

The kinetic properties of transcriptional bursting were comprehensively analyzed in mouse embryonic stem cells.
Abstract
Transcriptional bursting is the stochastic activation and inactivation of promoters, contributing to cell-to-cell heterogeneity in gene expression. However, the mechanism underlying the regulation of transcriptional bursting kinetics (burst size and frequency) in mammalian cells remains elusive. In this study, we performed single-cell RNA sequencing to analyze the intrinsic noise and mRNA levels for elucidating the transcriptional bursting kinetics in mouse embryonic stem cells. Informatics analyses and functional assays revealed that transcriptional bursting kinetics was regulated by a combination of promoter- and gene body–binding proteins, including the polycomb repressive complex 2 and transcription elongation factors. Furthermore, large-scale CRISPR-Cas9–based screening identified that the Akt/MAPK signaling pathway regulated bursting kinetics by modulating transcription elongation efficiency. These results uncovered the key molecular mechanisms underlying transcriptional bursting and cell-to-cell gene expression noise in mammalian cells.
INTRODUCTION
Stochasticity in gene expression, also known as gene expression noise, induces substantial cell-to-cell heterogeneity in gene expression and introduces phenotypic diversity in unicellular organisms, improving species fitness by hedging against sudden environmental changes (1, 2). Gene expression noise is observed in a wide range of multicellular organisms as well (1, 3). For example, to acquire color vision during fly eye development, stochastic expression of a single transcription factor, Spineless, results in the mosaic expression of photoreceptors in individual ommatidia that detect light of different wavelengths (4). Similarly, the mosaic expression of olfactory receptors is well characterized in the olfactory system of several organisms, including humans (5). There are two orthogonal sources of gene expression noise: (i) intrinsic noise associated with stochasticity in biochemical reactions (such as transcription and translation) and (ii) extrinsic noise induced by true cell-to-cell variation (such as differences in microenvironment, cell size, cell cycle phase, and cellular component concentration) (3, 6, 7).
Transcription is the first step in gene expression, and its stochasticity is believed to be a major gene expression noise source (3). Noise at the transcription level, or “transcription noise,” is reflected in the production of RNA polymerase II (Pol II)–mediated transcripts. Any transcription, by either a single Pol II or multiple Pol II complexes, so-called transcriptional bursting, could contribute to transcription noise. In a simplified model, transcriptional bursting is mediated by the stochastic switch between the “ON” and “OFF” states of a promoter (Fig. 1A), and multiple transcripts are produced only during the ON state (3, 8–11). The ON state typically occurs with short pulses between long periods of the OFF state, causing dynamic changes in gene expression and heterogeneity in gene expression between cells and even between two alleles in a diploid genome (Fig. 1, B and C). Transcriptional bursting is thus considered to be a major source of both transcription and intrinsic noises and has been observed in various organisms (3, 8–13). Transcriptional bursting kinetics can be expressed by the frequency of the promoter present in the active state (burst frequency, f) and the mean number of transcripts produced per burst (burst size, b). Assuming that transcriptional bursting is a main source of intrinsic noise, transcriptional bursting kinetics (f and b) can be estimated from intrinsic noise (), the mean mRNA expression level (μ), and RNA degradation rate (γm; see Materials and Methods) (12, 14). Transcriptional bursting kinetics has also been studied using single-molecule fluorescence in situ hybridization (smFISH), MS2 system, and destabilized reporter proteins (8–10, 15, 16). These reports have indicated that mammalian genes are transcribed with broadly different transcriptional bursting kinetics and that the bursting can be influenced by the local chromatin environment (14). As for the mechanism of transcriptional bursting, promoter reactivation suppression has been proposed to be essential for its control (17), while cis-regulatory elements (such as the TATA box) and chromatin accessibility at the core promoter can regulate transcriptional bursting kinetics (11, 13, 17, 18). Imaging and genome-wide analyses have suggested that promoter and enhancer elements regulate burst size and frequency, respectively (10, 11).
Fig. 1. Genome-wide analysis of the kinetic properties of transcriptional bursting.
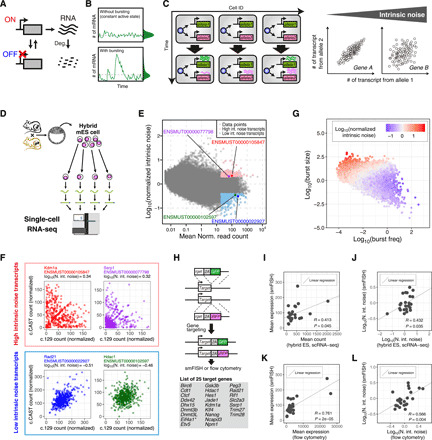
(A) Schematic diagram of gene expression with stochastic switching between ON and OFF states. (B) Schematic representations of the dynamics of transcript levels of a gene with or without transcriptional bursting. (C) Transcriptional bursting induces inter-allelic and intercellular heterogeneity in gene expression (left). Scatter plots of the individual allele-derived transcript numbers (right). (D) Schematic representation of scRNA-seq using hybrid mESCs. (E) Scatter plot of mean normalized read counts and normalized intrinsic noise of individual transcripts revealed by scRNA-seq. (F) Representative scatter plots of normalized individual allelic read counts of high and low intrinsic noise transcripts. N. int. noise, normalized intrinsic noise. (G) Scatter plot of burst size and burst frequency of individual transcripts. (H) Schematic representation of KI of GFP and iRFP gene cassette into individual alleles of mESC derived from inbred mice. Targeted genes are listed in the lower panel. Asterisks indicate genes in which KI cassettes were inserted immediately downstream of the start codon. (I to L) Scatter plots of the mean number of transcripts of targeted genes in KI cell lines counted by smFISH versus mean normalized read counts of corresponding genes in hybrid mESCs revealed by scRNA-seq (I) or versus mean expression levels of targeted genes in KI cell lines revealed by flow cytometry (K). Scatter plots of normalized intrinsic noise of targeted gene transcripts in KI cell lines revealed by smFISH versus that of corresponding genes in hybrid mESCs revealed by scRNA-seq (J) or versus that of targeted genes in KI cell lines revealed by flow cytometry (L).
Mouse embryonic stem cells (mESCs) are derived from the inner cell mass of the preimplantation embryo. A large number of genes in mESCs, cultured on gelatin in standard (Std) medium containing serum and leukemia inhibitory factor (LIF), show expression with cell-to-cell heterogeneity (19). For example, several genes encoding key transcription factors, including Nanog, display heterogeneous expression in the inner cell mass and mESCs (19). Heterogeneity in gene expression has been proposed to break the symmetry within the system and prime cells for subsequent lineage segregation (19). Previously, we have quantified Nanog transcriptional bursting kinetics in live cells using the MS2 system and determined intrinsic noise as a major cause of heterogeneous NANOG expression in mESCs (20). A recent study using intron-specific smFISH has revealed that most of the genes in mESCs are transcribed through bursting kinetics (21). However, a comprehensive understanding of how the kinetic properties of transcriptional bursting (burst size, frequency, and intrinsic noise) are regulated at the molecular level is still lacking.
In the present study, we performed single-cell RNA sequencing (scRNA-seq) (22) using hybrid mESCs to obtain the parameters for intrinsic noise and mean mRNA levels to investigate transcriptional bursting kinetics. We identified the genes with high intrinsic noise, and their promoters and gene bodies were positively associated with the polycomb repressive complex 2 (PRC2) and/or negatively associated with transcription elongation factors, based on informatics analysis using chromatin immunoprecipitation followed by sequencing (ChIP-seq) data. In addition, CRISPR library screening revealed that the Akt/mitogen-activated protein kinase (MAPK) pathway regulates transcriptional bursting via modulation of the transcription elongation efficiency.
RESULTS
Measuring intrinsic noise in hybrid mESCs with scRNA-seq
To study the genome-wide kinetic properties of transcriptional bursting, we analyzed allele-specific mRNA levels in 447 individual 129/CAST hybrid mESCs [grown on Laminin-511 (LN511) without feeder cells in the G1 phase] by single-cell (sc) random displacement amplification sequencing (RamDA-seq)—a highly sensitive RNA sequencing (RNA-seq) method (Fig. 1D and figs. S1A and S2, A to E) (22). A subset of genes have transcript variants with different transcription start sites (TSSs). Because the kinetic properties of transcriptional bursting may differ depending on the promoter, we mainly used transcript-level abundance, rather than gene-level abundance, to estimate the kinetic properties of transcriptional bursting (see Materials and Methods; fig. S1, B to H). Intrinsic noise, which is mainly induced by transcriptional bursting (9, 12, 13), was estimated from distribution of the number of mRNAs produced by the two alleles (see Materials and Methods) (6, 7). We also normalized intrinsic noise based on the expression level and transcript length of the gene (Fig. 1E and fig. S1, B to H). We excluded low abundance transcripts (with a mean read count of less than 20) from downstream analysis, as it is difficult to distinguish whether technical or biological noise contributed to the measured heterogeneity of allele-specific expression. We ranked the genes based on their normalized intrinsic noise and defined the top and bottom 5% transcripts as high and low intrinsic noise transcripts, respectively (Fig. 1E). As expected, high intrinsic noise transcripts showed larger inter-allelic expression heterogeneity than low intrinsic noise transcripts (Fig. 1F and tables S1 and S2).
Because mRNA degradation rate could affect intrinsic noise (see Materials and Methods), we checked the relationship between the published mRNA degradation rate in mESCs (23) and the normalized intrinsic noise that we measured; however, no correlation was observed (fig. S1H). Following this, we estimated the burst size and frequency of each transcript based on the published mRNA degradation rate, intrinsic noise, and mean expression levels (fig. S2, F to K; see Materials and Methods) (12, 14). As expected, transcripts with larger burst size and lower burst frequency tended to show higher normalized intrinsic noise and vice versa (Fig. 1G). Thus, normalized intrinsic noise was positively correlated with the ratio of burst size to the burst frequency (Spearman’s rho = 0.869).
To determine whether the intrinsic noise measured by scRNA-seq indicated true gene expression noise, we first chose 25 genes with medium expression levels and diverse intrinsic noise. Using CRISPR-Cas9 genome editing, we integrated a green fluorescent protein (GFP) and a near-infrared fluorescent protein (iRFP) reporter gene separately into both alleles of those genes in an inbred mESC line [knock-in (KI) mESC line; Fig. 1H and fig. S3]. It would be worth noting that the GFP and iRFP reporter cassettes were flanked by a 2A peptide and a degradation-promoting sequence, and were inserted immediately upstream of the stop codon of each allele (except one cell line, in which reporter cassettes were knocked in immediately downstream of the start codon; see Fig. 1H and fig. S3). The 2A peptide separated the reporter protein from the endogenous gene product. The degradation-promoting sequence ensured rapid degradation of GFP or iRFP reporter so that the amount of fluorescent protein produced in the cell would reflect the cellular mRNA levels. Using these cell lines, we measured mean expression levels and normalized intrinsic noise of the 25 genes with smFISH and found that the two parameters showed a significant correlation with scRNA-seq–based measurements (Fig. 1, I and J; fig. S1I; and table S3). Furthermore, flow cytometry analysis confirmed that the mean expression level and normalized intrinsic noise at the protein level also showed a significant correlation with the smFISH-based measurements (Fig. 1, K and L, and fig. S1J). There was a substantial correlation between expression level of the endogenous target protein and that of the knocked-in fluorescent protein in all tested genes as well (fig. S1, K and L). These validation experiments demonstrated the reliability of scRNA-seq in determining intrinsic noise and revealed that heterogeneity in expression of the tested genes largely originated from variation of the mRNA levels.
Recently, it has been reported that intrinsic noise can be buffered by nuclear retention of mRNA molecules (24). Here, we investigated the relationship between subcellular localization of mRNA and normalized intrinsic noise in KI mESC lines by smFISH (fig. S1M). We observed no correlation between nuclear retention rate of mRNA and intrinsic noise, total noise, or normalized intrinsic noise at either mRNA or protein level (fig. S1N). Thus, it is unlikely that nuclear retention of mRNA would play a role in buffering intrinsic noise for the 25 genes tested in mESCs.
