

Abstract
Obesity is associated with increased risk of many types of cancer and can be induced by various high-fat diets (HFD) from different fat sources. It remains unknown whether fatty acid composition in different HFD influences obesity-associated tumor development. Here we report that consumption of either a cocoa butter or fish oil HFD induced similar obesity in mouse models. While obesity induced by the cocoa butter HFD was associated with accelerated mammary tumor growth, consumption of the fish oil HFD uncoupled obesity from increased mammary tumor growth and exhibited a decrease in pro-tumor macrophages. Compared to FA components in both HFD, n-3 FA rich in the fish oil HFD induced significant production of reactive oxygen species (ROS) and macrophage death. Moreover, A-FABP expression in the pro-tumor macrophages facilitated intracellular transportation of n-3 FA and oxidation of mitochondrial FA. A-FABP deficiency diminished n-3 FA-mediated ROS production and macrophage death in vitro and in vivo. Together, our results demonstrate a novel mechanism by which n-3 FA induce ROS-mediated pro-tumor macrophage death in an A-FABP dependent manner.
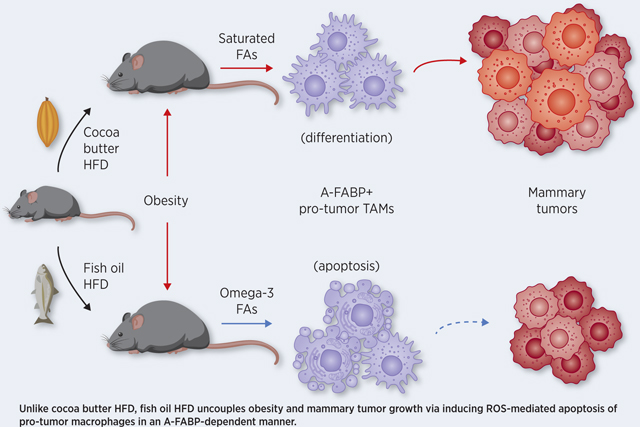
Graphical Abstract
Introduction
Obesity is associated with increased cancer risk and mortality (1;2). Specifically, recent epidemiologic studies confirmed that excess body fat significantly increases the risk of at least 13 types of cancer, including breast, colon, liver and ovary cancer (3). While obesity is usually defined as a body mass index (BMI) of 30 or more, BMI alone cannot predict cancer susceptibility in obese individuals (4;5).
Obesity occurs when excess energy intake is stored as different lipid species in adipose tissue. Thus, intake of different types of caloric components is critical in determining the composition of stored lipids and lipid-mediated responses in obesity. Recent studies have linked overconsumption of high-fat diets to the epidemic of obesity (6;7). Dietary fatty acids (FA) are either saturated (e.g. 16:0 palmitic acid, PA; 18:0 stearic acid, SA) or unsaturated (e.g. monounsaturated 18:1 oleic acid, OA; polyunsaturated 18:2 linoleic acid, LA). Polyunsaturated FA include n-6 (omega-6, e.g. LA) and n-3 (omega-3, e.g. docosapentaenoic acid, DPA; eicosapentaenoic acid, EPA) FA, depending on the double bond position counting from the methyl end of the carbon chain. Given the different FA components in various high-fat diets (HFD), it is of interest to determine whether and how FA composition in HFD influences obesity and obesity-associated immune regulation and tumor progression.
The family of fatty acid binding proteins (FABPs) consists of a group of intracellular protein members which bind and transport FA to different cellular compartments, thus regulating lipid metabolism and intracellular responses (8;9). Among FABP members, the role of A-FABP (adipose/macrophage FABP, also known as FABP4) has been studied extensively in obesity-associated diseases due to its characteristic expression profile in macrophages and adipocytes (10-13). Our recent studies demonstrated that A-FABP functions as a functional biomarker for pro-tumor macrophages (exhibiting CD11b+F4/80+Ly6C-MHCII-CD11c- phenotype) via induction of oncogenic IL-6/STAT3 signaling (14). Moreover, circulating A-FABP mechanistically links obesity-associated breast cancer development by enhancing tumor stemness and aggression (15). However, the role of A-FABP in regulating the metabolic activities of individual dietary FA during obesity and obesity-associated cancer remains unexplored.
While obesity is known to increase chronic inflammation, which is characterized by elevated free FA, proinflammatory macrophages and increased reactive oxygen species (ROS) (16-18), the mechanistic insights into how individual FA regulate ROS production in macrophages in obesity-associated metabolic diseases are largely unknown. In our previous studies, we showed that A-FABP enhanced obesity-linked chronic inflammation by promoting saturated FA-induced ceramide but not ROS production in macrophages (19). Of note, unsaturated FA did not induce ceramide production in macrophages, suggesting that individual FA exhibit unique response/function during HFD-induced obesity.
In the present study, we established obese mouse models with different HFD rich in either saturated FA (cocoa butter) or n-3 FA (fish oil). We found that both HFD were able to induce murine obesity, but obesity-associated mammary tumor growth in these HFD-induced obese mice was dramatically different. While the cocoa butter HFD-induced obesity promoted mammary tumor growth, the fish oil HFD uncoupled obesity from accelerated tumor growth. Moreover, we mechanistically identified that A-FABP was a key molecular sensor mediating n-3 FA-induced pro-tumor macrophage death through ROS production.
Material and Methods
Animals
C57BL/6 background A-FABP-deficient mice (A-FABP−/−) and their wild type (WT) littermates were bred and housed in the animal facility of the University of Louisville. All animal manipulations were carried out according to the protocol approved by the Institutional Animal Care and Use Committee of the University of Louisville. Weaned mice were fed one of three custom-made diets: low fat diet (LFD, 10% fat), fish oil HFD (45% fat), or cocoa butter HFD (45% fat), respectively, for 5 months before they were euthanized for immune cell phenotype analysis in various tissues or organs. Mouse E0771 cells were from CH3 BioSystems. MMT060562 cells were from ATCC. All cells were cultured less than 3 months in vitro for experiments. Cells were negative for mycoplasma testing without further authentication.
