

Abstract
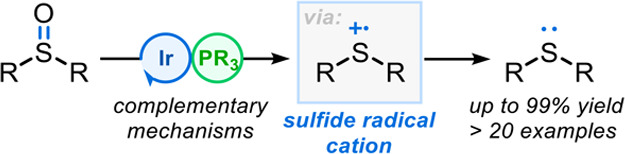
The photocatalytic deoxygenation of sulfoxides to generate sulfides facilitated by either Ir[(dF(CF3)ppy)2(dtbbpy)]PF6 or fac-Ir(ppy)3 is reported. Mechanistic studies indicate that a radical chain mechanism operates, which proceeds via a phosphoranyl radical generated from a radical/polar crossover process. Initiation of the radical chain was found to proceed via two opposing photocatalytic quenching mechanisms, offering complementary reactivity. The mild nature of the radical deoxygenation process enables the reduction of a wide range of functionalized sulfoxides, including those containing acid-sensitive groups, in typically high isolated yields.
Keywords: sulfoxide, deoxygenation, reduction, radical, visible light, photoredox catalysis, sulfide radical cation
The deoxygenation of sulfoxides to generate sulfides is a fundamental transformation in organic synthesis1 and biochemistry.2 Established methods to convert sulfoxides into sulfides3 involve the use of low-valent metallic species,4 metal hydride reagents,5 halide ions,6 and phosphorus compounds.7 However, these reaction systems can suffer from potential disadvantages, including the use of expensive and/or toxic reagents, difficult workup procedures, and use of harsh reaction conditions, which often limit their functional group tolerance. Consequently, this is an area of continued research, and new, efficient procedures for the reduction of sulfoxides into their corresponding sulfides are desirable.
Over the past decade, photoredox catalysis has evolved into a vitally important method able to address long-standing challenges in synthetic chemistry,8 in large part due to the mild conditions by which reactive radicals can be generated. However, photocatalytic methods for deoxygenation of sulfoxides have rarely been explored.9 The cleavage of C–O bonds via β-scission of phosphoranyl radicals was initially recognized in the early 1970s by Bentrude,10 and since then, the groups of Zhu and Xie,11Doyle,12 and Rovis12 have extended the synthetic application of this strategy to incorporate photoredox catalysis (Figure 1A), establishing valuable methods for deoxygenation of alcohols and carboxylic acids. More recently, this work was extended to include cleavage of N–O bonds by Yang et al.13 and also by Lardy and Schmidt,14 who employed more traditional radical initiation methods.
Figure 1.
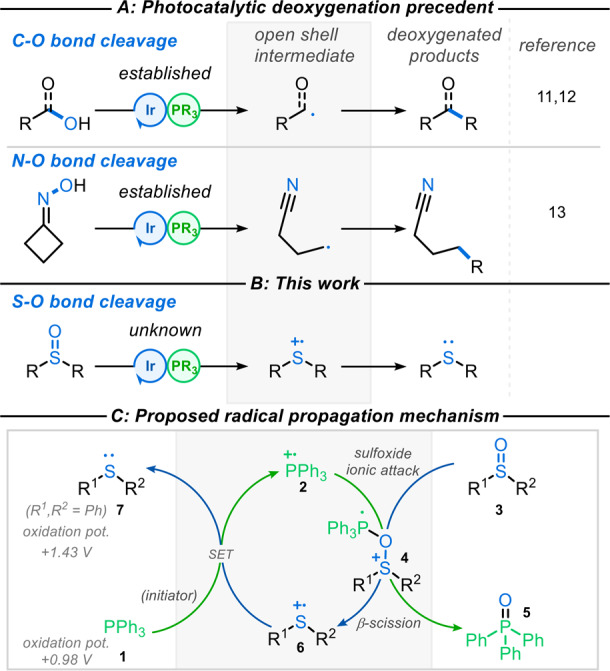
Photocatalytic deoxygenation methods.
Inspired by these works, we speculated that direct cleavage of S=O bonds could be accomplished via a polar/radical crossover process between phosphine radical cations, generated from a photocatalyst (PC) initiator, and sulfoxides, resulting in mild deoxygenation of sulfoxides (Figure 1B). Based on existing phosphoranyl radical studies (Figure 1A) and the reported oxidation potentials of sulfides (e.g., diphenyl sulfide {E1/2 = +1.43 V versus saturated calomel electrode [SCE]})15 relative to those of phosphines (e.g., PPh3 {E1/2 = +0.98 V versus SCE}),12 a radical chain mechanism was proposed16 (Figure 1C, see later for a description).
