

SUMMARY
Precise control of the RNA polymerase II (Pol II) cycle, including pausing and pause release, maintains transcriptional homeostasis and organismal functions. Despite previous work to understand individual transcription steps, we reveal a mechanism that integrates Pol II cycle transitions. Surprisingly, KAP1/TRIM28 uses a previously uncharacterized chromatin reader cassette to bind hypo-acetylated histone 4 tails at promoters thereby guaranteeing a continuous progression of Pol II entry to, and exit from, the pause state. Upon chromatin docking, KAP1 first associates with Pol II and then recruits a pathway-specific transcription factor (SMAD2) in response to cognate ligands thereby enabling gene-selective CDK9-dependent pause release. This coupling mechanism is exploited by tumor cells to aberrantly sustain transcriptional programs commonly dysregulated in cancer patients. The discovery of a factor integrating transcription steps expands the functional repertoire by which chromatin readers operate and provides mechanistic understanding of transcription regulation, offering alternative therapeutic opportunities to target transcriptional dysregulation.
Graphical Abstract
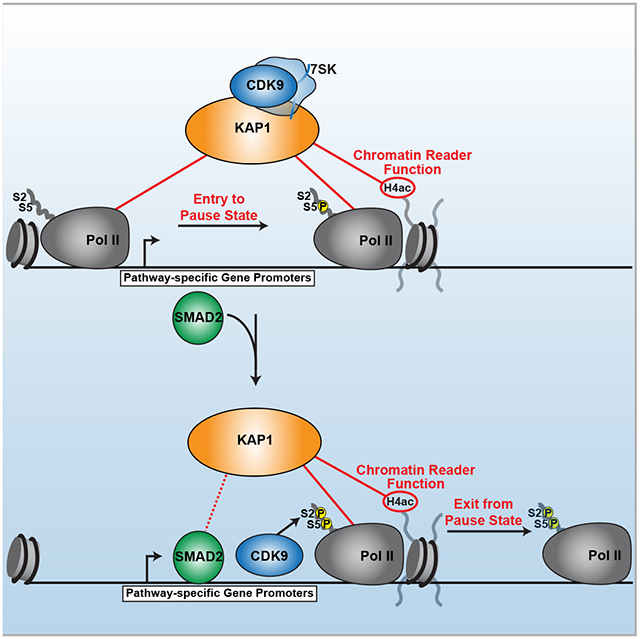
eTOC Blurb
Bacon et al. discover KAP1/TRIM28 utilizes a previously uncharacterized chromatin reader cassette to facilitate select gene promoter occupancy through direct engagement with hypo-acetylated H4 tails. KAP1 then orchestrates interactions with Pol II, a pathway-specific transcription factor (SMAD2) and CDK9 in response to cognate signaling to couple Pol II entry to, and exit from, the pause state thereby sustaining transcriptional programs involved in cancer maintenance.
INTRODUCTION
Precise regulation of transcriptional responses maintains cellular homeostasis and prevents the development and progression of diseases (Miller et al., 2017; Smith et al., 2011). Transcription is initiated by chromatin-remodeling, promoter accessibility, and pre-initiation complex (PIC)-mediated Pol II promoter recruitment (Grunberg and Hahn, 2013; Thomas and Chiang, 2006). Shortly after initiation Pol II transiently pauses downstream of the transcription start site (TSS) (Muse et al., 2007; Nechaev et al., 2010; Zeitlinger et al., 2007) through the action of Negative Elongation Factor (NELF) and DRB-sensitivity Inducing Factor (DSIF) (Gilchrist et al., 2008; Wada et al., 1998a; Wu et al., 2003; Yamaguchi et al., 1999). The release of promoter-proximal paused Pol II into productive elongation is partially dependent on the recruitment of Positive Transcription Elongation Factor (P-TEFb) (Gomes et al., 2006; Wada et al., 1998a; Yamada et al., 2006), which is composed of CDK9 (kinase subunit) and CycT1/T2 (regulatory subunit) (Peng et al., 1998). P-TEFb facilitates Pol II pause release and entry into productive elongation through phosphorylation of NELF, DSIF (Wada et al., 1998b; Yamaguchi et al., 1999), and the Pol II C-terminal domain (CTD) (Ahn et al., 2004; Cho et al., 2001; Eick and Geyer, 2013; Marshall et al., 1996; Suh et al., 2016; Yamada et al., 2006). This transient cycle of Pol II pausing and pause release has emerged as a key regulatory process for timing the activation of transcriptional programs required for signal-dependent biological processes including development, differentiation, and stress responses (Barboric et al., 2001; Gaertner et al., 2012; Lis et al., 2000; Liu et al., 2014; Rasmussen and Lis, 1993).
To maintain transcriptional homeostasis, P-TEFb is inactivated through incorporation into the 7SK small nuclear ribonucleoprotein (snRNP) (Nguyen et al., 2001; Yang et al., 2001), which is tethered to promoters to prime genes for activation (D’Orso and Frankel, 2010; Liu et al., 2013b; McNamara et al., 2016). Recent work has identified KRAB-associated protein 1/Tripartite Motif-containing 28/Transcription Intermediary Factor 1β (KAP1/TRIM28/TIF1β, hereafter referred to as KAP1) as a factor critical for the delivery of 7SK-bound P-TEFb to signal-regulated gene promoters thereby enabling on-site kinase activation (Gudipaty et al., 2015; McNamara et al., 2016).
Notably, KAP1 participates in several physiologic processes, including embryonic stem cell maintenance and restricting the production of induced pluripotent stem cells (Cheng et al., 2014; Miles et al., 2017; Seki et al., 2010). Given its importance in regulating a broad range of physiologic processes, it is not surprising that KAP1 has been co-opted in pathologic conditions. Indeed, KAP1 haplo-insufficiency facilitates obesity (Dalgaard et al., 2016), is amplified in cancer (Hector et al., 2012; Ho et al., 2009; Liu et al., 2013a; Wang et al., 2016; Wang et al., 2013), is a potential biomarker for colorectal cancer (CRC) (Gawel et al., 2019; Shiromizu et al., 2017), its expression negatively correlates with cancer patient survival (Cui et al., 2014; Liu et al., 2013a), is required for xenograft proliferation in nude mice (Chen et al., 2014; Wu et al., 2018), and increases TGF-β-induced epithelial-to-mesenchymal transition (Xu et al., 2009). Despite this compelling in vivo relevance, the molecular mechanisms underlying KAP1 transcriptional regulation in both physiologic and pathologic states remain poorly understood.
Remarkably, here we report that KAP1 sustains cell-intrinsic, oncogenic signaling programs commonly dysregulated in cancer patients. Mechanistically, we define an unprecedented function for the KAP1 chromatin reader domain in binding hypo-acetylated histone 4 (H4) tails. This binding activity positions KAP1 on chromatin where it can directly associate with the Pol II complex to maintain a continuous delivery of Pol II molecules to target gene promoters. KAP1 then tethers the sequence- and pathway-specific TGF-β effector (SMAD2) and CDK9 to induce a transcription factor (TF)-mediated, CDK9-dependent switch from Pol II pausing to pause release. Unlike factors that either regulate Pol II promoter levels or pause release, KAP1 couples both steps to promote efficient yet selective gene activation. The identification of a chromatin reader directly involved in the coupling of Pol II transcription steps provides a revised mechanistic understanding of transcriptional regulation critical for cancer maintenance and progression.
RESULTS
KAP1 Regulates the Growth and Transcriptional Output of Cancer Cells
Previous studies demonstrated that KAP1 is necessary for signal-regulated transcription of select CDK9-dependent target genes (McNamara et al., 2016). Given the lack of genome-wide studies interrogating KAP1 function in transcriptional activation, we asked whether KAP1 is a global or pathway-specific regulator and whether this transcription function may be required for critical functions such as cell proliferation. To address this, we utilized CRISPR-Cas9 and distinct gRNAs to generate two KAP1 knockout (KO) HCT116 CRC cell clones, and one non-targeting control (Ctrl) clone (Figure 1A). Targeting of either gRNA to KAP1 created a deletion (KO1) or insertion (KO2) in the first exon, thus generating premature stop codons (Figures S1A and S1B). Expectedly, we observed an almost identical growth defect in both clones (Figure 1B). Furthermore, a colony formation assay revealed a consistent, almost identical decrease of the clonogenic capability of both clones compared to the Ctrl (Figure 1C). Notably, the growth defect was further cross-validated by cell growth assays in HCT116 cells treated with two distinct siRNA’s to acutely silence KAP1 expression (Figures 1D and 1E).
Figure 1. KAP1 Regulates the Growth and Transcriptional Output of Cancer Cells.
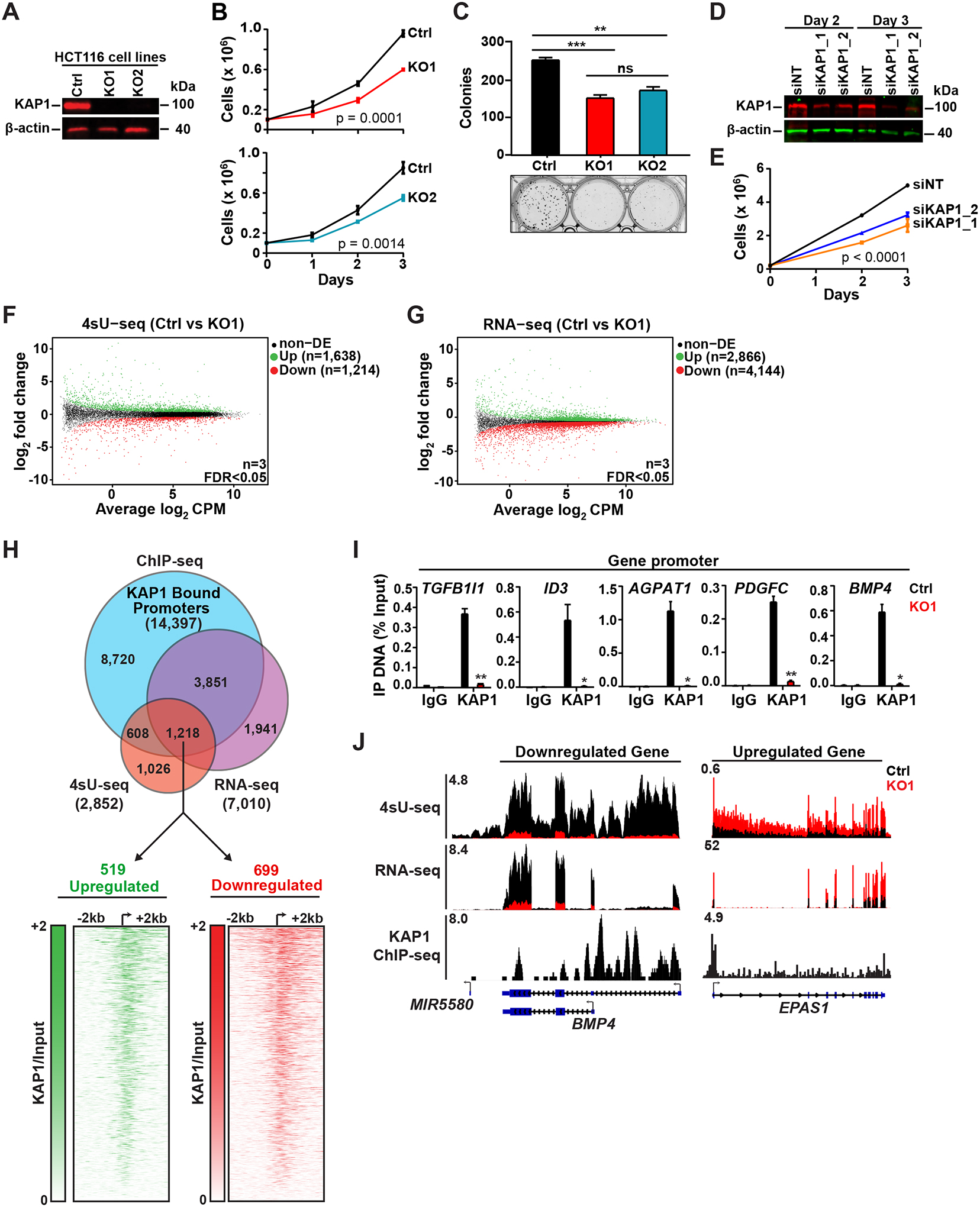
(A) Western blot verifying KAP1 KO.
