

Summary
A fundamental problem in studies on human microbiome-associated diseases (MADs) is to understand the relationships between microbiome structures and health status of hosts. For example, species diversity metrics have been routinely evaluated in virtually all studies on MADs, yet a recent meta-analysis revealed that, in only approximately one-third of the cases, diversity and diseases were related. In this study, we ask whether Hubbell's neutral theory (supplemented with the normalized stochasticity ratio [NSR]) or critical microbiome network structures may offer better alternatives. Whereas neutral theory and NSR focus on stochastic processes, we use core/periphery and high-salience skeleton networks to evaluate deterministic, asymmetrical niche effects, assuming that all species or their interactions were not “born” equal and focusing on non-neutral, critical network structures. We found that properties of critical network structures are more indicative of disease effects. Finally, seven findings (mechanisms, interpretations, and postulations) regarding medical ecology mechanisms underlying MADs were summarized.
Subject Areas: Ecology, Microbiology, Microbiome, Microbial Interactions
Graphical Abstract
Highlights
-
•
Seven findings (mechanisms/interpretations/postulations) of medical ecology proposed
-
•
Critical network structures more indicative of disease effects than ecology metrics
-
•
One-third seems ceiling of diversity-disease relations, half to two-thirds of network structures
-
•
Super resilience (unexplained one-third to half gap) is likely attributed to host genome
Ecology; Microbiology; Microbiome; Microbial Interactions
Introduction
Despite extensive studies on the human microbiome-associated diseases (MADs) during the last decade (HMP Consortium, 2012, Integrative HMP (iHMP) Research Network Consortium, 2019), there is still little consensus on the ecological mechanism underlying the etiology of MADs. For example, the routinely computed microbiome diversity indexes (Shannon entropy, Simpson index, and species richness) and associated diversity-disease relationships (DDRs) have revealed little insights on the ecological mechanisms, not to mention insights on the disease etiology. In a recent meta-analysis of the human MADs, it was discovered that, somewhat contrary to commonly perceived intuition, there was not a consistent DDR in the majority of studied cases (approximately two-third cases)—that the species or Operational Taxonomic Unit (OTU) diversity was not related with MAD (Ma et al., 2019). This DDR in the human MADs is in strong contrast with the DDR in zoonoses, where the dilution/amplification hypotheses have achieved wide recognition (Johnson et al., 2013, Johnson et al., 2015). Indeed, the disease systems and targets of diversity analysis in zoonoses and human MADs are rather different (e.g., diversity of alternative hosts or vectors in zoonoses versus diversity of microbiome). Sometimes, even more confusing is the lack of obvious pathogens and vectors in the case of human MADs, e.g., the “pathogen” of mastitis, which is related to the shift of species interactions in suppressing opportunistic pathogens (Ma et al., 2015, Ma et al., 2016a, Ma and Ye, 2017, Ma, 2018).
Multiple causes have contributed to the enormous challenges for the mechanistic studies on the human MADs. First, the problem itself is exceedingly complex because, the MAD, as a category of human diseases, whether the diversity change is the disease cause or consequence, may be different from case to case, and even more frustrating is that we do not yet have an answer for the cause-consequence question for many of the MADs (e.g., Castaño-Rodríguez et al., 2018, Duvallet et al., 2017, Zaneveld et al., 2017). Second, many existing studies have ignored perhaps the most important sub-system, the human immune system except for small number of studies (e.g., Zaneveld et al., 2017, Vogelzang et al., 2018, Lotter and Altfeld, 2019, Vemuri et al., 2019). Third, the etiologies of many MADs are not clear and most existing mechanistic studies were conducted with animal models (e.g., Turner, 2018). Particularly, how deeply microbiome is involved in a particular MAD can be rather different. In some cases, microbiome is likely deeply involved such as in the case of BV (bacterial vaginosis), where classic diversity-stability relationship (DSR) in ecology has been invoked to explain its etiology (Sobel, 1999, Ma et al., 2012, Li and Ma, 2019, Ma and Ellison, 2019). In other cases, microbiome may simply act as part of the host environment that influences the metabolism of hosts. Obesity may belong to such a case, where the role of gut microbiome, although extensively investigated in the last decade, is still hotly debated (Rastelli et al., 2018). In yet other cases, such as cancers and HIV, the involvement of microbiome may be indirect but can still be significant (Williams et al., 2016, Bandera et al., 2018). It is for these diverse scenarios we use the term “microbiome-associate disease” (MAD) in this study.
If the extensively investigated DDR in human MADs is inconsistent in the majority of cases (approximately two-thirds) (Ma et al., 2019, Ma, 2020a, Ma, 2020b), using diversity indexes as diagnostic indicators for MADs becomes minimally useful, not to mention its role in mechanistic/etiological studies of MADs. The present study therefore aims to search for possible alternatives, and here we focus on detecting the ecological/network “imprints” of MADs on the human microbiomes, which may help to reveal underlying ecological mechanisms of MADs and possibly act as diagnosis and risk prediction indicators for MADs (Figure 1). For example, the network assortativity is related to network resilience, and it was found that slightly disassocitative networks (negative assortativity but with small absolute value) are less resilient because network percolates less easily in such networks (Newman and Clauset, 2016). In this study, we test whether critical network structure and properties such as assortativity can be more indicative than standard diversity metrics such as Shannon entropy in detecting the effects of MADs. Besides identifying potentially more powerful network indicators for MAD effects, a second objective of the present study is to establish possible mechanistic links between the critical network structures (of microbiomes) and the processes that drive the microbiome dynamics, which is of obvious significance for deep understanding of the mechanisms and etiologies of MADs.