We postulated that initiation of a radical chain deoxygenation process could be promoted by single-electron oxidation of PPh31 using a suitable oxidizing photocatalyst (initiator)17 to afford a catalytic amount of phosphine radical cation 2. Polar nucleophilic addition of sulfoxide 3 to radical cation 2 would generate phosphoranyl radical 4, which upon β-scission, would afford sulfide radical cation 6 and triphenylphosphine oxide 5. Finally, reduction of the sulfide radical cation 6 by PPh31 would afford the desired sulfide 7, as well as propagating the radical chain via regeneration of phosphine radical cation 2. Herein, we describe the realization of this radical chain process for the high-yielding deoxygenation of sulfoxides under mild, visible light-driven reaction conditons.18
Studies began by surveying the ability of a series of photocatalyst initiators (PC1–PC8, Scheme 1) to promote the reduction of 4-bromophenyl methyl sulfoxide 8a into sulfide 8b using PPh3 as the terminal reductant and CH2Cl2 as the solvent, irradiating with a 60 W blue LED light19 under an argon atmosphere. In line with related literature,11,12 both PC1 and PC2, which have excited-state oxidation potentials (M*/M–) greater than that of PPh3 (E1/2 = +0.98 V versus SCE), afforded sulfide 8b in excellent yields.20 Moreover, PC3 and PC6, which have excited-state oxidation potentials lower than PPh3, resulted in much lower yields of sulfide 8b (11 and 5%, respectively) as expected. However, PCs possessing a far lower oxidation potential than PPh3 (e.g., PC7 and PC8; the PCs that we originally considered to be the least likely to promote effective deoxygenation of 8a) unexpectedly promoted the formation of sulfide 8b in high yields. We noticed that all four PCs (PC4, PC5, PC7, and PC8) are able to initiate the reaction effectively despite their low excited-state oxidation potentials and relatively high excited-state reduction potentials (M+/M*). In contrast, the PCs with relatively low excited-state oxidation and reduction potentials (i.e., PC3 and PC6, for which both potentials are within the white area in Scheme 1) did not perform well in the reaction. These observations suggested that two mechanistic pathways may be viable, based on either a reductive or oxidative photocatalyst quenching cycle, with the route taken dependent on the redox potentials of the PC initiator used.
Scheme 1. Photocatalyst Initiator Screening.

To probe this possibility further, comparative control reactions were conducted, with Ir[(dF(CF3)ppy)2(dtbbpy)]PF6 (PC2) chosen as a representative oxidizing photocatalyst and fac-Ir(ppy)3 (PC8) as a representative reducing photocatalyst. In the absence of PC and light (entries 2 and 3, Table 1), no reaction occurred in either system. In the absence of PPh3, no reaction occurred when employing the oxidizing PC2, although contrastingly, a small amount of conversion into sulfide 8b was observed when employing the reducing PC8. TEMPO drastically suppressed the efficiency of both reaction systems, supporting a free-radical reaction pathway (entry 5); triphenylphosphine oxide was the major product formed in these TEMPO reactions, presumably via the pathway described by Bentrude.10 Both reactions could be performed in other solvents or under an atmosphere of air but a reduction in yield was generally observed (entries 6–8). Other readily available phosphines, phosphites, and phosphinites were also able to promote sulfoxide reduction, albeit in reduced yields compared with PPh3 (entries 9–11).
Table 1. General Reaction Condition Optimizationa.

| entry | deviation from standard conditions | yield (PC2) (%) | yield (PC8) (%) |
|---|---|---|---|
| 1 | none | 99 | 99 |
| 2 | no PC | 0 | 0 |
| 3 | no light | 0 | 0 |
| 4 | no PPh3 | 0 | 9 |
| 5 | 3 equiv of TEMPO | 2 | 3 |
| 6 | under air | 69 | 46 |
| 7 | THF | 85 | 69 |
| 8 | toluene | 44 | 99 |
| 9 | PCy3 | 48 | 33 |
| 10 | PPh2OEt | 68 | 5 |
| 11 | P(OPh)3 | 28 | 8 |
Reaction conditions: 8a (0.20 mmol), PC2 or PC8 (1 mol %), PPh3 (0.24 mmol) in CH2Cl2 (1.0 mL) at RT, 24 h. 1H NMR yields reported based on a trimethoxybenzene internal standard.
Mechanistically, the single-electron oxidation of PPh3 by the most oxidizing catalysts (e.g., PC1 and PC2, Scheme 1) is an established concept.11,12 Consequently, initiation of the radical chain cycle when using such oxidizing PCs is proposed to occur via reductive quenching of the excited-state PC to generate the key phosphorus radical cation 2 required to initiate the proposed radical chain mechanism (Scheme 2A). To support this, Stern–Volmer quenching studies were conducted, confirming that the emission of the excited-state PC2 is quenched by PPh3 (Scheme 2B). Furthermore, when DDQ, an organic oxidant, was used in sub-stoichiometric amounts (≥5 mol %) in place of the PC, sulfide 8b was produced in excellent yields (Scheme 2C), further supporting the notion that the generation of phosphorus radical cation 2 promotes an efficient radical chain process, as depicted in Figure 1C.
Scheme 2. Initiation via Phosphine Oxidation (PC2).
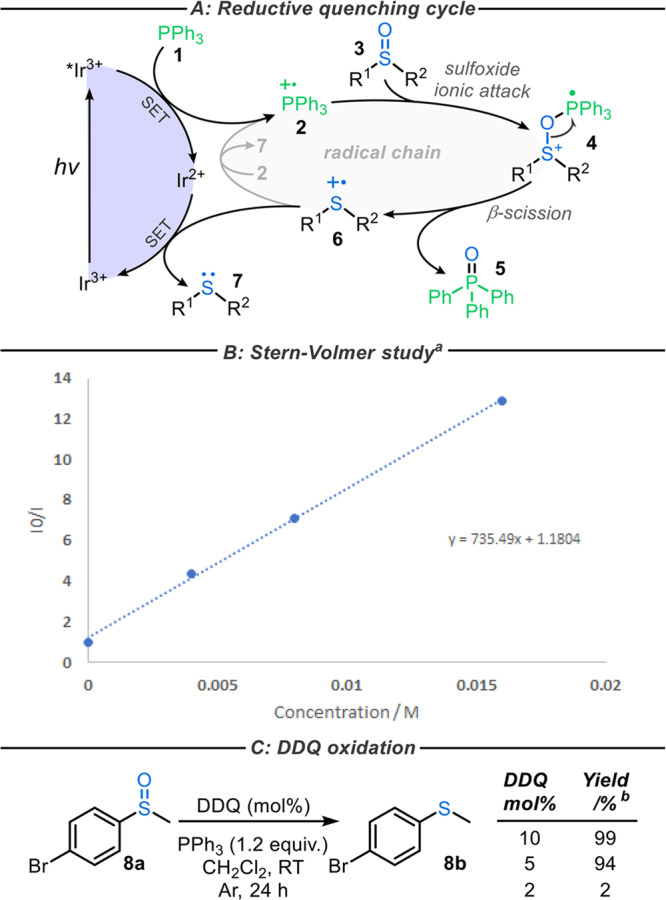
Stern–Volmer quenching study of Ir[(dF(CF3)ppy)2(dtbbpy)]PF6 with PPh3 in degassed CH2Cl2.
