

Abstract
We have previously shown that pancreatic islets engineered to transiently display a modified form of FasL protein (SA-FasL) on their surface survive indefinitely in allogeneic recipients without a need for chronic immunosuppression. Mechanisms that confer long-term protection to allograft are yet to be elucidated. We herein demonstrated that immune protection evolves in two distinct phases; induction and maintenance. SA-FasL-engineered allogeneic islets survived indefinitely and conferred protection to a second set of donor-matched, but not third party, unmanipulated islet grafts simultaneously transplanted under the contralateral kidney capsule. Protection at the induction phase involved a reduction in the frequency of proliferating alloreactive T cells in the graft-draining lymph nodes, and required phagocytes and TGF-β. At the maintenance phase, immune protection evolved into graft site-restricted immune privilege as the destruction of long-surviving SA-FasL-islet grafts by streptozotocin followed by the transplantation of a second set of unmanipulated islet grafts into the same site from the donor, but not third party, resulted in indefinite survival. The induced immune privilege required both CD4+CD25+Foxp3+ Treg cells and persistent presence of donor antigens. Engineering cell and tissue surfaces with SA-FasL protein provides a practical, efficient, and safe means of localized immunomodulation with important implications for autoimmunity and transplantation.
1. INTRODUCTION
Type 1 diabetes (T1D) is an autoimmune disease caused by the destruction of insulin-producing beta cells in the pancreas, resulting in long term hyperglycemia. Transplantation of allogeneic islets is effective in reversing hyperglycemia in patients with T1D (1). However, allogeneic islet grafts are subject to rejection initiated and perpetuated by T effector (Teff) cells (2, 3). Thus, approaches that specifically target alloreactive Teff cells for physical elimination or functional inactivation have the potential to support sustained graft survival as a curative therapy for T1D.
Teff cells upregulate Fas receptor on their surface following activation and become sensitive to FasL-mediated apoptosis, defined as activation-induced cell death (AICD) (4–6). AICD is critical for the establishment of immune homeostasis and tolerance to self-antigens (4). The pivotal role of Fas/FasL pathway in regulating T cell responses is emphasized by the emergence of autoimmunity in cases of Fas or FasL deficiencies (5, 7). The Fas pathway, therefore, has significant potential for the development of therapeutic approaches to treat autoimmune diseases and transplant rejection. However, the pursuit of tissue-targeted expression of FasL for immunomodulation in settings of autoimmunity and transplantation has produced conflicting observations (8–11), potentially arising from the complex nature of FasL expression, the existence of two different isoforms, and the pleiotropic and opposing functions performed by each isoform. FasL is expressed as a type II membrane-bound protein, which can be cleaved into a soluble form by matrix metalloproteinases in response to environmental cues (12). The membrane-bound form was reported to have apoptotic activity, while the soluble form lacks such activity and serves as a chemotactic factor for neutrophils (13, 14). These initial observations were further confirmed in transgenic mice expressing either a soluble or membrane-bound form of FasL (15). The membrane-bound form was shown to be apoptotic and essential for controlling autoimmunity, while the soluble form promoted autoimmunity and tumorigenesis via non-apoptotic functions. Therefore, the therapeutic application of FasL as an immunomodulator may require a form that primarily has apoptotic function.
We have previously reported the generation of a novel form of FasL chimeric with a modified form of core streptavidin (SA-FasL) that exists as oligomers with robust apoptotic activity on Fas-expressing lymphocytes (16). Importantly, SA-FasL can be positionally and transiently displayed on biotinylated biologic (cells, tissues, or organs) and nonbiologic surfaces (various biomaterials) in a rapid and efficient manner taking the advantage of high affinity interaction between biotin and streptavidin (17–19). Islets directly engineered to transiently display SA-FasL on their surface showed indefinite survival in allogeneic hosts (17). We herein investigated the mechanistic underlying of SA-FasL-induced tolerance.
2. MATERIALS AND METHODS
2.1. Mice and recombinant proteins
C57BL/6, B6.Cg-Foxp3tm2(EGFP)Tch/J, BALB/c, and C3H mice were obtained from Jackson Laboratories). C57BL/6.SJL and TCR transgenic OT-I and OT-II mice on Rag2−/− background were purchased from Taconic Farms. BALB/c.RIP-OVA mice were a gift from Dr. S. Webb, Scripps Research Institute, La Jolla, CA. Animal were kept in our SPF vivarium at the University of Louisville using protocols approved by the IACUC. Recombinant SA and SA-FasL proteins were produced in our laboratory using the Drosophila DES expression system (Invitrogen) as previously described (16).
2.2. Islet isolation and transplantation
Pancreatic islets isolation, engineering with SA-FasL, and transplantation were performed per published protocols (16, 17) and detailed in Supplementary Materials and Methods.
2.3. T Cell proliferation
For in vivo proliferation, OVA CD8+ T cells were isolated from spleen and mesenteric LNs of OT-I transgenic C57BL/6 mice, labeled with CFSE as described (20), and 15 x 106 cells/animal were transferred by tail vein injection into C57BL/B6.SJL congenic mice. One day later, these mice were transplanted with SA- or SA-FasL-engineered pancreatic islets isolated from RIP-mOVA transgenic BALB/c mice expressing a membranous form of OVA in pancreatic beta cells under the control of rat insulin promoter (21). Lymphocytes were harvested from kidney-draining LNs, mesenteric LNs, and spleens 5 days after islet transplantation and stained with antibodies against CD8-PerCp, Vβ-5-PE, and CD45.2-APC. Proliferation of OT-I cells were determined by gating on Vβ-5+CD8+CD45.2+ T cells. Details of in vivo alloreactive T cell proliferation in the F1 setting are provided in Supplementary Materials and Methods.
2.4. Phagocyte depletion and TGF-β blockade
The roles of phagocytes and TGF-β in the induction of tolerance were assessed by treatment with clodronate-loaded liposomes and a blocking Ab against TGF-β, respectively, as detailed in Supplementary Materials and Methods.
2.5. STATISTICS
Graph pad prism was used to perform statistical analyses (Welch’s t-test and unpaired one-tailed or two tailed t-tests) to determine difference between the groups where indicated. Graft survival was analyzed using log-rank test. Data are expressed as mean ± SEM where indicated. P <0.05 was considered significant.
3. RESULTS
3.1. Immunomodulation with FasL-engineered islet grafts results in reduced frequency of proliferating alloreactive t cells in graft-draining lymph nodes
We have previously reported that pancreatic islets engineered with SA-FasL survived indefinitely in allogeneic hosts (17). Survival was associated with intragraft depletion of alloreactive CD4+ and CD8+ Teff cells. To assess if the depletion of alloreactive Teff cells is systemic, CFSE-labeled OT-I CD8+ T cells (CD45.2) recognizing a dominant epitope of ovalbumin (OVA) were adoptively transferred into congenic C57BL/6.SJL mice (CD45.1) one day before the transplantation of islets from OVA transgenic BALB/c (RIP-OVA) donors. We chose this model system based on the demonstrated critical roles of CD8+ T cells and indirect allorecognition in islet allograft rejection (22, 23). The proliferation of alloreactive OT-I cells was assessed in graft-draining lymph nodes as well as the spleen and mesenteric lymph nodes as distant lymphoid tissues. There was a marked reduction in the frequency of proliferating OT-I cells in the kidney-draining lymph nodes of the group transplanted with SA-FasL-engineered islet grafts as compared with the control streptavidin (SA)-engineered islets group (Figures 1A and 1B; p < 0.05). OT-I cells also underwent proliferation, albeit to a lesser extent, in the spleen and mesenteric lymph nodes of both groups. This observation is consistent with the reported critical role of graft-draining lymph nodes in regulating alloreactive immune responses (2, 24, 25). Although the OT-I response of the SA-FasL group in these distant lymphoid tissues trended towards lower proliferation as compared with the SA control group, it did not reach statistical significance.
FIGURE 1.
