

TET2 deficiency reprograms B cell epigenome linked to lymphomagenesis.
Abstract
The TET2 DNA hydroxymethyltransferase is frequently disrupted by somatic mutations in diffuse large B cell lymphomas (DLBCLs), a tumor that originates from germinal center (GC) B cells. Here, we show that TET2 deficiency leads to DNA hypermethylation of regulatory elements in GC B cells, associated with silencing of the respective genes. This hypermethylation affects the binding of transcription factors including those involved in exit from the GC reaction and involves pathways such as B cell receptor, antigen presentation, CD40, and others. Normal GC B cells manifest a typical hypomethylation signature, which is caused by AID, the enzyme that mediates somatic hypermutation. However, AID-induced demethylation is markedly impaired in TET2-deficient GC B cells, suggesting that AID epigenetic effects are partially dependent on TET2. Last, we find that TET2 mutant DLBCLs also manifest the aberrant TET2-deficient GC DNA methylation signature, suggesting that this epigenetic pattern is maintained during and contributes to lymphomagenesis.
INTRODUCTION
Disruption of epigenetic programming has emerged as a hallmark of various types of hematological malignancies (1), including diffuse large B cell lymphomas (DLBCLs), which is the most common form of non-Hodgkin lymphomas (2). A number of studies have demonstrated disruption of cytosine methylation [5-methylcytosine (5mC)] patterning as a factor linked to the clinical outcome and biology of DLBCL (3). One manner in which aberrant 5mC contributes to the growth of these tumors is through silencing of tumor suppressors such as CDKN2A, a process that is linked to unfavorable clinical outcome in DLBCL and other hematological cancers (4). The degree to which 5mC patterning in DLBCL deviates from that in normal B cells is negatively correlated with survival time (5). Moreover, DLBCLs manifest substantial inter- and intratumor epigenetic heterogeneity, which has been linked to poorer clinical outcomes, likely due to increased population fitness (6). The importance of aberrant 5mC in DLBCL is further supported by data suggesting favorable response of newly diagnosed, high-risk DLBCL patients to DNA methyltransferase inhibitors (DNMTi) given in combination with standard chemoimmunotherapy (7). Nevertheless, little is still known about the molecular mechanisms underlying aberrant 5mC in lymphomagenesis. The fact that many patients with high-risk DLBCL will die of their disease underlies the clinical importance of understanding the mechanisms through which cytosine methylation patterning is affected during lymphomagenesis.
5mC is well established as an epigenetic mark associated with transcriptional silencing, especially when linked to promoter-associated CpG islands (8). The distribution and dynamic turnover of cytosine methylation are controlled by enzymes that modify or excise cytosine residues in DNA. The ten-eleven translocation (TET) family enzymes are involved in active DNA demethylation, catalyzing the oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine, or 5-carboxylcytosine (9). More recently, it has been appreciated that 5hmC also functions as an epigenetic mark, and when linked to gene enhancers, is associated with activation of nearby genes (10, 11). Of the three TET-family genes, TET2 is the one most often altered by somatic mutations in hematological malignancies, including in approximately 10% of patients with DLBCL (12–15). These mutations are similar to those observed in myeloid and T cell neoplasms and disrupt TET2 through various mechanisms, including accumulation of nonsense, missense, or frameshift mutations within the TET2 coding region, splicing sites, or other evolutionary conserved regions of the gene, which result in partial or total loss of function of the TET2 protein (16–18).
DLBCLs arise from B cells transiting the germinal center (GC) reaction. Programmed deletion of TET2 in hematopoietic cells or B cells disrupts the ability of GC B cells to undergo class switch recombination and terminal differentiation (14, 19, 20). Furthermore, GC-directed TET2 deletion in mice results in accelerated development of DLBCLs, thus confirming its role as a bona fide B cell tumor suppressor (14). One notable consequence of TET2 loss of function in GC B cells is focal loss of 5hmC at enhancers linked to B cell differentiation (14). Since TET2 deficiency in GC B cells leads to loss of 5hmC, it is reasonable to assume that TET2 deficiency could be connected with a consequent gain of 5mC levels. However, the impact of TET2 loss of function directly on cytosine methylation patterning in GC B cells is unknown. Using a 450 K DNA methylation microarray in a cohort of patients with DLBCL, Asmar et al. (12) observed evidence of relative hypermethylation in CpG-rich regions in TET2 mutant DLBCL cases, which raises the possibility that TET2 loss of function might alter the epigenome through this mechanism in GC B cells. Here, we investigated the impact of TET2 deficiency on cytosine methylation patterning in GC B cells in mice, how this links to disruption of transcriptional regulation, and whether and how these observations can be extended to primary human DLBCLs to illustrate the manner in which TET2 deficiency contributes to the tumor phenotype.
RESULTS
TET2 deficiency leads to hypermethylation in GC B cells
Given that TET2 mediates DNA demethylation, we hypothesized that TET2 deficiency in GC B cells might result in aberrant hypermethylation. To test this, we performed enhanced reduced representation bisulfite sequencing (ERRBS) on sorted naïve B (NB) cells (B220+GL7−FAS−DAPI−) and GC B cells (B220+GL7+FAS+DAPI−) from Vav-Cre/Tet2−/− (conditional knockout) and Vav-Cre/Tet2+/+ (control) mice. Principal components analysis and unsupervised hierarchal clustering yielded a clear separation of methylation profiles between Tet2−/−- and Tet2+/+-sorted mouse GC B cells (Fig. 1, A and B). In contrast, there was little difference between the DNA methylation profiles of Tet2−/− and Tet2+/+ NB cells (fig. S1, A and B). A supervised analysis of 5mC profiles revealed 10,730 differentially methylated cytosines (DMCs) in Tet2−/− GC B cells, compared to only 2091 DMCs in control Tet2−/− NB cells (q value < 0.01; methylation difference > 25%; table S1 and Fig. 1C). In Tet2−/− GC B cells, DMCs were distributed approximately uniformly across chromosomes (Fig. 1D), and a majority of these (9043 or 84.3%) were hypermethylated (Fig. 1E). Of the 9043 hypermethylated DMCs, 2126 (23.5%) were located within promoter regions [2 kb up- and downstream of the transcriptional starting sites (TSSs)], where they could potentially influence gene expression.
Fig. 1. Tet2 deficiency leads to hypermethylation in GC B cells and loss of hypomethylation during the NB-to-GCB transition.
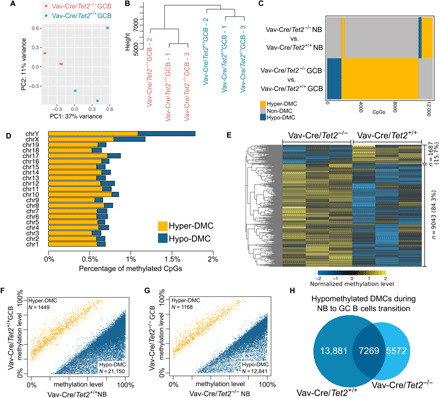
(A) Principal components analysis based on ERRBS data from three Tet2−/− and three Tet2+/+ GC B cell (GCBs) samples. (B) Hierarchal clustering based on ERRBS data from three Tet2−/− and three Tet2+/+ GCB samples. (C) Comparison of differential methylation events in NB cells and GCBs. (D) Differential methylation events per chromosome. (E) Row-normalized heatmap of the methylation levels of differential methylation events in GCBs (three Tet2−/− replicates and three Tet2+/+ replicates). (F) Differential methylation during transition of NB cells to GCBs in Tet2+/+ mice. Each dot on the scatter plot represents an individual CpG called as differentially methylated. Blue dots represent hypomethylation, and yellow represent hypermethylation. (G) Differential methylation during transition of NB cells to GCBs in Tet2−/− mice. Each dot on the scatter plot represents an individual CpG called as differentially methylated. Blue dots represent hypomethylation, and yellow represent hypermethylation. (H) Venn diagram of the overlap between hypomethylated DMCs in Tet2−/− and in Tet2+/+ mouse models.
The epigenetic signature of GC B cells is reported to be, for the most part, hypomethylated relative to that of NB cells (21, 22). Since TET2 is linked to demethylation, we next asked whether TET2 loss of function would affect formation of this characteristic GC B cell epigenetic signature. We first examined DMCs in TET2 wild-type (WT) NBs and GC B cells and observed that, of the total of 22,599 DMCs in the two types of cells, 93.6% were hypomethylated (Fig. 1F and table S1), in accordance with previous reports. Notably, when comparing Tet2−/− NBs to Tet2−/− GC B cells, we observed significantly fewer hypomethylated DMCs in Tet2−/− GC B cells than we observed in GC B cells when comparing TET2 WT NBs and GC B cells (12,841 versus 21,150; 8309 fewer; Fisher’s exact test, P value ≈ 0; Fig. 1, F and G, and table S1), suggesting that hypomethylation of these sites might be dependent on TET2 demethylating activity. Tet2−/− mice failed to demethylate 13,881 of the ~21,150 DMCs that were hypomethylated in Tet2+/+ mice (Fig. 1H). Nonetheless, more than half of the DMCs hypomethylated in Tet2−/− mice were also hypomethylated in WT Tet2 mice (7269 of 12,841; Fig. 1H), suggesting that demethylation of these 7269 residues is independent of TET2. Together, these results are consistent with the notion that TET2 loss of function might disrupt the normal biology of GC B cells in part through disruption of cytosine methylation patterning.
