

SUMMARY
Increasing evidence suggests that deficient immune modulation and microbial infections underline neurodegeneration, but the mechanisms remain obscure. Here, we show that the G-protein-coupled receptor (GPCR) SRBC-48, which belongs to the class BC serpentine receptors, has a protective role in Caenorhabditis elegans dendrite degeneration caused by Pseudomonas aeruginosa infection. Our results indicate that SRBC-48 functions in a cell-autonomous manner in AWC neurons to protect against infection-associated dendrite degeneration. The absence of SRBC-48 results in a reduced lifespan caused by a pathogen infection early in life that induces dendrite degeneration. The decreased longevity in animals deficient in SRBC-48 is due to uncontrolled activation of immune genes, particularly those regulated by the FOXO family transcription factor DAF-16 that is part of the insulin/insulin-like growth factor (IGF)-1 receptor homolog DAF-2. These results reveal how an infection early in life can not only induce dendrite degeneration but also reduce lifespan.
Graphical Abstract
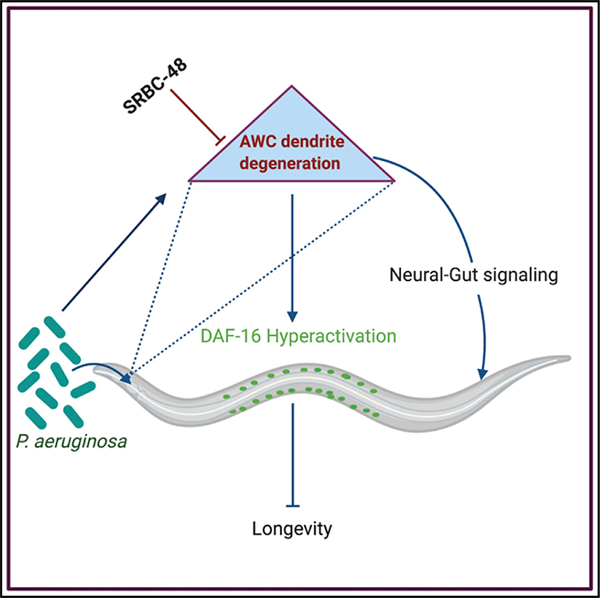
In Brief
Kaur and Aballay show that the G-protein-coupled receptor SRBC-48 functions cell autonomously in AWC neurons to protect from dendrite degeneration caused by infection with Pseudomonas aeruginosa. Dendrite degeneration in AWC results in decreased longevity that may be due, in part, to hyperactivation of the FOXO family transcription factor DAF-16.
INTRODUCTION
Neurodegenerative diseases are important medical problems characterized by the progressive degeneration or death of one or more types of neurons. Dendrites, the primary site for entry of neural signals into neurons, have been reported to get degenerated in various pathological conditions including neurodegenerative and cognitive disorders (Kweon et al., 2017; Kwon et al., 2018). Recent evidence indicates that degeneration involves not only cell-autonomous processes but also cell-non-autonomous mechanisms. The role of activated microglia in disease progression in various degenerative neurological conditions (Heneka et al., 2014) points toward the involvement of cell-non-autonomous factors. Increasing evidence suggests that immune modulators play a role in different neurodegenerative diseases, including Alzheimer’s and multiple sclerosis (Camara-Lemarroy et al., 2018; Cao and Zheng, 2018). The gut microbiome and pathogenic microbes like herpes simplex virus-1, human immunodeficiency virus (HIV), and hepatitis C virus (HCV) have been identified as potential candidates that play a role in Alzheimer’s disease (AD) (Cho and Blaser, 2012; Hill et al., 2014; Huang et al., 2014; Mancuso et al., 2014; Miklossy, 2011; Poole et al., 2017). Additionally, microbial infections have also been also linked to several of the changes seen in AD patients, like cognitive deficits, inflammation, brain cell atrophy, and altered gene expression (Heintz and Mair, 2014; Kim et al., 2013; Yatsunenko et al., 2012).
The nematode Caenorhabditis elegans has been extensively used to study age-associated neurodegeneration because of the simplicity of its nervous system and the mapping of its entire connectome, which make it easy to use as a model for neuroscience studies (Cook et al., 2019). Moreover, it was found that exposure to Streptomyces venezuelae caused dopaminergic neurodegeneration (Caldwell et al., 2009) and that exposure to Pseudomonas aeruginosa triggered changes in neural dendrites that are hallmarks of neurodegeneration (Wu et al., 2015). In nature, C. elegans is found in environments particularly rich in microbes and have evolved mechanisms to differentiate between non-pathogenic and pathogenic bacteria. Sensory neurons and G-protein-coupled receptors (GPCRs) play a role in controlling innate immunity against bacterial infections (Cao et al., 2017; Singh and Aballay, 2009; Styer et al., 2008; Sun et al., 2011, 2017). Indeed, GPCRs present in the sensory neurons play an essential role in protecting the nematode from pathogenic bacteria by activating a flight-and-fight response that involves activation of microbicidal mechanisms and pathogen avoidance (Singh and Aballay, 2019).
Herein, we took a forward genetic approach to uncover regulatory mechanisms involved in the control of the effects of P. aeruginosa infection on the degeneration of chemosensory neurons in C. elegans. Chemosensory neurons are one of the first cell types that encounter external environment stimuli via surface receptors. We previously found that during pathogen infection the dendrites of the sensory neurons displayed degeneration phenotypes, including dendrite blebbing and dendrite branching (Wu et al., 2015). To identify genes involved in the control of these degeneration phenotypes, we conducted a screen to isolate mutants exhibiting enhanced susceptibility to infection-associated dendrite degeneration. We identified a mutant carrying a mutation in the srbc-48 gene, which was susceptible to infection-associated dendrite degeneration. The gene srbc-48 belongs to a secretin-like (class B) family of GPCRs. GPCRs are the largest membrane-bound protein family and more than half of all drugs target these receptors (Lagerström and Schiöth, 2008). They have important roles in physiological processes, including pain sensation, tumorigenesis, inflammation, metabolic disorders, and neurotransmission. The secretin family of GPCRs, specifically, represents important drug targets for therapeutics against neurodegeneration, diabetes, and stress (Bortolato et al., 2014; Hollenstein et al., 2014).
We found that the srbc-48 gene is expressed in olfactory AWC neurons where it plays a protective role by preventing pathogen-mediated deleterious effects on both neural integrity and the lifespan of the animals. The mutation in this receptor resulted in enhanced infection-associated dendrite degeneration and the hyperactivation of the DAF-16/FOXO transcription factor. Our data suggest that hyperactivation of DAF-16 in srbc-48 animals, which resulted in the uncontrolled expression of immune genes, may ultimately decrease longevity. Thus, our findings uncover a role of SRBC-48 in protecting animals from infection-associated degeneration in a cell-autonomous manner. They also indicate that dendrite degeneration caused by an infection early in life may negatively impact the lifespan of animals.
RESULTS
SRBC-48 Protects against Infection-Associated Dendrite Degeneration
To identify the genes that play a role in the neurodegenerative changes induced by infection with P. aeruginosa, we conducted a forward genetic screen for mutants exhibiting an enhanced infection-associated dendrite degeneration (IADD) phenotype. The strain used, CX5974, takes advantage of the odr-1 promoter to express red fluorescent protein (RFP) in AWC, AWB, and I1 neurons (Chen et al., 2011). Each of the two AWC neurons, AWCL, and AWCR, are sensory neurons with ciliated sheet-like endings and association with amphid sheath. We screened approximately 80,000 mutagenized haploid genomes resulting in the selection of 17 mutants exhibiting changes in the morphology of the dendrites after 24 h of P. aeruginosa infection (Figure 1A). These mutants showed significant changes in the morphology of the dendrites in terms of the bead and blebbing like structures compared to CX5974 control animals (Figure 1B). Some of the mutants also exhibited waviness in the dendrite structure. Mutants showing strong IADD phenotypes after being backcrossed six times (Figure 1C) were sequenced.
Figure 1. Forward Genetic Screen to Isolate Mutants Susceptible to Infection-Associated Dendrite Degeneration.

(A) Schematic diagram of the forward genetic screen. Animals (P0) were treated with ethylmethanesulfonate (EMS) and the F1 animals were self-fertilized. The F2 animals were analyzed for dendrite degeneration, and animals exhibiting any changes were discarded. The remaining mutants were infected with P. aeruginosa for 24 h and screened for dendrite degeneration.