Reagents
M-CSF was purchased from Cell Signaling Technology (Danvers, MA). PA (S-1109, 5 mM), OA (S-1120, 5 mM), LA (S-1127, 5 mM), EPA (S-1144, 5 mM), and DPA (DPA n-3, S-1145, 5 mM) were purchased from Nu-Chek Prep and prepared with 2 mM of endotoxin-free BSA (Akron) in PBS, sonicated until dissolved, and filtered through 0.22 μm sterile filter as we previously described (19-22). For the cell culture, RPMI 1640 (Cellgro, 15–040-cv) was from Corning and Fetal Bovine Serum (FBS, S11550) from Atlanta Biologicals. ROS generation was detected using the DCFDA (2’,7’-dichlorofluorescin diacetate) cellular ROS detection assay kit (ab113851) from Abcam. The ROS inhibitor N-acetyl-L-cysteine (NAC, A7250) was purchased from Sigma-Aldrich. APC conjugated Annexin-V (640920) was purchased from Biolegend. Antibodies used for flow cytometry were obtained from Biolegend and BD Bioscience.
MRI body fat quantification
The MR imaging data of lean or obese mice were acquired using an Agilent 9.4 T horizontal bore MRI system equipped with an Agilent 205/120 HD gradient coil (Agilent Technologies, Santa Clara, CA, USA). After a quick 3-plane gradient echo based scout scan to verify animal positioning, both coronal and axial oriented T1-weighted (T1W) images were obtained. Body fat was shown as hyperintensity in these images. All imaging data were processed using Analyze12 data analysis software (AnalyzeDirect, Inc, Overland Park, KS, USA). Whole-body fat (overall fat including both visceral and subcutaneous fat) and visceral fat (fat inside abdominal cavity) volumes were segmented and calculated. Segmentation of fat regions of interest (ROI) were automatically generated by 3D signal thresholding and then manually inspected and corrected (if necessary) by ROI editing tools.
Syngeneic tumor models
Briefly, 5×105 E0771 cells derived from a C57BL/6 mouse mammary adenocarcinoma were orthotopically implanted into the fat pad of the 4th mammary gland of lean or obese mice on different diets, respectively. Tumor growth was monitored at 3-day intervals with calipers and tumor volume was calculated by the formula 0.5× (large diameter) × (small diameter)2. Tumors were weighed at the end point when mice were sacrificed. To detect tumor associated macrophages in tumor stroma, cells were separated in a digestion buffer including a final concentration of 500μg/mL collagenase A, 20μg/mL DNase I and 200μg/mL type V hyaluronidase at 37 ˚C for 45 minutes. Single cells from tumor tissue, bone marrow, peritoneum and spleen were collected for the biological assays using flow cytometry.
Cell culture and treatment
For culture of primary bone marrow-derived macrophages (BMMs), total bone marrow cells from WT and A-FABP−/− mice were respectively collected as previously described (20;22;23). Briefly, bone marrow cells were plated in 10 mL RPMI 1640 supplemented with 5% heat inactivated FBS, 10μg/mL gentamicin, 50μL glutamax (referred to as R5). Two hours after plating, non-adherent cells were collected and cultured at the concentration of 2×106/mL in the R5 medium supplemented with 10ng/mL M-CSF and L-929 soup (30% volume). On days 2 and 5, half of the medium was replaced with fresh medium supplemented with M-CSF and L929 soup as described above. On day 7, BMMs were collected for following experiments. For FA treatment, WT and A-FABP−/− macrophages were treated with different dietary FA, including PA, SA, OA, LA, EPA,n-3 DPA or BSA control, for 30 min, 6 or 20 hours, dependent on the experiments. Flow cytometry and quantitative PCR were used for analyses of macrophage cell death, ROS production and gene expression.
Flow cytometric analyses
For analyzing macrophage phenotype, BMMs and macrophages isolated from spleen, peritoneum or tumors were surface stained for 30 min at 4˚C in 1% BSA PBS containing different Abs (anti-CD11b, clone M1/70; anti-CD11c, clone HL3; anti-F4/80, clone BM8; anti-MHC class II, clone M5/114.15.2; anti-Ly6C, clone HK1.5). Macrophages with a phenotype of F4/80+CD11b+ were gated for flow cytometric analysis. For the apoptosis assay, cells were collected after treatment with different FA for 20 hours and stained with antibodies as mentioned above. Cells were washed with PBS and re-stained with Annexin-V-APC in the Annexin-V binding buffer for 20 minutes. To measure the ROS generation in macrophages, cells were collected in tubes and stained with 10 μM DCFDA for 45 min at 37°C. All samples for either Annexin-V or DCFDA staining were acquired without washing using FACS LSR Fortessa flow cytometer, and data were analyzed using FlowJo software.
Cellular ROS analysis
Macrophages were seeded (5×104 cells/well) overnight in a 96 well plate. Cells were washed once with 1×buffer and stained with 10 μM DCFDA (Cellular ROS Assay Kit, ab113851, Abcam) for 45 min at 37°C. Cells were washed and treated with different FA, including PA, OA, LA, EPA and DPA, and control BSA for indicated time (e.g. 30min, 1.5h, 6h). The fluorescent signal was measured using Ex/Em 485/535nm. Data are reported as mean ± SD after background subtraction.
Quantitative RT-PCR
For real-time PCR analysis, RNA was extracted from cells using PureLink™ RNeasy Mini Kit (Invitrogen, 12183025). Complementary DNA synthesis was performed with a QuantiTect Reverse Transcription Kit (Qiagen, 205314). Quantitative PCR was performed with PowerSYBR Green PCR Master Mix(Applied biosystems,4367659) using StepOnePlus™ Real-Time PCR Systems (Applied Biosystems) to analyze the expression of TNFα, IL-6, IL-1β, IL-12, IL-23 A-FABP, E-FABP, NOX1, Caspase-8, iNOS, Caspase-11, Cpt1b, PCX, UCP1, UCP2, RIPK3, NOX2 (see primer sequences in Supplementary Table 1). Relative mRNA levels were determined using the housekeeping HPRT1 gene as a reference.
