

Summary
The cardinal property of bone marrow (BM) stromal cells is their capacity to contribute to hematopoietic stem cell (HSC) niches by providing mediators assisting HSC functions. In this study we first contrasted transcriptomes of stromal cells at different developmental stages and then included large number of HSC-supportive and non-supportive samples. Application of a combination of algorithms, comprising one identifying reliable paths and potential causative relationships in complex systems, revealed gene networks characteristic of the BM stromal HSC-supportive capacity and of defined niche populations of perivascular cells, osteoblasts, and mesenchymal stromal cells. Inclusion of single-cell transcriptomes enabled establishing for the perivascular cell subset a partially oriented graph of direct gene-to-gene interactions. As proof of concept we showed that R-spondin-2, expressed by the perivascular subset, synergized with Kit ligand to amplify ex vivo hematopoietic precursors. This study by identifying classifiers and hubs constitutes a resource to unravel candidate BM stromal mediators.
Subject Areas: Cell Biology, Stem Cells Research, Bioinformatics
Graphical Abstract
Highlights
-
•
A correlation network with predictor genes for the BM HSPC-supportive stromal niche
-
•
An information theoretic network for the supportive perivascular stromal niche
-
•
Wnt facilitator Rspo2 together with SCF to amplify ex vivo hematopoietic precursors
-
•
Resource combining bioinformatics algorithms to search for novel stromal mediators
Cell Biology; Stem Cells Research; Bioinformatics
Introduction
It is now well established that stem cells reside in niches where they are associated with other cell types that control stem cell behavior. This is particularly true for hematopoietic stem cells (HSCs) whose niche include several cell types that express a large array of mediators that facilitate or impede the different stem cell attributes (self-renewal, lineage commitment, proliferation, and survival). During the last decade, in vivo studies, by means of transgenic mice and lineage tracing or transplantation of cells defined by phenotype, have started to unravel the complexity of the bone marrow (BM) HSC niches (Kfoury and Scadden, 2015, Mendelson and Frenette, 2014, Morrison and Scadden, 2014, Pinho and Frenette, 2019). Several types of microenvironmental cells are involved in HSC regulation, including neural, hematopoietic, and stromal cells. Stromal cell types essential for HSC maintenance include vascular endothelial cells and perivascular cells, peri-sinusoidal on the abluminal side of the endothelial lining of BM sinusoids, and peri-arteriolar in the media and adventitia of small BM arterioles (Acar et al., 2015, Asada et al., 2017, Chen et al., 2016, Ding and Morrison, 2013, Ding et al., 2012, Greenbaum et al., 2013, Kunisaki et al., 2013, Oguro et al., 2013, Sugiyama et al., 2006). Bone-forming osteoblasts also contribute to the HSC niche, in particular in irradiated animals transplanted with HSCs when perivascular and endothelial cells are impaired (Silberstein et al., 2016). Moreover, both perivascular and osteoblastic cell subsets are implicated in the regulation of distinctive, B-lymphoid and multipotent progenitors (Balzano et al., 2019, Cordeiro Gomes et al., 2016, Ding and Morrison, 2013, Greenbaum et al., 2013, Silberstein et al., 2016). Mesenchymal stromal cells (MSCs) may also be considered as niche components by virtue of their phenotype, perivascular location, pro-hematopoietic factor expression, and precursors of the osteoblastic lineage (Hu et al., 2016, Mendez-Ferrer et al., 2010, Morikawa et al., 2009, Pinho et al., 2013, Zhao et al., 2019, Zhou et al., 2014). MSCs with hematopoietic stem/progenitor cell (HSPC)-supportive ability have also been isolated from human fetal and adult BM by culture or by phenotype using different antigen membrane combinations, some of which are similar to those used for murine MSCs (Chan et al., 2018, Isern et al., 2013, Pinho et al., 2013, Sacchetti et al., 2007).
Recent transcriptomic and proteomic analyses at the single-cell level have helped define the gene sets characterizing the BM stromal populations under steady state, after stress, or in diseased conditions (Balzano et al., 2019, Baryawno et al., 2019, Tikhonova et al., 2019, Wolock et al., 2019). These works first explored the cell heterogeneity and then inferred the possible capacity of support of the cell sets by unraveling their hematopoietic regulator profiles. On the contrary, in this work we aim at developing a strategy to first define the genes that orchestrate a functional phenotype and then investigate the cell populations contributing to that phenotype. In a previous work we have attempted to provide such information, defining several networks characterizing HSC-supportive stromal cells (Charbord et al., 2014). However, our previous study suffered from the following limitations: (1) it included too few observations for statistically sound correlation studies; (2) it did not indicate which genes in the characteristic gene sets were the best classifier for the corresponding cell population; (3) the described networks characterized a core gene set for stromal cells from different developmental origin, and consequently could not apply to defined BM stromal populations; and (4) the described networks were based on the analysis of gene-to-gene correlations; consequently most of the inferred edges included a number of indirect paths resulting from transitive correlations, which did not indicate causal relationships.
We undertook the present work to remedy those limitations. We enlarged the number of genes of interest by filtering out a minimal number of genes. We also considerably enlarged the number of observations by including transcriptomes selected from available databases according to well-defined criteria. The distribution of the samples in the 3-dimensional space defined by principal-component analysis (PCA) then enabled to identify a population of BM HSPC-supportive stromal cells, subsequently dissected in clusters corresponding to stromal cell subsets. We applied a discriminant analysis pipeline to identify the genes predominantly expressed by the different cell sets and analyzed the different cell populations to unravel the gene relatedness (Horvath, 2011, Langfelder and Horvath, 2008). Moreover, we took advantage of the numerous observations provided by recently published single-cell transcriptomes (Tikhonova et al., 2019) to disclose in the perivascular network the direct gene-to-gene interactions and reveal oriented links based on the signature of causality (Sella et al., 2018, Verny et al., 2017). Finally, as proof of concept, we studied the protein expression and activity of the top most discriminant and connected gene, the Wnt signaling pathway facilitator R-spondin 2 (Rspo2), and validated that it was expressed by a small set of perivascular cells, and acted in concert with the Stem Cell Factor (SCF) (also known as Kit Ligand) to amplify ex vivo hematopoietic precursors.
Results
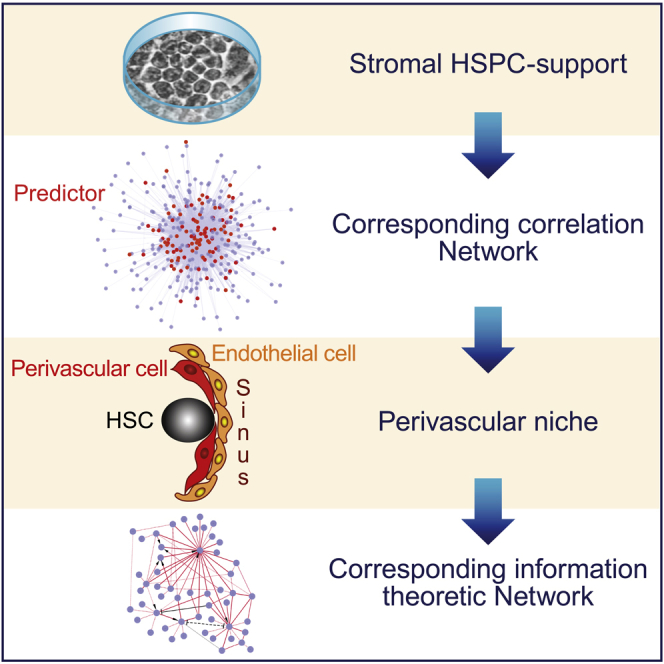
The major aim of our work was to unravel how genes expressed by the different stromal niche populations were related to one another, i.e., how genes were organized around central hubs serving as keystones of the network. To achieve this goal we devised a strategy of analysis consisting of a sequence of steps as indicated in the flowchart shown in Figure 1A.
Figure 1.

Study Flowchart and Cell Cluster Identification
(A) Study flowchart. Sup, supportive; Nsup, non-supportive; BM, bone marrow; O, osteoblast; V, perivascular cell; MSC, mesenchymal stromal cell; PCA, principal-component analysis; PAMr, Prediction Analysis for Microarrays; LIMMA, linear model for microarray analysis; SAM, Significance Analysis of Microarrays; WGCNA: Weighted Gene Correlation Network Analysis; miic, multivariate information-based inductive causation algorithm.