(B) Representative photomicrographs of dendrite degeneration in control animals expressing RFP under the control of odr-1 promoter (CX5974) (top panel) and two representative mutants (#23 and #15) (bottom panels) after exposed to P. aeruginosa for 24 h. The scale bars represent 20 µm. White asterisk indicates bead-like structures and wavy dendrites.
(C) Quantification of dendrite degeneration in control and different mutant animals after 24 h of infection.
After subtraction of the common variants, linkage maps of single-nucleotide polymorphisms (SNPs) were obtained (Figure S1). Further analysis of the mapped region of mutant number 23 revealed a single C→T mutation in the GPCR srbc-48 gene in the C. elegans genome that results in a serine-to-phenylalanine substitution at residue 280. As shown in Figure 2A, the AWC cells of srbc-48(ac23) animals exhibited blebbed and beaded dendrites, which are hallmarks of neurodegeneration, after only 24 h of exposure to P. aeruginosa, while those changes were not observed in control animals. Only animals exhibiting blebbing or bead-like structures along the length of the dendrite were scored as having infection-associated dendrite degeneration. While 76% of the srbc-48(ac23) animals exhibited the dendrite degeneration phenotype, only 8% of the control animals exhibited the phenotype (Figure 2B). To address whether the dendrite degeneration observed in the AWC cells of srbc-48(ac23) animals affected the function of the cells, we studied the chemotaxis of infected animals toward the chemoattractant benzaldehyde. As shown in Figure 2C, srbc-48(ac23) animals exhibited a significant reduction in chemotaxis compared to control animals, indicating that the function of AWC neurons is defective.
Figure 2. srbc-48(ac23) Animals Are Susceptible to Infection-Associated Dendrite Degeneration.

(A) Representative photomicrographs of dendrite degeneration in control (CX5974) and srbc-48(ac23) animals after exposure to P. aeruginosa for 24 h. The scale bars represent 20 µm.
(B) Quantification of dendrite degeneration in control and srbc-48(ac23) animals. Control versus srbc-48(ac23) (p < 0.0001) via t test (n = 5; animals per condition = 50).
(C) Quantification of chemotaxis index (CI) in control and srbc-48(ac23) animals in response to benzaldehyde. Control versus srbc-48(ac23) (p < 0.01) via t test (n = 5; animals per condition > 100).
(D) Quantification of dendrite degeneration in control and srbc-48(ac23) animals after exposure to E. faecalis for 24 h. Control versus srbc-48(ac23) (n.s.) via t test (n = 5; animals per condition = 20).
(E) Quantification of dendrite degeneration in control and srbc-48(ac23) animals in response to cold stress. Control versus srbc-48(ac23) (n.s.) via t test (n = 5; animals per condition = 20).
(F) Quantification of dendrite degeneration in control and srbc-48(ac23) animals in response to osmotic stress. Control versus srbc-48(ac23) (n.s.) (n = 5; animals per condition = 20) via t test.
(B–F) The rhomboids represent individual experiments.
We also studied the morphological changes in AWC dendrites in srbc-48(ac23) animals exposed to the Gram-positive pathogen Enterococcus faecalis or insults like cold and osmotic stress. We found that exposure to E. faecalis for 24 h did not induce any dendrite degeneration in the srbc-48(ac23) mutants compared to control animals (Figure 2D). To study whether cold stress induces dendrite degeneration, control and srbc-48(ac23) animals were exposed to 4°C for 24 h. As shown in Figure 2E, although the number of srbc-48(ac23) animals exhibiting morphological changes in the dendrites was higher than that of control animals, the differences were not significant. We also studied the susceptibility of srbc-48(ac23) and control animals to another non-pathogen stressor, osmotic stress. Mutants and control animals were exposed to 500 mM sodium chloride (NaCl) to cause osmotic stress and then were observed for changes in the morphology of the dendrites. As in the case of cold stress, srbc-48(ac23) mutants exhibited a trend toward higher dendrite degeneration than control animals (Figure 2F). However, the trend was neither significant nor was it nearly as strong as the dendrite degeneration induced by P. aeruginosa infection (Figure 2B). Overall, these results indicate that srbc-48(ac23) animals are not necessarily susceptible to dendrite degeneration caused by stress in general.
Gene srbc-48 Acts Cell Autonomously to Regulate Infection-Associated Dendrite Degeneration
To confirm that the absence of srbc-48 caused the srbc-48(ac23) animals to become susceptible to infection-associated dendrite degeneration, we created srbc-48(ac23) animals expressing srbc-48 under its own promoter (strain AY145). As shown in Figures 3A and 3B, the IADD phenotype of srbc-48(ac23) animals was completely rescued in these animals.
Figure 3. Expression of the Gene srbc-48 Rescues the Infection-Associated Dendrite Degeneration Phenotype of srbc-48(ac23) Mutants.

(A) Representative photomicrographs of dendrite degeneration after exposed to P. aeruginosa for 24 h in srbc-48(ac23) animals and animals expressing srbc-48 under its own promoter, strain AY145 (srbc-48(ac23);Psrbc-48::srbc-48). The scale bars represent 20 µm.
(B) Quantification of the dendrite degeneration phenotype in control, srbc-48(ac23), and strain AY145 (srbc-48(ac23);Psrbc-48::srbc-48) animals. The black symbols represent individual experiments (n = 5; animals per condition = 50). Control versus srbc-48(ac23) (p < 0.0001) and srbc-48(ac23) versus strain AY145 (p < 0.0001) via one-way ANOVA test.
(C) Representative photomicrographs of animals expressing RFP under the control of the odr-1 promoter and GFP under the srbc-48 promoter. The bottom panel corresponds to the merged images. The scale bars represent 20 µm.
To determine the expression pattern of the srbc-48 gene, we expressed GFP driven by the 4-kb promoter of the srbc-48 gene in control animals that expressed RFP under the odr-1 promoter. The transgene srbc-48::gfp was expressed in head neurons that were likely to be AWC neurons and one additional neuron (Figure 3C). Thus, we reasoned that SRBC-48 could have cell-autonomous or non-cell-autonomous effects in infection-associated dendrite degeneration. To distinguish between these possibilities, we used 4 kb of the odr-1 promoter to express srbc-48 in the AWC neurons but not in the additional neuron that also expresses srbc-48. As shown in Figures 4A and 4B, srbc-48(ac23) animals expressing srbc-48 under the odr-1 promoter (AY146) showed full rescue of the IADD phenotype.
Figure 4. srbc-48 Mutation Enhances that Animal Susceptibility to Infection-Associated Dendrite Degeneration in a Cell-Autonomous Manner.

(A) Representative photomicrographs of srbc-48 animals expressing srbc-48 and gfp under the control of the odr-1 promoter, strain AY146 (srbc-48(ac23);Podr-1::srbc-48::SL2::GFP), after 24 h of P. aeruginosa infection. The scale bars represent 20 µm.
(B) Quantification of the dendrite degeneration phenotype in control, srbc-48(ac23), and strain AY146 (srbc-48(ac23);Podr-1::srbc-48::SL2::GFP) animals. The black symbols represent individual experiments (n = 5; animals per condition = 50). Control versus srbc-48(ac23) (p < 0.0001) and srbc-48(ac23) versus strain AY146 (p < 0.0001) via one-way ANOVA test.
(C) Representative photomicrographs of strain NC1686 (wdIs51), which expresses GFP in PVD neurons, crossed with srbc-48(ac23) animals after exposure to P. aeruginosa for 24 h (n = 3; animals per condition = 20). The scale bars represent 100 µm.
(D) Representative photomicrographs of strain CX3553 (kyIs104), which expresses GFP in AWB neurons, crossed with srbc-48(ac23) animals after exposure to P. aeruginosa for 24 h (n = 3; animals per condition = 20). The scale bars represent 20 µm.
Next, we tested whether srbc-48 could function in a non-cell-autonomous manner to prevent infection-associated dendrite degeneration in different types of neurons. Thus, we analyzed dendrite degeneration in the PVD and AWB neurons of srbc-48(ac23) animals. PVD neurons are mechanosensory neurons that show age-related morphological changes in their dendrites (Kravtsov et al., 2017), and AWB are chemosensory neurons that are in close proximity to AWC neurons. We did not observe any signs of dendrite degeneration in the PVD and AWB neurons of srbc-48(ac23) animals after exposure to P. aeruginosa for 24 h (Figures 4C and 4D). Taken together, these results suggest srbc-48 functions cell autonomously to protect AWC neurons from infection-associated dendrite degeneration.
