) for at least 1 hr or overnight, washed thrice with TBST and then incubated with primary antibody: p53 DO-1, β-actin, histone H3, GAPDH (Santa Cruz Biotechnology, Dallas, TX) and mortalin, Tom20, V5 tag antibodies, p21, PARP, SLC7A11, caspase 3 (Cell Signaling Technology, Danvers, MA) for at least 1 hr or overnight. After three washes, membranes were incubated with anti-mouse HRP conjugated (Santa Cruz Biotechnology) or anti-Rabbit HRP conjugated (Cell Signaling Technology) for 40 min. Membranes then were washed and developed with ECL reagents (BioRad, Hercules, CA). Bands were imaged and quantified (ChemiDoc imaging system with Image Lab Software/BioRad).
Immunofluorescence
Chamber slides (Lab-Tek II) were seeded with 1 × 104 cells in 1 ml medium and incubated up to 50–70% confluence. Cells treated with SHetA2 or an equivalent volume of DMSO were fixed in 2% paraformaldehyde in PBS, pH 7.4 for 30 min at room temperature, washed with PBS three times and then permeabilized with 0.5% Triton X-100 in PBS, pH 7.4 for 5 min. After three PBS washes, cells were incubated in PBS containing 0.1% BSA in PBS for 30 min at 37°C and then with p53 DO-1 antibody for 1 hr at 37°C. After three PBS washes, cells were incubated with the secondary antibody tagged with fluorochrome (Alexa fluor 488 or 647) for 1 hr at 37°C and washed thrice in PBS. Nuclei were stained with 300 nM DAPI for 7 min at room temperature and cells were washed thrice in PBS. Cells were covered in Prolong Gold mounting medium (Life Technology, Carlsbad, CA) and 1 mm thick cover glasses and left to dry overnight at room temperature and then imaged with a confocal microscope Leica SP2 (Leica, Wetzlar, Germany) at 63×.
MTT cytotoxicity assay
Cells plated at 2,000 cells/well in 96-well microtiter plates were incubated for 36–48 hr and treated with a range of doses and combinations of SHetA2 and PRIMA-1 or PRIMA-1MET, or the same volume of DMSO solvent for 72 hr of incubation. The percentage of growth for single drug treatments or fold effect (1 – ratio of treated/untreated growth) was measured using the CellTiter 96 AQueous Non-Radioactive Cell Proliferation Assay (Promega, Madison, WI) and the BioTeK SYNERGY H1 (BioTeK, Winooski, VT). Each experiment was performed in triplicate and repeated 2–3 times.
Transfection of cells
About 1 × 106 cells were electroporated with plasmid (5–10 μg) using the Neon transfection unit (Life Technology, Waltham, MA) at 1,170 V, 30 width and 2 pulses. Transfected cells were seeded onto 6-well and 96-well plates, and incubated for 36–48 hr prior to evaluation by Western blot and MTT assays, respectively. A stable cell line of A2780 cells transfected with pLKO-p53-shRNA-427 was generated by transfecting A2780 cells using the electroporation conditions above. Transfected cells were titrated for the optimum puromycin concentration needed for the selection, which was determined to be 20 μg. Transfected cells were grown under puromycin pressure for 2 weeks and after that continued to be grown in growth medium without puromycin. The puromycin-resistant cell line was evaluated for p53 knockdown using Western blot.
p53 luciferase reporter assay
To determine the transcriptional activity of p53, MESOV cells were transfected with PG13-Luc plasmids. Approximately 5,000 transfected cells were seeded on to each well of 96-well plates and incubated overnight. These cells were treated with either control solvent, 5 μM SHetA2 or 60 μM PRIMA-1MET for 24 hr. Luciferase activity of the reporter was measured with the One-Glo luciferase reporter assay kit (Promega, Madison, WI) and a Biotek Synergy H1 plate reader (BioteK, Winooski, VT). The MTT assay was performed in parallel cultures and used to normalize the p53 reporter activity to the number of viable cells.
Drug interactions
The potencies (IC50’s) of SHetA2 or PRIMA-1 analogs were determined using GraphPad Prism 6 software analysis of average MTT Optical Density versus drug dose. The drugs were mixed at a 1:1 ratio of their IC50 concentrations and a series of two fold dilutions evaluated in the MTT assay. Single drug treatments at doses surrounding the IC50 values were evaluated in parallel. All treatments were performed in triplicate and averages used to draw isobolograms and calculate combination indices (CI) and dose-reduction indices (DRIs) using CompuSyn Software according to the Chou and Talalay method.28 All experiments were repeated at least twice for each cell line to confirm the results.
Quantitative reverse transcriptase-polymerase chain reaction
SKOV3 parental and SKOV3 R273H p53 mutant cells were grown to 80% confluency in a 100 mm tissue culture dish and then treated with SHetA2 10 μM or PRIMA-1 20 μM or both drugs combined for 16 hr after which cells were harvested and RNA was isolated using RNeasy Mini Kit (QIAGEN). RNA quality and concentration were evaluated by measuring the OD260/280 ratio. The cDNA was synthesized from 1 μg RNA using RT2 First Strand Kit (Qiagen, Hilden, Germany) and then Real-time PCR was performed by using RT2 SYBR Green master mix (GeneCopoeia, Rockville, MD) and the BioRad CFX96 Real-Time PCR detection system (Bio-Rad, Winooski, VT). The real-time expression values were analyzed by calculating ΔΔCT and mean fold change (2−ΔΔCT) values.
Colony formation assay
Ovarian cancer cells MESOV (500 cells/well), A2780 (1,000 cells/well), SKOV3 R273H p53 (5,000 cells/well), SKOV3 R248W p53 (5,000 cells/well) and SKOV3 p53 Null (5,000 cells/well) were seeded in six-well plates in triplicate. After 24 hr, the cells were treated with either 5 μM SHetA2, 12.5 μM PRIMA1MET or the combination of both drugs for 8 hr, and then cells were maintained in fresh complete growth medium until cells in the control well had formed sufficiently large clones. Cells were fixed with 4% paraformaldehyde and then stained with 0.5% crystal violet dye for 1 hr. The visible colonies were photographed and the colonies were counted using Optronix GelCount colony counter (Scintica Instrumentation Inc. London, UK).
Assays for reactive oxygen species and ATP
The reactive oxygen species (ROS)-Glo H2O2 Assay and CellTiter-Glo 2.0 Assay (Promega, Madison, WI) kits were used to measure ROS and ATP, respectively, in cells cultured on 96-well plates with various combinations of SHetA2 (5 and 10 μM) and PRIMA-1 or PRIMA-1MET (25 μM), and the luminescence endpoints were measured with a BioTeK SYNERGY H1. The ROS assay luminescence endpoints were normalized to total viable cells using the MTT assay. For ROS to ATP ratio determination, the ROS luminescence endpoint was normalized to total protein content (μg/μl) measured with the BCA reagent in parallel wells, and the ATP concentrations were derived from a standard curve and normalized to total protein content. All assays were performed in triplicate and repeated three times.
Animal model
Four-week-old female athymic nude mice (NCr-Foxn1nu) were obtained from Taconic Biosciences under an approved Institutional Animal Care and Use Committee (IACUC) protocol. Mice were acclimatized for 2 weeks, fed with a standard diet and ear-tagged during the acclimatization period. The timing of tumor development after intraperitoneal injection with various amounts of MESOV cells was determined in a prior pilot study. In the intervention experiment, 6-week-old mice were injected with 20 million MESOV human ovarian cancer cells in their intraperitoneal region. Drug treatment was initiated 4 weeks after the injection of cancer cells. Mice were randomized to four treatment groups before the start of treatment based on their body weight. There was no significant (one-way ANOVA, p > 0.999) difference in body weight between groups at the start of treatment. Twelve mice were allocated to each group based on the power analysis performed while designing the experiment. The sample size was determined to provide over 80% power to detect a 915 mm3 difference in tumor volume between PRIMA-1MET treatment alone and the drug combination treatment using a two-sided 0.05 level t-test. SHetA2 was administered by gavage at 60 mg/kg in 30% Kolliphor HS15 (Sigma). Controls were gavaged with Kolliphor HS15 (30% in water). PRIMA-1MET (10 mg/kg) was dissolved in phosphate-buffered saline and injected into the peritoneum of mice every other day. Initially, SHetA2 dosing was administered every day for 2 weeks in SHetA2 alone and drug combination groups, and then switched to every other day for the next 3 weeks due to the stress caused to the mice by daily gavage. Oral gavaging was performed with sterile disposable plastic feeding tubes 20 ga × 38 mm (Instech Laboratories, Inc., Plymouth Meeting, PA). Three mice in the drug combination group died after 2 weeks of drug treatment due to gavaging incidents. Body weights were recorded and animal health was monitored weekly. After the end of drug intervention period, all mice were euthanized by CO2 asphyxiation followed by exsanguination, their tumors and other vital organs were collected for further analysis. After measuring the total tumor weight, a portion of the tumors were snap-frozen in liquid nitrogen and another portion was formalin-fixed. The formalin-fixed tissue samples were paraffin-embedded, sectioned and stained with hematoxylin and eosins for histology studies. An experienced pathologist (S.L.) reviewed blinded histology slides for kidney and liver toxicity.
