

Abstract

Hydrogels’ hydrated fibrillar nature makes them the material of choice for the design and engineering of 3D scaffolds for cell culture, tissue engineering, and drug-delivery applications. One particular class of hydrogels which has been the focus of significant research is self-assembling peptide hydrogels. In the present work, we were interested in exploring how fiber–fiber edge interactions affect the self-assembly and gelation properties of amphipathic peptides. For this purpose, we investigated two β-sheet-forming peptides, FEFKFEFK (F8) and KFEFKFEFKK (KF8K), the latter one having the fiber edges covered by lysine residues. Our results showed that the addition of the two lysine residues did not affect the ability of the peptides to form β-sheet-rich fibers, provided that the overall charge carried by the two peptides was kept constant. However, it did significantly reduce edge-driven hydrophobic fiber–fiber associative interactions, resulting in reduced tendency for KF8K fibers to associate/aggregate laterally and form large fiber bundles and consequently network cross-links. This effect resulted in the formation of hydrogels with lower moduli but faster dynamics. As a result, KF8K fibers could be aligned only under high shear and at high concentration while F8 hydrogel fibers were found to align readily at low shear and low concentration. In addition, F8 hydrogels were found to fragment at high concentration because of the high aggregation state stabilizing the fiber bundles, resulting in fiber breakage rather than disentanglement and alignment.
Introduction
Hydrogels have attracted significant interest in the past decade because of their potential for use in a wide range of biomedical applications.1−5 Their highly hydrated fibrillar nature makes them the material of choice for the design and engineering of 3D scaffolds for cell culture, that is, cell niches.6 One particular class of hydrogels which has been the focus of significant research is self-assembling peptide hydrogels.7−9 Peptides are of particular interest as building blocks as they can be synthesized with high purity and high fidelity at reasonable costs, avoiding the batch-to-batch variation and high costs usually linked with the use of natural and synthetic proteins, respectively. A variety of peptide designs can be found in the literature that self-assemble into fibers and above a critical gelation concentration (CGC) form hydrogels.9−14 One of the most popular and successful designs, as far as hydrogel formation is concerned, was devised by Zhang’s group and is based on short peptides (4–20 amino acids long) with alternating hydrophilic and hydrophobic residues (Figure 1A).15,16 This family of amphipathic peptides is known to self-assemble into antiparallel β-sheet-rich fibers and form stable transparent hydrogels which have been shown to be suitable for the 3D culture of a variety of cells.3,7,17−19 They have also been shown to be biocompatible20,21 and suitable for use as bio-inks for 3D bioprinting.22
Figure 1.

Schematic representation of (A) the self-assembly and gelation pathway of β-sheet forming peptides and (B) the fibers formed (side and top views). The peptide illustrated here is FEFKFEFK (F8) (F: phenylalanine, E: glutamic acid, K: lysine). (C) Chemical structures of F8 and KF8K (KFEFKFEFKK) peptides presented in a schematic antiparallel β-sheet conformation (top view).
One particular feature of this peptide design is that when self-assembled into an antiparallel β-sheet, all hydrophobic residue side groups are located on one face of the β-sheet while all the hydrophilic residue side groups are located on the opposite face. As a result, it is thought that two β-sheets come together to bury their hydrophobic faces and form the “elemental” fibers of the network.13,23 The fibers formed are usually twisted and have a rectangular cross-section with a width ranging from 3 to 10 nm, depending on the length of the peptide used and a thickness of ∼1.5 nm. There are three remarkable structural features in these peptide fibers (Figure 1B): fiber core which contains all the hydrophobic residues and controls the fiber cohesion and morphology; fiber surface which contains all the hydrophilic residues and controls the fiber solubility and fiber–fiber associative interactions; and fiber edges where the hydrophobic residues can be exposed to water.
The final properties of the hydrogels will depend on three key factors: the intrinsic properties of the fibers, the network topology formed, and the fiber interaction with the media (saline solution). In our previous work, we have shown how the hydrophobic core and the hydrophilic surfaces of the fibers can be manipulated by the design to control the fiber morphology and the fiber–fiber interactions, respectively, allowing to control the type of network topology formed and ultimately the final bulk mechanical properties of the hydrogels.23−25 We have also shown how the media properties, in particular, pH and ionic strength, can affect the bulk properties of the hydrogels by changing the net charge carried by the peptides or via charge screening effects, respectively.24,26 In the present work, we were interested in exploring how the fiber edges affect the properties of the hydrogels. As illustrated in Figure 1C for the peptide FEFKFEFK (F8), the first hydrophobic residue, F, can be exposed to water at the fiber edges.27 As a result, it is thought that these edges promote fiber–fiber adhesions through hydrophobic contacts, resulting in fiber lateral association leading to fiber bundling and network cross-links. In order to explore this hypothesis, we designed a new sequence in which the edges of the fibers are covered with lysine residues. This was achieved by simply adding two lysine residues, one at each end, to the F8 peptide: KFEFKFEFKK (KF8K). As can be seen from Figure 1C, these two lysine residues will prevent the first hydrophobic residue in the sequence being exposed to water, making the fiber edges hydrophilic when the lysine residues are charged. As a result, KF8K fibers will be “covered” with lysine residues that are known to structure water,28,29 making the fibers more soluble and less prone to fiber–fiber associative interactions, bundling, and cross-link formation.
In order to understand the effect of changing the fiber edge properties on the gelation of these peptides, we first build the pH versus concentration phase diagrams. Subsequently, the structural and mechanical properties of the hydrogels at the selected pH, 3.5 for F8 and 6.0 for KF8K, were investigated using a range of techniques including transmission electron microscopy (TEM), Fourier transform infrared (FTIR) spectroscopy, small-angle X-ray scattering (SAXS), shear rheology, and shear-induced polarized light imaging (SIPLI).
Materials and Methods
Materials
The peptides used in this work were purchased as HCl salts from Biomatik LLC (Wilmington, Delaware, USA) and used as received. The peptide sequence purity was confirmed using reverse-phase high-performance liquid chromatography and mass spectroscopy and was >97% (Figure S1). All other chemicals were purchased from Sigma-Aldrich and used as received.
Titration
Peptide titrations experiments were performed by adding a 50 mM NaOH solution in 5 μL steps to a 1 mL double-deionized water (ddH2O) peptide solution with a 1 mg mL–1 starting concentration. After each NaOH addition, the samples were vigorously agitated using a vortexer to ensure homogenous mixing and the pH was measured 3 times using an Orion 3-Star Benchtop pH Meter (Thermo Scientific, Waltham, Massachusetts, USA).
Hydrogel Preparation
The required amount of peptide powder was dissolved in 400 or 800 μL of ddH2O. To ensure full and homogeneous dissolution, the samples were sonicated at 80 kHz for 60 min and regularly agitated using a vortexer then left overnight in a fridge (4 °C) to equilibrate. Hydrogels were then prepared the following day by adjusting the pH of the peptide solutions to the desired value through step-wise addition of a 1.0 M NaOH solution. After each addition, the samples were vigorously agitated using a vortexer to ensure homogeneous mixing. If bubbles were present after mixing, gentle centrifugation was used to remove them.
pH Versus Peptide Concentration Phase Diagrams
Samples (1 mL) were prepared directly, as described above, in 2 mL plastic cylindrical standard laboratory vials and stored in a fridge (4 °C) for 72 h before their physical state was examined. Samples were classified as liquid if they flowed freely upon inversion of vials, and as they were classified as gel if they did not freely flow.
Fourier Transform Infrared Attenuated Total Reflectance Spectroscopy
Fourier transform infrared attenuated total reflectance spectroscopy (FTIR–ATR) measurements were performed on a Bruker VERTEX 80 FTIR spectrometer equipped with a Diamond ATR accessory. The beam path was purged with dry CO2-scrubbed air. The spectra were an average of 128 scans collected using a 4 cm–1 resolution. A ddH2O spectrum was used as the background and subtracted from each samples spectrum. The spectra were normalized using the amide II peak area. For this purpose, a base line from 1480 to 1720 cm–1 was subtracted from each spectrum and the area of the amide II peak estimated using Origin Pro 9 software.