Confocal analysis
BMMs or peritoneal macrophages from WT or A-FABP−/− mice were cultured on poly-D-lysine coated coverslips (NeuVitro) in a 24-well plate and treated with different FA (e.g. n-3 DPA, PA) for 30 minutes. For ROS and mitochondrial potential staining, cells were stained with DCFDA or MitoSpy (Biolegend) as previously described (21-23). Nuclei were stained with 0.2μM DAPI (Invitrogen). Confocal slides were analyzed with a Nikon A1 laser scanning confocal microscope.
Analysis of oxygen consumption rate in macrophages
Mitochondrial oxygen consumption rate (OCR) was determined using an extracellular flux assay (Seahorse XF, Agilent). Briefly, 5×104 BMMs or peritoneal macrophages were plated in 96-wells with OCR medium in a non-CO2 incubator for 45min for the equilibrium of temperature and pH before FA or different drugs (including oligomycin, an ATP synthase inhibitor; FCCP, a proton ionophore, Rotenone/ Antimycin A, complex I/III inhibitors) were injected via the delivery chambers, at the designated time points, respectively. The analysis was performed according to the manufacturer’s instructions.
Statistical analysis
The student’s t test was performed to compare results from different treatments. A p value less than 0.05 was considered statistically significant.
Results
HFD with different FA components induce mouse obesity
As HFD can induce obesity (6;7), we utilized custom HFD with different fat sources (see diet ingredients in Supplementary Table 2) to observe if HFD with different FA components were able to induce similar rates of murine obesity. Notably, these low and high-fat diets had identical base ingredients such that only the fat content differed, avoiding confounding effects from other ingredients. In HFD supplemented with cocoa butter, saturated and monounsaturated FA accounted for 58.6% and 32.6%, respectively. In contrast, the fish oil HFD contained 35% n-3 FA besides saturated and monounsaturated FA. n-6 and other FA components were similar between cocoa butter and fish oil HFD (Figure 1A, 1B). Interestingly, during the first two months that C57/B6 mice were fed the different diets, the cocoa butter HFD appeared to increase body weight more than the fish oil HFD, but weight gains of the two HFD groups were similar by 3 months. Both HFD induced significant increases in body weight (p<0.01) as compared to the LFD group (Figure 1C). When we assessed food consumption rate (Figure 1D) and subcutaneous/visceral fat deposition (Figure 1E, 1F) in HFD-fed mice, we did not observe significant differences between cocoa butter- and fish oil-fed mice. These data indicate that lipids either from fish oil or cocoa butter are able to induce similar rates and levels of obesity in our mouse models.
Figure 1.

HFD with different FA components induce mouse obesity. A, B, fatty acids composition in cocoa butter diet (A) and fish oil diet (B). C, mouse body weight on LFD (low fat diet, 10% fat), HFD (high-fat diet, 45%) cocoa butter diet or fish oil diet for 5 months (n=10/group). D, measurement of average food consumption of each mouse per day. E, F, quantification of total, subcutaneous and visceral fat in cocoa butter HFD- or fish oil HFD-induced obese mice by the MRI scanning. Representative MR images are shown in panel E. Average fat weight is shown in panel F. Data are shown as mean ± SD (**p<0.01, n.s., not significant).
Fish oil HFD uncouples obesity and tumor growth via reduced pro-tumor macrophages
HFD-induced obesity is believed to be associated with increased macrophage accumulation and tumor progression (18;24). We wanted to determine whether obesity induced by HFD with different FA components might exhibit phenotypic differences due to their influence on immune cell function as previously shown (19;21). To this end, we injected same numbers of E0771 mammary tumor cells in LFD lean mice, cocoa butter-fed obese mice or fish oil-fed obese mice, and measured tumor growth. Whereas cocoa butter-induced obesity caused accelerated tumor growth as compared to control LFD mice, fish oil-induced obesity did not increase tumor growth (Figure 2A). Consistent with this finding, tumor size in fish oil-fed mice was significantly smaller than cocoa butter-fed mice, but not different than that in the LFD mice (Figure 2B). Similar observations were made when MMT060562 mammary tumor cells were implanted in these mice (Supplementary Figure 1). We previously demonstrated that TAM, particularly the Q4 CD11b+F4/80+Ly6C-MHCII- TAM subset promote tumor growth (14). In the current study, we observed that the pro-tumor Q4 TAM were significantly less prevalent in the fish oil- compared to the cocoa butter-fed mice (Figure 2C, 2D). Moreover, Q4 macrophages were also dramatically reduced in the spleens of the fish oil-fed mice (Figure 2E, 2F). These data suggest that FA components in the fish oil diet play a unique role in decreasing pro-tumor macrophages compared to those in the cocoa butter diet.
Figure 2.

Fish oil HFD uncouples obesity with E0771 mammary tumor growth with reduced pro-tumor macrophages. A, mice on LFD, cocoa butter or fish oil HFD (n=11/group) were orthotopically injected with E0771 mammary tumor cells (0.5×106). Tumor growth was monitored by measuring tumor size every 3 days. B, E0771 tumor weight in each group 21 days post tumor injection. C, D, analysis of phenotype (MHCII and Ly6C staining) of tumor infiltrating macrophages (F4/80+CD11b+) in E0771 tumors 21 days post tumor injection by flow cytometry. Average of Q4 macrophage subset is shown in panel D. E, F, analysis of TAM subsets with MHCII and Ly6C staining by gating on F4/80+CD11b+ macrophages in spleen of tumor-bearing lean or obese mice (E). Average percentage of pro-tumor Q4 TAM subset is shown in panel F. Data are shown as mean ± SD (**p<0.01, ***p<0.001, ****p<0.0001).