1H NMR yields reported based on a trimethoxybenzene internal standard.
In contrast, PCs possessing lower oxidation potentials (M*/M–) such as fac-Ir(ppy)3 (PC8) should not be able to oxidize PPh3, which is supported by the absence of emission quenching of the excited state of PC8 by PPh3 (see the Supporting Information). It has also been documented that sulfoxides such as DMSO are unable to quench the emission of PC8.21 Nonetheless, contrary to these observations, which suggest that no reaction should occur, it was found that DMSO can be reduced to DMS in high yields (99%) when reacted with PC8 and PPh3 under our standard reaction conditions (for conditions, see Table 1). We initially postulated that this may be a result of energy transfer from the excited state of PC8 to the sulfoxide,22 thus forming an excited-state sulfoxide species able to undergo deoxygenation. The low-yielding deoxygenation of 8a in the absence of PPh3 (see earlier control reactions, entry 4, Table 1) offers some support for this hypothesis, and the direct deoxygenation of sulfoxides under UV irradiation has also been reported.23 However, we could find no evidence of emission quenching of the excited state of PC8 by either sulfoxides 8a or 18a in Stern–Volmer quenching studies (in line with literature precedent).21 Furthermore, if energy transfer is involved, it is not clear why the redox properties of the various PCs would have such a pronounced influence on the observed reactivity. Based on these observations, it was considered more likely that initiation is mediated by a redox process. It is also clear from the synthetic results that phosphine plays a key role in the reaction. Thus, an alternative mechanism was postulated, in which PPh3 and the sulfoxide interact to form an adduct that can initiate the radical chain mechanism via an initial oxidative quench of the PC (Scheme 3A).
Scheme 3. Initiation via Adduct Reduction (PC8).
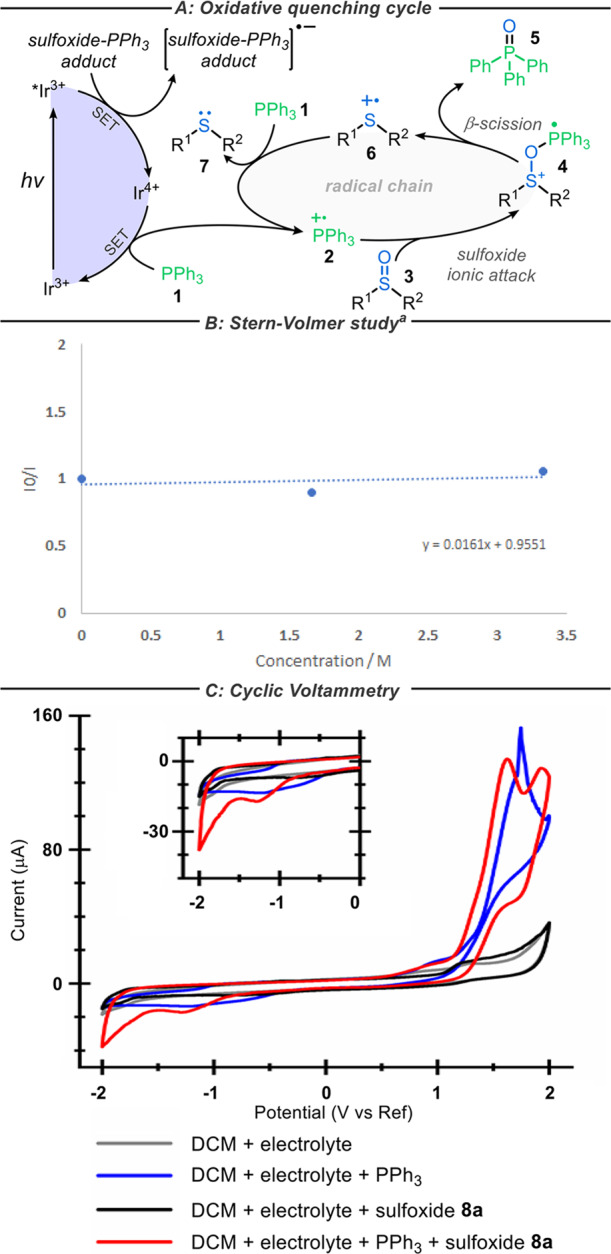
Stern–Volmer quenching study of fac-Ir(ppy)3 with 1:1 solution of sulfoxide 8a:PPh3 in degassed CH2Cl2.