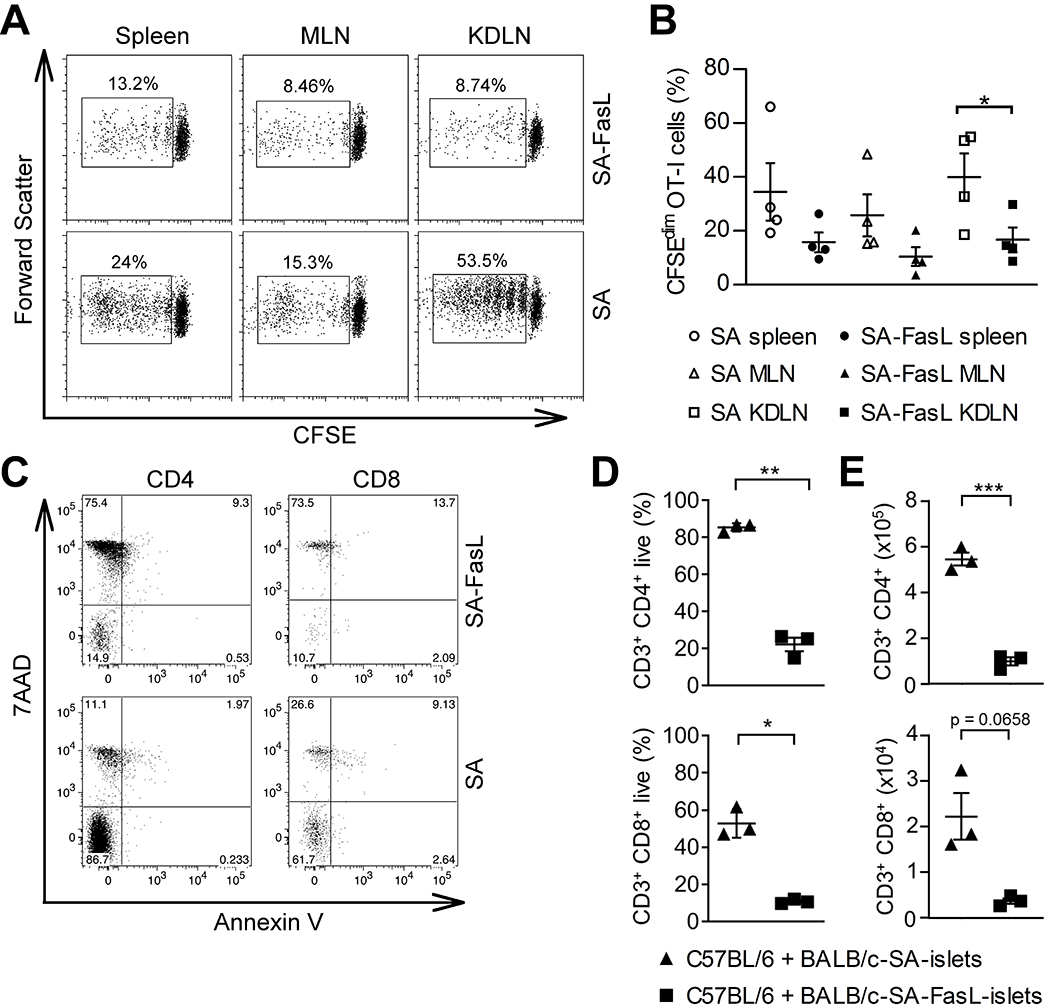
Graft milieu-localized modulation of alloreactive T cell responses as the prominent feature of tolerance established by SA-FasL-engineered allogeneic islets. A, Dot plots showing the frequency of proliferating CFSE-labeled OT-I CD8+ T cells (CD45.2) adoptively transferred into C57BL/6.SJL mice (CD45.1) one day prior to transplantation of BALB/c RIP-OVA islet graft engineered with SA-FasL or control SA proteins. Single cells harvested from the indicated lymphoid tissues 5 days post islet transplantation were analyzed for CFSE dilution using flow cytometry gating on the OT-I cell population. B, Tabulation of the data shown in (A). Points indicate values for individual mouse, bars indicate means ± SEM, and statistical significance was calculated using a one-tailed unpaired t-test *p <0.05, **p <0.01. C, Dot plots showing apoptosis of alloreactive C57BL/6 T cells to BALB/c SA-FasL-engineered islets in an in vitro co-culture assay. D and E, Tabulation of percentages and absolute number of cells in (C). Points indicate values for individual mouse, bars indicate means ± SEM, and statistical significance was calculated using a two-tailed unpaired Welch’s t-test *p <0.05, **p <0.01.
To provide evidence for SA-FasL-mediated apoptosis being responsible for the observed marked reduction in the number of proliferating alloreactive cells in kidney-draining lymph nodes, we assessed the impact of SA-FasL-engineered BALB/c allogeneic islets on the survival of alloreactive T cells in an in vitro co-culture system. SA-FasL-engineered islets induced robust death in both CD4+ and CD8+ T cells as compared with SA-engineered control islets (Figure 1C). Indeed, less than 25% of CD4+ and 15% of CD8+ T cells remained live as compared with > 80% and 40% of cells, respectively, in SA control cultures (Figure 1D and E). These data support the role of SA-FasL-induced apoptosis in reducing the frequency of alloreactive T cells within the graft-draining lymph nodes. Further, this finding is consistent with our previously reported study demonstrating local apoptosis of Teff, but not Treg cells within SA-FasL-engineered islet grafts (17).
3.2. Systemic donor-reactive responses persist despite long-term islet graft acceptance
The reduced proliferative response of donor-reactive T cells in SA-FasL-engineered islet graft recipients (Fig. 1) in the early phase of tolerance induction may evolve into two different outcomes at the maintenance phase; establishment of generalized tolerance and consequent unresponsiveness to donor antigens or localized immune privilege in spite of persistent donor-reactive responses. We first confirmed our previous observations (17) by demonstrating that SA-FasL-engineered allogeneic BALB/c islet grafts transplanted under transient cover of rapamycin (15 daily doses) into chemically diabetic C57BL/6 hosts show sustained survival (100-day observation period; Figure 2A). This tolerogenic effect was dictated by SA-FasL, as islet grafts engineered with streptavidin as control protein transplanted using the same rapamycin regimen were acutely rejected (MST 24 ± 2.3 days).
FIGURE 2.