n-3 FA induce macrophage cell death through ROS production
Since obesity is linked to increased levels of free FA and ROS production (17;25;26), we speculated that free FA upregulated during obesity induced ROS production in macrophages. We thus treated macrophages with individual FA found in cocoa butter and fish oil, and measured ROS production. We found that n-3 FA (e.g. DPA, EPA) that are present in fish oil induced significant ROS production in macrophages as early as 30 mins after treatment. In contrast, saturated (PA, SA) and unsaturated (OA) FA which existed in both diets did not induce obvious ROS production (Figure 3A). Given the well-known function of ROS in activating apoptosis (27), we further measured if n-3 FA treatment induced macrophage cell death. Indeed, macrophages treated with n-3 DPA exhibited enhanced levels of apoptosis-related transcripts including iNOS, RIPK3, Caspase 8 and 11 (Figure 3B-3E). Annexin V staining showed that DPA induced significant apoptosis of macrophages (Figure 3F, 3G). Notably, NAC, an effective ROS scavenger, inhibited DPA-induced ROS production in macrophages (Figure 3H, 3I). Moreover, NAC pretreatment prevented DPA-induced macrophage cell death (Figure 3J). Consistent with DPA treatment, EPA treatment also induced expression of ROS-related apoptosis genes, ROS production, and ROS-dependent macrophage apoptosis (Supplementary Figure 2). Thus, n-3 FA specifically induced ROS-mediated macrophage apoptosis, providing direct evidence for the reduced pro-tumor macrophages observed in fish oil-fed obese mice.
Figure 3.

n-3 FA induce macrophage cell death through ROS production. A, Kinetics of ROS levels (shown as relative fluorescent intensity of DCFDA) in bone marrow-derived macrophages (BMM) treated with different dietary FA (200μM) or BSA control at the indicated time points. B-E, analysis of the expression of n-3 DPA-induced iNOS (B), RIPK3 (C), Caspase 8 (D) and Caspase 11 (E) in BMMs by real time PCR. F, G, BMMs were treated with DPA (200μM) or carrier BSA control for 20h. Macrophage apoptosis was analyzed by annexin V staining with flow cytometry (F). Average percentage of annexin V+ macrophages is shown in panel G. H, I, analysis of ROS production in BMMs treated with 200μM DPA or BSA control for 30 mins in the presence or absence of 1mM NAC (an effective ROS scavenger) by flow cytometric staining of DCFDA. Relative mean fluorescent intensity (MFI) is shown in panel I. J, BMMs were treated with 200μM DPA for 20h in the presence or absence of NAC. Flow cytometric analysis of annexin V+ apoptotic macrophages. Experiments were repeated at least three times. Data are shown as mean ± SD (*p<0.05; **p<0.01, ***p<0.001 as compared to BSA group).
n-3 FA enhance A-FABP expression and lipid metabolism in macrophages
Given that FABPs facilitate FA metabolism/responses inside cells and that A-FABP is a functional biomarker for pro-tumor macrophages (14), we wondered if A-FABP expression in macrophages promoted the n-3 FA-mediated effects. Analysis of pro-inflammatory cytokine expression demonstrated that n-3 DPA treatment did not induce obvious production of TNFα, IL-6, IL-12 or IL-1β (Figure 4A-4D). However, A-FABP, as well as mitochondrial FA oxidation-related genes (e.g. Cpt1b, PCX, UCP2), were significantly upregulated in response to DPA treatment (Figure 4E-4H), suggesting that A-FABP mediates n-3 FA-induced changes in lipid metabolism. Moreover, DPA treatment induced increased mitochondrial potential (Figure 4I), which was accompanied by the increase of oxidative phosphorylation as measured by OCR at both basal and maximal levels (Figure 4J). As A-FABP was highly expressed in the Q4 pro-tumor TAM subsets, we purified the Q4 subsets from E0771 tumors in LFD and fish oil-fed mice and demonstrated that fish oil HFD induced upregulation of A-FABP and lipid oxidation genes in the Q4 pro-tumor TAMs (Supplementary Figure 3). Collectively, these data suggest that macrophages respond to n-3 FA treatment by increasing expression of A-FABP, which facilitates the transportation of DPA to mitochondria for oxidative phosphorylation.
Figure 4.

n-3 FA enhances A-FABP expression and lipid metabolism in macrophages. A-H, BMMs were treated with 200 μM DPA or BSA control overnight. Real-time PCR analysis of indicated gene expression of IL-6 (A), TNFα (B), IL-12 (C), IL-1β (D), A-FABP (E), PCX (F), UCP2 (G) and Cpt1b (H) in BMMs. I, confocal microscopy analysis of mitochondrial potential with MitoSpy (red) in BMMs (DAPI, blue for nuclei) treated with BSA or DPA for 30 mins. J, extracellular flux analysis of BMMs treated with BSA and DPA. OCR (oxygen consumption rate) was measured in the presence of injected drugs at the indicated time points. Experiments were repeated at least three times. Data are shown as mean ± SD (*p<0.05; **p<0.01 as compared to BSA group).
A-FABP is critical in n-3 FA-mediated ROS production and macrophage cell death
To further examine the critical role of A-FABP in mediating n-3 FA-induced effects in macrophages, we first utilized BMMs from WT and A-FABP−/− mice and observed reduced MitoSpy staining in A-FABP−/− BMMs (Figure 5A, 5B), indicating that A-FABP deficiency significantly reduced DPA-induced mitochondrial potential. Accordingly, DPA-induced mitochondrial OCR was also dramatically decreased in A-FABP−/− BMMs as compared to WT controls (Figure 5C). As A-FABP was also highly expressed in peritoneal macrophages (PMs), we used PMs from WT and A-FABP−/− mice and confirmed the role of A-FABP in inducing DPA-induced mitochondrial potential and lipid oxidation (Supplementary Figure 4A-4C). Consistent with reduced oxygen consumption, ROS generation, as measured by DCFDA staining using flow cytometry (Figure 5D) and confocal microscopy (Figure 5E), was significantly reduced in A-FABP−/− macrophages. Of note, A-FABP deficiency also impaired EPA-induced mitochondrial potential and ROS production in both BMMs and PMs (Supplementary Figure 4D-4G).
Figure 5.