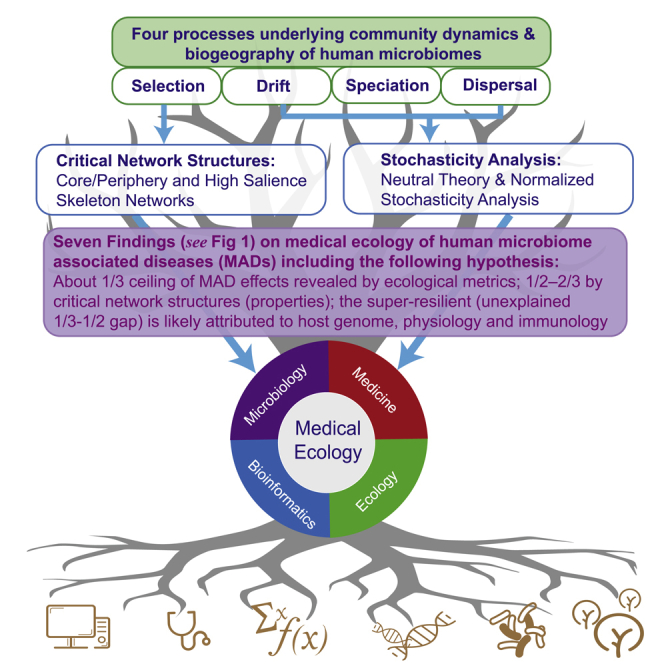
Figure 1.

Diagram for the Study Design and Conclusions
Illustration of the goal, objectives, approaches, and results (main findings) from investigating the critical network structures and ecological processes (mechanisms) underlying the human microbiome-associated diseases.
We resort to Hubbell (2001) unified neutral theory of biodiversity (UNTB) and network analyses (Figure 1). We dissect the influences of MADs on the community assembly and diversity maintenance by harnessing the power of UNTB, which covers the three processes (mechanisms) of Vellend-Hanson four-processes synthesis of community ecology and biogeography (Vellend, 2010, Vellend, 2016, Hanson et al., 2012), including drift, speciation (mutation), and dispersal (Rosindell et al., 2011) (the other is selection), and by integrative analyses with core/periphery network (CPN) (Csermely et al., 2013, Ma and Ellison, 2019) and high-salience skeleton network (HSN) (Grady et al., 2012, Shekhtman et al., 2014, Ma and Ellison, 2019) modeling. The four-processes (mechanism) synthesis stated that, similar to the modern synthesis of population genetics, drift, speciation, dispersal, and selection constitute the underlying processes (mechanisms) that drive community dynamics and shape the microbial biogeography (the spatial and temporal distribution patterns of community diversity) (Vellend, 2010, Vellend, 2016, Hanson et al., 2012). We argue that the CPN/HSN analyses possess the potential to assess and interpret the effects of selection in the four-processes synthesis as further elaborated below.
We realized that, since the UNTB optimizes the fit of the relative abundance to the predictions of the neutral model, it might over-estimate the true strength of neutral processes. Therefore, rather than examining the fit of the relative abundance data to the predictions (i.e., the passing rate of neutrality tests), we should focus on the estimated parameters of the neutral model (fundamental numbers of diversity and fundamental number of dispersal) and ask if they differ between healthy and diseased individuals. We also recognized that the four-processes synthesis is still largely conceptual in its current stage and refrained from inferring conclusions from it, particularly quantitative inferences. That said, in this article, we primarily depend on the analyses of critical network structures with the CPN and HSN (Csermely et al., 2013, Grady et al., 2012, Shekhtman et al., 2014, Ma and Ellison, 2019) and use neutral-theoretic approach and four-processes synthesis to cross-verify and supplement our findings (Figure 1).
According to Vellend, 2010, Vellend, 2016, selection refers to the deterministic fitness difference between individuals from different species, and it can also be treated as the deterministic interactions among species and between species and their environments. Selection represents asymmetric or unequal interactions; in other words, not all species or their interactions were “born” equal. From a conceptual perspective, we argue that critical network structures (core/periphery/skeleton/backbone) in the CPN/HSN can be considered as outcome of selection given their asymmetrical and heterogeneous nature. We argue that, conceptually, our integrated analyses (Figure 1) of the human MADs datasets with the UNTB (Hubbell, 2001), Ning et al. (2019) ecological stochasticity framework, and CPN/HSN networks (Csermely et al., 2013, Grady et al., 2012, Shekhtman et al., 2014, Ma and Ellison, 2019) cover the full spectrum of all four processes (drift, dispersal, speciation, and selection) of Vellend-Hanson synthesis of community ecology and biogeography (Vellend, 2016, Vellend, 2010, Hanson et al., 2012). Figure 1 summarizes the overall goal, objectives, datasets, approaches, as well as findings and conclusions of the present study.
Results
A brief description on the metagenomic datasets of the 27 MAD studies, which are used to generate the results here, is provided in Table S1 of the Supplemental Information, which are the same datasets used in Ma et al. (2019). Among the 27 datasets, there were 3 and 2 datasets that are not suitable for the neutral-theoretic analysis and CPN/HSN analyses, respectively, and therefore were excluded from the analyses of this study. The 27 case studies cover most of the high-profile MADs, including obesity, IBD, diabetes, BV, periodontitis, and neurodegenerative diseases. The datasets were in the abundance of 16s-rRNA reads clustered at the species level (97% similarity level), equivalent with the species abundance tables in macrobial ecology.
This section is organized as eight subsections covering (see the bottom section of Figure 1): (1) the first three subsections on neutral theoretic analysis—testing the MAD effects on the microbiome neutrality and on the neutral model parameters (fundamental biodiversity/dispersal numbers) as well as estimating the normalized stochasticity ratio (NSR) to cross-verify the neutral-theoretic analyses; (2) two consequent subsections on the CPN—shared core/periphery network analysis between the healthy (H) and diseased (D) treatments and further testing the MAD effects on the key CPN properties; (3) followed by two subsections on the HSN (high-salience skeleton network)—shared skeleton network analysis and further testing the MAD effects on the key HSN properties; (4) finally, the actual (observed) key CPN/HSN network properties for the 25 MAD case studies. The consequent discussion section further summarizes our findings from the results presented in this section.
Neutral-Theoretic Analysis: Testing MAD Effects on the Microbiome Neutrality
Tables S2A–S2E exhibited the detailed results of the neutrality tests for the 24 case studies of MADs. For each case study, the neutrality test was performed for each community sample (from each individual subject) of the H (healthy) and D (diseased) treatments, separately. For each community sample, five data pre-processing schemes (raw—no preprocessing, singleton—removal of singleton, minus–1, minus–2, minus–3, i.e., removal of 1, 2, or 3 individuals across all OTUs, respectively) were implemented to obtain robust test results. What were displayed in Tables S2A–S2E included the key parameters of the classic neutral model, such as the fundamental biodiversity number (θ), migration probability (m), and p value for the neutrality testFigure 1, Figure 2, Figure 3, Figure 4, Figure 5, Figure 6, Figure 7
Figure 2.