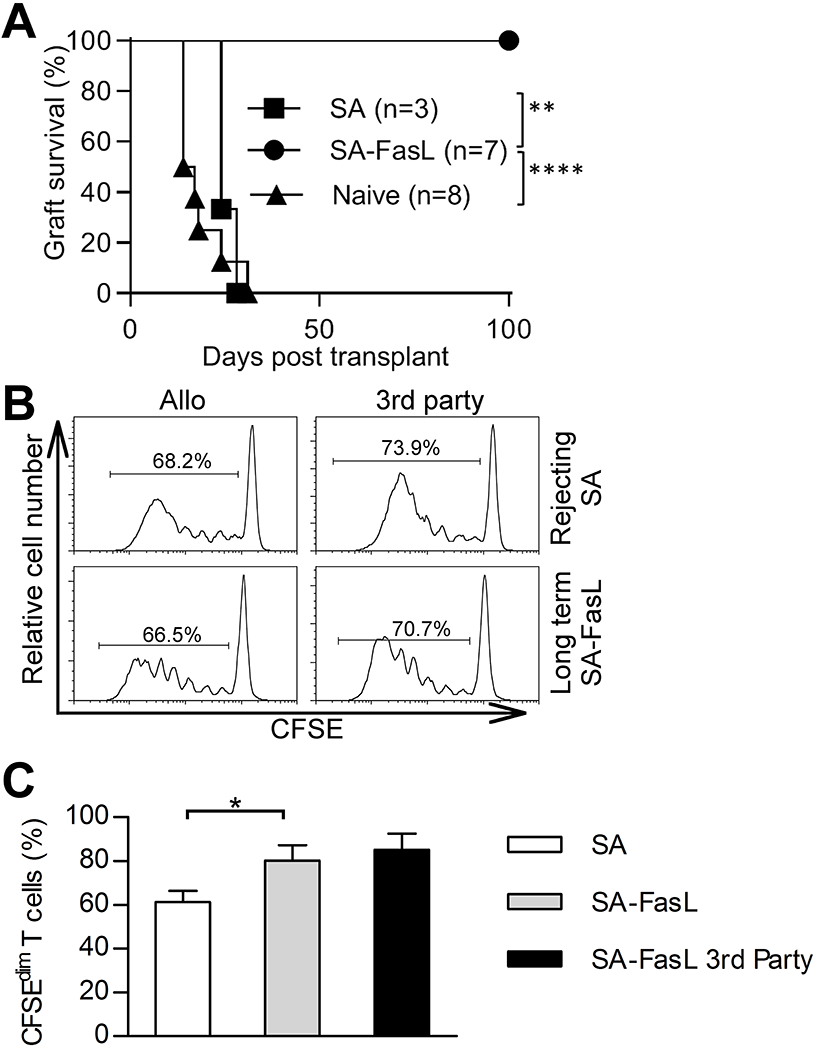
Survival of SA-FasL-engineered allogeneic islet grafts in the absence of systemic unresponsiveness to donor antigens. A, Survival of BALB/c allogeneic islet grafts engineered with SA-FasL or SA control protein under a short course of rapamycin (0.2 mg/kg daily for 15 doses starting on the day of transplantation). Recipients of unmodified allogeneic islet grafts without rapamycin served as controls. Data were analyzed using log-rank test (**p < 0.01 and ****p < 0.0001). B, In vivo proliferation assay. Splenocytes were harvested from recipients of allogeneic SA-engineered islet grafts at rejection or SA-FasL-engineered allogeneic islet grafts after 100 days post-transplantation. Cells were labeled with CFSE and injected i.v. into F1 (C57BL/6 x BALB/c, H-2b/d) or (C57BL/6 x C3H, H-2b/k) mice to test their proliferative response against donor and third-party antigens, respectively. C, Tabulation of the data shown in (B). Points indicate values for individual mouse, bars indicate means ± SEM, and statistical significance was calculated using a one-tailed unpaired t-test *p < 0.05.
To assess if tolerance is systemic and donor specific, an in vivo proliferation assay was performed. T cells harvested from the spleen of long-term (> 100 days) SA-FasL-islet graft recipients generated a strong in vivo proliferative response against both donor (BALB/c) and third party (C3H) antigens (Figures 2B and 2C). The donor-reactive response was of a similar magnitude to that generated by T cells of recipients rejecting control SA-engineered allograft. Lack of systemic tolerance, in spite of sustained survival of allogeneic islet grafts, implies localized tolerance, which is consistent with the demonstrated role of FasL in physiological immune privilege (26, 27).
3.3. Phagocytes and TGF-β are required for tolerance induction
Apoptotic lymphocytes were shown to have immunomodulatory features involving both phagocytes and TGF-β (28, 29). Thus, we investigated the role of phagocytes in the induction of tolerance in our model. Depletion of macrophages and immature DCs using clodronate loaded liposomes (Figure S1) administered one day before transplantation resulted in acute rejection of SA-FasL-engineered islet graft in all recipients (Figure 3A; MST = 24 ± 9.6 days). In marked contrast, all the mice treated with liposome without clodronate showed graft survival over an observation period of 100 days, supporting a critical role of phagocytes in the induction of tolerance. Clearance of apoptotic T cells by macrophages is associated with the secretion of TGF-β (28, 30). Consistent with these reports, macrophages cocultured with T cells undergoing SA-FasL-induced apoptosis engulfed apoptotic bodies and secreted TGF-β (Figure 3B).
FIGURE 3.
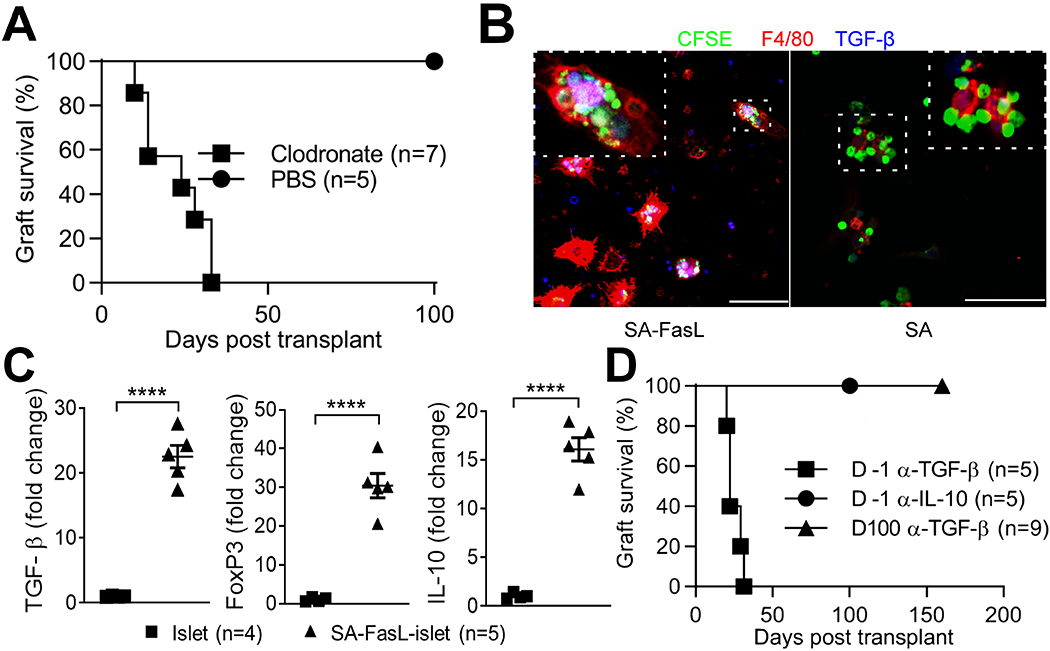
Depletion of phagocytes or blockade of TGF-β at the induction phase of SA-FasL-mediated tolerance results in prompt islet graft rejection. A, Survival of SA-FasL-engineered BALB/c islets in allogeneic C57BL/6 mice treated with liposomes encapsulated with PBS or clodronate injected intravenously (0.1 ml/10 g body weight) one day before islet transplantation (clodronate MST 24 ± 9.6 days; log-rank p = 0.0014). B, Production of TGF-β by macrophages digesting apoptotic bodies. OT-II TCR transgenic T cells recognizing ovalbumin were activated with ovalbumin in vitro and treated with SA-FasL for the induction of apoptosis. Apoptotic T cells were then cocultured with macrophages and analyzed using confocal microscopy for the uptake of apoptotic bodies (green) and production of TGF-β (blue). Mostly, macrophages (red) engulfing apoptotic bodies produce cytoplasmic TGF-β (pink) scale bar 50 μm. C, Increased intragraft expression of immunoregulatory factors in SA-FasL-engineered islet grafts on day 3 post-transplantation. Total RNA isolated form unmodified and SA-FasL-engineered islet grafts, both under rapamycin treatment, were used to assess the transcript levels for FoxP3, TGF-β, and IL-10 using Taqman probes based real time PCR in normalization to housekeeping gene GAPDH. Data are plotted as relative fold expression (2-ΔΔCT) as compared to control group (islet + rapamycin). Each dot represents one mouse; mean ± SD of two independent experiments. Median values are indicated by bars. ****p < 0.00001 (Mann-Whitney t test, two-tailed). D, Survival of SA-FasL-engineered BALB/c islets in allogeneic C57BL/6 mice treated with blocking antibodies to TGF-β or IL-10 cytokines. Graft recipients were treated intravenously with 4-5 doses of the indicated antibodies starting one day prior to or 100 days post-transplantation of SA-FasL-engineered allogeneic islet grafts. Treatment with the antibody to TGF-β at the induction, but not maintenance phase of tolerance results in islet graft rejection (MST = 22 ± 4.9 days vs > 100 days; log-rank p = 0.0006).