Infection-Associated Dendrite Degeneration Decreases Animal Longevity in srbc-48 Animals
Because it has been shown that inhibition of str-2, which is a GPCR expressed in the AWC neurons, extends lifespan (Alcedo and Kenyon, 2004), we decided to investigate whether srbc-48(ac23) animals exhibited any abnormal longevity. We did not find any differences in the lifespan of control and srbc-48(ac23) animals in the absence of infection or after 8 h of infection (Figures 5A and 5B). These results were expected as the AWC dendrites of srbc-48(ac23) animals do not exhibit degeneration in the absence of an infection. However, we reasoned that a short infection by P. aeruginosa that is insufficient to cause death but sufficient to cause dendrite degeneration might alter the longevity of srbc-48(ac23) animals. To test this hypothesis, we performed an acute P. aeruginosa infection by only exposing the animals to the pathogen for 12 and 24 h. To remove any live P. aeruginosa, the animals were rinsed with a buffer containing antibiotics before transferring them to plates that contained antibiotics and were seeded with killed E. coli (Head et al., 2017). As shown in Figures 5C and 5D, there was a significant decrease in the lifespan of srbc-48(ac23) animals infected for 12 and 24 h with P. aeruginosa compared to control animals.
Figure 5. Pathogen Infection in srbc-48 Animals Results in a Reduced Lifespan.

(A) Representative lifespan plots of control and srbc-48(ac23) without infection. Control versus srbc-48(ac23) (n.s.).
(B) Representative lifespan plots of control and srbc-48(ac23) animals after 8 h of P. aeruginosa exposure. Control versus srbc-48(ac23) (n.s.).
(C) Representative lifespan plots of control and srbc-48(ac23) after 12-h P. aeruginosa exposure. Control versus srbc-48(ac23) (p < 0.001).
(D) Representative lifespan plots of control, srbc-48(ac23), and rescued strains AY145(srbc-48(ac23);Psrbc-48::srbc-48) and AY146 (srbc-48(ac23);Podr-1::srbc-48::SL2::GFP) after 24 h of P. aeruginosa exposure. Control versus srbc48(ac23) (p < 0.0001), srbc-48(ac23) versus strain AY145 (p < 0.0001), srbc-48(ac23) versus strain AY146 (p < 0.0001), control versus strain AY145 (n.s.), and control versus strain AY146 (n.s.) (n = 3; animals per condition = 100).
We observed a decrease in the lifespan of srbc-48(ac23) animals infected for only 12 h with P. aeruginosa (Figure 5C), but we only observed significant dendrite degeneration 24 h after infection (Figures 2A and 2B). Therefore, we asked whether only 12 h of P. aeruginosa infection was sufficient to induce dendrite degeneration later in life, even in the absence of the pathogen. Animals exposed to P. aeruginosa for only 12 h developed dendrite degeneration 12 h later (Figures S2A and S2B), which is consistent with the reduced longevity of animals infected for only 12 h (Figure 5C) and suggests that infection-associated dendrite degeneration is irreversible. To test this hypothesis, we analyzed dendrite degeneration in srbc-48(ac23) animals at 1, 3, and 5 days after the infection with P. aeruginosa has been cleared and found that dendrite degeneration continued to increase (Figure S2C). These results indicate that the infection-associated dendrite degeneration caused by infection with P. aeruginosa for only 12 or 24 h results in a reduction of the longevity of srbc-48(ac23) animals.
Because expression of srbc-48 under the control of its own promoter or the odr-1 promoter rescued the dendrite degeneration phenotype induced by P. aeruginosa infection, we next sought to address whether srbc-48 expression could rescue the decrease in longevity caused by P. aeruginosa infection early in the life of srbc-48(ac23) animals. As shown in Figure 5D, expression of srbc-48 fully rescued the decreased longevity of srbc-48(ac23) animals caused by an infection early in life, indicating that defective AWC cells reduce lifespan. Consistent with this idea, we found that pathogen infection reduced the lifespan of both AWC ablated and srbc-48(ac23) animals to a similar extent (Figure S3A).
We then studied the susceptibility to P. aeruginosa of the animals. We observed that there was no significant change in the survival of the control and the srbc-48(ac23) animals constantly exposed to P. aeruginosa (Figure S4A). Similar results were obtained with animals infected with E. faecalis (Figure S4B). Taken together, these results indicate that srbc-48(ac23) animals are not susceptible to pathogen infection, suggesting that the deleterious effects of P. aeruginosa infection on dendrites is not a consequence of the premature death of the infected animals or due to the animals being sickly. The results also suggest that SRBC-48 and AWC control pathways play an important role in the control of longevity.
Infection-Associated Dendrite Degeneration Increases Gene Expression of Immune Genes
To elucidate the mechanism by which infection-associated dendrite degeneration decreases longevity in srbc-48(ac23) animals compared to control animals, we studied changes in gene expression after 24 h of infection with P. aeruginosa. We crossed both control and srbc-48(ac23) animals with sterile fer-1 animals to avoid the collection of RNA from the progeny. Only differentially expressed genes with p-adj < 0.05 were considered to avoid false discovery rate, and for the upregulated genes, only those having a fold change >2-fold were taken into account (Tables S1, S2, and S3). An unbiased gene enrichment analysis using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) (https://david.ncifcrf.gov/) (Huang et al., 2009) showed that the innate immune Gene Ontology (GO) cluster is among the groups with the most hits. Other GO clusters correspond to ion, acetylcholine, glutamate, and chloride transport genes, neuropeptide signaling pathway, and signal transduction (Figure 6A; Table S4).
Figure 6. RNA-Sequencing Analysis of the Upregulated Genes Showed an Enrichment for Immune Genes as well as Overlap with the Genes Regulated by DAF-16.

(A) Enrichment score of the upregulated genes in srbc-48(ac23) animals compared to control animals.
(B) Graphical representation of the representation factor of the upregulated genes that are controlled by the PMK-1, DAF-16, CEP-1, and SKN-1 pathways.
(C) Quantitative real-time PCR analysis of CEP-1 and DAF-16-dependent genes in srbc-48(ac23) compared with control animals (n = 3; three technical replicates in each case).
To analyze more in depth the innate immunity cluster, we compared this cluster of genes to previously identified gene sets that are known to be controlled by various signaling pathways and transcription factors involved in C. elegans response against pathogen infection (Table S5). We found a significant overlap between genes upregulated in srbc-48(ac23) animals infected with P. aeruginosa and gene sets regulated by DAF-16/FOXO, CEP-1, and SKN-1 (Figure 6B). We further confirmed the RNA-sequencing results by selecting the genes from pathways with maximum overlap and performing quantitative realtime PCR (Figure 6C). Our analysis suggests that the decreased longevity that srbc-48 animals exhibit after exposure to P. aeruginosa may be a consequence of uncontrolled activation of immune pathways.
Infection-Associated Dendrite Degeneration Possibly Decreases Lifespan of srbc-48 Animals by Hyperactivating DAF-16
The insulin/insulin-like growth factor (IGF)-1 receptor homolog DAF-2 and its main target, DAF-16, play an important role in the regulation of lifespan at least in part by controlling immune gene expression (Garsin et al., 2003; Lee et al., 2003; Murphy et al., 2003). Because our gene expression studies indicate that there is a significant enrichment of DAF-16 controlled genes in infected srbc-48(ac23) animals and excessive DAF-16 activity may have deleterious consequences (Singh and Aballay, 2009), we investigated DAF-16 activation. Like mammalian FOXO3a, the activity of DAF-16 is tightly regulated by a wide variety of external stimuli, including different stresses, that result in its nuclear localization (Brunet et al., 2004; Henderson and Johnson, 2001; Lin et al., 2001). As shown in Figures 7A and 7B, srbc-48(ac23) animals showed an extensive DAF-16 nuclear localization after 24 h of P. aeruginosa infection that was absent in control animals. To address whether the increased DAF-16 nuclear localization exhibited by srbc-48(ac23) animals is reversible, we studied DAF-16 localization 10 days after the animals were cleared from P. aeruginosa and transferred to E. coli. As shown in Figures S5A and S5B, DAF-16 remains in the nuclei of srbc-48(ac23) animals, which suggests that DAF-16 inactivation is deficient in these animals. Given that the decrease in longevity of srbc-48(ac23) animals was rescued by the expression of srbc-48 under the control of the odr-1 promoter that drives its expression to AWC (Figure 5D), we reasoned that we should also observe rescue of DAF-16 nuclear localization in srbc-48(ac23) animals expressing srbc-48 in AWC. We found that DAF-16 relocalized to the cytosol 5 days after infection with P. aeruginosa in the rescued animals (Figures S5C and S5D). These findings indicate that a defect in AWC neuron structure and function causes an irreversible DAF-16/FOXO nuclear localization induced by P. aeruginosa that results in a reduced lifespan long after the infection has been cleared.