Small-Angle X-ray Scattering
SAXS experiments were performed on beamline I22 at the diamond light source (DLS) synchrotron (Didcot, UK). The energy of the beam was 12.4 keV, corresponding to an X-ray wavelength of 0.1 nm. Quartz capillaries (1.5 mm outer diameter and 0.01 mm wall thickness) from Capillary Tube Supplies Ltd (Bodmin, UK) were used as the sample holders. The samples were prepared as described above and injected in the capillaries using a syringe. The sample-to-detector distance was fixed to 3.47 m, corresponding to an accessible momentum transfer vector range of 0.05 nm–1 < q = (4π/λ) sin(θ/2) < 3.0 nm–1, where θ is the scattering angle and λ the wavelength of the incident photons. Calibration of the SAXS detector (Pilatus P3-2M, Dectris, Switzerland) was performed using silver behenate powder. An empty capillary was used as the background and subtracted from all spectra, and data were reduced using the Dawn software suite available from DLS. The 2D scattering patterns were integrated using azimuthal integration to generate the 1D scattering patterns. Under these conditions, the samples coherent normalized scattering intensity IN(q) is30−32
| 1 |
where Ip(q) is the normalized intensity scattered using the sample, Is(q) is the normalized intensity scattered using the solvent in our case ddH2O, Cp is the peptide concentration in g cm–3, and Ib is the background scattering originating mainly from the incoherent scattering of the peptides. Ib was estimated using the Porod law which gives the scattered intensity of a two-phase system at high q values23,24,31,32
| 2 |
where Kp is the Porod constant. Ib was estimated by fitting the last 30 data points of the 1D scattering pattern using a Porod plot (q4I(q) vs q4). The flow SAXS experiments were performed on the same beamline using a Watson-Marlow (Wilmington, Massachusetts, USA) Sci Q-323 peristaltic pump. Samples were flowed continuously for 3 min through a 1 mm diameter quartz capillary and 300 ms spectra recorded every 30 s. The flow was then stopped, and spectra were recorded every 300 ms over 90 s. Four flow rates were used: 80, 90, 120, and 150 mL s–1.
Transmission Electron Microscopy
Hydrogels were prepared as described above and diluted 40-folds. A carbon-coated copper grid (400 mesh grid Electron Microscopy Sciences, Hatfield, Pennsylvania, USA) was placed sequentially on a 10 μL sample droplet for 60 s, a 10 μL droplet of ddH2O for 10 s, a 10 μL droplet of 1% uranyl acetate solution for 30 s, and finally on a 10 μL droplet of ddH2O for 10 s. After each step, excess liquid was drained off using a lint-free tissue. The grid was then left to air-dry for a day. TEM images were taken using a FEI Tecnai12 BioTwin transmission electron microscope running at 100 keV. A total of 700 fiber, and fiber bundle widths were measured manually using ImageJ software from multiple TEM images for each peptide system.
Oscillatory Shear Rheology
Rheological measurements were performed using a Discovery Hybrid 2 (DHR-2) rheometer from TA Instruments (New Castle, Delaware, USA) using a 20 mm parallel plate geometry and 500 μm gap. Samples were prepared as described above. Hydrogel (200 μL) was pipetted onto the rheometer’s static bottom plate and the rheometer top plate lowered to the desired gap size. Samples were left to equilibrate for 180 s and then covered with a solvent trap to avoid evaporation before starting the shear strain sweep experiments. The shear-thinning and recovery experiments were performed by applying sequentially low shear strain (0.2%) and high shear strain (100%) in 10 min intervals over 5 cycles. The fitting of the recovery curves were performed using Origin Pro 9 software suite. All measurements were performed at 25 °C and a frequency of 1 Hz and repeated at least three times to ensure reproducibility.
Shear-Induced Polarized Light Imaging
SIPLI measurements were performed at room temperature (21 °C) using a setup based on a reflective polariscope.33 The polarized light-imaging device was attached to an Anton Paar (Graz, Austria) Physica MCR 301 rotational rheometer. The SIPLI setup consisted of a parallel plate geometry with a bottom (static) transparent plate and a top (rotating) reflective plate (polished steel mirror), allowing reflected polarized light images (PLI) of sheared samples to be recorded during shear.34 Samples were prepared as described above and placed on the rheometer using a positive displacement pipette. The top plate was then lowered to set the geometry gap (d = 1 mm) and the sample left to equilibrate for 180 s. The samples were then sheared sequentially for 120 s using three angular speeds ω: 0.08, 0.8, and 8.0 rad s–1. After each shearing experiments, the samples were left to rest for 60 s (no shear applied). In a rotational parallel plate geometry, the shear rate γ̇ across the sample increases linearly from the axis of rotation (0 s–1) to a maximum at the outer edge of the plate (γ̇max), according to the following equation
| 3 |
where r is radial distance from the axis of rotation. At the angular speeds used here, the calculated edge shear rates, γ̇max, are as follows: 1, 10, and 100 s–1. Time-resolver PLI were recorded at a frame rate of 1 and 0.2 s–1 during shearing and quiescent conditions, respectively.
Results and Discussion
As shown in the literature, electrostatic interactions play a key role in the self-assembly and gelation properties of this family of β-sheet forming peptides.24,35,36 The overall charge carried by a peptide can be calculated using the following equation
| 4 |
where Ni/j are the numbers and pKai/j are the pKa values of the basic (i—pKa > 7) and acidic (j—pKa < 7) groups present, respectively. The ionic groups present on the peptides under investigation here are as follows: carboxylic acid (COOH/COO–) at the C-terminus (theoretical pKa 2.18) and on the glutamic acid side chains (theoretical pKa 4.25) and amine (NH3+/NH2) at the N-terminus (theoretical pKa 9.13 and 8.95 on F and K side, respectively) and on the lysine side chains (theoretical pKa 10.53)37 (Figure 1C). In Figure 2A, the theoretical charge carried by the peptides under consideration as a function of pH is presented, and the pKa of the different ionic groups is indicated. These calculations are based on the assumption that the self-assembly of the peptides does not affect pKa of the different ionic groups. In order to investigate this point, the peptides were titrated at low concentration, 1 mg mL–1. The titration curves obtained are presented in Figure 2B. The peptides used for this study were purchased as HCl salts, and as a result, when solubilized in water, the pH of the solutions were acidic: 3.0 ± 0.2 for F8 and 3.3 ± 0.2 for KF8K. At these pH values, it can be assumed, from the theoretical pKa, that the carboxylic acid C-terminus end groups are deprotonated while the carboxylic acid group on the glutamic acid side chains is still protonated. As can be seen from the titration curve, as soon as NaOH is added, a pKa-like transition is observed between pH 3.0 and 4.5 for F8 and pH 3.3 and 5.0 for KF8K. These transitions correspond to the deprotonation of the carboxylic acid groups on the glutamic acid side chains. This interpretation is supported by the requirement of adding 2 mol of NaOH for each mole of peptide present to complete the transition. Indeed, each peptide has two glutamic acid residues in its sequence. The pH at which this transition is observed suggests a shift of roughly 0.5 pH unit toward lower values for the pKa of the carboxylic acid side groups in F8. At this stage of the titration experiment, F8 will be neutral while KF8K will carry a charge of +2. The next pKa-like transition is observed around pH 7.5 and 9.3 for KF8K and F8, respectively. One mole of NaOH is required for each mole of peptide present to complete this transition, suggesting that it corresponds to the deprotonation of the amine N-terminus group. The pH at which this transition is observed suggests a shift of 2 pH units toward lower values in the pKa of the amine N-terminus group for KF8K, while for F8, the transition is observed at the expected pH, in good agreement with the theoretical pKa. The difference in experimental pKa observed between these two peptides is linked to differences in the chemical environment in which the ionic groups sit.38 Indeed, as can be seen from Figure 1C, the amine N-termini are placed in very different environments upon the self-assembly of these two peptides into antiparallel β-sheets. In KF8K, the N-terminus is surrounded by lysine amine side groups and therefore is placed, at this pH, in a strongly positive environment, probably leading to the destabilization of the NH3+ form and its deprotonation at a lower pH than expected. At this stage of the titration experiment, F8 will carry a charge of −1, while KF8K will carry a charge of +1. Finally, the deprotonation of the amine groups on the lysine side chains is observed, as expected, for both peptides at around pH 10.5, in good agreement with their theoretical pKa.