Tet2 deficiency links to transcriptional repression via promoter hypermethylation and loss of enhancer 5hmC
TET2 was shown to play a role in gene activation by demethylation of enhancers (23). Tet2-deficient GC B cells manifest an aberrant transcriptional signature featuring widespread gene repression that is associated with loss of gene enhancer (but not promoter) 5hmC peaks (14). It is possible that aberrant 5mC hypermethylation might also be linked to these enhancer effects. Alternatively, Tet2 deficiency might result in aberrant promoter methylation that could repress genes in cooperation with enhancer loss of 5hmC. To address these questions, we performed an integrative analysis of ERRBS DNA methylation profiles, genome-wide 5hmC profiles [hydroxymethylated DNA immunoprecipitation sequencing (hMeDIP-seq)] (14), and expression profiles [RNA sequencing (RNA-seq)] (14), all obtained from Tet2−/− versus Tet2+/+ GC B cells. We organized this analysis based on functional annotation of the genome into promoters (TSS ± 2 kbp), exons, introns, putative enhancers (defined as intergenic or intronic H3K27ac peaks in splenic B cells, excluding promoters), intergenic regions, and regions losing 5hmC signal [hypo–DHMRs (differentially hydroxymethylated regions)] (Fig. 2A). Notably, the number of hyper-DMCs overlapping with hypo-DHMRs, equal to 562 CpGs, is 7.69 higher than expected by chance (hypergeometric test, P value ≈ 0), considering sites covered by both ERRBS and hMeDIP-Seq reads. Moreover, hyper-DMCs are also overrepresented at putative enhancers [fold change (FC) = 3.51; hypergeometric test, P value ≈ 0]. These results are visualized in the UpSet plot (24) in Fig. 2B, where the number of hyper-DMCs overlapping with each region (or intersection of regions; e.g., intergenic enhancer) was normalized to the number of hyper-DMC sites per 100 CpGs covered by at least 10 ERRBS reads. The number of hyper-DMCs and of reference CpGs covered in each region is additionally shown in table S2. Last, the number of hyper-DMCs overlapping with promoter regions was significantly underrepresented (FC = 0.41; hypergeometric test, P value ≈ 0). These results support the notion that TET2 is primarily responsible for the control of enhancers and, to a lesser degree, for control of promoter activity. Nevertheless, despite the underrepresentation of TET2 loss-of-function–related hypermethylation in promoters, 23.5% of hyper-DMCs were located in these elements (2126 of 9043; table S2), highlighting their potential functional relevance.
Fig. 2. Cooperation of 5hmC loss in enhancers and 5mC gain in promoters enhances gene silencing.
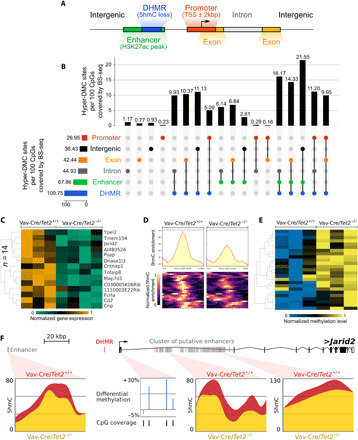
(A) Genomic features within which hyper-DMC distribution was studied. They include promoters, exons, introns, enhancers (defined as H3K27ac peaks from WT GC B cells, excluding promoters), DHMRs losing 5hmC signal, and intergenic regions. (B) This panel depicts an UpSet intersection diagram, showing the total set size and overlaps between the 9043 hyper-DMCs and genomic elements including DHMRs, enhancers (defined as H3K27ac peaks, excluding promoters), exons, promoters, and intergenic and intronic regions. The shaded circles connected by solid black lines in the bottom show the intersecting combinations of genomic elements. This panel illustrates all feature combinations with at least one hyper-DMC. The number of hyper-DMCs overlapping in each region (or regions intersection, e.g., intergenic enhancer) was normalized, by the total number of CpGs covered by ERRBS reads in this region (or intersection of regions), and multiplied the value by 100. (C) Heatmap of the expression levels (row normalized) of 15 down-regulated genes with 5hmC loss in enhancers and 5mC gain in promoters. (D) Heatmap and signal distribution of the 5hmC enrichment at 24 DHMR regions overlapping with enhancer regions of the 15 genes for which expression was visualized in (C). (E) Heatmap of 5mC enrichment at 25 hyper-DMCs overlapping with the promoter regions of 15 genes for which expression (row normalized) was visualized in (C). (F) Genome browser view of the example Jarid2 gene locus, visualizing 5hmC loss in two of its enhancers and 5mC gain in its promoter.
As an orthogonal approach, we focused on the previously defined 1977 differentially expressed genes in Tet2−/− versus Tet2+/+ GC B cells (14). We mapped putative enhancers to the nearest genes, within 100 kb of the TSS, which effectively narrowed down the analysis to a set of 584 differentially expressed genes with known intronic or intergenic enhancers (25) in B cells. Of these 584 genes, 395 were down-regulated and 189 were up-regulated in Tet2−/− GC B cells (Table 1). These 584 genes were separated into four categories based on the presence or absence of promoter hyper-DMCs and enhancer hypo-DHMRs. Fifteen of these genes showed both hyper-DMCs in promoter regions and hypo-DHMRs in enhancer regions (Table 1). Of these 15 genes, 14 showed down-regulated expression in Tet2−/− GC B cells (down-regulated:up-regulated ratio = 14:1; Table 1), which is significantly higher than expected by chance [hypergeometric test, false discovery rate (FDR) = 0.0094; Fig. 2C]. The respective enhancer DHMRs and promoter DMCs are shown in Fig. 2 (D and E). For example, we show that Jarid2, a gene that is associated with Polycomb complex functions and that was affected by hypermethylation in its promoter region, combined with an intergenic enhancer hypo-DHMR and two hypo-DHMRs overlapping with a cluster of putative intronic enhancers (Fig. 2F). Twenty-eight genes manifested promoter hyper-DMCs without enhancer loss of 5hmC, and 22 of these 28 genes were down-regulated (down-regulated:up-regulated ratio = 3.7:1; hypergeometric test, FDR = 0.0407; Table 1). One hundred and fifty-four genes manifested decreased levels of enhancer 5hmC without promoter hyper-DMCs, and these genes were biased toward repression (down-regulated:up-regulated ratio = 3:1; hypergeometric test, FDR = 0.0016; Table 1). Collectively, these data suggest that either impaired enhancer 5hmC or promoter 5mC patterning could be associated with transcriptional repression. This effect was most consistent when both marks were perturbed, which occurred, however, at only a subset of TET2-dependent genes. Strengthening the argument that promoter hypermethylation due to TET2 deficiency is linked to transcriptional repression, we first narrowed down the list of 930 genes with hypermethylated promoters to 755 genes expressed at a threshold of at least 20 reads per gene mapped in all samples. Next, we confirmed that a set of 755 genes with hypermethylated promoters is significantly overlapping with a list of down-regulated genes in TET2-deficient GC B cells (n = 69 genes; hypergeometric test, P value = 0.0016; fig. S2, B and C). Our work identified significantly differentially expressed genes with absolute FC of >1.2 and FDR less than 0.05. We next performed gene set enrichment analysis (GSEA) using the set of 755 expressed genes with hypermethylated promoters in Tet2−/− versus Tet2+/+ GC B cells and observed the significant enrichment for repression of these genes in the absence of TET2 (FDR = 0.02; fig. S2A). Moreover, this analysis revealed a list of 141 leading-edge genes, the expression of which was negatively affected to the highest degree with promoter hypermethylation (table S3). Leading-edge genes are core genes that contribute to the gene set’s enrichment signal.
Table 1. The number and ratio of down-DEGs and up-DEGs with 5hmC loss in enhancer or 5mC gain in promoter.
|
5hmC loss in enhancer |
5mC gain in promoter |
Number of |
Ratio (down to up-DEGs) |
|
|
Down- DEGs |
Up-DEGs | |||
| Yes | Yes | 14 | 1 | 14.0:1 |
| No | Yes | 22 | 6 | 3.7:1 |
| Yes | No | 116 | 38 | 3.0:1 |
| No | No | 243 | 144 | 1.7:1 |
Tet2 loss of function is associated with the repression of key B cell pathway genes
The above data illustrate how promotor hypermethylation, as a result of aberrant promoter demethylation, is associated with transcriptional repression in Tet2−/− GC B cells. To gain a sense of the biological functions that might be perturbed by promoter hypermethylation in TET2-deficient GC B cells, we next performed a hypergeometric gene pathway enrichment analysis of 930 genes with hyper-DMCs in their promoter regions. This procedure yielded highly significant enrichment for genes induced in centrocytes as they exit the GC reaction, CD40-induced genes, and genes involved in antigen presentation (Fig. 3A and table S4). This is consistent with the light zone expansion and differentiation blockade observed in immunized Tet2−/− mice (14). Mechanistically, genes repressed and hypermethylated in Tet2−/− mice include genes that are normally only transiently poised during the GC reaction and that become aberrantly repressed in patients with somatic mutations of related histone acetyltransferase encoding genes CREBBP and EP300, as well as de novo bivalent genes, i.e., genes that were repressed in GC B cells through promoter H3K27me3 bivalent domains, modified by the histone methyltransferase EZH2 (Fig. 3A). These CREBBP, EP300, and EZH2 target genes are similar to those linked to CD40 signaling, GC exit, and antigen presentation (25, 26), suggesting that Tet2 might normally oppose EZH2 while enhancing the actions of CREBBP and EP300. Furthermore, taking together all of the hypermethylated genes linked to the seven gene sets shown in Fig. 3A (n = 163), we again observed significant enrichment for repression of these genes in Tet2−/− GC B cells (FDR = 1.7×10−4; Fig. 3B and table S5). Further examination of the leading edge of this GSEA analysis (Fig. 3C and table S5) yielded genes including Tapbp and H2-Q7. Tapbp encodes tapasin, which is a subunit of the antigen processing (TAP) complex, responsible for binding of TAP1 and major histocompatibility complex (MHC) class I molecules, and which is required for an efficient peptide-TAP interaction (27), as well as for quality control of human leukocyte antigen-G (HLA-G) molecules (28). H2-Q7 is an ortholog of the human leukocyte antigen-A (HLA-A) gene, which is one of the major types of MHC class I heavy chain molecules. Down-regulation of these two genes might potentially destabilize MHC class I complexes, thus impairing signals required for GC exit and helping GC B cells to escape from immune recognition mechanisms.
Fig. 3. Hypermethylation affects key pathways in B cell biology.