Figure 7. Inactivation of DAF-16 Rescues the Decreased Lifespan of srbc-48(ac23) Animals that Suffered an Infection Early in Life.

(A) Representative mosaic photomicrographs of DAF-16::GFP nuclear localization in control [daf-16p::daf-16a/b::gfp+rol-6(su1006)] and mutant srbc-48(ac23);[daf-16p::daf-16a/b::gfp+rol-6(su1006)] animals after exposed to P. aeruginosa for 24 h. The scale bars represent 100 µm.
(B) Quantification of DAF-16 nuclear localization in control [daf-16p::daf-16a/b::gfp+rol-6(su1006)] and mutant srbc-48(ac23);[daf-16p::daf-16a/b::gfp+rol-6(su1006)] animals (n = 3; animals per condition = 20). Nuclear translocation of control versus srbc-48(ac23) (p < 0.0001), one-way ANOVA test.
(C) Representative lifespan plots of control and srbc-48(ac23) animals after knockdown of daf-16 via RNAi followed by 24-h exposure to P. aeruginosa (n = 3; animals per condition = 100). srbc-48(ac23) control RNAi versus srbc-48(ac23);daf-16 RNA (p < 0.0001).
(D) Representative lifespan plots of control and srbc-48(ac23) animals after knockdown of cep-1 via RNAi followed by 24-h exposure to P. aeruginosa (n = 3; animals per condition = 100). srbc-48(ac23) control RNAi versus srbc-48(ac23);cep-1 RNAi (p < 0.01).
(E) Representative lifespan plots of control, srbc-48(ac23), control;daf-16(mu86), and srbc-48(ac23);daf-16(mu86) animals after 24-h P. aeruginosa exposure (independent replicates, n = 3; animals per condition, n = 100). srbc-48(ac23) versus srbc-48(ac23);daf-16(mu86) (p < 0.0001), control versus control;daf-16(mu86) (p < 0.0001), control;daf-16(mu86) versus srbc-48(ac23);daf-16(mu86) (p < 0.0001), and control versus srbc-48(ac23);daf-16(mu86) (n.s.).
We hypothesized that if the infection-associated dendrite degeneration in srbc-48 animals that results in the activation of DAF-16 is responsible for their reduced longevity, DAF-16 inhibition should improve their longevity. Consistent with this idea, we found that RNAi inactivation of daf-16 suppressed the reduced lifespan of srbc-48(ac23) animals that were infected with P. aeruginosa as young adults (Figure 7C). In contrast, RNAi inactivation of cep-1 decreased the lifespan of both srbc-48(ac23) and control animals (Figure 7D). The rescue in lifespan after daf-16 knockdown was further confirmed by using daf-16(mu86) mutant animals (Figure 7E). The rescue in lifespan by daf-16 inactivation suggests that the dendrite degeneration suffered by srbc-48(ac23) animals due to infection early in their life may result in a decreased longevity due to hyperactivation of DAF-16.
DISCUSSION
Although there are differences between the C. elegans and mammalian nervous system, several studies have shown that genes linked to neurodegenerative diseases in humans have C. elegans homologs. These genes when mutated or deleted result in strong degeneration phenotypes in C. elegans neurons, demonstrating the remarkable similarity between the two systems in terms of connectivity and functionality (Alexander et al., 2014; Faber et al., 2002; Fang et al., 2019; Harrington et al., 2010). Here, we show that P. aeruginosa infection induces dendrite degeneration in the AWC neurons of C. elegans. Dendrites have an essential role in the function of neuronal circuits, and dendrite degeneration is reported in neurodegenerative and cognitive disorders (Kweon et al., 2017; Kwon et al., 2018). Studies have also shown that microbial pathogens can trigger neurodegenerative diseases and associated symptoms, including inflammation and memory deficits in Alzheimer patients (Itzhaki et al., 2016).
The underlying mechanisms responsible for infection-associated dendrite degeneration are unclear. To gain insight into the mechanisms responsible for this phenomenon, we performed a forward genetics screen to discover genes involved in infection-associated dendrite degeneration. We studied dendrite degeneration in the sensory neurons because chemoperception is used in C. elegans to distinguish pathogenic from non-pathogenic bacteria. Sensory neurons are the first neural cells to encounter external cues and they play an essential role in regulating immune responses during pathogen exposure. Our forward genetic screen identified animals that carry a mutation in the gene srbc-48, which makes them susceptible to dendrite degeneration induced by P. aeruginosa infection at earlier time points after infection. The lack of pathogen susceptibility of srbc-48(ac23) animals indicates that the observed dendrite degeneration is not a consequence of the animals being sickly or their premature death. While we did not observe dendrite degeneration in srbc-48(ac23) animals infected with E. faecalis for 24 h, other pathogens should be studied to address the pathogen specificity of the susceptibility of srbc-48(ac23) animals to infection-associated dendrite degeneration. However, the infection-associated dendrite degeneration due to srbc-48 mutation seems to be specific to AWC neurons as no dendrite degeneration was observed in AWB neurons, which are also chemosensory. Neither did we observe dendrite degeneration in PVD neurons, which undergo age-related degeneration. This suggests that srbc-48 is involved in infection-associated dendrite degeneration in AWC neurons, and that this occurs independently of aging. Additionally, dendrite degeneration of AWC neurons in the absence of functional SRBC-48 suggests that this GPCR has a neuroprotective role during P. aeruginosa infection.
We also found that infection-associated dendrite degeneration negatively impacted the lifespan of the animals. Sensory perception plays an important role in the control of lifespan in C. elegans (Alcedo and Kenyon, 2004; Apfeld and Kenyon, 1999), as GPCRs present in the sensory neurons detect olfaction and taste cues to activate other G protein-mediated signaling cascades (Troemel et al., 1995). To decipher the relationship between dendrite degeneration and decreased lifespan, we studied changes in gene expression in animals susceptible to infection-associated dendrite degeneration. We found that the immune response genes were among the most enriched cluster of genes upregulated in srbc-48(ac23) animals and that many of them are controlled by DAF-16, which plays an essential role in lifespan regulation. We also found that there was enhanced nuclear translocation of DAF-16 in srbc-48(ac23) animals after P. aeruginosa infection and that these animals were deficient in DAF-16 inactivation. Inhibition of daf-16 through RNAi or mutation suppressed the reduced lifespan of srbc-48(ac23) animals that suffered pathogen-mediated dendrite degeneration, suggesting that DAF-16 hyperactivity may be responsible for their decreased lifespan. Even though srbc-48(ac23) animals exhibit higher expression of immune genes, they are not more resistant to pathogen-mediated killing. Our lifespan studies suggest that DAF-16 hyperactivity is deleterious. Thus, it is possible that the overall activation of immune genes exhibited by srbc-48(ac23) animals is dampened by the harmful effects of excessive DAF-16 activity.
In conclusion, our study suggests a cell-autonomous role of the neuronal GPCR, srbc-48, in protecting against infection-associated dendrite degeneration. Our study provides insights into the integration between the chemosensory system and the insulin-signaling pathway. The protective function of SRBC-48 against dendrite degeneration caused by pathogen infection was found to be critical to prevent the DAF-16 hyperactivation, establishing a link between infection-associated dendrite degeneration and longevity.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagent should be directed to and will be fulfilled by the lead contact, Alejandro Aballay (aballay@ohsu.edu)
Materials Availability
All reagents generated in this study are available from the Lead contact without restriction.