The Neutrality Passing Rates for Each of the 24 Case Studies
The neutrality passing rates for each of the 24 case studies: the cases with significant differences in the passing rates between the healthy and diseased treatments are marked with “∗”; for each case study, the average percentage across five data-preprocessing schemes was computed and utilized to represent the neutrality of a case study.
Figure 3.

The Key Parameters from Neutral-theoretic Modeling
The fundamental diversity number (θ) and immigration probability (m) for each of the 24 case studies, and the cases with significant differences in the parameters between the healthy and diseased treatments are marked with “∗”; for each case study, the average parameters across five data-preprocessing schemes are used to plot the figure.
Figure 4.

The NSR (Normalized Stochasticity Ratio)
The NSR (normalized stochasticity ratio) for each of the 27 case studies: the treatments with significant differences in NSR were marked with star (∗).
Figure 5.

Network Graphs Illustrated with BV (Bacterial Vaginosis) Study
The core/periphery/skeleton network for the healthy (A) and BV (B) treatment, respectively, and the legends used include: the core nodes, circle in cyan; the periphery nodes, circle in dodger blue; regular positive links, regular lines in green; high-salience positive skeleton (links), thick lines in green; regular negative links, regular lines in red; high-salience negative skeleton (links), thick lines in red.
Figure 6.

Share Core/Periphery Node Analysis
The numbers of shared core (the left graph) or periphery (the right graph) species (nodes) between the healthy (H) and diseased (D) treatments: the permutation test was performed with 1,000 pairs (pair = H and D) of permutated networks (“∗”: represents the cases with significant difference between the H and D). The figure is plotted based on Table S6.
Figure 7.

Share High-Salience Skeleton Analysis
The numbers of shared high-salience skeletons (HSS>0) between the healthy (H) and diseased (D) treatments: the permutation test was performed with 1,000 pairs (pair = H and D) of permutated networks (“∗”: represents the treatments with significant difference between H and D). The figure is plotted based on Table S8.
Table S3, summarized from Table S2, exhibited the neutrality passing rates for each of the 24 case studies. Table S4 exhibited the results of Fisher's exact probability test for determining the effects of MAD diseases on the passing rates of neutrality.
Figure 2, drawn based on Tables S2, S3, and S4, illustrated the passing rates for each case study. Note that Figure 2 was plotted with the average passing rates across the results obtained from five data pre-processing schemes. In Figure S1 (see Supplemental Information), the passing rate corresponding to each of the five data pre-processing schemes was plotted. Hence, Figure 2 here is a summary version of Figure S1.
From Figures 2 and S1, and Tables S2, S3, and S4, we summarize the following findings:
-
(1)
Across the five data pre-processing schemes, the neutrality passing rates ranged from 26.2% to 78.3% with an average of 57% for the H treatments and ranged from 27.1 to 80.1 with an average of 60% for the D treatments. Given that the three schemes with minus 1, 2, or 3 across all OTUs achieved neutrality rates higher than the raw and singleton-removed schemes, obviously, the results from using raw datasets should be more robust if the objective is to obtain more conservative (reliable) estimation of the neutrality level. These results indicate that the human microbiome, regardless of the health/disease status, exhibited significant level of neutrality that cannot be ignored. In other words, at least, in close to one-third (H = 26.2% D = 27.1%) of the tested samples, the neutrality plays a predominant role in community assembly and diversity maintenance, and furthermore, the neutrality passing rates can be as high as 80% (78.3%–80.1%).
-
(2)
Fisher's exact probability test showed that, in absolute majority cases, diseases did not exert a significant effect on the neutrality passing rates (Table S4 and Figure 3). Specifically, in only 5 of 165 comparisons (3%), there were significant differences in the neutrality passing rates between the H and D microbiome samples.
Neutral-theoretic analysis: testing disease effects on the fundamental biodiversity number (θ) and fundamental dispersal numbers (m)
Table S5 listed the results of the significance tests for θ (the fundamental biodiversity numbers), which is a measure of speciation, and for m (the migration probability or fundamental dispersal numbers), a measure for dispersal limitation, between the H and D samples. The Wilcoxon test was performed to determine whether or not MAD could have significant influence on the parameters (θ and m). Figure 3 displayed the average values of m and θ (across five data pre-processing schemes) for each case study, and it also marked the cases with significant differences between the H & D treatments. Note that Figure 3 (a summary version of Figures S2 and S3) was plotted with the average parameters across five data pre-processing schemes. In Figures S2 and S3, the parameters for each of the five data pre-processing scheme were plotted.
The findings from the significance tests (Table S5 and Figures 3, S2, and S3) seem rather consistent across five different data-preprocessing schemes. In approximately one-third (30%–33%) of the comparisons, the fundamental biodiversity numbers (θ) were significantly different between the H and D treatments. In only approximately 15% of the comparisons (9.1%–21.2%), the migration probabilities (m) were significantly different between the H and D treatments. These findings suggest that diseases have significant influences on the speciation in approximately one-third of the comparisons, whereas significant influences exist in only 15% of the comparisons in term of the dispersal limitation.
Cross-Verification of Neutral Theoretic-Analyses with Normalized Stochasticity Ratio
As explained previously, the optimized fitting of UNTB is likely to lead to overestimation of the stochastic neutral levels. We apply Ning et al. (2019) framework for estimating community stochasticity level, specifically NSR (Table S6 and Figure 4), for cross-verifying the previous UNTB results. Table S6 included the mean NSRs for the intra-H (intra-healthy), intra-D (intra-diseased), and inter-HD (healthy versus diseased) treatments, respectively, as well as the p values from the Wilcoxon tests of the differences in the NSR among the three treatments. Figure 4 illustrated the contents listed in Table S6, i.e., the NSRs for each treatment in the 27 MAD cases, and the treatments with statistically significant differences were marked with star (∗). From Table S6 and Figure 4, we summarize the following four findings, which can be used to verify/calibrate the previous UNTB results.