Identifying molecular determinants of transcriptional bursting
It has been reported that promoters with a TATA box tend to show higher burst size and gene expression noise than those without in yeast and mESCs (11, 13, 17, 18). To confirm whether our results were consistent with the previous findings, we compared the kinetic properties of transcriptional bursting between genes with and without a TATA box. Although no significant difference was observed in burst frequency, both burst size and normalized intrinsic noise were significantly higher in the promoters with TATA box than in those without (Fig. 2, A to C). These data, which are consistent with those of previous reports, validated the quality of our results and supported the involvement of TATA box in burst size and gene expression noise (Fig. 2, A to C).
Fig. 2. Exploring molecular determinants of transcriptional bursting.
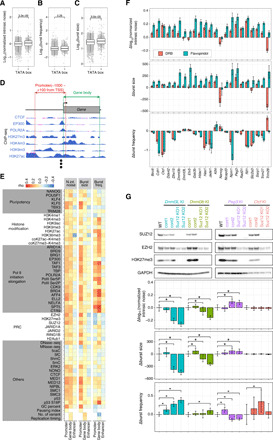
(A to C) Kinetic properties of transcriptional bursting of genes either with or without a TATA box. (D) Schematic representation of calculating reads per million (RPM) at the promoter and gene body from ChIP-seq data. In addition, similar calculations were also performed for enhancers (see Materials and Methods). (E) Heat maps of Spearman’s rank correlation between promoter-, gene body–, or enhancer-associated factors and either normalized intrinsic noise (N. int. noise), burst size, or burst frequency (burst freq.). (F) Effect of the Pol II pause release inhibitor, DRB, and flavopiridol treatment on the kinetic properties of transcriptional bursting. Δnormalized intrinsic noise, Δburst size, and Δburst frequency are residuals of normalized intrinsic noise, burst size, and frequency of inhibitor-treated cells from that of control cells, respectively. Error bars indicate 95% confidence interval. (G) Effect of Suz12 K/O on normalized intrinsic noise. Suz12 K/O cell lines derived from Dnmt3l, Dnmt3b, Peg3, and Ctcf KI cell lines were established. Upper panel represents the result of Western blotting. In the lower part of the panel, the Δnormalized intrinsic noise, Δburst size, and Δburst frequency compared with the control (cont1) are shown. Error bars indicate 95% confidence interval. Asterisks indicate significance at P < 0.05.
We next compared the kinetic properties of transcriptional bursting to genome-wide transcription factor–binding patterns (Fig. 2D; see Materials and Methods). Specifically, we calculated Spearman’s rank correlations between the kinetic properties of transcriptional bursting and ChIP-seq enrichment in the promoter, gene body, or enhancer elements (Fig. 2E). We found that the localization of several transcription regulators (such as EP300, ELL2, and MED12) in the promoter showed substantial positive correlations with burst size. However, the correlation coefficients between the burst size and transcription regulators bound to enhancers were overall relatively low. This was consistent with the findings of a report showing that burst size is mainly controlled by the promoter region (11). Distal enhancers are reported to be important for regulating burst frequency (10). In our analysis, the localization of several factors (such as BRD9, TAF3, AFF4, and CTR9) in enhancers showed relatively lower yet positive correlations with burst frequency.
We found that localization of transcription elongation factors [such as trimethylated histone H3 at lysine 36 (H3K36me3), BRD4, AFF4, SPT5, and CTR9] on the gene body was positively correlated with burst frequency (Fig. 2E). Transcription is well known to occur in at least three stages: initiation, elongation, and termination. During early elongation, Pol II often pauses near the promoter region, a phenomenon known as Pol II promoter-proximal pausing (25). The paused Pol II transitions into productive elongation by the activity of positive transcription elongation factor b (P-TEFb), which phosphorylates serine-2 in the heptaptide (Tyr-Ser-Pro-Thr-Ser-Pro-Ser) repeats of the C-terminal domain. The extent of Pol II pausing, estimated by the pausing index, had a negative correlation with burst frequency (Fig. 2E).
To dissect the link between Pol II pause release and burst frequency, we inhibited P-TEFb with 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB) and flavopiridol in KI mESC lines cultured in 2i conditions (26). Two days after DRB and flavopiridol treatment, cells were subjected to flow cytometry analysis (fig. S4, A and B). DRB and flavopiridol treatment increased the normalized intrinsic noise and burst size in most of the cell lines (Fig. 2F). However, the effects of DRB and flavopiridol treatment on burst frequency were highly gene specific, suggesting that Pol II pause release likely contributes to the regulation of both burst size and frequency, whereas the regulation of burst frequency by Pol II pause release is more context dependent.
Promoter localization of PRC2 subunits (EZH2, SUZ12, and JARID2) correlated inversely with burst frequency, while they correlated positively with burst size and normalized intrinsic noise (Fig. 2E), thus suggesting a possible link between PRC2 and intrinsic noise. To test how PRC2 regulates transcriptional bursting, we inactivated PRC2 functionality by knocking out SUZ12 (27) in Dnmt3l, Dnmt3b, Peg3, and Ctcf KI cell lines (Fig. 2G). These targeted genes showed relatively high trimethylated histone 3 at lysine residue 27 (H3K27me3) enrichment in the promoter compared to the other available KI-targeted genes. Loss of H3K27me3 modification in Suz12 knockout (K/O) cell lines was confirmed by Western blotting (Fig. 2G). Next, we quantified GFP and iRFP expression levels by flow cytometry in the Suz12 K/O and control cell lines and found that normalized intrinsic noise and burst size of Dnmt3l and Dnmt3b were significantly reduced by Suz12 K/O (Fig. 2G). In contrast, Suz12 K/O significantly increased normalized intrinsic noise and burst size of Peg3. No significant change was observed for Ctcf. While the burst frequency of Dnmt3l was increased significantly, that of Peg3 was markedly reduced by Suz12 K/O. These results suggest that PRC2-mediated control of the kinetic properties of transcriptional bursting is also possibly context dependent.
Combination of promoter- and gene body–binding factors regulates transcriptional bursting
To study the combinatorial regulations underlying the kinetic properties of transcriptional bursting, we first classified the genetic and epigenetic features, based on the sequence and transcription regulatory factor binding patterns at the promoter and gene body of high intrinsic noise transcripts, into 10 clusters (Fig. 3). To identify the features that can distinguish a cluster of high intrinsic noise transcripts from low intrinsic noise transcripts, we performed orthogonal partial least squares discriminant analysis (OPLS-DA) modeling, which is a useful method for identifying features that contribute to class differences (28). In 8 of the 10 clusters, the model successfully separated the high intrinsic noise transcripts from the low intrinsic noise transcripts (Fig. 3). Specifically, we obtained the top three positively and negatively contributing factors using S-plot (Fig. 3). For example, in cluster 3, promoter binding of PRC2-related factors (SUZ12, EZH2, and H3K27me3) was a positive contributor to intrinsic noise, while the gene body localization of TAF1, BRD4, and CTR9 was a negative contributor. This result suggested that promoter localization of PRC2-related factors influences bursting properties in a gene-specific manner.
Fig. 3. Normalized intrinsic noise is determined by combinations of promoter- and gene body–binding factors.
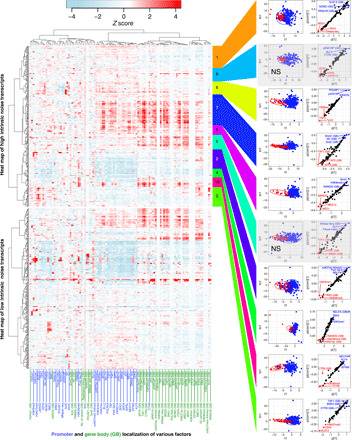
The left side of the panel shows a heat map of promoter and the gene body (GB) localization of various factors with high and low intrinsic noise transcripts. The high intrinsic noise transcripts were classified into 10 clusters, and each cluster of high intrinsic noise transcripts and low intrinsic noise transcripts was subjected to OPLS-DA modeling. The right side of the panel represents score plots of OPLS-DA [the first predictive component (t1) versus the first orthogonal component (to1)] and S-plots constructed by presenting the modeled covariance (p[1]) against modeled correlation {p(corr)[1]} in the first predictive component. In clusters 5 and 6, the first orthogonal component was not significant (NS).
In cluster 10, promoter localization of H3K36me3, a histone mark associated with transcriptional elongation, and promoter and gene body localization of CTR9, a subunit of the PAF1 complex involved in Pol II pausing and transcription elongation, were the positive contributors (Fig. 3). In contrast, promoter localization of negative elongation factor complex member A (NELFA) was a strong negative contributor (Fig. 3). These results implied that transcriptional elongation is involved in the regulation of normalized intrinsic noise in this cluster. We similarly identified the factors regulating burst size and frequency and found them to be also affected by a combination of promoter- and gene body–binding factors (fig. S5). Collectively, the kinetic properties of transcriptional bursting in mammalian cells appear to be regulated by a combinatory suite of promoter- and gene body–binding factors in a context-dependent manner.
Genome-wide CRISPR library screening identified genes involved in the regulation of intrinsic noise
To identify genes regulating intrinsic noise in an unbiased manner, we performed high-throughput screening with the CRISPR K/O library (29). The lentiviral CRISPR library targeting genes in the mouse genome was introduced into Nanog, Dnmt3l, and Trim28 KI cell lines. Although genes with high intrinsic noise showed a larger variation in the expression levels of one allele (such as GFP) and the other allele (such as iRFP) perpendicular to the diagonal line (Fig. 1, C and F), we found that the loss of genomic integrity (such as by loss of function of p53) induced instability in the number of alleles, resulting in an unintended increase in intrinsic noise levels in a pilot study. Therefore, to reduce false negatives and selectively enrich cell populations with suppressed intrinsic noise, we first sorted out cells showing expression levels close to the diagonal line of GFP and iRFP expression by fluorescence-activated cell sorting (FACS; Fig. 4A). After expanding the sorted cells for a week, the cells were sorted again. These sorting and expansion procedures were repeated four times in total to selectively enrich cell populations with suppressed intrinsic noise. Even for genes with high intrinsic noise, a large fraction of cells showed a smaller variation in the expression levels of one allele (such as GFP) and the other allele (such as iRFP) perpendicular to the diagonal line (Fig. 1, C and F). Therefore, enrichment of cells with low intrinsic noise by repeated sorting procedures appeared to reduce false positives. Last, we compared the targeted K/O gene profile in the sorted cells with that in an unsorted control by high-throughput genomic DNA sequencing (Fig. 4A). To gain a comprehensive picture of the genes involved in intrinsic noise regulation, we performed Kyoto Encyclopedia of Genes and Genomes (KEGG) (30) pathway enrichment analysis of the enriched (top 100) and depleted targeted genes (bottom 100) in the three cell lines (Fig. 4, B and C). We found that the mammalian target of rapamycin (mTOR) and MAPK signaling pathways were involved in promoting intrinsic noise in all three cell lines (Fig. 4C). Previous studies demonstrated that mTOR and MAPK pathways are involved in “Proteoglycans in Cancer” and “Sphingolipid signaling” pathways and cross-talk with each other via the PI3/Akt pathway (Fig. 4D) (31). To test whether these signaling pathways are involved in intrinsic noise regulation, we conditioned Nanog, Trim28, and Dnmt3l KI cells with inhibitors for the MAPK, mTOR, and Akt pathways (Fig. 4, D to F). When treated with the Akt inhibitor MK-2206 alone, normalized intrinsic noise decreased in all three cell lines (Fig. 4F). Furthermore, treatment with MK-2206 and MAPK kinase (MEK) inhibitor PD0325901 (PD-MK condition) resulted in a substantial decrease in the normalized intrinsic noise in all three cell lines (Fig. 4F). In addition, normalized intrinsic noise was reduced in most of the other KI cell lines under the PD-MK condition (Fig. 4G), while mRNA degradation rates were largely unaltered (fig. S6). Under PD-MK and 2i culture conditions, one and three genes, respectively, showed Δlog10 (normalized intrinsic noise) > −0.05 (Fig. 4G), hence suggesting that more genes showed reduced normalized intrinsic noise under the PD-MK condition than under the 2i condition. It should be noted that the PD-MK condition, which decreased the burst size, increased the burst frequency for most genes, although the extent of changes varied depending on the genes. Therefore, decrease in normalized intrinsic noise under the PD-MK condition is likely caused by changes in both burst size and burst frequency.
Fig. 4. CRISPR library screening of genes involved in intrinsic noise regulation.