(B) Cell growth assay (cell counts ± SEM; n=3).
(C) Colony formation assay (colonies ± SEM; n=3).
(D) Western blot verifying KAP1 KD of cells of panel (E).
(E) Cell growth assay (cell counts ± SEM; n=3).
(F-G) Scatter plot of the differentially expressed genes (n=3, FDR<0.05).
(H) Top: Differentially expressed genes identified in 4sU-seq/RNA-seq (FDR<0.05) overlaid with KAP1 promoter ChIP-seq peaks. Bottom: Heatmaps of KAP1 ChIP-seq signal over input.
(I) Validation of KAP1 ChIP-seq promoter peaks by ChIP-qPCR (mean % Input ± SEM; n=3).
(J) Genome browser tracks for select genes.
Because of the virtually identical growth defects in both KAP1 KO clones, for the rest of the study we primarily focused on the KO1 clone and cross-validated key genomic experiments in the KO2 clone and with acute KAP1 silencing. Using Ctrl and KO1 cells we first defined KAP1-regulated transcriptional programs by performing a combination of 4sU-seq (to measure nascent RNA synthesis (Rabani et al., 2011)) and RNA-seq (to measure steady-state RNA levels) (Figures 1F and 1G) to identify key pathways promoting KAP1-dependent cell proliferation. In order to rigorously identify genes directly regulated by KAP1, we overlaid the highly similar replicate 4sU-seq and RNA-seq datasets in KO1 (Figures S1C and S1D) with our previously generated KAP1 chromatin immunoprecipitation (ChIP)-seq dataset (McNamara et al., 2016). This analysis identified upregulated (n=519) and downregulated (n=699) genes bound by KAP1 at their promoters and differentially expressed in both 4sU-seq and RNA-seq (Figure 1H, top; Tables S1 and S2). Importantly, the majority of the direct KAP1 target genes in the KO1 clone are also differentially expressed in KO2, as determined by 4sU-seq (70%) and RNA-seq (73%) (Figures S1E–S1G; Table S1) and after acute depletion of KAP1 by siRNA (Figure 1D, siKAP1_1), as determined by RNA-seq (~70%) (Figures S1H and S1I). These data suggest that the most critical transcriptional changes upon KAP1 loss, potentially contributing to the observed cell growth defects, are not a consequence of long-term clone-specific changes.
Importantly, heatmap analysis of upregulated and downregulated genes centered on the TSS and ranked based on KAP1 levels illustrated KAP1 occupies the promoter-proximal region just downstream of the TSS (Figure 1H, bottom). Furthermore, ChIP-qPCR revealed the expected enrichment of KAP1 at target gene promoters in Ctrl but not in KO1 (Figure 1I). Finally, inspection of genome browser tracks further illustrated the strong gene expression changes observed upon KAP1 loss and KAP1 target gene enrichment (Figure 1J).
Together, these data indicate that KAP1 is required for CRC cell growth, localizes to target gene promoters, and regulates the transcription of a subset of genes. The observed gene upregulation and downregulation upon KAP1 depletion is consistent with its dual role as repressor and activator, respectively (Bunch et al., 2014; Cheng et al., 2014; Fong et al., 2018; Li et al., 2017). In this study, we focus on KAP1’s activating function because its repressive role has been described elsewhere (Friedman et al., 1996; Ivanov et al., 2007; Nielsen et al., 1999; Schultz et al., 2001; Sripathy et al., 2006).
KAP1 Directly Associates with Pol II and Facilitates Pol II Target Gene Promoter Occupancy
Given the transcriptional changes upon KAP1 loss and its previously defined role in regulating Pol II elongation, we asked how loss of KAP1 selectively impacts Pol II occupancy at target genes. To this end, we examined the log2 ratio of total Pol II signal in KO1 over Ctrl (ΔPol II KO1/Ctrl) for downregulated genes that contain a significant Pol II promoter-proximal peak in Ctrl cells, and used k-means clustering based on Pol II density loss to classify KAP1 target genes yielding three gene clusters (C1, C2, C3). Surprisingly, we found decreased promoter-proximal Pol II levels in all three clusters (Figures 2A and S2A), extending previous studies suggesting KAP1 only regulates Pol II pause-release.
Figure 2. Pol II Promoter Levels and Pause-release are Dependent on KAP1.
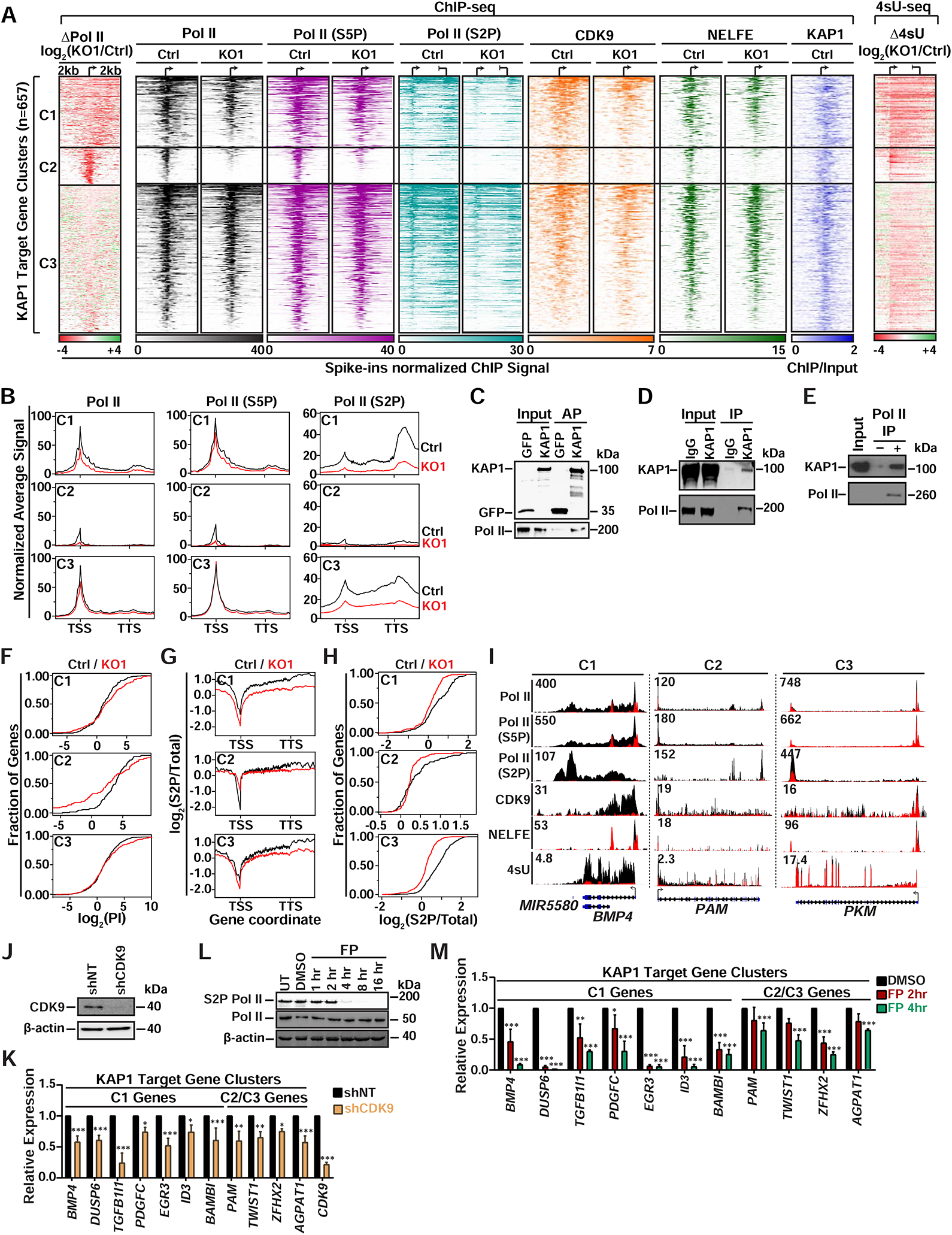
(A) Left: K-means clustered log2(KO1/Ctrl) heatmap of Pol II. Middle: Heatmaps of factor promoter occupancy. Right: Heatmap of Δ4sU-seq. ChIP-seq signal was normalized to Drosophila spike-ins. Individual clusters (C1 n=177, C2 n=98, C3 n=382) are sorted by decreasing Pol II occupancy.
(B) Metagene plots (± 2 kb flanking regions) of Pol II at downregulated KAP1 target gene clusters.
(C) STREP AP of ectopically expressed STREP-tagged KAP1 from HCT116 nuclear extracts.
(D) Endogenous IP of KAP1 from HCT116 nuclear extracts.
(E) In vitro binding assay between purified KAP1 and Pol II.
(F) Empirical cumulative density function plots of Pol II PI at genes from each cluster.
(G) Metagene plots (± 2 kb flanking regions) representing the log2(S2P/total) signal at genes from each cluster.
(H) Empirical cumulative density function plots of log2(S2P/total) signal at genes from each cluster.
(I) Genome browser tracks of ChIP-seq and 4sU-seq for genes from each cluster.
(J) Western blot of CDK9 KD in HCT116.
(K) RT-qPCR analysis after CDK9 KD (mean ± SEM; normalized to RPL19; n=3).
(L) Western blots at different time points after Flavopiridol (FP) treatment. Pol II (RPB3).
(M) RT-qPCR analysis after FP treatment (mean expression relative to DMSO ± SEM; normalized to U6; n=3).
Intriguingly, we found gene-specific differences in both the levels and distribution of Pol II promoter-proximal signal reduction upon KAP1 loss. Particularly, C1 and C2 genes displayed more severe decreases in Pol II promoter levels compared to C3, and C1 and C3 genes showed “dispersed” Pol II losses compared to the more “focused” decreases observed at C2 genes (Figure 2A). Expectedly, metagene plots for all three clusters demonstrated that the observed reductions in promoter-proximal Pol II lead to reduced gene body levels (Figure 2B), consistent with reductions in nascent transcript levels seen by log2 fold changes in 4sU-seq (Δ4sU KO1/Ctrl) (Figure 2A). Importantly, we observed nearly identical Pol II and 4sU-seq phenotypes for all three gene clusters in KO2 (Figures S2B–S2D), again illustrating the phenotypes observed in KO1 are not clonal artifacts.
Interestingly, KAP1 has previously been found to activate a subset of genes in MCF7 breast cancer cells (Li et al., 2017). Upon KAP1 silencing in MCF7 cells, we also observed ≥ 1 log2 fold decreases in Pol II promoter levels and associated expression decreases at genes enriched for processes regulating cancer cell growth, including cell cycle and DNA replication (Figures S2E–S2H and Table S3). Together, these data suggest KAP1 plays an activating role in breast cancer cells by controlling Pol II promoter levels, potentially similar to CRC cells.
The Pol II promoter defects in the absence of KAP1 in various cancer cell types prompted us to examine if KAP1 maintains Pol II promoter levels through direct interaction. Supporting this idea, we observed that the RPB1 subunit of Pol II co-purifies with both ectopically expressed and endogenous KAP1 from MNase-digested nuclear extracts (Figures 2C and 2D). Strikingly, we observed an in vitro interaction between purified components (Figures 2E and S2I), demonstrating KAP1 directly recognizes core Pol II. Collectively, the biochemical and genetic evidence signifies an undescribed role for KAP1 in mediating transcriptional activation by directly associating with Pol II.
KAP1 Regulates CDK9-dependent Pol II Pause Release
Given that Pol II promoter-proximal levels are not completely diminished in KAP1 depleted cells and because KAP1 regulates pause release, we reasoned that the Pol II that still occupies promoters without KAP1 may display pause-release defects. To test this, we first determined if KAP1 target genes contain paused Pol II by calculating the ratio of promoter to gene body Pol II signal, known as “Pausing Index (PI)”, for each gene cluster in Ctrl cells. This analysis revealed that C2 genes displayed higher levels of pausing (median PI ≈ 4) compared to C1 and C3 (median PI < 2) (Figure S2J), consistent with the more “focused” Pol II promoter-proximal profile in C2 genes.