-
(1)
The average NSR for intra-H, intra-D, and inter-HD is 0.342, 0.334, and 0.287, respectively. The slightly decreased NSR in this order suggests that diseases might cause the decline of stochasticity. Among the three NSRs, the difference between intra-H (0.342) and inter-HD (0.287) should be a more objective indicator for disease effects, which is about 16% roughly in terms of the “face” values of NSRs. Further rigorous statistical tests with Wilcoxon tests indicate that, in 30% of the MAD cases (12 of 40 possible comparisons between H and D) comparing the intra-H and intra-D treatments exhibited significant differences in their NSRs. The percentage is slightly lower (27.5% or 11 of 40) after FDR (false discovery rate) control is applied.
-
(2)
Since the NSR was normalized to the range between 0 and 1 and is a ratio (hence dimensionless), the range of 0.297–0.334 also indicates the general stochasticity level in the human microbiome, regardless of diseases status, is rather tight, i.e., around one-third. Compared with the findings from previous UNTB modeling, i.e., the range between 26.2% and 78.3% for the H treatments, and the range between 27.1 and 80.1 for the D treatments, the NSR range is rather close to the floor limits of the neutral modeling. If we accept the conjecture that UNTB tends to overestimate the stochastic neutral forces, it should be safe to conclude that the neutrality-passing rate or stochasticity level in the human microbiome should be about one-third, and the upper range (up to 80%) may indeed be unduly overestimated.
-
(3)
As to the MAD effects, the previous tests with fundamental dispersal numbers (migration probability) and fundamental biodiversity numbers (speciation rate) indicated 15% (dispersal) to 33% (biodiversity) of the comparisons, and the NSR here revealed a similar effect size (30%–32.5%) based on Wilcoxon tests. Therefore, we conclude that the MAD effects on the neutral processes are in the range between 15% and 33% (=1/3) approximately, depending on whether dispersal (migration) or biodiversity is considered, with the former receiving a lower impact than the latter.
Core/Periphery Network Analysis: Shared Core/periphery Analysis
Shared core/periphery nodes analyses (SCA/SPA) aim to detect the effects of MADs on the numbers of shared core/periphery species (nodes) between the H and D treatments because the decline of shared nodes may signal the effects of MADs. Table S7 listed the results from shared core species (nodes) (the left side) and periphery species (nodes) (the right side) analyses between the H and D treatments. The randomization tests were performed based on 1,000 sets of CPNs built with FDR (false discovery rate) control and p value = 0.001, which ensures only significant correlations based on Spearman's correlation coefficient are admitted into the species correlation networks (SCN). From the SCNs (species correlation networks), the corresponding CPNs were constructed based on the algorithms previously introduced. Figure 5 illustrates the graphs of CPN and HSN (high-salience skeleton network) with BV case study as examples.
In Table S7, both the observed (actual) shared nodes and permutated (simulated) nodes were listed for the core and periphery, respectively. The p value from comparing the observed- and permutated-shared species (nodes) is used to determine whether or not the shared species (core or periphery) between the H and D treatments decreased more than by chance. When the p value <0.05, the shared core or periphery species were reduced more than the decrease by chance; in other words, disease caused the reduction of shared nodes (core or periphery species). Otherwise, the reduction of shared nodes was not caused by disease, instead by chance.
The bottom section of Table S7 exhibited the percentages (%) of MAD cases with decreased shared core (24%) or periphery (18%) species, respectively. Figure 6 illustrated the same information listed in Table S7. The cases with decreased shared species (core or periphery) were marked with star (∗).
Core/Periphery Network Analysis: Testing the Differences in CPN Properties
Table S8 listed the p values from testing the differences in the CPN properties (parameters) between the H and D treatments. The CPN properties tested include core strength (ρ), the fraction of core nodes, the four components of core density matrix, and the network nestedness. The randomization tests were performed again based on the same 1,000 sets of permutated networks used in the shared core/periphery analysis above, but the algorithm used was slightly different (see the Supplemental Information for the related algorithms).
Table S8 shows that the disease effects on the CPN properties ranged from 45% to 64%; in all but two properties [C/(C + P) and B12/B21], the percentage with significant differences exceeded 50%. The parameter with the highest percentage in the difference between the H and D is B22, which represents the interactions among the periphery nodes. This should be expected since periphery nodes are weakly connected and more likely influenced by perturbations such as diseases.
High-Salience Skeleton Network Analysis: Shared Skeleton Analysis
Table S9 listed the results from shared skeleton analysis between the H and D treatments. The randomization test was performed based on 1,000 sets of permutated HSNs built with FDR control and p value = 0.001. In Table S9, both the observed (actual) shared skeletons and permutated (simulated) skeletons were listed. The p value from comparing the observed- and permutated-shared skeletons is used to determine whether or not the shared skeletons between the H and D treatments decreased more than the decrease by chance. When p value <0.05, the shared skeleton was reduced more than the decline by chance; in other words, disease caused statistically significant reduction of shared skeletons. Otherwise, the reduction of shared skeletons was not caused by disease, instead by chance.
The bottom section of Table S9 exhibited the percentages of MAD cases with decreased shared skeletons (40%). Figure 7 illustrated the same information listed in Table S9. The cases with declined shared skeletons were marked with star (∗). The percentage with significant decline in shared skeleton is higher than the percentage of shared core (24%) and shared periphery (18%) in the previous subsection. This difference suggests that diseases have more far reaching influences on the interactions (skeletons) than on the species per se (nodes).
High-Salience Skeleton Network Analysis: Testing the Differences in HSN Properties
Table S10 listed the p values from testing the differences in the HSN properties between the H and D treatments. The HSN properties tested include links% (the percentage of high salience skeletons), the max, mean, skewness, and kurtosis of the salience values, as well as assortativity (a measure of network resilience). The randomization tests were performed based on the same 1,000 sets of permutated networks used for the previous shared skeleton analysis, but the algorithm is slightly different from that used in the shared skeleton analysis and is the same as that used for testing the CPN properties previously (see the Supplemental Information).
Table S10 shows that the effects of diseases on the HSN properties ranged from 47% to 74%, and in all but one property (skewness), the percentages with significant differences exceeded 50%. The highest percentage in the difference between the H and D occurred in assortativity (rHSS = 74%), which represents for the assortativity of HSN and measures the resilience of the network. This suggests that, in approximately three-fourth (74%) studied cases, diseases significantly impact the network resilience. This finding cross-validated the previous finding from shared skeleton analysis, i.e., diseases exert more far-reaching effects on species interactions (skeletons) than on the species per se (nodes).