TGF-β as an immunoregulatory cytokine has been implicated in tolerance involving T cell apoptosis in various models (28, 29, 31). Quantitative RT-PCR studies have demonstrated significantly higher levels of FoxP3, TGF-β, and IL-10 transcripts in SA-FasL-engineered islet grafts on day 3 post-transplantation as compared with controls (Figure 3C). Importantly, in vivo neutralization of TGF-β by intravenous injection of a blocking antibody at the peri-transplant period resulted in acute rejection of all SA-FasL engineered islet grafts (Figure 3D; MST = 22 ± 4.9 days). In marked contrast, antibody treatment at the maintenance phase of tolerance (100 days post-transplantation) had no impact on graft survival (MST > 160 days). Also, treatment with a blocking antibody against IL-10, as another immunoregulatory cytokine showing increased levels of intragraft transcripts, at the induction phase did not impact long-term acceptance of SA-FasL-engineered islet grafts (Figure 3D; MST > 100 days). These results demonstrate the critical role of TGF-β in the induction, but not maintenance phase of tolerance.
3.4. Tolerance is systemic at the induction phase and shows both donor and tissue specificity
We have previously shown that long-term tolerance achieved by SA-FasL-engineered islet grafts is localized to the graft and requires Treg cells for maintenance (17). Treg cells were shown to traffic to allogeneic pancreatic islets immediately post-transplantation in response to inflammatory cues, where they manifest their immunoregulatory function within the graft microenvironment (2, 25). Therefore, we assessed if SA-FasL-mediated long-term, localized tolerance is systemic at the induction phase using a simultaneous two-islet graft model (Figure 4A). C57BL/6 mice were transplanted with BALB/c SA-FasL-engineered islets under the right kidney capsule and unmanipulated islets from BALB/c or C3H third party donors under the left kidney capsule. These animals were also subjected to a short course of rapamycin (0.2 mg/kg) administered daily for 15 days starting the day of transplantation. Surgical removal of the kidney harboring SA-FasL-engineered islet graft around 60 days post-transplantation did not result in hyperglycemia in recipients transplanted with donor-matched, unmodified islet grafts, demonstrating the survival and function of unmodified donor grafts (Figure 4B; MST > 100 days). In marked contrast, surgical removal of the SA-FasL-engineered BALB/c islet graft resulted in prompt hyperglycemia in recipients transplanted with the unmodified C3H third party islet graft (Figure 4C). This observation indicates that the third-party graft had already been rejected and the euglycemia was maintained by SA-FasL-engineered BALB/c graft, demonstrating the antigen-specificity of the induced tolerance. Importantly, tolerance to unmanipulated BALB/c islets was maintained by Treg cells as their depletion using an antibody to CD25 resulted in rejection of 3/4 grafts within 22 days (Fig. 4D). The mouse that did not reject the graft had minimal depletion of Treg cells (Figure S2).
FIGURE 4.
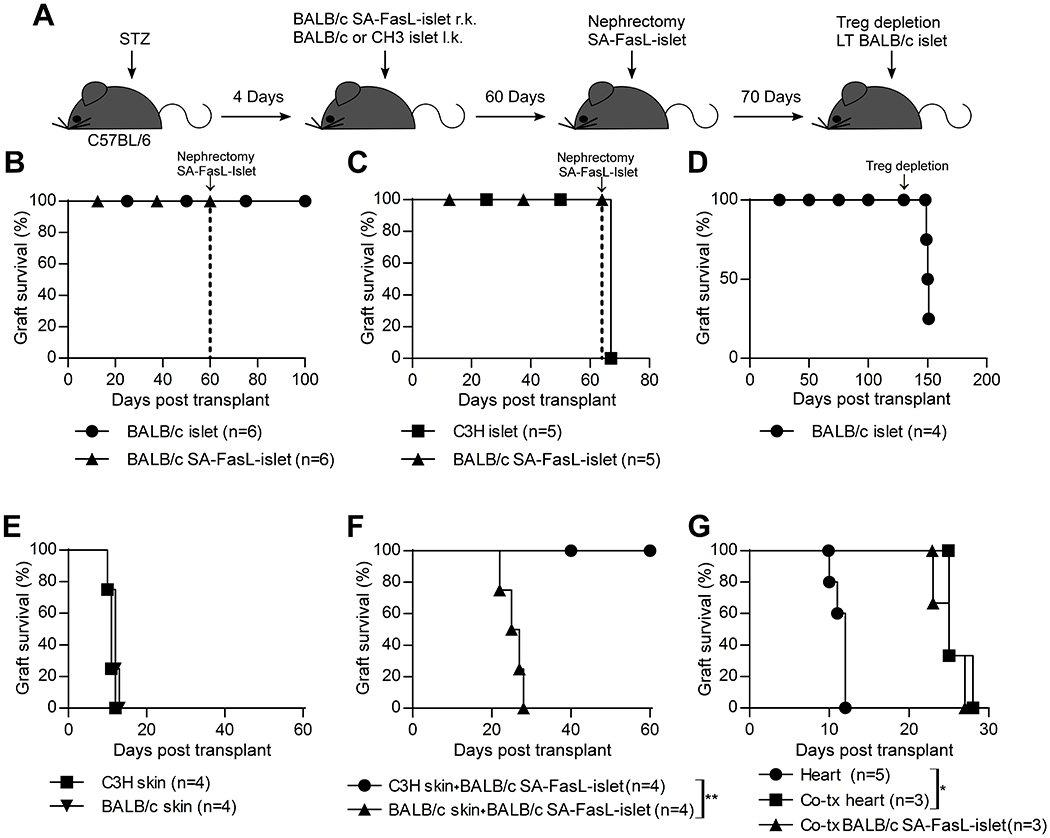
Tolerance to SA-FasL-engineered islet grafts is donor-specific and systemic at the induction phase. A, Schematic diagram showing the study design. Chemically diabetic C57BL/6 mice were transplanted with SA-FasL-engineered BALB/c islets under the right kidney capsule and unmodified BALB/c (B) or C3H (C) islet grafts under the contralateral kidney capsule. Mice were subjected to a short course of rapamycin treatment (0.2 mg/kg daily for 15 doses starting the day of transplantation). B, Surgical removal of the kidney harboring SA-FasL-engineered BALB/c islet graft 60 days post-transplantation (arrow) did not result in hyperglycemia, demonstrating the survival and function of unmodified donor-matched islets. C, Surgical removal of SA-FasL-engineered BALB/c islet graft (arrow) results in prompt hyperglycemia in the cohort harboring unmodified C3H third party islet graft under the contralateral kidney capsule, demonstrating rejection. D, Ablation of Treg cells in mice shown in panel B (after surgical removal of the kidney harboring the SA-FasL-engineered islets) using an antibody to CD25 (PC.61) on day 130 post-transplantation (arrow) resulted in rejection of 3 out of 4 grafts, demonstrating the role of Treg cells in maintaining survival of the unmodified islet graft. E, Survival of BALB/c donor and C3H third party skin grafts in C57BL/6 recipients simultaneously transplanted with BALB/c SA-FasL-engineered islet grafts. F, Rejection of BALB/c, but not third party C3H, skin results in prompt rejection of SA-FasL-engineered islets (MST = 26 ± 2.6 days, log-rank comparison of islet survival p = 0.0067). G, SA-FasL-engineered islets do not induce tolerance when co-transplanted with donor-matched heart grafts. Chemically diabetic C57BL/6 were transplanted with SA-FasL-engineered islets under the kidney capsule and donor-matched heart graft in the abdomen. A separate group was transplanted with heart only under a brief course of rapamycin to serve as control. Both graft types are rejected with heart graft showing prolonged survival as compared with the control (MST 25 ± 1.7 vs 12 ± 0.9 days, log-rank comparison of heart survival p = 0.0074).
We next used a simultaneous two-graft model to determine the tissue-specific nature of tolerance at the induction phase. C57BL/6 mice were transplanted with BALB/c SA-FasL-engineered islets and donor-matched or C3H third party skin grafts under the transient cover of rapamycin. Both donor and third-party skin grafts were acutely rejected (Fig. 4E). Rejection of BALB/c skin also triggered rejection of SA-FasL-engineered BALB/c islets, causing development of hyperglycemia within 30 days (Fig. 4F; MST 26 ± 2.6 days). In marked contrast, the rejection of C3H skin did not interfere with long-term acceptance of BALB/c SA-FasL-engineered islets as all mice remained euglycemic for an observation period of 60 days (Figure 4F; MST > 60 days).