Data and Code Availability
The accession number for the RNA seq data reported in this paper is Gene Expression Omnibus (GEO: GSE137112).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The C. elegans strains used in the study wild-type N2 Bristol, CX5974 (kyIs262), NC1686 (wdIs51), HH142 [fer-1(b232ts)], CF1038 [daf-16(mu86)], CX3553 (kyIs104) and TJ356 (zIs356) were procured from the Caenorhabditis Genetics Center (University of Minnesota, Minneapolis, MN). The following lines were constructed using standard genetic manipulation techniques: AY145[Psrbc-48::srbc-48], AY146[Podr-1p::srbc-48::GFP], AY147[odr-1::RFP], AY148[Psrbc-48p::GFP], AY149[odr-1::RFP; Psrbc-48p::GFP], AY151 [AY-23; wdIs51], AY152[kyIs262; mu86], AY153 [AY-23; mu86], AY154 [kyIs262; zIs356], and AY155 [AY-23; zIs356]. The rescue strain AY145 was constructed by injecting Psrbc-48::srbc-48 and co-injecting the marker Punc-122::GFP into srbc-48(ac23) animals. All strains were cultured on plates containing nematode growth medium (NGM) and maintained at 20°C. fer-1 animals were grown at the 15°C permissive temperature. All experiments were performed using hermaphrodites at young adult stage.
The bacterial strains used were Escherichia coli OP50, E. coli HT115 (DE3), Enterococcus faecalis (OG1RF), and Pseudomonas aeruginosa PA14. The cultures for these bacteria were grown in Luria-Bertani (LB) broth overnight at 37°C. E. faecalis was grown in brain-heart infusion (BHI) broth.
METHOD DETAILS
Forward Genetic Screen for Mutants Susceptible to Pathogen-Mediated Dendrite Degeneration
Ethyl-methanesulfonate (EMS) mutagenesis was performed using the CX5974 strain, which expresses red fluorescent protein (RFP) in AWC and AWB neurons. Approximately 3,000 synchronized late L4 larvae of CX5974 were treated with 50mM EMS for 4h followed by washing with M9 buffer three times for 2 hours. The washed animals (P0 generation) were then transferred to large Petri dishes (10 cm) containing E. coli OP50, and these animals were allowed to lay eggs (F1 progeny) overnight. The animals corresponding to the P0 generation were then washed away with M9 buffer, and the F1 eggs that remained attached to the plates were allowed to grow until adulthood. After one day, the F1s and any hatched F2 progeny were removed by washing. The eggs of the F2s, which remained attached to the plates after washing, were allowed to develop to young adults on E. coli OP50. These animals were observed under a fluorescent microscope and mutants exhibiting any morphological changes in the dendrites were removed. The remaining mutants were transferred to plates containing PA14 and incubated for 24 hours at 25°C. The animals that showed any morphological changes in dendrites at 24 hours were transferred to individual plates. Approximately 80,000 haploid genomes were screened. Animals showing above 65% phenotype were backcrossed six times to the parental CX5974 strain before further analysis.
Fluorescence Imaging
Confocal fluorescent images of AWC, AWB and PVD neurons were taken with a Zeiss confocal microscope system (LSM-780) and Apotome Zeiss microscope with grid-based optical sectioning using a 20X and 40X objective water-immersion lens. Animals were immobilized with 20 mM sodium-azide in M9 buffer, mounted on a 2% agar pad, and covered with a glass coverslip. The screening of the mutants was done using a Leica M165 FC fluorescence microscope.
Quantification of Dendrite Degeneration
Animals were visualized for morphological changes in the dendrites, which are considered as hallmarks of neurodegeneration, including soma branching, wavy dendrites, dendrite branching, and beaded dendrites. Animals exhibiting the aforementioned morphological changes were considered as having dendrite degeneration. Briefly, the bacterial lawns used for the degeneration assay were prepared by spreading 300ul of the 8–10 hours culture of P. aeruginosa and E. faecalis over the entire surface of modified NGM or BHI agar media, respectively, in 6-cm diameter plates and incubated at 37°C. Young adult nematodes were transferred to P. aeruginosa or E. faecalis plates and grown for 24 hours at 25°C (Wu et al., 2015). For microscopy, the animals were anesthetized using 20 mM sodium-azide in M9 buffer, mounted onto agar pads, and visualized using a Zeiss microscope or Leica M165 FC microscope.
This assay was done five times with 50 srbc-48(ac23) and control animals. The percentage of the animals exhibiting dendrite degeneration phenotypes was calculated by dividing the number of animals showing dendrite degeneration by the total number of animals in each group. To analyze PVD and AWB dendrite degeneration, the animals expressing GFP in the PVD and AWB neurons were crossed to control and srbc-48(ac23) animals and then checked for the dendrite degeneration phenotypes after 24 hours of P. aeruginosa and E. faecalis infection. Animals displaying bead dendrites, broken dendrites, or bubble-like structures were scored as exhibiting dendrite degeneration. Animals were only considered exhibiting the dendrite degeneration phenotype if the number of beads was more than 3–4 or along the whole length of the dendrite (Oren-suissa et al., 2010). The number of animals observed in each group was 20 and the magnification used was 40X and was repeated three times.
Whole-Genome Sequencing and Data Analysis
For whole-genome sequencing, the DNA of the mutant animals was extracted. Briefly, the mutants were grown at 20°C on 10 cm NGM plates seeded with E. coli OP50. These animals were grown until starvation, and then the plates were rinsed thrice with M9 buffer to remove any bacteria. Then, the animals were incubated in M9 buffer with rotation for 2h and washed three times with M9 buffer to remove bacteria from the intestine. The genomic DNA was extracted using the Gentra Puregene kit (QIAGEN, Netherlands). The DNA was subjected to whole-genome sequencing (WGS) on an Illumina HiSeq 4000 sequencing platform using 50 single-end nucleotide reads, and DNA libraries were prepared according to a standard Illumina (San Diego, CA) protocol. Library preparation and WGS were performed at Novogene Genomic Services & Solutions Company, USA.
For analyzing the whole-genome sequence data, the EMS density mapping workflow from the Cloud Map program of the Galaxy web platform was used (Minevich et al., 2012). A list of single nucleotide polymorphisms (SNPs) in the mutant was generated by comparing it with the reference C. elegans (WS220). After that, the common SNPs were subtracted, and linkage maps for the mutants were created.
C. elegans Killing Assay on P. aeruginosa and E. faecalis
Synchronized animals were obtained by allowing gravid animals to lay eggs on NGM plates for 4–6 hours at 20°C. The bacterial lawns used for C. elegans killing assays were prepared by spreading 300 ul of 8–10 hours grown culture of P. aeruginosa and E. faecalis on the complete surface of modified NGM agar medium (0.35% peptone) and BHI (brain heart infusion) agar media, respectively. The plates were subsequently incubated overnight at 37°C and then cooled to room temperature for at least 1 hour before seeding them with synchronized young adult animals. The killing assays were performed at 25°C, and live animals were transferred daily to fresh pathogen seeded plates. Animals were scored at the indicated times and were considered dead when they failed to respond to the touch of a platinum wire. Each experiment was done in triplicate with 20 worms in each group.
Osmotic Stress Assay
The plates were prepared by adding 29.22 g/L of NaCl instead of 3.0 g/L into the NGM plates as previously described (Solomon et al., 2004). Synchronized 30 young adult animals were washed with the M9 buffer thrice to remove any bacteria. Then, the pellet of the worms was added into the center of a 500 mM NaCl plate seeded with E. coli. Mutant and control animals were observed after 12 hours of exposure to check for any morphological changes in their dendrites. Each experiment was done with 20 worms in each group and repeated three times.
Cold tolerance Assay
Synchronized young adult animals were added on standard NGM plates seeded with OP50 bacteria. These plates were then transferred to a 4°C for 24 hours as previously described (Ohta et al., 2014). After 24 hours, the plates were stored at 20°C for 4 hours to allow the animals to recover from the cold stress. Then the animals were scored for changes in their dendrites. The experiment was repeated three times with 20 worms in each group.