Statistics
For tissue culture studies, the normality of data and statistical significance were analyzed using Prism 8 Software (GraphPad, San Diego, CA). Comparison of drug effects in the presence and absence of p53 knockdown or mortalin overexpression were done using T-tests corrected for multiple comparisons using the Holm–Sidak method. Comparison of drug combinations compared to single-drug treatments were done using a two-way ANOVA with a Tukey’s multiple comparison test. Values of p < 0.05 were considered significant.
The presence or absence of tumor at the end of the treatment period was used as the primary outcome measure. A binary variable (no tumor) was created which equaled 1 if the tumor weight was zero and equaled 0 otherwise. Two by two logistic regression was used to compare the four treatment groups (Untreated control, SHetA2, PRIMA-1MET, SHetA2 + PRIMA-1MET combination) in the study. Mice in the first three groups had complete data, while three mice in the combo group died early and had zero tumor weights. To assess the synergistic effects of SHetA2 and PRIMA-1MET, a logistic regression model was used to analyze the no tumor variable with two factors SHetA2 (indicating whether SHetA2 was part of the treatment) and PRIMA-1MET (indicating whether PRIMA-1MET was part of the treatment) and their interaction. The interaction coefficient was not included in the model because it was not significant, which resulted in an additive model. Statistical Analysis (SAS 9.4) was used to perform the analysis. All of the tumors found and excised during necropsy were weighed simultaneously to obtain the tumor burden, which was considered a continuous variable. Tumor burdens between the different treatment groups were compared by the Kruskal–Wallis test using Prism 8.0 (GraphPad Software, San Diego, CA).
Data availability
The data that support the findings of our study are available from the corresponding author upon reasonable request.
Results
Alterations of p53 and Mortalin in ovarian cancer specimens
The basis of our hypothesis suggests that ovarian cancer patient response to our maintenance strategy will depend on the type of TP53 mutation present in their tumors. Various missense p53 mutations commonly present in cancer exert different functions that could cause dissimilar consequences on responses of tumors to therapy, and therefore have potential to predict treatment response when evaluated individually.29 To evaluate the profile of TP53 mutations in TCGA HGSOC data, Ensembl exons of p53 genomic regions corresponding to various domains of the p53 protein were evaluated individually (Fig. 1a). TP53 exon mutations in TCGA HGSOC data occurred at a 94.4% (405/429) frequency. The majority (58%, 234/405) were missense mutations within the DNA binding domain (aa 102–292). Another 3% (12/405) were in regions outside of the DNA binding domain. The types of mutations known, or likely, to result in loss of full-length p53 protein expression from one allele (frameshift deletions and insertions, nonsense mutations and splice site mutations) totaled 39% (157/405). The most frequently mutated amino acids in p53 protein were R248, R273, R175, G245, R249 and R282. HSPA9 (mortalin) gene mutations were infrequent.
Figure 1.
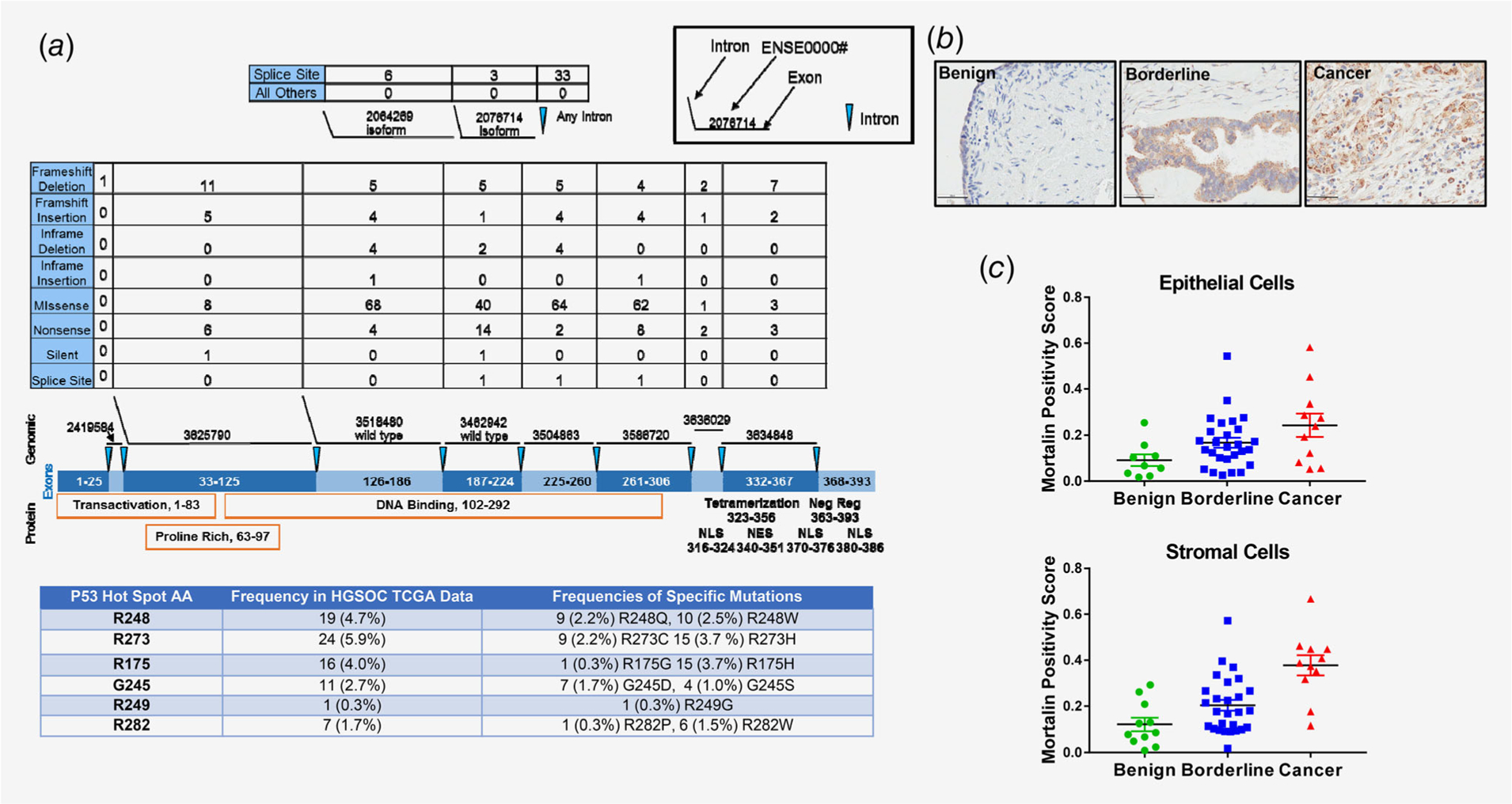
p53 and mortalin alterations in serous ovarian cancer tissues. (a) Data analysis for detection of p53 mutation status in HGSOC tumors in the TCGA database. The numbers of each type of mutation present are listed above the exons and protein domains depicted. The table shows the most frequently mutated p53 amino acids. (b) Representative images of mortalin immunohistochemical staining from the GOG TMA. (c) Comparison of average mortalin immunohistochemical positivity stain scores for epithelial and stromal cells in benign, borderline and serous tissues. *p ≤ 0.05, ***p ≤ 0.001.