Because skin grafts elicit vigorous allogeneic immune responses, we next assessed the survival of heart allografts. Similar to skin grafts, BALB/c heart grafts transplanted simultaneously with SA-FasL-engineered islets were rejected, albeit at a delayed tempo as compared with control heart allografts alone (Figure 4G; MST = 25 ± 1.7 vs 12 ± 0.9 days for controls, p = 0.0169). Heart graft rejection also caused hyperglycemia, an indication of SA-FasL-engineered islet graft rejection (Figure 4G; MST = 25 ± 2.0 days). Thus, these results demonstrate that localized immunomodulation with SA-FasL-engineered islets evolves into systemic tolerance at the induction phase that is both donor- and tissue-specific.
3.5. SA-FasL-engineered islets attain donor-specific immune privilege
Although FasL has been implicated in physiological immune privilege (26, 27), the direct evidence for such a role in induced immune privilege remains to be provided. To assess if the SA-FasL-engineered islet graft induces an immune privileged site, long-term (> 60 days) recipients were treated with streptozotocin to destroy the graft (Figure 5A). A group of mice were transplanted with a second set of unmodified donor matched or C3H third party islets into the same primary graft site 4 days after the confirmation of hyperglycemia. The second set of BALB/c islet grafts restored euglycemia in all recipients for an observation period exceeding 70 days (Figure 5B). In marked contrast, third party islet allografts were rejected within 20 days (Figure 5B; MST = 19 ± 2.7 days).
FIGURE 5.
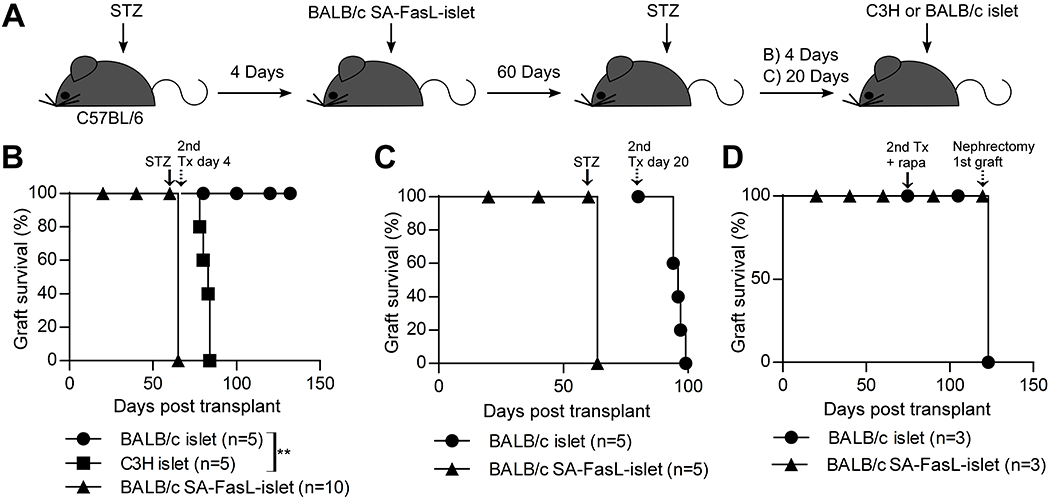
Tolerance induced by SA-FasL-engineered islet grafts evolves into donor-specific immune privilege at the maintenance phase sustained by the presence of graft and Treg cells. A, Schematic diagram showing the study design. B, C57BL/6 mice with functional SA-FasL-engineered BALB/c islet grafts were treated with streptozotocin a second time on day 60 post transplantation (solid arrow) to destroy the graft. After confirmation of hyperglycemia, a cohort of mice were re-transplanted immediately (4 days post-streptozotocin treatment) with a second set of unmodified third party C3H or donor-matched islet grafts under the same kidney capsule harboring the original graft (dotted arrow). C3H third party, but not BALB/c donor-matched, islet grafts were rejected acutely (MST = 19 ± 2.7 days; log-rank p = 0.0027). C, Delayed re-transplantation of a second set of donor-matched unmodified islets (dotted arrow) 20 days post streptozotocin treatment (arrow) results in acute islet rejection (MST = 16 ± 2.1 days), demonstrating that the induced graft site-immune privilege requires the presence of alloantigens (graft) for maintenance. D, Rapamycin treatment does not confer protection to a second set of islet graft transplanted at a distant site. Long-term (80 days) C57BL/6 recipients of BALB/c SA-FasL-engineered islet grafts were transplanted with a second set, donor-matched unmanipulated islet graft under the contralateral kidney capsule (arrow), and treated with rapamycin (0.2 mg/kg daily for 15 days starting the day of second islet transplantation). Surgical removal of the kidney harboring the original islet graft on day 120 post-transplantation (dotted arrow) resulted in prompt hyperglycemia, demonstrating that treatment with rapamycin does not extend the immune privilege status of the primary SA-FasL-engineered islets to a second set unmodified graft.
To test whether alloantigens expressed by the graft are the driving force for the maintenance of induced immune privilege, a second set of long-term (> 60 days) graft recipients were injected with streptozotocin and maintained for 20 days with exogenous insulin, a period considered to be sufficient for clearance of the injured graft. Transplantation of unmodified donor-matched second set of islet grafts under the same kidney capsule that harbored the primary graft resulted in temporary euglycemia and rejection of all grafts in an acute fashion (Figure 5C; MST = 16 ± 2.1 days). These data demonstrate a requirement for the continued presence of islet alloantigens for the maintenance of induced immune privilege site.
3.6. Immune privilege cannot be extended to a second set of unmanipulated islet grafts transplanted to a distant site under the cover of rapamycin
The two-islet graft model showed that peripheral tolerance is systemic at the induction phase and requires a short course of rapamycin. This agent works in synergy with Fas-mediated apoptosis to eliminate Teff cells (32, 33) and also has a positive effect on the generation, maintenance, and function of Treg cells (34–36). In as much as tolerance in our model is maintained by Treg cells (Figure 4D) that infiltrate into the graft site and localize to the periphery of the long surviving grafts (17), we tested if rapamycin can expand and/or mobilize Treg cells from long-term surviving primary grafts into the second set of donor graft transplanted at a distal site. Long-term (80 days) acceptors of SA-FasL-engineered BALB/c islets were transplanted with a second set of unmodified, donor-matched islet grafts under the contralateral kidney capsule under the same rapamycin regimen used for the induction of tolerance. Surgical removal of the primary graft 40 days after transplantation of the secondary graft resulted in prompt hyperglycemia (Figure 5D), showing that the secondary graft had already been rejected. These data confirm our previously published studies demonstrating that tolerance at maintenance phase is localized (17), and further show that rapamycin at this phase cannot mobilize/extend tolerance to a secondary graft placed in a distal site.
4. DISCUSSION
We report four novel findings in this manuscript. First, tolerance is biphasic and spatiotemporal; systemic at the induction phase and restricted to the transplant site at the maintenance phase. Second both phagocytes and TGF-β play direct roles in SA-FasL-induced tolerance, as depletion of phagocytes or neutralization of TGF-β negates tolerance. Third, our studies provide direct evidence that SA-FasL-engineered islet grafts achieve a bona fide induced immune privilege status as demonstrated by permanent survival of an unmodified second set graft transplanted to the same site after streptozotocin destruction of the original graft. Fourth, both the islet graft and Treg cells are required for the maintenance of SA-FasL-induced immune privilege.