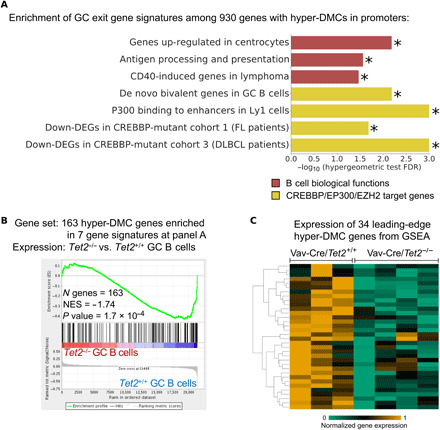
(A) Result of pathway enrichment analysis of the 930 genes with hyper-DMCs in their promoter regions. Plot shows minus log10 of the FDR scores for the enrichment of genes in seven functional categories important in GC or DLBCL biology. FDR scores were computed using hypergeometric test for these 930 genes. (B) GSEA enrichment plots in Vav-Cre/Tet2−/− versus Vav-Cre/Tet2+/+ GC B cells, against 163 genes with hyper-DMCs in their promoter regions, which were enriched for pathways listed in (A). (C) Row-normalized gene expression of 34 genes, which were identified as “leading-edge genes” in the GSEA analysis shown in (B). Yellow represents high expression, and green represents low expression. NES, normalized enrichment score; Down-DEGs, down-regulated genes; FL, follicular lymphoma. Star (*) indicates the value of FDR below 0.05.
TET2-deficient hypermethylated regions are enriched for binding by key transcription factors essential for B cell development
During the humoral immune response, phenotypic transitions of GC B cells in and out of the GC reaction are controlled by transcription factors (TFs) with stage-specific functions. To determine whether the binding sites for these TFs might be affected by aberrant 5mC patterning in Tet2−/− GC B cells, we performed a motif enrichment analysis using 2126 hyper-DMCs (±50-bp flanking regions) within the promoters of the 930 genes shown to have methylated promoters in Tet2−/− versus Tet2+/+ GC B cells (Fig. 2B). This analysis showed that 47 different TFs and regulators were enriched by this analysis; a number of these 47 proteins are relevant to controlling the GC reaction (q value < 10%; Fig. 4A and table S6). These include BATF (B-cell-activating transcription factor), which is a basic leucine zipper TF that activates expression of activation-induced cytidine deaminase (AID) through the recruitment of the TET2 and TET3 proteins (29); interferon regulatory factor 4 (IRF4), which is the master regulator of the GC exit program (30); and nuclear factor κB 1 (NF-κB1) and NF-κB2, which are downstream of the B cell–activating pathways induced by B cell receptor and CD40 (31). We also observed enrichment of PU.1:IRF8 hybrid sites; notably, PU.1 has been shown to activate gene expression via recruitment of TET proteins in normal pro–B cells (29, 32). Other motifs for B cell TFs, of note, included c-MYC and MAX, FOXM1, RARγ, E2A, PAX5, and MEF2C. Disruption of binding sites for any of these factors could lead to aberrant transcriptional states in Tet2−/− GC B cells.
Fig. 4. Hypermethylated regions are enriched for binding motives of key TFs in B cell biology.
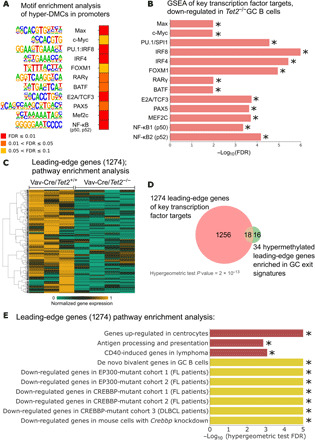
(A) Heatmap of the FDR scores of the motif enrichment analysis conducted using Homer for hyper-DMCs (±50 bp) located in promoter regions. Each TF name is accompanied by a logo sequence of the binding site. (B) Minus log10 of the FDR scores for enrichment of the target genes of 13 TFs, the binding sites of which were identified as hyper-methylated in Tet2−/− GC B cells. FDR scores were computed using GSEA, as described in Materials and Methods. Direction of the enrichment is biased toward Vav-Cre/Tet2+/+ GC B cells, when compared with Vav-Cre/Tet2−/− GC B cells (i.e., target genes are down-regulated in Tet2−/− GC B cells). (C) Normalized gene expression of the 1274 genes identified as leading-edge genes in at least 1 of 13 significantly enriched gene sets shown in (B). (D) Overlap between the 1274 leading-edge genes and 34 hypermethylated leading-edge genes of seven gene signatures important in GC exit or B cell biology. (E) Minus log10 of the FDR scores for the enrichment of 10 gene sets important in B cell biology. FDR scores were computed using hypergeometric test for leading-edge genes presented in (C) and (D). Star (*) indicates the value of FDR below 0.05.
In accordance with these binding-motif findings, previous studies measuring TF affinity to methylated versus unmethylated DNA elements using either DNA methylation–sensitive site selection in vitro or ChIP-seq (chromatin immunoprecipitation sequencing) coupled to methylome analysis suggest that the TFs MAX, c-MYC, IRF4, FOXM1, and MEF2C might be biased toward binding with unmethylated motifs (33–35). However, the actual extent of aberrant TF motif DNA methylation is likely not fully captured by the ERRBS method used in our studies, as it is designed to enrich for CpG-rich regions. Hence, we conducted a GSEA analysis for the target gene sets of the key TFs shown in Fig. 4A, using the gene expression profiles of Tet2−/− versus Tet2+/+ GC B cells. This analysis indeed showed significant down-regulation of the target genes of all 13 TFs in Tet2−/− GC B cells (Fig. 4B and fig. S3). To gain insight into the biological functions of these target genes, we first identified the genes contained in the leading-edge of the GSEA analysis of the target genes of the 13 TFs shown in Fig. 4B, which yielded 1274 genes (Fig. 4C). Notably, as visualized in Fig. 4D, these 1274 genes are overlapped significantly with 34 previously identified hypermethylated leading-edge genes (hypergeometric test, P value ≈ 0), which, as demonstrated in Fig. 3, are enriched for pathways essential in the exit from the GC reaction. This gene overlap prompted us to check whether these 1274 leading-edge genes are enriched for the same gene signatures. Hypergeometric analysis of these genes again identified significant enrichment of key GC exit genes including genes up-regulated in centrocytes, genes induced by CD40 genes in lymphoma, and genes involved with antigen processing and presentation (Fig. 4E and table S7). This analysis also showed enrichment for genes repressed due to loss of function of CREBBP or EP300, as well as de novo bivalent chromatin genes regulated by EZH2 in the GC reaction (Fig. 4E). Collectively, the data suggest that aberrant cytosine methylation induced by Tet2 loss of function might disrupt expression of genes that are targets of TFs that play critical roles in GC exit, which, in turn, might contribute to the differentiation blockade observed in Tet2−/− GCs (14), which should be further validated experimentally. These hypothesis-generating analyses may be useful to guide functional studies exploring the manner in which these TFs might contribute to the TET2-deficient phenotype in GC B cells.
TET2-deficient GC B cells manifest an AID loss-of-function signature
Somatic hypermutation of immunoglobulin genes during the GC reaction is mediated by AID (Aicda gene), through cytosine deamination. The effects of AID are not limited to immunoglobulin loci, and there is extensive bystander mutagenesis throughout the accessible genome in GC B cells (36). The effect of AID on non-immunoglobulin sites is markedly underlined by the characteristic DNA hypomethylation signature observed in normal GC B cells, which is largely mediated by AID, as GC B cells from Aicda−/− mice fail to manifest this hypomethylation (21). Several lines of evidence suggest that TET enzymes cooperate with AID cytosine demethylation (37–40). These considerations prompted us to examine whether the aberrant hypermethylation observed in Tet2−/− GC B cells might, in part, reflect disruption of Aicda-mediated hypomethylation.
To explore this question, we compared and contrasted ERRBS methylation profiles obtained in Tet2−/− versus Aicda−/− GC B cells and NBs. We focused the analysis on CpGs with at least 10 ERRBS reads in all samples from both mouse models to ensure a quantitatively meaningful comparison. First, we identified 19,111 CpGs that normally become hypomethylated in GC B cells versus NBs (Fig. 5A). Next, we determined how many of these 19,111 CpGs failed to become hypomethylated in Aicda−/− or Tet2−/− GC B cells. In the case of Aicda−/− mice, there was failure to hypomethylate 16,048 CpGs (84%), and in the case of Tet2−/− mice, there was failure to hypomethylate 12,756 CpGs (66.7%; Fig. 5A). Notably, 12,002 of these Aicda-dependent CpGs were also Tet2-dependent, indicating a highly significant overlap between these factors (hypergeometric test, P value ≈ 0; Fig. 5B). The CpGs that failed to demethylate in Aicda−/− GC B cells were associated with 1238 gene promoters. GSEA analysis on this Aicda-associated promoter gene set in Tet2−/− GC B cells indicated a significant trend for these genes to be expressed at lower levels in Tet2−/− GC B cells (FDR = 0.037; Fig. 5C).
Fig. 5. TET2-deficient GC B cells manifest AID loss-of-function signature.
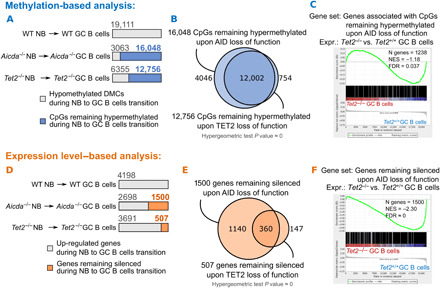
(A) Examination of 5mC accumulation during NB to GC B cell differentiation in TET2- and AID-deficient mouse models. CpGs (19,111) that are hypomethylated during normal NB to GC B cell differentiation in a WT mouse model were taken as a reference. (B) Venn diagram showing the overlap between CpGs that accumulate 5mC in Aicda−/− and in Tet2−/− NB to GC B cell differentiation. (C) GSEA enrichment analysis of 1238 genes accumulating 5mC during Aicda−/− NB to GC B cell transition, in Tet2−/− versus Tet2+/+ GC B cells. (D) Examination of gene inactivation during NB to GC B cell differentiation in TET2- and AID-deficient mouse models. Genes (4198) up-regulated during normal NB to GC B cell differentiation in a WT mouse model were taken as a reference. (E) Venn diagram showing the overlap between genes that remain silenced during Aicda−/− and Tet2−/− NB to GC B cell differentiation. (F) Enrichment of the 1500 genes that remain silenced during Aicda−/− NB to GC B cells transition, in Tet2−/− versus Tet2+/+ GC B cells.