A-FABP is critical in n-3 FA-mediated ROS production and macrophage cell death. A, B, BMMs from WT and A-FABP−/− mice were treated with DPA for 30mins. Mitochondrial potential was measured by flow cytometry with MitoSpy staining (A). Relative MFI of MitoSpy is shown in panel B. C, extracellular flux analysis of OCR in DPA-treated BMMs from WT and A-FABP−/− mice in the presence of injected drugs at the indicated time points. D, measurement of fluorescent intensity of DCFDA in WT or A-FABP−/− BMMs treated with DPA (200μM) for 30 mins by flow cytometry. E, confocal microscopy analysis of ROS production (green) in WT and A-FABP−/− BMMs treated with DPA for 30 mins. F-G, WT and A-FABP−/− BMMs were treated with DPA 12h, respectively. Real-time PCR analysis of expression of Cpt1b (F), iNOS (G), Caspase 8 (H) and Caspase 11 (I) in these cells. J, flow analysis of ROS levels in A-FABP−/− BMMs treated with 200 μM DPA in the presence or absence of 1mM NAC for 30 mins. K, flow cytometric analysis of DPA-induced A-FABP−/− BMM cell death with or without NAC treatment. Experiments were repeated three times. Data are shown as mean ± SD (*p<0.05; **p<0.01 as compared to BSA group).
Reduced mitochondrial lipid oxidation and ROS production were associated with reduced expression of Cpt1b, an essential mitochondrial enzyme responsible for FA mitochondrial transportation and β-oxidation, in A-FABP−/− macrophages (Figure 5F). Moreover, ROS-related signaling pathway markers, including iNOS, Caspase 8 and 11, were also inhibited by A-FABP deficiency (Figure 5G-5I). As n-3 FA-induced ROS production was responsible for macrophage cell death in WT macrophages (Figure 3), we determined if these effects were dependent on A-FABP expression. Indeed, A-FABP deficiency diminished DPA-induced ROS production and cell death irrespective of the presence or absence of an ROS inhibitor (Figure 5J, 5K). Altogether, these results indicate that A-FABP is essential in n-3 FA-induced ROS production and cell death in macrophages in vitro.
A-FABP deficiency diminishes fish oil-induced changes in macrophage metabolism in obese mice
To assess the role of A-FABP in vivo, we fed A-FABP deficient mice the fish oil HFD same as their WT littermates. A-FABP deficiency did not influence fish oil HFD-induced obesity (Figure 6A). However, fish oil HFD-induced ROS upregulation was observed in macrophages from WT but not in A-FABP−/− obese mice (Figure 6B). Accordingly, WT mice exhibited reduced numbers of macrophages in bone marrow when fed the fish oil HFD. In contrast, there were no differences in macrophage numbers in lean and obese A-FABP−/− mice (Figure 6C), confirming an A-FABP-dependent effect through which the fish oil HFD-induced ROS elevation promoted macrophage death in vivo. Similar effects were observed in peritoneal macrophages, which were reduced in fish oil-fed WT mice (Figure 6D, 6E), but not in A-FABP−/− mice (Figure 6F, 6G). Moreover, when we implanted E0771 mammary tumor cells in these mice and analyzed tumor infiltrating macrophage phenotype, we found that ROS production from apoptotic TAMs (Annexin V+ DCFDA+ cells) was significantly higher in WT mice on the fish oil HFD as compared to the LFD mice. However, there was no difference in ROS production between lean and obese A-FABP−/− mice (Figure 6H, 6I). Accordingly, obesity-associated E0771 tumor growth was diminished in WT mice fed the fish oil HFD (Figure 6J). Of note, fish oil HFD appeared to increase tumor growth when compared to LFD in A-FABP−/− mice, but the increase did not reach statistical significance (Figure 6J), suggesting possible non-A-FABP-mediated effects involved in obesity/tumor associations. Collectively, these data provided convincing evidence that high levels of ROS induced by the fish oil HFD promoted apoptosis of pro-tumor macrophages in an A-FABP-dependent manner, thus uncoupling obesity associated pro-tumor effects.
Figure 6.

A-FABP deficiency diminishes fish oil-induced changes in macrophage metabolism in obese mice. A, measurement of the body weight of WT and A-FABP−/− mice fed with fish oil HFD at the indicated time points (n=9/group). B, bone marrow macrophages collected from fish oil HFD-fed WT or A-FABP−/− were analyzed for ROS production by DCFDA staining. Bone marrow macrophages from LFD-fed mice were used as controls. C, quantification of bone marrow macrophage numbers in WT or A-FABP−/− mice fed with fish oil HFD or LFD control diet. D, E, analysis of peritoneal macrophage percentage (D) and actual numbers (E) in fish oil HFD- or LFD-fed WT mice. F, G, analysis of peritoneal macrophage percentage (F) and actual numbers (G) in fish oil HFD- or LFD-fed A-FABP−/− mice. H-K, E0771 tumor cells were orthotopically injected into LFD- or fish oil HFD-fed WT and A-FABP−/− mice, respectively (n=7/group). ROS-induced apoptotic macrophages (DCFDA+ Annexin V+) in tumors of WT and A-FABP−/− mice (H, I) were analyzed on 21 days post tumor injection by flow cytometric staining. E0771 tumor weight in each group was shown in panel J. Data are shown as mean ± SD (*p<0.05; **p<0.01 as compared to LFD group. n.s., not significant).
Discussion
Obesity is known to increase the risk of many types of cancer (3). During our studies investigating how obesity was mechanistically linked to cancer development, we noticed that while murine obesity could be induced by consumption of various HFD with different FA components, these seemingly “equally obese” mice exhibited dramatic differences in immune regulation and tumor progression. Specifically, whereas the cocoa butter HFD-induced obesity was associated with accelerated tumor growth and enhanced differentiation of pro-tumor TAMs, the fish oil HFD-induced obesity uncoupled obesity-associated tumor growth via reduction of pro-tumor TAMs. In dissecting the underlying mechanisms by which consumption of the fish oil HFD reduced pro-tumor TAMs, we demonstrated that n-3 FA treatment enhanced pro-tumor macrophage cell death through A-FABP-ROS dependent axis. Furthermore, A-FABP deficiency diminished fish oil-mediated effects both in vitro and in vivo. Thus, our studies support a new concept that dietary lipid components, via their differential effects on host immunity, influence obesity-associated tumor progression independent of BMI.