(B) PCA highlighting the different clusters. Left panel: The 3D score plot (PC1, PC1, PC3) using the entire sample set (224 samples) as observations and the Set1 (1,859 genes) as variables. Right panel: The 3D score plot using the entire sample set as observations and the entire set of protein-coding genes (15,056 genes) as variables. Sup-BM, supportive BM samples; Sup-EM, supportive extra-medullary samples; Nsup-BM, non-supportive BM samples; Nsup-Other, non-supportive samples of other origin than BM.
(C) PCA highlighting the cell sources (cell lines versus primary cells).
(D) Hierarchical clustering highlighting the different clusters. Euclidian distances and Ward's linkage were used.
see also Figure S1.
Configuring a Dataset Representative of Stromal Cells of Mesenchymal Origin
Our starting hypothesis was the same as that published in 2014 (Charbord et al., 2014), considering that essential genes for HSC support would be those differentially expressed by at least two of the major developmental sites of definitive hematopoiesis. Thus, we used the same stromal cell lines as before, from the aorta-gonad-mesonephros (AGM), fetal liver (FL), and BM, either with supportive activity in ex vivo co-culture experiments with HSPCs or with reportedly less or non-supportive ability. To initiate the present study, we filtered out a minimal number of genes as the subsequent analyses (Weighted Gene Correlation Analysis [WGCNA], discriminant) constituted by themselves dimensional reduction methods. Thus, for comparing supportive with less supportive lines we now took into account all differentially expressed genes given by ANOVA (2-way with support, site and interaction as grouping variables) with a low-stringency p value < 0.06. The different genes sets were then intersected, which resulted in more than a 4-fold increase in the number of retained genes when compared with our previous study (2,327 and 481 genes in the present and previous work, respectively). The Venn diagram is shown in Figure S1A. The resulting gene set was designated as Set 1.
To account for the diversity of stromal cells, we selected in the publicly available databases datasets corresponding to articles wherein the reported supportive capacity had been evidenced by one or several of the following criteria (Table S1): (1) generation of hematopoietic colonies in co-culture of HSPCs with stromal cells, (2) hematopoietic differentiation of embryonic stem cells co-cultured with the stromal cells, (3) in vivo co-localization of stromal cells expressing specific markers with HSCs, (4) lineage reconstitution in vivo by HSCs co-cultured with stromal cells for variable time spans, (5) reduction of the HSC pool in vivo after depletion of the stromal cell population, and (5) characteristic phenotype (e.g., expression of the major hematopoietic regulators Kitl and Cxcl12). The relative relevance of the six criteria in the ensemble of articles is shown in Figure S1B. The supportive datasets included mesenchymal cells from the AGM, spleen, FL, and BM, either derived as continuous cell lines or primary cells isolated from the different tissues by phenotype (and in one case by culture) from transgenic or wild-type mice. The non-supportive datasets included not only stromal cells unable to support HSPCs according to the aforementioned criteria but also pluripotent stem cells (induced or embryonic), either undifferentiated or differentiated into the neural-ectodermal or endodermal lineages, and adult cardiac cells. In all, by analyzing 30 datasets we extracted 224 samples. A normalization step was necessary to get rid of the batch effect (Figure S1C). When genes corresponding to Set 1 were mapped in all the datasets, a gene count of 1,869 genes remained. The final expression matrix was therefore 224 × 1,869 (observations × variables). Details on samples and the list of genes belonging to Set 1 are given in Tables S1 and S2, respectively.
PCA of the Dataset Allows Identifying Four Distinct Cell Clusters
Using the 224 samples and the 1,869 Set 1 genes PCA revealed four major clusters (Figure 1B, left panel). The p value of group discrimination evaluated by correlation on the first principal component (PC) axis was highly significant (p = 2.45 × 10−190). The BM stromal cells segregated in two homogeneous clusters (colored green and magenta) with 69 and 9 samples of supportive cells and non-supportive cells, respectively (Sup-BM and Nsup-BM). The blue cluster included 47 samples of supportive extra-medullary (Sup-EM) cells (26 AGM, 18 FL, and 3 spleen samples). Finally, the red cluster included 99 samples of non-supportive cells (Nsup-Other). The covariate cell source (primary cells or continuous cell lines) was not relevant in the context of this study because it did not generate sub-groups within the clusters (Figure 1C). As expected, the four clusters were not discriminated from one another when including all 15,056 genes, although a gradient from non-supportive to supportive samples was apparent (Figure 1B, right panel). Hierarchical clustering confirmed the four major clusters (Figure 1D).
One Gene Network Characterizes the BM Stromal Supportive Capacity
Analysis of the score plots in Figure 1B indicated that the PC1 representing 35% of the variance corresponded to the contrast between Sup-BM and Nsup-Other. To understand how Set 1 genes correlated to this trait were inter-related, we applied WGCNA (Horvath, 2011, Langfelder and Horvath, 2008), which enabled identifying one module (sub-network) named turquoise whose eigengene positively correlated to the trait (Figures 2A and S2). This module included 758 genes with significant (p < 0.05) gene significance and module membership. This gene set characteristic of the Sup-BM cell population was designated as Set 2 (Table S3). Gene Ontology analyses (Huang da et al., 2009a, Huang da et al., 2009b) indicated that Set 2 genes included molecules of the extracellular matrix (ECM) and implicated in the ECM-to-cell receptor interaction, and genes implicated in protein processing in the endoplasmic reticulum (glycosyl transferases and members of the glycosaminoglycan and N-glycan biosynthesis pathways) (Figure 2B). Importantly, genes belonging to the canonical Wnt signaling pathway (such as Fzd1, Porcn) were also enriched in the turquoise module. The most connected genes of the turquoise module are represented on the connectivity plots (Figure 2C). Of the two major hematopoietic regulators Kitl and Cxcl12, the former was included in the gene set, but not the latter due to its high expression in FL non-supportive lines and in some pluripotent stem cell lines. Among the other molecules some were soluble mediators (e.g., Rspo2, Srpx2, Fam20a, Il7), membrane receptors (e.g., Lpl), ECM components (e.g., Col1a2, Efemp1, Tgfbi), adhesion molecules (e.g., Svep1), and transcription factors (e.g., Ebf3, Snai2, Prrx1).
Figure 2.

One Gene Network Characterizes the BM Stromal Supportive Capacity
(A) Module detection by clustering using the Topological Overlap Matrix (TOM) and merged dynamic tree cut method. The merged dynamic hybrid branch cutting method was used to define the module named by color. Parameters: power: 14, adjacency: signed; correlation: Pearson's; linkage: average; minimum module size: 30. The gene Set 2 corresponds to the set of 758 genes belonging to the turquoise module.
(B) Major Gene Ontology (GO) categories corresponding to Set 2 genes. ER, endoplasmic reticulum; ECM, extracellular matrix; GAG, glycosaminoglycan.
(C) Connectivity plots of genes belonging to Set 2. Left panel: Intra-modular connectivity bar plot of the top 30 genes. Right panel: Cytoscape connectivity plot of the 249 most connected genes; genes were selected for representation above a threshold weight ≥ 0.1228.
(D) GSEA plot using as reference the gene Set 2, as expression dataset the matrix of all genes and samples, and as phenotype labels Sup versus Nsup.
(E) Misclassification error plot using the PAMr algorithm.
(F) Volcano plot obtained using the LIMMA algorithm with Rspo2 expression as quantitative linear predictor (only genes linearly dependent on Rspo2 are represented).
(G) Last step of the discriminant analysis (118 genes, “Discriminant 2” predictors). Scatterplot of the Sup-BM PAMr score versus the fold change (FC) given by the SAM algorithm (FC of gene expression in Sup-BM versus all other populations).
(H) Hierarchical clustering (Euclidian distance, average linkage) and heatmap of the 118 ′Discriminant 2′ genes.
(I) Gene intra-modular connectivity versus Sup-BM PAMr score scatterplot for the 28 most discriminant genes.
See also Figures S2 and S3.
To further confirm that the turquoise module gene set clearly discriminated the Sup-BM samples from all others, we performed gene set enrichment analyses (GSEA) (Liberzon et al., 2015, Subramanian et al., 2005). Using the Sup-BM gene set as reference, the enrichment plot was clearly shifted to the left corresponding to up-regulated genes in the Sup-BM cell population (Figure 2D).