The status of the SHetA2 drug target mortalin in a patient’s tumor is also likely to affect treatment outcome. Our analysis of TCGA data found no association of mortalin mutations with ovarian cancer. Immunohistochemical analysis of mortalin expression in a series of serous benign, borderline and cancerous tissues on a TMA demonstrated punctate expression of mortalin in both epithelial and cancer cells consistent with its primary localization in mitochondria (Fig. 1b). Mortalin positivity scores in epithelial and stromal cells significantly increased in a step-wise manner from benign to borderline to cancerous tissues (Fig. 1c; Kruskal–Wallis test p = 0.0282 for epithelial cells; p = 0.0002 for stromal cells).
Effects of SHetA2 on p53 cellular localization
We predicted that SHetA2 disruption of mortalin/p53 complexes,14,19 would cause p53 to translocate from the cytoplasm where it is sequestered by mortalin to accumulate in its nuclear and mitochondrial sites of action. Immunofluorescent imaging confirmed SHetA2 dose-dependent elevation of both wild type and mutant p53 in nuclei of ovarian cancer cell lines, but not in hFTSECs (Fig. 2a). Very little p53 was detectable in the cytoplasm likely due to masking of the epitope by binding proteins and the short p53 half-life. Western blots demonstrated SHetA2 dose-dependent decreases in cytoplasmic fractions and increases in nuclear (Supporting Information Figs. S1a and S1b) and mitochondrial (Supporting Information Figs. S2a and S2b) fractions of the A2780 wild type p53 and OV90 S215R or OVCAR4 m-p53 cell lines. SHetA2 caused minimal effects on p53 cellular localization in hFTSECs (Supporting Information Figs. S1c and S2c). In contrast to p53, mortalin cellular or nuclear levels were not affected by SHetA2 treatment in these cell types except for one instance of nuclear accumulation in A2780 cells treated with 5 μM SHetA2 (Supporting Information Figs. S1a–S1c).
Figure 2.
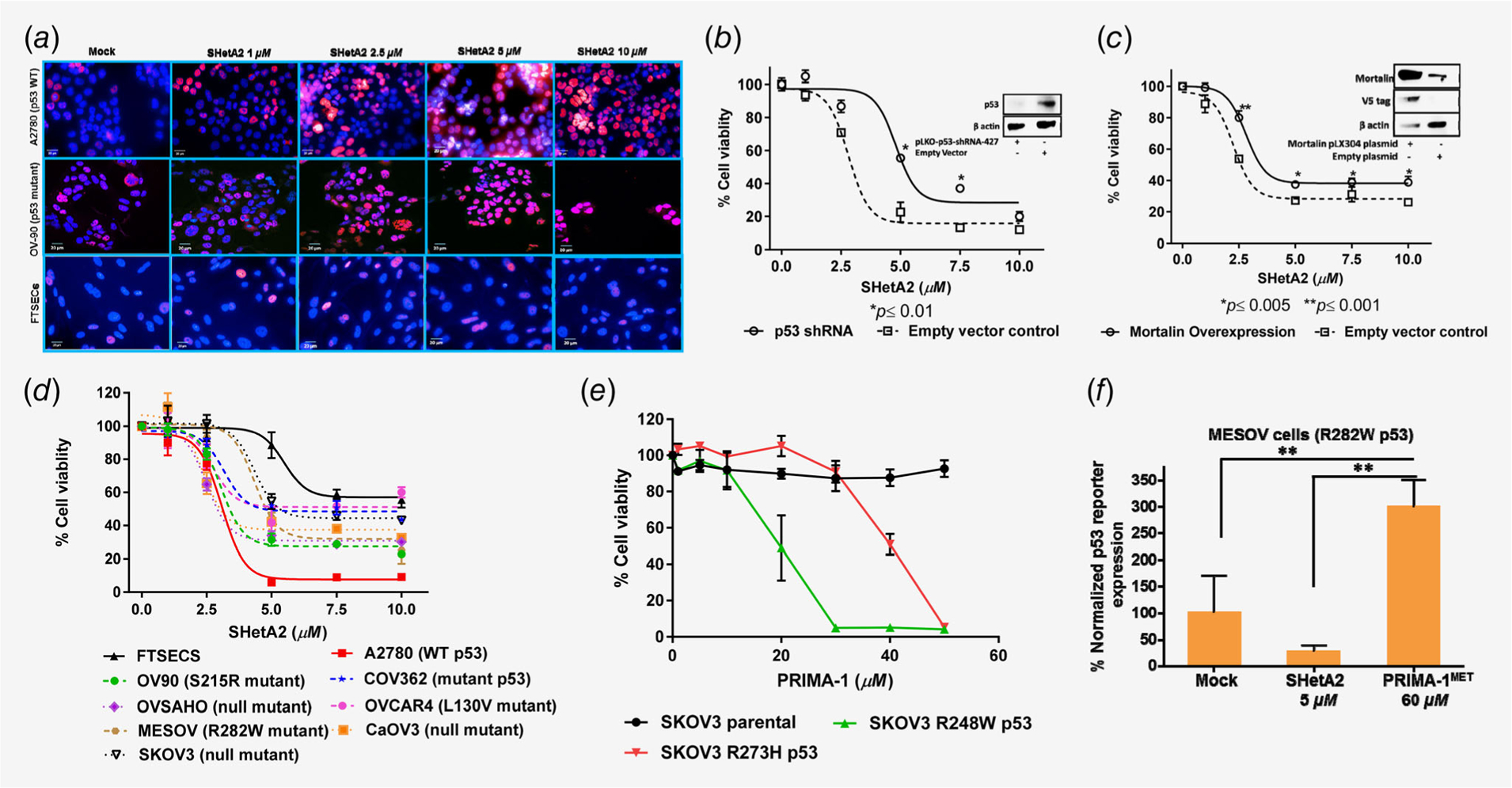
Involvement of p53 in SHetA2 or PRIMA-1 growth inhibition and apoptosis. (a) Immunofluorescence assay was performed on A2780, OV90 cells and FTSECs by detecting p53 with Alexa fluor 647 (Red) fluorochrome-conjugated antibody and nuclei were stained with DAPI (blue). Imaging was performed with a confocal microscope at 63X. (b) Comparison of SHetA2 sensitivity in A2780 cells transfected with p53 shRNA or empty vector using an MTT assay. Insert is a Western blot confirming p53 reduction. (c) Effect of mortalin overexpression on SHetA2 cytotoxicity in A2780 cells evaluated with an MTT assay. Insert is a Western blot confirming mortalin overexpression. (d). SHetA2 sensitivities of hFTSECs and ovarian cancer cell lines with various p53 status were compared using an MTT cytotoxicity assay. (e) MTT assay to compare PRIMA-1 cytotoxicity in SKOV3 parental cell line and SKOV3 sublines stably transfected with p53 mutant (R248W and R273H). (f). MESOV cells transfected with PG13-Luc plasmid were evaluated with a luciferase reporter assay to measure the effects of SHetA2 and PRIMA-1MET on p53 transcriptional activity. ANOVA: **p ≤ 0.01.
Effects of SHetA2 and PRIMA-1 as single agents
shRNA knockdown of p53 reduced SHetA2 potency in A2780 cells from 2.8 to 4.8 μM IC50 (Fig. 2b). Overexpression of mortalin in A2780 cells also reduced SHetA2-induced cytotoxicity (Fig. 2c), likely due to mortalin sequestering SHetA2 away from mortalin/p53 complexes. Ovarian cancer cell lines expressing m-p53 were more resistant to SHetA2 in comparison to the wild-type p53 A2780 cell line, while hFTSECs were the least sensitive (Fig. 2d). SKOV3 p53 null cells permanently transfected to express m-p53 exhibited increased PRIMA-1 sensitivity in comparison to PRIMA-1-resistant parental SKOV-3 cells to different extents depending on the mutant (R248W: 19.98 μM IC50; R273H:IC50 39.96 μM IC50; Fig. 2e). PRIMA-1MET reactivation of R282W m-p53 in MESOV was observed using a p53 reporter plasmid (Fig. 2f).