This alternative mechanism would proceed via an electron transfer from the excited-state PC to a sulfoxide–PPh3 adduct, thus accessing a ground-state Ir4+ complex (M+/M, Scheme 1). This Ir4+ species (which is considerably more oxidizing than the corresponding *Ir3+ state) could afford the key phosphine radical cation 2 via phosphine oxidation (1 → 2), thus enabling the earlier proposed radical chain reaction to proceed. Following sulfoxide attack (2 → 4) and β-scission (4 → 6), the resultant sulfide radical cation 6 could then undergo reduction in a number of ways: (1) reaction with PPh3, thus regenerating phosphine radical cation 2 and propagating the radical chain (depicted in Scheme 3A); (2) reaction with the reduced sulfoxide–PPh3 adduct; and (3) reaction with the excited-state PC to form the oxidizing ground-state Ir4+ complex, which would then go on to propagate initiation via phosphine oxidation (1 → 2) (2 and 3 are not depicted in Scheme 3A). We first sought to identify the formation of the proposed sulfoxide–PPh3 adduct spectroscopically, but regrettably, no evidence for phosphine–sulfoxide interaction was evident using 1H/31P NMR or UV–vis spectroscopy (see the Supporting Information). Stern–Volmer quenching studies also revealed that a 1:1 mixture of PPh3 and sulfoxide 8a did not quench the emission of excited-state fac-Ir(ppy)3 (PC8) even at concentrations far greater than that found in the reaction (Scheme 3B). We therefore turned to cyclic voltammetry to see if we could observe a reduction potential consistent with oxidative quenching of the excited state of PC8. More encouragingly, a unique reduction process was observed (with an onset potential of approximately −0.8 V vs Ag/AgCl) when both the sulfoxide and PPh3 were present in solution, which was absent when either of these reagents was omitted (Scheme 3C). This electrochemical data certainly suggests that the redox chemistry of the sulfoxide and PPh3 is affected by the presence/absence of the other. At present, these findings still leave some questions unanswered (most pertinently, what the structure of the hypothetical sulfoxide–PPh3 adduct could be), but the synthetic and mechanistic results do support the notion that an alternative mechanism for deoxygenation operates when a PC with a sufficiently reductive potential is used.
Next, attention was turned to probing the synthetic utility of the deoxygenation. A preliminary substrate screen was conducted from which the relatively oxidizing photocatalyst PC2 was identified as the most broadly effective PC (see the Supporting Information) and was taken into further substrate scoping studies (Scheme 4). Diaryl sulfoxides 9a–11a were all well tolerated; notably, sulfoxide 11a, incorporating an acid-sensitive Boc group, was converted into its sulfide 11b in 99% yield. Various sulfoxides bearing a single functionalized aryl group also worked well, including halogenated systems (e.g., 8b and 15b). Sulfoxide 14a, which contains an acid-labile silyl ether, was also an excellent substrate for this transformation, providing the corresponding sulfide 14b in 90% yield.
Scheme 4. Substrate Scope of Sulfoxide-to-Sulfide Reduction.

Reaction conditions: sulfoxide (0.30 mmol), Ir[(dF(CF3)ppy)2(dtbpy)]PF6 (1 mol %), PPh3 (0.36 mmol) in CH2Cl2 (1.5 mL) at RT, 24 h.
1H NMR yields reported based on a trimethoxybenzene internal standard; isolated yields of products after column chromatography are reported in parentheses.
1 mol % Ir[(dF(CF3)ppy)2(d(CF3)bpy)]PF6 (PC1) and 4 day reaction time employed.
1 mol % fac-Ir(ppy)3 (PC8).
48 h reaction time employed.
Yields of the corresponding sulfide observed by 1H NMR spectroscopy based on a trimethoxybenzene internal standard are presented.
Importantly, the freedom to vary the PC (and in particular, to vary its redox properties) allows deoxygenation to be performed on a wide range of substrates. For example, when using the most oxidizing photocatalyst Ir[(dF(CF3)ppy)2(d(CF3)bpy)]PF6 (PC1), we were pleased to discover that sulfoxide 13a, which contains an unprotected alcohol, afforded sulfide 13b in 78% yield, which was a significant improvement upon the yield using PC2.24 Sulfoxide 17a also reacted poorly with PC2 under optimized conditions (34% conversion), with this attributed to competing oxidation of the benzothiazole moiety in this substrate. To address this, we tested the deoxygenation of sulfoxide 17a using the less oxidizing fac-Ir(ppy)3 photocatalyst PC8, and gratifyingly, the corresponding sulfide 17b was isolated in near-quantitative yield, further demonstrating the value of having complementary synthetic protocols based on both oxidizing and reducing catalysts (see the Supporting Information for more comparisons between the reactivities of PC2 and PC8).
Sulfoxide reduction was also performed on a wide range of dialkyl sulfoxides with varying alkyl chain lengths; all reactions progressed cleanly to furnish the desired linear (18b, 20b, and 21b) and cyclic (19b) sulfide products in excellent yields. Acetal protecting groups are also well tolerated by this procedure, with sulfide 24b generated in 90% yield. Complete reduction of sulfoxide 25a derived from N-Boc-protected methionine was also achieved, furnishing the corresponding sulfide 25b in 99% isolated yield. A list of low-yielding or unreactive substrates is presented at the bottom of Scheme 4 (26a–32a). We believe that the low reactivities of these substrates can generally be attributed to poor solubility of the sulfoxide starting material in CH2Cl2 or low nucleophilicity of the sulfoxide/sulfone starting material. Interestingly, aryl carboxylic acid-containing sulfoxide 29a undergoes deoxygenation when using PC2 (38% yield), but incomplete conversion of the sulfoxide starting material is observed alongside the formation of a side product.25 When performing the deoxygenation reaction using PC8, the corresponding sulfide is formed cleanly in 67% yield, with the remaining mass balance composed of unreacted sulfoxide. In all the above scoping studies, the only byproduct formed is triphenylphosphine oxide, and no discernible side products were isolated except where explicitly stated.