To uncover the best classifiers in the Sup-BM cluster we followed several steps through a customized pipeline. First, using the Prediction Analysis for Microarrays (PAM) algorithm (Tibshirani et al., 2002), supervised machine learning analysis was carried out on the four clusters observed in PCA. Leave-one-out with cross-validation indicated that the gene Set 1 enabled effective discrimination between each cluster because the misclassification error was nil when a threshold value of 1,401 genes was reached (Figure 2E). The best 939 genes predictive of BM support were then selected and applied to a Random Forest algorithm (Breiman, 2001). After performing learning on training dataset (n = 168 samples randomly selected) the model was built on 100 trees with optimization of the number of features needed to perform the tree split (the “mtry” parameter) for a minimal error of tree aggregations (bagging). No misclassifications of samples were found using the validation set (n = 52 samples randomly selected) when testing the Sup-BM cluster versus other clusters pooled in one single class. ANOVA on the best 720 discriminating genes (“Discriminant1” gene set) indicated that Rspo2 was the best predictive as it was up-regulated uniquely in the Sup-BM cluster. This gene was therefore used as quantitative predictor in linear models such as “linear model for microarray analysis” (LIMMA) (Figure 2F) (Ritchie et al., 2015). Significance Analysis for Microarray (SAM) (Tusher et al., 2001) was performed in parallel by supervised analysis between independent clusters (Sup-BM versus others). The intersection between SAM and LIMMA using Rspo2 as predictor allowed obtaining a gene set of 118 genes called “Discriminant 2” (Figure 2G and Table S3). By unsupervised classification, this gene set achieved a perfect discrimination of Sup-BM when compared with all others (Figure 2H). Remarkably, the “Discriminant 2” gene set was included within the Set 2 (Table S3). The combination of the results given by the discriminant analysis on the one hand and WGCNA and GSEA on the other enabled identifying the most discriminative and most connected genes, foremost Rspo2 (Figure 2I).
A Similar Gene Network Characterizes the BM Stromal Supportive Capacity after Inclusion of Human Samples
To investigate whether the most discriminative and connected genes found in mouse BM supportive stromal cells were also characteristic of human samples we included in the study five datasets (24 samples) reporting transcriptomes of supportive or non-supportive MSCs, either primary layers or cell “strains” pooling a few clones. Details are given in Table S1. The genes corresponding to Set 1 were selected in all databases, giving a final gene count of 1,619 (instead of 1,871 due to difference in species, some of the murine genes, such as Ly6a, having no corresponding human homolog). The final expression matrix was therefore 248 × 1,619 (observations × variables). In the PCA the human samples merged well with the murine ones, and the clusters were similar to those obtained with murine samples alone (Figure S3A). WGCNA allowed identifying the turquoise module whose eigengene was positively and most significantly positively correlated to PC1 (p = 10−137). Most connected genes in the mouse turquoise network were among the most connected genes in the human + mouse blue module (e.g., Kitl, Rspo2, Ebf3) (Figure S3B). In conclusion, the inclusion of human BM samples had not introduced major modifications of the gene organization, suggesting its inter-species conservation.
The Supportive Capacity Is Implemented in Four BM Niche Cell Populations
As cultured stromal cell lines do not necessarily represent uncultured primary cells directly isolated from the in vivo BM niches, we selected the 51 samples corresponding to primary HSPC-supportive stromal cells isolated by phenotype (and in one case by culture) from transgenic or wild-type mice (Table S1). To investigate in which cell types the genes characteristic of supportive stromal cells were expressed and how they were organized, we analyzed the gene expression in these 51 primary cell samples, taking into account the 758 Set 2 genes up-regulated in the Sup-BM turquoise module described earlier. In the PCA the samples segregated into three clusters (Figure 3A, upper panel), which were confirmed by hierarchical clustering (Figure S4A). In the PCA the cluster on the right included 23 samples designated perivascular cells (V) as they were defined by the expression of the markers Nestin, Leptin receptor, Chondroitin sulfate proteoglycan, Glutamyl aminopeptidase, the cytokine SCF, or the chemokine Stromal cell-derived factor 1 (Asada et al., 2017, Balzano et al., 2019, Chan et al., 2013, Decker et al., 2017, Ding et al., 2012, Greenbaum et al., 2013, Kunisaki et al., 2013, Mendez-Ferrer et al., 2010). The cluster on the left included 15 samples designated osteoblastic cells (O) as they were defined as Sca-1neg, but CD166pos/ne, Pdgfra or CD51pos cells, or derived from Col2.3 mice (Eash et al., 2010, Nakamura et al., 2010, Schepers et al., 2013). The cluster in the middle included 11 samples designated MSCs as they were defined as Sca-1pos, but Pdgfrapos or CD166neg cells, or as adherent cells from a clone cultured at high density (Greenbaum et al., 2013, Mead et al., 2017, Nakamura et al., 2010, Shoshani et al., 2014); it also included two samples of cells expressing Myosin-11 (Asada et al., 2017), which was probably related to their expression of the Sca-1 membrane antigen (Kunisaki et al., 2013) that also specifies PαS MSCs (Morikawa et al., 2009). PC1 (39.4% of the variance) corresponded to the contrast between the V and O clusters (PC1 trait), whereas PC2 (13.5% of the variance) corresponded to the contrast between the MSC cluster (including the two Myosin-11-positive cells) and the two others (PC2 trait).
Figure 3.

The Supportive Capacity Is Implemented in Four BM Niche Cell Populations
(A) PCA and module eigenvectors corresponding to samples of osteoblasts (O), perivascular cells (V), and MSCs. Upper panel: PC1 versus PC2 score plot using 51 samples and the 758 Set 2 genes. Lower panel: plot of the module eigenvectors versus PC1 and PC2 traits for the four modules identified by WGCNA.
(B) Module detection by clustering using the Topological Overlap Matrix (TOM) and merged dynamic tree cut method. See legend of Figure 2A. Parameters: power: 6; adjacency: signed; correlation: Pearson's; linkage: complete; minimum module size: 30.
(C) Connectivity plots of genes belonging to the V module. Right panel: Intra-modular connectivity bar plot of the top 30 genes. Left panel: Cytoscape connectivity plot of the 109 most connected genes; genes were selected for representation above a threshold weight ≥0.4623.
(D) Discriminant analysis identifying the O-, V-, and MSC-specific genes. Hierarchical clustering classification (Euclidean distance, Ward linkage) performed on the 61 best predictors.
See also Figure S4.
In accord with the merged dynamic tree cut, WGCNA identified four modules named red, blue, magenta, and black (Figures 3B, S4B–S4D and Tables S4 and S5). The module eigengene correlations (Figure 3A, lower panel) to PC1 and PC2 traits and the analysis of the genes expressed in each module enabled characterizing the red and magenta modules as specific to V and O cells, respectively. Similarly, the black and blue modules were specified as MSC osteolineage-primed (MSC-Op) and perivascular-primed (MSC-Vp), respectively. The V module (Figure 3C) included the major hematopoietic regulator Kitl and the transcription factor Ebf3 reported to maintain the supportive capacity and the architecture of the HSC niche (Seike et al., 2018). Its core of 109 highly correlated genes (out of 167) was designated as Set 3 (Table S4). The O module (Figure S4F) included the skeletal mineralization-inducing enzyme Alpl and the osteogenic transcription factor Runx2. The MSC-Op module (not shown) included the ECM components Col1a1, Col1a2, and Sparc, all implicated in ossification. Finally the MSC-Vp module (not shown) included the transcription factor Prrx1 essential for the development of vascular and perivascular matrix (Bergwerff et al., 2000), and the ECM components Emilin2 and Thsd7a implicated in vessel assembly and endothelial cell migration. KEGG pathway analysis confirmed the characterization of the V and MSC-Vp modules, canonical Wnt signaling and focal adhesion pathways being preeminent for genes in V and MSC-Vp modules, respectively (Figure S4E).
The proportion of the sum of genes in the niche cell-specific networks (V, MSC-Vp, O, MSC-Op) to the total number of genes in the Sup-BM network was 78.5%. However, the hubs in niche cell-specific networks were not necessarily hubs in the Sup-BM network. This result showed that the gene components found in the niche cell-specific networks were rewired in a different circuitry in the Sup-BM network.