Figure 2.

(A) Theoretical charge carried by each peptide vs pH (dotted lines indicate the theoretical pKa of the different ionic groups present on the peptides); (B) molar ratio of added NaOH to peptide vs pH (shadowed regions indicate the protonation/deprotonation transition regions of the different ionic groups); (C,D) concentration vs pH phase diagrams describing the samples’ physical appearance/state.
In order to investigate the gelation behavior of the two peptides, the concentration versus pH phase diagrams were built and are presented in Figure 2C,D. For F8, which has two cationic and two anionic residues, the phase diagram is symmetric around pH 7, suggesting that the modulus rather than the sign of the charge carried by the peptide is the key parameter. At pH below 4 and above 10, clear hydrogels are obtained. In the pH ranges 4–6 and 8–10, cloudy hydrogels form, and for pH 6–8, bulk precipitation is observed. For KF8K, which has two more cationic than anionic residues, the phase diagram loses its symmetry. Clear hydrogels are obtained up to pH 7.5, and above that pH, cloudy hydrogels form. A number of authors have shown that the transition from clear to cloudy hydrogel in these systems correlates with the charge modulus of the peptide becoming 1 or less.24,26,39 In our case too, the phase diagrams in combination with the titration experiments seem to confirm this observation. Indeed, through the titration experiments, we have shown that for F8, the peptide charge goes from +2 to 0 between pH 3 and 4.5 and from +1 to +2 between pH 10 and 10.5, while for KF8K, the peptide charge goes from +2 to +1 between pH 7 and 8. In both cases, these changes in peptide charge moduli correlate well with the transition from clear to cloudy hydrogel observed in the phase diagrams.
In order to compare in a meaningful way, the properties of the hydrogels, it is essential to have similar charges carried by the peptides. We, therefore, chose pH values which were 0.5 unit lower than the clear to cloudy gel boundary and corresponding to the peptides carrying charges between +2 and +1 for both systems. From this point onward, unless stated otherwise, the hydrogels investigated were prepared at pH 3.5 ± 0.2 and 6.0 ± 0.2 for F8 and KF8F, respectively. At these pH values, both peptides form clear hydrogels with similar CGC: ∼7 mM for F8 and ∼10 mM for KF8K.
The formation of β-sheet rich fibers was confirmed using FTIR–ATR and TEM. As can be seen from Figure 3A, in both peptides, FTIR–ATR spectra has a strong absorption band at 1624 cm–1 and a weaker band at 1694 cm–1, characteristic of the adoption by the peptides of β-sheet conformations, are observed.40,41 Small differences in the amide II region (1500–1580 cm–1) related to the presence in KF8K of two additional lysine residues are also observed.42 Nevertheless, the similar relative overall intensities between the two β-sheet bands and the amide II region in both peptides suggests that they both have a similar propensity to form β-sheets at the selected pH.
Figure 3.

(A) FTIR–ATR normalized spectra obtained for F8 and KF8K hydrogels prepared at a concentration of 26.8 mM (dotted lines indicate the position of the two bands characteristic of adoption by peptides of β-sheet conformations); (B) TEM images obtained for F8 and KF8K diluted hydrogels and bottom characteristic examples of topological features observed across the fibrillar networks; (C) measured fiber and fiber bundle width distributions and lognormal fits obtained (fitting parameters: F8: μ = 8.4 ± 0.1, σ = 3.5 ± 0.1 and KF8K: μ = 5.8 ± 0.1, σ = 1.3 ± 0.1).
TEM images obtained and presented in Figure 3B clearly show the presence of thin fibers in both peptide hydrogels, with the thinnest fibers having diameters ranging from 3 to 5 nm in good agreement with the formation of β-sheet-rich fibers. The images also suggest a more pronounced tendency for F8 to form straight fibers that aggregate laterally into bundles and are more difficult to disassemble compared to KF8K. This was confirmed by the distribution of fiber and fiber bundle widths measured (Figure 3C) that clearly show a significantly broader distribution for F8 (center: 7.7 ± 0.1 nm and fwhm 6.5 ± 0.1 nm) compared to KF8K (center: 4.7 ± 0.1 nm and fwhm 2.8 ± 0.1 nm). It should be kept in mind that sample preparation for TEM requires dilution and agitation to disentangle the fibrous network and allow observation of the isolated fibers. The resulting images, and in particular, the apparent network topologies, are affected by the “easiness” of this process, giving indirect information on the fiber interactions and “adhesion” strength rather than the actual topology of the native fibrillar network. Nevertheless, the TEM images clearly show the three key topological features present in these networks (Figure 3B): branches (one fiber giving rise to two “daughter” fibers with same widths); entanglements (fibers crossing on top of each other); and bundles (multiple fibers coming together to form a larger fiber bundle). Branching in these systems was discussed in details by Pochan and co-workers and is thought to result from the miss-assembly of the peptides. Entanglement will be present as in any fibrillar network. At constant concentration, branches and entanglements will contribute in a similar fashion toward the mechanical properties of both the F8 and KF8K hydrogels. Fiber bundles on the other hand and, in particular, the degree of bundling occurring will, as shown in our previous work, significantly influence the properties of these hydrogels, as we will see below.
SAXS was used to confirm the presence of fibers and their dimensions. In Figure 4A,B, the SAXS patterns obtained for F8 and KF8K at various concentrations are presented as double logarithmic plots. As can be seen, q–1 behavior typical of the scattering of fibers can be observed at low q for all the samples. For F8, above the CGC, a structure factor peak is observed, suggesting the formation of a homogeneous network.23,43 An estimation of the hydrogel mesh size, dm, can be obtained through the Bragg law dm = 2π/q*, q* being the maxima of the scattering peak.30,31,43 Mesh sizes of 23 ± 2 and 18 ± 2 nm were obtained for the 8.9 and 17.8 mM samples, respectively. As expected, the hydrogel mesh size was found to decrease with increasing peptide concentration, in other words, with increasing fiber density. For the KF8K peptide, no scattering peak was observed, suggesting the presence of either a more disordered heterogeneous network or a larger mesh size. It has been previously shown that for infinitely long rodlike objects, in the dilute regime and for qRσ < 1, Rσ being the cross-section radius of gyration of the rod-like object, the normalized scattering intensity, IN, can be written as30−32
| 5 |
where K is the scattering
contrast factor, μL is the fiber linear mass in g
mol–1 nm–1, and Cp the peptide concentration in g cm–3. If the scattering is of the form described by eq 5, then at low q, a linear
behavior should be observed in a ln q IN(q) versus q2 plot.
This is indeed the case for peptide concentrations below the CGCs
(4.5 and 2.2 mM) at which dilute regime conditions can be assumed
(Figure 4C,D). From
the slope of the linear section of the 4.5 mM sample SAXS pattern, Rσ was estimated and 1.1 ± 0.2 and
1.4 ± 0.2 nm were obtained for F8 and KF8K, respectively. If
it is assumed that the fibers can be modeled by infinitely long homogeneous
cylinders, Rσ is related to the
diameter of the fiber, df, through the
following equation: 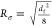 ,
leading to estimated diameters of 3.1
± 0.2 and 4.0 ± 0.2 nm for F8 and KF8K, respectively, in
good agreement with the fiber diameters estimated from TEM. The results
also agree well with the molecular dimensions of the peptides, indeed,
the fully extended conformation width of F8 and KF8K were calculated
to be 2.8 and 3.5 nm, respectively.
,
leading to estimated diameters of 3.1
± 0.2 and 4.0 ± 0.2 nm for F8 and KF8K, respectively, in
good agreement with the fiber diameters estimated from TEM. The results
also agree well with the molecular dimensions of the peptides, indeed,
the fully extended conformation width of F8 and KF8K were calculated
to be 2.8 and 3.5 nm, respectively.
Figure 4.