The tolerogenic efficacy of FasL reported here is consistent with several studies reporting the utility of FasL for immunomodulation (8, 26, 37–40). However, these observations are at variance with others studies showing the inefficacy of FasL to induce tolerance to allografts (9, 10, 41). The complex transcriptional control, post-translational regulation and processing of FasL, the existence of two distinct forms (soluble and membrane-bound) having opposite functions, the reverse signaling observed for FasL, as well as the complication of continuous expression of wild type FasL in target tissues, in case of gene therapy, are some of the variables that may explain the observed conflicting results (8–10, 15, 42–46). In particular, wild type FasL is cleaved from the cell surface by matrix metalloproteinases into a soluble form that has minimal apoptotic function, blocks apoptosis by competing with membrane-bound FasL for binding Fas, and also has chemotactic function on neutrophils (13, 15, 47). Thus, ectopic expression of FasL in grafts may accelerate rejection (10, 46).
The lack of chemotactic function for neutrophils, potent apoptotic activity on Fas-expressing lymphocytes, and transient display on islet grafts are some important features that distinguish the SA-FasL used in the present study from the wild type FasL and may account for its demonstrated tolerogenic efficacy (16, 17, 48). Islet grafts ectopically expressing wild type FasL were shown to undergo acute rejection mediated by neutrophils (10, 46). Similarly, tumors expressing the wild type FasL were rejected as a result of neutrophil influx (44). Co-expression of TGF-β with FasL was effective in overcoming tumor rejection by blocking neutrophil infiltration (44). Furthermore, the tolerogenic function of FasL may be regulated by the extent and duration of FasL expression in the target tissues. Sustained expression may cause excessive apoptosis that not only negatively affect pathogenic, but also regulatory immune responses. Excessive apoptosis was shown to result in secondary necroptosis that precipitates proinflammatory responses (49). Lastly, overexpression of the soluble form of FasL in tissues rich in metalloproteinases may program an anti-apoptotic and proinflammatory cascade that results in destructive, rather than protective immune responses against allografts (45). In this context, the transient display of SA-FasL having primarily apoptotic function on the surface of allogeneic islets for localized immunomodulation overcomes various shortcomings of the wild type FasL.
Induction of tolerance in our model required phagocytes and TGF-β, as ablation of phagocytes or in vivo blockade of TGF-β resulted in acute graft rejection. Clearance of apoptotic cells by dendritic cells and macrophages has been shown to reprogram these cells to acquire a tolerogenic phenotype characterized by reduced capacity to produce proinflammatory cytokines and heightened ability to secrete regulatory cytokines, including TGF-β (50–52). Treatment of mice with an anti-CD3 antibody was shown to prevent and treat induced experimental autoimmune encephalomyelitis by inducing apoptosis in T cells. Clearance of apoptotic bodies by phagocytes resulted in the production of TGF-β and induction of CD4+CD25+Foxp3+ T cells that conferred autotolerance (28). Similarly, systemic immunomodulation with ECDI-fixed donor splenocytes were shown to induce tolerance to auto and allografts through a complex circuit of immunoregulation that also involves phagocytosis of apoptotic bodies by phagocytes, requirement for TGF-β, dendritic cells, and Treg cells for tolerance induction (52, 53). FasL-mediated tumor immune evasion was also shown to require TGF-β (44). Thus in our model, TGF-β secreted by T cells undergoing apoptosis and/or macrophages engulfing apoptotic bodies within the graft milieu may act to restrict Teff cell function and also contribute to the generation and suppressor function of Treg cells (29–31, 54). The systemic nature of tolerance at the induction phase may involve the trafficking of some of Treg cells generated within SA-FasL-engineered graft milieu to the unmanipulated islet grafts transplanted at a distant site in response to inflammatory cues. In marked contrast, the lack of apoptosis and immunoregulatory loop containing phagocytes and TGF-β at the maintenance phase may present a limitation for the generation of new Treg cells and/or mobilization to a distant site to confer tolerance to unmanipulated second set of islet grafts.
In variance to the induction phase where tolerance is systemic, at maintenance phase tolerance evolved into graft-localized immune privilege that required the presence of Treg cells as well as the islet allograft. Although FasL has been implicated in acquired immune privilege (8, 26, 27), to our knowledge this is the first study to provide direct evidence for such a role by demonstrating that a naïve, unmanipulated second set islet grafts survived rejection following transplantation into the same site that had previously supported the long-term survival of SA-FasL-engineered grafts. The immune privilege was antigen-specific and could not be extended to an unmanipulated islet graft transplanted at a distal site, even in the presence of rapamycin potentially expanding and mobilizing Treg cells (34–36) as required for immune privilege in our model. The established immune privilege required the persistence of alloantigens in the form of graft, as delayed (4 vs 20 days) transplantation of a second set of unmanipulated islet grafts following the destruction of original grafts with streptozotocin resulted in acute rejection. These observations are consistent with the reported role of FasL in physiological immune privileged sites (27, 55). FasL in the eye contained herpes simplex virus-induced inflammation by eliminating activated T cells (27). Corneal grafts expressing FasL under normal physiological conditions from wild type, but not mutant mice lacking this molecule, showed long-term survival in allogeneic recipients (26). Our findings are also consistent with studies using tissues with ectopic expression of FasL. Syngeneic myoblasts transfected to express FasL protected unmodified allogenic islets from rejection when co-transplanted under the same kidney capsule (8).
Tolerance was antigen and tissue specific, as SA-FasL-engineered islets failed to protect third party islet and donor-matched skin and heart grafts. These observations are consistent with the established nature of tolerance specificity and split tolerance dictated by the nature of tissue-specific antigens (56). However, rejection of donor heart and skin, but not the third-party skin, also culminated in the rejection of SA-FasL-engineered islet grafts. These data do not fully support the tissue-specific nature of tolerance, but are more compatible with a model whereby tolerance varies with the potency of induced alloreactive responses, which depends on site of immune priming as well as vascularized nature of the graft, and immunoregulatory mechanisms. Non-vascularized skin grafts have been shown to generate robust alloreactive T cell responses induced in the draining LNs by both direct and indirect recognition pathways, whereas vascularized heart grafts mainly generate a rapid and vast repertoire of alloreactive CD4+ T cells primed in the spleen by the direct alloantigen recognition pathway (22, 57, 58). Such activated CD4+ T cells reject heart graft without a contribution form CD8+ T cells (57). In marked contrast, rejection of neovascularized allogenic islet grafts requires a collaboration between CD4+ T and CD8+ T cells (23, 59). Thus, we speculate that a large repertoire of activated alloreactive T cells generated by heart graft provides help to alloreactive T cells responding to the islet graft and that have not been eliminated by SA-FasL due to their expression of low affinity TCR or insufficient concentration of SA-FasL arising from its short half-life (~3. 5 days) in vivo on biological surfaces (16). We have previously shown that systemic immunomodulation with SA-FasL-engineered splenocytes induces tolerance to cardiac allograft in rats in the absence of any conjunctive immunosuppression (60). This observation demonstrates the robust nature of SA-FasL-based immunomodulation for tolerance induction to allografts, and further suggests that the potency of tolerance will depend on the setting, systemic versus localized, on the nature of the target tissue engineered with SA-FasL, and ultimately on the treatment scheme and/or dose of SA-FasL. For example, immunomodulation with SA-FasL-engineered splenocytes utilized repeated systemic treatments with higher doses of SA-FasL that proved to be more robust in tolerance induction to cardiac grafts (60).
The transient display of immunomodulatory ligands on the surface of grafts holds the potential to induce permanent graft acceptance in the absence of chronic immunosuppression. This approach of localized immunomodulation also has the added benefit of safety, particularly for biologics with robust pleiotropic functions that may have off-target effects when used systemically, and improved efficacy, presentation of biologics where they are needed, i.e. at the target site. As such, this concept The marked advantage of an SA-FasL-based immunomodulatory approach is donor antigen and site specificity that may allow for generation of competent systemic immune responses against pathogens without a deleterious effect on the survival of immune privileged transplant. Furthermore, given that newly activated and memory Teff cells express Fas on their surface and are sensitive to FasL-mediated apoptosis (4–6), SA-FasL-based immunomodulation may also have utility for the prevention and treatment of various autoimmune diseases, including type 1 diabetes by inducing immune privilege through modulation of Teff and Treg cells. Lastly, this technology allows for simultaneous display of more than one immunomodulatory proteins on the surface of target cells or tissues at desired concentrations, thereby providing an opportunity to exploit functional synergy between biologics for improved efficacy (61).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in parts by grants from National Institutes of Health (R56AI121281, R21AI113348-01, R01AI121281, U01AI132817), Juvenile Diabetes Research Foundation (17-2012-527), American Diabetes Association (1-12-BS-191), the Commonwealth of Kentucky Research Challenge Trust Fund, Keck Foundation, William Marvin Petty Gift for Type 1 Diabetes, and National Institutes of Health Training Grant 5T32 HL076138 to H.Z.