To develop murine obese models with HFD, we designed various custom HFD with different fat compositions. Unlike human diet supplementation studies, these well-controlled murine diets have identical base ingredients and defined FA components, thus eliminating the possible influence of unknown confounding factors associated with human studies. We chose cocoa butter and fish oil fat in the current studies due to their common usage in human diets and clear difference in the n-3 FA component. Intake of n-3 FA has long been believed to be associated with inhibition of inflammation and protection of cancer and cardiovascular disease (28;29), but data from recent n-3 FA clinical trials did not show any significant benefits of n-3 FA supplements (30-33). These negative results, which contradicted multiple earlier findings, reflect the mechanistic knowledge gap in how consumption of n-3 FA induces anti-tumor effects beyond the known anti-inflammatory effects.
As the most dominant leukocytes in the tumor stroma, TAMs play an essential role in tumor initiation and progression (34-36). Targeting TAMs for cancer immunotherapy has been proposed for many years, but has been difficult to implement due to the heterogeneity of macrophage subsets in the tumor microenvironment (37;38). In our recent studies, we demonstrated that a subset of TAMs, which exhibited CD11b+F4/80+Ly6C-MHCII-CD11c- phenotype, accumulated in tumor stroma and promoted tumor progression through high expression of A-FABP (14). Thus, it is very likely that environmental factors (e.g. dietary FA, tumor-derived signaling) which are able to promote A-FABP activity will enhance pro-tumor TAM differentiation and tumor development. Indeed, pro-tumor macrophages were enhanced in obese mice that were fed the cocoa butter HFD. Cocoa butter is rich in saturated FA (e.g. palmitic acid), which have been demonstrated to induce ER stress and A-FABP expression in macrophages (39). By contrast, n-3 FA in the fish oil diet exerted a unique effect on pro-tumor TAMs in obese mice. As compared to common FA (e.g. PA, OA) in the cocoa butter diet, n-3 FA induced remarkable upregulation of ROS production in A-FABP+ macrophages. Given the known mechanisms of ROS-mediated activation of cell death (27;40), we found that n-3 FA-induced ROS was associated with increased mitochondrial oxygen consumption and upregulation of caspase-8 and −11 signaling. Importantly, a scavenger of ROS production inhibited n-3 FA-mediated macrophage cell death. Therefore, our studies demonstrated a new mechanism by which fish oil diets induce pro-tumor macrophage cell death through n-3 FA-induced ROS production.
Due to the high expression of A-FABP in pro-tumor macrophages, we further investigated whether A-FABP mediated n-3 FA-induced effect. First, we observed that n-3 FA-induced ROS production as well as oxidative phosphorylation in macrophages was attenuated in the absence of A-FABP. As an intracellular lipid chaperone, A-FABP has high binding affinity for n-3 FA as compared to saturated FA (e.g. palmitic acid and stearic acid) (41). Thus, A-FABP deficiency compromised n-3 FA intracellular transportation across cellular compartments. Second, as a rate-limiting enzyme, CPT1 is located on the outer mitochondrial membrane and responsible for FA transport into mitochondria for FA oxidation (42). We noticed that A-FABP deficiency remarkably reduced CPT1 expression, leading to reduced n-3 FA oxidation in mitochondria. Moreover, n-3 FA-induced cell death-related signaling (e.g. caspases, RIPKs) was also dampened when A-FABP was depleted in macrophages. Using A-FABP deficient mice, we further confirmed that A-FABP expression in macrophages promoted fish oil HFD-induced macrophage cell death via ROS-mediated pathways in vivo. Collectively, our data suggest A-FABP as a new molecular sensor mediating n-3 FA-induced effects.
It is worth noting that obesity induced by various HFD is associated with complex signaling events and biological changes at multiple levels. First, different dietary FA compositions in various HFD exert diverse regulatory effects due to their chemical and structural differences. Second, individual cell populations or subsets exhibit different metabolic needs under basal and activating status. Third, FABP members exhibit distinct expression profile in different cells, tissues and organs, which have unique binding affinity to dietary FA, coordinating different FA-mediated intracellular effects. Therefore, consumption of a particular HFD will have multiple systemic or local effects. As such, it is likely that the physical effects of n-3 FA diet (e.g. fish oil fat) will extend beyond the induction of A-FABP/ROS-mediated macrophage cell death. Indeed, other studies have shown that intake of n-3 FA can modulate tumor-infiltrating T cells or reduce circulating levels of A-FABP (43;44). Our findings complement these studies, and provide new evidence of how consumption of fish-oil diet uncouples obesity-associated tumor growth.
In addition, it should be noted that whereas fish oil-induced obesity did not promote tumor growth, it also did not inhibit tumor growth compared to the LFD mice. In line with our observations, recent VITAL studies reported that while supplementation with fish oil did not lower cancer incidence (32), it did not increase the cancer risk either. Due to the paucity of n-3 FA in a typical western diet, it is most likely that the beneficial effects of n-3 FA observed in animal studies are confounded by adverse effects mediated by other FA components in western diets. Thus, fish oil supplementation should be combined with reduced intake of other saturated FA to acquire the expected benefits of cancer prevention and dietary control of obesity-associated diseases.
In summary, we report, for the first time, that unlike the cocoa butter HFD, obesity induced by the fish oil HFD is not associated with accelerated mammary tumor growth. Moreover, we demonstrate that n-3 FA in the fish oil diet induce ROS-mediated pro-tumor macrophage cell death in an A-FABP dependent manner. This study provides mechanistic insight into n-3 FA-mediated effects and introduces a new concept that not all HFD leading to obesity are tumorigenic.
Supplementary Material
Statement of Significance.
This study provides mechanistic insight into dietary supplementation with fish oil for breast cancer prevention and advances a new concept that not all HFD leading to obesity are tumorigenic.
Acknowledgements
We thank Dr. Hyeran Jang from Research Diets for assistance with the design of custom rodent diets. This study was supported by NIH R01CA180986 (to B. Li), NIH R01CA177679 (to B. Li), and R01AI137324 (to B. Li).