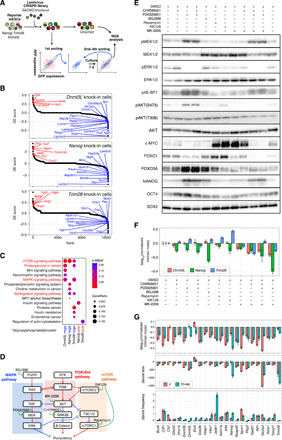
(A) Schematic diagram of CRISPR lentivirus library screening. Screening was performed independently for each of the three (Nanog, Trim28, and Dnmt3l) KI cell lines. (B) Ranked differentially expressed (DE) score plots obtained by performing CRISPR screening on three cell lines. The higher the DE score, the more the effect of enhancing intrinsic noise. (C) KEGG pathway enrichment analysis. KEGG pathway enrichment analysis was performed using clusterProfiler (see Materials and Methods), with the upper or lower 100 genes of DE score obtained from the CRISPR screening (referred as posi and nega, respectively). The pathways shown in red indicate hits in multiple groups of genes. Genes corresponding to these pathways are labeled in (B). (D) Simplified diagram of MAPK, Akt, and mTOR signaling pathways. These pathways are included in the pathways highlighted in red in (C) and cross-talk with each other. (E) Western blot of cells treated with signal pathway inhibitors. (F) Δnormalized intrinsic noise of cells treated with signal pathway inhibitors against control [dimethyl sulfoxide (DMSO)–treated] cells. Error bars indicate 95% confidence interval. (G) Twenty-four KI cell lines were conditioned to 2i or PD-MK conditions and subjected to flow cytometry analysis. Δnormalized intrinsic noise, Δburst size, and Δburst frequency against control (DMSO-treated) cells are shown. Error bars indicate 95% confidence interval.
The phenotype observed under PD-MK treatment could be due to its effects on cell viability and/or pluripotency. We observed that PD-MK treatment substantially reduced the proliferation rates of mESCs (Fig. 5A), consistent with the function of Akt or MAPK pathways in cell cycle progression (31). However, we did not observe increased cell apoptosis after the PD-MK treatment (Fig. 5B). Cell cycle distribution was also unaffected by the PD-MK treatment (Fig. 5C), suggesting a global slowdown of individual cell cycle phases under the PD-MK condition. Thus, the reduced intrinsic noise caused by PD-MK does not appear to be the result of cell death or cell cycle arrest. We also analyzed the expression of pluripotent markers and found that they were largely unaffected under the PD-MK condition (Figs. 4E and 5D). Furthermore, we were able to generate chimeric mice using mESCs cultured in the PD-MK condition, indicating that PD-MK treatment does not affect mESC pluripotency (Fig. 5E).
Fig. 5. PD-MK conditioned mESCs retain pluripotency.
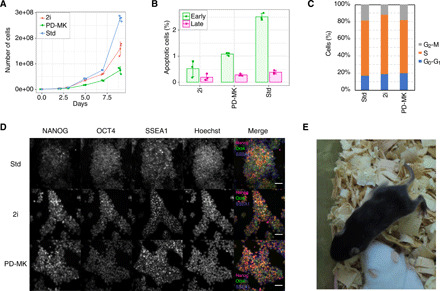
(A) Growth curve of mESC conditioned to Std, 2i, and PD-MK conditions. Error bars indicate SD (n = 3). (B) Percentage of apoptotic cells of mESC conditioned to Std, 2i, and PD-MK conditions. Error bars indicate SD (n = 3). (C) Cell cycle distribution of mESCs conditioned to Std, 2i, and PD-MK conditions. (D) Immunofluorescence of pluripotency markers (NANOG, OCT4, and SSEA1) of mESCs conditioned to Std, 2i, and PD-MK conditions. The images show maximum intensity projections of stacks. Scale bar, 50 μm. (E) Chimeric mice with black coat color generated from C57BL/6NCr ES cells conditioned to the PD-MK condition and then to the Std condition before injection into albino host embryos. Photo credit: T. Okamura (National Center for Global Health and Medicine).
To further characterize how the PD-MK condition affects mESC gene expression programs, we performed RNA-seq analysis of mESCs cultured in Std, 2i, and PD-MK conditions (Fig. 6A). Flow cytometry analysis revealed that more genes showed decreased normalized intrinsic noise under the PD-MK condition than under the 2i condition (Fig. 4G). To obtain a comprehensive view of the genes involved in intrinsic noise suppression under the PD-MK condition compared to that under the 2i condition, we performed gene ontology (GO) analysis and found that the “transcription elongation factor complex”–related genes were significantly enriched in the up-regulated genes under PD-MK (Fig. 6, B and C). Furthermore, the up-regulated genes were enriched in factors involved in transcriptional regulation (Fig. 6B), consistent with our observation of a positive correlation between gene body localization of transcription elongation factor and burst frequency (Fig. 2C). Thus, it is possible that up-regulation of transcription elongation factors promotes burst frequency, thereby reducing the intrinsic noise under the PD-MK condition. We also found that the expression of Aff1 and Aff4, which encode subunits of the super elongation complex (SEC) that regulates Pol II pause release and transcription elongation rate (32), was significantly up-regulated under the PD-MK condition (Fig. 6C).
Fig. 6. Increase in the expression level of transcription elongation factors in the PD-MK condition.
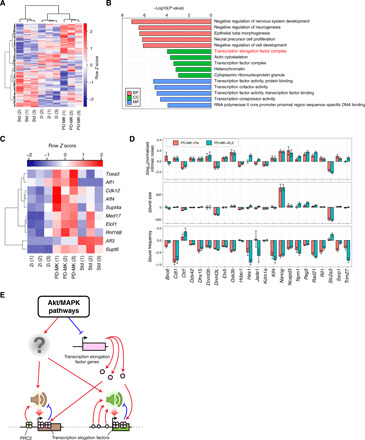
(A) Comparison of transcriptome of cells conditioned to Std, 2i, and PD-MK conditions. (B) GO analysis of genes whose expression significantly increased in the PD-MK condition against the 2i condition. BP, biological process; CC, cellular components; MF, molecular function. (C) Expression levels of genes encoding transcriptional elongation factors were elevated under the PD-MK condition as compared to the 2i condition. (D) Effect of RNA Pol II pause release inhibitor flavopiridol and SEC inhibitor KL-2 treatment on normalized intrinsic noise, burst size, and frequency. The KI cell lines conditioned to PD-MK were treated with flavopiridol or KL-2 for 2 days and analyzed by flow cytometry, and normalized intrinsic noise, burst size, and frequency were calculated. Δnormalized intrinsic noise, Δburst size, and Δburst frequency are residuals of normalized intrinsic noise, burst size, and frequency of inhibitor-treated cells from that of control cells (PD-MK condition), respectively. Error bars indicate 95% confidence interval. (E) Schematic summary of the determination of kinetic properties of transcriptional bursting (including intrinsic noise, burst size, and burst frequency) by a combination of promoter- and gene body–binding proteins, including PRC2 and transcription elongation factors.
To examine the contribution of SEC in the regulation of bursting, we treated cells cultured under the PD-MK condition with the P-TEFb inhibitor flavopiridol or AFF1/4 inhibitor KL-2 (32) for 2 days and then compared the intrinsic noise in these cells with that in control cells under the PD-MK condition (Fig. 6D and fig. S4, C and D). We found that the normalized intrinsic noise was significantly increased in most of the genes, although a decrease was also observed in a small fraction of genes (Fig. 6D). We next examined how the chemical inhibitors could affect the burst size and frequency. For most of the genes, Δburst sizes, which are residuals of burst size of inhibitor-treated cells from that of control cells, were small, while the burst frequency was substantially reduced overall. This suggested that PD-MK treatment enhances Pol II pause release and transcription elongation efficiency without strongly affecting the burst size, at least in the genes tested here. Because P-TEFb and SEC inhibitors affected Δburst frequency, which is a residual of burst frequency of inhibitor-treated cells from that of control cells, in a similar fashion in most genes analyzed, SEC is probably responsible for regulating Pol II pause release in these genes. It should be noted here that most genes displayed reduced burst sizes under the PD-MK condition (Fig. 4G), suggesting that Pol II pause release is not the only downstream effector of Akt and MAPK pathways regulating the normalized intrinsic noise.
DISCUSSION
scRNA-seq has been used to detect transcriptome-wide cell-to-cell heterogeneity in gene expression in inbred (33, 34) and hybrid mESC lines (11, 35). However, most of these analyses considered the overall heterogeneity in gene expression and/or cell cycle–dependent effects, rather than focusing on the regulatory mechanism of intrinsic noise generation. To exclude the heterogeneity due to cell cycle differences, we performed scRNA-seq using 447 hybrid mESCs only in the G1 phase for measuring genome-wide kinetic properties of transcriptional bursting. The larger the number of analyzed cells, the more reliable was the calculated data regarding the kinetic properties of transcriptional bursting (fig. S2, D and E). Thus, by measuring intrinsic noise and heterogeneity in gene expression in a genome-wide fashion, our study enabled a detailed investigation of potential factors contributing to the regulation of transcriptional bursting and intrinsic noise.
The promoter region of a gene has been reported to be mainly involved in regulating the burst size, while enhancers have been reported to be associated with regulation of burst frequency (11). We confirmed that the presence of a TATA box at the promoter (Fig. 2, A to C) and the promoter localization of several factors (such as EP300, ELL2, and MED12) positively correlated with the burst size (Fig. 2E). Consistent with previous reports, enhancer localization of several factors (including transcription elongation factors) was positively correlated with burst frequency (11). Here, we found that PRC2 and transcription elongation factors also regulate the kinetic properties of transcriptional bursting. However, our data indicated that the kinetic properties of transcriptional bursting of individual genes were not driven by a type of “master regulator”; they are rather determined by a combination of multiple factors that bind to the promoter and gene body (Figs. 2, F and G, 3, 4G, and 6, D and E, and fig. S5).
In addition to transcriptional bursting, other downstream processes, including posttranscriptional regulation (24) or translation (36), also contribute to generating heterogeneity in gene expression as intrinsic noise (3). Nuclear retention of mRNA could suppress intrinsic noise at the protein level (24); however, at least in the 25 genes analyzed in this study in mESCs, suppression of intrinsic noise by nuclear retention was not observed (fig. S1N), suggesting that noise suppression via nuclear mRNA retention is not a general phenomenon across cell types and genes.
In 25 KI-mESC lines, we observed a significant correlation between mRNA and protein expression levels (Fig. 1K). On one hand, simultaneous measurement of mRNA and protein levels in single mammalian cells showed a low correlation between them, suggesting the possibility of a cause for gene expression noise even at the translation level (36); on the other hand, the average expression level of mRNA and protein was reported to be significantly correlated in mammalian cells (37). Such a conflict is probably related to the difference in expression kinetics over time between mRNA and protein, that is, there is a time lag between the transcription of mRNA and translation of the protein, thus suggesting a deviation between the temporal peaks of mRNA and protein expression amount. In contrast to mRNAs that are transcribed from only one or two copies of the gene, which generates intrinsic noise during the process, proteins are translated from a large number of mRNAs under typical conditions. Hence, the noise arising from translation is expected to be much smaller than that arising from transcription.
In conclusion, we observed that the kinetic properties of transcriptional bursting were regulated by a combination of promoter- and gene body–binding proteins, including transcription elongation factors and PRC2. In addition, using lentiviral CRISPR library screening, we observed that the Akt/MAPK signaling pathway was involved in this process by modulating the efficiency of Pol II pause release. Pluripotent cells in the inner cell mass share a similar transcriptomic gene expression profile with mESCs. However, how heterogeneous gene expression might contribute to the differentiation of pluripotent cells into an epiblast and primitive endoderm is still poorly understood (19). We envision future investigations to explore the involvement of intrinsic noise in cell fate decisions of the inner cell mass. We believe that our study provides important information for understanding the molecular basis of transcriptional bursting, which underlies cellular heterogeneity.