Since genes in all three clusters displayed varying decreases in promoter and gene body Pol II, we compared the PI in both Ctrl and KO1 cells and observed an increase in PI for ~70% of C1 genes (mean log2(fold change) = 1.23) and ~50% of C3 genes (mean log2(fold change) = 0.957) (Figure 2F), indicating that a large fraction of the residually bound promoter-proximal Pol II in KO1 cannot efficiently escape into elongation. In contrast, we observed a PI decrease for ~70% of C2 genes (mean log2(fold change) = 3.04), suggesting C2 genes predominantly display Pol II promoter-proximal level defects. Interestingly, decreased paused Pol II at promoter-proximal regions of all three gene clusters mirrors the loss of negative transcription elongation factors involved in the establishment of Pol II pausing (NELFE) (Figures 2A and S2A), consistent with the idea that proper establishment of pausing is critical to maintain Pol II promoter stability (Henriques et al., 2013).
To start defining the mechanisms by which KAP1 selectively controls levels of promoter-proximal and -distal Pol II, we focused on the role of Pol II site-specific phosphorylation. Specifically, phosphorylation of Ser5 (S5P) in the RPB1 subunit occurs shortly after initiation but prior to pause-release, while phosphorylation of Ser2 (S2P) marks elongating Pol II (Eick and Geyer, 2013). Using previously validated phospho-specific antibodies (Chapman et al., 2007) we observed decreases in both S5P and S2P Pol II at promoter-proximal and -distal regions across all three gene clusters upon KAP1 loss (Figures 2A and 2B and S2A). Interestingly, we observed large decreases in the ratio of S2P to total Pol II signals in the C1 and C3 genes (mean log2(fold change) = 1 and 0.5, respectively) and a ~2-fold increase for the C2 genes in KO1 compared to Ctrl cells (Figure 2G). Additionally, cumulative distribution plots illustrate that the decrease in the S2P/total Pol II promoter-proximal ratio occurs at 80%, 40%, and ~90% of C1, C2, and C3 genes, respectively (Figure 2H). Together, these data suggest that losses in total Pol II do not account for all S2P losses and further demonstrates that, in addition to Pol II promoter level reductions, loss of KAP1 yields pause-release defects, at least at C1 and C3 genes. Of note, the Pol II changes in the gene body of C2 genes may not be readily apparent due to the low Pol II levels relative to other clusters (Figure 2B), thereby limiting our measurements. Importantly, the described Pol II phenotypes are also evident from genome browser signal tracks (Figure 2I).
Given the observed decreases in S2P Pol II at KAP1 target gene promoters upon KAP1 loss, the previously reported functional interplay between KAP1 and 7SK-bound P-TEFb, and P-TEFb’s role in depositing S2P, we explored if CDK9 recruitment to KAP1 target gene promoters is also defective upon KAP1 loss. Indeed, CDK9 promoter occupancy at downregulated genes decreased in KO1 compared to Ctrl cells (average of ~8-fold) (Figures 2A and S2A), thus demonstrating that S2P Pol II is dependent on KAP1’s ability to facilitate CDK9 promoter recruitment for gene activation. Furthermore, KAP1 target gene promoters are occupied by 7SK snRNP (Figures S2K and S2L), supporting previously proposed models that KAP1 “pre-loads” target genes with 7SK-bound P-TEFb (McNamara et al., 2016).
The dual decrease of CDK9 and S2P Pol II at gene promoters upon KAP1 loss prompted us to examine if KAP1 target genes are CDK9-dependent as originally predicted. Indeed, inducible CDK9 silencing and short-term inhibition with flavopiridol dampened expression of KAP1 target genes (Figures 2J–2M). Overall, these data support a model in which KAP1 contributes to CDK9-dependent activation of a select gene class by directly engaging Pol II to regulate both Pol II promoter levels and pause release.
KAP1 Stimulates Pol II Elongation of a Select Subset of Genes Through Recruitment of a Pathway-Specific Pause-release Factor
Given the various degrees of Pol II promoter loss observed across the three gene clusters upon KAP1 loss in both KO clones (Figures 2A and S2B), we performed KEGG pathway analysis for each gene cluster separately to test if they enriched for different functional processes. Interestingly, we found enrichment of several distinct pathways such as RAS, TGF-β, and WNT (C1), mTOR (C2), and metabolic processes (C3) in the KO1 (Figures 3A–C), KO2 (data not shown), and after acute KAP1 silencing (Figures S3A–C), indicating that the mode of KAP1 regulation of Pol II promoter occupancy and activity may differ at groups of genes controlling different functional processes.
Figure 3. KAP1 Recruits a Pathway Specific Factor to Stimulate Pol II Pause-release.
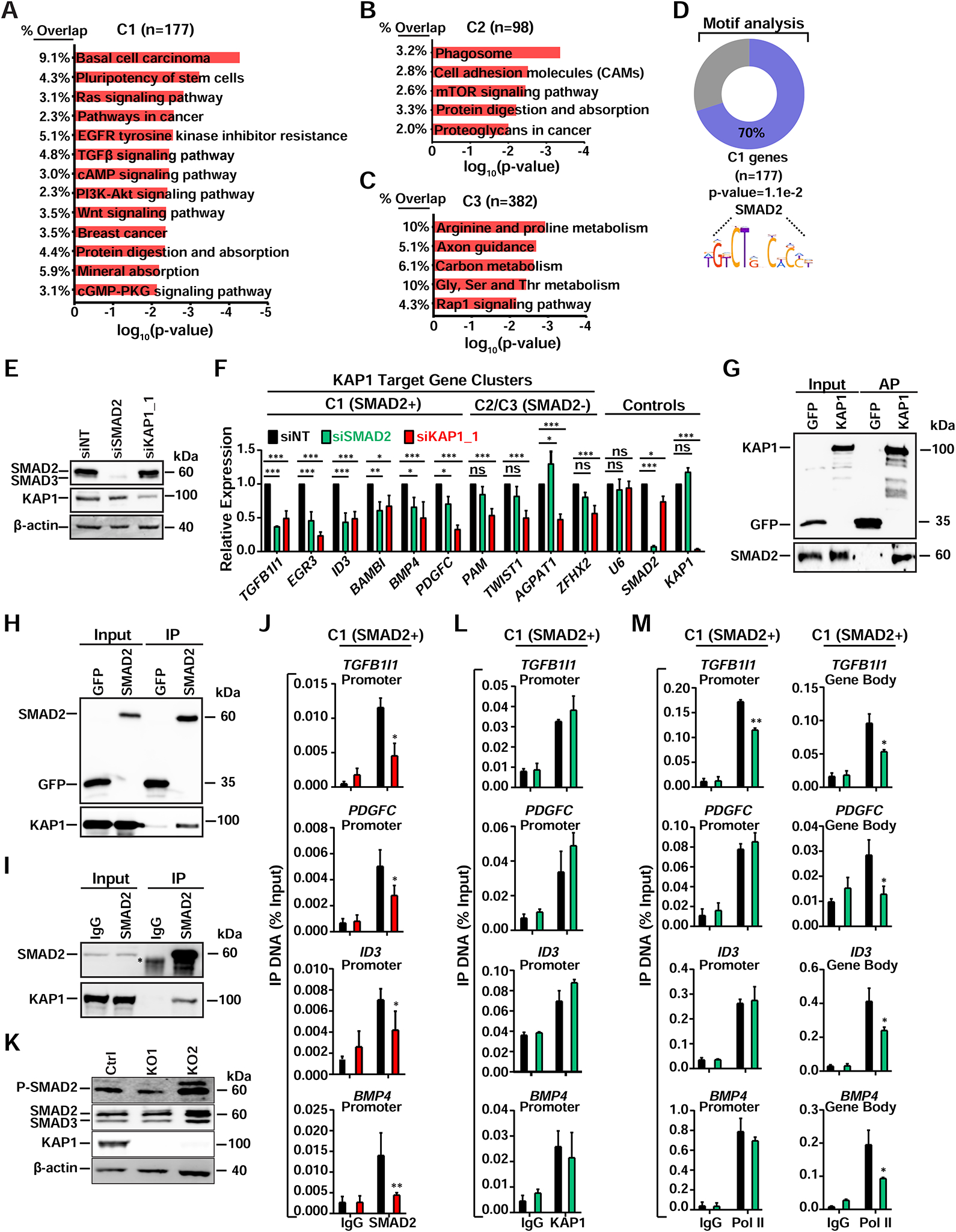
(A–C) KEGG pathway analysis for the three gene clusters. Pathways with P<0.01 are shown.
(D) Enriched SMAD2 DNA motifs in promoter regions for C1 genes.
(E) Western blot validating KD efficiency in HCT116.
(F) Expression levels of genes after RNAi in HCT116 (mean expression relative to siNT ± SEM; normalized to RPL19; n=3).
(G) STREP AP of ectopically expressed STREP-tagged KAP1 (same AP from Figure 2C)
(H) FLAG IP of ectopically expressed FLAG-tagged SMAD2 from HCT116 nuclear extract.
(I) Endogenous IP of SMAD2 from HCT116 nuclear extracts. *IgG.
(J) ChIP-qPCR of SMAD2 at the indicated gene promoters (mean ± SEM; n=3).
(K) Western blots of the indicated factors.
(L) ChIP-qPCR of KAP1 at the indicated gene promoters (mean ± SEM; n=2).
(M) ChIP-qPCR of Pol II at the indicated gene regions (mean ± SEM; n=2).
Notably, aberrant, chronic signaling of several of the pathways found in C1 genes for both the KAP1 KO and KD systems (RAS, TGF-β, WNT) are known to promote CRC cell growth, dictate poor patient prognosis (Calon et al., 2012; Cancer Genome Atlas, 2012; Massague, 2008; Saxton and Sabatini, 2017), and is in agreement with sustained TGF-β signaling in several CRC cell lines, including HCT116 (Berg et al., 2017). Additionally, the observations that KAP1 sustains the transcription of gene programs conferring a cell growth advantage are consistent with the growth defects in cells lacking KAP1 (Figures 1B–E), highlighting KAP1’s oncogenic transcriptional potential.
To determine if the KAP1 target gene signature was clinically relevant, we mined datasets from The Cancer Genome Atlas (TCGA) which revealed KAP1 is upregulated in tumor relative to matched non-tumor tissue samples in most cancer types, including CRC (Figures S3D and S3E; Tables S4 and S5). Furthermore, higher expression of 49 of the 177 C1 genes correlates with poor prognosis among patients with CRC (either shorter overall or disease-free, survival), 39 of 177 C1 genes upregulated in CRC patients positively correlate with KAP1 expression and enrich for processes similar to those in Figure 3A, and less than 5% of C1 genes are deleted in more than 2% of CRC patient tumors, signifying they are regulated transcriptionally, and potentially in a KAP1-dependent fashion (Figures S3F–S3I; Tables S4 and S5).
Since KAP1 target genes are enriched for signal-regulated responses, we reasoned that KAP1 might regulate these programs through a coactivator function with pathway-/sequence-specific TFs. Therefore, we performed motif analysis (±100 bp relative to the TSS) and found that ~70% of C1 genes (but not C2 or C3) contain binding motifs for two TFs at their promoters: SMAD2 (Figure 3D) and KLF12. Additionally, this search did not retrieve any enriched motifs for C2 genes and a lower enrichment (~6.5%) for ZNF331 in C3 genes. Since KLF12 is a known transcriptional repressor (Imhof et al., 1999), we instead focused our attention on SMAD2 because it is known to activate many of the signal-regulated transcriptional programs enriched in C1 genes (Figure 3A) (Massague, 2012). Based on these findings, we refer to C1 genes as “C1 (SMAD2+)” and C2/C3 genes as “C2/C3 (SMAD2-)”.
In order to determine if SMAD2 co-activates C1 (SMAD2+) genes, we silenced SMAD2, and KAP1 as positive control, from HCT116 using RNAi, which did not affect their reciprocal protein levels (Figure 3E), and observed that while genes from all three clusters were sensitive to KAP1 loss, only C1 (SMAD2+) genes were co-dependent on SMAD2 and KAP1 (Figure 3F), demonstrating that SMAD2 acts as a specific activator for KAP1 target genes containing SMAD2 motifs at their promoters.