The Key Properties of Critical Network Structures (Core/Periphery/Skeletons)
The core/periphery nodes and high-salience skeletons represent for the critical structures of the species interactions in the community (network), from either node or link perspective. In the previous sub-sections, we focused on statistical (permutation) tests for the potential differences between the H and D treatments in terms of either shared critical network structures (core/periphery/skeleton) or their network properties. In this sub-section, we briefly introduce the key properties of those critical network structures per se, for example, the list of shared or unique core species in the H or D treatment in the SI.
Table S11 listed the key network properties of the CPN for each of the 25 MAD case studies, including the core strength (ρ), fraction of core nodes, density matrix of core/periphery structure, and network nestedness. For example, the average core strength (ρ) for the healthy and diseased treatments is 0.175 versus 0.216, with standard error of 0.026 versus 0.033. Theoretically, ρ ranges from 0 to 1 and represents for the relative strength of core structure. The range indicates that the strength of human microbiome networks is relatively loose. As shown in Table S8, ρ is significantly different between the H and D treatments in approximately 60% of the MAD cases. Similarly, Table S12 listed the key network properties of the HSN for each of the 25 MAD cases. As exhibited in Table S10 previously, almost all HSN properties showed significant difference between the H and D individuals in more than half of the 26 MAD cases, and in particular, the assortativity of high-salience skeletons differed in 74% of the cases.
Tables S13–S15 further listed the actually observed number of shared core nodes, observed number of shared periphery nodes, and observed number of shared skeletons, respectively, shared between the H and D treatments in each of the 25 MAD datasets. Those three tables exhibited the actually observed levels of similarity (shared core/periphery/skeletons) between the H and D treatments and deserve further investigations aimed to understand specific disease mechanisms/etiologies.
Conclusions and Discussion
Ecologists often seek to understand the relationships between the structure and function (process) of ecological systems to deepen their understanding to ecological mechanisms. Nonetheless, this relationship is rarely straightforward, and what is being seen (i.e., community or network structure) must be filtered and analyzed carefully before committing a belief on what is going on in the system (e.g., community stability). This is because structure is usually only an imperfect and often ambiguous manifestation of the process (Ma, 2015). In the case of this study, we aimed to detect the relationships between critical community (network) structures and disease effects (states) beyond the findings revealed in our previous DDR, in which we found that in only approximately one-third of the MAD cases disease states were related to diversity measures, but in the majority of cases (approximately two-thirds) there was not a consistent DDR pattern (Ma et al., 2019). In the present study, we focus on detecting the ecological/network “imprints” of MADs on the human microbiomes, which may help to reveal underlying ecological mechanisms of MADs and possibly act as diagnosis and risk prediction indicators for MADs. Such explorations are important because revealing the relationships (imprints) is critical not only for understanding the disease mechanisms/etiologies but also for developing diagnosis and/or risk prediction techniques. Methodologically, we introduced three approaches to looking into the structure-mechanism paradigms in the medical ecology of the human MADs, including Hubbell (2001) UNTB, Ning et al. (2019) framework for stochasticity assessment, and CPN/HSN networks (Csermely et al., 2013, Grady et al., 2012, Shekhtman et al., 2014, Ma and Ellison, 2019). Here, we further summarize the following seven findings from the previous sections and discuss their implications. It should be noted that the findings are supported by the analyses introduced in previous sections, but the implications may include postulations or even speculations.
Finding (1)
The stochasticity or neutrality level, in the human microbiomes of the MAD case studies we analyzed, was approximately one-third, which is revealed by Hubbell (2001) UNTB testing, cross-verified by Ning et al. (2019) stochasticity analysis framework. The disease status of MADs did not significantly influence the stochasticity/neutrality level. The implications of this finding are as follows:
Since the neutrality level is a measure of stochastic drift, speciation, and dispersal (i.e., three of the four processes of Vellend-Hanson synthesis) (Rosindell et al., 2011, Vellend, 2010, Hanson et al., 2012), it indicates the extent or level (i.e., one-third) that the microbial community is driven by the stochastic neutral forces. We might further postulate that the deterministic selection (i.e., the other process in the four process of Vellend-Hanson synthesis) (Vellend, 2010, Vellend, 2016, Hanson et al., 2012) could be at the most. However, this postulation is contingent on the additivity of the four processes of the synthesis, which is still an open question.
Finding (2)
In approximately one-third of the cases analyzed, MADs had significant effects on the fundamental biodiversity numbers (θ), whereas the MAD effects on the fundamental dispersal numbers (m) were approximately 15%. Therefore, although MADs did not significantly influence the mode (nature of mechanism) of microbiome assembly and diversity maintenance—whether the mechanism is stochastic or deterministic, as indicated by Finding (1) above—MADs may indeed have certain level of effects on the microbiome diversity and the upper bound (ceiling) seems to be one-third as indicated by the disease effects on θ.
This “one-third MAD effects on θ (the fundamental biodiversity number)” offers a mechanistic interpretation for the “one-third DDR pattern” (i.e., in only approximately one-third of the MAD cases, diversity is related to disease status) from our previous DDR analysis (Ma et al., 2019).
Finding (3)
The critical network structures detected with CPN/HSN analyses revealed that MADs can lead to significant reductions of shared (common) core/periphery/skeleton structures, approximately 24%, 18%, and 40% respectively. That is, there should be disease-specific core/periphery (species) and skeletons (species interactions). First principles suggest that disease may lead to divergence between the H (healthy) and D (diseased) individuals, consequently the reduction of shared (common) critical structures. Therefore, the reduction of shared critical structures should be expected.
Finding (4)
Besides influencing the shared critical network structures (which ranged between 18% and 40%), as stated above, the MAD effects on the CPN properties ranged between 45% and 64% depending on specific CPN properties and the MAD effects on the HSN properties ranged between 47% and 74%. The difference between the “shared critical network structures” and “network (CPN/HSN) properties” lies in that the former is holistic and structural and the latter is a collection of network properties that may reflect either global or local network characteristics.
Summarizing Findings (3) and (4), we conclude that, for diagnostic and/or risk assessment purposes, the properties (particularly network assortativity) of critical network structures can be more promising than the numbers of network structures per se, and also more promising than the neutral theory parameters, given that the MAD effects on most of the network properties exceeded half. The most promising network property reflecting the microbiome resilience against MADs seems to be network assortativity, which reached 74% of differences between the H and D treatments, which is discussed further below in Finding (6).