C. elegans Longevity Assay
Lifespan assays were performed without uninfected animals and animals infected with P. aeruginosa infection for 8, 12, and 24 hours at 25°C. After infection, mutant or control animals grown on P. aeruginosa were rinsed by transferring them into 100 µL M9 containing 300 µg/ml concentration of streptomycin on NGM plates seeded with E. coli OP50. To remove any bacteria attached to the body wall, the animals were allowed to crawl out of the solution toward the OP50 lawn. Animals were further transferred onto new plates containing dead E. coli OP50 prepared by concentrating the overnight cultures at a 1:20 ratio and heat-killed at 100°C for 60 min. These plates were also supplemented with streptomycin (100 µg/ml), kanamycin (50 µg/ml) and Nystatin (10 µg/ml) to avoid contamination. A 350 µL drop of the killed bacteria was plated on a 6-cm plate and incubated at 25°C. Animals were scored at the indicated times for survival, and live animals were transferred to fresh plates if needed. Each experiment was repeated three times with 50 worms in each group and triplicate sets. For the lifespan assay after RNAi, second generation RNAi worms were used. These RNAi animals were exposed to PA14 for 24 hours at 25°C and analyzed as described above. This experiment was repeated twice with 50 worms in each group and duplicate sets.
Cloning and Generation of Transgenic C. elegans Strains
For srbc-48 rescue, plasmid pSK1(pPD95.77_Psrbc-48::srbc-48) was constructed by cloning gene srbc-48 with its 4kb promoter into the KpnI and SmaI sites of the pPD95.77 vector (Fire Lab C. elegans Vector Kit; Addgene, Cambridge, MA). To prevent gene products to fuse with GFP, plasmid pRK1 (pPD95.77_SL2::GFP) was used (Cao et al., 2017). For the AWC-specific srbc-48 rescue, plasmid pSK2 (pRK1_pPD95.77_Podr-1::srbc-48_SL2::GFP) were constructed by inserting 4kb of the odr-1 promoter sequence upstream into the SphI and SalI sites and 1101bp of the srbc-48 sequence into the SalI and BamHI sites of the plasmid pRK1. To identify the foci of srbc-48 expression, plasmid pSK4 was constructed by cloning the 4kb promoter of srbc-48 upstream into the KpnI and SmaI of the pRK1 vector. Transgenic strains were created by injecting 25 ng/µl of the plasmids together with 50 ng/µl of the co-injection marker Punc-122::GFP.
RNA Interference (RNAi)
RNA Interference was used for generating loss-of-function phenotypes by feeding worms E. coli strain HT115(DE3) expressing double-stranded RNA (dsRNA) homologous to a target gene. E. coli with the appropriate vectors were grown overnight at 37°C in LB broth containing ampicillin (100 µg/ml). The bacteria were then plated onto RNAi plates, which were NGM plates containing ampicillin (100 µg/ml) and 3 mM isopropyl β-D-thiogalactoside (IPTG). RNAi-expressing bacterial clones were allowed to grow overnight at 37°C. Gravid adults were transferred to RNAi-expressing bacterial lawns and allowed to lay eggs for 2 hours. The gravid adults were removed, and the eggs were allowed to develop at 20°C for 2 days. Gravid adults were then transferred to fresh RNAi-expressing bacterial lawns and allowed to lay eggs for 2 hours to synchronize a second-generation RNAi population. The gravid adults were removed, and the eggs were allowed to develop at 20°C to reach the young adult stage for experimental use. In all experiments, unc-22 RNAi was included as a positive control to account for the RNAi efficiency.
Assay for DAF-16 Nuclear Localization
After 24 hours of pathogen infection, mutant and control animals were washed with M-9 buffer followed by washing with the antibiotic streptomycin and transferred to plates seeded with heat-killed OP50. The animals were mounted in M9 onto microscope slides after 24 hours, 5 days, and 10 days. The slides were viewed using a Zeiss fluorescence microscope. The DAF-16 localization was studied as cytosolic, nuclear and intermediate localization as described (Oh et al., 2005). This assay was done in triplicate with 20 animals in each group.
Chemotaxis Assay
The assay for odor chemotaxis was performed as previously described (Leinwand et al., 2015) to check the function of the AWC neurons. AWC olfactory neurons play an important role in chemotaxis to volatile odorants like isoamyl alcohol, 2, 3 pentanedione, 2, 4,5 trimethylthiazole, benzaldehyde, and butanone. Briefly, synchronized young adult nematodes were transferred to plates seeded with P. aeruginosa for 24 hours at 25°C. After the incubation, the animals were washed three times with S basal buffer to remove any bacteria, suspended in a smaller volume of S basal, and added onto the center of the chemotaxis plate. Washed animals were placed on the plate and allowed to move freely for one hour. Then, 1ul of sodium azide was added to the odor spots to anesthetize animals reaching the end points. The underside of a chemotaxis plate was divided into 4 equal quadrants and a circle of 0.5 cm around the origin of the plate. Each quadrant was marked with either ‘‘BZ’’ for ‘‘Benzaldehyde’’(0.2% vol/vol, in ethanol) or ‘‘Ctrl’’ for ‘‘95% Ethanol’’ and marks were equidistant from the center and each other. Five assays were performed in triplicates and on different days. The chemotaxis index was computed as the number of animals in the odor region minus the number of animals in the control region. The chemotactic index (CI) was calculated by the equation below:
RNA Isolation and Quantitative Real-Time PCR
Synchronized animals were placed on NGM plates seeded with E. coli OP50 and grown at 20°C until the animals reached the young adult stage. Animals were collected and washed with M9 buffer three times to remove any bacteria and then were transferred to modified NGM plates containing E. coli OP50 or P. aeruginosa PA14 for 24 hours at 25°C. The P. aeruginosa plates were prepared by spreading the 300 µl of P. aeruginosa culture on the modified NGM plates and then incubated overnight at 37°C. These animals were then washed off the plates with M9 buffer 3–4 times and frozen in TRIzol (Life Technologies, Carlsbad, CA). Total RNA extraction was done using the RNeasy Plus Universal Kit (QIAGEN, Netherlands). Residual genomic DNA was removed using TURBO DNase (Life Technologies, Carlsbad, CA). A total of 2 µg of total RNA was reverse transcribed with random primers using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Quantitative RT-PCR was conducted using Power SYBR PCR Master Mix (Applied Biosystems, Foster City, CA) on an Applied Biosystems 7900HT real-time PCR machine in 96-well plate format using 25 µL in each reaction. The relative fold-changes of the transcripts were calculated using the comparative CT (2−ΔΔCT) method and normalized to pan-actin values obtained by using the Step-OnePlus Software (Life Technologies, Carlsbad, CA). Three technical replicates of samples were used, and the experiment was repeated three times.
RNA Sequencing and Gene Expression Analysis
RNA Sequencing (RNA-Seq) was done for gene-expression analysis and transcriptomic studies. The total RNA of fer-1(b232ts) and srbc-48(ac23); fer-1(b232ts) animals was isolated from three biological replicates as described above. The sample integrity and purity were checked using Agilent 2100. The RNA was sequenced on HiseqX sequencing platform-using 150bp paired-end nucleotide reads. The RNA sequencing libraries were prepared using a NEBNext® Ultra RNA Library Prep Kit for Illumina® (NEB#E7530L). Library preparation and sequencing were performed at the Novogene Genomic Services & Solutions Company, USA.