Effects of SHetA2 and PRIMA-1/PRIMA-1MET in combination
SHetA2 and PRIMA-1 showed synergism in ovarian cancer cells harboring wild-type p53 or m-p53, additive interaction in Caov3 p53 null cells and antagonism in hFTSECs (Figs. 3a and 3c). SHetA2 and PRIMA-1MET also showed synergism in m-p53 MESOV cells and antagonism in hFTSECs (Figs. 3b and 3c). DRIs of SHetA2 and PRIMA-1/PRIMA-1MET combinations indicated that the combination allows lower doses of each drug to be used without reducing activity (Supporting Information Table S1). The SHetA2 + PRIMA-1MET drug combination also reduced survival compared to untreated controls or single-drug treatments in a clonogenic assay (Supporting Information Fig. S4a). Quantification of triplicate experiments showed significant reduction in the colony formation of A2780, MESOV and SKOV3 R273H p53 cell lines treated with the drug combination in comparison to untreated controls (Supporting Information Fig. S4b). SKOV3 parental and SKOV3 R248W cell lines treated with the drug combination showed decreased colony counts in three independent experiments, but did not show significance, as there was wide variation in colony numbers due to dense growth patterns leading to inaccurate counting by the GelCount colony counter.
Figure 3.
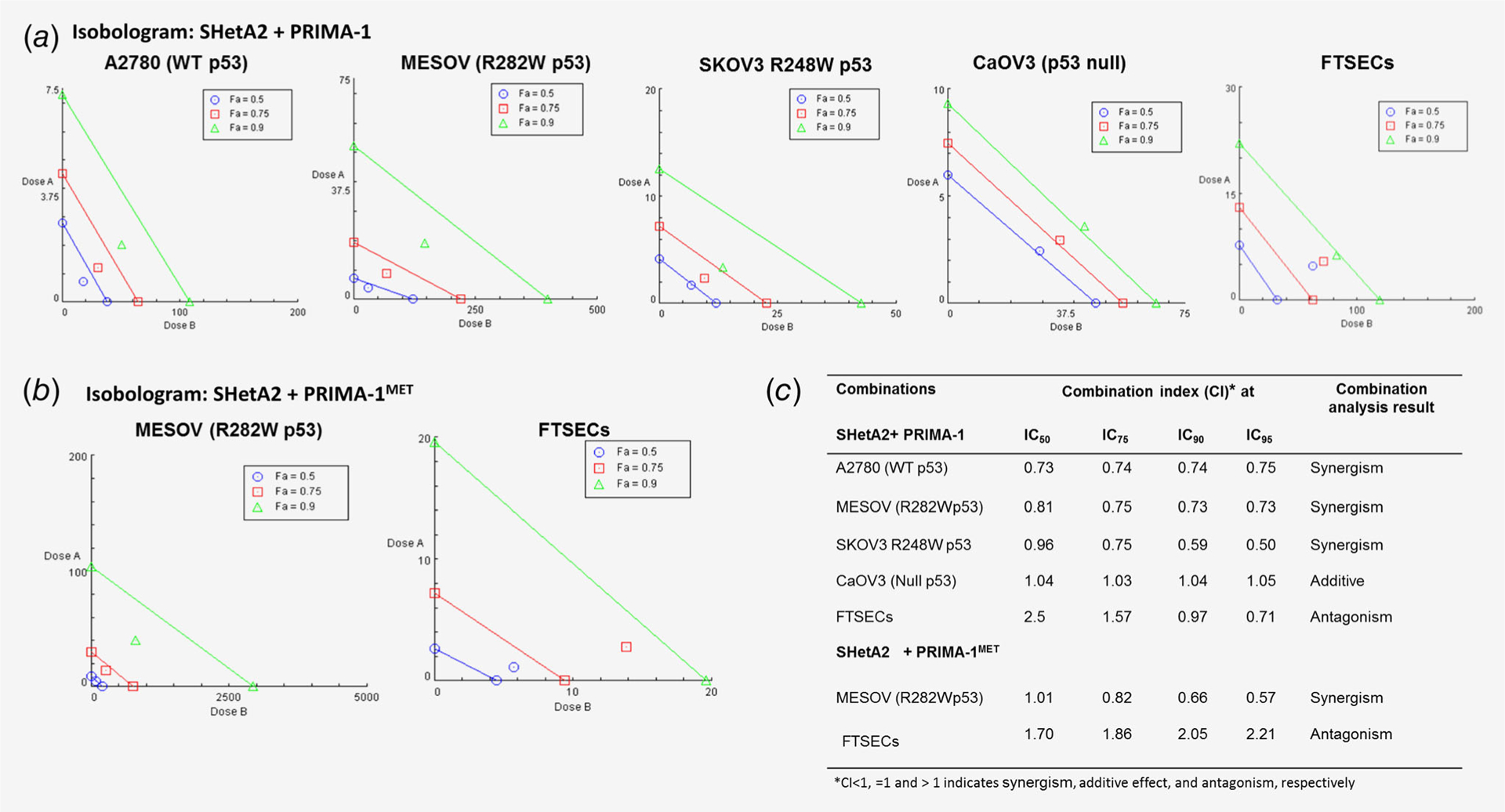
Isobolograms of SHetA2 and PRIMA-1/PRIMA-1MET combinations. (a) Isobolograms of SHetA2 and PRIMA-1 on the indicated culture types. (b) Isobolograms of SHetA2 and PRIMA-1MET on the indicated culture types. (c) Combination index values at various effective doses (EDs) as calculated by CompuSyn using the Chou–Talalay method. CI < 1, =1 and >1 indicate synergism, additive effect and antagonism, respectively.
Evaluation of SHetA2 and PRIMA-1 synergy mechanism
The TP21 gene was evaluated as a marker of p53 transcriptional activity. SHetA2 induced TP21 transcription in p53 null and m-p53-expressing SKOV3 cells (Fig. 4a), and increased p21 protein in all cell lines evaluated regardless of p53 status (Fig. 4b). Interestingly, PRIMA-1 did not induce TP21 transcription in m-p53-expressing SKOV3 cells (Fig. 4a) and only induced p21 protein expression in A2780 (wt p53) (Fig. 4b). PRIMA-1 prevented SHetA2 induction of p21 only in the presence of R248W m-p53, but did not prevent PARP-1 cleavage as an indicator of apoptosis (Fig. 4b).
Figure 4.
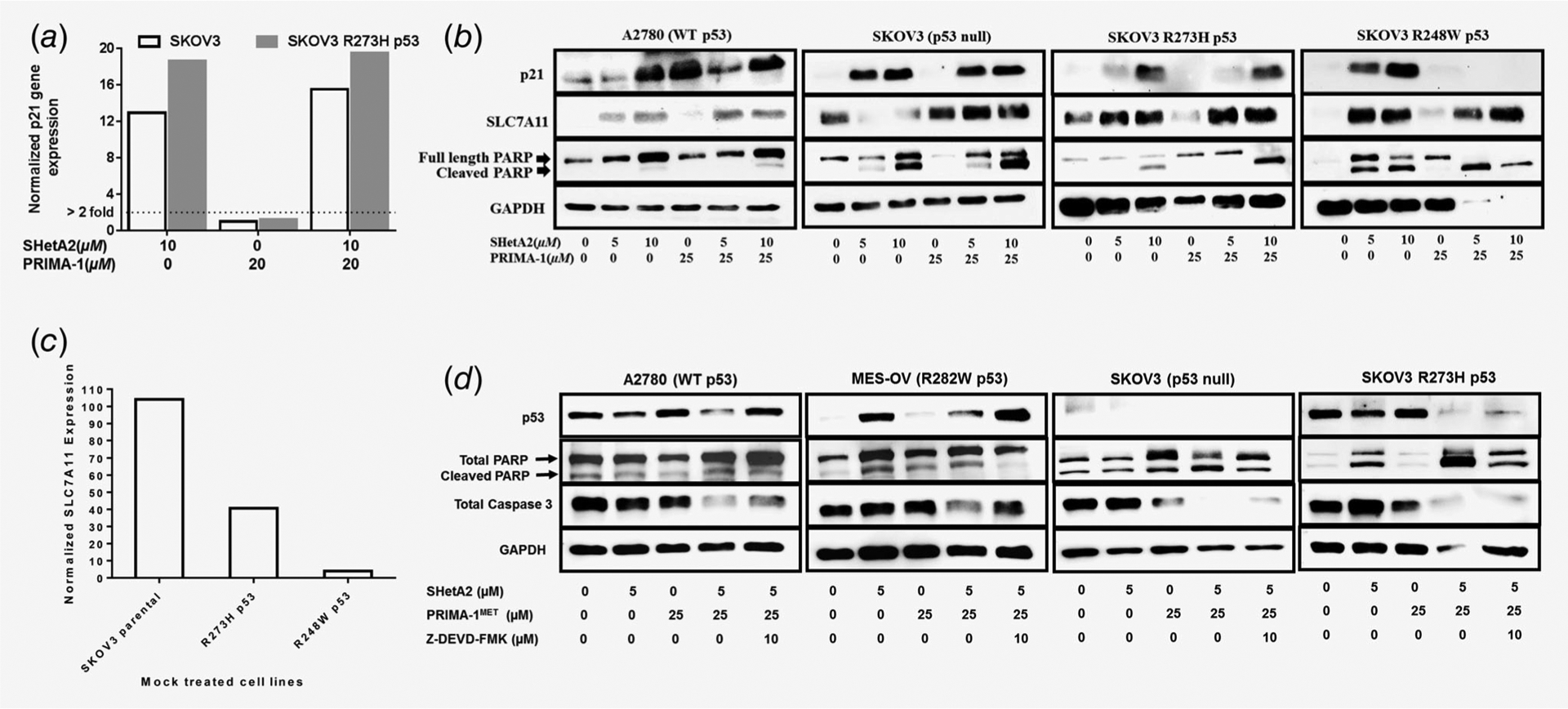
Evaluation of SHetA2 and PRIMA-1 synergy mechanism. (a) p21 expression determined by qRT-PCR in SKOV3 parental and stably transfected p53 R273H mutant cells treated with solvent only, 20 μM PRIMA-1, 10 μM SHetA2 or the combination of 20 μM PRIMA-1 + 10 μM SHetA2. Expression values were normalized to GAPDH, and then the treated were normalized to the solvent only control samples. (b) Western blots of p21, SLC7A11 and PARP in the indicated cell lines after treatment with either SHetA2 or PRIMA-1 or both combined at the indicated concentrations. (c) Basal expression of SLC7A11 in untreated SKOV3 parental and p53 mutant transfected sublines (R273H, R248W) after normalization with GAPDH. (d) Western blots of p53, PARP-1 and caspase 3 in cultures with SHetA2 and/or PRIMA-1MET in the absence or presence of the caspase 3 inhibitor Z-DEVD-FMK.