Interestingly, we found KAP1 is necessary for basal expression and induction of TGF-β/SMAD2 regulated target genes in non-cancerous, Human Colonic Epithelial Cells (HCEC) after TGF-β1 stimulation (Figures S3J and S3K). Importantly, KAP1 is not necessary for SMAD2 S456/467 phosphorylation (P-SMAD2), which is critical for SMAD2 cytoplasmic-nuclear translocation and promoter binding in response to cognate signaling for activation of the TGF-β/SMAD axis (Massague, 2012) (Figure S3J), suggesting KAP1 is required for activation of SMAD2-regulated genes in response to physiologic ligands. Consistent with a potential KAP1-SMAD2 regulatory axis, we observed interactions between ectopically expressed and endogenous proteins (Figures 3G–3I).
In agreement with the interaction data, loss of KAP1 partially (TGFB1I1, PDGFC, ID3) or completely (BMP4) reduced (~1.5–3–fold) SMAD2 occupancy at C1 (SMAD2+) gene promoters (Figure 3J), while loss of SMAD2 did not affect KAP1 recruitment (Figure 3L), demonstrating a key role for KAP1 in facilitating SMAD2 recruitment to activate signal-regulated transcriptional programs. Importantly, C1 (SMAD2+), but not C2/C3 (SMAD2-), genes are occupied by SMAD2 in HCT116 Ctrl, KO1, and parental cells (Figures S3L and S3M) confirming the SMAD2 ChIP data is specific. Additionally, loss of KAP1 in CRC cells does not decrease the cell-intrinsic TGF-β signaling that activates SMAD2 (Figure 3K), suggesting KAP1 regulates SMAD2 function(s) on chromatin in both normal and cancer cells.
Since SMAD2 depletion does not dampen KAP1 levels at C1 (SMAD2+) gene promoters, and because KAP1 is found in complex with Pol II and required for its promoter occupancy (Figures 2A–E), we predicted that SMAD2 would promote Pol II pause release rather than promoter recruitment. Supporting this model, while Pol II promoter-proximal levels remain constant at 3 of the 4 genes tested upon SMAD2 depletion, promoter-distal levels decrease (~2-fold) at all 4 genes (Figure 3M), indicating SMAD2 mainly functions through a pause-release mechanism.
Collectively, these data not only demonstrate that KAP1 interacts with, and assists in, SMAD2 recruitment to target gene promoters but also identify a previously uncharacterized role for SMAD2 in facilitating Pol II pause release in response to oncogenic signaling.
The KAP1 Chromatin Reader Cassette Directly Recognizes Hypo-acetylated H4 Tails
Since KAP1 occupies target gene promoters independently of SMAD2, we aimed at defining the mechanism of KAP1 promoter recruitment to coordinate establishment of promoter-bound Pol II and the SMAD2-mediated, CDK9-dependent switch to pause release. Intriguingly, the role of KAP1’s tandem plant homeodomain (PHD)-bromodomain (BD) cassette in chromatin binding and transcription activation has not been previously elucidated. Because PHD and BD domains in chromatin readers typically interact with histone tails, we asked whether the KAP1 PHD-BD does so directly to mediate gene activation (Allis and Jenuwein, 2016; Taverna et al., 2007). To test this idea, we performed in vitro pull-down assays with mono-nucleosomes and found that GST-tagged KAP1PHD-BD (Figure S4A), but not GST, indeed binds mono-nucleosomes (Figure 4A) and contacts the H4 tail with high specificity (Figure 4B). Furthermore, the PHD alone is sufficient for H4 recognition (Figures 4B and S4A).
Figure 4. The KAP1 Chromatin Reader Cassette Directly Recognizes Hypo-acetylated H4 Tails.
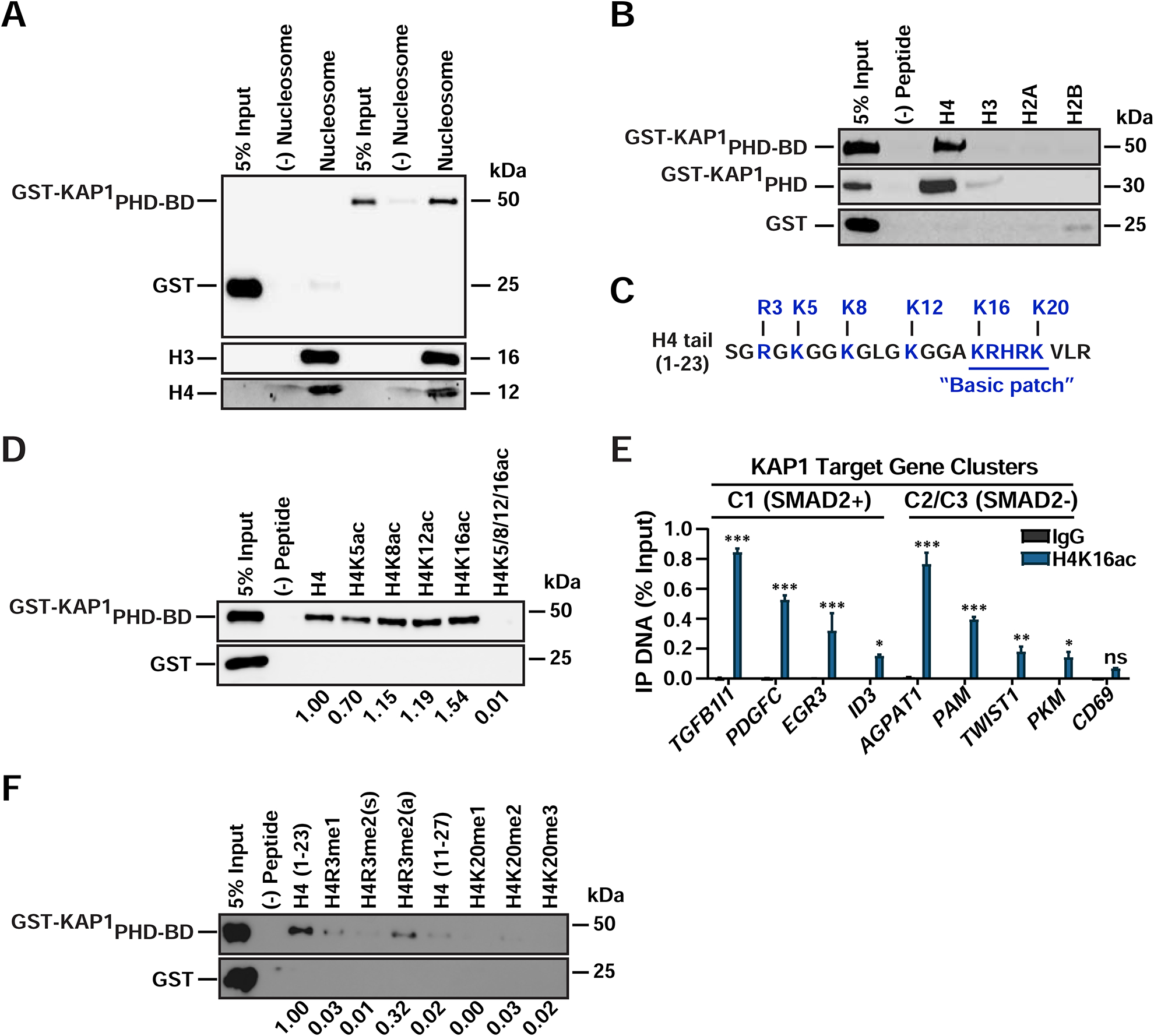
(A-B) In vitro binding assay between recombinant proteins and (A) biotinylated mono-nucleosomes or (B) biotinylated histone peptides.
(C) H4 tail sequence with commonly modified residues highlighted.
(D) In vitro binding assay between recombinant proteins and acetylated H4 peptides.
(E) ChIP-qPCR of H4K16ac in HCT116 (mean ± SEM; n=3).
(F) In vitro binding assay between recombinant proteins and methylated H4 histone peptides. For panels (D) and (F) quantifications relative to unmodified H4 are shown. All western blots were probed with anti-GST.
Histone tails are post-translationally modified to establish regulatory codes that facilitate or prevent factor-chromatin interactions (Allis and Jenuwein, 2016). Therefore, we next asked whether H4 tail residues that can be modified (Figure 4C) could function as a histone code to enhance and/or regulate KAP1 binding strength. Unexpectedly, we observed similar PHD-BD binding levels to H4 and hypo-acetylated H4 peptides containing a single acetylated Lys (K5, K8, K12, or K16), although binding to H4K5ac was ~30% lower, indicating KAP1 tolerates the presence of a single acetylated Lys (Figure 4D). However, H4 hyper-acetylation virtually abolished PHD-BD binding, consistent with the idea that the BD does not contain the required pocket typically involved in binding acetylated Lys in histone tails (e.g., in BRD4) (Jung et al., 2014; Zeng et al., 2008), potentially indicating that it may not contribute to H4 binding. However, because the recombinant BD was unstable in solution, we were unable to assess if the BD alone bound hypo- or hyper-acetylated H4 peptides.
H4 acetylation is strongly associated with gene activation (Allis and Jenuwein, 2016). Given the unexpected KAP1-binding mode to both unmodified and hypo-acetylated H4 tails, and because H4 may not be found unmodified at KAP1 target genes, we predicted that these promoters should contain some level of acetylation. Indeed, we found that H4K16ac, which is known to promote chromatin decompaction and transcriptional activation (Shogren-Knaak et al., 2006), is present at KAP1 target gene promoters, while a non-expressed gene in HCT116 (CD69) contains ~2–8—fold less (Figure 4E). These data further illustrate that even though H4 hypo-acetylation alone does not increase KAP1’s binding affinity, KAP1 is still able to occupy gene promoters containing some level of H4 acetylation.
PHDs often show increased affinity towards methylated Lys or Arg (Musselman et al., 2012). Surprisingly, we observed an ~70–100% decrease in PHD-BD binding to all methylated H4 peptides including R3 mono- and di-methylation (symmetric and asymmetric) and K20 mono-, di-, tri-methylation (Figure 4F), indicating that KAP1 cannot accommodate any methyl marks on the H4 tail. These results are consistent with data suggesting that the KAP1 PHD is atypical given it lacks the aromatic cage required for methylated Lys recognition (Li et al., 2006; Pena et al., 2006).
KAP1 belongs to the TIF1 protein family, which contains two additional members whose tandem PHD-BD domains recognize modified H3 peptides (TIF1α binds H3K4me0/K23ac and TIF1γ binds H3K4me0/K9me3/K18ac (Agricola et al., 2011; Tsai et al., 2010; Xi et al., 2011)), and thus we asked whether KAP1 interacts with these marks. Consistent with the H4 tail binding data, KAP1 did not interact with the marks recognized by TIF1α and TIF1γ (Figures S4B–S4D); however, despite several attempts we could not reproduce the previously reported interaction between TIF1γ and its H3 substrate. Consistent with these findings, residues required for TIF1α’s (F979 and N980) and TIF1γ’s (W889 and E981) interaction with histone tails are either shifted or substituted with other residues in KAP1 PHD-BD (Figure S4E). These observations support the model that the KAP1 PHD-BD cassette interacts with histone tails in a unique way that is divergent from the binding mode of other TIF1 family members.
Direct H4 Tail Binding is Necessary for KAP1 Promoter Occupancy and Target Gene Activation
To further delineate the KAP1-H4 interaction, we titrated the H4 peptide to 15N-uniformly labeled KAP1PHD-BD and monitored chemical shift perturbations in the protein by NMR spectroscopy. Notably, we observed changes in backbone amides of several PHD residues (e.g., G661 and W664) (Figures 5A and 5B). Interestingly, using the previously determined KAP1PHD-BD structure (Zeng et al., 2008) and our NMR chemical shifts as restraints, we performed molecular dynamics (MD) simulations which predicted strong interactions between negatively charged residues (E662 and E663) in the PHD and several basic residues in the H4 tail basic patch (R17-K20) (Figures 5C and S5A). Furthermore, after mapping the NMR chemical shift perturbations onto the predicted KAP1-H4 model, we found that the residues most perturbed in the NMR titration experiment (G661, W664, and S665) surround the residues (E662/E663) predicted to interact with H4 (Figure 5C), highlighting the importance of this region in the PHD for H4 tail recognition. Remarkably, the PHD predicted residues (N641, D658, E662, E663) are evolutionarily conserved among mammals, and one of them (E662) is identical to an evolutionary distant ortholog in Drosophila (Figure S5B), suggesting that the KAP1-chromatin interaction may be critical for KAP1 function across species.