Finally, to further demonstrate the functional group tolerance and utility of the procedure, the deoxygenation of a sulfoxide-containing agrochemical (33a) and drug molecule (34a) was investigated (Scheme 5). In these examples, low to moderate yields of sulfide products were observed when using PC2 under a range of conditions. However, upon switching to PC8 (with a greater reduction potential), far superior reactivity was observed. Thus, agrochemical agent fensulfothion 33a, which contains a phosphorothioate moiety, was cleanly reduced to its corresponding sulfide 33b in 96% isolated yield. Furthermore, sulmazole 34a, a cardiotonic drug containing an imidazopyridine ring, was converted into sulfide 34b in 62% yield under the same conditions; this was a more challenging substrate due to its limited solubility in a range of solvents.
Scheme 5. Biologically Active Sulfoxide Reduction,
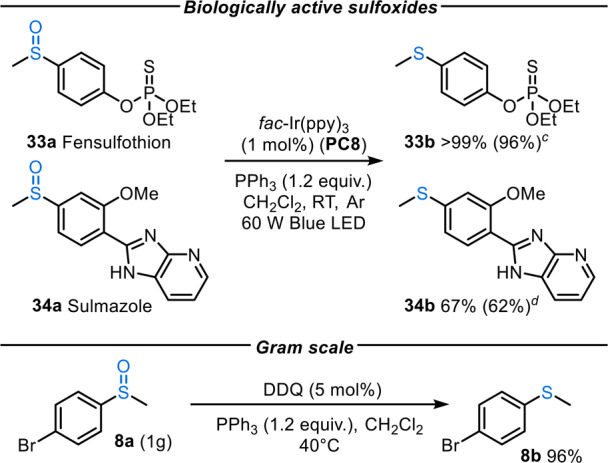
Reaction conditions: sulfoxide (0.30 mmol), fac-Ir(ppy)3 (1 mol %), PPh3 (0.36 mmol) in CH2Cl2 (1.5 mL) at RT.
1H NMR yields reported based on a trimethoxybenzene internal standard and isolated yields of products after column chromatography are shown in parentheses.
48 h reaction time.
24 h reaction time.
Scalability can be a concern in photoredox-catalyzed processes, with an increased photon flux needed for large-scale photochemical reactions. Advances in flow chemistry technology have come a long way in addressing this problem; however, larger-scale photochemical reactions are still typically less straightforward to achieve experimentally compared to the scale-up of thermal reactions.26 To demonstrate that this phosphine radical cation strategy can easily be adopted by researchers who do not have access to the necessary equipment to perform photochemical flow reactions, deoxygenation of sulfoxide 8a was performed on a 1 g scale, using DDQ as the radical chain initiator, affording sulfide 8b in 96% yield, with the rest of the mass balance consisting of unreacted sulfoxide 8a.
Acknowledgments
The authors would like to thank the EPSRC (A.K.C., EP/R013748/1), the University of York (J.A.R.-A. and W.P.U.), and the Leverhulme Trust (W.P.U., for an Early Career Fellowship, ECF-2015-013) for financial support. We are also grateful for the provision of Eleanor Dodson Fellowship (W.P.U.) by the Department of Chemistry, University of York. We also thank Dr. James R. Donald, Dr. Michael J. James, Prof. Robin Perutz, Dr. Richard E. Douthwaite, and Prof. Anne-Kathrin Duhme-Klair for insightful discussions and Mr. Alex Derry for help with preliminary work.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.0c00690.
Experimental procedures and characterization data for all new compounds (PDF)
Author Contributions
‡ A.K.C. and J.A.R.-A. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- a Kunieda N.; Nokami J.; Kinoshita M. Stereoselective reactions of (+)-(R)-α(p-tolylsulfinyl)acetophenone with alkyl Grignard reagents in ethyl ether. Chem. Lett. 1974, 3, 369–372. 10.1246/cl.1974.369. [DOI] [Google Scholar]; b Solladié G. Asymmetric synthesis using nucleophilic reagents containing a chiral sulfoxide group. Synthesis 1981, 185–196. 10.1055/s-1981-29378. [DOI] [Google Scholar]; c Kosugi H.; Konta H.; Uda H. Highly diastereoselective reduction of chiral β-ketosulphoxides under chelation control: application to the synthesis of (R)-(+)-n-hexadecano-1,5-lactone. J. Chem. Soc., Chem. Commun. 1985, 211–213. 10.1039/C39850000211. [DOI] [Google Scholar]; d Solladié G.; Demailly G.; Greck C. Reduction of β-hydroxysulfoxides: Application to the synthesis of optically active epoxides. Tetrahedron Lett. 1985, 26, 435–438. 10.1016/S0040-4039(00)61904-4. [DOI] [Google Scholar]; e Kosugi H.; Watanabe Y.; Uda H. Lewis acid-mediated carbon–carbon bond forming reaction using the pummerer rearrangement products from chiral β-hydroxy sulfoxides. Chem. Lett. 1989, 18, 1865–1868. 10.1246/cl.1989.1865. [DOI] [Google Scholar]; f Carreño M. C. Applications of sulfoxides to asymmetric synthesis of biologically active compounds. Chem. Rev. 1995, 95, 1717–1760. 10.1021/cr00038a002. [DOI] [Google Scholar]