MATERIALS AND METHODS
Cell culture and cell lines
The hybrid mESC line F1-21.6 (129Sv-Cast/EiJ, female), a gift from J. Gribnau, was grown on either LN511 (BioLamina, Stockholm, Sweden) or gelatin-coated dish in either Std medium [15% fetal bovine serum (FBS; Gibco), 0.1 mM β-mercaptoethanol (Wako Pure Chemicals, Osaka, Japan), and LIF (1000 U/ml; Wako Pure Chemicals, Osaka, Japan)] or 2i medium [StemSure Dulbecco’s modified Eagle’s medium (DMEM; Wako Pure Chemicals, Osaka, Japan), 15% FBS, 0.1 mM β-mercaptoethanol, 1× MEM nonessential amino acids (Wako Pure Chemicals), a 2 mM l-alanyl-l-glutamine solution (Wako Pure Chemicals), LIF (1000 U/ml; Wako Pure Chemicals), gentamicin (20 mg/ml; Wako Pure Chemicals), 1 μM PD0325901 (CS-0062, ChemScene), and 3 μM CHIR99021 (034-23103, Wako Pure Chemicals)]. This cell line was previously described in (38).
Wild-type (WT) mESCs derived from inbred mouse (Bruce 4 C57BL/6J, male, EMD Millipore, Billerica, MA) and other KI derivatives were cultured on either LN511 or gelatin-coated dish under either Std, 2i, or PD-MK medium [StemSure DMEM (Wako Pure Chemicals, Osaka, Japan), 15% FBS, 0.1 mM β-mercaptoethanol, 1× MEM nonessential amino acids (Wako Pure Chemicals), a 2 mM l-alanyl-l-glutamine solution (Wako Pure Chemicals), LIF (1000 U/ml; Wako Pure Chemicals), gentamicin (20 mg/ml; Wako Pure Chemicals), 1 μM PD0325901 (CS-0062, ChemScene), and 4 μM MK-2206 2HCl (S1078, Selleck Chemicals, Houston TX)]. Inhibitors were added at the following concentrations: 40 μM DRB (D1916, Sigma-Aldrich, St. Louis, MO), 0.25 μM flavopiridol (CS-0018, ChemScene, Monmouth Junction, NJ) in the 2i condition, 0.125 μM flavopiridol in the PD-MK condition, 3 μM CHIR99021 (034-23103, Wako Pure Chemicals) in Std medium, 1 μM PD0325901 (CS-0062, ChemScene) in Std medium, 5 μM BGJ398 (NVP-BGJ398; S2183, Selleck Chemicals) in Std medium, 1 μM rapamycin (R-5000, LC Laboratories, MA, USA) in Std medium, 0.2 μM INK128 (11811, Cayman Chemical Company, MI, USA) in Std medium, and 4 μM MK-2206 2HCl (S1078, Selleck Chemicals) in Std medium. C57BL/6NCr mESCs (male) were cultured on gelatin-coated dish under the PD-MK condition.
Establishment of KI mESC lines
To quantify the intrinsic noise level of a particular gene, it is necessary to establish a cell line with the GFP and iRFP reporter genes individually knocked into both alleles of the target genes. Therefore, on the basis of the scRNA-seq data, 25 genes (Fig. 1H) with medium expression levels and variable intrinsic noise levels were manually selected.
GFP/iRFP KI cell lines were established using CRISPR-Cas9 or transcription activator-like effector nuclease (TALEN) expression vectors and targeting vectors [with about 1-kbp (kilo–base pair) homology arms]. Vectors used in this study are listed in table S4. C57BL/6J mESCs (5 × 105) conditioned to 2i medium were plated onto gelatin-coated six-well plates. After 1 hour, the cells were then transfected with 1 μg each of GFP and iRFP targeting vectors (table S4), 1 μg total of nuclease vectors (table S4), and pKLV-PGKpuro2ABFP (puromycin resistant, Addgene, plasmid #122372) using Lipofectamine 3000 (catalog no. L3000015, Life Technologies, Gaithersburg, MD), according to the manufacturer’s instructions. Cells were selected by adding puromycin (1 μg/ml) to the 2i medium 24 hours after transfection. After another 24 hours, the medium was exchanged. The medium was exchanged every 2 days. At 5 days after transfection, cells were treated with 25 μM biliverdin (BV). BV is used for forming a fluorophore by iRFP670. Although BV is a molecule ubiquitous in eukaryotes, the addition of BV to culture medium increases the fluorescence intensity. Twenty-four hours later, cells were trypsinized and subjected to FACS analysis, and GFP/iRFP double-positive cells were sorted and seeded on a gelatin-coated 6-cm dish. The medium was exchanged every 2 days. One week after sorting, 16 colonies were picked for downstream analysis and checking gene targeting. Polymerase chain reaction (PCR) was carried out using primers outside the homology arms, and cells that seemed to be successfully knocked into both alleles were selected. Thereafter, candidate clones were further analyzed by Southern blotting as described before (fig. S3) (20). Restriction enzymes and genomic regions used for Southern blot probes are listed in table S4. Probes were prepared using the PCR DIG Probe Synthesis Kit (Roche Diagnostics, Mannheim, Germany).
Mice
ICR mice were purchased from CLEA Japan (Tokyo, Japan). All mice were housed in an air-conditioned animal room under specific pathogen–free conditions, with a 12-hour light/12-hour dark cycle. All mice were fed a standard rodent CE-2 diet (CLEA Japan, Tokyo, Japan) and had ad libitum access to water. All animal experiments were approved by the President of the National Center for Global Health and Medicine, following consideration by the Institutional Animal Care and Use Committee of the National Center for Global Health and Medicine (approval ID no. 17043), and were carried out in accordance with the institutional procedures, national guidelines, and the relevant national laws on the protection of animals.
Plasmid construction
To construct the lentiCRISPRv2-sgSuz12_1, lentiCRISPRv2-sgSuz12_2, lentiCRISPRv2-sgSuz12_3, and lentiCRISPRv2_sgMS2_1 plasmids, which are single-guide RNA (sgRNA) expression vectors, we performed inverse PCR using R primer (5′-GGTGTTTCGTCCTTTCCACAAGAT-3′) and either of F primers (5′-AAAGGACGAAACACCGCGGCTTCGGGGGTTCGGCGGGTTTTAGAGCTAGAAATAGCAAGT-3′, 5′-AAAGGACGAAACACCGGCCGGTGAAGAAGCCGAAAAGTTTTAGAGCTAGAAATAGCAAGT-3′, 5′-AAAGGACGAAACACCGCATTTGCAACTTACATTTACGTTTTAGAGCTAGAAATAGCAAGT-3′, or 5′-AAAGGACGAAACACCGGGCTGATGCTCGTGCTTTCTGTTTTAGAGCTAGAAATAGCAAGT-3′), respectively, and lentiCRISPR v2 (Addgene, plasmid #52961) as a template, followed by self-circularization using the In-Fusion HD Cloning Kit (catalog no. 639648, Clontech Laboratories, Mountain View, CA, USA).
RNA-seq of mESCs cultured on LN511 and serum-LIF medium
129/CAST hybrid mESCs need to be maintained on feeder cells in the gelatin/Std condition. To eliminate the need for feeder cells, we decided to maintain the hybrid mESCs on dishes coated with LN511, enabling maintenance of mESCs without feeder cells in the Std condition. To compare the transcriptomes of mESCs cultured on gelatin-coated dish and those cultured on LN511-coated dish, we performed RNA-seq analysis as follows. First, C57BL/6J WT mESCs were conditioned on either gelatin- or LN511-coated dish in either Std or 2i medium for 2 weeks. Next, RNA was recovered from 1 × 106 cells using the NucleoSpin RNA Kit (Macherey-Nagel, Düren, Germany). The RNA was sent to Eurofins for RNA-seq analysis. RNA-seq reads were aligned to the mouse reference genome (mm10) using TopHat (version 2.1.1) (https://ccb.jhu.edu/software/tophat/index.shtml). Fragments per kilobase per million mapped reads (FPKM) values were quantified using Cufflinks (version 2.1.1) (http://cole-trapnell-lab.github.io/cufflinks/) to generate relative gene expression levels. Hierarchical clustering analyses were performed on FPKM values using CummeRbund (v2.18.0) (https://bioconductor.org/packages/release/bioc/html/cummeRbund.html). In comparison, the transcriptomes of mESCs cultured on gelatin-coated dish and those cultured on LN511-coated dishes showed no considerable difference in expression patterns (fig. S1A).
Sequencing library preparation for RamDA-seq
Library preparation for single-cell RamDA-seq was performed as described previously (22). Briefly, hybrid mESC line F1-21.6 (129Sv-Cast/EiJ) conditioned to the LN511/Std condition was dissociated with 1× trypsin (Thermo Fisher Scientific, Rochester, NY) with 1 mM EDTA at 37°C for 3 min. The dissociated cells were adjusted to 1 × 106 cells/ml and stained with Hoechst 33342 dye (10 μg/ml; Sigma-Aldrich) in phosphate-buffered saline (PBS) at 37°C for 15 min to identify the cell cycle. After Hoechst 33342 staining, the cells were washed once with PBS and stained with propidium iodide (PI; 1 μg/ml; Sigma-Aldrich) to remove dead cells. Single-cell sorting was performed using MoFlo Astrios (Beckman Coulter; table S4). Recent studies of scRNA-seq using mESCs have suggested that genes related to the cell cycle demonstrate considerable heterogeneity in expression (35). Therefore, to minimize this variation, 474 cells only in the G1 phase were collected (table S4). Single cells were collected in 1 μl of cell lysis buffer [1 U of RNasin Plus (Promega, Madison, WI), RealTime ready Cell Lysis Buffer (10%; catalog no. 06366821001, Roche), 0.3% NP-40 (Thermo Fisher Scientific), and ribonuclease (RNase)–free water (Takara, Japan)] in a 96-well PCR plate (BIOplastics).
The cell lysates were denatured at 70°C for 90 s and held at 4°C until the next step. To eliminate genomic DNA contamination, 1 μl of genomic DNA digestion mix [0.5× PrimeScript Buffer, 0.2 U of DNase I Amplification Grade, and 1:5,000,000 ERCC RNA Spike-In Mix I (Thermo Fisher Scientific) in RNase-free water] was added to 1 μl of the denatured sample. The mixtures were agitated for 30 s at 2000 rpm using ThermoMixer C at 4°C, incubated in a C1000 thermal cycler at 30°C for 5 min, and held at 4°C until the next step. One microliter of RT-RamDA mix [2.5× PrimeScript Buffer, 0.6 pmol of oligo(dT)18 (catalog no. SO131, Thermo Fisher Scientific), 8 pmol of 1st-NSRs (22), 100 ng of T4 gene 32 protein (New England Biolabs), and 3× PrimeScript enzyme mix (catalog no. RR037A, TAKARA Bio Inc.) in RNase-free water] was added to 2 μl of the digested lysates. The mixtures were agitated for 30 s at 2000 rpm and 4°C and incubated at 25°C for 10 min, 30°C for 10 min, 37°C for 30 min, 50°C for 5 min, and 94°C for 5 min. Then, the mixtures were held at 4°C until the next step. After RT (reverse transcription), the samples were added to 2 μl of second-strand synthesis mix [2.5× NEBuffer 2 (New England Biolabs), 0.625 mM each dNTP mixture (Takara), 40 pmol of 2nd-NSRs (22), and 0.75 U of Klenow Fragment (3′ → 5′ exo-; New England Biolabs) in RNase-free water]. The mixtures were agitated for 30 s at 2000 rpm and 4°C and incubated at 16°C for 60 min, 70°C for 10 min, and then 4°C until the next step. Sequencing library DNA preparation was performed using the Tn5 tagmentation-based method with one-fourth volumes of the Nextera XT DNA Library Preparation Kit (catalog nos. FC-131-1096, FC-131-2001, FC-131-2002, FC-131-2003, and FC-131-2004, Illumina, San Diego, CA) according to the manufacturer’s protocol. The above-described double-stranded complementary DNAs (cDNAs) were purified by using 15 μl of AMPure XP SPRI beads (catalog no. A63881, Beckman Coulter) and a handmade 96-well magnetic stand for low volumes. Washed AMPure XP beads attached to double-stranded cDNAs were directly eluted using 3.75 μl of 1× diluted Tagment DNA Buffer (Illumina) and mixed well using a vortex mixer and pipetting. Fourteen cycles of PCR were applied for the library DNA. After PCR, sequencing library DNA was purified using 1.2× the volume of AMPure XP beads and eluted into 24 μl of TE buffer.