Figure 5. Direct H4 Tail Binding is Necessary for KAP1 Promoter Occupancy and Target Gene Activation.
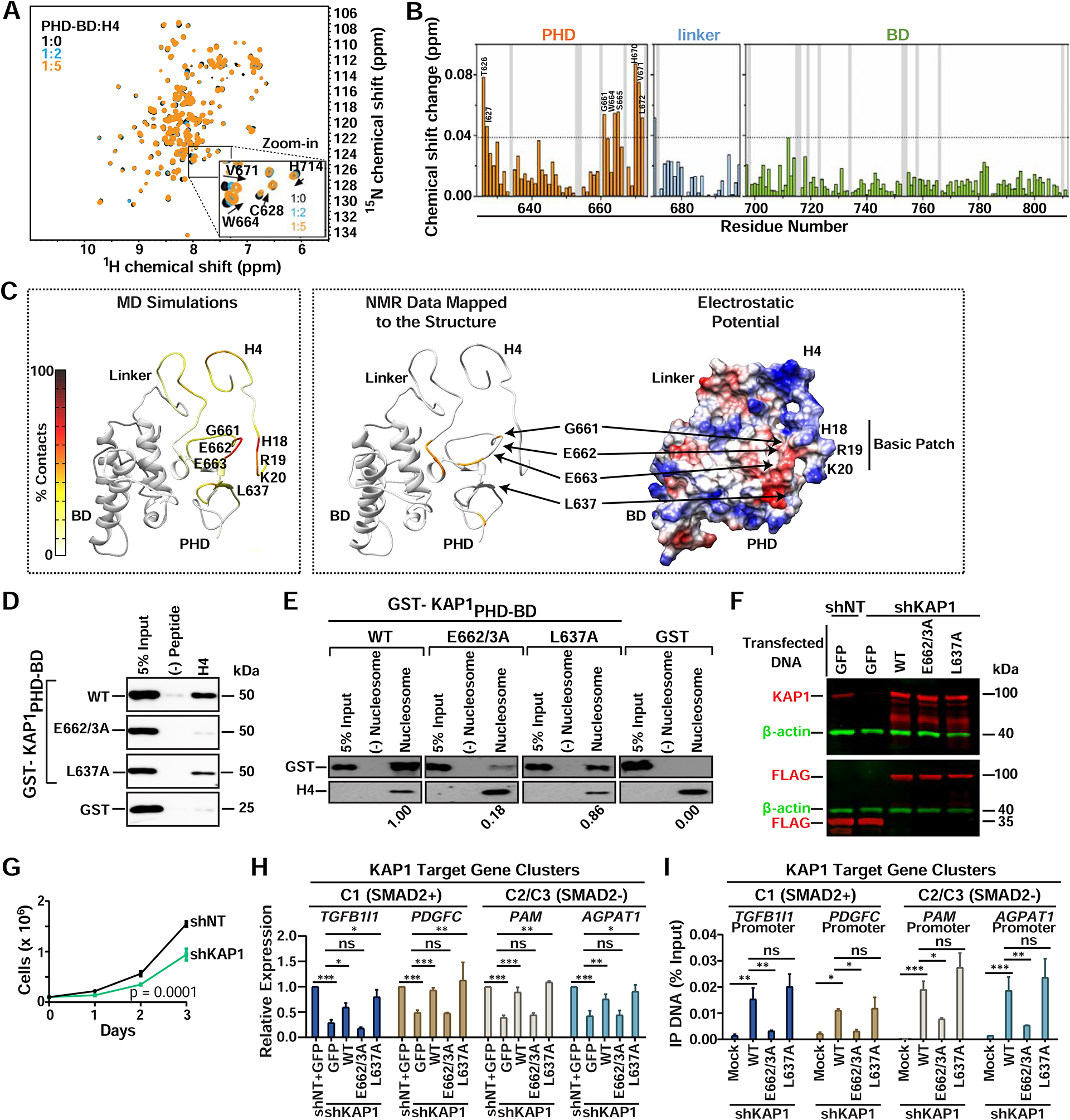
(A) The 1H, 15N HSQC spectra of KAP1PHD-BD collected upon titration with H4 peptide. Inset shows residues displaying chemical shifts. Spectra are color-coded according to the PHD-BD:H4 molar ratio.
(B) Histogram showing chemical shift perturbations. Vertical grey bars indicate Pro and unmapped residues. The dash line indicates a threshold value of average + 3×SD. Chemical shift assignments of the apo-PHD-BD state were from BMRB (ID 11036).
(C) Left: Predicted model depicting the frequency of residue specific contacts between KAP1PHD and H4 in the production run of the MD simulations using backbone amide resonances in panel (A) as restraints. Middle: Residues exhibiting H4-induced resonance perturbations above threshold are mapped in orange onto the structure of the predicted complex. Right: The electrostatic surface potential of the predicted complex is colored blue and red for positive and negative charges, respectively.
(D-E) In vitro binding assay between recombinant proteins and (D) H4 peptide or (E) biotinylated mono-nucleosomes. Western blots probed with anti-GST. Quantitation relative to WT KAP1 of (E) are shown.
(F) Western blot of KAP1 proteins in the indicated HCT116 cell lines.
(G) Cell growth assay (cell counts ± SEM; n=3).
(H) RT-qPCR of KAP1 target genes after reconstitution of the indicated proteins in shKAP1 HCT116 (mean expression relative to shNT+GFP ± SEM; normalized to RPL19; n=3).
(I) FLAG ChIP-qPCR of the indicated proteins at KAP1 target gene promoters (mean % Input DNA ± SEM; n=3).
In light of the predicted KAP1-H4 binding model, we mutated E662/E663 to perform binding assays, and observed that the E662/3A mutation virtually abolished H4 peptide binding and largely (~80%) reduced KAP1’s ability to interact with mono-nucleosomes (Figures 5D, 5E, and S5C). Importantly, mutation of a control residue (L637) located in the flexible loop between the PHD and BD, which does not disrupt PHD folding (Capili et al., 2001), is not perturbed in the NMR titrations and is not predicted to interact with H4 (Figures 5B and 5C), retains H4 and mono-nucleosome binding activity (Figures 5D and 5E). Interestingly, the fact that E662/3A did not completely abolish mono-nucleosome binding suggests that there may be additional contacts between KAP1 and other parts of the nucleosome (e.g., DNA) that contribute to KAP1’s binding specificity and/or affinity.
To test whether the KAP1-H4 interaction is functionally linked to KAP1 target gene activation, we reconstituted KAP1-depleted cells with FLAG-tagged wild-type (WT) KAP1 and point mutants. Because the KO1 cells were highly resistant to plasmid DNA transfection compared to Ctrl cells (data not shown), we performed the reconstitution assays in HCT116 cells stably expressing a short-hairpin RNA targeting the 3’-UTR of KAP1 (shKAP1) or a non-targeting shRNA (shNT) as control (Figure 5F). Importantly, shKAP1 cells displayed both the expected growth defect (Figure 5G) and decreases in target gene expression (Figure 5H) consistent with phenotypic and genetic data upon KAP1 KO and acute KD (Figure 1), thus strongly indicating the shRNA system can be used for KAP1 reconstitution purposes to study KAP1 chromatin interactions and gene activation.
Remarkably, and in agreement with the in vitro H4 peptide-binding assay, WT KAP1 and L637A, but not E662/3A, restored KAP1 target gene expression in shKAP1 cells (Figure 5H) signifying that KAP1 binding to histones is required for KAP1’s transcription activation function. Additionally, ChIP-qPCR assays revealed that WT KAP1 and L637A, but not E662/3A, localized to all tested gene promoters (Figure 5I), highlighting the importance of the KAP1-H4 interaction for KAP1 promoter occupancy and subsequent gene activation.
Given the H4 interaction was required for both KAP1 promoter occupancy and gene activation, one possible model for KAP1-mediated maintenance of Pol II promoter levels (Figure 2A) would be that KAP1 binds chromatin to maintain the nucleosome-free region (NFR) around the gene TSS. To test this idea, we performed MNase-seq in Ctrl and KO1 cells and found that the NFR width (distance between nuc−1 to nuc+1) and nucleosome array spacing (nucleosome repeat length) at all downregulated genes did not apparently change upon KAP1 loss, suggesting that promoter nucleosome positions remain virtually identical (Figures S5D–S5I). The slight decrease (median ~20%) in nucleosome density at most downregulated genes in all three gene clusters (Figures S5G and S5I) does not explain decreased transcription given that this is a phenomenon also observed in most upregulated genes (data not shown). Thus, while the chromatin-binding activity tethers KAP1 to target promoters to stimulate gene transcription, this function does not appear to be required to maintain nucleosome positioning and promoter accessibility at target genes. These observations, along with the physical association between KAP1 and Pol II, and the decreased Pol II loading at target genes upon KAP1 loss, strongly suggest that KAP1’s chromatin reader activity regulates Pol II function.
Collectively, we have identified a previously uncharacterized function for the KAP1 chromatin reader cassette, which tethers KAP1 to promoters through a direct interaction between its PHD and hypo-acetylated H4 tails, thereby facilitating KAP1-mediated transcriptional activation. While this data does not fully address how KAP1 promoter specificity is achieved, it does reveal an undescribed chromatin interaction that is critical for KAP1 to activate gene transcription through coordination of Pol II early and late steps in the transcription cycle.
Upon Chromatin Binding KAP1 Scaffolds Pol II and Pause-release Factors at Select Gene Promoters Through Different Protein Domains
In addition to the chromatin binding and target gene activation role of the C-terminal PHD-BD cassette, KAP1 contains several domains that could engage in multivalent interactions including a RING E3 ubiquitin ligase domain (Doyle et al., 2010), two B-boxes and a coiled-coil (CC) required for protein-protein interactions and dimerization (Fonti et al., 2019; Peng et al., 2000), a linker involved in heterochromatin protein (HP1) binding (Ryan et al., 1999) and the PHD-BD (Figure 6A).
Figure 6. The KAP1 Chromatin Reader Function is Necessary to Scaffold Pol II and Pause-release Factors at Promoters Through Different Protein Domains.