SHetA2 also induced SLC7A11, a cysteine/glutamate anti-porter shown to be inversely associated with m-p53 expression and PRIMA-1MET sensitivity by others30 and confirmed in our study (compare Figs. 2e and 4b–4d). The extents of SLC7A11 repression and ROS generation were associated with PRIMA-1 sensitivity (compare Figs. 2e and 5a and 5b). The involvement of ROS in the synergy mechanism was suggested by higher levels of ROS generation in SKOV3 cells treated with SHetA2 and PRIMA-1 in comparison to single-drug treatments, which occurred in an additive manner in p53 null cells and in a synergistic manner in m-p53 expressing cells (Fig. 5a). Levels of cellular ATP were decreased as levels of ROS were increased in SKOV3 p-53 null and m-p53 cells treated with SHetA2 and PRIMA-1 (compare Figs. 2e and 5b). ATP levels were also reduced in association with increased ROS by combined treatment with SHetA2 and PRIMA-1MET in A2780 wild type p53 cells, and this effect was antagonized by p53 shRNA (Figs. 5c and 5d). The involvement of apoptosis in the synergy mechanism was verified by caspase 3-inhibitor, Z-DEVD-FMK, reduction of caspase 3 and PARP-1 cleavage across all cell lines treated with SHetA2 and PRIMA-1MET (Fig. 4d).
Figure 5.
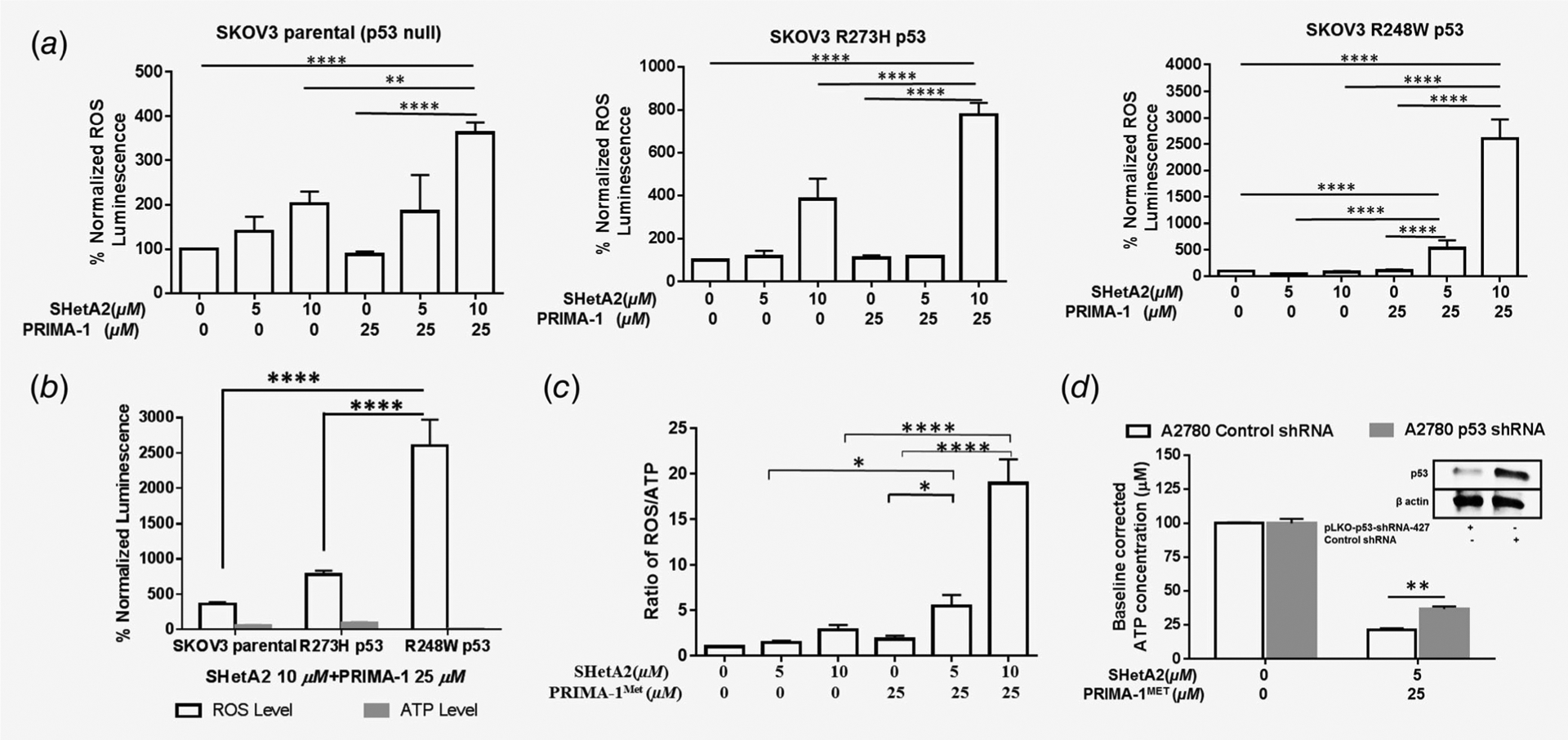
Evaluation of ROS and ATP in the mechanism of SHetA2 and PRIMA-1/PRIMA-1MET synergy. (a) Induction of ROS in SKOV3 parental, SKOV3 R273H p53 and SKOV3 R248W p53 cells after treatment with SHetA2 or PRIMA-1 alone or in combinations **p ≤ 0.01, ***p ≤ 0.001 ****p ≤ 0.0001. (b) ROS and ATP levels in the SKOV3 parental and SKOV3 R248W p53, R73H p53 cells after treatment with drug combination. ****p < 0.0001. (c) Ratio of ROS/ATP (each normalized to protein concentration). ANOVA with Tukey’s multiple comparison test: *p ≤ 0.05, **p ≤ 0.01, ****p ≤ 0.0001. (c) Ratio of ROS/ATP in A2780 cells after treatment with SHetA2 or PRIMA-1MET or both combined. (d) Change in ATP concentration in A2780 cells stably transfected with control shRNA or p53 shRNA after treatment with SHetA2 and PRIMA-1MET drug combination.