Quality control and sequencing of library DNA
All the RamDA-seq libraries prepared with Nextera XT DNA Library Preparation were quantified and evaluated using a MultiNA DNA-12000 kit (Shimadzu, Kyoto, Japan) with a modified sample mixing ratio (1:1:1; sample, marker, and nuclease-free water) in a total of 6 μl. The length and yield of the library DNA were calculated in the range of 161 to 2500 bp. The library DNA yield was estimated as 0.5 times the value quantified from the modified MultiNA condition. Subsequently, we pooled each 110 fmol of library DNA in each well of a 96-well plate. The pooled library DNA was evaluated on the basis of the averaged length and concentration using the Bioanalyzer Agilent High-Sensitivity DNA Kit (catalog no. 5067-4626) in the range of 150 to 3000 bp and the KAPA Library Quantification Kit (catalog no. KK4824, Kapa Biosystems, Wilmington, MA). Pooled library DNA (1.5 pM) was sequenced using Illumina HiSeq2000 (single-read 50-cycle sequencing).
Single-molecule fluorescence in situ hybridization
Trypsinized cells (2 × 105) were transferred onto LN511-coated round coverslips and cultured for 1 hour at 37°C and 5% CO2. Cells were washed with PBS, fixed with 4% paraformaldehyde in PBS for 10 min, and washed with PBS two times. Then, cells were permeabilized in 70% ethanol at 4°C overnight. Following a wash with 10% formamide dissolved in 2× saline sodium citrate buffer, the cells were hybridized to probe sets in 60 μl of hybridization buffer containing 2× saline sodium citrate, 10% dextran sulfate, 10% formamide, and each probe set (table S4). Hybridization was performed for 4 hours at 37°C in a moist chamber. The coverslips were washed with 10% formamide in 2× saline sodium citrate solution and then with 10% formamide in 2× saline sodium citrate solution with Hoechst 33342 (1:1000). Hybridized cells were mounted in catalase/glucose oxidase containing mounting media [0.4% glucose in 10 mM tris, 2× saline sodium citrate, glucose oxidase (37 μg/ml), and 1/100 catalase (Sigma-Aldrich, C3155)]. Images were acquired using a Nikon Ti-2 microscope with a CSU-W1 confocal unit, a 100× Nikon oil-immersion objective of 1.49 numerical aperture (NA), and an iXon Ultra EMCCD camera (Andor, Belfast, UK), with laser illumination at 405, 561, and 637 nm, and were analyzed using NIS-elements software (version 5.11.01, Nikon, Tokyo, Japan); 101 z planes per site spanning 15 μm were acquired. Images were filtered with a one-pixel-diameter three-dimensional median filter and subjected to background subtraction via a rolling ball radius of 5 pixels, using FIJI software. Detection and counting of smFISH signals were performed using FISH-quant software version 3 (https://bitbucket.org/muellerflorian/fish_quant/src/master/). FISH-quant quantifies the number of mRNAs in the cell nucleus and cytoplasm. Mixtures of mNeonGreen and iRFP670 probes conjugated with CAL Fluor Red 590 and Quasar 670 were obtained from Biosearch Inc. (Novato, CA) and used at 0.25 μM. Probe sequences are shown in table S4. Intrinsic noise was calculated as described in the “Estimation of the kinetic properties of transcriptional bursting using transcript-level count data” section. Because smFISH has almost the same average value, correction between alleles was not carried out. The count normalized log ratios of intrinsic noise (normalized intrinsic noise) were calculated as the residuals of the regression line (fig. S1I). Normalization by gene length had not been applied for the smFISH data.
Flow cytometry analysis for calculation of intrinsic noise
On the day before flow cytometry, cells were treated with 25 μM BV. Cells that became 80% confluent were washed with PBS, trypsinized, inactivated with FluoroBrite DMEM (Thermo Fisher Scientific) containing 10% FBS, and centrifuged to collect the cells. Cells were suspended in PBS to be 1 × 106 to 5 × 106 cells/ml. Fluorescence data of side scatter (SSC), forward scatter (FSC), GFP, and iRFP were obtained with BD FACSAria III. Cells were gated on the basis of FSC and SSC using a linear scale to gate out cellular debris. Among GFP and iRFP data, extreme values indicating 20 * interquartile range or more were excluded from analysis. The mean value of the negative control data of WT mESC was subtracted from the data to be analyzed, and the data that fell below zero were excluded. We confirmed that the mean number of GFP and iRFP mRNAs in the KI cell lines are almost exactly matched that obtained in the smFISH analysis, suggesting that the expression levels of GFP and iRFP proteins are also similar. Therefore, we applied a correction using the following equations so that the mean fluorescence intensity between GFP and iRFP was consistent
Here, the ith element of vectors GFP and iRFP contains the fluorescence intensities of GFP and iRFP, respectively, of the ith cell in the sample. GFPn and iRFPn represent mean normalized GFP and iRFP, respectively. Then, intrinsic noise is calculated as described in the “Estimation of the kinetic properties of transcriptional bursting using transcript-level count data” section. The relationship between mean fluorescence intensities and intrinsic noise was plotted (fig. S1J). The fluorescence intensity normalized log ratios of intrinsic noise (normalized intrinsic noise) were calculated as the residuals of the regression line (fig. S1J).
Immunofluorescence
On the day before immunostaining, Trim28, Dnmt3l, Klf4, Peg3, Npm1, Dnmt3b, Nanog, Rad21, and Hdac1 KI cell lines at ~70% confluence were treated with 25 μM BV. After 24 hours, 1 × 105 cells were plated onto the eight-well Lab-Tek II chambered coverglass (Thermo Fisher Scientific) coated with LN511. For immunostaining of C57BL/6J WT mESCs conditioned to Std/LN511, 2i/LN511, and PD-MK/LN511 conditions, cells were plated 1 × 105 onto an eight-well Lab-Tek II chambered coverglass coated with LN511. After 1 hour, cells were washed once with PBS and fixed with 4% paraformaldehyde for 10 min at room temperature. Fixed cells were washed with BBS buffer [50 mM N,N-bis(2-hydroxyethyl)-2-aminoethanesulfonic acid (BES), 280 mM NaCl, 1.5 mM Na2HPO4·2H2O, and 1 mM CaCl2] two times and blocked for 30 min in BBT-BSA buffer [BBS with 0.5% bovine serum albumin (BSA), 0.1% Triton, and 1 mM CaCl2] at room temperature. Cells with primary antibodies were incubated overnight at 4°C at the following dilutions: anti-TRIM28 (1:500; GTX102227, GeneTex, RRID:AB_2037323), anti-DNMT3L (1:250; ab194094, Abcam, Cambridge, MA, RRID:AB_2783649), anti-KLF4 (1:250; ab151733, Abcam, RRID:AB_2721027), anti-PEG3 (1:500; BS-1870R, Bioss Antibodies, RRID:AB_10855800), anti-NPM1 (1:100; A302-402A, Bethyl Laboratories Inc., RRID:AB_1907285), anti-DNMT3B (1:500; 39207, Active Motif, RRID:AB_2783650), anti-NANOG (1:500; 14-5761-80, eBioscience, RRID:AB_763613), anti-RAD21 (1:500; GTX106012, GeneTex, RRID:AB_763613), anti-HDAC1 (1:500; GTX100513, GeneTex, RRID:AB_1240929), anti–OCT-4A (1:400; 2840, Cell Signaling Technology, RRID:AB_2167691), and anti-SSEA1 (1:1000; 4744, Cell Signaling Technology, RRID:AB_1264258). Cells were washed and blocked in BBT-BSA. Then, for KI cell lines, cells were incubated with Alexa Fluor 594–conjugated secondary antibodies (1:500; Life Technologies). For C57BL/6J WT mESCs, cells were incubated with Alexa Fluor 488 goat anti-mouse immunoglobulin G (IgG), Alexa Fluor 594 goat anti-rabbit IgG, and Alexa Fluor 647 goat anti-rat IgG secondary antibodies (1:500; Life Technologies). Images were acquired using a Nikon Ti-2 microscope with a CSU-W1 confocal unit, a 100× Nikon oil-immersion objective of 1.49 NA, and an iXon Ultra EMCCD camera (Andor, Belfast, UK).
Suz12 K/O
Dnmt3l, Dnmt3b, Peg3, and Ctcf KI cell lines conditioned to the gelatin/2i condition were trypsinized and plated onto a 24-well plate at 5 × 105 cells per 500 μl each. One hour later, for Suz12 K/O, 330 ng each of lentiCRISPRv2-sgSuz12_1, lentiCRISPRv2-sgSuz12_2, and lentiCRISPRv2-sgSuz12_3 and 300 ng of pCAG-mTagBFP2 (Addgene, plasmid #122373) plasmids or, for control, 1000 ng of lentiCRISPRv2_sgMS2_1 and 300 ng of pCAG-mTagBFP2 (Addgene, plasmid #122373) plasmids were transfected using Lipofectamine 3000 into each cell line. Two days later, blue fluorescent protein (BFP)–positive cells were sorted by FACS and plated onto a 6-cm dish. After 1 week, we picked up eight colonies for Suz12 K/O and four colonies for control for downstream analysis. We checked the expression of PRC2-related proteins by Western blotting (see below). Then, cells were conditioned to LN511/Std medium for at least 2 weeks. As described above, flow cytometry analysis was performed to calculate normalized intrinsic noise, burst size, and burst frequency.
Western blotting
Cells are washed twice with PBS, trypsinized, and collected by centrifugation. Cells were counted and then washed twice with PBS. Last, cells were lysed in the lysis buffer [0.5% Triton X-100, 150 mM NaCl, and 20 mM tris-HCl (pH 7.5)] to obtain 1 × 106 cells per 100 μl. Then, the lysates were incubated at 95°C for 5 min and filtered by QIAshredder homogenizer (Qiagen). The extracted proteins were analyzed by 5 to 20% gradient SDS–polyacrylamide gel electrophoresis and transferred onto Immobilon Transfer Membranes (Millipore, Billerica, MA, USA) for immunoblotting analyses. The primary antibodies used were anti-SUZ12 (1:1000; 3737, Cell Signaling Technology, RRID:AB_2196850), anti-EZH2 (1:1000; 5246, Cell Signaling Technology, RRID:AB_10694683), anti–histone H3K27me3 (1:1000; 39155, Active Motif, RRID:AB_2561020), anti-GAPDH (1:5000; 5174, Cell Signaling Technology, RRID:AB_10622025), anti–phospho-MEK1/2 (Ser217/Ser221; 1:1000; 9154, Cell Signaling Technology, RRID:AB_2138017), anti-MEK1/2 (1:1000; 8727, Cell Signaling Technology, RRID:AB_10829473), anti-p44/42 MAPK (Erk1/2; 1:1000; 4695, Cell Signaling Technology, RRID:AB_390779), anti–phospho-p44/42 MAPK (Erk1/2; Thr202/Tyr204; 1:2000; 4370, Cell Signaling Technology, RRID:AB_2315112), anti–phospho-4E-BP1 (Thr37/Thr46; 1:1000; 2855, Cell Signaling Technology, RRID:AB_560835), anti–phospho-Akt (Ser473; 1:1000; 4060, Cell Signaling Technology, RRID:AB_2315049), anti–phospho-Akt (Thr308; 1:1000; 13038, Cell Signaling Technology, RRID:AB_2629447), anti-Akt (pan; 1:1000; 4691, Cell Signaling Technology, RRID:AB_915783), anti–c-Myc (1:1000; ab32072, Abcam, RRID:AB_731658), anti-FoxO1 (1:1000; 14952, Cell Signaling Technology, RRID:AB_2722487), anti-FOXO3A (1:2500; ab12162, Abcam, RRID:AB_298893), anti-Nanog (1:500; 14-5761-80, eBioscience, RRID:AB_763613), anti–OCT-4A (1:500; 2840, Cell Signaling Technology, RRID:AB_2167691), and anti-SOX2 (1:1000; ab97959, Abcam, RRID:AB_2341193).