- a Debaun J. R.; Menn J. J. Sulfoxide reduction in relation to organophosphorus insecticide detoxification. Science 1976, 191, 187–188. 10.1126/science.1246606. [DOI] [PubMed] [Google Scholar]; b Ejiri S.-I.; Weissback H.; Brot N. Reduction of methionine sulfoxide to methionine by Escherichia coli. J. Bacteriol. 1979, 139, 161–164. 10.1128/JB.139.1.161-164.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Yoneyama K.; Matsumura F. Reductive metabolism of heptachlor, parathion, 4,4′-dichlorobenzophenone, and carbophenothion by rat liver systems. Pestic. Biochem. Physiol.. 1981, 15, 213–221. 10.1016/0048-3575(81)90003-1. [DOI] [Google Scholar]; d Brot N.; Weissbach H. The biochemistry of methionine sulfoxide residues in proteins. Trends Biochem. Sci. 1982, 7, 137–139. 10.1016/0968-0004(82)90204-3. [DOI] [Google Scholar]; e Surur A. S.; Schulig L.; Link A. Interconnection of sulfides and sulfoxides in medicinal chemistry. Arch. Pharm.Chem. Life Sci. 2019, 325, 1800248. 10.1002/ardp.201800248. [DOI] [PubMed] [Google Scholar]
- a Madesclaire M. Reduction of sulfoxides to thioethers. Tetrahedron 1988, 44, 6537–6580. 10.1016/S0040-4020(01)90096-1. [DOI] [Google Scholar]; b Shiri L.; Kazemi M. Deoxygenation of sulfoxides. Res. Chem. Intermed. 2017, 43, 6007–6041. 10.1007/s11164-017-2976-6. [DOI] [Google Scholar]
- a Mikami K.; Noujima A.; Mitsudome T.; Mizugaki T.; Jitsukawa K.; Kaneda K. Highly efficient gold nanoparticle catalyzed deoxygenation of amides,sulfoxides, and pyridine N-oxides. Chem. – Eur. J. 2011, 17, 1768–1772. 10.1002/chem.201003109. [DOI] [PubMed] [Google Scholar]; b García N.; García-García P.; Fernández-Rodríguez M. A.; Rubio R.; Pedrosa M. R.; Arnáiz F. J.; Sanz R. Pinacol as a new green reducing agent: molybdenum-catalyzed chemoselective reduction of sulfoxides and nitroaromatics. Adv. Synth. Catal. 2012, 354, 321–327. 10.1002/adsc.201100877. [DOI] [Google Scholar]; c García N.; García-García P.; Fernández-Rodríguez M. A.; García D.; Pedrosa M. R.; Arnáiz F. J.; Sanz R. An unprecedented use for glycerol: chemoselective reducing agent for sulfoxides. Green Chem. 2013, 15, 999–1005. 10.1039/c3gc36908k. [DOI] [Google Scholar]
- a Chasar D. W. Quantitative reduction of sulfoxides. J. Org. Chem. 1971, 36, 613–614. 10.1021/jo00803a033. [DOI] [Google Scholar]; b Drabowicz J.; Mikolajczyk M. A mild and efficient reduction of sulphoxides by means of lithium alanate and titanium(IV) chloride. Synthesis 1976, 527–528. 10.1055/s-1976-24107. [DOI] [Google Scholar]; c Harrison D. J.; Tam N. C.; Vogels C. M.; Langler R. F.; Baker R. T.; Decken A.; Westcott S. A. A gentle and efficient route for the deoxygenation of sulfoxides using catecholborane (HBcat; cat = 1,2-O2C6H4). Tetrahedron Lett. 2004, 45, 8493–8496. 10.1016/j.tetlet.2004.09.068. [DOI] [Google Scholar]; d Zhang J.; Gao X.; Zhang C.; Zhang C.; Luan J.; Zhao D. Mild and efficient deoxygenation of sulfoxides to sulfides using HfCl4/KBH4 system. Synth. Commun 2010, 40, 1794–1801. 10.1080/00397910903161819. [DOI] [Google Scholar]
- a Aida T.; Furukawa N.; Oae S. Catalytic reduction of dimethyl sulfoxide and other sulfoxides with bromine - hydrobromic acid system. Tetrahedron Lett. 1973, 14, 3853–3856. 10.1016/S0040-4039(01)87056-8. [DOI] [Google Scholar]; b Landini D.; Maia A. M.; Rolla F. Racemization of sulphoxides with halide ions anchimerically assisted by a carboxy-group. The different behaviour of chloride and bromide ions. J. Chem. Soc., Perkin Trans. 2 1976, 2, 1288–1291. 10.1039/P29760001288. [DOI] [Google Scholar]; c Firouzabadi H.; Karimi B. Efficient deoxygenation of sulfoxides to thioethers and reductive coupling of sulfonyl chlorides to disulfides with tungsten hexachloride. Synthesis 1999, 500–502. 10.1055/s-1999-3414. [DOI] [Google Scholar]; d Shimizu M.; Shibuya K.; Hayakawa R. Chemoselective deoxygenation of sulfoxides with titanium tetraiodide. Synlett 2000, 1437–1438. [Google Scholar]; e Yoo B. W.; Choi K. H.; Kim D. Y.; Choi K. I.; Kim J. H. Mild and efficient reduction of sulfoxides to sulfides with titanium tetrachloride–indium system. Synth. Commun. 2003, 33, 53–57. 10.1081/SCC-120015558. [DOI] [Google Scholar]; f Iranpoor N.; Firouzbadi H.; Shaterian H. R. A new approach to the reduction of sulfoxides to sulfides with 1,3-dithiane in the presence of electrophilic bromine as catalyst. J. Org. Chem. 2002, 67, 2826–2830. 10.1021/jo016027y. [DOI] [PubMed] [Google Scholar]