Finding (5)
Selection is the difference in the deterministic fitness between individuals from different species, and it can also be treated as the deterministic interactions among species and between species and their environments (Vellend, 2010, Vellend, 2016). The former definition emphasizes the outcome of selection at the individual level, and the latter definition emphasizes the process selection occurs at the species level. According to these definitions, what CPN/HSN analyses reveal, including the asymmetrical and heterogeneous network structures (i.e., core/periphery/skeleton), can be considered as the outcome of selection.
Therefore, the selection effects should range from 18% to 74% but range between 40% and 60% in terms of the most metrics, depending on the kinds of critical network structures or their properties. We consider that the selection effects ranged between 40% and 60% in terms of the most metrics are moderate and non-extreme. According to Vellend, 2010, Vellend, 2016, selection may vary across space or time, with potentially important consequences for community dynamics. Furthermore, when selection is relatively strong and the community size is large, any effects of drift may be canceled by selection. However, when selection is relatively weak and the community size is small, the opposite can occur, i.e., drift can override selection effects. Between the two previously conceived extremes, i.e., being moderate or non-extreme, selection could make some community states more likely than others, but it does not guarantee any particular state (outcome) (Nowak, 2006, Vellend, 2010, Vellend, 2016). As we argued previously, the selection effects in human microbiomes should be non-extreme or moderate. We postulate that, according to Nowak (2006) and Vellend, 2010, Vellend, 2016, it is expected that, in the majority of cases, selection forces may not override the neutral forces nor be overridden by neutral forces. There is no guarantee that the community will be in any particular state (outcome). This uncertainty (unpredictability) may explain the lack of a consistent DDR pattern in the majority (approximately two-thirds) of MAD cases analyzed by Ma et al. (2019).
Finding (6)
The network assortativity is related to network resilience. It was found that slightly disassocitative networks (negative assortativity but with small absolute value) are less resilient (Newman and Clauset, 2016). Table S12 showed that the average assortativity of H and D is −0.041 and −0.022, respectively. Hence, the D treatments should be less resilient than the H treatments, which were the case in 74% of the 25 MAD cases, as revealed by the randomization tests exhibited in Table S10. In fact, the assortativity of HSN is the property that exhibited the highest divergence (74%) between the H and D treatments we discovered in this study. This finding may not be incidental given that assortativity can be considered as a measure of network resilience and loss of resilience or dysbiosis is well recognized as a hallmark of MADs.
Finding (7)
The last finding here is indirectly inferred from the previous six findings and is more speculative. Throughout the study, it appears that we could not break the ceiling limits of “one-third” in the case of neutral theory modeling or “half to two-thirds” in the case of network analysis regarding disease effects (also see Ma et al., 2019, Ma, 2020a, Ma, 2020b). The reality that we could not break the limits should not be surprising. Besides the possible imperfectness of the “structure-process (mechanism)” strategy our approaches followed, we expect a major source for the unexplained gap in disease effects or lack of MAD effects should be the intrinsic stability (resilience) of human microbiome against disturbances including diseases. The intrinsic resilience should primarily be due to host genomes, which should be rather stable at ecological timescale, the scale MADs occur. Therefore, we conjecture that the genome effects on the MADs should be approximately one-third or even more. Alternatively, we conjecture that the remaining unexplained MAD effects could be due to nearly neutral forces (Ohta, 2008, He et al., 2012). Compared with animal and plants, bacterial species seem to resemble each other closely in their demographic parameters but may not be the exactly same as the neutral theory assumes; consequently, nearly neutral effects could be significant. Finally, Figure 1 further summarized the above-discussed seven findings.
Limitations of the Study
The present study was aimed to investigate the critical network structures and medical ecology mechanisms underlying the human MADs, and it was a direct extension to our previous works in this field, based on the same MAD datasets but was approached with different objectives, analytic methods, and ecological theories (Ma et al., 2019, Ma, 2020a, Ma, 2020b). The most important limitations of our works have been discussed in Finding (7) in the previous section, which summarized the issues and proposed a conjectural hypothesis for further investigations. An additional limitation is that the scope of MAD datasets was limited to the marker gene (16s-rRNA) sequencing reads obtained from amplicon sequencing technology, and the metagenome datasets from the whole-genome sequencing (shotgun sequencing) were not implicated in this serious of studies (Ma et al., 2019, Ma, 2020a, Ma, 2020b). Both types of metagenomics studies (datasets) have not only different data structures (matrices of OTU abundances versus matrices of metagenomic gene abundances) but also very different bioinformatics and subsequent analytic approaches. In particular, there have been relatively few ecological approaches to the metagenomic gene abundance (GA) datasets. We argue that the lag in the applications of ecological theories for GA studies is not because of the applicability or importance of ecological theories; instead it is because of the enormous difficulties in analyzing the big data of metagenomic genes (the number of which is orders of magnitude larger than the number of OTUs in the case of human microbiomes), which made subsequent ecological analyses rather difficult. To deal with the difficulty, we have been developing the applications of major ecological theories for the metagenomic GA datasets in another series of studies (Ma and Li, 2018, Ma, 2020c, Ma and Ellison, 2020). We argue that the medical ecology of human microbiomes, which can be considered as a cross-disciplinary field of microbiology, medicine, ecology, and bioinformatics (Ma, 2017, Ma et al., 2016), is complete only if both microbial OTUs and metagenomic genes are included. Therefore, future studies to complement the present study based on metagenomic GA datasets are certainly worthy of further explorations.
Resource Availability
Lead Contact
Sam Ma: ma@vandals.uidaho.edu.
Material Availability
N/A.
Data and Code Availability
All datasets analyzed in this study are available in public domain, and see Table S1 for the access information. All software codes are available in public domain as cited in the paper.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
I appreciate Prof. Nicholas Gotelli, University of Vermont, USA, for his review, suggestions, and comments, for an early version of this manuscript, which played an important role in improving this manuscript. I am indebted to the anonymous reviewers and Dr. Simona Fiorani, the iScience Lead Editor, for their insightful comments and suggestions, which helped to improve my manuscript significantly. I thank Mr. L.W. Li and Miss Wendy Li of the Chinese Academy of Sciences for their computational support to this study. This study received funding from the following sources: A National Natural Science Foundation of China (NSFC) Grant (No. 31970116) on Medical Ecology of Human Microbiome; The Cloud-Ridge Industry Technology Leader Award; An International Cooperation Grant (YNST) on Genomics & Metagenomics Big Data. The funders played no roles in interpreting the results.