The RNA sequence data was analyzed using the Galaxy. The sequenced reads obtained for each sample were mapped to the C. elegans genome (WS255) using the aligner STAR. For sequencing depth, an RNA composition counts were normalized across all samples. Differential gene expression analysis was then performed on normalized samples using DESeq2. Genes exhibiting at least two-fold change and a false-discovery rate (FDR) of 0.05 were considered differentially expressed. Analysis for gene enrichment, gene ontology (GO), or biological processes was performed using the Database for Annotation, Visualization, and Integrated Discovery (DAVID) (https://david.ncifcrf.gov/). The overlap of the upregulated genes in previously studied pathways, including DAF-16, PMK-1, CEP-1/p53 and SKN-1 regulated genes (Derry et al., 2007; Murphy et al., 2003; Oliveira et al., 2009; Troemel et al., 2006), was calculated. Statistical significance of the overlap between two gene sets was calculated using the following on-line program: nemates.org/MA/progs/overlap_stats.html. Representation Factor represents the number of overlapping genes divided by the expected number of overlapping genes drawn from 2 independent groups.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using Graph Pad Prism 8 (Graph Pad Software, La Jolla, CA). Survival curves were considered statistically significant when p values were < 0.05. The Kaplan-Meier method was used to calculate the survival fractions, and statistical significance between survival curves was determined using the log-rank test. Dendrite degeneration was studied in groups of 20 animals and the results are shown as mean ± SEM, treating each experiment as one data point. Significance was calculated by two-sample t test, and assuming a normal distribution. Data normality was confirmed using the D’Agostino-Pearson and Shapiro-Wilk tests. Each experiment was repeated at least three times and each comparison was done with the corresponding control group individually. In figures, all bars represent mean ± SEM and asterisks (*) denote p value as *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001, NS, not significant.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Escherichia coli OP50 | Caenorhabditis Genetics Center | Wormbase ID: OP50 |
| Pseudomonas aeruginosa PA14 | Laboratory of Frederick M. Ausubel | PA14 |
| Ahringer RNAi Libraries in E. coli HT115 (DE3) | Source BioScience | N/A |
| Enterococcus faecalis OGIRF | Laboratory of Danielle A Garsin | OGIRF |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ethyl-methanesulfonate (EMS) | Millipore Sigma | Catalog No. M0880 |
| Isopropyl-β-D-thiogalactoside | Anatrace | Catalog No. I1003 |
| Proteinase K | QIAGEN | Catalog No. 19131 |
| KpnI Restriction enzyme | NEW ENGLAND Biolabs | Catalog No. R0142S |
| SalI-HF Restriction enzyme | NEW ENGLAND Biolabs | Catalog No. R3138S |
| SphI-HF Restriction enzyme | NEW ENGLAND Biolabs | Catalog No. R3182S |
| BamHI Restriction enzyme | NEW ENGLAND Biolabs | Catalog No. R3136S |
| T4 DNA Ligase | NEW ENGLAND Biolabs | Catalog No. M0202S |
| Phusion High-Fidelity PCR Kit | NEW ENGLAND Biolabs | Catalog No. M0531S |
| Dream Taq Green PCR Master Mix (2X) | Thermo Fisher Scientific | Catalog No. K1081 |
| Critical Commercial Assays | ||
| Gentra Puregene kit | QIAGEN | Catalog No. 158667 |
| RNeasy Plus Universal Kit | QIAGEN | Catalog No. 73404 |
| TURBO DNase | Life Technologies | Catalog No. AM1907 |
| High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | Catalog No. 4368814 |
| Power SYBR PCR Master Mix | Applied Biosystems | Catalog No. 4367659 |
| Deposited Data | ||
| Raw and analyzed mRNA-Seq data | This study | GEO: GSE137112 |
| Experimental Models: Organisms/Strains | ||
| N2 C.elegans wild isolate | Caenorhabditis Genetics Center | WormBase ID: N2 |
| unc-86::myr GFP + odr-1::RFP | Caenorhabditis Genetics Center | Strain CX5974 |
| F49H12.4::GFP + unc-119(+) | Caenorhabditis Genetics Center | Strain NC1686 |
| fer-1(b232ts) | Caenorhabditis Genetics Center | Strain HH142 |
| kyIs104 | Caenorhabditis Genetics Center | Strain CX3553 |
| daf-16 (mu86) | Caenorhabditis Genetics Center | Strain CF1038 |
| daf-16p::daf-16a/b::GFP + rol-6(su1006) | Caenorhabditis Genetics Center | Strain TJ356 |
| unc-86::myr GFP + odr-1::RFP;F49H12.4::GFP + unc-119(+) | This study | N/A |
| srbc-48(ac23);F49H12.4::GFP + unc-119(+) | This study | N/A |
| unc-86::myr GFP + odr-1::RFP; daf-16p::daf-16a/b::GFP + rol-6(su1006) | This study | N/A |
| srbc-48(ac23); daf-16p::daf-16a/b::GFP + rol-6(su1006) | This study | N/A |
| odr-1p::srbc-48::GFP;daf-16p::daf-16a/b::GFP + rol-6(su1006) | This study | N/A |
| srbc-48p::srbc-48;daf-16p::daf-16a/b::GFP + rol-6(su1006) | This study | N/A |
| srbc-48(ac23);daf-16 (mu86) | This study | N/A |
| Oligonucleotides | ||
| See Table S6 for the primers used in the study | Integrated DNA Technologies | N/A |
| Recombinant DNA | ||
| srbc-48p::srbc-48 | This study | N/A |
| odr-1p::srbc-48::GFP | This study | N/A |
| odr-1p::RFP | This study | N/A |
| srbc-48p::GFP | This study | N/A |
| odr-1p::RFP; Psrbc-48::GFP | This study | N/A |
| Software and Algorithms | ||
| Prism7 | Graph Pad Software, La Jolla, CA | https://www.graphpad.com/scientificsoftware/prism/ |
| Snapgene | GSL Biotech | https://www.snapgene.com |
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Galaxy | N/A | https://usegalaxy.org |
| Database for Annotation, Visualization, and Integrated Discovery (DAVID) | N/A | https://david.ncifcrf.gov/ |
Highlights.
SRBC-48 is a GPCR that protects C. elegans against P. aeruginosa-induced degeneration
SRBC-48 functions in AWC neurons to protect against dendrite degeneration
Dendrite degeneration hyperactivates DAF-16 in srbc-48 animals and decreases longevity
DAF-16 inhibition rescues the reduced lifespan caused by degeneration in srbc-48 animals
ACKNOWLEDGMENTS
This work was supported by NIH grants AI117911 and GM070977 (to A.A.). C. elegans strains used in the study were provided by the Caenorhabditis Genetics Center (CGC), which is funded by the NIH Office of Research Infrastructure Programs (P40 OD010440).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107662.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alcedo J, and Kenyon C (2004). Regulation of C. elegans longevity by specific gustatory and olfactory neurons. Neuron 41, 45–55. [DOI] [PubMed] [Google Scholar]
- Alexander AG, Marfil V, and Li C (2014). Use of C. elegans as a model to study Alzheimer’s disease and other neurodegenerative diseases. Front. Genet 5, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apfeld J, and Kenyon C (1999). Regulation of lifespan by sensory perception in Caenorhabditis elegans. Nature 402, 804–809. [DOI] [PubMed] [Google Scholar]
- Bortolato A, Doré AS, Hollenstein K, Tehan BG, Mason JS, and Marshall FH (2014). Structure of class B GPCRs: new horizons for drug discovery. Br. J. Pharmacol 171, 3132–3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet A, Sweeney LB, Sturgill JF, Chua KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, et al. (2004). Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 303, 2011–2015. [DOI] [PubMed] [Google Scholar]
- Caldwell KA, Tucci ML, Armagost J, Hodges TW, Chen J, Memon SB, Blalock JE, DeLeon SM, Findlay RH, Ruan Q, et al. (2009). Investigating bacterial sources of toxicity as an environmental contributor to dopaminergic neurodegeneration. PLoS ONE 4, e7227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camara-Lemarroy CR, Metz LM, and Yong VW (2018). Focus on the gut-brain axis: multiple sclerosis, the intestinal barrier and the microbiome. World J. Gastroenterol 24, 4217–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao W, and Zheng H (2018). Correction to: peripheral immune system in aging and Alzheimer’s disease. Mol. Neurodegener 13, 58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Kajino-Sakamoto R, Doss A, and Aballay A (2017). Distinct roles of sensory neurons in mediating pathogen avoidance and neuropeptide-dependent immune regulation. Cell Rep 21, 1442–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Fu Y, Ren M, Xiao B, and Rubin CS (2011). A RasGRP, C. elegans RGEF-1b, couples external stimuli to behavior by activating LET-60 (Ras) in sensory neurons. Neuron 70, 51–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho I, and Blaser MJ (2012). The human microbiome: at the interface of health and disease. Nat. Rev. Genet 13, 260–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook SJ, Jarrell TA, Brittin CA, Wang Y, Bloniarz AE, Yakovlev MA, Nguyen KCQ, Tang LT-H, Bayer EA, Duerr JS, et al. (2019). Whole-animal connectomes of both Caenorhabditis elegans sexes. Nature 571, 63–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derry WB, Bierings R, van Iersel M, Satkunendran T, Reinke V, and Rothman JH (2007). Regulation of developmental rate and germ cell proliferation in Caenorhabditis elegans by the p53 gene network. Cell Death Differ 14, 662–670. [DOI] [PubMed] [Google Scholar]