(A,B) Double logarithmic plots of SAXS patterns obtained for F8 and KF8K samples prepared at different concentrations in In(q) vs q representation. (C,D) SAXS patterns of F8 and KF8K samples plotted for low q in a ln q I(q) vs q2 representation (red lines show the linear fits used to calculate the fiber cross-section radii of gyration, Rσ).
According to eq 5 from the intercept of the linear fit with the y-axis, the quantity KπμLCp can be estimated. Cp is actually the concentration of the peptide participating in the fiber formation as any peptide molecularly dissolved in the water phase will not contribute to the overall coherent scattering. A number of authors have shown that peptides with this particular design are the strong β-sheet former and that the amount of the peptide dissolved in the water phase is relatively small.44,45 As a result, Cp can be assumed to be the same as the nominal sample concentration: 5.0 and 6.2 mg cm–3 for the 4.5 mM F8 and KF8K samples, respectively. As the two peptides have similar chemical structures and form similar fibers, it is reasonable to assume that they both have similar contrast factors and fiber linear masses. As a result, if both peptides have the same propensity to form β-sheet fibers, as suggested by the FTIR results (Figure 3A), the ratio between the intercept values should be equal to the concentration ratio. This is indeed the case as the intercept ratio (F8/KF8K) was found to be 0.80 ± 0.03, while the concentration ratio was calculated to be 0.79.
Finally, it can also be noted that above the CGC for F8, a pronounced “roll-over” usually associated with fiber–fiber associative interactions (i.e., aggregation)23,30,45 is observed, while for KF8K, a significantly smaller “roll-over” is present (Figure 4C,D). These observations are in good agreement with the TEM results (Figure 3B,C), suggesting that the F8 fibers tend to interact and associate/aggregate laterally and form larger fiber bundles compared to the KF8K fibers.
The results discussed above clearly show that the addition of the two lysine residues does not affect the ability of the peptides to form β-sheet fibers but does affect the ability of these fibers to aggregate/associate and form larger fiber bundles. As one would expect, this will have a significant effect on the topology of the resulting networks and, therefore, the rheological behavior of the hydrogels. To explore this point, we investigated next the mechanical properties of F8 and KF8F hydrogels as well as their rheological behavior under shear.
The hydrogel storage shear moduli (G′) was found to be typically an order of magnitude larger than the storage shear moduli (G″), confirming the solid-like nature of these materials at low strain (Figures 5A and S2). One of the key challenges with this family of hydrogels is to understand the correlations between the network structure and bulk mechanical properties. Mechanical properties of self-assembled peptide hydrogels are quite challenging to measure consistently as they depend on several factors including the formulation and measurement methodologies. As a result, significant variability in mechanical property measurements can be found across the literature including from the same research groups as hydrogel preparation and measurement methodologies evolve with time.46,47
Figure 5.

(A) Storage (G′—closed symbols) and loss (G″—open symbols) shear moduli vs strain (ε) curves obtained at 1 Hz frequency for F8 and KF8K hydrogels prepared at 26.8 mM concentration. (B) Storage modulus (G′) vs peptide concentration (C) plots obtained for F8 and KF8K hydrogels measured at 0.2% strain and 1 Hz frequency. (C,D) Shear thinning and recovery behavior of F8 and KF8K hydrogels prepared at 26.8 mM peptide concentration. Experiments were performed at 1 Hz and shear strain of 0.2 and 100% were applied alternatingly in 10 min intervals.
A number of models can be found in the literature that correlates hydrogel mechanical properties to network topologies. Usually, these models link storage modulus to concentration through power laws: G′ ∝ Cα.32 Similar power laws with α = 1.5 ± 0.1 and 1.3 ± 0.2 were obtained for F8 and KF8K, respectively (Figure 5B). The suitability of a theory to describe a specific hydrogel system is related to the assumption made in terms of type of network present. This family of peptides tends to form relatively rigid β-sheet rich fibers/fiber bundles that are connected at fixed junction points. In this context, as discussed by Guenet,48,49 one of the most interesting theories was developed by Jones and Marques.50 These authors described the mechanical properties of these types of networks through the concepts of enthalpic elasticity, which relates to the elastic deformation of the rigid fibers/fiber bundles, and entropic elasticity, which relates to conformational rearrangements occurring either within the fibers or at the junctions (hinged junctions) of the network. Depending on the dominant elasticity, these authors derived two power law expressions
| 6 |
| 7 |
where Df is the
fractal dimension of the fiber long axis and is defined as: 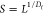 , where S is the
end-to-end
distance of the fiber and L its contour length. For
rigid straight fibers, Df = 1, leading
to power laws of 2 and 1.5 for enthalpic and entropic elasticity-dominated
networks, respectively. As mentioned above, in our case, power laws
close to 1.5 were obtained, suggesting that most of the elasticity
is related to the conformational rearrangement occurring within the
fibers or at the junctions of the network. Considering the rigid nature
of the β-sheet fibers/fiber bundles formed in these systems,
conformational rearrangement within the fibers is unlikely. These
types of conformational rearrangement are expected for polymer chains
for which this theory was originally developed. Therefore, our rheological
results would suggest that the network elasticity originates from
the network junction and their deformation. As discussed above for
F8 hydrogels, the mesh size was found to be in the 20 nm range, pointing
toward relatively short distances between junctions. As a result,
when deformed, it is not unrealistic to assume that the network struts
will not bend and that most of the deformation and elasticity will
originate at the network junctions.
, where S is the
end-to-end
distance of the fiber and L its contour length. For
rigid straight fibers, Df = 1, leading
to power laws of 2 and 1.5 for enthalpic and entropic elasticity-dominated
networks, respectively. As mentioned above, in our case, power laws
close to 1.5 were obtained, suggesting that most of the elasticity
is related to the conformational rearrangement occurring within the
fibers or at the junctions of the network. Considering the rigid nature
of the β-sheet fibers/fiber bundles formed in these systems,
conformational rearrangement within the fibers is unlikely. These
types of conformational rearrangement are expected for polymer chains
for which this theory was originally developed. Therefore, our rheological
results would suggest that the network elasticity originates from
the network junction and their deformation. As discussed above for
F8 hydrogels, the mesh size was found to be in the 20 nm range, pointing
toward relatively short distances between junctions. As a result,
when deformed, it is not unrealistic to assume that the network struts
will not bend and that most of the deformation and elasticity will
originate at the network junctions.
One additional observation made by Guenet using the Jones and Marques theory is that thicker fibers will lead to higher moduli.48 In our case, F8 hydrogels were found to have 1 order of magnitude higher moduli than KF8K hydrogels, in agreement with TEM and SAXS results that showed F8 fibers having a more pronounced tendency to aggregate/associate laterally and form thicker fiber bundles.
Another key property of these hydrogels is their injectability or, in other words, their ability to instantaneously shear thin and recover upon application and removal of high shear. In order to examine this aspect, we disrupted the 26.8 mM hydrogels and allowed them to recover in situ, in the rheometer, by applying low shear strain (0.2%) in alternation with high shear strain (100%) over 10 min intervals at a fixed frequency of 1 Hz (Figure 5C,D). As can be seen for both peptide hydrogels, when the high shear strain is applied (10–20; 30–40; 50–60; 70–80; and 90–100 min time intervals), G′ decreases immediately and becomes smaller than G″, clearly showing that under high shear, the hydrogels become liquid-like, that is, shear-thin. Once the high shear strain is removed (0–10, 20–30, 40–50, 60–70, and 80–90 min time intervals), G′ recovers immediately and becomes once again larger than G″, clearly showing that the hydrogels re-acquire their solid-like properties upon removal of high shear. It is evident from the results presented in Figure 5C,D that following the initial instantaneous recovery, G′ continues to increase over the 10 min recovery cycles, pointing toward further structural evolution of the hydrogels taking place over time. As discussed by a number of authors, peptide hydrogels, like most self-assembled materials, tend to be kinetically trapped and not reach their thermodynamic equilibrium.51,52 As a result, long-term aging of this type of materials is not uncommon and is usually dependent on the molecular/structural mobility of the system. In order to extract characteristic dynamics times for our hydrogels, we decided to fit the recovery curves using the classical mechanical model (one spring element in parallel with two spring + dash pot elements)53
| 8 |
were G0′ is the instantaneous shear modulus, that is, the shear modulus measured immediately after removing the high shear strain, G1′ and G2′ are the storage shear moduli related to the two dynamic recovery processes used in our model, and τ1 and τ2 their characteristic times. It should be stated here that using a single dynamic process did not allow to fit adequately the shear moduli recovery curves.