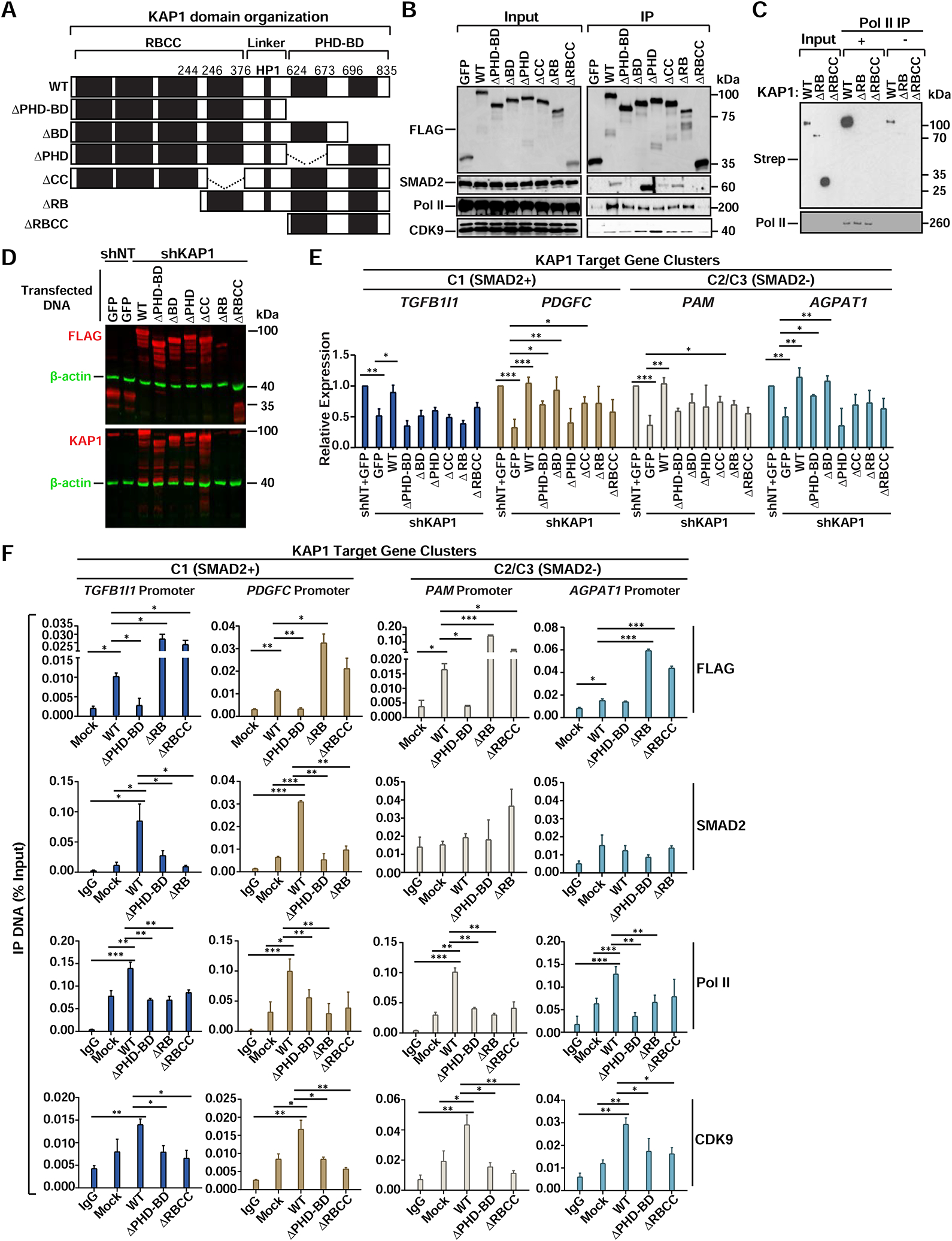
(A) FLAG-tagged KAP1 constructs.
(B) Co-IP of FLAG-tagged KAP1 constructs from HCT116 nuclear extracts.
(C) In vitro binding assay between core Pol II and KAP1 constructs. Pol II (RPB1).
(D) Expression of FLAG-tagged KAP1 constructs in shKAP1 HCT116. Note that N-terminal deletion constructs are not recognized by the KAP1 antibody.
(E) Expression levels of KAP1 target genes after reconstitution of HCT116 shKAP1 cells with KAP1 constructs (mean expression relative to shNT+GFP ± SEM; normalized to RPL19; n=3).
(F) ChIP-qPCR of the indicated factors at KAP1 target gene promoters after reconstitution of shKAP1 HCT116 with the indicated constructs (mean % Input DNA ± SEM; n=3).
To test if KAP1 uses its modular organization to first bind chromatin and then engage in multivalent interactions with Pol II, SMAD2, and CDK9, we built FLAG-tagged KAP1 deletion constructs and performed co-IPs to identify the necessary binding surfaces. While WT KAP1 associates with all three factors (Figure 6B), ΔRB displays an ~80% and ~30% decrease in SMAD2 and Pol II binding, respectively, suggesting the RING–B-box domain is most critical for SMAD2 association and contributes to Pol II association. Intriguingly, ΔBD showed increased (~2–3—fold) SMAD2 interaction, indicating a possible regulatory role of the BD in preventing SMAD2 recognition or timing its recruitment to chromatin. Additionally, ΔRBCC could not interact with any of the three factors, suggesting the linker is the binding surface for Pol II and CDK9. Finally, in vitro Pol II binding assays with purified WT KAP1 and mutants further confirmed the inability of ΔRB and ΔRBCC to directly engage Pol II (Figures 6C, S6, and S2I).
While the above data provided evidence of a domain-specific requirement for co-factor interaction, it did not inform about domain-specific function in gene activation. Reconstitution of KAP1 and deletions in shKAP1 cells (Figure 6D), revealed that KAP1 restored expression of all tested genes (~2-fold over GFP-expressing cells), while ΔPHD-BD and ΔPHD either completely (TGFB1I1 and PAM) or partially (PDGFC and AGPAT1) failed to restore target gene expression (Figure 6E), consistent with the newly identified role for the PHD in H4 tail binding. However, the ΔBD restored expression of 2 out of the 4 genes tested (PDGFC and AGPAT1), suggesting that in some circumstances the BD is dispensable for KAP1 activation. Additionally, the ΔCC, ΔRB, and ΔRBCC constructs failed to fully restore both C1 (SMAD2+) and C2/C3 (SMAD2-) gene expression (Figure 6E), consistent with the ΔRB and ΔRBCC’s inability to interact with Pol II and SMAD2 (Figure 6B). While ΔRB and ΔRBCC express ~35% less compared to KAP1 this does not appear to affect their promoter-bound levels at the tested target genes (Figure 6F).
Consistent with the above data, ChIP-qPCR assays revealed that reconstituted KAP1 was able to localize to both C1 (SMAD2+) and C2/C3 (SMAD2-) gene promoters to facilitate recruitment of Pol II and CDK9 (Figure 6F). However, KAP1 only facilitated SMAD2 recruitment to C1 (SMAD2+) gene promoters, confirming C2/C3 (SMAD2-) genes are regulated in a SMAD2-independent manner. In contrast to KAP1, ΔPHD-BD displayed compromised promoter interaction at all genes tested (except for AGPAT1), thereby virtually abolishing promoter recruitment of Pol II, SMAD2, and CDK9, consistent with decreased gene activation and the PHD’s role in binding H4. Intriguingly, ΔRB was able to interact with gene promoters, due to the intact PHD-BD cassette, but at levels even higher (~2–10—fold) than KAP1, suggesting that the N-terminal domains (RING and/or B-boxes) could regulate KAP1 promoter occupancy; however, it did not facilitate Pol II nor SMAD2 promoter recruitment, consistent with compromised Pol II and SMAD2 binding as well as its decreased ability to activate gene expression. Finally, like ΔRB, ΔRBCC was able to localize to all tested KAP1 target gene promoters above KAP1 levels; however, it failed to facilitate Pol II and CDK9 promoter recruitment, consistent with its inability to bind these factors and to activate gene expression.
Collectively, KAP1 requires its direct chromatin binding activity to occupy target gene promoters and its multidomain organization to engage in multivalent factor interactions thereby coordinating the assembly of a pathway-specific TF complex to couple the establishment of promoter-bound Pol II and pause release.
DISCUSSION
Current models of transcriptional regulation posit that there are separate factors controlling Pol II promoter recruitment, establishment of pausing, and pause release (Bacon and D’Orso, 2018; Buratowski, 2012; Chen et al., 2018; Rahl et al., 2010). Here we provide evidence that KAP1 links the establishment of Pol II promoter occupancy with pause release, a mechanism reminiscent of the extensive coupling of steps in the eukaryotic gene expression cycle (Maniatis and Reed, 2002). These results lead to an “integrated model” of transcriptional control. First, establishment of the nucleosome promoter landscape occurs in a KAP1-independent manner (Figure 7A). Second, KAP1 occupies target promoters through direct interactions between its PHD and hypo-acetylated H4 tails (Figure 7B), then directly engages with, and facilitates, Pol II promoter recruitment and/or stabilizes promoter-engaged Pol II thereby establishing pausing (Figure 7C). KAP1 also tethers CDK9, presumably as part of the 7SK snRNP, suggesting Pol II and CDK9 recruitment to gene promoters may be temporally linked, consistent with previous findings (D’Orso and Frankel, 2010). Finally, KAP1 recruits SMAD2 in the presence of constitutive TGF-β signaling to induce selective CDK9-dependent pause release at SMAD2-regulated genes (Figure 7D).
Figure 7. KAP1 Couples Establishment of Pol II Promoter Levels with Pause-release to Sustain Oncogenic Transcriptional Programs.
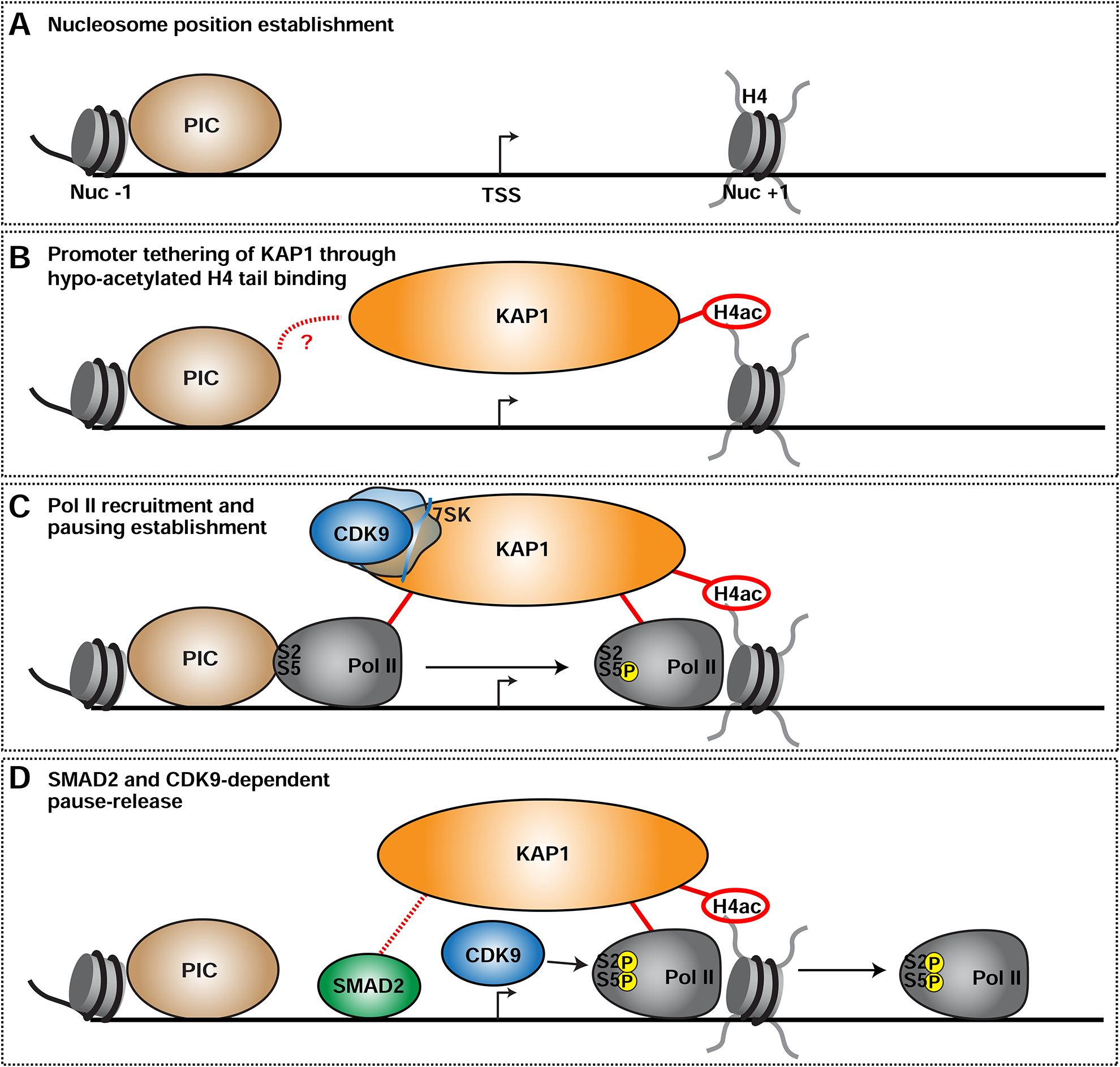
(A) Establishment of the NFR around promoters.
(B) KAP1 is tethered to gene promoters through direct interactions with the hypo-acetylated H4 tail. Specific promoter recognition may occur through yet unknown interactions with other TFs such as PIC subunits. See Discussion for complete details.
(C) Upon promoter binding, KAP1 directly stimulates Pol II recruitment and/or pausing and tethers 7SK-bound CDK9.