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed
Reference List
- 1.Calle EE, Kaaks R. Overweight, obesity and cancer: epidemiological evidence and proposed mechanisms. Nat.Rev.Cancer 2004. August;4(8):579–91 [DOI] [PubMed] [Google Scholar]
- 2.Bianchini F, Kaaks R, Vainio H. Overweight, obesity, and cancer risk. Lancet Oncol. 2002. September;3(9):565–74 [DOI] [PubMed] [Google Scholar]
- 3.Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, Straif K. Body Fatness and Cancer-Viewpoint of the IARC Working Group. N.Engl.J.Med. 2016. August 25;375(8):794–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee KR, Seo MH, Do HK, Jung J, Hwang IC. Waist circumference and risk of 23 site-specific cancers: a population-based cohort study of Korean adults. Br.J.Cancer 2018. October;119(8):1018–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Purcell SA, Elliott SA, Kroenke CH, Sawyer MB, Prado CM. Impact of Body Weight and Body Composition on Ovarian Cancer Prognosis. Curr.Oncol.Rep. 2016. February;18(2):8. [DOI] [PubMed] [Google Scholar]
- 6.Buckley J Availability of high-fat foods might drive the obesity epidemic. Nat.Rev.Endocrinol. 2018. Oct;14(10):574–5 [DOI] [PubMed] [Google Scholar]
- 7.Hu S, Wang L, Yang D, Li L, Togo J, Wu Y, Liu Q, Li B, Li M, Wang G, et al. Dietary Fat, but Not Protein or Carbohydrate, Regulates Energy Intake and Causes Adiposity in Mice. Cell Metab 2018. September 4;28(3):415–31 [DOI] [PubMed] [Google Scholar]
- 8.Furuhashi M, Hotamisligil GS. Fatty acid-binding proteins: role in metabolic diseases and potential as drug targets. Nat.Rev.Drug Discov. 2008. June;7(6):489–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Storch J, Corsico B. The emerging functions and mechanisms of mammalian fatty acid-binding proteins. Annu.Rev.Nutr. 2008;28:73–95 [DOI] [PubMed] [Google Scholar]
- 10.Cao H, Sekiya M, Ertunc ME, Burak MF, Mayers JR, White A, Inouye K, Rickey LM, Ercal BC, Furuhashi M, et al. Adipocyte lipid chaperone AP2 is a secreted adipokine regulating hepatic glucose production. Cell Metab 2013. May 7;17(5):768–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furuhashi M, Tuncman G, Gorgun CZ, Makowski L, Atsumi G, Vaillancourt E, Kono K, Babaev VR, Fazio S, Linton MF, et al. Treatment of diabetes and atherosclerosis by inhibiting fatty-acid-binding protein aP2. Nature 2007. June 21;447(7147):959–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maeda K, Cao H, Kono K, Gorgun CZ, Furuhashi M, Uysal KT, Cao Q, Atsumi G, Malone H, Krishnan B, et al. Adipocyte/macrophage fatty acid binding proteins control integrated metabolic responses in obesity and diabetes. Cell Metab 2005. February;1(2):107–19 [DOI] [PubMed] [Google Scholar]
- 13.Makowski L, Brittingham KC, Reynolds JM, Suttles J, Hotamisligil GS. The fatty acid-binding protein, aP2, coordinates macrophage cholesterol trafficking and inflammatory activity. Macrophage expression of aP2 impacts peroxisome proliferator-activated receptor gamma and IkappaB kinase activities. J.Biol.Chem. 2005. April 1;280(13):12888–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hao J, Yan F, Zhang Y, Triplett A, Zhang Y, Schultz DA, Sun Y, Zeng J, Silverstein KAT, Zheng Q, et al. Expression of Adipocyte/Macrophage Fatty Acid-Binding Protein in Tumor-Associated Macrophages Promotes Breast Cancer Progression. Cancer Res. 2018. May 1;78(9):2343–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hao J, Zhang Y, Yan X, Yan F, Sun Y, Zeng J, Waigel S, Yin Y, Fraig MM, Egilmez NK, et al. Circulating adipose fatty acid binding protein is a new link underlying obesity-associated breast/mammary tumor development. Cell Metab 2018. November 6; 28; 689–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Steen KA, Xu H, Bernlohr DA. FABP4/aP2 Regulates Macrophage Redox Signaling and Inflammasome Activation via Control of UCP2. Mol.Cell Biol. 2017. January 15;37(2):e00282–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Obesity Boden G. and free fatty acids. Endocrinol.Metab Clin.North Am. 2008. September;37(3):635-ix. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW, Jr. Obesity is associated with macrophage accumulation in adipose tissue. J.Clin.Invest 2003. December;112(12):1796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Y, Rao E, Zeng J, Hao J, Sun Y, Liu S, Sauter ER, Bernlohr DA, Cleary MP, Suttles J, et al. Adipose Fatty Acid Binding Protein Promotes Saturated Fatty Acid-Induced Macrophage Cell Death through Enhancing Ceramide Production. J.Immunol. 2017. January 15;198(2):798–807. PMCID:PMC5225136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Sun Y, Rao E, Yan F, Li Q, Zhang Y, Silverstein KA, Liu S, Sauter E, Cleary MP, et al. Fatty acid-binding protein E-FABP restricts tumor growth by promoting IFN-beta responses in tumor-associated macrophages. Cancer Res. 2014. June 1;74(11):2986–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Li Q, Rao E, Sun Y, Grossmann ME, Morris RJ, Cleary MP, Li B. Epidermal Fatty Acid binding protein promotes skin inflammation induced by high-fat diet. Immunity. 2015. May 19;42(5):953–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Hao J, Sun Y, Li B. Saturated Fatty Acids Induce Ceramide-associated Macrophage Cell Death. J.Vis.Exp. 2017. October 31;(128) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zeng J, Zhang Y, Hao J, Sun Y, Liu S, Bernlohr DA, Sauter ER, Cleary MP, Suttles J, Li B. Stearic acid induces CD11c expression in proinflammatory macrophages via epidermal fatty acid binding protein. J.Immunol. 