To investigate which genes were predominantly expressed in each population we used the PAMr algorithm identifying 61 discriminant genes (designated “Discriminant 3”), including Ebf3 and Cd302 (also known as Clec13a or Dcl1) for V and Alpl and Runx2 for O (Figure 3D and Tables S4 and S5). The combination of results given by discriminant analysis and WGCNA enabled identifying the most discriminative and most connected genes in V and MSC-Vp modules (Figure S4H).
Single-Cell-Level Analysis Identifies High-Confidence Paths in the Perivascular Cell Network
To refine the gene network characterizing the perivascular cell population, we analyzed a recent database (GSE108891) providing the transcriptome of the BM microenvironment at the single-cell level (Tikhonova et al., 2019). This publication compared three cell populations of vascular endothelial cells expressing VE-cadherin, perivascular cells expressing Leptin receptor, and osteoblasts expressing Col2.3. Different sub-populations were identified, some of which expressed the major pro-hematopoietic mediators implicated in cell-to-cell communication.
First, we confirmed that Set 3 including the 109 most connected genes of the V module specified the perivascular population. We randomly selected 100 cells from each of the three populations and applied PCA using either the whole transcriptome or the gene Set 3. The three cell populations were not segregated when including the entire transcriptome, whereas they were clearly resolved when using the Set3 (Figure 4A). The genes with the highest negative PC2 loadings were Kitl, Cd302, and Ebf3. Second, we applied t-distributed stochastic neighbor embedding (t-SNE) to the entire population of perivascular cells (1,712 cells) (Figure 4B). Some genes were expressed at high level in almost all cells (e.g., Marcks), whereas other genes were expressed in only a fraction of the cells. It was roughly possible to distinguish two regions, a large cluster A (circled red) including cells expressing genes of the V module (e.g., Rspo2 and high amounts of Kitl) and a smaller cluster B (circled green) including cells expressing genes of the O module (e.g., Alpl and Runx2).
Figure 4.

Single-Cell-Level Analysis Using a Recently Described Algorithm Identifies High-Confidence Paths in the Perivascular Cell Network
(A) PCA using either the whole transcriptome (left) or the Set 3 gene set (right) and 100 randomly selected genes from each of the three populations (VE-cadherin-positive, Leptin receptor-positive, and Col2.3-positive).
(B) t-SNE of example genes expressed in the Leptin receptor-positive population at baseline. All 1,712 cells were included. Negative cells are in gray, and scaled positivity for a given gene is in different hues of blue.
(C) Miic plot of the Set3 genes expressed in the Leptin receptor-positive cell population at baseline. The matrix consisting of 1,712 observations (cells of the Leptin receptor-positive population at baseline) and 109 variables (the Set 3 genes) was submitted to the miic algorithm. 52 genes (nodes) were retained. Edge width increases with degree of confidence in the link as given by miic. The edge line is continuous if the link was found both by miic and WGCNA, dotted if the link was found only by miic, and dashed if there is a common latent regulator found by miic. The edge color in red corresponds to a positive correlation by miic; in black, to a negative correlation by miic; and in light blue, to a positive correlation by WGCNA. The edge head pointed corresponds to an activation, and the edge head blunted corresponds to an inhibition as indicated by miic.
(D) Miic plot of the Set3 genes expressed in the Leptin receptor-positive cell population after stress (treatment with 5-fluorouracil [5-FU]). The matrix consisted of 3,467 observations (cells of the Leptin receptor-positive population after 5-FU treatment) and 109 variables. The new hub and linked targets appearing in condition of stress is encircled.
As the Set 3 genes specified the perivascular population corresponding to the major cell cluster A we took advantage of the large number of Leptin receptor-positive (LEPR) single cells to refine the V gene network. To identify direct paths between genes including causal relationships and inferring latent common regulators of expressed genes we applied the multivariate information-based inductive causation algorithm (miic) (Sella et al., 2018, Verny et al., 2017), submitting to it the matrix consisting of 1,712 observations and 109 variables (the Set 3 genes). As expected only a fraction of nodes (52) defining direct high-confidence paths were retained (Figure 4C). There were two major hubs, Kitl and Cd302. Remarkably, Kitl was linked to Rspo2, Il7, Ebf3, and Snai2. Moreover, the miic algorithm uncovered a number of “v-structures” (e.g., Gja1-- > Ctsk < -- Ostc) suggesting cause-and-effect interactions and the presence of latent regulators (e.g., a latent negative regulator upstream of Srpx2 and Kitl).
Tikhonova et al. studied the transcriptome of LEPR cells not only under steady-state condition (baseline) but also after stress, i.e., after treatment of the mice with the drug 5-fluorouracil. We introduced therefore in our study this dataset including 3,467 cells. As shown by miic analysis the network was largely modified with a large increase in the number of nodes (89) and appearance of new hubs such as Marcks (Figure 4D). However, the hubs observed in baseline condition, in particular Cd302 and Kitl, were still present and Kitl retained its direct links with Rspo2, Il7, Ebf3, and Snai2. There was also a large increase of new (not found in cells at baseline) “v-structures” (e.g., Marcks-- > Ncam < -- Srpx2 or Ltbp1-- > Ncam < -- Srpx2). In short, by means of miic we were able to prune and remodel the gene network generated by WGCNA, which confirmed Kitl as major hub for the perivascular cell network, as well as identify possible causal interactions and show major network remodeling in cells responding to stress.
The Wnt Signaling Pathway Facilitator R-Spondin 2 Acts in Concert with the Kit Ligand to Amplify Ex Vivo Hematopoietic Precursors
In our analysis, Rspo2 appeared as the best predictor of the Sup-BM capacity and was directly linked related to Kitl coding for the SCF widely recognized as a major stromal regulator of HSC attributes. R-spondin 2 (encoded by Rspo2) is known as a Wnt signaling pathway facilitator (Niehrs, 2012). Thus, to validate the aforementioned integrative analyses to define candidate stromal regulators of hematopoiesis, we studied the cell distribution of R-spondin 2 on BM biopsies and its effect on HSPC in vitro.
Histology of murine BM showed that R-spondin 2 was expressed in a subset of the LEPR peri-sinusoidal cell population, which confirmed the results of the single-cell transcriptome analysis described earlier (Figure 5A). It was also expressed in some LEPR-negative hematopoietic cells.
Figure 5.

The Wnt Signaling Pathway Facilitator R-Spondin 2 Acts in Concert with the Stem Cell Factor (SCF) to Amplify Ex Vivo Hematopoietic Precursors
(A) Expression of R-spondin 2 in peri-sinusoidal cells. R-spondin 2-positive/Leptin receptor-positive (LEPR) peri-sinusoidal cells: continuous arrows; R-spondin 2-negative/LEPR-positive peri-sinusoidal cells: discontinuous arrows; S, sinusoids; IsoB4, isolectin B4 labeling of endothelial cells and macrophages. Scale bar, 10 μm.
(B) Correlation of Rspo2 to Kitl expression values in all studied samples.
(C) Effect of R-spondin 2 on Linneg/Sca-1pos/Kitlpos(LSK) cell number. SCF (10 ng/mL) was present in all tested conditions, whereas Wnt-3a (100 ng/mL) and R-spondin 2 (20 ng/mL) were as indicated. N = 60 per condition (12 experiments with 5 wells per condition). Data are presented as mean ± SEM. Paired t test ∗∗∗p < 0.0001, ∗∗p < 0.001 in paired comparisons.
(D) Effect of R-spondin 2 on LSK cell phenotype (one representative experiment out of 8). Flow cytometry analysis was carried out on cells collected at day 12 and cultured in the presence of 10 ng/mL SCF alone (upper panels), 100 ng/mL SCF + Wnt-3a (middle), or 20 ng/mL SCF + R-spondin 2 (lower). To obtain a more precise evaluation of the Sca-1pos/Kitpos cells we first selected the Ter119neg/Gr-1neg cells before evaluating in this gate the % of Sca-1pos/Kitpos cells.