(D) In response to cognate signals (TGF-β), KAP1 recruits a pathway-specific TF (SMAD2) to select gene promoters thereby promoting CDK9 activation and Pol II pause-release.
Previous observations are consistent with the proposed “integrated model” including: (i) the block of new initiation by paused Pol II (Gressel et al., 2017; Shao and Zeitlinger, 2017), suggesting that pause release may augment initiation frequency to allow for the coordinated recruitment of additional Pol II molecules; (ii) the dynamic behavior of Pol II pausing, indicating that Pol II molecules recruited to a large proportion of promoters undergo abortive termination within minutes (Chen et al., 2015; Erickson et al., 2018; Shao and Zeitlinger, 2017); and (iii) the idea that Pol II promoter stability is decreased when pausing is not properly established (Henriques et al., 2013). Together, we envision KAP1 regulates a switch from Pol II promoter recruitment and/or establishment of transiently paused Pol II to pause release, thereby preventing premature termination, consistent with previous ideas (Core et al., 2012; Gilchrist et al., 2010; Gilchrist et al., 2008; Williams et al., 2015).
In our “integrated model”, KAP1 pathway selectivity is initiated by cell-intrinsic events that culminate with the activity of the cognate, sequence-specific TF complex becoming activated in response to the signaling cascade. Consistent with our model, KAP1 maintains the tolerogenic potential of CD4+ T regulatory cells through interactions with the master regulator FOXP3 in response to unknown ligands (Tanaka et al., 2018), and helps translate a diverse range of physiologic inputs, such as pro-inflammatory cytokines (McNamara et al., 2016) and androgen receptor agonists (Fong et al., 2018), into select transcriptional outputs.
Surprisingly, SMAD2 is dispensable for the early phases of the Pol II cycle (namely promoter recruitment and/or establishment of pausing) at least at the genes interrogated. These results were unanticipated given that SMAD2 was proposed to be required for Pol II promoter recruitment after nucleosome displacement and chromatin remodeling (Hill, 2016; Ross et al., 2006). The indication that SMAD2 activates transcription combinatorially through lineage-specific TFs to elicit select biological responses (David and Massague, 2018; Hill, 2016), suggests that these factors, in addition to KAP1, could be required to poise the promoter landscape for subsequent SMAD2 gene activation.
Strikingly, like KAP1, TIF1γ interacts with and recruits SMAD2 to enhancers of key ES cell differentiation genes in response to nodal TGF-β signals (Xi et al., 2011). Given the discoveries that KAP1-SMAD2 and TIF1γ-SMAD2/3 cooperate for gene activation in different cell types (this study and Xi et al., 2011), and that KAP1-TIF1γ associate (Herquel et al., 2011), it is likely that TIF1 family members cooperate in the normal regulation of SMAD2 target gene expression for controlling unknown physiologic responses and/or to favor tumor progression. The intrinsic dimerization property of TIF1 family members through their coiled-coil domain could thus represent a built-in mode to potentially assemble homo- or hetero-dimers (Peng et al., 2002) thereby expanding the functional repertoire by which these chromatin readers modulate target gene selectivity and/or to enable promoter-enhancer communication in response to physiologic and oncogenic signaling.
The unique histone tail binding activity facilitates KAP1 occupancy at genomic loci thereby enabling target gene activation. While PHDs typically bind modified (e.g., methylated) H3 and H4 tails through aromatic cages and BDs recognize acetylated Lys pockets (Musselman et al., 2012) to regulate unique functions, an acidic patch in KAP1’s PHD engages H4 with no apparent BD contributions, indicating that KAP1 is the first example of a PHD-containing chromatin reader stimulating transcription through the coupling of Pol II regulatory steps.
Given that the interrogated H4 PTMs do not increase KAP1’s binding ability, we propose KAP1 may rely on other critical interactions to increase target gene specificity, such as recognition of unknown histone PTMs, additional heterotypic interactions within the same or different nucleosomes (Ruthenburg et al., 2011), with PIC components, sequence-specific TFs, and/or nucleic acids. The KAP1 chromatin reader domain tolerates hypo-acetylated H4 tails and co-occupies target gene promoters with H4K16ac, suggesting that the KAP1-H4 complex can potentially accommodate unknown H4 interactions to increase the combinatorial potential of much larger chromatin reader complexes that remain to be defined. Moreover, given the inability of KAP1 to bind hyper-acetylated H4, extensive acetylation may function as a switch from KAP1 promoter binding to eviction, which may allow the recruitment of factors required for downstream functions.
Besides directly interacting with chromatin, the KAP1PHD appears to function as an intramolecular E3 sumo ligase of the adjacent BD to enable gene repression (Ivanov et al., 2007). However, given that the only known mutation that abolishes the sumo-ligase activity (C651A) also disrupts metal coordination, PHD folding, and obliterates H4 recognition (Figure S4F), further studies are required to uncouple these two functions to study their contribution to gene activation.
In addition to the future work proposed above, this study opens exciting avenues for other forthcoming investigations. It will be interesting: (i) to interrogate whether the atypical BD establishes additional heterotypic interactions; (ii) to determine the role(s) of the KAP1-HP1 interaction for the various Pol II steps; and (iii) to investigate if KAP1 communicates with specialized PIC components as a way to control pathway-restrained program activation.
Finally, it is possible that several other proteins/complexes use similar mechanisms to control the switch between Pol II initiation, stabilization at the pause site, and pause release. For example, the Mediator complex interacts with transcription initiation and elongation complexes to stimulate gene activation (Takahashi et al., 2011). Notably, our studies provide generalizable principles by which scaffolding factors orchestrate selective transcriptional programs through the coupling of Pol II regulatory steps to ensure the appropriate cellular responses.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Requests for further information and reagents may be directed to the Lead Contact, Dr. Iván D’Orso, at The University of Texas Southwestern Medical Center (ivan.dorso@utsouthwestern.edu)
Materials Availability
Plasmids and cell lines generated in this study may be requested from the lead contact.
Data and Code Availability
Original western blot data has been deposited at Mendeley Data (http://dx.doi.org/10.17632/fc56g3m8ff.1). Raw and analyzed NGS datasets have been deposited at NCBI GEO under accession number GSE132705. Detailed scripts and commands used for all NGS analysis can be found on the D’Orso Lab public GitHub repository (https://git.biohpc.swmed.edu/ivandorsolab/Curtis_Dissertation).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture
HCT116 parental (male colorectal carcinoma), HEK 293T (embryonic kidney cells, gender unknown), and MCF7 (female breast adenocarcinoma) cell lines were obtained from the American Type Culture Collection. HCT116 and 293T cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) (HyClone, SH30022.FS) supplemented with 10% Fetal Bovine Serum (FBS) (Millipore Sigma, H9268) and 1% Penicillin/Streptomycin (MP Biomedicals, 091670049). MCF7 cells were cultured in Minimum Essential Medium (MEM) supplemented with 10% FBS, 1% Penicillin/Streptomycin, and 0.01 mg/mL of insulin from bovine pancreas (Sigma-Aldrich, I6634). HCT116, HEK293T, and MCF7 cells were grown at 37°C with 5% CO2 at an optimal density of 1 × 106 cells/mL. Cells were cultured for 20 passages and then discarded. HCEC 1CT cells were cultured in Primaria flasks (Fisher Scientific, #08-772-45) in basal X media (4 parts of DMEM to 1 part of Medium 199, (HyClone, SH30253.01)) supplemented with 25 ng/mL EGF (PeproTech, AF-100–15), 1 μg/mL hydrocortisone (Sigma, H4001), 10 μg/mL insulin (Sigma, I6634), 2 μg/mL Apo-transferrin (Sigma, T1147), 5 nM sodium selenite (Sigma, S5261), 2% cosmic calf serum (HyClone, SH3008703), and 50 μg/mL gentamicin sulfate (Gemini Bio-Products, 400–108), as previously described (Roig et al., 2010). HCEC cells were incubated in 2% oxygen and 7% carbon dioxide as previously described (Wright and Shay, 2006).
Bacterial Strains
DH5α, STBL3, and BL21 cells were obtained from Thermo Fisher, stored at −80°C, grown in Luria Broth (LB) media at 37°C, and used to propagate plasmid DNAs.
METHOD DETAILS
Western Blot
All western blots were transferred on 0.45 μM nitrocellulose membranes (Bio-Rad, #1620115) using the Bio-Rad Trans-Blot Turbo Transfer System, blocked for 1 hr in 5% Milk + Tris-buffered saline-Tween-20 (TBS-T), probed with primary antibody in 5% non-fat dry milk + TBS-T. The primary antibody concentration and time of incubation is indicated below in the specific method that each antibody was used. Unless otherwise noted, all blots were also probed with either α-mouse-HRP (Cell Signaling Technology, #7076) or α-rabbit-HRP (Cell Signaling Technology, #7074) secondary antibody in 5% non-fat dry milk TBS-T at 1:10,000 antibody dilution for 30 min. Blots were exposed using Clarity Western ECL (Bio-Rad, 1705061). All western blot images were acquired using the Chemidoc (Bio-Rad).
RT-qPCR
RNA was extracted from mammalian cells using the Zymo Quick-RNA MiniPrep Kit (Zymo Research, R1055). RNA was quantified using the DeNovix DS-II FX+ Spectrophotometer. 2 μg of total RNA was incubated with 2.5 μM Oligo dT(18) and 0.5 mM dNTP mix (NEB, N0447L) for 5 min at 70°C. Samples were spun down and immediately placed on ice for 2 min. Next, 1X M-MuLV Reverse Transcriptase buffer (NEB, M02553L) and 10 U/μL of M-MuLV Reverse Transcriptase (NEB, M02553L) was added to each sample and incubated at 42°C for 1 hr. The reaction was inactivated at 70°C for 10 min and samples were then diluted 1:5 with H2O. For qPCR, 5 μL SYBR Green, 1 μL of each primer (10 μM stock), 0.5 μL of cDNA, and 3.5 μL of H2O was used for each per well in a 96-well plate. Samples were amplified 40 cycles using the Applied Biosystems 7500 Fast Real-Time PCR System. All primers used for qPCR analysis are listed in Table S6.
Generation of KO Cell Lines with CRISPR-Cas9
gRNAs targeting exon 1 of KAP1 with BsmB1 overhangs (Table S6) were designed using the CRISPR Design Tool created by the Zhang Lab (http://crispr.mit.edu/) and obtained from Sigma-Aldrich. The lentiCRISPRv2 plasmid encoding SpCas9 (Addgene, 52961) was digested with BsmB1 (NEB, R0580) and the annealed oligos were then inserted by ligation using Quick Ligase (NEB, M2200). The ligated products were then transformed into Stbl3 E. coli and plasmids isolated using the Plasmid MiniPrep Kit (Qiagen, 27104). The plasmids containing the gRNAs were verified by sequencing at the UTSW McDermott Sanger Sequencing Core. HEK 293T were transfected with either the lentiCRISPRv2 containing only Cas9 (Ctrl) or the gRNA-containing lentiCRISPRv2 vectors along with gag/pol (pSPAX, Addgene, 12260) and VSV-G (pMD2.G, Addgene, 12259) for expression of competent lentiviruses. Cell supernatants were collected two days post-transfection. Viral transduction was performed by spinoculation using 2×105 cells, 200 μL of lentivirus at a concentration of 84 ng/mL, 8 ug/mL polybrene (Hexadimethrine Bromide) (Millipore Sigma, H9268), and serum-free DMEM to a final volume of 500 μL at 18°C for 2 hr at 400 g. Transduced cells were selected with puromycin (1 mg/mL) for 3 days post-infection. After puromycin selection, cells were single cell sorted by FACs at the UTSW Flow Cytometry Core into 96-well plates with 100 μL of DMEM + 20% FBS. Cells were expanded to 48-well plates and harvested to confirm KAP1 KO by western blot. Blots were probed with anti-KAP1 (Abcam #Ab22553, 1:5000 dilution) and Rhodamine conjugated anti-Actin (Bio-Rad #12004166, 1:10,000 dilution) for 1 hr. Blots were then probed for 30 min and exposed with secondary anti-mouse Starbright Blue 700 (Bio-Rad, #12004159, 1:10,000 dilution). Two clones (KO1 and KO2) were selected for RNA-seq analysis (see RNA-seq). RNA-seq alignment files confirmed a 7-bp deletion in the KO1 cell line and a single C insertion in the KO2 cell line. Both mutations result in a frame shift and creation of an early stop codon (see Figure S1).