- Faber PW, Voisine C, King DC, Bates EA, and Hart AC (2002). Glutamine/proline-rich PQE-1 proteins protect Caenorhabditis elegans neurons from huntingtin polyglutamine neurotoxicity. Proc. Natl. Acad. Sci. USA 99, 17131–17136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan-Olive MM, Caponio D, Dan X, et al. (2019). Mitophagy inhibits amyloid-b and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci 22, 401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garsin DA, Villanueva JM, Begun J, Kim DH, Sifri CD, Calderwood SB, Ruvkun G, and Ausubel FM (2003). Long-lived C. elegans daf-2 mutants are resistant to bacterial pathogens. Science 300, 1921. [DOI] [PubMed] [Google Scholar]
- Harrington AJ, Hamamichi S, Caldwell GA, and Caldwell KA (2010). C. elegans as a model organism to investigate molecular pathways involved with Parkinson’s disease. Dev. Dyn 239, 1282–1295. [DOI] [PubMed] [Google Scholar]
- Head BP, Olaitan AO, and Aballay A (2017). Role of GATA transcription factor ELT-2 and p38 MAPK PMK-1 in recovery from acute P. aeruginosa infection in C. elegans. Virulence 8, 261–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heintz C, and Mair W (2014). You are what you host: microbiome modulation of the aging process. Cell 156, 408–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson ST, and Johnson TE (2001). daf-16 integrates developmental and environmental inputs to mediate aging in the nematode Caenorhabditis elegans. Curr. Biol 11, 1975–1980. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, and Latz E (2014). Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol 14, 463–477. [DOI] [PubMed] [Google Scholar]
- Hill JM, Clement C, Pogue AI, Bhattacharjee S, Zhao Y, and Lukiw WJ (2014). Pathogenic microbes, the microbiome, and Alzheimer’s disease (AD). Front Aging Neurosci 16, 127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenstein K, de Graaf C, Bortolato A, Wang MW, Marshall FH, and Stevens RC (2014). Insights into the structure of class B GPCRs. Trends Pharmacol. Sci 35, 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Huang WS, Yang TY, Shen WC, Lin CL, Lin MC, and Kao CH (2014). Association between Helicobacter pylori infection and dementia. J. Clin. Neurosci 21, 1355–1358. [DOI] [PubMed] [Google Scholar]
- Itzhaki RF, Lathe R, Balin BJ, Ball MJ, Bearer EL, Braak H, Bullido MJ, Carter C, Clerici M, Cosby SL, et al. (2016). Microbes and Alzheimer’s disease. J. Alzheimers Dis 51, 979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B-S, Jeon Y-S, and Chun J (2013). Current status and future promise of the human microbiome. Pediatr. Gastroenterol. Hepatol. Nutr 16, 71–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kravtsov V, Oren-Suissa M, and Podbilewicz B (2017). The fusogen AFF-1 can rejuvenate the regenerative potential of adult dendritic trees by self-fusion. Development 144, 2364–2374. [DOI] [PubMed] [Google Scholar]
- Kweon JH, Kim S, and Lee SB (2017). The cellular basis of dendrite pathology in neurodegenerative diseases. BMB Rep 50, 5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon MJ, Han MH, Bagley JA, Hyeon DY, Ko BS, Lee YM, Cha IJ, Kim SY, Kim DY, Kim HM, et al. (2018). Coiled-coil structure-dependent interactions between polyQ proteins and Foxo lead to dendrite pathology and behavioral defects. Proc. Natl. Acad. Sci. USA 115, E10748–E10757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagerström MC, and Schiöth HB (2008). Structural diversity of G proteincoupled receptors and significance for drug discovery. Nat. Rev. Drug Discov 7, 339–357. [DOI] [PubMed] [Google Scholar]
- Lee SS, Kennedy S, Tolonen AC, and Ruvkun G (2003). DAF-16 target genes that control C. elegans life-span and metabolism. Science 300, 644–647. [DOI] [PubMed] [Google Scholar]
- Leinwand SG, Yang CJ, Bazopoulou D, Chronis N, Srinivasan J, and Chalasani SH (2015). Circuit mechanisms encoding odors and driving aging-associated behavioral declines in Caenorhabditis elegans. eLife 4, e10181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin K, Hsin H, Libina N, and Kenyon C (2001). Regulation of the Caenorhabditis elegans longevity protein DAF-16 by insulin/IGF-1 and germline signaling. Nat. Genet 28, 139–145. [DOI] [PubMed] [Google Scholar]
- Mancuso R, Baglio F, Cabinio M, Calabrese E, Hernis A, Nemni R, and Clerici M (2014). Titers of herpes simplex virus type 1 antibodies positively correlate with grey matter volumes in Alzheimer’s disease. J. Alzheimers Dis 38, 741–745. [DOI] [PubMed] [Google Scholar]
- Miklossy J (2011). Emerging roles of pathogens in Alzheimer disease. Expert Rev. Mol. Med 13, e30. [DOI] [PubMed] [Google Scholar]
- Minevich G, Park DS, Blankenberg D, Poole RJ, and Hobert O (2012). CloudMap: a cloud-based pipeline for analysis of mutant genome sequences. Genetics 192, 1249–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy CT, McCarroll SA, Bargmann CI, Fraser A, Kamath RS, Ahringer J, Li H, and Kenyon C (2003). Genes that act downstream of DAF-16 to influence the lifespan of Caenorhabditis elegans. Nature 424, 277–283. [DOI] [PubMed] [Google Scholar]
- Oh SW, Mukhopadhyay A, Svrzikapa N, Jiang F, Davis RJ, and Tissenbaum HA (2005). JNK regulates lifespan in Caenorhabditis elegans by modulating nuclear translocation of forkhead transcription factor/DAF-16. Proc. Natl. Acad. Sci. USA 102, 4494–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta A, Ujisawa T, Sonoda S, and Kuhara A (2014). Light and pheromone-sensing neurons regulates cold habituation through insulin signalling in Caenorhabditis elegans. Nat. Commun 5, 4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira RP, Porter Abate J, Dilks K, Landis J, Ashraf J, Murphy CT, and Blackwell TK (2009). Condition-adapted stress and longevity gene regulation by Caenorhabditis elegans SKN-1/Nrf. Aging Cell 8, 524–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oren-Suissa M, Hall DH, Treinin M, Shemer G, and Podbilewicz B (2010). The fusogen EFF-1 controls sculpting of mechanosensory dendrites. Science 328, 1285–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole S, Singhrao SK, Kesavalu L, Curtis MA, and Crean S (2017). Determining the presence of periodontopathic virulence factors in shortterm postmortem Alzheimer’s disease brain tissue. J. Alzheimers Dis 36, 665–677. [DOI] [PubMed] [Google Scholar]
- Singh V, and Aballay A (2009). Regulation of DAF-16-mediated innate immunity in Caenorhabditis elegans. J. Biol. Chem 284, 35580–35587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh J, and Aballay A (2019). Microbial colonization activates an immune fight-and-flight response via neuroendocrine signaling. Dev. Cell 49, 89–99.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon A, Bandhakavi S, Jabbar S, Shah R, Beitel GJ, and Morimoto RI (2004). Caenorhabditis elegans OSR-1 regulates behavioral and physiological responses to hyperosmotic environments. Genetics 167, 161–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Styer KL, Singh V, Macosko E, Steele SE, Bargmann CI, and Aballay A (2008). Innate immunity in Caenorhabditis elegans is regulated by neurons expressing NPR-1/GPCR. Science 322, 460–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Singh V, Kajino-Sakamoto R, and Aballay A (2011). Neuronal GPCR controls innate immunity by regulating noncanonical unfolded protein response genes. Science 332, 729–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Chen WD, and Wang YD (2017). DAF-16/FOXO transcription factor in aging and longevity. Front. Pharmacol 8, 548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troemel ER, Chou JH, Dwyer ND, Colbert HA, and Bargmann CI (1995). Divergent seven transmembrane receptors are candidate chemosensory receptors in C. elegans. Cell 83, 207–218. [DOI] [PubMed] [Google Scholar]
- Troemel ER, Chu SW, Reinke V, Lee SS, Ausubel FM, and Kim DH (2006). p38 MAPK regulates expression of immune response genes and contributes to longevity in C. elegans. PLoS Genet 2, e183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Cao X, Yan D, Wang D, and Aballay A (2015). Genetic screen reveals link between the maternal effect sterile gene mes-1 and pseudomonas aeruginosa-induced neurodegeneration in Caenorhabditis elegans. J. Biol. Chem 290, 29231–29239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, et al. (2012). Human gut microbiome viewed across age and geography. Nature 486, 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA seq data reported in this paper is Gene Expression Omnibus (GEO: GSE137112).