Evaluation of SHetA2 and PRIMA-1MET in a model of secondary chemoprevention
To model maintenance therapy for ovarian cancer, effects of SHetA2 and PRIMA-1MET on the establishment of tumors from MESOV cells injected into the peritoneum of immunocompromised mice were evaluated. Treatment was initiated 28 days postinjection and continued for 38 days. Treated mice appeared normal, however, the average body weights of the PRIMA-1MET group were significantly higher than the mock group (Fig. 6d). This was attributed to reduced gavage-associated stress and development of ascites in the PRIMA-1MET group (Fig. 6d). At necropsy, all control group mice had solid tumor nodules with variable numbers and sizes (Fig. 6a). Tumors were commonly observed on uterine horns, intestine, chest cavity, stomach, near the spleen and below the kidney and pancreas. Tumor-free rates were 0% in the untreated controls, 25% in the PRIMA-1MET treatment group, 42% in the SHetA2 treatment group and 67% in the combination treatment group (Fig. 6b). Logistic regression indicated that the interaction between SHetA2 and PRIMA-1MET was not significantly synergistic (p = 0.973). A subsequent additive model showed that both SHetA2 (p = 0.004, OR = 10.384, 95% confidence interval: 2.158, 48.965) and PRIMA-1MET (p = 0.048, OR = 4.464, 95% confidence interval: 1.014, 19.655) significantly prevented tumor development, and that these two drugs acted additively in preventing tumor establishment.
Figure 6.
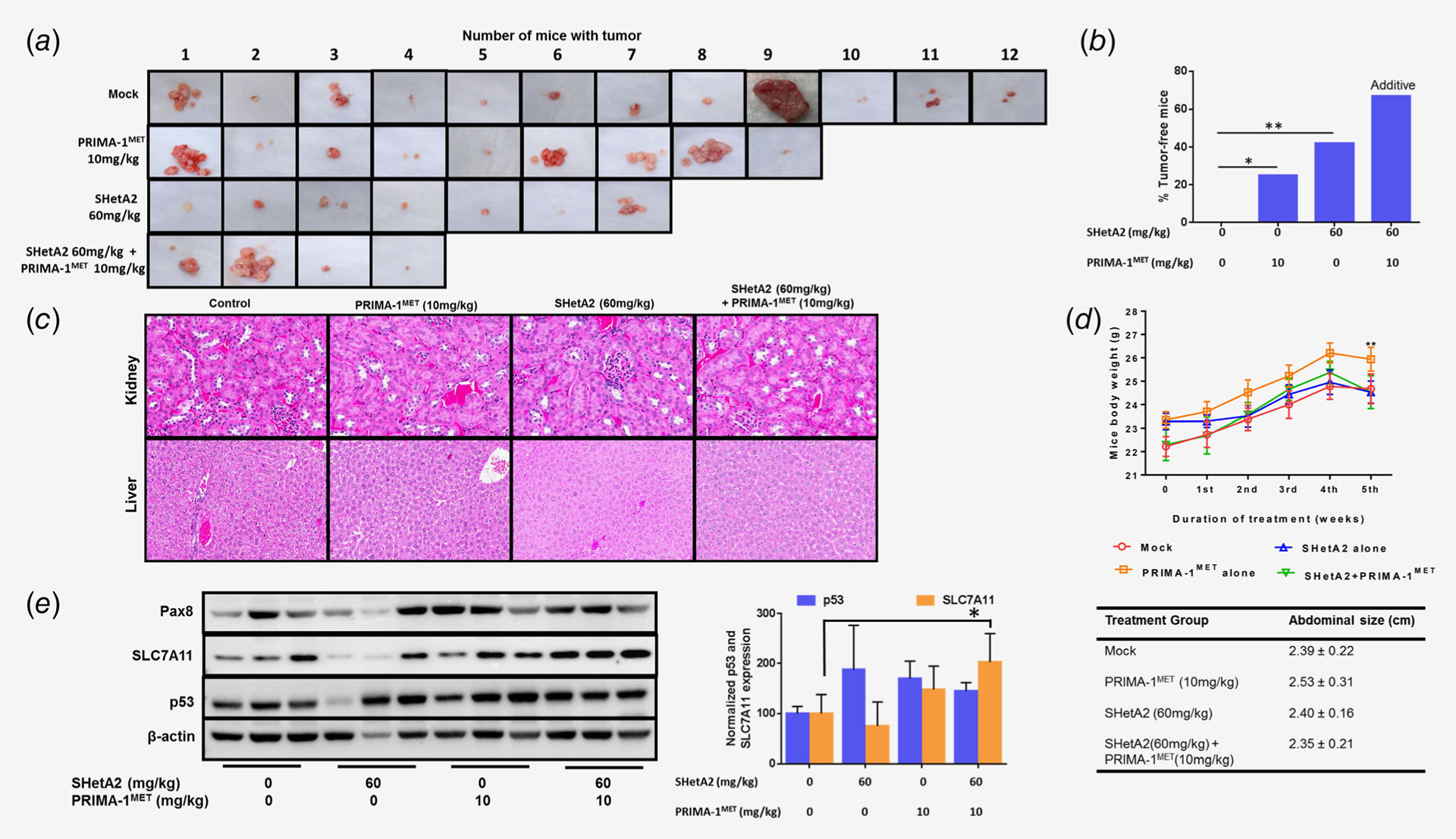
Evaluation of SHetA2 and PRIMA-1MET in model of secondary chemoprevention. (a) Peritoneal tumors harvested from MESOV-injected athymic nude mice after the treatment period. (b) Tumor free rates in the various treatment groups, linear regression model: SHetA2 (p = 0.004, OR = 10.384, 95% CI: 2.158, 48.965) and PRIMA1MET (p = 0.048, OR = 4.464, 95% CI: 1.014, 19.655) functioned additively in preventing tumor development. (c) 20× imaging of H&E staining of liver and kidney specimens of the mouse model to determine the toxicity of drug combination. The kidneys exhibited glomeruli that were well-formed without inflammation and the tubules were normal with no necrosis, apoptosis or inflammation. Livers exhibited no necrosis, fat deposition or inflammation. (d) Mice body weight during the duration of treatment duration and abdominal size observed at the end of the treatment period. ANOVA: ** p < 0.01. (e) Western blot analysis was performed on tumors from three randomly chosen mice in each treatment group to analyze expression of Pax8, SLC7A11 and p53 in mice tumor specimens. Graph shows normalized expression level of p53 and SLC7A11. ANOVA: *p < 0.05.
Histologic analysis of kidneys and livers collected upon necropsy showed no evidence of toxicity (Fig. 6c). Western blot analysis of tumors confirmed expression of Pax8, a marker of high-grade serous ovarian cancer (Fig. 6d). There was no association of p53 expression with treatment (Fig. 6d). SLC7A11 was increased significantly in the drug combination group compared to control group (Fig. 6d).
Discussion
Our results provide justification for targeting m-p53 and mortalin in the development of ovarian cancer maintenance therapy. TCGA data analysis revealed a 58% m-p53 rate in HGSOC. TMA analysis demonstrated elevated mortalin expression in serous cancer. Two drugs, SHetA2 and PRIMA-1MET, which wield complementary mechanisms targeted at m-p53 and mortalin, exerted synergistic cytotoxicity against ovarian cancer cell lines expressing m-p53 and additive activity in a p53 null cell line. The antagonism observed in hFTSEC cells suggests that this combination will not be toxic in clinical trials. The significant in vivo prevention of tumor establishment by each drug alone and the additive activity, when used in combination, validate the tissue culture results.
We predicted that SHetA2 would increase p53 activity because it releases p53 from binding to mortalin,31 and mortalin can sequester p53 in the cytoplasm away from its apoptosis sites of action in stressed cancer cells.32,33 In this study, SHetA2 caused p53 accumulation in nuclei and mitochondria in ovarian cancer cells, but not in hFTSECs. This differential effect may explain the lack of observed toxicity for SHetA2.21,23 Targeting mortalin/p53 has limitations in HGSOC, due to high m-p53 frequency and our observation that cell lines expressing endogenous or exogenous m-p53 exhibited increased SHetA2 resistance compared to wild type or p53 null cell lines.