2018. May 15;200(10):3407–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Park J, Morley TS, Kim M, Clegg DJ, Scherer PE. Obesity and cancer--mechanisms underlying tumour progression and recurrence. Nat.Rev.Endocrinol. 2014. August;10(8):455–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fernandez-Sanchez A, Madrigal-Santillan E, Bautista M, Esquivel-Soto J, Morales-Gonzalez A, Esquivel-Chirino C, Durante-Montiel I, Sanchez-Rivera G, Valadez-Vega C, Morales-Gonzalez JA. Inflammation, oxidative stress, and obesity. Int.J.Mol.Sci. 2011;12(5):3117–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marseglia L, Manti S, D’Angelo G, Nicotera A, Parisi E, Di RG, Gitto E, Arrigo T. Oxidative stress in obesity: a critical component in human diseases. Int.J.Mol.Sci. 2014. Dec 26;16(1):378–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Redza-Dutordoir M, Averill-Bates DA. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim.Biophys.Acta 2016. December;1863(12):2977–92 [DOI] [PubMed] [Google Scholar]
- 28.Bordeleau L, Yakubovich N, Dagenais GR, Rosenstock J, Probstfield J, Chang YP, Ryden LE, Pirags V, Spinas GA, Birkeland KI, et al. The association of basal insulin glargine and/or n-3 fatty acids with incident cancers in patients with dysglycemia. Diabetes Care 2014;37(5):1360–6 [DOI] [PubMed] [Google Scholar]
- 29.Wu JH, Mozaffarian D. omega-3 fatty acids, atherosclerosis progression and cardiovascular outcomes in recent trials: new pieces in a complex puzzle. Heart 2014. April;100(7):530–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bowman L, Mafham M, Wallendszus K, Stevens W, Buck G, Barton J, Murphy K, Aung T, Haynes R, Cox J, et al. Effects of n-3 Fatty Acid Supplements in Diabetes Mellitus. N.Engl.J.Med. 2018. October 18;379(16):1540–50 [DOI] [PubMed] [Google Scholar]
- 31.Manson JE, Cook NR, Lee IM, Christen W, Bassuk SS, Mora S, Gibson H, Albert CM, Gordon D, Copeland T, et al. Marine n-3 Fatty Acids and Prevention of Cardiovascular Disease and Cancer. N.Engl.J.Med. 2019. January 3;380(1):23–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Keaney JF, Jr., Rosen CJ. VITAL Signs for Dietary Supplementation to Prevent Cancer and Heart Disease. N.Engl.J.Med. 2019. January 3;380(1):91–3 [DOI] [PubMed] [Google Scholar]
- 33.Manson JE, Cook NR, Lee IM, Christen W, Bassuk SS, Mora S, Gibson H, Gordon D, Copeland T, D’Agostino D, et al. Vitamin D Supplements and Prevention of Cancer and Cardiovascular Disease. N.Engl.J.Med. 2019. January 3;380(1):33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Richards DM, Hettinger J, Feuerer M. Monocytes and macrophages in cancer: development and functions. Cancer Microenviron. 2013. August;6(2):179–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pawelek J, Chakraborty A, Lazova R, Yilmaz Y, Cooper D, Brash D, Handerson T. Co-opting macrophage traits in cancer progression: a consequence of tumor cell fusion? Contrib.Microbiol. 2006;13:138–55 [DOI] [PubMed] [Google Scholar]
- 36.Carron EC, Homra S, Rosenberg J, Coffelt SB, Kittrell F, Zhang Y, Creighton CJ, Fuqua SA, Medina D, Machado HL. Macrophages promote the progression of premalignant mammary lesions to invasive cancer. Oncotarget. 2017. August 1;8(31):50731–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Movahedi K, Laoui D, Gysemans C, Baeten M, Stange G, Van den Bossche J, Mack M, Pipeleers D, In’t VP, De BP, et al. Different tumor microenvironments contain functionally distinct subsets of macrophages derived from Ly6C(high) monocytes. Cancer Res. 2010. July 15;70(14):5728–39 [DOI] [PubMed] [Google Scholar]
- 38.Stout RD, Jiang C, Matta B, Tietzel I, Watkins SK, Suttles J. Macrophages sequentially change their functional phenotype in response to changes in microenvironmental influences. J.Immunol. 2005. July 1;175(1):342–9 [DOI] [PubMed] [Google Scholar]
- 39.Hoo RL, Shu L, Cheng KK, Wu X, Liao B, Wu D, Zhou Z, Xu A. Adipocyte Fatty Acid Binding Protein Potentiates Toxic Lipids-Induced Endoplasmic Reticulum Stress in Macrophages via Inhibition of Janus Kinase 2-dependent Autophagy. Sci.Rep. 2017. January 17;7:40657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ryter SW, Kim HP, Hoetzel A, Park JW, Nakahira K, Wang X, Choi AM. Mechanisms of cell death in oxidative stress. Antioxid.Redox.Signal. 2007. January;9(1):49–89 [DOI] [PubMed] [Google Scholar]
- 41.Lee CW, Kim JE, Do H, Kim RO, Lee SG, Park HH, Chang JH, Yim JH, Park H, Kim IC, et al. Structural basis for the ligand-binding specificity of fatty acid-binding proteins (pFABP4 and pFABP5) in gentoo penguin. Biochem.Biophys.Res.Commun. 2015. September 11;465(1):12–8 [DOI] [PubMed] [Google Scholar]
- 42.Briant LJB, Dodd MS, Chibalina MV, Rorsman NJG, Johnson PRV, Carmeliet P, Rorsman P, Knudsen JG. CPT1a-Dependent Long-Chain Fatty Acid Oxidation Contributes to Maintaining Glucagon Secretion from Pancreatic Islets. Cell Rep. 2018. June 12;23(11):3300–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Song M, Nishihara R, Cao Y, Chun E, Qian ZR, Mima K, Inamura K, Masugi Y, Nowak JA, Nosho K, et al. Marine omega-3 Polyunsaturated Fatty Acid Intake and Risk of Colorectal Cancer Characterized by Tumor-Infiltrating T Cells. JAMA Oncol. 2016. September 1;2(9):1197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Furuhashi M, Hiramitsu S, Mita T, Omori A, Fuseya T, Ishimura S, Watanabe Y, Hoshina K, Matsumoto M, Tanaka M, et al. Reduction of circulating FABP4 level by treatment with omega-3 fatty acid ethyl esters. Lipids Health Dis. 2016. January 12;15:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.