In Figure 6A,C, fits obtained for the 60–70 min recovery cycles for both 26.8 mM hydrogels using the above equation are presented. As can be seen, very good fits were obtained, and this was indeed the case for all the recovery cycles with R-square values typically higher than 0.98. In the tables of Figure 6B,D, the fitting parameters derived for each recovery cycle for both peptide hydrogels are listed alongside the estimated “equilibrium” (t = +∞) storage moduli: G∞′ = G0′ + G1′ + G2′. From the fitting parameters obtained, it can be noted that the first recovery cycle (0–10 min) differs significantly from the subsequent four cycles. This is not a surprise as the mechanical recovery curves will strongly depend on the disruption methodology used. The first cycle is linked to the recovery of the material following its deposition on the rheometer plate. In addition, there is a lag time between the setting up of the sample and the start of the experiment, resulting in the early stages of the recovery process not being observed. As a result, the dynamic parameters extracted from this cycle are not reliable. The fitting parameters of the second cycles (20–30 min) are more in line with the following three cycles, although still some differences can be observed for both samples, suggesting that two disruption cycles are required in order to reach a pseudo “steady state”. Nevertheless, the first and second recovery cycle curves were still fitted adequately using our model allowing estimating G∞′. The 3rd (40–50 min), 4th (60–70 min), and 5th (80–90 min) recovery cycle fitting parameters are consistent with each other and were the ones considered for further analysis and discussion. For both peptides, the results obtained suggest the presence of two recovery processes with two different characteristic times: a fast recovery process, similar for both peptides, with a characteristic τ1 time of ∼0.35 min and an associate shear modulus G0′ of ∼0.27 kPa and a slower recovery process which is peptide-dependent. For KF8K hydrogel, the characteristic τ2 time was found to be ∼4.0 min with an associated G2′ of ∼0.24 kPa, while for F8 hydrogel, this second recovery process was found to have a significant larger characteristic τ2 time of ∼7.8 min and a higher associated shear modulus G2′ of ∼0.72 kPa. As discussed by Pochan and coworkers, this type of hydrogels tends to break up under shear into globular hydrogel particulates that “roll-over” each other.54 Upon removal of high shear, a fast percolation of the globular hydrogel domains is observed, akin to the percolation event observed in colloidal gels.54,55 In our case, this process occurs within the first 90 s of removing the high shear. This initial fast recovery process is then followed by a slower process, during which further structural rearrangements occur over time, leading to a slow increase in G′. This second process characteristic time is linked to the molecular and structural mobility of the peptides and fibers present in the network. As can be seen from our results, this second process is faster for KF8K, suggesting a higher mobility in this system in agreement with our starting hypothesis, and the results above suggesting that KF8K fibers have a lower tendency to associate/aggregate, resulting in higher mobility in comparison with F8 fibers. An interesting observation is that for KF8K, the estimated “equilibrium” (t = +∞) storage modulus increases after each disruption cycle, while for F8, it was found to be constant. The increase in G∞′ is due to the increase in G0′, while the overall recovery curve does not change, and the fitting parameters G1′, G2′, τ1, and τ2 remain the same, indicating that the overall dynamics of the recovery processes remains unchanged. The origin of this effect is unclear, but it is thought to be linked to the fast percolation step, following the removal of the high shear strain and therefore to the hydrogel disruption methodology used.
Figure 6.
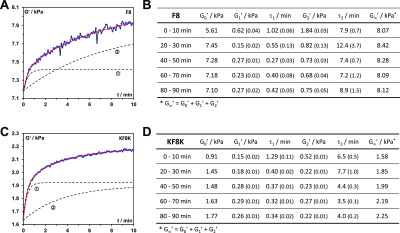
(A,C) Shear storage modulus (G′) vs time (t) curves obtained for the 60–70 min recovery cycle (4th recovery cycle, Figure 5C,D); black dotted lines: the two dynamic recovery processes curves derived from eq 8: ① “fast” process, ② “slow” process (curves have been shifted upwards by G0 for ease of visualization); red line: curve’s fit obtained using eq 8. (B,D) fitting parameters obtained by fitting each recovery cycle curves using eq 8 for F8 and KF8K hydrogels prepared at 26.8 mM peptide concentration. The standard fitting errors are shown in brackets. All fits had R-square values of 0.98 or higher.
A key question when shearing these hydrogels is whether any level of the alignment is observed.54,56 For this purpose, we performed two set of experiments. First, we investigated the alignment of these systems under linear flow using SAXS. Because of the limitation on the viscosity of the materials imposed using the peristatic pump used only samples below or around the CGC could be investigated in this case. The samples were made to flow through a 1 mm diameter capillary at varying flow rates from 80 to 150 mL s–1, while the 2D SAXS patterns were recorded. As can be seen from Figure 7A, the alignment was observed for F8 samples only and the degree of alignment (lengths ratio of meridian axis, y, over the equatorial axis, x, of the 2D SAXS patterns) was similar for all concentrations and flow rates probed. Once the flow was stopped the F8 sample alignment was lost relatively rapidly, within 60 s (Figure 7B). These results suggest once again a high tendency of associative interaction for the F8 fibers. Indeed, fiber–fiber contacts promote the transfer of the shear forces applied to the fibers facilitating their alignment. On the other hand, for KF8K, no alignment was observed pointing once again toward weak associative fiber–fiber interactions and higher mobility for these fibers (Figure 7A).
Figure 7.

(A) Degree of peptide fiber alignment taken as the ratio of the meridian (y) over the equatorial axis (x) lengths of the 2D SAXS pattern observed vs sample concentration. Flow rates: 80 (△), 90 (◊), 120 (○), 150 mL s–1 (□). Inset shows the 2D scattering patterns obtained for the F8 and KF8K sample prepared at 8.9 mM; (B) 2D scattering patterns obtained for the F8 8.9 mM sample after stopping the flow: time elapsed from stopping the flow is shown in the top left corner and the degree of fiber alignment is shown in the bottom left corner; (C) PLIs of the sample taken after loading the samples in the SIPLI rheometer (⊥ indicates that the polarizer and analyzer are oriented at 90° and // indicates that the polarizer and analyzer are oriented at 0°).
In order to promote alignment of the KF8K peptide fibers, two approaches can be used. The first option is to increase the concentration of the sample in order to increase the level of entanglement and fiber–fiber interactions through spatial confinement.57 The second option is to increase the shear forces applied using higher shear rates. For this purpose, we performed SIPLI experiments which enable sample birefringent properties to be monitored under a broad range of shear conditions in situ using the parallel-plate rotational geometry and PLI.33,34 The F8 hydrogels showed strong birefringence straight after placing the samples between the shearing plates at quiescent conditions (Figure 7C). As the hydrogel concentration was increased, granular patterns were observed, suggesting the presence at high concentration of micron-sized liquid-crystalline domains throughout the sample. These domains are thought to be linked to the formation in this system of associated/aggregated fiber bundles that behave like rigid rods and show liquid crystalline properties. For KF8K, no birefringence was observed for the 8.9 mM sample which is below the CGC. Small increasing amounts of birefringence were observed when the sample concentration was increased above the CGC. These small birefringent domains are thought to have the same origin as for the F8 hydrogels, suggesting that for the KF8K, increasing the peptide concentration also promotes the formation of fiber bundles but to a significantly lower level than F8.