(E) Effect of R-spondin 2 and Wnt-3a on Wnt downstream targets Tcf7, Bmp4, and Axin2 and on the R-spondin receptor Lgr6. Expression level as 2-ΔCq, ΔCq = Cq(gene of interest) - Cq(housekeeping gene), housekeeping gene being Gapdh. N = 5 per condition. Box limits are the 25th and 75th percentages, center lines are mean, and the whiskers are the mean SD values. Data comparison by paired t test. ∗∗p < 0.01, ∗p < 0.05 in paired comparisons.
(F) Effect of the ß-catenin signaling inhibitor increasing concentrations of PKF118-310 (ng/mL) in the presence (left) or absence (right) of R-spondin 2 on LSK cell number. N = 14 per condition. Legend as in (E). ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 in paired comparisons.
Over the whole range of our 224 samples, the expression level of Rspo2 was positively correlated, in a highly significant way, to that of Kitl, being the seventh ranked gene among the top correlated ones (Figure 5B). To validate the functional consequences of Rspo2 expression, we studied the effect of the recombinant R-spondin 2 on hematopoietic precursors in the presence or not of recombinant SCF and compared the effect with that of the canonical Wnt pathway effector Wnt-3a. Linneg/Sca-1pos/Kitpos (LSK) cells were sorted from the BM of C57BL/6 mice and seeded in methylcellulose for colony-forming unit (CFU) evaluation in the presence of a range of R-spondin 2 concentrations (10, 20, and 50 ng/mL) with and without hematopoietic cytokines and Wnt-3a (100 ng/mL). CFUs were observed only in the presence of cytokines, without additional effect of Wnt-3a or R-spondin 2 (not shown). In a second set of experiments sorted LSK cells were seeded in 96-well plates (200 cells per well) in the presence of 10 ng/mL SCF, 20 ng/mL R-spondin 2, or 100 ng/mL Wnt-3a or a combination of these. After 12 days in culture in the presence of SCF alone the number of cells increased by 19-fold (Figure 5C) and cells differentiated into a majority of Gr-1pos granulocytes and monocytes (≈49% of the events), CD41pos megakaryocytes (≈2%), and Ter119pos erythroblasts (≈12%), whereas the percentage of Sca-1pos/Kitpos dropped to ≈1.9% (Figure 5D). No growth was observed in the presence of R-spondin 2 or Wnt-3a without SCF (not shown). When R-spondin 2 and Wnt-3a were added to SCF the number of cells increased by 32- and 27-fold, respectively (Figure 5C) and the pattern of myeloid differentiation was unchanged (Figure 5D). On the contrary, there was no amplification in cultures supplemented with the three factors (SCF + R-spondin 2 + Wnt-3a) when compared with cultures with SCF alone. When compared with cultures with SCF alone, increases in amplification factor were highly significant in cultures with SCF + R-spondin 2 or SCF + Wnt-3a (Figure 5C). The percentage of Sca-1pos/Kitpos increased by 3- to 4-fold in cultures with SCF + R-spondin 2, but only in five of eight cases (Figure 5D).
To verify that the proliferative effect of R-spondin 2 was mediated through the Wnt canonical pathway we carried out two sets of experiments. First, using qRT-PCR, the levels of the major Wnt downstream targets Axin2, Tcf7, and Bmp4 were measured after 24 h of treatment with R-spondin 2, Wnt-3a, and their combination (Figure 5E). These experiments showed that R-spondin 2 had no detectable effect by itself, contrarily to Wnt-3a that induced, as expected, a significant increase. Importantly, the combination of Wnt-3a and R-spondin 2 significantly increased the effect even more when compared with Wnt-3a alone, confirming the facilitating role of R-spondin 2. Interestingly, in our culture system Wnt-3a significantly increased the expression of the R-spondin receptor Lgr6 without additional effect of R-spondin 2 (Figure 5E), whereas the levels of the other receptors Lgr4 and Lgr5 remained low to nil (not shown). Second, sorted LSK cells were cultured in the presence of R-spondin 2 and with the β-catenin signaling chemical inhibitor PKF118-310 at increasing concentrations (Figure 5F). The inhibitor in the presence of R-spondin 2 induced a significant dose-response inhibition of LSK proliferation, which was not observed in the absence of R-spondin 2.
Taken together, these data indicate that, in the presence of SCF, R-spondin 2 has a direct proliferative effect on LSK cells, but does not modify the myeloid pattern of differentiation. This effect is mediated by the Wnt canonical pathway, which confirms the facilitating role of R-spondin 2 in our experimental setting.
Discussion
In this study on the BM microenvironment using a combination of algorithms, one of which recently applied to identify reliable paths and network v-structures in complex systems (Sella et al., 2018, Verny et al., 2017), we have identified a gene network characteristic of the HSC-supportive capacity of BM stromal cells and then defined gene networks for the different supportive niche cell populations contributing to that phenotype, i.e., perivascular cells (V), osteoblasts (O), and MSCs including perivascular-primed (MSC-Vp) or osteoblast-primed (MSC-Op) cells. Comparison of the gene networks specific to the niche cell populations with the network characterizing the HSC-supportive capacity as a whole made clear that the supportive phenotype resulted from the coordinated effect of several niche cell populations. The major hematopoietic regulator Kitl was a major hub in the network characterizing the supportive perivascular stromal cells, and the facilitator of the Wnt signaling pathway Rspo2 appeared as the best predictor of the supportive capacity and was directly linked to Kitl in the perivascular cell network. We have therefore validated the biological relevance of R-spondin 2 by showing that this protein enhanced the proliferation of hematopoietic precursors in the presence of SCF. This study by identifying many classifiers and hubs beside Rspo2 and Kitl constitutes a resource to unravel candidate BM stromal mediators.
In recently published analyses of transcriptomes where a large number of stromal cells were studied at the single-cell level the primary purpose has been to identify stromal cell sets, and the secondary purpose to infer possible capacity of support of the cell sets by unraveling their hematopoietic regulator profiles. Bearing in mind the opposite difference of approaches as here we first defined the genes that organize a functional phenotype and then investigate the cell populations contributing to that phenotype, one may attempt to compare how subsets resulting from our analysis relate to subpopulations resulting from single-cell analysis. Comparison of our gene sets with data reported by Tikhonova et al. indicates that the V and O clusters in our analysis broadly relate to adipocyte-biased P1 and P2 and osteo-primed P3 and P4 clusters of the LEPR population, respectively (Tikhonova et al., 2019). Further comparisons indicate that our V cluster is related to the MSC cluster 1 expressing Lepr in Baryawno et al. (Baryawno et al., 2019), to the MSC P3 cluster in Wolock et al. (Wolock et al., 2019), and to the cluster of peri-sinusoidal stromal cells in Balzano et al. (Balzano et al., 2019). In our work, MSCs, prospectively specified using a distinct phenotype or selected by growth in culture, were found to constitute a heterogeneous population including cells expressing genes implicated either in ossification (MSC-Op) or in vascularization (MSC-Vp). Taken together our work and the works using single-cell analysis identify a perivascular cell population with HSC-supportive capacity, in part due to the expression of known hematopoietic regulators. Moreover, whereas single-cell analyses help establish subsets within a mixed population and unravel differentiation trajectories between these subsets, network analysis gives important insight in the organization of gene expression within the population as a whole and indicates how networks in different cell sub-populations are coordinated to orchestrate the global functional phenotype.
As analyses, such as WGCNA, are optimally suited to identify correlated networks, but do not identify direct gene-to-gene interactions or potential causal links, we used miic analysis (Sella et al., 2018, Verny et al., 2017), recently extended to analyze continuous or mixed-type (e.g., continuous-categorical) data (Cabeli et al., 2020). The miic algorithm identifies direct paths with high confidence, and, as such, reveals potential cause-and-effect links as well as latent regulators, which may not show up when looking only at gene expression data. However, to yield significant results the miic algorithm requires a large number of observations. Consequently, we took advantage of the recently published transcriptomes of single LEPR cells (Tikhonova et al., 2019) to apply the algorithm using a subset of genes characterizing the perivascular V population. This approach enabled pruning and remodeling the network defined by WGCNA, thus providing a clearer picture by clearly individualizing the major hubs in different conditions.