We examined the link between, on the one hand, Aicda−/− and Tet2−/− failure to demethylate and, on the other hand, gene expression, using a second approach. We first identified 4198 genes that were up-regulated during the NB to GC B cells transition (FDR < 0.05 and FC ≥ 1.2; Fig. 5D). Next, we identified the genes from this list that were not induced in Aicda−/− (n = 1500) or Tet2−/− (n = 507) GC B cells, respectively (Fig. 5D). Notably, there was significant overlap (n = 360 genes) between these lists (hypergeometric test, P value ≈ 0; Fig. 5E). Moreover, GSEA using the set of genes that were not induced in Aicda−/− GC B cells revealed significant enrichment among genes that were also relatively repressed in Tet2−/− GC B cells (FDR ≈ 0; Fig. 5F).
We then performed an integrative analysis of DNA methylation and gene expression patterns in Aicda−/− and Tet2−/− GC B cells by merging the lists of genes that failed to demethylate their promoters with genes that failed to up-regulate their expression in each mouse model, which yielded 3111 and 1949 genes in Aicda−/− and Tet2−/− GC B cells, respectively. As would be expected from the previous analyses, there was significant overlap between these two integrated gene sets (hypergeometric test, P value ≈ 0; Fig. 6A). Moreover, the Aicda−/− integrated gene set was enriched for down-regulation in Tet2−/− GC B cells (FDR ≈ 0; Fig. 6B). We then examined biological functions linked to either the Aicda−/− or Tet2−/− GC B cell–integrated gene sets. We identified a total of 187 gene sets significantly enriched in at least one phenotype and observed significant overlap between these two pathway lists (n = 130; hypergeometric test, P value ≈ 0; Fig. 6C and table S8). Among pathways enriched for down-regulation in both cases were antigen processing and presentation, PRDM1-repressed genes, and genes that were aberrantly silenced in CREBBP or EP300 loss-of-function DLBCLs in mice (Fig. 6D). However, repression of genes linked to CD40 signaling and NF-κB, as well as of genes normally induced in centrocytes, were enriched only in Tet2−/− mice, which, together with the preceding data, suggests that these two genes have partially but not fully overlapping functions in the GC reaction.
Fig. 6. TET2- and AID-deficient cells are enriched for the same gene signatures.
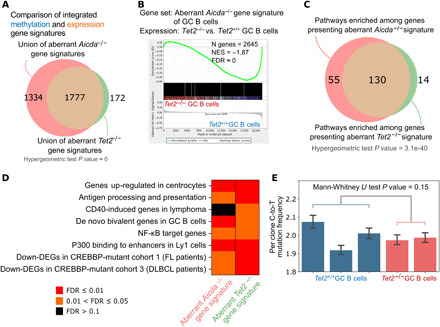
(A) Venn diagram showing the overlap between the integrated methylation- and expression-based aberrant gene signatures in Aicda−/− and Tet2−/− mouse models. (B) Enrichment of the aberrant Aicda−/− gene signature (3111 genes) in Tet2−/− versus Tet2+/+ GC B cells. Enrichment plot was generated using GSEA and shows down-regulation in Tet2−/− GC B cells. (C) Result of pathway enrichment analysis of the 3111 and 1949 genes with aberrant Aicda−/− and Tet2−/− signatures, respectively. FDR scores were computed using hypergeometric test. (D) Heatmap shows the FDR score of eight selected gene signatures from (C). FDR scores were computed using hypergeometric test. (E) Per-clone C-to-T mutation frequency at WRC motif, where W = A or T and R = G or A. Mann-Whitney U test was computed between Tet2−/− and Tet2+/+ conditions, combining mutation frequency values from each replicate accordingly to condition type.
Recent reports indicated that Tet2/Tet3 double knockout results in down-regulation of Aicda expression and a corresponding defect in Aicda-induced mutagenesis (19). However, here, in the setting of a Tet2 single knockout, Aicda expression was not significantly perturbed (FDR = 0.83; fig. S4A). Tet2/Tet3 double-knockout B cells were reported to show significantly reduced 5hmC at the Aicda enhancers. In contrast, with Tet2 single knockout, we see only small differences in 5hmC and also relatively little differential 5mC at Aicda enhancers (fig. S4B). Therefore, the notable failure to demethylate Aicda regulated CpGs in Tet2 single-knockout mice is more likely due to disruption of Aicda-mediated demethylation of these residues than to effects on Aicda expression itself.
Last, one of the key functions of AID is generation of C-to-U (Cytosine-to-Uracil) mutations during somatic hypermutation (41). Since Aicda expression is not affected by Tet2 deficiency in our mice model (fig. S4A), it was not clear whether these GC B cells would manifest impaired immunoglobulin somatic hypermutation. For this, we performed targeted sequencing of the immunoglobulin locus heavy-chain joining region 4 (JH4) and Sμ variable regions, which are known AID mutagenesis targets (42, 43), from Vav-cre/Tet2+/+ and Vav-cre/Tet2−/− GC B cells (B220+Fas+GL7+). We examined the percentage of clones per replicate, with at least one C-to-T (Cytosine-to-Thymine) (i.e., U) mutations at the WRC motif, which is the preferred site for AID-induced C-to-U deamination (44). This comparison did not yield a significant difference between Tet2 WT and Tet2-deficient cells (Wilcoxon rank sum test, P value = 0.24; table S9). There was also no significant difference in the frequency of clones with at least one C-to-T mutation (Mann-Whitney U test, P value = 0.15; Fig. 6E). Last, an orthogonal analysis revealed no significant difference in the C-to-T allele frequency in the JH4 or Sμ regions (Mann-Whitney U test P value = 0.09; fig. S4C). Together, these results suggest that TET2 loss of function does not negatively influence the process of somatic hypermutation and that the impact of Tet2 deficiency on Aicda in GC B cells is mostly restricted to DNA demethylation.
Tet2−/− 5mC signatures are reflected in human TET2 mutant DLBCL patients
We wondered whether the effects of Tet2 deficiency in mouse GC B cells on 5mC would carry through to primary human DLBCLs. Along these lines, using DNA methylation arrays, a previous study identified 578 hypermethylated DMCs present in TET2-mutated DLBCL primary samples and corresponding to 315 genes (12). To directly compare orthologous genes between these two species, we first removed from consideration any genes with promoters that were not covered by both platforms (methylation array in human and ERRBS in mouse). Together, 241 of 315 hypermethylated genes in human TET2MUT DLBCL and 614 of 930 hypermethylated genes in Tet2−/− GC B cells were considered for further analysis. Despite the cross-species comparison and the well-known heterogeneity among human DLBCLs (14), the overlap between the lists of hypermethylated genes in both species was still statistically significant (n = 18 genes; hypergeometric test, P value = 0.02). Moreover, the 241 hypermethylated human genes were significantly aberrantly repressed in murine Tet2−/− GC B cells as shown by GSEA (NES = −1.54, FDR = 0.01; Fig. 7A). In addition, running GSEA in the opposite direction, that is, testing of 614 murine Tet2−/− hypermethylated genes in the expression data in human TET2 mutant versus WT DLBCLs, showed a trend toward negative regulation of the murine Tet2-deficient gene signature in human DLBCLs (NES = −1.12, FDR = 0.53; Fig. 7B). To further probe similarities between signatures directly downstream of Tet2 in mouse and in humans, we examined gene pathways linked to the sets of hypermethylated genes in each species. Among hypermethylated genes in both human and mouse, we show enrichment of genes up-regulated in centrocytes, of de novo bivalent genes in GC B cells, and of terminally differentiated genes in GC B cells (Fig. 7C and table S10). Moreover, using GSEA, we additionally show that in both human and mouse, the expression of most of these gene signatures was skewed toward down-regulation (Fig. 7C). Collectively, the data indicate that TET2 loss of function results in an aberrant cytosine methylation pattern in GC B cells, leading to a state of aberrant epigenetic programming and silencing of critical gene pathways that is maintained in primary human DLBCLs, suggesting that these epigenetic effects are selected by and contribute to the disease phenotype.
Fig. 7. TET2 loss of function in human DLBCL manifest Tet2-deficient GC DMC and gene signature.
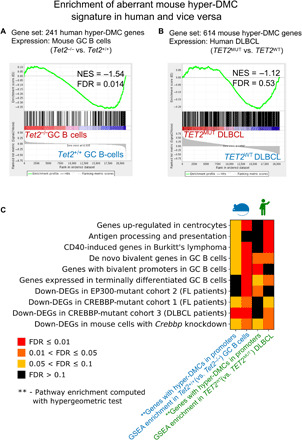
(A) Enrichment of the aberrant human TET2MUT gene signature (241 genes with hyper-DMCs in their promoters) in mouse Tet2−/− versus Tet2+/+ GC B cells. (B) Enrichment of the aberrant mouse Tet2−/− gene signature (614 genes with hyper-DMCs in their promoters) in human TET2MUT versus TET2WT DLBCL. (C) First and third columns of the heatmap represent the results of pathway enrichment analysis of 614 mouse and 241 human genes with hyper-DMCs in their promoters, respectively. FDR scores for these results were computed using hypergeometric test. Second and fourth columns represent the results of pathway enrichment analysis of the same gene sets, using GSEA in mouse Tet2−/− versus Tet2+/+ GC B cells and human TET2MUT versus TET2WT DLBCL, respectively. NES scores of all pathways in mouse and human are biased toward Tet2−/−/TET2WT.