Author Contributions
Z.S.M. designed and performed the study and wrote the manuscript.
Declaration of Interests
The author declares no competing interests.
Published: June 26, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101195.
Supplemental Information
Table S2A. The parameters of fitting Hubbell's (2001) classic neutral theory model with raw data∗, Related to Figures 2 and 3
Table S2B. The parameters of fitting Hubbell's (2001) classic neutral theory model with singleton removed∗, Related to Figures 2 and 3
Table S2C. The parameters of fitting Hubbell's (2001) classic neutral theory with data of minus-1∗, Related to Figures 2 and 3
Table S2D. The parameters of fitting Hubbell's (2001) classic neutral theory model with data of minus-2∗, Related to Figures 2 and 3
Table S2E. The parameters of fitting Hubbell's (2001) classic neutral theory with data of minus-3∗, Related to Figures 2 and 3
Table S6A. The means of Ning et al. (2019) normalized stochasticity ratio (NSR) for the intra-healthy treatment, intra-diseased treatment, and inter healthy-versus-diseased treatments, as well as the Wilcoxon tests among them (without using FDR control), Related to Figure 4.
Table S6B. The means of Ning et al. (2019) normalized stochasticity ratio (NSR) for the intra-healthy treatment, intra-diseased treatment, and inter healthy-versus-diseased treatments, as well as the Wilcoxon tests among them (after FDR control is applied), Related to Figure 4.
References
- Bandera A., Benedetto D.I., Bozzi G., Gori A. Altered gut microbiome composition in HIV infection: causes, effects and potential intervention. Curr. Opin. HIV AIDS. 2018;13:73–80. doi: 10.1097/COH.0000000000000429. [DOI] [PubMed] [Google Scholar]
- Castaño-Rodríguez N., Underwood A.P., Merif J., Riordan S.M., Rawlinson W.D., Mitchell H., Mitchell H.M., Kaakoush N.O. Gut microbiome analysis identifies potential etiological factors in acute gastroenteritis. Infect. Immun. 2018;86 doi: 10.1128/IAI.00060-18. e00060–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csermely P., London A., Wu L.Y., Uzzi B. Structure and dynamics of core/periphery networks. J. Complex Netw. 2013;1:93–123. [Google Scholar]
- Duvallet C., Gibbons S.M., Gurry T., Irizarry R.A., Alm E.J. Meta-analysis of gut microbiome studies identifies disease-specific and shared responses. Nat. Commun. 2017;8:1784. doi: 10.1038/s41467-017-01973-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grady D., Thiemann C., Brockmann D. Robust classification of salient links in complex networks. Nat. Commun. 2012 doi: 10.1038/ncomms1847. [DOI] [PubMed] [Google Scholar]
- Hanson C.A., Fuhrman J.A., Horner-Devine M.C., Martiny J.B.H. Beyond biogeographic patterns: processes shaping the microbial landscape. Nat. Rev. Microbiol. 2012;10:497–506. doi: 10.1038/nrmicro2795. [DOI] [PubMed] [Google Scholar]
- He F.L., Zhang D.Y., Lin K. Coexistence of nearly neutral species. J. Plant Ecol. 2012;5:72–81. [Google Scholar]
- HMP Consortium Structure, function and diversity of the healthy human microbiome. Nature. 2012;486:207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbell S.P. Princeton University Press; 2001. The Unified Neutral Theory of Biodiversity and Biogeography. [DOI] [PubMed] [Google Scholar]
- Integrative HMP (iHMP) Research Network Consortium The integrative human microbiome project. Nature. 2019;569:641–648. doi: 10.1038/s41586-019-1238-8. https://www.nature.com/articles/s41586-019-1238-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson P.T.J., Preston D.L., Hoverman J.T., Richgels K.L.D. Biodiversity decreases disease through predictable changes in host community competence. Nature. 2013;494:230–234. doi: 10.1038/nature11883. [DOI] [PubMed] [Google Scholar]
- Johnson P.T.J., Ostfeld R.S., Keesing F. Frontiers in research on biodiversity and disease. Ecol. Lett. 2015;18:1119–1133. doi: 10.1371/journal.pone.0041606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Ma Z.S. Diversity scaling of human vaginal microbial communities. Zoolog. Res. 2019;40:587–594. doi: 10.24272/j.issn.2095-8137.2019.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotter H., Altfeld M. Sex differences in immunity. Semin. Immunopathol. 2019;41:133–135. doi: 10.1007/s00281-018-00728-x. [DOI] [PubMed] [Google Scholar]
- Ma Z.S. Power law analysis of the human microbiome. Mol. Ecol. 2015;24:5425–5428. doi: 10.1111/mec.13394. [DOI] [PubMed] [Google Scholar]
- Ma Z.S. Science Press; 2017. Bioinformatics: Computing and Software. [Google Scholar]
- Ma Z.S. The P/N (Positive-to-Negative links) ratio in complex networks—a promising in silico biomarker for detecting changes occurring in the human microbiome. Microb. Ecol. 2018;75:1063–1073. doi: 10.1007/s00248-017-1079-7. [DOI] [PubMed] [Google Scholar]
- Ma Z.S. Testing the Anna Karenina principle in human microbiome-associated diseases. iScience. 2020 doi: 10.1016/j.isci.2020.101007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z.S. Heterogeneity-disease relationship in the human microbiome associated diseases. FEMS Microbiol. Ecol. 2020 doi: 10.1093/femsec/fiaa093. [DOI] [PubMed] [Google Scholar]
- Ma Z.S. Assessing and interpreting the metagenome heterogeneity with power law. Front. Microbiol. 2020 doi: 10.3389/fmicb.2020.00648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z.S., Ellison A.M. Dominance network analysis provides a new framework for studying the diversity-stability relationship. Ecol. Monogr. 2019;89:e01358. https://esajournals.onlinelibrary.wiley.com/doi/full/10.1002/ecm.1358 [Google Scholar]
- Ma Z.S., Ellison A.M. Towards a unifying diversity-area relationship (DAR) of species- and gene-diversity. bioRxiv. 2020 doi: 10.1101/2020.05.16.099861. [DOI] [Google Scholar]
- Ma Z.S., Li L.W. Measuring metagenome diversity and similarity with Hill numbers. Mol. Ecol. Resour. 2018;18:1339–1355. doi: 10.1111/1755-0998.12923. [DOI] [PubMed] [Google Scholar]