The combined WGCNA and miic analysis confirmed that Kitl was a major hub. Kit ligand is a well-established major hematopoietic regulator; its in vivo deletion in perivascular stromal cells leads to dramatic depletion of the BM HSC pool (Asada et al., 2017, Ding et al., 2012). Moreover, in our miic analysis, we found that Kitl was directly linked to Rspo2, Il7, Ebf3, and Snai2, all genes known as hematopoietic regulators (this study and Balzano et al., 2019, Charbord et al., 2014, Seike et al., 2018). Furthermore, we also identified the C-type lectin Cd302 as another major hub. CD302 has been reported as implicated in dendritic cell migration (Lo et al., 2016), but it remains to be studied how this lectin is involved in the stromal niche regulation of HSCs.
In cells exposed to stress, we observed remodeling of the gene networks with a large increase in the number of direct paths and the appearance of new hubs, such as the cytoskeletal Ca2+ regulator Marcks, suggesting that the cells had modified their actin cytoskeleton pattern with possible effect on migration and adhesion. However, the hubs observed in baseline condition, in particular Cd302 and Kitl, were still present with maintenance of their major connections, suggesting that the gene organization observed at baseline is not replaced, but augmented under condition of stress.
In our analysis Rspo2 appeared as the best predictor of the Sup-BM capacity and was directly linked to Kitl in the perivascular cell network. R-spondin 2 is a facilitator of the Wnt signaling pathway. By binding to one of its Leucine-rich repeat-containing G-protein-coupled Receptors (LGR4-6) it forms a complex that hampers the degradation of the Wnt frizzled receptors through the RNF43 and ZNFR3 transmembrane ubiquitin ligases, thereby boosting Wnt signaling strength (de Lau et al., 2014, Niehrs, 2012). Both Wnt and R-spondin ligands are essential to impede the differentiation of Lgr5-positive intestinal stem cells and maintain their self-renewal capacity (Yan et al., 2017). The reported effects of Wnt signaling and stromal Wnt inhibitors have underscored the dose and environmental context dependency of the different mediators on HSC cycling and their regenerative capacity (Fleming et al., 2008, Renstrom et al., 2009, Ruf et al., 2016, Staal et al., 2016). We found that R-spondin 2 enhanced the proliferation of hematopoietic precursors in the presence of SCF. This effect was Wnt dependent, which is in agreement with similar studies on R-spondin in other cell types (Kazanskaya et al., 2004, van Andel et al., 2017). The effect might be mediated by the binding of R-spondin 2 to LGR6, which, we found, is the only receptor whose transcript is expressed at a significant level by the hematopoietic precursors. Alternatively, R-spondin 2 may regulate frizzled receptor stability through the RNF43/ZNFR3 ligases independently of the LGRs, through an as yet undefined co-receptor (Lebensohn and Rohatgi, 2018, Szenker-Ravi et al., 2018). By analyzing the recently published transcriptomes of single LEPR cells we found that the R-spondin 2-expressing cell population was a subset of LEPR perivascular cells, which was confirmed by the examination of BM biopsies showing a subset of R-spondin 2-positive cells on the abluminal side of sinusoidal endothelial cells. R-spondin 2 was also expressed in some Leptin receptor-negative hematopoietic cells, which is in agreement with the expression, mostly in the erythroblastic series, reported in Gene Expression Commons (https://gexc.riken.jp). Taken together, our analysis identifies R-spondin 2 as a yet unreported Wnt signaling pathway-associated stromal regulator of hematopoiesis.
Second to Rspo2, the genes coding for the soluble mediators Fam20a and Srpx2 were highly connected best predictors of the Sup-BM capacity. Fam20a has been isolated from a hematopoietic cell line treated with SCF and is expressed in myeloid progenitors (Nalbant et al., 2005). This gene whose mutations are responsible for amelogenesis imperfecta and nephrocalcinosis codes for a secreted pseudokinase implicated in biomineralization (Ohyama et al., 2016). Srpx2, an X-linked gene acting as a ligand for the urokinase plasminogen activator surface receptor, plays a role in angiogenesis by inducing endothelial cell migration and the formation of vascular network (Miljkovic-Licina et al., 2009). Srpx2 is upregulated in certain types of telocytes that are interstitial cells probably of mesenchymal origin found in multiple tissues (Zhu et al., 2015). Of note, another gene belonging to the Sup-BM module, Svep1, a ligand for integrin α9β1 that has been isolated from the BM stromal line MS-5 (Gilges et al., 2000), is also upregulated in certain types of telocytes (Song et al., 2019).
In conclusion, this study by combining different bioinformatics algorithms constitutes a resource to identify candidate BM stromal mediators whose effect on the different HSC attributes should be demonstrated in detail by upcoming experimental work.
Limitations of the Study
The present analysis of transcriptomes relies on published datasets, and, as such, unpublished transcriptomes of relevant cell types could not be taken into account. In addition, this work indicates that R-spondin 2 amplifies in the presence of SCF the proliferation of murine hematopoietic precursors in vitro. However, in vitro studies do not faithfully recapitulate HSC self-renewal as studied in in vivo transplantation assays. Finally, although Wnt signaling is mostly preserved across species, and the identified gene network included human samples, the effect of R-spondin 2 on murine cells may not entirely reproduce the response of human precursors to this Wnt mediator.
Resource Availability
Lead Contact
Further information should be directed to and will be fulfilled by the Lead Contact, Pierre Charbord (pierre.charbord@inserm.fr).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
This study did not generate new dataset, but analyzed datasets in public repositories. Accession numbers for all datasets analyzed are given in Table S1.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
Work supported by grants of the Fondation pour la Recherche Médicale (DEQ20100318258), Institut national de la santé et de la recherche médicale (Inserm), Centre national de la recherche scientifique (CNRS), and German Research Foundation (RO: DFG OO8/16). We thank Sophie Gournet for revising the illustrations.
Author Contributions
Conceptualization: P.C. and C. Desterke; Methodology: P.C., H.I., and C. Desterke; Software: C. Desterke, N.S., and V.C.; Formal analysis: P.C., H.I., and C. Desterke; Investigation: L.P., L.C., and N.C.; Resources: T.J.; Writing -Original Draft: P.C.; Writing -Review and Editing: P.C., R.A.J.O., T.J., C. Durand; Funding Acquisition: T.J.
Declaration of Interests
The authors declare no competing interests.
Published: June 26, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101222.