Abbreviations
- AICD
Activation induced cell death
- SA
Streptavidin
- SA-FasL
Fas ligand chimeric with streptavidin
- DC
Dendritic cells
- TGF-β
Transforming growth factor-β
- DMDP
Dichloromethylenediphosphonic acid
- Teff
T effector cells
- Treg
Regulatory T cells
- CFSE
Carboxyfluorescein succinimidyl ester
- OVA
Ovalbumin
- HPRT
Hypoxanthine guanine phosphoribosyl transferase
- MST
Mean Survival time
Footnotes
DISCLOSURE
The authors of this manuscript have conflicts of interest to disclose as described by the American Journal of Transplantation. The SA-FasL molecule and method of cell surface display technology described in this manuscript are subject to a license from the University of Louisville by FasCure Therapeutics, LLC, Louisville, KY, founded by H.S. H.S. and E.S.Y. have significant equity interest in the Company. The other authors have no conflicts of interest to disclose.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- 1.Hering BJ, Clarke WR, Bridges ND, Eggerman TL, Alejandro R, Bellin MD et al. Phase 3 trial of transplantation of human Islets in Type 1 diabetes complicated by severe hypoglycemia. Diabetes Care 2016;39(7):1230–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fan Z, Spencer JA, Lu Y, Pitsillides CM, Singh G, Kim P et al. In vivo tracking of ‘color-coded’ effector, natural and induced regulatory T cells in the allograft response. Nat Med 2010;16(6):718–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gill RG, Rosenberg AS, Lafferty KJ, Singer A. Characterization of primary T cell subsets mediating rejection of pancreatic islet grafts. J Immunol 1989;143:2176–2178. [PubMed] [Google Scholar]
- 4.Ju S-T, Panka DJ, Cul H, Ettinger R, El-Khatib M, Sherr DH et al. Fas(CD95)/FasL interactions required for programmed cell death after T-cell activation. Nature 1996;373:444–448. [DOI] [PubMed] [Google Scholar]
- 5.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature 1992;356:314–317. [DOI] [PubMed] [Google Scholar]
- 6.Dhein J, Walczak H, Bäumler C, Debatin K-M, Krammer PH. Autocrine T-cell suicide mediated by APO-1/(Fas/CD95). Nature 1995;373(6513):438–441. [DOI] [PubMed] [Google Scholar]
- 7.Rieux-Laucat F, Le DF, Hivroz C, Roberts IA, Debatin KM, Fischer A et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science 1995;268(5215):1347–1349. [DOI] [PubMed] [Google Scholar]
- 8.Lau HT, Yu M, Fontana A, Stoeckert CJ Jr. Prevention of islet allograft rejection with engineered myoblasts expressing FasL in mice. Science 1996;273:109–112. [DOI] [PubMed] [Google Scholar]
- 9.Allison J, Georgiou HM, Strasser A, Vaux DL. Transgenic expression of CD95 ligand on islet beta cells induces a granulocytic infiltration but does not confer immune privilege upon islet allografts. Proc Natl Acad Sci U S A 1997;94:3943–3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kang SM, Schneider DB, Lin Z, Hanahan D, Dichek DA, Stock PG et al. Fas ligand expression in islets of Langerhans does not confer immune privilege and instead targets them for rapid destruction. Nat Med 1997;3:738–743. [DOI] [PubMed] [Google Scholar]
- 11.Zhang H, Yang Y, Horton JL, Samoilova EB, Judge TA, Turka LA et al. Amelioration of collagen-induced arthritis by CD95 (Apo-1/Fas)-ligand gene transfer. J Clin Invest 1997;100:1951–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tanaka M, Itai T, Adachi M, Nagata S. Downregulation of Fas ligand by shedding. Nat Med 1998;4:31–36. [DOI] [PubMed] [Google Scholar]
- 13.Schneider P, Holler N, Bodmer JL, Hahne M, Frei K, Fontana A et al. Conversion of membrane-bound Fas(CD95) ligand to its soluble form is associated with downregulation of its proapoptotic activity and loss of liver toxicity. J Exp Med 1998;187:1205–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Seino K, Iwabuchi K, Kayagaki N, Miyata R, Nagaoka I, Matsuzawa A et al. Chemotactic activity of soluble Fas ligand against phagocytes. J Immunol 1998;161:4484–4488. [PubMed] [Google Scholar]
- 15.O’ Reilly LA, Tai L, Lee L, Kruse EA, Grabow S, Fairlie WD et al. Membrane-bound Fas ligand only is essential for Fas-induced apoptosis. Nature 2009;461(7264):659–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yolcu ES, Askenasy N, Singh NP, Cherradi SE, Shirwan H. Cell membrane modification for rapid display of proteins as a novel means of immunomodulation: FasL-decorated cells prevent islet graft rejection. Immunity 2002;17(6):795–808. [DOI] [PubMed] [Google Scholar]
- 17.Yolcu ES, Zhao H, Bandura-Morgan L, Lacelle C, Woodward KB, Askenasy N et al. Pancreatic islets engineered with SA-FasL protein establish robust localized tolerance by inducing regulatory T cells in mice. J Immunol 2011;187(11):5901–5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Headen DM, Woodward KB, Coronel MM, Shrestha P, Weaver JD, Zhao H et al. Local immunomodulation with Fas ligand-engineered biomaterials achieves allogeneic islet graft acceptance. Nat Mater 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skoumal M, Woodward KB, Zhao H, Wang F, Yolcu ES, Pearson RM et al. Localized immune tolerance from FasL-functionalized PLG scaffolds. Biomaterials 2019;192:271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Elpek KG, Yolcu ES, Franke DD, Lacelle C, Schabowsky RH, Shirwan H. Ex vivo expansion of CD4+CD25+FoxP3+ T regulatory cells based on synergy between IL-2 and 4–1BB signaling. J Immunol 2007;179(11):7295–7304. [DOI] [PubMed] [Google Scholar]
- 21.Kurts C, Heath WR, Carbone FR, Allison J, Miller JF, Kosaka H. Constitutive class I-restricted exogenous presentation of self antigens in vivo. J Exp Med 1996;184(3):923–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kant CD, Akiyama Y, Tanaka K, Shea S, Connolly SE, Germana S et al. Primary vascularization of allografts governs their immunogenicity and susceptibility to tolerogenesis. J Immunol 2013;191(4):1948–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Makhlouf L, Yamada A, Ito T, Abdi R, Ansari MJ, Khuong CQ et al. Allorecognition and effector pathways of islet allograft rejection in normal versus nonobese diabetic mice. J Am Soc Nephrol 2003;14(8):2168–2175. [DOI] [PubMed] [Google Scholar]
- 24.Miska J, Abdulreda MH, Devarajan P, Lui JB, Suzuki J, Pileggi A et al. Real-time immune cell interactions in target tissue during autoimmune-induced damage and graft tolerance. J Exp Med 2014;211(3):441–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang N, Schroppel B, Lal G, Jakubzick C, Mao X, Chen D et al. Regulatory T cells sequentially migrate from inflamed tissues to draining lymph nodes to suppress the alloimmune response. Immunity 2009;30(3):458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stuart PM, Griffith TS, Usui N, Pepose J, Yu X, Ferguson TA. CD95 ligand (FasL)-induced apoptosis is necessary for corneal allograft survival. J Clin Invest 1997;99(3):396–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Griffith TS, Brunner T, Fletcher SM, Green DR, Ferguson TA. Fas ligand-induced apoptosis as a mechanism of immune privilege. Science 1995;270(5239):1189–1192. [DOI] [PubMed] [Google Scholar]
- 28.Perruche S, Zhang P, Liu Y, Saas P, Bluestone JA, Chen W. CD3-specific antibody-induced immune tolerance involves transforming growth factor-beta from phagocytes digesting apoptotic T cells. Nat Med 2008;14(5):528–535. [DOI] [PubMed] [Google Scholar]