We then applied a sequence of shear pulses to the studied samples using a range of angular speeds (0.08, 0.8 and 8 rad s–1) for a duration of 120 s each followed by 60 s rest, and observed whether fiber alignment could be detected (Figure 8A—Videos S1–S4 available in the Supporting Information). For the F8 samples below the CGC, 4.7 mM, a clear regular Maltese cross pattern could be seen at 0.08 rad s–1, indicating strong alignment of the fibers within the rotational flow with their optical axis either parallel or perpendicular to the polarizer or analyzer polarization plane. When the concentration was increased above the CGC, the cross pattern became less obvious and was lost for the 26.8 mM sample for which a granular pattern was observed. At low concentrations, the fibers are more diluted and less entangled/associated, allowing them to align within the flow, while at higher concentrations, the high density of the fiber and fiber bundles result in the fragmentation of the hydrogel, as suggested by Pochan and coworkers,54 leading to the formation of micron-sized liquid-crystalline domains “tumbling” over each other under shear, leading to the granular pattern observed (Figure 8B). Maltese cross patterns were once again observed for the 8.9 and 17.6 mM F8 samples at the higher shear rate of 0.8 rad s–1. In this case, the patterns were quite irregular, suggesting that alignment was occurring but in a complex turbulent flow. Clear regular Maltese cross patterns, similar to the one observed for the 4.9 mM sample, were observed at the highest shear rate of 8.0 rad s–1. These results suggest that increasing the shear rate allows the fibers in F8 hydrogels to disentangle and align within the flow. Disentanglements in these self-assembled systems can occur through two processes: the fiber can simply slide past each other or de-assemble and re-assemble under flow. The latter process is common in self-assembled fibrillar systems in which entanglements have been shown to have a limited life time linked to the opening/closing rate of the self-assembled fibers.58,59 In our case, it is thought that when higher shear rates are used, the increased shear forces applied promoted these two processes resulting in the fiber alignment within the rotational flow (Figure 8B). At the highest concentration of F8 peptides, 26.8 mM, fiber alignment was not observed at the probed shear rates. This observation indicates that the high fibrillar density does not allow the fiber to align along the flow direction. For the 0.8 rad s–1 angular speed, a faint “shadow” of the Maltese cross pattern could be recognized, which was probably due to some domains orienting within the flow. Finally, at the highest angular speed for the 17.8 and 26.8 mM samples, banding-like features were observed, suggesting that at this high angular speed, a more complex flow behavior occurred. The elucidation of such a behavior, although of interest, is beyond the scope of this work.
Figure 8.

(A) PLIs of the F8 and KF8K peptides at different concentrations after 120 s shearing (⊥ indicates that the polarizer and analyzer are oriented at 90°). The red dotted line indicates the CGC of the peptides. (B) Schematic representation of the different morphological transformations taking place when shearing peptides are assembled in fibers.
For KF8K samples, no alignment was observed at ω of 0.08 and 0.8 rad s–1. For the 8.9 mM KF8K sample, sheared at ω of 8.0 rad s–1, no alignment was observed either. Again these results point toward a low level of KF8K fiber–fiber associative interactions, allowing the fibers to “slide” past each other and/or open and close rapidly and retaining random conformation. On the other hand, some alignment was observed for the 17.6 and 26.8 mM samples at the highest angular speed used, suggesting that at these two higher concentrations, the shear forces applied become strong enough to force the KF8K fibers to align under shear flow. Once again, the results obtained point to a clear difference between these two studied systems dictated by fiber–fiber associative interactions, leading to very different mechanical behaviors.
Conclusions
We have investigated the role that β-sheet edge interactions play in the self-assembly and gelation properties of β-sheet forming peptides. For this purpose, we designed a new peptide sequence by adding two lysine residues at each end of the FEFKFEFK (F8) peptide to prevent the first hydrophobic amino acid residue, F, to be exposed to water and promote fiber–fiber associative interactions and network cross-links through hydrophobic edge interactions. The addition the two lysine residues did not affect the ability of the new peptide, KFEFKFEFKK (KF8K), to form β-sheet-rich fibers and hydrogels provided that the overall charges carried by the peptide were kept the same, +2 in our case. The addition of the two lysine residues did result in a decrease in fiber–fiber edge associative interactions and result in a lower propensity of KF8K fibers to associate/aggregate laterally and form larger fiber bundles.
The differences in network topologies resulting from preventing fiber–fiber association/aggregation were shown to have a significant effect on the rheological behavior of the two peptides. KF8K hydrogels were found to have significantly lower shear moduli and to align only at high shear rates and concentrations. It is thought that the introduction of the two lysine residues, and therefore the covering of the fiber edges with lysine residues that structure water, results in a continuous water shell forming around the fibers, increasing their solubility and mobility. This high fiber mobility is thought to allow fast dynamics, resulting in the needs for high concentration and high shear rates to force alignment in KF8K.
On the other hand, F8 fibers were found to align under simple flow even at low shear rate and low concentration. The higher propensity for fiber to interact and associate through hydrophobic contacts in this system is thought to promote the transfer of the shear forces to the fibers and as a result to promote their alignment. When the concentration was increased, F8 hydrogels were found to fragment. At high concentrations, the strong fiber–fiber associative interactions are thought to stabilize the fibers and fiber bundles, resulting in fiber breaking rather than disentangling and aligning, whether through fiber slipping past each other or molecular de-assembling and re-assembling. The prevalence of these two processes over each other was shown to be linked to the concentration of the sample as well as to the shear forces applied.
Our work clearly shows how controlling edge interactions, in addition to the other structural features of these peptides (core and surface), is key to controlling the final mechanical properties of the hydrogels formed and thus their use as shear-thinning injectable tissue engineering and drug-delivery platforms.
Acknowledgments
The authors would like to thank the EPSRC (Fellowship grant no: EP/K016210/1 and Northwest Nanoscience Doctoral Training Centre (NOWNANO DTC) grant no: EP/G03737X/1 and the EPSRC and MRC Centre for Doctoral Training (CDT) in Regenerative Medicine grant no: EP/L014904/1) for providing financial support to this project. The authors thank the staff in the EM facility in the Faculty of Life Sciences of the University of Manchester, in particular, Aleksandr Mironov, for their assistance, and the Welcome Trust for equipment grant support to the EM facility. The authors also thank Owen Marshall for his kind assistance with the FTIR equipment. The authors are also grateful to DLS (Didcot, UK) for awarding beam time (SM12950 and SM15246) to this project and to the staff, in particular, Olga Shebanova on beamline I22 for their support with the SAXS experiments.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biomac.0c00229.
Author Present Address
⊥ (J.K.W.) School of Chemistry, University College Dublin, Belfield, Dublin 4, Ireland.
Author Present Address
# (A.F.M.) Manchester BIOGEL, 19F4 Mereside, Alderley Park, Alderley Edge, Cheshire SK10 4TG, UK.
The authors declare no competing financial interest.
Notes
All research data supporting this work are directly available within this publication and the linked Supporting Information.