Infection of CRISPR lentivirus library
Nanog, Trim28, and Dnmt3L KI cells were transduced with the Mouse CRISPR K/O Pooled Library (GeCKO v2; Addgene, #1000000052) (29) via spinfection as previously described. We used only Mouse library A gRNA. Briefly, 3 × 106 cells per well (a total of 1.2 × 107 cells) were plated into an LN511-coated 12-well plate in the Std media supplemented with polybrene (8 μg/ml; Sigma-Aldrich). Each well received a virus amount equal to a multiplicity of infection (MOI) of 0.3. The 12-well plate was centrifuged at 1000g for 2 hours at 37°C. After the spin, media were aspirated and fresh media (without polybrene) were added. Cells were incubated overnight. Twenty-four hours after spinfection, cells were detached with trypsin and replated into four of LN511-coated 10-cm dishes with puromycin (0.5 μg/ml) for 3 days. Media were refreshed daily. At 6 days after transduction, cells were treated with 25 μM BV. After 24 hours, at least 1.75 × 105 cells showing GFP/iRFP expression ratio close to 1 were sorted by FACS and plated on 12-well plates (LN511/Std condition). Unsorted cells were passaged to 10-cm plates, 5 × 105 each. After the expansion of these sorted cells for 1 week, cells with GFP/iRFP expression ratio close to 1 were sorted again. These sorting and expansion procedures were repeated four times in total. At 3 days after the fourth sorting, 2 × 105 cells were collected and genomic DNA was extracted. PCR of the virally integrated sgRNA coding sequence was performed on genomic DNA at the equivalent of approximately 2000 cells per reaction in 48 parallel reactions using KOD FX Neo (TOYOBO, Japan). Amplification was carried out with 22 cycles. Primers are listed as follows: forward primer, AATGATACGGCGACCACCGAGATCTACACTCTTTC CCTACACGACGCTCTTCCGATCTNNNNNNNN(1–8-bp stagger) GTGGAAAGGACGAAACACCG; reverse primer, CAAGCAGAAGACGGCATACGAGATNNNNNNNN GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTTGTGGGCGATGTGCGCTCTG (8-bp index read barcode indicated in italics). PCR products from all 48 reactions were pooled, purified using a PCR purification kit (Qiagen, Hilden, Germany), and gel-extracted using the Gel Extraction Kit (Qiagen, Hilden, Germany). The resulting libraries were deep-sequenced on Illumina HiSeq platform with a total coverage of >8 million reads passing filter per library.
Cell cycle analysis
Cells (4 × 105) were seeded on LN511-coated six-well plates. After overnight culture, the cells were incubated for 1 hour with 5-ethynyl-2-deoxyuridine (EdU) diluted to 10 μM in the indicated embryonic stem (ES) cell media. All samples were processed according to the manufacturer’s instructions (Click-iT Plus EdU Alexa Fluor 647 Flow Cytometry Assay Kit, catalog no. C10634, Thermo Fisher Scientific). EdU incorporation was detected by Click-iT chemistry with an azide-modified Alexa Fluor 647. Cells were resuspended in EdU permeabilization/wash reagent and incubated for 30 min with Vybrant DyeCycle Violet (Thermo Fisher Scientific). Flow cytometry was performed on FACSAria III (BD Biosciences) and analyzed with Cytobank (www.cytobank.org; Cytobank Inc., Santa Clara, CA).
Analysis of apoptosis
Annexin V staining was performed using Annexin V Apoptosis Detection Kit APC (catalog no. 88-8007-72, Thermo Fisher Scientific) as described in the manufacturer’s manual. Briefly, cells were trypsinized and centrifuged, and then the supernatant was removed. The remaining cells were resuspended in PBS and counted. Cells were washed once with PBS and then resuspended in 1× Annexin V binding buffer at 1 × 106 to 5 × 106 cells/ml. Pellets were resuspended in 100 μl of Annexin V buffer to which 5 μl of fluorochrome-conjugated Annexin V was added. Cells were incubated in the dark at room temperature for 15 min, washed in 1× Binding Buffer, and resuspended in 200 μl of 1× Binding Buffer. PI Staining Solution (5 μl) was added and immediately analyzed by flow cytometry.
Bulk RNA-seq
RNA was extracted from either Std/LN511, 2i/LN511, or PD-MK/LN511 conditioned cells at 70% confluency in a well of a six-well plate using RNeasy Plus Mini (Qiagen). Three biological replicates were prepared. Bulk RNA-seq was performed by CEL-Seq2 method (39), with total RNA amounts used in the range of 30 to 60 ng. The resulting reads were aligned to the reference genome (GRCm38) using HISAT (v.2.1.0; https://ccb.jhu.edu/software/hisat/index.shtml). The software HTSeq (version 0.6.1; https://htseq.readthedocs.io/en/release_0.11.1) was used in calculating gene-wise unique molecular identifier (UMI) counts that were converted into transcript counts after collision probability correction. The counts were input to the R library DESeq2 (version 1.14.1; https://bioconductor.org/packages/release/bioc/html/DESeq2.html) for differentially expressed (DE) analysis. The genes that increased significantly (adjusted P < 0.05) in the PD-MK condition against the 2i condition were subjected to GO analysis using an R package, clusterProfiler (v3.9.2) (https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html).
RNA degradation rate determination using 4sU pulse labeling
C57BL/6J WT mESCs conditioned to LN511/Std or LN511/PD-MK conditions were treated with 400 μM 4-thiouridine (4sU) for 20 min. Then, RNA was extracted from more than 1 × 107 cells using the RNeasy Plus Mini Kit (Qiagen, Valencia, CA). Three biological replicates were prepared for each condition. We synthesized mRuby2 RNA for spike-in RNA by standard PCR, in vitro transcription using the T7 High Yield RNA Synthesis Kit (catalog no. E2040, New England Biolabs), and purification with RNeasy Plus Mini Kit (Qiagen, Valencia, CA). Biotinylation of 4sU-labeled RNA was carried out in RNase-free water with 10 mM tris-HCl (pH 7.4), 1 mM EDTA, and Biotin-HPDP (0.2 mg/ml; catalog no. 341-09101, Dojindo) at a final RNA concentration of 1 μg/μl extracted RNA (a total of 125 μg) with spike-in RNA (125 ng/μl) for 3 hours in the dark at room temperature. To purify biotinylated RNA from an excess of Biotin-HPDP, a phenol:chloroform:isoamylalcohol (v/v = 25:24:1; Nacalai Tesque, Kyoto, Japan) extraction was performed. Phenol:chloroform:isoamylalcohol was added to the reaction mixture in a 1:1 ratio, followed by vigorous mixing, and centrifuged at 20,000g for 5 min at 4°C. The RNA containing aqueous phase was removed and transferred to a fresh, RNase-free tube. To precipitate RNA, 1/10 reaction volume of 5 M NaCl and an equal volume of 2-propanol were added and incubated for 10 min at room temperature. Precipitated RNA was collected through centrifugation at 20,000g for 30 min at 4°C. The pellet was washed with an equal volume of 75% ethanol and precipitated again at 20,000g for 20 min. Last, RNA was reconstituted in 25 to 50 μl of RNase-free water. For removing of biotinylated 4sU-RNA, streptavidin-coated magnetic beads (Dynabeads MyOne Streptavidin C1 beads, Thermo Fisher Scientific) were used according to the manufacturer’s manual. To avoid unfavorable secondary RNA structures that potentially impair the binding to the beads, the RNA was first denatured at 65°C for 10 min followed by rapid cooling on ice for 5 min. Dynabeads magnetic beads (200 μl per sample) were transferred to a new tube. An equal volume of 1× B&W [5 mM tris-HCl (pH 7.5), 0.5 mM EDTA, 1 M NaCl] was added to the tube and mixed well. The tube was placed on a magnet for 1 min, and the supernatant was discarded. The tube was removed from the magnet. The washed magnetic beads were resuspended in 200 μl of 1× B&W. The bead washing step was repeated for a total of three times. The beads were washed twice in 200 μl of solution A [diethyl pyrocarbonate (DEPC)–treated 0.1 M NaOH and DEPC-treated 0.05 M NaCl] for 2 min. Then, the beads were washed once in 200 μl of solution B (DEPC-treated 0.1 M NaCl). Washed beads were resuspended in 400 μl of 2× B&W Buffer. An equal volume of 20 μg of biotinylated RNA in distilled water was added. The mixture was incubated for 15 min at room temperature with gentle rotation. The biotinylated RNA-coated beads were separated with a magnet for 2 to 3 min. Unbound (unbiotinylated) RNA from the flow-through was recovered using the RNeasy MinElute Kit (Qiagen) and reconstituted in 25 μl of RNase-free water. cDNA was synthesized with the ReverTra Ace qPCR RT Kit (catalog no. FSQ-101, TOYOBO, Japan) from both total RNA and unbound (unbiotinylated) RNA. The relative amount of existing RNA (unbiotinylated RNA)/total RNA was quantified by quantitative PCR (qPCR) with THUNDERBIRD SYBR qPCR Mix (catalog no. QPS-201, TOYOBO). cDNAs were derived from total and unbound RNA, and primers used are listed in table S4.
Generation of chimeras
C57BL/6NCr ES cells derived from C57BL/6NCr (Japan SLC, Hamamatsu, Japan) were cultured in PD-MK medium on a gelatin-coated dish for 2 weeks. The day before injection, the culture medium was changed to Std medium. mESCs were microinjected into eight-cell–stage embryos from ICR strain (CLEA Japan, Tokyo, Japan). The injected embryos were then transferred to the uterine horns of appropriately timed pseudopregnant ICR mice. Chimeras were determined by the presence of black eyes at birth and by coat color around 10 days after birth.
Quantification of gene and allelic expression level
For each scRamDA-seq library, the FASTQ files of sequencing data with 10 pg of RNA were combined. Fastq-mcf (version 1.04.807) (https://github.com/ExpressionAnalysis/ea-utils/blob/wiki/FastqMcf.md) was used to trim adapter sequences and generate read lengths of 50 nucleotides with the parameters “-L 42 -l 42 -k 4 -q 30 -S.” The reads were mapped to the mouse genome (mm10) using HISAT2 (version 2.0.4) (https://ccb.jhu.edu/software/hisat2/index.shtml) with default parameters. We confirmed that there was no large difference in the number of reads and mapping rates across the cell samples (table S4). We removed 27 abnormal samples showing abnormal gene body coverage of sequencing reads by human curation. Using the remaining data derived from 447 cells, allelic gene expressions were quantified using EMASE (version 0.10.11) with default parameters (https://github.com/churchill-lab/emase). 129 and CAST genomes by incorporating single-nucleotide polymorphisms and indels into reference genome and transcriptome were created by Seqnature (https://github.com/jaxcs/Seqnature). Bowtie (version 1.1.2) (http://bowtie-bio.sourceforge.net/index.shtml) was used to align scRamDA-seq reads against the diploid transcriptome with the default parameters.
Estimation of the kinetic properties of transcriptional bursting using transcript-level count data
To calculate intrinsic noise using the equations indicated below, we used three normalization steps. First, the global allelic bias in expression level was subjected to the Trimmed Means of M values (TMM) normalization method implemented in the R package “edgeR” (https://bioconductor.org/packages/release/bioc/html/edgeR.html; see the next section). This normalization removes global allelic bias in expression level as well as differences in sequencing depth in each cell. The total noise () for each transcript was calculated using the following equation (6)
Here, the ith element of vectors a1 and a2 contains the read counts of transcript from allele 1 or allele 2, respectively, of the ith cell in the sample. Global normalization did not substantially change the shape of the read count–total noise distribution (fig. S1B). Second, the read counts were subjected to quantile normalization between alleles at each transcript by the “normalize.quantiles.robust” method using the Bioconductor “preprocessCore” package (version 1.38.1; https://bioconductor.org/packages/release/bioc/html/preprocessCore.html). Third, we performed a correction using the following equations so that the mean read counts among the alleles were consistent
Here, the ith element of vectors ag1 and ag2 contains the globally and allelically normalized read counts of transcript from allele 1 or allele 2, respectively, of the ith cell in the sample. agn1 and agn2 represent the mean normalized ag1 and ag2, respectively. Angled brackets denote means over the cell population. From these read count matrices, the intrinsic noise () for each transcript was calculated using the following equation (6, 7)
Transcripts showing a relatively large difference in expression level between alleles before correction (the average prenormalized expression level between alleles was >100 read counts) were excluded from subsequent analysis. A large fraction of transcripts (25,481 transcripts) showed intrinsic noise below Poisson noise (fig. S1C). Theoretically, intrinsic noise cannot be below Poisson noise (17). These transcripts are extremely similar in expression between the alleles, resulting in very low intrinsic noise values (fig. S1D). The expression level of each allele was originally calculated using the polymorphisms contained in the sequencing reads (see the previous section). However, if the sequencing reads for a particular transcript does not contain polymorphisms, the expression levels for each allele cannot be accurately calculated, and the expression levels for each allele are considered equal. Thus, transcripts with intrinsic noise below Poisson noise were excluded from the downstream analysis. A decrease in intrinsic noise was observed as the expression level increased, as theoretically expected (fig. S1C). To investigate the factors involved in the intrinsic noise and bursting properties independent of expression level, the count normalized log ratios of intrinsic noise were calculated as the residuals of a regression line that was calculated using a dataset with more than 1 mean read count (fig. S1E). In addition, a global correlation was found between the length of the transcript and the count normalized intrinsic noise (fig. S1F). Thus, the count and transcript length normalized log ratios of intrinsic noise were calculated as the residuals of a regression line (fig. S1, F and G). We call these read count and transcript length normalized intrinsic noise simply normalized intrinsic noise. For transcripts with low expression levels, it is difficult to distinguish whether their heterogeneity in expression level is due to technical or biological noise. Therefore, transcripts with read counts less than 20 were excluded from the downstream analysis (remaining 5992 transcripts).