- 29.Chen W, Frank ME, Jin W, Wahl SM. TGF-beta released by apoptotic T cells contributes to an immunosuppressive milieu. Immunity 2001;14(6):715–725. [DOI] [PubMed] [Google Scholar]
- 30.Belghith M, Bluestone JA, Barriot S, Megret J, Bach JF, Chatenoud L. TGF-beta-dependent mechanisms mediate restoration of self-tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med 2003;9(9):1202–1208. [DOI] [PubMed] [Google Scholar]
- 31.Fadok VA, Bratton DL, Konowal A, Freed PW, Westcott JY, Henson PM. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest 1998;101(4):890–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li Y, Li XC, Zheng XX, Wells AD, Turka LA, Strom TB. Blocking both signal 1 and signal 2 of T-cell activation prevents apoptosis of alloreactive T cells and induction of peripheral allograft tolerance. Nat Med 1999;5(11):1298–1302. [DOI] [PubMed] [Google Scholar]
- 33.Algeciras-Schimnich A, Griffith TS, Lynch DH, Paya CV. Cell cycle-dependent regulation of FLIP levels and susceptibility to Fas-mediated apoptosis. J Immunol 1999;162(9):5205–5211. [PubMed] [Google Scholar]
- 34.Delgoffe GM, Kole TP, Zheng Y, Zarek PE, Matthews KL, Xiao B et al. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 2009;30(6):832–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de la Rosa M, Rutz S, Dorninger H, Scheffold A. Interleukin-2 is essential for CD4+CD25+ regulatory T cell function. Eur J Immunol 2004;34(9):2480–2488. [DOI] [PubMed] [Google Scholar]
- 36.Battaglia M, Stabilini A, Roncarolo MG. Rapamycin selectively expands CD4+CD25+FoxP3+ regulatory T cells. Blood 2005;105(12):4743–4748. [DOI] [PubMed] [Google Scholar]
- 37.George JF, Sweeney SD, Kirklin JK, Simpson EM, Goldstein DR, Thomas JM. An essential role for Fas ligand in transplantation tolerance induced by donor bone marrow. Nat Med 1998;4:333–335. [DOI] [PubMed] [Google Scholar]
- 38.Matsue H, Matsue K, Walters M, Okumura K, Yagita H, Takashima A. Induction of antigen-specific immunosuppression by CD95L cDNA-transfected ‘killer’ dendritic cells. Nat Med 1999;5:930–937. [DOI] [PubMed] [Google Scholar]
- 39.Motz GT, Santoro SP, Wang LP, Garrabrant T, Lastra RR, Hagemann IS et al. Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat Med 2014;20(6):607–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Takeda Y, Gotoh M, Dono K, Nishihara M, Grochowiecki T, Kimura F et al. Protection of islet allografts transplanted together with Fas ligand expressing testicular allografts. Diabetologia 1998;41(3):315–321. [DOI] [PubMed] [Google Scholar]
- 41.Takeuchi T, Ueki T, Nishimatsu H, Kajiwara T, Ishida T, Jishage K et al. Accelerated rejection of Fas ligand-expressing heart grafts. J Immunol 1999;162:518–522. [PubMed] [Google Scholar]
- 42.Batteux F, Tourneur L, Trebeden H, Charreire J, Chiocchia G. Gene therapy of experimental autoimmune thyroiditis by in vivo administration of plasmid DNA coding for Fas ligand. J Immunol 1999;162(1):603–608. [PubMed] [Google Scholar]
- 43.Bossi G, Griffiths GM. Degranulation plays an essential part in regulating cell surface expression of Fas ligand in T cells and natural killer cells. Nat Med 1999;5:90–96. [DOI] [PubMed] [Google Scholar]
- 44.Chen JJ, Sun Y, Nabel GJ. Regulation of the proinflammatory effects of Fas ligand (CD95L). Science 1998;282(5394):1714–1717. [DOI] [PubMed] [Google Scholar]
- 45.Lau HT, Stoeckert CJ. FasL--too much of a good thing? Transplanted grafts of pancreatic islet cells engineered to express Fas ligand are destroyed not protected by the immune system. Nat Med 1997;3(7):727–728. [DOI] [PubMed] [Google Scholar]
- 46.Ottonello L, Tortolina G, Amelotti M, Dallegri F. Soluble Fas ligand is chemotactic for human neutrophilic polymorphonuclear leukocytes. J Immunol 1999;162:3601–3606. [PubMed] [Google Scholar]
- 47.Suda T, Hashimoto H, Tanaka M, Ochi T, Nagata S. Membrane Fas ligand kills human peripheral blood T lymphocytes, and soluble Fas ligand blocks the killing. J Exp Med 1997;186:2045–2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Askenasy N, Yolcu ES, Wang Z, Shirwan H. Display of Fas Ligand protein on cardiac vasculature as a novel means of regulating allograft rejection. Circulation 2003;107:41–47. [DOI] [PubMed] [Google Scholar]
- 49.Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL et al. Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 2001;411(6834):207–211. [DOI] [PubMed] [Google Scholar]
- 50.Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest 2002;109(1):41–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sen P, Wallet MA, Yi Z, Huang Y, Henderson M, Mathews CE et al. Apoptotic cells induce Mer tyrosine kinase-dependent blockade of NF-kappaB activation in dendritic cells. Blood 2007;109(2):653–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Luo X, Pothoven KL, McCarthy D, DeGutes M, Martin A, Getts DR et al. ECDI-fixed allogeneic splenocytes induce donor-specific tolerance for long-term survival of islet transplants via two distinct mechanisms. Proc Natl Acad Sci U S A 2008;105(38):14527–14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen G, Kheradmand T, Bryant J, Wang S, Tasch J, Wang JJ et al. Intragraft CD11b(+) IDO(+) cells mediate cardiac allograft tolerance by ECDI-fixed donor splenocyte infusions. Am J Transplant 2012;12(11):2920–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N et al. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med 2003;198(12):1875–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Griffith TS, Ferguson TA. The role of fasl-induced apoptosis in immune privilege. Immunol Today 1997;18(5):240–244. [DOI] [PubMed] [Google Scholar]
- 56.Chan WF, Razavy H, Luo B, Shapiro AM, Anderson CC. Development of either split tolerance or robust tolerance along with humoral tolerance to donor and third-party alloantigens in nonmyeloablative mixed chimeras. J Immunol 2008;180(8):5177–5186. [DOI] [PubMed] [Google Scholar]
- 57.Pietra BA, Wiseman A, Bolwerk A, Rizeq M, Gill RG. CD4 T cell-mediated cardiac allograft rejection requires donor but not host MHC class II. J Clin Invest 2000;106(8):1003–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jones ND, Turvey SE, Van Maurik A, Hara M, Kingsley CI, Smith CH et al. Differential susceptibility of heart, skin, and islet allografts to T cell-mediated rejection. J Immunol 2001;166(4):2824–2830. [DOI] [PubMed] [Google Scholar]
- 59.Coulombe M, Yang H, Wolf LA, Gill RG. Tolerance to antigen-presenting cell-depleted islet allografts is CD4 T cell dependent. J Immunol 1999;162(5):2503–2510. [PubMed] [Google Scholar]
- 60.Yolcu ES, Gu X, Lacelle C, Zhao H, Bandura-Morgan L, Askenasy N et al. Induction of tolerance to cardiac allografts using donor splenocytes engineered to display on their surface an exogenous fas ligand protein. J Immunol 2008;181(2):931–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sharma RK, Yolcu ES, Elpek KG, Shirwan H. Tumor cells engineered to codisplay on their surface 4-1BBL and LIGHT costimulatory proteins as a novel vaccine approach for cancer immunotherapy. Cancer Gene Ther 2010;17(10):730–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.