Supplementary Material
References
- Sun J. E. P.; Stewart B.; Litan A.; Lee S. J.; Schneider J. P.; Langhans S. A.; Pochan D. J. Sustained release of active chemotherapeutics from injectable- solid - hairpin peptide hydrogel. Biomater. Sci. 2016, 4, 839–848. 10.1039/c5bm00538h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada Y.; Chowdhury A.; Schneider J. P.; Stetler-Stevenson W. G. Macromolecule- network electrostatics controlling delivery of the biotherapeutic cell modulator TIMP- 2. Biomacromolecules 2018, 19, 1285. 10.1021/acs.biomac.8b00107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mujeeb A.; Miller A. F.; Saiani A.; Gough J. E. Self-assembled octapeptide scaffolds for in vitro chondrocyte culture. Acta Biomater. 2013, 9, 4609–4617. 10.1016/j.actbio.2012.08.044. [DOI] [PubMed] [Google Scholar]
- Qi Y.; Min H.; Mujeeb A.; Zhang Y.; Han X.; Zhao X.; Anderson G. J.; Zhao Y.; Nie G. Injectable hexapeptide hydrogel for localized chemotherapy prevents breast cancer recurrence. ACS Appl. Mater. Interfaces 2018, 10, 6972–6981. 10.1021/acsami.7b19258. [DOI] [PubMed] [Google Scholar]
- Tang J. D.; Mura C.; Lampe K. J. Stimuli-responsive, pentapeptide, nanofiber hydrogel for tissue engineering. J. Am. Chem. Soc. 2019, 141, 4886–4899. 10.1021/jacs.8b13363. [DOI] [PubMed] [Google Scholar]
- Madl C. M.; Heilshorn S. C. Engineering hydrogel microenvironments to recapitulate the stem cell niche. Annu. Rev. Biomed. Eng. 2018, 20, 21–47. 10.1146/annurev-bioeng-062117-120954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haines-Butterick L.; Rajagopal K.; Branco M.; Salick D.; Rughani R.; Pilarz M.; Lamm M. S.; Pochan D. J.; Schneider J. P. Controlling hydrogelation kinetics by peptide design for three-dimensional encapsulation and injectable delivery of cells. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 7791–7796. 10.1073/pnas.0701980104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D.; Workman V. L.; O’Brien M.; McLaren J.; White L.; Ragunath K.; Rose F.; Saiani A.; Gough J. E. Peptide hydrogels-A tissue engineering strategy for the prevention of oesophageal strictures. Adv. Funct. Mater. 2017, 27, 1702424. 10.1002/adfm.201702424. [DOI] [Google Scholar]
- Saiani A.; Mohammed A.; Frielinghaus H.; Collins R.; Hodson N.; Kielty C. M.; Sherratt M. J.; Miller A. F. Self-assembly and gelation properties of alpha-helix versus beta-sheet forming peptides. Soft Matter 2009, 5, 193–202. 10.1039/b811288f. [DOI] [Google Scholar]
- Schneider J. P.; Pochan D. J.; Ozbas B.; Rajagopal K.; Pakstis L.; Kretsinger J. Responsive hydrogels from the intramolecular folding and self-assembly of a designed peptide. J. Am. Chem. Soc. 2002, 124, 15030–15037. 10.1021/ja027993g. [DOI] [PubMed] [Google Scholar]
- Mart R. J.; Osborne R. D.; Stevens M. M.; Ulijn R. V. Peptide-based stimuli-responsive biomaterials. Soft Matter 2006, 2, 822–835. 10.1039/b607706d. [DOI] [PubMed] [Google Scholar]
- Pappas C. G.; Shafi R.; Sasselli I. R.; Siccardi H.; Wang T.; Narang V.; Abzalimov R.; Wijerathne N.; Ulijn R. V. Dynamic peptide libraries for the discovery of supramolecular nanomaterials. Nat. Nanotechnol. 2016, 11, 960–967. 10.1038/nnano.2016.169. [DOI] [PubMed] [Google Scholar]
- Li J.; Du X.; Hashim S.; Shy A.; Xu B. Aromatic-aromatic interactions enable alpha-helix to beta-sheet transition of peptides to form supramolecular hydrogels. J. Am. Chem. Soc. 2017, 139, 71–74. 10.1021/jacs.6b11512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decandio C. C.; Silva E. R.; Hamley I. W.; Castelletto V.; Liberato M. S.; Oliveira V. X.; Oliveira C. L. P.; Alves W. A. Self-assembly of a designed alternating arginine/phenylalanine oligopeptide. Langmuir 2015, 31, 4513–4523. 10.1021/acs.langmuir.5b00253. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Holmes T.; Lockshin C.; Rich A. Spontaneous assembly of a self-complementary oligopeptide to form a stable macroscopic membrane. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 3334–3338. 10.1073/pnas.90.8.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Altman M. Peptide self-assembly in functional polymer science and engineering. React. Funct. Polym. 1999, 41, 91–102. 10.1016/s1381-5148(99)00031-0. [DOI] [Google Scholar]
- Castillo Diaz L. A.; Elsawy M.; Saiani A.; Gough J. E.; Miller A. F. Osteogenic differentiation of human mesenchymal stem cells promotes mineralization within a biodegradable peptide hydrogel. J. Tissue Eng. 2016, 7, 2041731416649789. 10.1177/2041731416649789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligorio C.; Zhou M.; Wychowaniec J. K.; Zhu X.; Bartlam C.; Miller A. F.; Vijayaraghavan A.; Hoyland J. A.; Saiani A. Graphene oxide containing self-assembling peptide hybrid hydrogels as a potential 3D injectable cell delivery platform for intervertebral disc repair applications. Acta Biomater. 2019, 92, 92–103. 10.1016/j.actbio.2019.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faroni A.; Workman V. L.; Saiani A.; Reid A. J. Self-assembling peptide hydrogel matrices improve the neurotrophic potential of human adipose-derived stem cells. Adv. Healthcare Mater. 2019, 8, 1900410. 10.1002/adhm.201900410. [DOI] [PubMed] [Google Scholar]
- Morris O.; Elsawy M. A.; Fairclough M.; Williams K. J.; McMahon A.; Grigg J.; Forster D.; Miller A. F.; Saiani A.; Prenant C. In vivo characterisation of a therapeutically relevant self-assembling F-18-labelled -sheet forming peptide and its hydrogel using positron emission tomography. J. Labelled Compd. Radiopharm. 2017, 60, 481–488. 10.1002/jlcr.3534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markey A.; Workman V. L.; Bruce I. A.; Woolford T. J.; Derby B.; Miller A. F.; Cartmell S. H.; Saiani A. Peptide hydrogel in vitro non-inflammatory potential. J. Pept. Sci. 2017, 23, 148–154. 10.1002/psc.2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raphael B.; Khalil T.; Workman V. L.; Smith A.; Brown C. P.; Streuli C.; Saiani A.; Domingos M. 3D cell bioprinting of self-assembling peptide-based hydrogels. Mater. Lett. 2017, 190, 103–106. 10.1016/j.matlet.2016.12.127. [DOI] [Google Scholar]
- Gao J.; Tang C.; Elsawy M. A.; Smith A. M.; Miller A. F.; Saiani A. Controlling self-assembling peptide hydrogel properties through network topology. Biomacromolecules 2017, 18, 826–834. 10.1021/acs.biomac.6b01693. [DOI] [PubMed] [Google Scholar]
- Roberts D.; Rochas C.; Saiani A.; Miller A. F. Effect of peptide and guest charge on the structural, mechanical and release properties of beta-sheet forming peptides. Langmuir 2012, 28, 16196–16206. 10.1021/la303328p. [DOI] [PubMed] [Google Scholar]
- Elsawy M. A.; Smith A. M.; Hodson N.; Squires A.; Miller A. F.; Saiani A. Modification of β-sheet forming peptide hydrophobic face: effect on self-assembly and gelation. Langmuir 2016, 32, 4917–4923. 10.1021/acs.langmuir.5b03841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boothroyd S.; Miller A. F.; Saiani A. From fibres to networks using self-assembling peptides. Faraday Discuss. 2013, 166, 195–207. 10.1039/c3fd00097d. [DOI] [PubMed] [Google Scholar]
- Bowerman C. J.; Liyanage W.; Federation A. J.; Nilsson B. L. Tuning beta-sheet peptide self-assembly and hydrogelation behavior by modification of sequence hydrophobicity and aromaticity. Biomacromolecules 2011, 12, 2735–2745. 10.1021/bm200510k. [DOI] [PubMed] [Google Scholar]