We hypothesized that a combination of SHetA2 with a p53 reactivator drug would be synergistic in m-p53 cell lines because SHetA2 would increase the amount of p53 available to induce apoptosis while the p53-reactivator would restore apoptosis function to released m-p53. Our results support this hypothesis in that SHetA2 and PRIMA-1 or PRIMA-1MET acted synergistically in cytotoxicity assays of ovarian cancer cell lines expressing m-p53, but only additively in p53 null lines, and antagonistically in hFTSECs. A first-in-human clinical trial of PRIMA-1MET infusion in cancer patients found that PRIMA-1MET reached its target in tumors and was relatively safe with fully reversible central nervous system-related side effects.13 The DRI’s generated in our study suggest that these side effects could be reduced or eliminated by combination treatment with SHetA2 allowing lower doses of PRIMA-1MET to be used. Infusion of drugs is an imperfect approach for maintenance therapy, thus alternative formulations and routes of administration of PRIMA-1MET need to be developed.
The design and interpretation of SHetA2 and PRIMA-1MET clinical trials and translational endpoints can be optimized by greater knowledge of their mechanisms of action as individual drugs and when combined. Contribution of wild type p53 to the mechanism of SHetA2 as a single agent is indicated by p53 shRNA reduction of SHetA2 sensitivity in A2780 wild-type p53 cells. Wild-type p53 also contributes to the mechanism of synergy as evidenced by the documented synergy of SHetA2 and PRIMA-1, and the p53 shRNA interference with SHetA2 and PRIMA-1MET regulation of ROS/ATP ratios in A2780 cells.
The type of p53 mutation present in a patient’s tumor could be used to identify patients who would most benefit from these drugs. Various m-p53s exert different functions that could cause dissimilar therapy responses and therefore have potential to predict treatment response.29 Although PRIMA-1 was shown to restore wild type function to two m-p53s (R273H, R175H),34 the ability of PRIMA-1 or PRIMA-1MET to reactivate other p53 mutations is unknown and these drugs may be effective against only a subset of p53 mutations. Our study demonstrated PRIMA-1MET reactivation of p53 R282, and that endogenous or exogenous m-p53 expression increased sensitivity to the SHetA2 and PRIMA-1 or PRIMA-1MET combination. Further studies profiling sensitivities of various m-p53s to SHetA2 and PRIMA-1MET alone, and in combination, are needed.
Patients with p53 null tumors might also benefit from SHetA2 and PRIMA-1MET combination treatment. In this and other studies, SHetA2 was cytotoxic in p53 null SKOV3 and Caov3 cells.35 While some studies found selective cytotoxicity of PRIMA-1 toward cells harboring m-p53 in comparison to wild type or null p53 cells,34,36–40 others reported that PRIMA-1 induces apoptosis in ovarian cancer cell lines irrespective of their p53 status through ROS generation.41,42 In vivo models of the SHetA2/PRIMA-1MET combination in p53 null ovarian cancers are needed to justify inclusion of patients with p53 null tumors in a drug combination clinical trial.
Our study indicates that SHetA2 and PRIMA-1/PRIMA-1MET drug combinations are inducing apoptotic cell death by pushing cells past the “point of no return” to a ratio of ROS/ATP that is increased beyond a point at which recovery is impossible as defined by the Nomenclature Committee on Cell Death.43 Both wild-type and m-p53 proteins appear to be involved in this mechanism as ovarian cancer cells expressing wild-type p53, or endogenous or exogenous m-p53, exhibited greater ROS/ATP ratio elevation upon drug combination treatment compared to cells that were p53 null or had reduced p53. SLC7A11 levels were inversely associated with PRIMA-1 sensitivity, but not SHetA2/PRIMA-1 sensitivity, possibly due to the SHetA2 induction of SLC7A11 expression. In esophageal cancer, m-p53 increased PRIMA-1MET sensitivity, m-p53/NRF2 complexes repressed SLC7A11 expression and SLC7A11 overexpression decreased, while knockdown increased, PRIMA-1MET sensitivity in m-p53 cells.30 Taken together, these data suggest that SLC7A11 inhibits PRIMA-1MET sensitivity, but not SHetA2/PRIMA-1MET synergy.
In summary, these preclinical results justify further study and development of SHetA2 and PRIMA-1MET for ovarian cancer maintenance therapy. Initially, either drug alone or in combination could be tested when a patient can no longer tolerate bevacizumab maintenance therapy. Promising results could justify randomized studies in comparison to bevacizumab. m-p53 genotyping could be used to identify patients most likely to benefit from the therapies. The ultimate goal is to prolong ovarian cancer patients overall survival and improve quality of life.
Supplementary Material
What’s new?
Ovarian cancer patients need an effective maintenance therapy with minimal toxicity to prevent deadly recurrence of the disease. In this paper, the authors focused on reactivating missense mutant p53. Most high grade serous ovarian cancer contains mutations in p53, which can confer resistance to therapy. The authors tested SHetA2, a drug that frees p53 from sequestration by the protein mortalin, combined with a p53 activating drug, PRIMA-1MET. The two drugs combined achieved a 67% tumor-free rate in mice, higher than either alone. This success fuels further testing of the two agents as combination therapy for ovarian cancer.
Acknowledgements
This grant was supported by the National Cancer Institute of the National Institutes of Health (R01 CA196200 to D.M.B.), Foundation for Women’s Cancer-Ovarinnovate: Ovarcome Research Excellence Award to S.R., and COBRE grant number 1 P30 GM110766-01 and grant P30 CA225520 for availability of core services. We thank Lucila Garcia-Contreras, PhD, for her guidance in drug formulation and Gopal Pathuri, PhD and the Center for Cancer Prevention and Drug Development at the Stephenson Cancer Center (SCC) at the University of Oklahoma Health Sciences Center for synthesis of PRIMA-1MET. We appreciate free use of the SCC Cancer Functional Genomics Core for Operetta microscope imaging service and the SCC Cancer Pathology Core for histology service. The OUHSC Flow cytometry and Imaging core provided use of the confocal microscope. We thank Dr Jeremy Chien, University of Kansas Medical Center (currently University of New Mexico) for sharing SKOV3 cells stably transfected with p53 mutant plasmids and Dr. Francois Moisan, Stanford University for the MESOV/GFP-luc cell line.
Abbreviations:
- CI
combination index
- DRI
dose-reduction index
- FDA
Food and Drug Administration
- GOG
Gynecologic Oncology Group
- hFTSECs
human fallopian tube secretory epithelial cells
- HGSOC
high-grade serous ovarian cancer
- IC50
half-maximal inhibitory concentration
- m-p53
missense mutant p53
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- OS
overall survival
- RAID
rapid access to intervention development
- RAPID
rapid access to preventive intervention development
- ROS
reactive oxygen species
- TCGA
The Cancer Genome Atlas
- TMA
tumor microarray
Footnotes
Additional Supporting Information may be found in the online version of this article.
Conflict of interest
The authors do not have conflicts of interest with this study.