Supplemental Information
References
- Acar M., Kocherlakota K.S., Murphy M.M., Peyer J.G., Oguro H., Inra C.N., Jaiyeola C., Zhao Z., Luby-Phelps K., Morrison S.J. Deep imaging of bone marrow shows non-dividing stem cells are mainly perisinusoidal. Nature. 2015;526:126–130. doi: 10.1038/nature15250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asada N., Kunisaki Y., Pierce H., Wang Z., Fernandez N.F., Birbrair A., Ma'ayan A., Frenette P.S. Differential cytokine contributions of perivascular haematopoietic stem cell niches. Nat. Cell Biol. 2017;19:214–223. doi: 10.1038/ncb3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzano M., De Grandis M., Vu Manh T.P., Chasson L., Bardin F., Farina A., Serge A., Bidaut G., Charbord P., Herault L. Nidogen-1 contributes to the interaction network involved in pro-B cell retention in the peri-sinusoidal hematopoietic stem cell niche. Cell Rep. 2019;26:3257–3271.e8. doi: 10.1016/j.celrep.2019.02.065. [DOI] [PubMed] [Google Scholar]
- Baryawno N., Przybylski D., Kowalczyk M.S., Kfoury Y., Severe N., Gustafsson K., Kokkaliaris K.D., Mercier F., Tabaka M., Hofree M. A cellular taxonomy of the bone marrow stroma in homeostasis and leukemia. Cell. 2019;177:1915–1932.e6. doi: 10.1016/j.cell.2019.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergwerff M., Gittenberger-de Groot A.C., Wisse L.J., DeRuiter M.C., Wessels A., Martin J.F., Olson E.N., Kern M.J. Loss of function of the Prx1 and Prx2 homeobox genes alters architecture of the great elastic arteries and ductus arteriosus. Virchows Arch. 2000;436:12–19. doi: 10.1007/pl00008193. [DOI] [PubMed] [Google Scholar]
- Breiman L. Random forests. Mach. Learn. 2001;45:5–32. [Google Scholar]
- Cabeli V., Verny L., Sella N., Uguzzoni U., Verny M., Isambert H. Learning clinical networks from medical records based on information estimates in mixed-type data. PLoS Comput. Biol. 2020 doi: 10.1371/journal.pcbi.1007866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C.K., Lindau P., Jiang W., Chen J.Y., Zhang L.F., Chen C.C., Seita J., Sahoo D., Kim J.B., Lee A. Clonal precursor of bone, cartilage, and hematopoietic niche stromal cells. Proc. Natl. Acad. Sci. U S A. 2013;110:12643–12648. doi: 10.1073/pnas.1310212110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C.K.F., Gulati G.S., Sinha R., Tompkins J.V., Lopez M., Carter A.C., Ransom R.C., Reinisch A., Wearda T., Murphy M. Identification of the human skeletal stem cell. Cell. 2018;175:43–56.e21. doi: 10.1016/j.cell.2018.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charbord P., Pouget C., Binder H., Dumont F., Stik G., Levy P., Allain F., Marchal C., Richter J., Uzan B. A systems biology approach for defining the molecular framework of the hematopoietic stem cell niche. Cell Stem Cell. 2014;15:376–391. doi: 10.1016/j.stem.2014.06.005. [DOI] [PubMed] [Google Scholar]
- Chen J.Y., Miyanishi M., Wang S.K., Yamazaki S., Sinha R., Kao K.S., Seita J., Sahoo D., Nakauchi H., Weissman I.L. Hoxb5 marks long-term haematopoietic stem cells and reveals a homogenous perivascular niche. Nature. 2016;530:223–227. doi: 10.1038/nature16943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordeiro Gomes A., Hara T., Lim V.Y., Herndler-Brandstetter D., Nevius E., Sugiyama T., Tani-Ichi S., Schlenner S., Richie E., Rodewald H.R. Hematopoietic stem cell niches produce lineage-instructive signals to control multipotent progenitor differentiation. Immunity. 2016;45:1219–1231. doi: 10.1016/j.immuni.2016.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lau W., Peng W.C., Gros P., Clevers H. The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev. 2014;28:305–316. doi: 10.1101/gad.235473.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker M., Martinez-Morentin L., Wang G., Lee Y., Liu Q., Leslie J., Ding L. Leptin-receptor-expressing bone marrow stromal cells are myofibroblasts in primary myelofibrosis. Nat. Cell Biol. 2017;19:677–688. doi: 10.1038/ncb3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L., Morrison S.J. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. 2013;495:231–235. doi: 10.1038/nature11885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding L., Saunders T.L., Enikolopov G., Morrison S.J. Endothelial and perivascular cells maintain haematopoietic stem cells. Nature. 2012;481:457–462. doi: 10.1038/nature10783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eash K.J., Greenbaum A.M., Gopalan P.K., Link D.C. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. J. Clin. Invest. 2010;120:2423–2431. doi: 10.1172/JCI41649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming H.E., Janzen V., Lo Celso C., Guo J., Leahy K.M., Kronenberg H.M., Scadden D.T. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell. 2008;2:274–283. doi: 10.1016/j.stem.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilges D., Vinit M.A., Callebaut I., Coulombel L., Cacheux V., Romeo P.H., Vigon I. Polydom: a secreted protein with pentraxin, complement control protein, epidermal growth factor and von Willebrand factor A domains. Biochem. J. 2000;352(Pt 1):49–59. [PMC free article] [PubMed] [Google Scholar]
- Greenbaum A., Hsu Y.M., Day R.B., Schuettpelz L.G., Christopher M.J., Borgerding J.N., Nagasawa T., Link D.C. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. 2013;495:227–230. doi: 10.1038/nature11926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S. Springer; 2011. Weighted Network Analysis. Applications in Genomics and Systems Biology. [Google Scholar]
- Hu X., Garcia M., Weng L., Jung X., Murakami J.L., Kumar B., Warden C.D., Todorov I., Chen C.C. Identification of a common mesenchymal stromal progenitor for the adult haematopoietic niche. Nat. Commun. 2016;7:13095. doi: 10.1038/ncomms13095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W., Sherman B.T., Lempicki R.A. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W., Sherman B.T., Lempicki R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- Isern J., Martin-Antonio B., Ghazanfari R., Martin A.M., Lopez J.A., del Toro R., Sanchez-Aguilera A., Arranz L., Martin-Perez D., Suarez-Lledo M. Self-renewing human bone marrow mesenspheres promote hematopoietic stem cell expansion. Cell Rep. 2013;3:1714–1724. doi: 10.1016/j.celrep.2013.03.041. [DOI] [PubMed] [Google Scholar]
- Kazanskaya O., Glinka A., del Barco Barrantes I., Stannek P., Niehrs C., Wu W. R-Spondin2 is a secreted activator of Wnt/beta-catenin signaling and is required for Xenopus myogenesis. Dev. Cell. 2004;7:525–534. doi: 10.1016/j.devcel.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Kfoury Y., Scadden D.T. Mesenchymal cell contributions to the stem cell niche. Cell Stem Cell. 2015;16:239–253. doi: 10.1016/j.stem.2015.02.019. [DOI] [PubMed] [Google Scholar]
- Kunisaki Y., Bruns I., Scheiermann C., Ahmed J., Pinho S., Zhang D., Mizoguchi T., Wei Q., Lucas D., Ito K. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. 2013;502:637–643. doi: 10.1038/nature12612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder P., Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebensohn A.M., Rohatgi R. R-spondins can potentiate WNT signaling without LGRs. Elife. 2018;7:e33126. doi: 10.7554/eLife.33126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A., Birger C., Thorvaldsdottir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo T.H., Silveira P.A., Fromm P.D., Verma N.D., Vu P.A., Kupresanin F., Adam R., Kato M., Cogger V.C., Clark G.J. Characterization of the expression and function of the C-type lectin receptor CD302 in mice and humans reveals a role in dendritic cell migration. J. Immunol. 2016;197:885–898. doi: 10.4049/jimmunol.1600259. [DOI] [PubMed] [Google Scholar]
- Mead A.J., Neo W.H., Barkas N., Matsuoka S., Giustacchini A., Facchini R., Thongjuea S., Jamieson L., Booth C.A.G., Fordham N. Niche-mediated depletion of the normal hematopoietic stem cell reservoir by Flt3-ITD-induced myeloproliferation. J. Exp. Med. 2017;214:2005–2021. doi: 10.1084/jem.20161418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelson A., Frenette P.S. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat. Med. 2014;20:833–846. doi: 10.1038/nm.3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez-Ferrer S., Michurina T.V., Ferraro F., Mazloom A.R., Macarthur B.D., Lira S.A., Scadden D.T., Ma'ayan A., Enikolopov G.N., Frenette P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. 2010;466:829–834. doi: 10.1038/nature09262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miljkovic-Licina M., Hammel P., Garrido-Urbani S., Bradfield P.F., Szepetowski P., Imhof B.A. Sushi repeat protein X-linked 2, a novel mediator of angiogenesis. FASEB J. 2009;23:4105–4116. doi: 10.1096/fj.09-135202. [DOI] [PubMed] [Google Scholar]