Intrinsic noise is a function of the mRNA degradation rate (9, 12, 14). The mRNA degradation rate in mESC has been genome-wide analyzed (23). Genes whose degradation rate is unknown were provisionally assigned a median value. The burst size (b) and burst frequency (f) of each transcript can be estimated by the mRNA degradation rate (γm), intrinsic noise (), and mean number of mRNA (μ) according to the following equations (9, 12, 14)
Previous studies have reported the estimation of the burst size and burst frequency for each allele using a Poisson-Beta hierarchical model with scRNA-seq data of hybrid cells (11, 40). To evaluate the validity of the parameters derived from the abovementioned equations, we used our hybrid mESC scRNA-seq data and the SCALE software (version 1.3.0) that enables the estimation of the burst frequency and burst size per allele using a Poisson-Beta hierarchical model (40). Because the SCALE software always sets the RNA degradation rate to 1, the resulting parameters can be considered as RNA degradation rate–normalized parameters. Therefore, for comparison, the burst frequency calculated from intrinsic noise was divided by the RNA degradation rate to obtain RNA degradation rate–normalized burst frequency (see above formula). The SCALE- and intrinsic noise–based parameters were well correlated (R > 0.8; fig. S2, F to K), suggesting that the burst size and burst frequency calculated using intrinsic noise are valid. In hybrid cells, as the expression levels of alleles can vary depending on the polymorphisms present in the genome (41), a three-step normalization was used before the calculation as mentioned above. To determine whether the intrinsic noise measured by scRNA-seq of hybrid mESCs indicates true gene expression noise, we integrated GFP and iRFP reporter genes separately into both alleles of 25 genes in an inbred mESC line (KI mESC lines; Fig. 1H and fig. S3). Using these cell lines, the mean expression levels and normalized intrinsic noise of the 25 genes were measured by smFISH, resulting in a significant correlation with scRNA-seq–based measurements (Fig. 1, I and J; table S3). These validation experiments also confirmed the conclusions derived from the intrinsic noise calculation.
Comparison of tools for global normalization
As noted above, TMM normalization, commonly used in bulk RNA-seq analysis, was used to normalize the global allelic bias in expression levels of scRNA-seq. TMM normalization is based on the construction of the size factor, which represents the ratio at which each cell is normalized by a reference cell constructed by some kind of averaging across all other cells per cell. scRNA-seq generally has fewer reads per sample and is prone to generate dropout events, where expressed transcripts stochastically appear to have zero reads due to technical limitations. Therefore, the size factors of TMM may be inappropriately large or equal to zero when applied to scRNA-seq. Hence, normalization methods optimized for scRNA-seq have been developed (42). To validate our use of TMM normalization on scRNA-seq data, we normalized scRNA-seq data using scran (http://bioconductor.org/packages/release/bioc/html/scran.html; version 1.14.5), a normalization tool optimized for scRNA-seq data. We then used the scran-normalized data to calculate intrinsic noise and compare the results with those previously obtained through TMM normalization. Among the transcripts with an average expression level of more than 20, the average expression (R = 0.97), intrinsic noise (R = 0.95), and normalized intrinsic noise (R = 0.94) derived from TMM-normalized dataset were highly correlated with those from scran-normalized dataset. TMM, scran, and other scRNA-seq–optimized normalization methods have been reported to not show large differences in performance when there are relatively few DE genes among samples (42). In this case, the target dataset for normalization is derived from cells of the same cell type in the G1 phase; therefore, the difference in expression levels between samples is considered to be relatively small. Hence, we consider the use of TMM normalization appropriate to calculate intrinsic noise in this case.
Estimation of the kinetic properties of transcriptional bursting using gene-level count data
It is thought that the RNA detected by smFISH is not a specific transcript and contains multiple transcript variants. Therefore, intrinsic noise data calculated using transcript-level count data could not be compared to those from smFISH data. To solve this problem, scRamDA-seq data for each transcript were summed up for each gene, and intrinsic noise was recalculated. For this purpose, global allelic bias in expression level was first normalized as described above. Then, data of each transcript were summed up for each gene at this time point. Next, the read counts were normalized between alleles at each gene by the normalize.quantiles.robust method using the Bioconductor preprocessCore package. Furthermore, correction was made so that the mean read counts among the alleles were consistent as described above. From these read count matrices, the intrinsic noise for each gene can be calculated as described above. Data with intrinsic noise below Poisson noise were excluded from the downstream analysis. To investigate the factors involved in the intrinsic noise and bursting properties independent of expression level, the count normalized log ratios of intrinsic noise were calculated as the residual of a regression line that is calculated using a dataset with more than 1 mean read counts. Then, the count and gene length normalized log ratios of intrinsic noise were calculated as the residual of a regression line. We call these read count and gene length normalized intrinsic noise simply normalized intrinsic noise. The burst size (b) and burst frequency (f) of each gene can be estimated as described above.
TATA box identification
We used FindM (https://ccg.epfl.ch/ssa/findm.php) to determine whether a sequence of 50 bp upstream from the TSS of transcripts, with an average read count of more than 20 in our scRNA-seq, contained a TATA box.
Correlation analysis
We used bioinformatics tools freely available on Galaxy Project platform (https://galaxyproject.org/). Various ChIP-seq data were obtained from the bank listed in table S4. Then, we mapped them to mm10 genome with Bowtie (Galaxy version 1.1.2) and converted them to bam file with SAM-to-BAM tool (Galaxy version 2.1). Reads per million mapped reads (RPM) data from −1000 to +100 from TSS and gene body of individual transcripts were analyzed by ngs.plot (version 2.61; https://github.com/shenlab-sinai/ngsplot). Of these, extreme outliers (100 times the average value) were excluded from analysis. In addition, we also considered the replication timing, promoter proximal pausing of RNA Pol II, considered to be related to the characteristics of transcriptional bursting. To determine the pausing index of Pol II, GRO-seq (global run-on sequencing) data in mESCs were used [Gene Expression Omnibus (GEO) ID: GSE48895]. We obtained the fastq file from the bank [ENA (European Nucleotide Archive) accession number (fastq.gz): PRJNA 21123]. As described previously, after removing the adapter sequence with the Cutadapt tool (version 2.4; https://cutadapt.readthedocs.io/en/stable/index.html), reads were mapped to mm10 genome with Bowtie (Galaxy version 1.1.2) and converted to bam file with SAM-to-BAM tool (Galaxy version 2.1). These data were analyzed with the pausingIndex function of the groHMM tool (size, 500; up, 250; down, 250; http://bioconductor.org/packages/release/bioc/html/groHMM.html; version 1.10.0). Data of replication timing of mESCs were obtained from the following source (GEO ID: GSM450272). Spearman’s rank correlation coefficient between either normalized intrinsic noise, burst size, or burst frequency and either promoter or gene body localization degree (RPM) of various factors at the upper and lower 5% transcripts of normalized intrinsic noise, burst size, and burst frequency was calculated. Next, the promoter-interacting distal enhancers were considered. Enhancers are believed to regulate gene expression by physical interaction with the promoter (10). Candidate distal cis-regulatory elements that interact with specific genes have been identified using capture Hi-C in mESCs (https://genomebiology.biomedcentral.com/articles/10.1186/s13059-015-0727-9). These data contain regions that interact with promoters and may include insulators and other elements in addition to enhancers. To identify the enhancers from regions that interact with the promoter of a particular gene, we manually screened for enhancers with relatively high RPM of H3K27ac ChIP-seq (RPM > 1.5; fig. S2L). Using these data, the RPM of other ChIP-seq data was calculated in the same manner as mentioned above in the candidate enhancers. Extreme outliers (with values 100 times the average) were excluded from the analysis. These enhancer data do not correspond to each transcript; instead, they rather correspond to each gene. Thus, the intrinsic noise, burst size, and burst frequency calculated using the gene-level count data were applied at this stage. Spearman’s rank correlation coefficient of normalized intrinsic noise, burst size, and burst frequency with localization degree (RPM) of various factors in the upper and lower 5% enhancer of normalized intrinsic noise, burst size, and burst frequency of corresponding genes were calculated.
Orthogonal partial least squares discriminant analysis
First, we classified promoter- and gene body–associated features of high (either intrinsic noise, burst size, or burst frequency) transcripts into 10 clusters. Then, to identify the most contributing features for characterization of a cluster of high transcripts (either intrinsic noise, burst size, or burst frequency) against low transcripts (either intrinsic noise, burst size, or burst frequency), we performed OPLS-DA modeling using ropls R package with 500 random permutations (version 1.8.0; https://bioconductor.org/packages/release/bioc/html/ropls.html). One predictive component and one orthogonal component were used. To find the most influential variables for separation of high groups (either intrinsic noise, burst size, or burst frequency) against low groups (either intrinsic noise, burst size or burst frequency), an S-plot with loadings of each variable on the x axis and correlation of scores to modeled x matrix [p(corr)[1]=Corr(t1,X),t1 = scores in the first predictive component] on the y axis was constructed. Three each of the top and bottom variables with absolute value of loadings were selected.
NGS (next generation sequencing) and analysis of CRISPR library screening
After primer trimming with the Cutadapt software (https://cutadapt.readthedocs.io/en/stable/guide.html), read counts were generated and statistical analysis was performed using MAGeCK (v0.5.5) (https://sourceforge.net/p/mageck/wiki/Home/). DE scores were calculated from the gene-level significance returned by MAGeCK with the following formula: DE score = log10(gene-level depletion P value) – log10(gene-level enrichment P value). Genes with allelically normalized mean read count less than 10 from scRamDA-seq analysis were excluded from the downstream analysis. Then, genes were ranked by DE score. Subsequently, the top and bottom 100 genes were subjected to KEGG pathway enrichment analysis using an R package, clusterProfiler (v3.9.2; https://github.com/GuangchuangYu/clusterProfiler).
RNA degradation rate quantification
mRNA half-life can be determined using the following equation (37)
where t, existing, new, and total indicate the 4sU treatment time and amounts of existing, newly synthesized, and total RNA, respectively. Here, t is 1/3; new/total is 1 − (existing/total)
| (1) |
All samples contained spike-in RNA. Because they are unlabeled by 4sU and biotin, they are not trapped by streptavidin beads, except for nonspecific adsorption and technical loss. Therefore, by normalization with the amount of spike-in RNA in total and unbound (existing), the true ratio of total and unbound transcript can be obtained using the following equation
Unbound (target)/total (target) and unbound (spike-in)/total (spike-in) can be obtained by qPCR. Although most of the genes showed Norm.Ratio(existing/total) of more than 1, this is theoretically impossible (fig. S6B). It is possible that reverse transcription efficiency is drastically decreased by biotinylation of RNA. Here, we assumed that the presence of biotinylated RNA during reverse transcription may trap reverse transcriptase and that the efficiency of reverse transcription is further reduced globally. We assume that the global suppression effect of reverse transcriptase trapping is Ig (global inhibitory effect). Moreover, the reverse transcription inhibitory effect of biotinylated RNA itself is defined as Is. Also, we defined N, E, T, and Reff as the amount of biotinylated (newly synthesized) RNA, the amount of existing unbiotinylated RNA, the amount of reverse transcriptase, and reverse transcription efficiency of reverse transcriptase, respectively. From these definitions, the cDNA amount derived from total and existing RNA can be determined by the following equations
| (2) |
Next, a known value is introduced into Eq. 1 to solve coefficients. The half-life of Nanog mRNA under Std conditions has been reported to be approximately 4.7 hours (20). Therefore, the ideal ratio of existing/total Nanog mRNA amount is approximately 0.95203. In this case, the ideal relationship between newly synthesized and existing RNA is as follows
The mean ratio of existing/total Nanog cDNA revealed by qPCR was 3.436867. Therefore, the relationship between Is and Ig is as follows from Eq. 2
To determine the appropriate value of Is, several values were assigned to Is, and mRNA half-lives in the Std condition were compared with the previously reported mRNA half-lives (fig. S6C) (23). We found that the scaling of mRNA half-lives in the Std condition and that of previously reported mRNA half-lives were quite similar when Is is 0.1 and Ig is 0.289. Using Eqs. 1 and 2, the half-lives of mRNA can be obtained on the basis of the data using the value obtained from qPCR (fig. S6D). No significant difference in mRNA half-life was observed between Std and PD-MK conditions for the genes examined.