DISCUSSION
Among hematologic malignancy disease alleles, TET2 somatic mutations are unique in that they occur in tumors arising from multiple hematopoietic lineages (15). Although DLBCLs arise from mature GC B cells, they seem to inherit TET2 mutations from hematopoietic stem cells (HSCs), which can give rise to additional hematologic malignancies harboring the same TET2 allele (15, 45). Therefore, we chose to use the Vav-cre/Tet2−/− model here as the most likely to be physiologically relevant to human lymphomagenesis, although formal proof via mutational analysis on matched HSC and lymphoma samples is still pending. TET2 is normally required for GC B cells to exit the GC reaction and undergo plasma cell differentiation (14). Although the pathways leading to TET2 activation in the GC are not yet defined, it is plausible that cytokines produced by T cells such as interleukin (IL)–2, IL-4, IL-10, and IL-21 (46, 47), which signal to GC B cells through JAK-STAT (Janus kinase–signal transducers and activators of transcription), induce JAK2-mediated TET2 phosphorylation, as has been recently described for stem cells in the bone marrow (48). TET2-deficient GC B cells cannot up-regulate the plasma cell master regulator PRDM1 due, at least in part, to reduction in 5hmC at its locus (14). Tet2−/− GC B cells feature disruption of many enhancers linked to GC exit signaling pathways, antigen presentation, and terminal differentiation genes. This role of Tet2 in GC B cells is conceptually similar to the functions of the histone modifiers KMT2D, CREBBP, and EP300, which are also commonly affected by loss-of-function mutations in DLBCL and result in enhancer dysfunction (2). The TET2-regulated transcriptome overlaps substantially with that regulated by CREBBP, and mutation of these two factors is generally mutually exclusive (14). However, whereas the role of enhancer loss of function in lymphoma pathogenesis is well established (2), very little is known about how disruption of 5mC patterning might contribute to these diseases. Although aberrant 5mC distribution has been shown to occur in DLBCL, it is not clear whether this is an early/causal or late event inherent to transformed cells (49). Here, we show that Tet2 loss of function in GC B cells leads to disruption in 5mC patterning largely associated with gene promoters, with down-regulation of the respective transcripts. This effect is at least partially retained in primary human TET2 mutant DLBCLs, thus providing evidence that aberrant 5mC patterning can be an early event disrupting key gene regulatory pathways during lymphomagenesis. Genes affected by aberrant DNA hypermethylation in Tet2−/− GC B cells are involved in similar pathways as those repressed by loss of enhancer 5hmC. The fact that genes with both loss of enhancer 5hmC and gain of promoter 5mC are particularly strongly affected is suggestive of a dual mechanism of action of gene disruption by TET2 loss-of-function alleles.
DNA hypermethylation can lead to gene silencing through a variety of mechanisms, including direct repression due to recruitment of methyl-binding repressor proteins or indirect repression by reducing the affinity of TFs such as MAX, c-MYC, IRF4, FOXM1, RelA, and MEF2C, for their DNA binding sites (33). Target genes for these factors were among those aberrantly methylated and repressed in Tet2−/− GC B cells. DNA hypermethylation at TF binding sites could occur through loss of recruitment of TET2 to convert 5mC to 5hmC. Along these lines, we observed the enrichment for the binding sites of PU.1, E2A, and BATF in hypermethylated regions, which is notable because these TFs have previously been linked with gene activation via recruitment of TET proteins in B cells (29, 32). PU.1 and E2A have been shown to physically interact with the TET2 and TET3 proteins, recruiting them to the enhancers, where they contribute to increasing chromatin accessibility (32). Similarly, over 81% of BATF peaks are colocalized with 5hmC in murine B cells, which is lost upon conditional Tet2 and Tet3 knockout (29). Loss of 5hmC in Tet2-deficient GC B cells might therefore lead to a relative decrease in chromatin accessibility and the inability of other downstream TFs to regulate gene expression, which is also consistent with our observation of DNA hypermethylation at these sites. Collectively, these considerations underscore the notion that precise regulation of gene expression in the humoral immune response requires cross-talk between DNA and histone modifications, both of which are severely disrupted in Tet2-deficient GC B cells.
GC B cells typically undergo extensive AID-dependent DNA hypomethylation (21). We show that this effect of AID is severely impaired in the absence of TET2, without impairment of AID mutability on WRC sequence motif at the immunoglobulin variable region, during somatic hypermutation. This effect could be due to loss of AID expression, which has been reported to occur with double knockout of Tet2 and Tet3 (19). However, we show that in mice with Tet2 knockout alone, there is no reduction in AID expression and relatively little perturbation of AID gene regulatory elements, suggesting that residual Tet3 can still maintain AID expression without Tet2. Therefore, loss of hypomethylated cytosines in Tet2−/− mice is more likely due to impairment of AID-mediated deamination, consistent with studies that suggest interdependence between TET2 and AID in DNA demethylation (37–40). Cortellino et al. (37) suggested that AID can deaminate modified cytosine residues, which are then repaired to unmodified cytosines through excision repair. Moreover, Guo et al. (38) reported that AID preferentially mediates DNA demethylation of 5hmCs but not of 5mCs. This notion is challenged by studies emphasizing low levels of AID-mediated deamination of hydroxymethyl cytosines due to the size of the hydroxymethyl modification (50, 51). However, it might also be possible that AID first deaminates 5mC to thymidine, which is then oxidized by TET2 to 5-hydroxymethyluracil (40), which can be further excised by base excision repair machinery to unmethylated cytosine (52). Together, our data suggest that in the GC B cell context, Tet2 plays a critical role in AID-mediated deamination of methylated cytosines.
TET2 is the only highly recurrently mutated member in the TET family in lymphomas, with somatic mutations occurring in 6 to 12% of DLBCL (12–15). Our data point to aberrant DNA hypermethylation as a contributor to the malignant phenotype of TET2 mutant DLBCLs, since aberrant repression of genes affected in this way by loss of TET2 (e.g., antigen presentation genes or interferon pathway) is strongly linked to DLBCL pathogenesis (2). This warrants consideration of DNMTi for the treatment of TET2 mutant patients. DNMTi are showing promising activity in high-risk DLBCLs, and perhaps, mutation of TET2 could serve as a biomarker to select patients for such treatment (7). However, it is important to emphasize that DNMTi alone would not likely fully reverse the aberrant silencing of Tet2 target genes caused by loss of 5hmC. The concept of targeting different layers of the epigenome has recently been shown to be particularly effective in TET2 mutant patients with acute myeloid leukemias (53). An equivalent strategy in DLBCL could include the use of HDAC3-selective inhibitors to rescue the effect of loss of 5hmC at gene enhancers (14), together with DNMTi to rescue the effect of DNA hypermethylation at promoters, and to more fully restore the expression of aberrantly silenced genes and thus the result in greater therapeutic efficacy.
MATERIALS AND METHODS
Animal models
Vav-Cre/Tet2f/f mice were obtained as a gift from R. Levine, Memorial Sloan Kettering Cancer Center (54). Experiments with conditional knockout of Tet2 (Tet2−/−) were conducted according to Gustave Roussy institutional guidelines and were authorized by the Direction Départementale des Services Vétérinaires du Val de Marne, as described previously (14). Aicda−/− mice were a gift from T. Honjo (Kyoto University Graduate School of Medicine), as described earlier (21). All mice were maintained according to the Weill Cornell Medicine Institutional Animal Care and Use Committee–approved protocol (ID no. 2011-0031) and guidelines of the Research Animal Resource Center of Weill Cornell Medicine.
DLBCL patient samples
Analysis of the influence of TET2 mutations in DLBCL on expression levels was conducted on the same samples, as described previously. Briefly, a cohort of 128 tumors from patients with pathologic diagnosis of DLBCL were interrogated by targeted sequencing, as described in García-Ramírez et al. (55). The TET2 mutations in patients with DLBCL for this cohort was published and obtained in (14). Affymetrix U133 plus 2 gene expression microarrays were performed on 84 matched DLBCL tumors in previous work, which were stored in the Gene Expression Omnibus (GEO) database (accession number GSE10846) (56).
Analysis of the influence of TET2 mutations in DLBCL samples on methylation levels was conducted on the methylation array data from Asmar et al. (12), which is accessible from the GEO database (accession number GSE37362). Samples include 12 patients with TET2 mutations. Briefly, the diagnoses were based on standard histology and immunophenotyping according to the 2008 World Health Organization lymphoma classification. Samples comprising more than 50 and 80% tumor cells were selected for DNA and RNA extraction, respectively. Genomic DNA was isolated after proteinase K digestion using the Purescript DNA Isolation Kit (Gentra Systems). Research on all human subjects was approved by the respective institutional review boards.
Mouse B cell isolation
To induce GC formation, Vav-Cre/Tet2−/− and Aicda−/− mice, and their corresponding controls, were immunized with sheep red blood cells (1 × 108 cells per mouse) or NP-CGG ratio of 20 to 25 (from Biosearch Technologies) in alum (1:1). Mice were euthanized at day 10 after immunization, spleens were dissected, and mononuclear cells were purified using Histopaque gradient configuration (Sigma). Isolation of NB cells and GC B cells was conducted from cell suspensions enriched in B cells by positive selection with anti-B220 magnetic microbeads (Miltenyi Biotech, Germany). B cells were separated into NB cell (B220+GL7−FAS−DAPI−) and GC B cell (B220+GL7+FAS+DAPI−) using a BD FACSAria II sorter, as described previously (14, 21).