- a Castrillón J. P. A.; Szmant H. H. Reduction of sulfoxides by triphenylphosphine and carbon tetrachloride. J. Org. Chem. 1965, 30, 1338–1339. 10.1021/jo01015a565. [DOI] [Google Scholar]; b Szmant H. H.; Cox O. The acid-catalyzed reaction of triphenylphosphine with sulfoxides. J. Org. Chem. 1966, 31, 1595–1598. 10.1021/jo01343a065. [DOI] [Google Scholar]; c Kikuchi S.; Konishi H.; Hashimoto Y. The deoxygenation of sulfoxide mediated by the Ph3P/Lewis acid combination and the application to the kinetic resolution of racemic phosphines using optically active sulfoxide. Tetrahedron 2005, 61, 3587–3591. 10.1016/j.tet.2005.01.127. [DOI] [Google Scholar]; d Hua G.; Woollins J. D. The syntheses of sulfides by deoxygenation of sulfoxides using Woollins’ reagent. Tetrahedron Lett. 2007, 48, 3677–3679. 10.1016/j.tetlet.2007.03.132. [DOI] [Google Scholar]; e Bahrami K.; Khodaei M. M.; Sheikh Arabi M. TAPC-promoted oxidation of sulfides and deoxygenation of sulfoxides. J. Org. Chem. 2010, 75, 6208–6213. 10.1021/jo1011784. [DOI] [PubMed] [Google Scholar]; f Jang Y.; Kim K. T.; Jeon H. B. Deoxygenation of sulfoxides to sulfides with thionyl chloride and triphenylphosphine: Competition with the Pummerer reaction. J. Org. Chem. 2013, 78, 6328–6331. 10.1021/jo4008157. [DOI] [PubMed] [Google Scholar]; g Zhao X.; Zheng X.; Yang B.; Sheng J.; Lu K. Deoxygenation of sulphoxides to sulphides with trichlorophosphane. Org. Biomol. Chem. 2018, 16, 1200–1204. and references therein 10.1039/C7OB02834B. [DOI] [PubMed] [Google Scholar]
- a Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible light photoredox catalysis with transition metal complexes: applications in organic synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Romero N. A.; Nicewicz D. A. Organic photoredox catalysis. Chem. Rev. 2016, 116, 10075–10166. 10.1021/acs.chemrev.6b00057. [DOI] [PubMed] [Google Scholar]; c Shaw M. H.; Twilton J.; MacMillan D. W. C. Photoredox catalysis in organic chemistry. J. Org. Chem. 2016, 81, 6898–6926. 10.1021/acs.joc.6b01449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki K.; Jeong J.; Yamaguchi K.; Mizuno N. Photoredox catalysis for oxygenation/deoxygenation between sulfides and sulfoxides by visible-light-responsive polyoxometalates. New J. Chem. 2016, 40, 1014–1021. 10.1039/C5NJ01045D. [DOI] [Google Scholar]
- Bentrude W. G. Phosphoranyl radicals: Their structure, formation, and reactions. Acc. Chem. Res. 1982, 15, 117–125. 10.1021/ar00076a004. [DOI] [Google Scholar]
- a Zhang M.; Xie J.; Zhu C. A general deoxygenation approach for synthesis of ketones from aromatic carboxylic acids and alkenes. Nat. Commun. 2018, 9, 3517–3527. 10.1038/s41467-018-06019-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ruzi R.; Ma J.; Yuan X.-A.; Wang W.; Wang S.; Zhang M.; Dai J.; Xie J.; Zhu C. Deoxygenative arylation of carboxylic acids by aryl migration. Chem. – Eur. J. 2019, 25, 12724–12729. 10.1002/chem.201903816. [DOI] [PubMed] [Google Scholar]
- Stache E. E.; Ertel A. B.; Rovis T.; Doyle A. G. Generation of phosphoranyl radicals via photoredox catalysis enables voltage–independent activation of strong C–O bonds. ACS Catal. 2018, 8, 11134–11139. 10.1021/acscatal.8b03592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia P.-J.; Ye Z.-P.; Hu Y.-Z.; Song D.; Xiang H.-Y.; Chen X.-Q.; Yang H. Photocatalytic, phosphoranyl radical-mediated N-O cleavage of strained cycloketone oximes. Org. Lett. 2019, 21, 2658–2662. 10.1021/acs.orglett.9b00651. [DOI] [PubMed] [Google Scholar]
- Lardy S. W.; Schmidt V. A. Intermolecular radical mediated anti-markovnikov alkene hydroamination using N-hydroxyphthalimide. J. Am. Chem. Soc. 2018, 140, 12318–12322. 10.1021/jacs.8b06881. [DOI] [PubMed] [Google Scholar]
- Fukuzumi S.; Shimoosako K.; Suenobu T.; Watanabe Y. Mechanisms of hydrogen-, oxygen-, and electron-transfer reactions of cumylperoxyl radical. J. Am. Chem. Soc. 2003, 125, 9074–9082. 10.1021/ja035156o. [DOI] [PubMed] [Google Scholar]
- Studer A.; Curran D. P. The electron is a catalyst. Nature Chem. 2014, 6, 765–773. 10.1038/nchem.2031. [DOI] [PubMed] [Google Scholar]