Supplementary Material
Acknowledgments
We would like to thank T. Miyamoto and T. Fukaya for helpful discussions and Y. Ochiai for technical assistance. We would also like to thank J. Gribnau for providing the hybrid mouse ES cell line F1-21.6 (129Sv-Cast/EiJ, female). We thank A. Matsushima and M. Ishii for their assistance with the infrastructure for the data analysis. Funding: This work was supported by the JST PRESTO program (Japan) to H.O. (JPMJPR15F2) and JSPS KAKENHI (Japan) to H.O. (JP18H05531 and JP19K06612), N.S. (JP18H05531), and H.K. and Y.O. (JP18H05527) and partially by JST CREST (Japan) to I.N. (JPMJCR16G3) and H.K. and Y.O. (JPMJCR16G1). The sequence operations of RamDA-seq using HiSeq2500 were supported by the Platform Project for Supporting Drug Discovery and Life Science Research (Platform for Drug Discovery, Informatics, and Structural Life Science) from the Japan Agency for Medical Research and Development (AMED). Author contributions: Conceptualization: H.O.; methodology: H.O., T.H., M.U., M.Y., and I.N.; investigation: H.O., T.H., M.U., M.Y., I.N., A.H., Y.O., Y.S., K.N., and T.O.; writing (original draft): H.O. and I.N.; writing (review and editing): H.O., I.N., N.S., T.Y., H.K., Y.O., and Z.L.; funding acquisition: H.O. and I.N.; supervision: H.O. and I.N. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The datasets supporting the conclusions of this article are available in the GEO (GEO: GSE132593; this SuperSeries is composed of the SubSeries listed as follows: GSE132589, GSE132590, GSE132591, and GSE132592). Source data generating figures are available at http://dx.doi.org/10.17632/5rchtsps3z.1. All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/25/eaaz6699/DC1
REFERENCES AND NOTES
- 1.Raj A., van Oudenaarden A., Nature, nurture, or chance: Stochastic gene expression and its consequences. Cell 135, 216–226 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fraser D., Kaern M., A chance at survival: Gene expression noise and phenotypic diversification strategies. Mol. Microbiol. 71, 1333–1340 (2009). [DOI] [PubMed] [Google Scholar]
- 3.Chalancon G., Ravarani C. N. J., Balaji S., Martinez-Arias A., Aravind L., Jothi R., Babu M. M., Interplay between gene expression noise and regulatory network architecture. Trends Genet. 28, 221–232 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wernet M. F., Mazzoni E. O., Çelik A., Duncan D. M., Duncan I., Desplan C., Stochastic spineless expression creates the retinal mosaic for colour vision. Nature 440, 174–180 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Buck L., Axel R., A novel multigene family may encode odorant receptors: A molecular basis for odor recognition. Cell 65, 175–187 (1991). [DOI] [PubMed] [Google Scholar]
- 6.Elowitz M. B., Levine A. J., Siggia E. D., Swain P. S., Stochastic gene expression in a single cell. Science 297, 1183–1186 (2002). [DOI] [PubMed] [Google Scholar]
- 7.Raser J. M., O'Shea E. K., Control of stochasticity in eukaryotic gene expression. Science 304, 1811–1814 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Golding I., Paulsson J., Zawilski S. M., Cox E. C., Real-time kinetics of gene activity in individual bacteria. Cell 123, 1025–1036 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Raj A., Peskin C. S., Tranchina D., Vargas D. Y., Tyagi S., Stochastic mRNA synthesis in mammalian cells. PLOS Biol. 4, e309 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fukaya T., Lim B., Levine M., Enhancer control of transcriptional bursting. Cell. 166, 358–368 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Larsson A. J. M., Johnsson P., Hagemann-Jensen M., Hartmanis L., Faridani O. R., Reinius B., Segerstolpe Å., Rivera C. M., Ren B., Sandberg R., Genomic encoding of transcriptional burst kinetics. Nature 565, 251–254 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.So L.-H., Ghosh A., Zong C., Sepúlveda L. A., Segev R., Golding I., General properties of transcriptional time series in Escherichia coli. Nat. Genet. 43, 554–560 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hornung G., Bar-Ziv R., Rosin D., Tokuriki N., Tawfik D. S., Oren M., Barkai N., Noise-mean relationship in mutated promoters. Genome Res. 22, 2409–2417 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dar R. D., Razooky B. S., Singh A., Trimeloni T. V., McCollum J. M., Cox C. D., Simpson M. L., Weinberger L. S., Transcriptional burst frequency and burst size are equally modulated across the human genome. Proc. Natl. Acad. Sci. U.S.A. 109, 17454–17459 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bahar Halpern K., Tanami S., Landen S., Chapal M., Szlak L., Hutzler A., Nizhberg A., Itzkovitz S., Bursty gene expression in the intact mammalian liver. Mol. Cell 58, 147–156 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Suter D. M., Molina N., Gatfield D., Schneider K., Schibler U., Naef F., Mammalian genes are transcribed with widely different bursting kinetics. Science 332, 472–474 (2011). [DOI] [PubMed] [Google Scholar]
- 17.Zoller B., Nicolas D., Molina N., Naef F., Structure of silent transcription intervals and noise characteristics of mammalian genes. Mol. Syst. Biol. 11, 823 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Newman J. R. S., Ghaemmaghami S., Ihmels J., Breslow D. K., Noble M., DeRisi J. L., Weissman J. S., Single-cell proteomic analysis of S. cerevisiae reveals the architecture of biological noise. Nature 441, 840–846 (2006). [DOI] [PubMed] [Google Scholar]
- 19.Simon C. S., Hadjantonakis A.-K., Schröter C., Making lineage decisions with biological noise: Lessons from the early mouse embryo. Wiley Interdiscip. Rev. Dev. Biol. 7, e319 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ochiai H., Sugawara T., Sakuma T., Yamamoto T., Stochastic promoter activation affects Nanog expression variability in mouse embryonic stem cells. Sci. Rep. 4, 7125 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shah S., Takei Y., Zhou W., Lubeck E., Yun J., Eng C.-H. L., Koulena N., Cronin C., Karp C., Liaw E. J., Amin M., Cai L., Dynamics and spatial genomics of the nascent transcriptome by intron seqFISH. Cell 174, 363–376.e16 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hayashi T., Ozaki H., Sasagawa Y., Umeda M., Danno H., Nikaido I., Single-cell full-length total RNA sequencing uncovers dynamics of recursive splicing and enhancer RNAs. Nat. Commun. 9, 619 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sharova L. V., Sharov A. A., Nedorezov T., Piao Y., Shaik N., Ko M. S. H., Database for mRNA half-life of 19 977 genes obtained by DNA microarray analysis of pluripotent and differentiating mouse embryonic stem cells. DNA Res. 16, 45–58 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bahar Halpern K., Caspi I., Lemze D., Levy M., Landen S., Elinav E., Ulitsky I., Itzkovitz S., Nuclear retention of mRNA in mammalian tissues. Cell Rep. 13, 2653–2662 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen F. X., Smith E. R., Shilatifard A., Born to run: Control of transcription elongation by RNA polymerase II. Nat. Rev. Mol. Cell Biol. 19, 464–478 (2018). [DOI] [PubMed] [Google Scholar]
- 26.Ying Q.-L., Wray J., Nichols J., Batlle-Morera L., Doble B., Woodgett J., Cohen P., Smith A., The ground state of embryonic stem cell self-renewal. Nature 453, 519–523 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pasini D., Bracken A. P., Hansen J. B., Capillo M., Helin K., The polycomb group protein Suz12 is required for embryonic stem cell differentiation. Mol. Cell. Biol. 27, 3769–3779 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cloarec O., Dumas M. E., Trygg J., Craig A., Barton R. H., Lindon J. C., Nicholson J. K., Holmes E., Evaluation of the orthogonal projection on latent structure model limitations caused by chemical shift variability and improved visualization of biomarker changes in 1H NMR spectroscopic metabonomic studies. Anal. Chem. 77, 517–526 (2005). [DOI] [PubMed] [Google Scholar]
- 29.Sanjana N. E., Shalem O., Zhang F., Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kanehisa M., Goto S., KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Manning B. D., Toker A., AKT/PKB signaling: Navigating the network. Cell 169, 381–405 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liang K., Smith E. R., Aoi Y., Stoltz K. L., Katagi H., Woodfin A. R., Rendleman E. J., Marshall S. A., Murray D. C., Wang L., Ozark P. A., Mishra R. K., Hashizume R., Schiltz G. E., Shilatifard A., Targeting processive transcription elongation via SEC disruption for MYC-induced cancer therapy. Cell 175, 766–779.e17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klein A. M., Mazutis L., Akartuna I., Tallapragada N., Veres A., Li V., Peshkin L., Weitz D. A., Kirschner M. W., Droplet barcoding for single-cell transcriptomics applied to embryonic stem cells. Cell 161, 1187–1201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Guo G., Pinello L., Han X., Lai S., Shen L., Lin T.-W., Zou K., Yuan G.-C., Orkin S. H., Serum-based culture conditions provoke gene expression variability in mouse embryonic stem cells as revealed by single-cell analysis. Cell Rep. 14, 956–965 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kolodziejczyk A. A., Kim J. K., Tsang J. C. H., Ilicic T., Henriksson J., Natarajan K. N., Tuck A. C., Gao X., Bühler M., Liu P., Marioni J. C., Teichmann S. A., Single cell RNA-sequencing of pluripotent states unlocks modular transcriptional variation. Cell Stem Cell 17, 471–485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Albayrak C., Jordi C. A., Zechner C., Lin J., Bichsel C. A., Khammash M., Tay S., Digital quantification of proteins and mRNA in single mammalian cells. Mol. Cell 61, 914–924 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Schwanhäusser B., Busse D., Li N., Dittmar G., Schuchhardt J., Wolf J., Chen W., Selbach M., Global quantification of mammalian gene expression control. Nature 473, 337–342 (2011). [DOI] [PubMed] [Google Scholar]
- 38.Jonkers I., Barakat T. S., Achame E. M., Monkhorst K., Kenter A., Rentmeester E., Grosveld F., Grootegoed J. A., Gribnau J., RNF12 is an X-encoded dose-dependent activator of X chromosome inactivation. Cell 139, 999–1011 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Hashimshony T., Senderovich N., Avital G., Klochendler A., de Leeuw Y., Anavy L., Gennert D., Li S., Livak K. J., Rozenblatt-Rosen O., Dor Y., Regev A., Yanai I., CEL-Seq2: Sensitive highly-multiplexed single-cell RNA-Seq. Genome Biol. 17, 77 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jiang Y., Zhang N. R., Li M., SCALE: Modeling allele-specific gene expression by single-cell RNA sequencing. Genome Biol. 18, 74 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu J., Carter A. C., Gendrel A.-V., Attia M., Loftus J., Greenleaf W. J., Tibshirani R., Heard E., Chang H. Y., Landscape of monoallelic DNA accessibility in mouse embryonic stem cells and neural progenitor cells. Nat. Genet. 49, 377–386 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vieth B., Parekh S., Ziegenhain C., Enard W., Hellmann I., A systematic evaluation of single cell RNA-seq analysis pipelines. Nat. Commun. 10, 4667 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/25/eaaz6699/DC1