Enhanced reduced representation bisulfite sequencing
Genomic DNA from GC B cells of Aicda−/− and Aicda+/+ mice was bisulfite-converted using the EZ DNA Methylation Kit (Zymo Research), as described previously in Dominguez et al. (21). Base-pair–resolution DNA methylation analysis was performed in Aicda−/− mice (n = 6, three males and three females) and Aicda+/+ mice (n = 7, three males and four females) following the ERRBS protocol previously described (57). The same protocol was applied to genomic DNA from a total of 12 samples of NBs and GC B cells in Vav-Cre/Tet2−/− and Vav-Cre/Tet2+/+ mice, with three replicates for each condition. DMCs were identified on the basis of logistic regression test with the following thresholds: q value < 0.01; methylation percentage difference of at least 25% (calculateDiffMeth function in R package methylKit) (58). Specifically, the logistic regression test was used to compare the fraction of methylated cytosines across the test and the control groups. The χ2 test was used to determine the methylation differences. Further, the sliding linear model method was used to correct the P values for multiple testing. In associating genes with hypermethylated CpGs (hyper-DMCs), we considered all genes with promoter regions containing hyper-DMC. Here, promoter regions are defined as up/down to a distance of 2 kb from the TSSs in the mm10 reference annotation.
Motif enrichment analysis
Identification of known TF binding sites overrepresented among DHMRs in Vav-Cre/Tet2−/− and Vav-Cre/Tet2+/+ samples was conducted using “findMotifsGenome.pl” from Homer (59). More specifically, the analysis was conducted for hypo-DHMR regions, identified as described above, overlapping with promoter regions. Moreover, the analysis was also conducted for hypermethylated regions, defined as regions 50 bp upstream or downstream of hyper-DMCs located within promoter regions. Motif enrichment analysis for these hyper-DMCs was conducted with a background of all reference promoters from the gencode (vM3) reference annotation.
Accession numbers
Raw sequence data of Vav-Cre RNA-seq, ERRBS, and hMeDIP-seq from the experiments conducted in GC B cells are stored under accession number GSE111700. Sequence data from Vav-Cre RNA-seq and ERRBS NBs are stored under accession numbers GSE132595 and GSE132596. Raw sequence data of Vav-Cre/Tet2−/− and Vav-Cre/Tet2+/+ SHM (somatic hypermutation) analysis are stored under accession number GSE140086.
Supplementary Material
Acknowledgments
We thank S. Sampson from The Jackson Laboratory for editing this manuscript. We thank members of Li Lab and Melnick Lab for discussion, and we thank the Jackson Laboratory Computer Sciences team and the Research Informatics Technology group, both for technical support. Funding: A.M. is funded by R35 CA220499, LLS TRP 6572-19, LLS SCOR 7012-16, the Follicular Lymphoma Consortium, Samuel Waxman Cancer Research Foundation, and the Chemotherapy Foundation. S.L. is supported by the National Institute of General Medical Sciences of the NIH under award number R35 GM133562, Leukemia Research Foundation New Investigator Grant, The Jackson Laboratory Director’s Innovation Fund 19000-17-31, and The Jackson Laboratory Cancer Center New Investigator Award. Research reported in this publication was partially supported by the National Cancer Institute of the NIH under award number P30 CA034196. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. P.M.D. was supported by a Lymphoma Research Foundation Postdoctoral Fellowship. Author contributions: A.M. and S.L. conceived the project; W.R., A.M., and S.L. designed the research; and W.R., X.C., and S.L. performed computational analysis. P.M.D. performed the experiments. P.M.D., H.G., S.A., and O.A.B. discussed analysis of the results. W.R., X.C., A.M., and S.L. wrote the manuscript; and W.R., X.C., P.M.D., A.M., and S.L. edited the manuscript. Competing interests: A.M. receives research funding from Janssen Pharmaceuticals and does consulting for Constellation and Epizyme. The other authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/25/eaay5872/DC1
REFERENCES AND NOTES
- 1.Hu D., Shilatifard A., Epigenetics of hematopoiesis and hematological malignancies. Genes Dev. 30, 2021–2041 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mlynarczyk C., Fontán L., Melnick A., Germinal center-derived lymphomas: The darkest side of humoral immunity. Immunol. Rev. 288, 214–239 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jiang Y., Dominguez P. M., Melnick A. M., The many layers of epigenetic dysfunction in B-cell lymphomas. Curr. Opin. Hematol. 23, 377–384 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Alhejaily A., Day A. G., Feilotter H. E., Baetz T., Lebrun D. P., Inactivation of the CDKN2A tumor-suppressor gene by deletion or methylation is common at diagnosis in follicular lymphoma and associated with poor clinical outcome. Clin. Cancer Res. 20, 1676–1686 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Chambwe N., Kormaksson M., Geng H., De S., Michor F., Johnson N. A., Morin R. D., Scott D. W., Godley L. A., Gascoyne R. D., Melnick A., Campagne F., Shaknovich R., Variability in DNA methylation defines novel epigenetic subgroups of DLBCL associated with different clinical outcomes. Blood 123, 1699–1708 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De S., Shaknovich R., Riester M., Elemento O., Geng H., Kormaksson M., Jiang Y., Woolcock B., Johnson N., Polo J. M., Cerchietti L., Gascoyne R. D., Melnick A., Michor F., Aberration in DNA methylation in B-cell lymphomas has a complex origin and increases with disease severity. PLOS Genet. 9, e1003137 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clozel T., Yang S., Elstrom R. L., Tam W., Martin P., Kormaksson M., Banerjee S., Vasanthakumar A., Culjkovic B., Scott D. W., Wyman S., Leser M., Shaknovich R., Chadburn A., Tabbo F., Godley L. A., Gascoyne R. D., Borden K. L., Inghirami G., Leonard J. P., Melnick A., Cerchietti L., Mechanism-based epigenetic chemosensitization therapy of diffuse large B-cell lymphoma. Cancer Discov. 3, 1002–1019 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deaton A. M., Bird A., CpG islands and the regulation of transcription. Genes Dev. 25, 1010–1022 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu X., Zhang Y., TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 18, 517–534 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Rampal R., Alkalin A., Madzo J., Vasanthakumar A., Pronier E., Patel J., Li Y., Ahn J., Abdel-Wahab O., Shih A., Lu C., Ward P. S., Tsai J. J., Hricik T., Tosello V., Tallman J. E., Zhao X., Daniels D., Dai Q., Ciminio L., Aifantis I., He C., Fuks F., Tallman M. S., Ferrando A., Nimer S., Paietta E., Thompson C. B., Licht J. D., Mason C. E., Godley L. A., Melnick A., Figueroa M. E., Levine R. L., DNA hydroxymethylation profiling reveals that WT1 mutations result in loss of TET2 function in acute myeloid leukemia. Cell Rep. 9, 1841–1855 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hon G. C., Song C.-X., du T., Jin F., Selvaraj S., Lee A. Y., Yen C. A., Ye Z., Mao S.-Q., Wang B.-A., Kuan S., Edsall L. E., Zhao B. S., Xu G.-L., He C., Ren B., 5mC oxidation by Tet2 modulates enhancer activity and timing of transcriptome reprogramming during differentiation. Mol. Cell 56, 286–297 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Asmar F., Punj V., Christensen J., Pedersen M. T., Pedersen A., Nielsen A. B., Hother C., Ralfkiaer U., Brown P., Ralfkiaer E., Helin K., Grønbæk K., Genome-wide profiling identifies a DNA methylation signature that associates with TET2 mutations in diffuse large B-cell lymphoma. Haematologica 98, 1912–1920 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Reddy A., Zhang J., Davis N. S., Moffitt A. B., Love C. L., Waldrop A., Leppa S., Pasanen A., Meriranta L., Karjalainen-Lindsberg M. L., Nørgaard P., Pedersen M., Gang A. O., Høgdall E., Heavican T. B., Lone W., Iqbal J., Qin Q., Li G., Kim S. Y., Healy J., Richards K. L., Fedoriw Y., Bernal-Mizrachi L., Koff J. L., Staton A. D., Flowers C. R., Paltiel O., Goldschmidt N., Calaminici M., Clear A., Gribben J., Nguyen E., Czader M. B., Ondrejka S. L., Collie A., Hsi E. D., Tse E., Au-Yeung R. K. H., Kwong Y.-L., Srivastava G., Choi W. W. L., Evens A. M., Pilichowska M., Sengar M., Reddy N., Li S., Chadburn A., Gordon L. I., Jaffe E. S., Levy S., Rempel R., Tzeng T., Happ L. E., Dave T., Rajagopalan D., Datta J., Dunson D. B., Dave S. S., Genetic and functional drivers of diffuse large B cell lymphoma. Cell 171, 481–494.e15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dominguez P. M., Ghamlouch H., Rosikiewicz W., Kumar P., Béguelin W., Fontán L., Rivas M. A., Pawlikowska P., Armand M., Mouly E., Torres-Martin M., Doane A. S., Calvo Fernandez M. T., Durant M., Della-Valle V., Teater M., Cimmino L., Droin N., Tadros S., Motanagh S., Shih A. H., Rubin M. A., Tam W., Aifantis I., Levine R. L., Elemento O., Inghirami G., Green M. R., Figueroa M. E., Bernard O. A., Aoufouchi S., Li S., Shaknovich R., Melnick A. M., TET2 deficiency causes germinal center hyperplasia, impairs plasma cell differentiation, and promotes B-cell lymphomagenesis. Cancer Discov. 8, 1632–1653 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Quivoron C., Couronné L., Della Valle V., Lopez C. K., Plo I., Wagner-Ballon O., Do Cruzeiro M., Delhommeau F., Arnulf B., Stern M.-H., Godley L., Opolon P., Tilly H., Solary E., Duffourd Y., Dessen P., Merle-Beral H., Nguyen-Khac F., Fontenay M., Vainchenker W., Bastard C., Mercher T., Bernard O. A., TET2 inactivation results in pleiotropic hematopoietic abnormalities in mouse and is a recurrent event during human lymphomagenesis. Cancer Cell 20, 25–38 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Delhommeau F., Dupont S., Valle V. D., James C., Trannoy S., Massé A., Kosmider O., Le Couedic J.-P., Robert F., Alberdi A., Lécluse Y., Plo I., Dreyfus F. J., Marzac C., Casadevall N., Lacombe C., Romana S. P., Dessen P., Soulier J., Viguié F., Fontenay M., Vainchenker W., Bernard O. A., Mutation in TET2 in myeloid cancers. N. Engl. J. Med. 360, 2289–2301 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Lemonnier F., Couronné L., Parrens M., Jaïs J.-P., Travert M., Lamant L., Tournillac O., Rousset T., Fabiani B., Cairns R. A., Mak T., Bastard C., Bernard O. A., de Leval L., Gaulard P., Recurrent TET2 mutations in peripheral T-cell lymphomas correlate with TFH-like features and adverse clinical parameters. Blood 120, 1466–1469 (2012). [DOI] [PubMed] [Google Scholar]