- Williams P. A.; Hughes C. E.; Martin J.; Courvoisier E.; Buanz A. B. M.; Gaisford S.; Harris K. D. M. Understanding the solid-state hydration behavior of a common amino acid: identification, structural characterization, and hydration/dehydration processes of new hydrate phases of l-lysine. J. Phys. Chem. C 2016, 120, 9385–9392. 10.1021/acs.jpcc.5b12420. [DOI] [Google Scholar]
- Deshmukh S. A.; Solomon L. A.; Kamath G.; Fry H. C.; Sankaranarayanan S. K. R. S. Water ordering controls the dynamic equilibrium of micelle–fibre formation in self-assembly of peptide amphiphiles. Nat. Commun. 2016, 7, 12367. 10.1038/ncomms12367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilbaud J.-B.; Saiani A. Using small angle scattering (SAS) to structurally characterise peptide and protein self-assembled materials. Chem. Soc. Rev. 2011, 40, 1200–1210. 10.1039/c0cs00105h. [DOI] [PubMed] [Google Scholar]
- Higgins J. S.; Benoit H. C.. Polymer and Neutron Scattering; Clarendon Press: Oxford, 1994. [Google Scholar]
- Guenet J.-M.Thermoreversible Gelation of Polymers and Biopolymers; Academic Press: London, 1992. [Google Scholar]
- Mykhaylyk O. O.; Warren N. J.; Parnell A. J.; Pfeifer G.; Laeuger J. Applications of shear-induced polarized light imaging (SIPLI) technique for mechano-optical rheology of polymers and soft matter materials. J. Polym. Sci., Part B: Polym. Phys. 2016, 54, 2151–2170. 10.1002/polb.24111. [DOI] [Google Scholar]
- Mykhaylyk O. O.; Parnell A. J.; Pryke A.; Fairclough J. P. A. Direct imaging of the orientational dynamics of block copolymer lamellar phase subjected to shear flow. Macromolecules 2012, 45, 5260–5272. 10.1021/ma3004289. [DOI] [Google Scholar]
- Caplan M. R.; Moore P. N.; Zhang S.; Kamm R. D.; Lauffenburger D. A. Self-assembly of a beta-sheet protein governed by relief of electrostatic repulsion relative to van der Waals attraction. Biomacromolecules 2000, 1, 627–631. 10.1021/bm005586w. [DOI] [PubMed] [Google Scholar]
- Aggeli A.; Bell M.; Boden N.; Carrick L. M.; Strong A. E. Self-assembling peptide polyelectrolyte beta-sheet complexes form nematic hydrogels. Angew. Chem., Int. Ed. 2003, 42, 5603–5606. 10.1002/anie.200352207. [DOI] [PubMed] [Google Scholar]
- Nelson D. L.; Lehninger A. L.; Cox M. M.. Lehninger Principles of Biochemistry. 7th ed.; Macmillan Higher Education: Basingstoke, 2017. [Google Scholar]
- Tang C.; Smith A. M.; Collins R. F.; Ulijn R. V.; Saiani A. Fmoc-diphenylalanine self-assembly mechanism induces apparent pKa shifts. Langmuir 2009, 25, 9447–9453. 10.1021/la900653q. [DOI] [PubMed] [Google Scholar]
- Aggeli A.; Bell M.; Carrick L. M.; Fishwick C. W. G.; Harding R.; Mawer P. J.; Radford S. E.; Strong A. E.; Boden N. pH as a trigger of peptide beta-sheet self-assembly and reversible switching between nematic and isotropic phases. J. Am. Chem. Soc. 2003, 125, 9619–9628. 10.1021/ja021047i. [DOI] [PubMed] [Google Scholar]
- Barth A. Infrared spectroscopy of proteins. Biochim. Biophys. Acta, Rev. Bioenerg. 2007, 1767, 1073–1101. 10.1016/j.bbabio.2007.06.004. [DOI] [PubMed] [Google Scholar]
- Barth A.; Zscherp C. What vibrations tell us about proteins. Q. Rev. Biophys. 2002, 35, 369–430. 10.1017/s0033583502003815. [DOI] [PubMed] [Google Scholar]
- Barth A. The infrared absorption of amino acid side chains. Prog. Biophys. Mol. Biol. 2000, 74, 141–173. 10.1016/s0079-6107(00)00021-3. [DOI] [PubMed] [Google Scholar]
- Yan H.; Frielinghaus H.; Nykanen A.; Ruokolainen J.; Saiani A.; Miller A. F. Thermoreversible lysozyme hydrogels: properties and an insight into the gelation pathway. Soft Matter 2008, 4, 1313–1325. 10.1039/b716966c. [DOI] [PubMed] [Google Scholar]
- Hamley I. W. Peptide fibrillization. Angew. Chem., Int. Ed. 2007, 46, 8128–8147. 10.1002/anie.200700861. [DOI] [PubMed] [Google Scholar]
- Ramachandran S.; Trewhella J.; Tseng Y.; Yu Y. B. Coassembling peptide-based biomaterials: effects of pairing equal and unequal chain length oligopeptides. Chem. Mater. 2006, 18, 6157–6162. 10.1021/cm061071l. [DOI] [Google Scholar]
- Sathaye S.; Mbi A.; Sonmez C.; Chen Y.; Blair D. L.; Schneider J. P.; Pochan D. J. Rheology of peptide- and protein-based physical hydrogels: Are everyday measurements just scratching the surface?. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol. 2015, 7, 34–68. 10.1002/wnan.1299. [DOI] [PubMed] [Google Scholar]
- Draper E. R.; Adams D. J. How should multicomponent supramolecular gels be characterised?. Chem. Soc. Rev. 2018, 47, 3395–3405. 10.1039/c7cs00804j. [DOI] [PubMed] [Google Scholar]
- Guenet J.-M.Organogels: Thermodynamics, Structure, Solvent Role, and Properties; Springer: Strasbourg, 2016. [Google Scholar]
- Ramzi M.; Rochas C.; Guenet J.-M. Structure-properties relation for agarose thermoreversible gels in binary solvents. Macromolecules 1998, 31, 6106–6111. 10.1021/ma9801220. [DOI] [Google Scholar]
- Guenet J.-M. Structure versus rheological properties in fibrillar thermoreversible gels from polymers and biopolymers. J. Rheol. 2000, 44, 947–960. 10.1122/1.551121. [DOI] [Google Scholar]
- Wang J.; Liu K.; Xing R.; Yan X. Peptide self-assembly: thermodynamics and kinetics. Chem. Soc. Rev. 2016, 45, 5589–5604. 10.1039/c6cs00176a. [DOI] [PubMed] [Google Scholar]
- Cardoso A. Z.; Alvarez Alvarez A. E.; Cattoz B. N.; Griffiths P. C.; King S. M.; Frith W. J.; Adams D. J. The influence of the kinetics of self-assembly on the properties of dipeptide hydrogels. Faraday Discuss. 2013, 166, 101–116. 10.1039/c3fd00104k. [DOI] [PubMed] [Google Scholar]
- Oyen M. L. Mechanical characterisation of hydrogel materials. Int. Mater. Rev. 2014, 59, 44–59. 10.1179/1743280413y.0000000022. [DOI] [Google Scholar]
- Yan C.; Altunbas A.; Yucel T.; Nagarkar R. P.; Schneider J. P.; Pochan D. J. Injectable solid hydrogel: mechanism of shear-thinning and immediate recovery of injectable beta-hairpin peptide hydrogels. Soft Matter 2010, 6, 5143–5156. 10.1039/c0sm00642d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coniglio A.; Arcangelis L. D.; Gado E. D.; Fierro A.; Sator N. Percolation, gelation and dynamical behaviour in colloids. J. Phys.: Condens. Matter 2004, 16, S4831–S4839. 10.1088/0953-8984/16/42/002. [DOI] [Google Scholar]
- Hamley I. W.; Burholt S.; Hutchinson J.; Castelletto V.; da Silva E. R.; Alves W.; Gutfreund P.; Porcar L.; Dattani R.; Hermida-Merino D.; Newby G.; Reza M.; Ruokolainen J.; Stasiak J. Shear alignment of bola-amphiphilic arginine-coated peptide nanotubes. Biomacromolecules 2017, 18, 141–149. 10.1021/acs.biomac.6b01425. [DOI] [PubMed] [Google Scholar]
- Cui H.; Pashuck E. T.; Velichko Y. S.; Weigand S. J.; Cheetham A. G.; Newcomb C. J.; Stupp S. I. Spontaneous and X-ray–triggered crystallization at long range in self-assembling filament networks. Science 2010, 327, 555. 10.1126/science.1182340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cates M. E. Reptation of living polymers: dynamics of entangled polymers in the presence of reversible chain-scission reactions. Macromolecules 1987, 20, 2289–2296. 10.1021/ma00175a038. [DOI] [Google Scholar]
- Semenov A. N.; Rubinstein M. Dynamics of entangled associating polymers with large aggregates. Macromolecules 2002, 35, 4821–4837. 10.1021/ma0117965. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.