- Ma Z.S., Ye D. Trios—promising in silico biomarkers for differentiating the effect of disease on the human microbiome network. Sci. Rep. 2017;7:13259. doi: 10.1038/s41598-017-12959-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma B., Forney L.J., Ravel J. The vaginal microbiome: rethinking health and disease. Annu. Rev. Microbiol. 2012;66:371–389. doi: 10.1146/annurev-micro-092611-150157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z.S., Guan Q., Ye C., Zhang C., Foster J.A., Forney L.J. Network analysis suggests a potentially ‘evil’ alliance of opportunistic pathogens inhibited by a cooperative network in human milk bacterial communities. Sci. Rep. 2015;5 doi: 10.1038/srep08275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z.S., Li L., Li W., Li J., Chen H. Integrated network-diversity analyses suggest suppressive effect of Hodgkin’s lymphoma and slightly relieving effect of chemotherapy on human milk microbiome. Sci. Rep. 2016;6:28048. doi: 10.1038/srep28048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z.S., Li L.W., Gotelli N.J. Diversity-disease relationships and shared species analyses for human microbiome-associated diseases. ISME J. 2019 doi: 10.1038/s41396-019-0395-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Z.S., Zhang C., Zhang Q., Li J., Li W., Qi L., Yang X. A brief review on the ecological network analysis with applications in the emerging medical ecology. In: McGenity T., et al, editors. Hydrocarbon and Lipid Microbiology Protocols, Springer Protocols Handbooks. Springer; 2016. pp. 7–41. [Google Scholar]
- Ning D., Deng Y., Tiedje J.M., Zhou J. A general framework for quantitatively assessing ecological stochasticity. Proc. Natl. Acad. Sci. U S A. 2019;116:16892–16898. doi: 10.1073/pnas.1904623116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman M.E.J., Clauset A. Structure and inference in annotated networks. Nat. Commun. 2016;7:11863. doi: 10.1038/ncomms11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak M.A. Evolutionary Dynamics: Exploring the Equations of Life. Belknap Press of Harvard University Press; 2006. p. 377. [Google Scholar]
- Ohta, T. (2008) Molecular Evolution: Nearly Neutral Theory. Encyclopedia of Life Sciences.
- Rastelli M., Knauf C., Cani P.D. Gut microbes and health: a focus on the mechanisms: linking microbes, obesity, and related disorders. Obesity. 2018;26:792–800. doi: 10.1002/oby.22175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosindell J., Hubbell S.P., Etienne R.S. The unified neutral theory of biodiversity and biogeography at age ten. Trends Ecol. Evol. 2011;26:340–348. doi: 10.1016/j.tree.2011.03.024. [DOI] [PubMed] [Google Scholar]
- Shekhtman L.M., Bagrow J.P., Brockmann D. Robustness of skeletons and salient features in networks. J. Complex Netw. 2014 doi: 10.1093/comnet/cnt019. [DOI] [Google Scholar]
- Sobel J.D. Is there a protective role for vaginal flora? Curr. Infect. Dis. Rep. 1999;1:379–383. doi: 10.1007/s11908-999-0045-z. [DOI] [PubMed] [Google Scholar]
- Turner P.V. The role of the gut microbiota on animal model reproducibility. Anim. Model. Exp. Med. 2018;1:109–115. doi: 10.1002/ame2.12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellend M. Conceptual synthesis in community ecology. Q. Rev. Biol. 2010;85:183–206. doi: 10.1086/652373. [DOI] [PubMed] [Google Scholar]
- Vellend M. Princeton University Press; 2016. The Theory of Ecological Communities. [Google Scholar]
- Vemuri R., Sylvia K.E., Klein S.L., Forster S.C., Plebanski M., Eri R., Flanagan K.L. The microgenderome revealed: sex differences in bidirectional interactions between the microbiota, hormones, immunity and disease susceptibility. Semin. Immunopathol. 2019;41:265–275. doi: 10.1007/s00281-018-0716-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelzang A., Guerrini M., Minato N., Fagarasan S. Microbiota—an amplifier of autoimmunity. Curr. Opin. Immunol. 2018;55:15–21. doi: 10.1016/j.coi.2018.09.003. [DOI] [PubMed] [Google Scholar]
- Williams B., Landay A., Presti R.M. Microbiome alterations in HIV infection a review. Cell. Micobiol. 2016;18:645–651. doi: 10.1111/cmi.12588. [DOI] [PubMed] [Google Scholar]
- Zaneveld J.R., Mcminds R., Vega Thurber R. Stress and stability: applying the Anna Karenina principle to animal microbiomes. Nat. Microbiol. 2017;2:17121. doi: 10.1038/nmicrobiol.2017.121. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2A. The parameters of fitting Hubbell's (2001) classic neutral theory model with raw data∗, Related to Figures 2 and 3
Table S2B. The parameters of fitting Hubbell's (2001) classic neutral theory model with singleton removed∗, Related to Figures 2 and 3
Table S2C. The parameters of fitting Hubbell's (2001) classic neutral theory with data of minus-1∗, Related to Figures 2 and 3
Table S2D. The parameters of fitting Hubbell's (2001) classic neutral theory model with data of minus-2∗, Related to Figures 2 and 3
Table S2E. The parameters of fitting Hubbell's (2001) classic neutral theory with data of minus-3∗, Related to Figures 2 and 3
Table S6A. The means of Ning et al. (2019) normalized stochasticity ratio (NSR) for the intra-healthy treatment, intra-diseased treatment, and inter healthy-versus-diseased treatments, as well as the Wilcoxon tests among them (without using FDR control), Related to Figure 4.
Table S6B. The means of Ning et al. (2019) normalized stochasticity ratio (NSR) for the intra-healthy treatment, intra-diseased treatment, and inter healthy-versus-diseased treatments, as well as the Wilcoxon tests among them (after FDR control is applied), Related to Figure 4.
Data Availability Statement
All datasets analyzed in this study are available in public domain, and see Table S1 for the access information. All software codes are available in public domain as cited in the paper.