- 18.Patnaik M. M., Zahid M. F., Lasho T. L., Finke C., Ketterling R. L., Gangat N., Robertson K. D., Hanson C. A., Tefferi A., Number and type of TET2 mutations in chronic myelomonocytic leukemia and their clinical relevance. Blood Cancer J. 6, e472 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lio C.-W. J., Shukla V., Samaniego-Castruita D., González-Avalos E., Chakraborty A., Yue X., Schatz D. G., Ay F., Rao A., TET enzymes augment activation-induced deaminase (AID) expression via 5-hydroxymethylcytosine modifications at the Aicda superenhancer. Sci. Immunol. 4, eaau7523 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mouly E., Ghamlouch H., Della-Valle V., Scourzic L., Quivoron C., Roos-Weil D., Pawlikowska P., Saada V., Diop M. K., Lopez C. K., Fontenay M., Dessen P., Touw I. P., Mercher T., Aoufouchi S., Bernard O. A., B-cell tumor development in Tet2-deficient mice. Blood Adv. 2, 703–714 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dominguez P. M., Teater M., Chambwe N., Kormaksson M., Redmond D., Ishii J., Vuong B., Chaudhuri J., Melnick A., Vasanthakumar A., Godley L. A., Papavasiliou F. N., Elemento O., Shaknovich R., DNA methylation dynamics of germinal center B cells are mediated by AID. Cell Rep. 12, 2086–2098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaknovich R., Cerchietti L., Tsikitas L., Kormaksson M., de S., Figueroa M. E., Ballon G., Yang S. N., Weinhold N., Reimers M., Clozel T., Luttrop K., Ekstrom T. J., Frank J., Vasanthakumar A., Godley L. A., Michor F., Elemento O., Melnick A., DNA methyltransferase 1 and DNA methylation patterning contribute to germinal center B-cell differentiation. Blood 118, 3559–3569 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang L., Ozark P. A., Smith E. R., Zhao Z., Marshall S. A., Rendleman E. J., Piunti A., Ryan C., Whelan A. L., Helmin K. A., Morgan M. A., Zou L., Singer B. D., Shilatifard A., TET2 coactivates gene expression through demethylation of enhancers. Sci. Adv. 4, eaau6986 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lex A., Gehlenborg N., Strobelt H., Vuillemot R., Pfister H., UpSet: Visualization of intersecting sets. IEEE Trans. Vis. Comput. Graph. 20, 1983–1992 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang Y., Ortega-Molina A., Geng H., Ying H.-Y., Hatzi K., Parsa S., McNally D., Wang L., Doane A. S., Agirre X., Teater M., Meydan C., Li Z., Poloway D., Wang S., Ennishi D., Scott D. W., Stengel K. R., Kranz J. E., Holson E., Sharma S., Young J. W., Chu C.-S., Roeder R. G., Shaknovich R., Hiebert S. W., Gascoyne R. D., Tam W., Elemento O., Wendel H.-G., Melnick A. M., CREBBP inactivation promotes the development of HDAC3-dependent lymphomas. Cancer Discov. 7, 38–53 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Béguelin W., Popovic R., Teater M., Jiang Y., Bunting K. L., Rosen M., Shen H., Yang S. N., Wang L., Ezponda T., Martinez-Garcia E., Zhang H., Zheng Y., Verma S. K., McCabe M. T., Ott H. M., Van Aller G. S., Kruger R. G., Liu Y., McHugh C. F., Scott D. W., Chung Y. R., Kelleher N., Shaknovich R., Creasy C. L., Gascoyne R. D., Wong K.-K., Cerchietti L., Levine R. L., Abdel-Wahab O., Licht J. D., Elemento O., Melnick A. M., EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell 23, 677–692 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li S., Paulsson K. M., Chen S., Sjögren H. O., Wang P., Tapasin is required for efficient peptide binding to transporter associated with antigen processing. J. Biol. Chem. 275, 1581–1586 (2000). [DOI] [PubMed] [Google Scholar]
- 28.Park B., Ahn K., An essential function of tapasin in quality control of HLA-G molecules. J. Biol. Chem. 278, 14337–14345 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Lio C.-W. J., Shukla V., Samaniego-Castruita D., González-Avalos E., Chakraborty A., Yue X., Schatz D. G., Ay F., Rao A., TET enzymes augment AID expression via 5hmC modifications at the Aicda superenhancer. bioRxiv, 438531 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mittrücker H. W., Matsuyama T., Grossman A., Kündig T. M., Potter J., Shahinian A., Wakeham A., Patterson B., Ohashi P. S., Mak T. W., Requirement for the transcription factor LSIRF/IRF4 for mature B and T lymphocyte function. Science 275, 540–543 (1997). [DOI] [PubMed] [Google Scholar]
- 31.Kennedy R., Klein U., Aberrant activation of NF-κB signalling in aggressive lymphoid malignancies. Cell 7, 189 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lio C.-W., Zhang J., González-Avalos E., Hogan P. G., Chang X., Rao A., Tet2 and Tet3 cooperate with B-lineage transcription factors to regulate DNA modification and chromatin accessibility. eLife 5, e18290 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yin Y., Morgunova E., Jolma A., Kaasinen E., Sahu B., Khund-Sayeed S., Das P. K., Kivioja T., Dave K., Zhong F., Nitta K. R., Taipale M., Popov A., Ginno P. A., Domcke S., Yan J., Schübeler D., Vinson C., Taipale J., Impact of cytosine methylation on DNA binding specificities of human transcription factors. Science 356, eaaj2239 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xuan Lin Q. X., Sian S., An O., Thieffry D., Jha S., Benoukraf T., MethMotif: An integrative cell specific database of transcription factor binding motifs coupled with DNA methylation profiles. Nucleic Acids Res. 47, D145–D154 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang G., Luo X., Wang J., Wan J., Xia S., Zhu H., Qian J., Wang Y., MeDReaders: A database for transcription factors that bind to methylated DNA. Nucleic Acids Res. 46, D146–D151 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jiang Y., Soong T. D., Wang L., Melnick A. M., Elemento O., Genome-wide detection of genes targeted by non-Ig somatic hypermutation in lymphoma. PLOS ONE 7, e40332 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cortellino S., Xu J., Sannai M., Moore R., Caretti E., Cigliano A., le Coz M., Devarajan K., Wessels A., Soprano D., Abramowitz L. K., Bartolomei M. S., Rambow F., Bassi M. R., Bruno T., Fanciulli M., Renner C., Klein-Szanto A. J., Matsumoto Y., Kobi D., Davidson I., Alberti C., Larue L., Bellacosa A., Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell 146, 67–79 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo J. U., Su Y., Zhong C., Ming G.-L., Song H., Hydroxylation of 5-methylcytosine by TET1 promotes active DNA demethylation in the adult brain. Cell 145, 423–434 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kumar S., Chinnusamy V., Mohapatra T., Epigenetics of modified DNA bases: 5-methylcytosine and beyond. Front. Genet. 9, 640 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pfaffeneder T., Spada F., Wagner M., Brandmayr C., Laube S. K., Eisen D., Truss M., Steinbacher J., Hackner B., Kotljarova O., Schuermann D., Michalakis S., Kosmatchev O., Schiesser S., Steigenberger B., Raddaoui N., Kashiwazaki G., Müller U., Spruijt C. G., Vermeulen M., Leonhardt H., Schär P., Müller M., Carell T., Tet oxidizes thymine to 5-hydroxymethyluracil in mouse embryonic stem cell DNA. Nat. Chem. Biol. 10, 574–581 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Muramatsu M., Sankaranand V. S., Anant S., Sugai M., Kinoshita K., Davidson N. O., Honjo T., Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. J. Biol. Chem. 274, 18470–18476 (1999). [DOI] [PubMed] [Google Scholar]
- 42.Reina-San-Martin B., Difilippantonio S., Hanitsch L., Masilamani R. F., Nussenzweig A′., Nussenzweig M. C., H2AX is required for recombination between immunoglobulin switch regions but not for intra-switch region recombination or somatic hypermutation. J. Exp. Med. 197, 1767–1778 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zheng S., Vuong B. Q., Vaidyanathan B., Lin J.-Y., Huang F.-T., Chaudhuri J., Non-coding RNA Generated following Lariat debranching mediates targeting of AID to DNA. Cell 161, 762–773 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pham P., Bransteitter R., Petruska J., Goodman M. F., Processive AID-catalysed cytosine deamination on single-stranded DNA simulates somatic hypermutation. Nature 424, 103–107 (2003). [DOI] [PubMed] [Google Scholar]
- 45.Rasmussen K. D., Helin K., Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 30, 733–750 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zotos D., Coquet J. M., Zhang Y., Light A., D'Costa K., Kallies A., Corcoran L. M., Godfrey D. I., Toellner K.-M., Smyth M. J., Nutt S. L., Tarlinton D. M., IL-21 regulates germinal center B cell differentiation and proliferation through a B cell-intrinsic mechanism. J. Exp. Med. 207, 365–378 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Turqueti-Neves A., Otte M., da Costa O. P., Höpken U. E., Lipp M., Buch T., Voehringer D., B-cell-intrinsic STAT6 signaling controls germinal center formation. Eur. J. Immunol. 44, 2130–2138 (2014). [DOI] [PubMed] [Google Scholar]