- Morikawa S., Mabuchi Y., Kubota Y., Nagai Y., Niibe K., Hiratsu E., Suzuki S., Miyauchi-Hara C., Nagoshi N., Sunabori T. Prospective identification, isolation, and systemic transplantation of multipotent mesenchymal stem cells in murine bone marrow. J. Exp. Med. 2009;206:2483–2496. doi: 10.1084/jem.20091046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison S.J., Scadden D.T. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505:327–334. doi: 10.1038/nature12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y., Arai F., Iwasaki H., Hosokawa K., Kobayashi I., Gomei Y., Matsumoto Y., Yoshihara H., Suda T. Isolation and characterization of endosteal niche cell populations that regulate hematopoietic stem cells. Blood. 2010;116:1422–1432. doi: 10.1182/blood-2009-08-239194. [DOI] [PubMed] [Google Scholar]
- Nalbant D., Youn H., Nalbant S.I., Sharma S., Cobos E., Beale E.G., Du Y., Williams S.C. FAM20: an evolutionarily conserved family of secreted proteins expressed in hematopoietic cells. BMC Genomics. 2005;6:11. doi: 10.1186/1471-2164-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niehrs C. The complex world of WNT receptor signalling. Nat. Rev. Mol. Cell Biol. 2012;13:767–779. doi: 10.1038/nrm3470. [DOI] [PubMed] [Google Scholar]
- Oguro H., Ding L., Morrison S.J. SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell. 2013;13:102–116. doi: 10.1016/j.stem.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohyama Y., Lin J.H., Govitvattana N., Lin I.P., Venkitapathi S., Alamoudi A., Husein D., An C., Hotta H., Kaku M. FAM20A binds to and regulates FAM20C localization. Sci. Rep. 2016;6:27784. doi: 10.1038/srep27784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho S., Frenette P.S. Haematopoietic stem cell activity and interactions with the niche. Nat. Rev. Mol. Cell Biol. 2019;20:303–320. doi: 10.1038/s41580-019-0103-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinho S., Lacombe J., Hanoun M., Mizoguchi T., Bruns I., Kunisaki Y., Frenette P.S. PDGFRalpha and CD51 mark human Nestin+ sphere-forming mesenchymal stem cells capable of hematopoietic progenitor cell expansion. J. Exp. Med. 2013;210:1351–1367. doi: 10.1084/jem.20122252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renstrom J., Istvanffy R., Gauthier K., Shimono A., Mages J., Jardon-Alvarez A., Kroger M., Schiemann M., Busch D.H., Esposito I. Secreted frizzled-related protein 1 extrinsically regulates cycling activity and maintenance of hematopoietic stem cells. Cell Stem Cell. 2009;5:157–167. doi: 10.1016/j.stem.2009.05.020. [DOI] [PubMed] [Google Scholar]
- Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K. Limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruf F., Schreck C., Wagner A., Grziwok S., Pagel C., Romero S., Kieslinger M., Shimono A., Peschel C., Gotze K.S. Loss of Sfrp2 in the niche amplifies stress-induced cellular responses, and impairs the in vivo regeneration of the hematopoietic stem cell pool. Stem Cells. 2016;34:2381–2392. doi: 10.1002/stem.2416. [DOI] [PubMed] [Google Scholar]
- Sacchetti B., Funari A., Michienzi S., Di Cesare S., Piersanti S., Saggio I., Tagliafico E., Ferrari S., Robey P.G., Riminucci M. Self-renewing osteoprogenitors in bone marrow sinusoids can organize a hematopoietic microenvironment. Cell. 2007;131:324–336. doi: 10.1016/j.cell.2007.08.025. [DOI] [PubMed] [Google Scholar]
- Schepers K., Pietras E.M., Reynaud D., Flach J., Binnewies M., Garg T., Wagers A.J., Hsiao E.C., Passegue E. Myeloproliferative neoplasia remodels the endosteal bone marrow niche into a self-reinforcing leukemic niche. Cell Stem Cell. 2013;13:285–299. doi: 10.1016/j.stem.2013.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seike M., Omatsu Y., Watanabe H., Kondoh G., Nagasawa T. Stem cell niche-specific Ebf3 maintains the bone marrow cavity. Genes Dev. 2018;32:359–372. doi: 10.1101/gad.311068.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sella N., Verny L., Uguzzoni G., Affeldt S., Isambert H. MIIC online: a web server to reconstruct causal or non-causal networks from non-perturbative data. Bioinformatics. 2018;34:2311–2313. doi: 10.1093/bioinformatics/btx844. [DOI] [PubMed] [Google Scholar]
- Shoshani O., Ravid O., Massalha H., Aharonov A., Ovadya Y., Pevsner-Fischer M., Leshkowitz D., Zipori D. Cell isolation induces fate changes of bone marrow mesenchymal cells leading to loss or alternatively to acquisition of new differentiation potentials. Stem Cells. 2014;32:2008–2020. doi: 10.1002/stem.1719. [DOI] [PubMed] [Google Scholar]
- Silberstein L., Goncalves K.A., Kharchenko P.V., Turcotte R., Kfoury Y., Mercier F., Baryawno N., Severe N., Bachand J., Spencer J.A. Proximity-based differential single-cell analysis of the niche to identify stem/progenitor cell regulators. Cell Stem Cell. 2016;19:530–543. doi: 10.1016/j.stem.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song D., Xu M., Qi R., Ma R., Zhou Y., Wu D., Fang H., Wang X. Influence of gene modification in biological behaviors and responses of mouse lung telocytes to inflammation. J. Transl Med. 2019;17:158. doi: 10.1186/s12967-019-1870-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staal F.J., Chhatta A., Mikkers H. Caught in a Wnt storm: complexities of Wnt signaling in hematopoiesis. Exp. Hematol. 2016;44:451–457. doi: 10.1016/j.exphem.2016.03.004. [DOI] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V.K., Mukherjee S., Ebert B.L., Gillette M.A., Paulovich A., Pomeroy S.L., Golub T.R., Lander E.S. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama T., Kohara H., Noda M., Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. 2006;25:977–988. doi: 10.1016/j.immuni.2006.10.016. [DOI] [PubMed] [Google Scholar]
- Szenker-Ravi E., Altunoglu U., Leushacke M., Bosso-Lefevre C., Khatoo M., Thi Tran H., Naert T., Noelanders R., Hajamohideen A., Beneteau C. RSPO2 inhibition of RNF43 and ZNRF3 governs limb development independently of LGR4/5/6. Nature. 2018;557:564–569. doi: 10.1038/s41586-018-0118-y. [DOI] [PubMed] [Google Scholar]
- Tibshirani R., Hastie T., Narasimhan B., Chu G. Diagnosis of multiple cancer types by shrunken centroids of gene expression. Proc. Natl. Acad. Sci. U S A. 2002;99:6567–6572. doi: 10.1073/pnas.082099299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tikhonova A.N., Dolgalev I., Hu H., Sivaraj K.K., Hoxha E., Cuesta-Dominguez A., Pinho S., Akhmetzyanova I., Gao J., Witkowski M. The bone marrow microenvironment at single-cell resolution. Nature. 2019;569:222–228. doi: 10.1038/s41586-019-1104-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusher V.G., Tibshirani R., Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc. Natl. Acad. Sci. U S A. 2001;98:5116–5121. doi: 10.1073/pnas.091062498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Andel H., Ren Z., Koopmans I., Joosten S.P., Kocemba K.A., de Lau W., Kersten M.J., de Bruin A.M., Guikema J.E., Clevers H. Aberrantly expressed LGR4 empowers Wnt signaling in multiple myeloma by hijacking osteoblast-derived R-spondins. Proc. Natl. Acad. Sci. U S A. 2017;114:376–381. doi: 10.1073/pnas.1618650114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verny L., Sella N., Affeldt S., Singh P.P., Isambert H. Learning causal networks with latent variables from multivariate information in genomic data. PLoS Comput. Biol. 2017;13:e1005662. doi: 10.1371/journal.pcbi.1005662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolock S.L., Krishnan I., Tenen D.E., Matkins V., Camacho V., Patel S., Agarwal P., Bhatia R., Tenen D.G., Klein A.M. Mapping distinct bone marrow niche populations and their differentiation paths. Cell Rep. 2019;28:302–311.e5. doi: 10.1016/j.celrep.2019.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan K.S., Janda C.Y., Chang J., Zheng G.X.Y., Larkin K.A., Luca V.C., Chia L.A., Mah A.T., Han A., Terry J.M. Non-equivalence of Wnt and R-spondin ligands during Lgr5(+) intestinal stem-cell self-renewal. Nature. 2017;545:238–242. doi: 10.1038/nature22313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M., Tao F., Venkatraman A., Li Z., Smith S.E., Unruh J., Chen S., Ward C., Qian P., Perry J.M. N-Cadherin-Expressing bone and marrow stromal progenitor cells maintain reserve hematopoietic stem cells. Cell Rep. 2019;26:652–669.e6. doi: 10.1016/j.celrep.2018.12.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.O., Yue R., Murphy M.M., Peyer J.G., Morrison S.J. Leptin-receptor-expressing mesenchymal stromal cells represent the main source of bone formed by adult bone marrow. Cell Stem Cell. 2014;15:154–168. doi: 10.1016/j.stem.2014.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y., Zheng M., Song D., Ye L., Wang X. Global comparison of chromosome X genes of pulmonary telocytes with mesenchymal stem cells, fibroblasts, alveolar type II cells, airway epithelial cells, and lymphocytes. J. Transl Med. 2015;13:318. doi: 10.1186/s12967-015-0669-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate new dataset, but analyzed datasets in public repositories. Accession numbers for all datasets analyzed are given in Table S1.