References
- 1.Ledermann JA, Raja FA, Fotopoulou C, et al. Newly diagnosed and relapsed epithelial ovarian carcinoma: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol 2018;29:iv259. [DOI] [PubMed] [Google Scholar]
- 2.Moore K, Colombo N, Scambia G, et al. Maintenance Olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med 2018;379: 2495–505. [DOI] [PubMed] [Google Scholar]
- 3.Burger R, Brady M, Bookman M, et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N Engl J Med 2011;365:2473–83. [DOI] [PubMed] [Google Scholar]
- 4.Perren TJ, Swart AM, Pfisterer J, et al. A phase 3 trial of bevacizumab in ovarian cancer. N Engl J Med 2011;365:2484–96. [DOI] [PubMed] [Google Scholar]
- 5.Burger RA, Enserro D, Tewari KS, et al. Final overall survival (OS) analysis of an international randomized trial evaluating bevacizumab (BEV) in the primary treatment of advanced ovarian cancer: a NRG oncology/Gynecologic oncology group (GOG) study. J Clin Oncol 2018;36:5517. [Google Scholar]
- 6.Vang R, Levine DA, Soslow RA, et al. Molecular alterations of TP53 are a defining feature of ovarian high-grade serous carcinoma: a Rereview of cases lacking TP53 mutations in the cancer genome atlas ovarian study. Int J Gynecol Pathol 2016;35:48–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011;474:609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Reade CJ, McVey RM, Tone AA, et al. The fallopian tube as the origin of high grade serous ovarian cancer: review of a paradigm shift. J Obstet Gynaecol Can 2014;36:133–40. [DOI] [PubMed] [Google Scholar]
- 9.Gurpinar E, Vousden KH. Hitting cancers’ weak spots: vulnerabilities imposed by p53 mutation. Trends Cell Biol 2015;25:486–95. [DOI] [PubMed] [Google Scholar]
- 10.Zawacka-Pankau J, Selivanova G. Pharmacological reactivation of p53 as a strategy to treat cancer. J Intern Med 2015;277:248–59. [DOI] [PubMed] [Google Scholar]
- 11.Lambert JM, Gorzov P, Veprintsev DB, et al. PRIMA-1 reactivates mutant p53 by covalent binding to the core domain. Cancer Cell 2009;15: 376–88. [DOI] [PubMed] [Google Scholar]
- 12.Bykov VJN, Zache N, Stridh H, et al. PRIMA-1MET synergizes with cisplatin to induce tumor cell apoptosis. Oncogene 2005;24:3484–91. [DOI] [PubMed] [Google Scholar]
- 13.Lehmann S, Bykov VJ, Ali D, et al. Targeting p53 in vivo: a first-in-human study with p53-targeting compound APR-246 in refractory hematologic malignancies and prostate cancer. J Clin Oncol 2012;30:3633–9. [DOI] [PubMed] [Google Scholar]
- 14.Benbrook DM, Nammalwar B, Long A, et al. SHetA2 interference with mortalin binding to p66shc and p53 identified using drug-conjugated magnetic microspheres. Invest New Drugs 2013; 32:412–23. [Selected for inclusion into GLOBAL MEDICAL DISCOVERY [ISSN 1929–8536] [https://globalmedicaldiscovery.com]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guruswamy S, Lightfoot S, Gold M, et al. Effects of retinoids on cancerous phenotype and apoptosis in organotypic culture of ovarian carcinoma. J Natl Cancer Inst 2001;93:516–25. [DOI] [PubMed] [Google Scholar]
- 16.Liu T, Masamha CP, Chengedza S, et al. Development of flexible-heteroarotinoids for kidney cancer. Mol Cancer Ther 2009;8:1227–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masamha CP, Benbrook DM. Cyclin D1 degradation is sufficient to induce G1 cell cycle arrest despite constitutive expression of cyclin E2 in ovarian cancer cells. Cancer Res 2009;69:6565–72. [DOI] [PubMed] [Google Scholar]
- 18.Liu S, Brown CW, Berlin KD, et al. Synthesis of flexible sulfur-containing heteroarotinoids that induce apoptosis and reactive oxygen species with discrimination between malignant and benign cells. J Med Chem 2004;47:999–1007. [DOI] [PubMed] [Google Scholar]
- 19.Liu T, Hannafon B, Gill L, et al. Flex-Hets differentially induce apoptosis in cancer over normal cells by directly targeting mitochondria. Mol Cancer Ther 2007;6:1814–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Benbrook DM, Lightfoot S, Ranger-Moore J, et al. Karyometric and gene expression analysis in an Organotypic model of endometrial carcinogenesis and chemoprevention. Gene Regul Syst Biol 2008; 2:21–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benbrook DM, Guruswamy S, Wang Y, et al. Chemoprevention of colon and Small intestinal tumorigenesis in APCmin/+ mice by SHetA2 (NSC721689) without toxicity. Cancer Prev Res 2013;6:908–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doppalapudi RS, Riccio ES, Davis Z, et al. Genotoxicity of the cancer chemopreventive drug candidates CP-31398, SHetA2, and phosphoibuprofen. Mutat Res 2012;746:78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kabirov KK, Kapetanovic IM, Benbrook DM, et al. Oral toxicity and pharmacokinetic studies of SHetA2, a new chemopreventive agent, in rats and dogs. Drug Chem Toxicol 2012;36:284–95. [DOI] [PubMed] [Google Scholar]
- 24.Zhang Y, Hua Y, Benbrook DM, et al. High performance liquid chromatographic analysis and preclinical pharmacokinetics of the heteroarotinoid antitumor agent, SHetA2. Cancer Chemother Pharmacol 2006;58:561–9. [DOI] [PubMed] [Google Scholar]
- 25.Liu Z, Zhang Y, Hua YF, et al. Metabolism of a sulfur-containing heteroarotionoid antitumor agent, SHetA2, using liquid chromatography/tandem mass spectrometry. Rapid Commun Mass Spectrom 2008;22:3371–81. [DOI] [PubMed] [Google Scholar]
- 26.Ramraj SK, Smith KM, Janakiram NB, et al. Correlation of clinical data with fallopian tube specimen immune cells and tissue culture capacity. Tissue Cell 2018;52:57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nielsen AT. Systems with bridgehead nitrogen. β-Ketols of the 1-Azabicyclo [2.2. 2] octane series. J Org Chem 1966;31:1053–9. [Google Scholar]
- 28.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984;22:27–55. [DOI] [PubMed] [Google Scholar]
- 29.Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol 2013;15:2–8. [DOI] [PubMed] [Google Scholar]
- 30.Liu DS, Duong CP, Haupt S, et al. Inhibiting the system xC−/glutathione axis selectively targets cancers with mutant-p53 accumulation. Nat Commun 2017;8:14844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Benbrook DM, Janakiram NB, Chandra V, et al. Development of a dietary formulation of the SHetA2 chemoprevention drug for mice. Invest New Drugs 2017;36:561–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lu WJ, Lee NP, Kaul SC, et al. Mortalin-p53 interaction in cancer cells is stress dependent and constitutes a selective target for cancer therapy. Cell Death Differ 2011;18:1046–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wadhwa R, Yaguchi T, Hasan MK, et al. Hsp70 family member, mot-2/mthsp70/GRP75, binds to the cytoplasmic sequestration domain of the p53 protein. Exp Cell Res 2002;274:246–53. [DOI] [PubMed] [Google Scholar]
- 34.Bykov VJN, Issaeva N, Shilov A, et al. Restoration of the tumor suppressor function to mutant p53 by a low-molecular-weight compound. Nat Med 2002;8:282–8. [DOI] [PubMed] [Google Scholar]
- 35.Chengedza S, Benbrook DM. NF-kappaB is involved in SHetA2 circumvention of TNF-alpha resistance, but not induction of intrinsic apoptosis. Anticancer Drugs 2010;21:297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zandi R, Selivanova G, Christensen CL, et al. PRIMA-1Met/APR-246 induces apoptosis and tumor growth delay in small cell lung cancer expressing mutant p53. Clin Cancer Res 2011;17: 2830–41. [DOI] [PubMed] [Google Scholar]
- 37.Izetti P, Hautefeuille A, Abujamra AL, et al. Prima-1, a mutant p53 reactivator, induces apoptosis and enhances chemotherapeutic cytotoxicity in pancreatic cancer cell lines. Invest New Drugs 2014;32:783–94. [DOI] [PubMed] [Google Scholar]
- 38.Li XL, Zhou J, Chan ZL, et al. PRIMA-1met (APR-246) inhibits growth of colorectal cancer cells with different p53 status through distinct mechanisms. Oncotarget 2015;6:36689–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Messina RL, Sanfilippo M, Vella V, et al. Reactivation of p53 mutants by prima-1 [corrected] in thyroid cancer cells. Int J Cancer 2012;130: 2259–70. [DOI] [PubMed] [Google Scholar]
- 40.Zhang W, Yi B, Wang C, et al. Silencing of CD24 enhances the PRIMA-1-induced restoration of mutant p53 in prostate cancer cells. Clin Cancer Res 2015;22:2545–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoshikawa N, Kajiyama H, Nakamura K, et al. PRIMA-1MET induces apoptosis through accumulation of intracellular reactive oxygen species irrespective of p53 status and chemo-sensitivity in epithelial ovarian cancer cells. Oncol Rep 2016;35:2543–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tessoulin B, Descamps G, Moreau P, et al. PRIMA-1Met induces myeloma cell death independent of p53 by impairing the GSH/ROS balance. Blood 2014;124:1626–36. [DOI] [PubMed] [Google Scholar]
- 43.Galluzzi L, Bravo-San Pedro JM, Vitale I, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ 2015;22:58–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of our study are available from the corresponding author upon reasonable request.