- Teegardin K.; Day J. I.; Chan J.; Weaver J. Advances in Photocatalysis: A Microreview of Visible Light Mediated Ruthenium and Iridium Catalyzed Organic Transformations. Org. Process Res. Dev. 2016, 20, 1156–1163. 10.1021/acs.oprd.6b00101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For pre-existing methods to generate sulfide radical cations, see:; a Fasani E.; Freccero M.; Mella M.; Albini A. The role of SET in the deprotection of (thio)ketals under photosensitization by π-acceptors. Tetrahedron 1997, 53, 2219–2232. 10.1016/S0040-4020(96)01123-4. [DOI] [Google Scholar]; b Yokoi H.; Hatta A.; Ishiguro K.; Sawaki Y. Formation of σ- and π-type dimer radical cations by the photochemical one-electron oxidation of aromatic sulfides. J. Am. Chem. Soc. 1998, 120, 12728–12733. 10.1021/ja982595s. [DOI] [Google Scholar]; c Vath P.; Falvey D. E.; Barnhurst L. A.; Kutateladze A. G. Photoinduced C–C bond cleavage in dithiane–carbonyl adducts: A laser flash photolysis study. J. Org. Chem. 2001, 66, 2887–2890. 10.1021/jo010102n. [DOI] [PubMed] [Google Scholar]; d Korzeniowska-Sobczuk A.; Hug G. L.; Carmichael I.; Bobrowski K. Spectral, kinetics, and theoretical studies of radical cations derived from thioanisole and its carboxylic derivative. J. Phys. Chem. A 2002, 106, 9251–9260. 10.1021/jp021039o. [DOI] [Google Scholar]; e Baciocchi E.; Giacco T. D.; Elisei F.; Gerini M. F.; Lapi A.; Liberali P.; Uzzoli B. Steady-state and laser flash photolysis study of the carbon–carbon bond fragmentation reactions of 2-arylsulfanyl alcohol radical cations. J. Org. Chem. 2004, 69, 8323–8330. 10.1021/jo0486544. [DOI] [PubMed] [Google Scholar]; f Baciocchi E.; del Giacco T.; Elisei F.; Lapi A. Sulfur radical cations. kinetic and product study of the photoinduced fragmentation reactions of (phenylsulfanylalkyl)trimethylsilanes and phenylsulfanylacetic acid radical cations. J. Org. Chem. 2006, 71, 853–860. 10.1021/jo051145x. [DOI] [PubMed] [Google Scholar]; g Bettoni M.; del Giacco T.; Stradiotto M.; Elisei F. Photoinduced one-electron oxidation of benzyl methyl sulfides in acetonitrile: Time-resolved spectroscopic evidence for a thionium ion intermediate. J. Org. Chem. 2015, 80, 8001–8008. 10.1021/acs.joc.5b01052. [DOI] [PubMed] [Google Scholar]
- When sulfoxide 8a was irradiated with a 3 W blue LED light using PC2 with the conditions seen in Scheme 1, sulfide 8b was formed in 65% yield based on a trimethoxybenzene internal standard.
- Examination of the reaction mixture for the conversion of 8a into 8b using PC2 revealed that 1 mol % PC2 remained, which was determined using 1H NMR spectroscopy with a trimethoxybenzene internal standard. Furthermore, 19F NMR spectroscopy revealed no new 19F signals after the reaction, suggesting that the catalyst did not degrade or form aggregates during the course of the reaction (see the Supporting Information).
- Liu W.-Q.; Lei T.; Song Z.-Q.; Yang X.-L.; Wu C.-J.; Jiang X.; Chen B.; Tung C.-H.; Wu L.-Z. Visible Light Promoted Synthesis of Indoles by Single Photosensitizer under Aerobic Conditions. Org. Lett. 2017, 19, 3251–3254. 10.1021/acs.orglett.7b01367. [DOI] [PubMed] [Google Scholar]
- Strieth-Kalthoff F.; James M. J.; Teders M.; Pitzer L.; Glorius F. Energy transfer catalysis mediated by visible light: principles, applications, directions. Chem. Soc. Rev. 2018, 47, 7190–7202. 10.1039/C8CS00054A. [DOI] [PubMed] [Google Scholar]
- a Archer R. A.; Kitchell B. S. The Photochemical rearrangement of a sulfoxide. J. Am. Chem. Soc. 1966, 88, 3462–3463. 10.1021/ja00966a071. [DOI] [Google Scholar]; b Schultz A. G.; DeBoer C. D.; Schlessinger R. H. Photodesulfurization of a Sulfoxide. J. Am. Chem. Soc. 1968, 90, 5314–5315. 10.1021/ja01021a070. [DOI] [Google Scholar]; c Gurria G. M.; Posner G. H. Photochemical deoxygenation of aryl sulfoxides. J. Org. Chem. 1973, 38, 2419–2420. 10.1021/jo00953a034. [DOI] [Google Scholar]
- When PC2 was used to reduce sulfoxide 13a, a diminished isolated yield of 59% was observed.
- When using PC2, an aldehyde-containing side product was observed in 10% yield by 1H NMR spectroscopy (using a trimethoxybenzene internal standard). Presumably, this aldehyde side product forms through a similar mechanism as that described in ref (12).
- Cambié D.; Bottecchia C.; Straathof N. J. W.; Hessel V.; Noël T. Applications of continuous-flow photochemistry in organic synthesis, material science, and water treatment. Chem. Rev. 2016, 116, 10276–10341. 10.1021/acs.chemrev.5b00707. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.