- 48.Jeong J. J., Gu X., Nie J., Sundaravel S., Liu H., Kuo W.-L., Bhagat T. D., Pradhan K., Cao J., Nischal S., McGraw K. L., Bhattacharyya S., Bishop M. R., Artz A., Thirman M. J., Moliterno A., Ji P., Levine R. L., Godley L. A., Steidl U., Bieker J. J., List A. F., Saunthararajah Y., He C., Verma A., Wickrema A., Cytokine-regulated phosphorylation and activation of TET2 by JAK2 in hematopoiesis. Cancer Discov. 9, 778–795 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dominguez P. M., Teater M., Shaknovich R., The new frontier of epigenetic heterogeneity in B-cell neoplasms. Curr. Opin. Hematol. 24, 402–408 (2017). [DOI] [PubMed] [Google Scholar]
- 50.Nabel C. S., Jia H., Ye Y., Shen L., Goldschmidt H. L., Stivers J. T., Zhang Y., Kohli R. M., AID/APOBEC deaminases disfavor modified cytosines implicated in DNA demethylation. Nat. Chem. Biol. 8, 751–758 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rangam G., Schmitz K.-M., Cobb A. J. A., Petersen-Mahrt S. K., AID enzymatic activity is inversely proportional to the size of cytosine C5 orbital cloud. PLOS ONE 7, e43279 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dominguez P. M., Shaknovich R., Epigenetic function of activation-induced cytidine deaminase and its link to lymphomagenesis. Front. Immunol. 5, 642 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Duy C., Teater M., Garrett-Bakelman F. E., Lee T. C., Meydan C., Glass J. L., Li M., Hellmuth J. C., Mohammad H. P., Smitheman K. N., Shih A. H., Abdel-Wahab O., Tallman M. S., Guzman M. L., Muench D., Grimes H. L., Roboz G. J., Kruger R. G., Creasy C. L., Paietta E. M., Levine R. L., Carroll M., Melnick A. M., Rational targeting of cooperating layers of the epigenome yields enhanced therapeutic efficacy against AML. Cancer Discov. 9, 872–889 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moran-Crusio K., Reavie L., Shih A., Abdel-Wahab O., Ndiaye-Lobry D., Lobry C., Figueroa M. E., Vasanthakumar A., Patel J., Zhao X., Perna F., Pandey S., Madzo J., Song C., Dai Q., He C., Ibrahim S., Beran M., Zavadil J., Nimer S. D., Melnick A., Godley L. A., Aifantis I., Levine R. L., Tet2 loss leads to increased hematopoietic stem cell self-renewal and myeloid transformation. Cancer Cell 20, 11–24 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.García-Ramírez I., Tadros S., González-Herrero I., Martín-Lorenzo A., Rodríguez-Hernández G., Moore D., Ruiz-Roca L., Blanco O., Alonso-López D., Rivas J. D. L., Hartert K., Duval R., Klinkebiel D., Bast M., Vose J., Lunning M., Fu K., Greiner T., Rodrigues-Lima F., Jiménez R., Criado F. J. G., Cenador Ma. B. G., Brindle P., Vicente-Dueñas C., Alizadeh A., Sánchez-García I., Green M. R., Crebbp loss cooperates with Bcl2 overexpression to promote lymphoma in mice. Blood 129, 2645–2656 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lenz G., Wright G., Dave S. S., Xiao W., Powell J., Zhao H., Xu W., Tan B., Goldschmidt N., Iqbal J., Vose J., Bast M., Fu K., Weisenburger D. D., Greiner T. C., Armitage J. O., Kyle A., May L., Gascoyne R. D., Connors J. M., Troen G., Holte H., Kvaloy S., Dierickx D., Verhoef G., Delabie J., Smeland E. B., Jares P., Martinez A., Lopez-Guillermo A., Montserrat E., Campo E., Braziel R. M., Miller T. P., Rimsza L. M., Cook J. R., Pohlman B., Sweetenham J., Tubbs R. R., Fisher R. I., Hartmann E., Rosenwald A., Ott G., Muller-Hermelink H.-K., Wrench D., Lister T. A., Jaffe E. S., Wilson W. H., Chan W. C., Staudt L. M.; Lymphoma/Leukemia Molecular Profiling Project , Stromal gene signatures in large-B-cell lymphomas. N. Engl. J. Med. 359, 2313–2323 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Akalin A., Garrett-Bakelman F. E., Kormaksson M., Busuttil J., Zhang L., Khrebtukova I., Milne T. A., Huang Y., Biswas D., Hess J. L., Allis C. D., Roeder R. G., Valk P. J. M., Löwenberg B., Delwel R., Fernandez H. F., Paietta E., Tallman M. S., Schroth G. P., Mason C. E., Melnick A., Figueroa M. E., Base-pair resolution DNA methylation sequencing reveals profoundly divergent epigenetic landscapes in acute myeloid leukemia. PLOS Genet. 8, e1002781 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Akalin A., Kormaksson M., Li S., Garrett-Bakelman F. E., Figueroa M. E., Melnick A., Mason C. E., methylKit: A comprehensive R package for the analysis of genome-wide DNA methylation profiles. Genome Biol. 13, R87 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., Glass C. K., Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liao Y., Smyth G. K., Shi W., The Subread aligner: Fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ignatiadis N., Klaus B., Zaugg J. B., Huber W., Data-driven hypothesis weighting increases detection power in genome-scale multiple testing. Nat. Methods 13, 577–580 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Love M. I., Huber W., Anders S., Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bourgon R., Gentleman R., Huber W., Independent filtering increases detection power for high-throughput experiments. Proc. Natl. Acad. Sci. U.S.A. 107, 9546–9551 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ritchie M. E., Phipson B., Wu D., Hu Y., Law C. W., Shi W., Smyth G. K., limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martin M., Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17, 10–12 (2011). [Google Scholar]
- 67.Langmead B., Salzberg S. L., Fast gapped-read alignment with bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhang Y., Liu T., Meyer C. A., Eeckhoute J., Johnson D. S., Bernstein B. E., Nussbaum C., Myers R. M., Brown M., Li W., Liu X. S., Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Mootha V. K., Lindgren C. M., Eriksson K.-F., Subramanian A., Sihag S., Lehar J., Puigserver P., Carlsson E., Ridderstråle M., Laurila E., Houstis N., Daly M. J., Patterson N., Mesirov J. P., Golub T. R., Tamayo P., Spiegelman B., Lander E. S., Hirschhorn J. N., Altshuler D., Groop L. C., PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34, 267–273 (2003). [DOI] [PubMed] [Google Scholar]
- 70.Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., Paulovich A., Pomeroy S. L., Golub T. R., Lander E. S., Mesirov J. P., Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Armstrong S. A., Staunton J. E., Silverman L. B., Pieters R., den Boer M. L., Minden M. D., Sallan S. E., Lander E. S., Golub T. R., Korsmeyer S. J., MLL translocations specify a distinct gene expression profile that distinguishes a unique leukemia. Nat. Genet. 30, 41–47 (2002). [DOI] [PubMed] [Google Scholar]
- 72.Ortega-Molina A., Boss I. W., Canela A., Pan H., Jiang Y., Zhao C., Jiang M., Hu D., Agirre X., Niesvizky I., Lee J.-E., Chen H.-T., Ennishi D., Scott D. W., Mottok A., Hother C., Liu S., Cao X.-J., Tam W., Shaknovich R., Garcia B. A., Gascoyne R. D., Ge K., Shilatifard A., Elemento O., Nussenzweig A., Melnick A. M., Wendel H.-G., The histone lysine methyltransferase KMT2D sustains a gene expression program that represses B cell lymphoma development. Nat. Med. 21, 1199–1208 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hatzi K., Jiang Y., Huang C., Garrett-Bakelman F., Gearhart M. D., Giannopoulou E. G., Zumbo P., Kirouac K., Bhaskara S., Polo J. M., Kormaksson M., MacKerell A. D. Jr., Xue F., Mason C. E., Hiebert S. W., Prive G. G., Cerchietti L., Bardwell V. J., Elemento O., Melnick A., A hybrid mechanism of action for BCL6 in B cells defined by formation of functionally distinct complexes at enhancers and promoters. Cell Rep. 4, 578–588 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shaker S., Bernstein M., Momparler R. L., Antineoplastic action of 5-aza-2′-deoxycytidine (Dacogen) and depsipeptide on Raji lymphoma cells. Oncol. Rep. 11, 1253–1256 (2004). [PubMed] [Google Scholar]
- 75.Lai L., Hennessey J., Bares V., Son E. W., Ban Y., Wang W., Qi J., Jiang G., Liberzon A., Ge S. X., GSKB: A gene set database for pathway analysis in mouse. bioRxiv, 082511 (2016). [Google Scholar]
- 76.Aibar S., González-Blas C. B., Moerman T., Huynh-Thu V. A., Imrichova H., Hulselmans G., Rambow F., Marine J.-C., Geurts P., Aerts J., van den Oord J., Atak Z. K., Wouters J., Aerts S., SCENIC: Single-cell regulatory network inference and clustering. Nat. Methods 14, 1083–1086 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bult C. J., Blake J. A., Smith C. L., Kadin J. A., Richardson J. E.; Mouse Genome Database Group , Nucleic Acids Res. 47, D801–D806 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.H. Li, Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997, (2013).
- 79.Wilm A., Aw P. P. K., Bertrand D., Yeo G. H. T., Ong S. H., Wong C. H., Khor C. C., Petric R., Hibberd M. L., Nagarajan N., LoFreq: A sequence-quality aware, ultra-sensitive variant caller for uncovering cell-population heterogeneity from high-throughput sequencing datasets. Nucleic Acids Res. 40, 11189–11201 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li H., A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gupta N. T., Vander Heiden J. A., Uduman M., Gadala-Maria D., Yaari G., Kleinstein S. H., Change-O: A toolkit for analyzing large-scale B cell immunoglobulin repertoire sequencing data. Bioinformatics 31, 3356–3358 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vander Heiden J. A., Yaari G., Uduman M., Stern J. N. H., O’Connor K. C., Hafler D. A., Vigneault F., Kleinstein S. H., pRESTO: A toolkit for processing high-throughput sequencing raw reads of lymphocyte receptor repertoires. Bioinformatics 30, 1930–1932 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/25/eaay5872/DC1