Cell Growth Assay
For growth assays using CRISPR-Cas9–depleted and shRNA-silenced KAP1, 100,000 cells were seeded in triplicate into 12-well plates at day 0. Every 24 hr, cells were trypsinized and then counted using a hemocytometer. For growth assays using siRNAs, 200,000 cells were seeded in triplicate into 6-well plates for 24 hr and then transfected with siNT or siRNA targeting KAP1 as described below (siRNA Knockdown). This was considered day 0. Cells were then trypsinized and counted at days 2 and 3 using a hemocytometer. Western blots for siNT and siKAP1 treated cells were probed with anti-KAP1 (1:5000 dilution) and Rhodamine conjugated anti-Actin (1:10,000 dilution) for 1 hr. Blots were then probed with secondary anti-mouse Starbright Blue 700 (1:10,000 dilution) for 30 min and exposed with Clarity Western ECL.
Colony Formation Assay
For foci assays, 300 HCT116 Ctrl, KO1, and KO2 cells were plated in 6-well plates and incubated in DMEM supplemented with 10% FBS and 1% Penicillin/Streptomycin for 8 days. After the 8-day incubation, cells were washed 1X with PBS, fixed with 2% paraformaldehyde for 10 min at room temperature, washed 1X with PBS, and stained with 0.5% crystal violet. Colonies were counted manually. Images were acquired using the Chemidoc system (Bio-Rad).
4sU-seq
The same number of cells (HCT116 Ctrl and KO1) was seeded into 3×10 cm plates per biological replicate. An extra plate was grown for each condition and counted. Samples were then normalized to cell count for the remainder of the experiment. Cells were pulsed with DMEM (10% FBS, 1% Penicillin/Streptomycin) + 500 μM 4-thio Uridine (4sU) (Sigma-Aldrich, T4509). At the 9 min mark, the DMEM + 4sU media was removed and cells were washed one time with 1X phosphor-buffered saline (PBS). The PBS was removed, and cells were lysed on-plate with 1 mL of TRizol (Thermo Fisher Scientific, 15596026). Nucleic acid was extracted by adding 200 μL chloroform followed by centrifugation at 12,000 g for 15 min at 4°C. The aqueous layer was transferred to new tubes and nucleic acid precipitated by adding 1.5 mL of EtOH and incubating at room temperature for 10 min. Precipitated material was collected by centrifugation at 12,000 g for 10 min at 4°C, followed by one wash with 1 mL of 100% EtOH, and re-centrifugation. Pellets were dried and resuspended in 300 μL of RNAse-free water. Samples were DNase treated with 2 U/uL of DNase (NEB, M0303) for 15 min at room temperature and then extracted with acid-phenol:chloroform pH 4.5 (Thermo Fisher Scientific, AM9720) followed by an additional chloroform (Fisher Scientific, C606SK-1) extraction. RNA was precipitated with 1/10 volume of NaCl (5M), 1 μL glycoblue, and 2.5 volumes of 100% EtOH. RNA pellets were washed with 1 mL of 75% EtOH and air-dried. RNA pellets (~300 μg) were then resuspended in RNase-free water to a final concentration of 0.4 mg/mL. A separate aqueous master mix (20 mM NaOAC pH 5.2, 1 mM EDTA pH 8.0, 0.1% SDS) was added to the diluted RNA mix followed by the addition of 0.2 mg/mL Biotin-HPDP (Thermo Fisher Scientific, 21341) in 100% dimethylformamide (Acros Organics, 279600010). The biotinylation reaction was incubated at room temperature for 2 hr followed by acid-phenol:chloroform extraction. Biotinylated RNA pellets were resuspended in 500 μL RPBS (10 mM Tris-HCl pH 7.5, 1 mM EDTA pH 8.0, 300 mM NaCl). Dynabeads MyOne Streptavidin T1 (200 uL/sample; Thermo Fisher Scientific, 65601) were blocked at room temperature in beads wash buffer + 1% polyvinylpyrrolidone (Fisher Scientific, BP431) for 10 min. After blocking, beads were washed 1X with 1 mL beads wash buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA pH 8.0, 50 mM NaCl) and then resuspended in RPBS. Biotinylated, 4sU RNA was denatured at 65°C for 5 min and then placed on ice for 2 min. 200 μL of blocked beads were added to each sample and incubated for 30 min with rotation at room temperature. Beads were washed 5X with 4sU wash buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA pH 8.0, 1 M NaCl, 0.1% Tween-20). Biotinylated, 4sU-RNA was eluted from beads 2 times in 75 μL of 0.1 M DTT for 15 min at room temperature with rotation, giving a final elution volume of 150 μL. 4sU-RNA was further purified and concentrated using the Zymo RNA Clean and Concentrator Kit (Zymo Research, R1013). RNA concentration was measured using the Qubit RNA HS assay (Thermo Fisher Scientific, Q32852) and quality was determined using the Agilent Tapestation RNA ScreenTape (Agilent, 5067–5576). The KAPA Stranded RNA-seq kit with RiboErase (HMR) (KAPA Biosystems, KK8483) was used to prepare sequencing libraries according to manufacturer’s instructions. Briefly, equal amounts of ERCC spike-ins (Thermo Fisher Scientific, 4456740) were added to Ctrl and KO1 samples according to the manufacturer’s instructions. Libraries were rRNA depleted followed by DNase treatment. RNA was fragmented and primed followed by first strand cDNA synthesis. Second strand synthesis was then performed with the incorporation of dUTP. Libraries were then A-tailed and ligated with Illumina Adapters (NEB, E7335L). After adapter ligation, samples were PCR amplified using Illumina indexed primers (NEB, E7335L) for 12 cycles. Size distribution and quality of libraries was determined using the Agilent Tapestation DNA ScreenTape (Agilent, 5067–5582). Libraries were quantified using the Qubit dsDNA HS Assay (Thermo Fisher Scientific, Q32851), submitted to the McDermott Center Sequencing Core at UTSW, and sequenced on the NextSeq 500 (Illumina) with 75-nt paired-end sequencing reads per sample.
RNA-seq
The same number of cells (HCT116 Ctrl, KO1, and KO2) were seeded into 1 well of a 6-well plate per biological replicate. An extra well was grown for each condition and counted. Samples were then normalized to cell count for the remainder of the experiment. RNA was extracted from HCT116 Ctrl, KO1, and KO2 cell lines in biological triplicates using the Zymo Quick-RNA MiniPrep Kit (Zymo Research, R1055). RNA concentration and quality were determined as described for 4sU-seq. 1 μg of total RNA was used to prepare and sequence whole transcriptome sequencing libraries as described for 4sU-seq. ERCC spike-ins were only added to the Ctrl and KO1 cell lines.
ChIP-qPCR and ChIP-seq
HCT116 Ctrl and KO1 cells were plated on one 15 cm plate per ChIP and grown to 90% confluency, yielding ~20×106 cells per plate. Cells were crosslinked on plate with 0.5% methanol-free formaldehyde (Thermo Fisher Scientific, 28908) added directly to the media at room temperature for 10 min with slow rotation. Crosslinking was quenched by adding glycine to a final concentration of 150 mM directly to the plate and incubating 5 min at room temperature with rotation. Media was removed and cells washed on plate 1 time with cold 1X PBS. Cells were then scraped off the plate and counted. Cells were pelleted at 1000 g for 5 min at 4°C and then washed one time with PBS and pelleted. Cells were resuspended to a final concentration of 10×106 cells/mL in Farnham Lysis Buffer (5 mM PIPES pH 8.0, 85 mM KCl, 0.5% NP-40, 1 mM PMSF, Protease Inhibitor Tablet (Millipore Sigma, 4693159001)). Supernatant was removed and nuclei were resuspended to a final concentration of 25×106 nuclei/mL in Szak’s RIPA Buffer (50 mM Tris-HCl pH 8.0, 1% NP-40, 150 mM NaCl, 0.5% Na Deoxycholate, 0.1% SDS, 2.5 mM EDTA, 1 mM PMSF, and Protease Inhibitor Tablet). Chromatin was sheared to a range of 200–500-bp by sonicating for 45 cycles (30 sec on, 30 sec off) on a Bioruptor UCD-300 water bath (Diagenode) set at high power. To reduce background, sheared chromatin was pre-cleared by incubating with 50 μL of Szak’s RIPA equilibrated Protein G Dynabeads (Thermo Fisher Scientific, 10003D). For immunoprecipitation of ChIP-seq samples, 100 μL of Protein G Dynabeads were equilibrated with 1X PBS+0.05% Tween-20 and resuspended to a final volume of 250 μL per ChIP. For ChIP-qPCR samples, 50 μL of Protein G Dynabeads was used. 5 μg of Pol II (anti-RPB3 (Millipore, #ABE999)) and anti-SMAD2 (Thermo Fisher Scientific #PA5–29237) antibody and 10 μg of all other antibodies was then added to each tube (see Key Resources Table for antibody catalogue numbers). Note that two different CDK9 antibodies were used for sequencing (For Replicate 1: Santa Cruz Biotechnology #Sc-484 (C-20) and For Replicate 2: Abcam #ab236045) due to the C-20 antibody being discontinued while completing these studies. Each antibody was conjugated to the Protein G Dynabeads in 1X PBS + 0.05% Tween-20 for 1 hr at 4°C with rotation. For samples that were sequenced an additional 2.5 μg of antibody specific for Drosophila H2Av (Active Motif, 61686) was added to each tube, allowing for specific enrichment of Drosophila H2Av from spike-ins chromatin and normalization of data to Drosophila spike-ins (see NGS Data Analysis). Beads were then washed 1 time with 1X PBS + Tween-20 and 2 times with Szak’s RIPA Buffer. Antibody bound beads then blocked in Szak’s RIPA Buffer + 5% BSA for 1 hr at 4°C with rotation. Pre-cleared sheared chromatin was then added to beads and incubated overnight at 4°C with rotation. For samples that were sequenced, 50 ng of Drosophila spike-ins chromatin (Active Motif, 53083) was added to each sample. Beads from each sample were washed 2 times with 1 mL of Szak’s RIPA Buffer, Low Salt Buffer (0.1% SDS, 1% NP-40, 2 mM EDTA, 20 mM Tris-HCl pH 8.0, 150 mM NaCl), High Salt Buffer (same as low salt but with 500 mM NaCl), LiCl buffer (250 mM LiCl, 1% NP-40, 1% sodium deoxycholate, 1 mM EDTA, 20 mM Tris-HCl pH 8.0), and TE Buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA). For anti-FLAG ChIP-qPCR samples were washed 4 times with each buffer to further reduce background. Samples were eluted from beads in 100 μL of elution buffer (100 mM NaHCO3, 1% SDS) for 30 min at 65°C while vortexing every 5 min. Eluents were transferred to new tubes and de-crosslinked for 4 hr at 65°C with an equal volume of de-crosslinking buffer (500 mM NaCl, 2 mM EDTA, 20 mM Tris-HCl pH 6.8, 0.5 mg/mL Proteinase K (Millipore Sigma, 3115879001)). ChIP DNA was purified and concentrated with the Zymo ChIP DNA Clean & Concentrator (Zymo Research, D5201). DNA was either sequenced (for ChIP-seq) or analyzed by qPCR (for ChIP-qPCR). For samples that were sequenced, libraries were prepared using the KAPA Hyper Prep Kit (KAPA Biosystems, KK8502) according to the manufacturer’s instructions. Libraries were quantified, and quality was ensured as described above for 4sU-seq. Samples were submitted to the McDermott Center Sequencing Core at UTSW and sequenced on the NextSeq 500 (Illumina) with 75-nt paired-end sequencing reads per sample. For qPCR, 10 μL SYBR Green (Thermo Fisher Scientific, 4385612), 0.5 μL of each primer (10 μM stock), 0.5 μL (out of 20 μL total) of ChIP DNA, and 8.5 μL of H2O was used per well in a 96-well plate in triplicate. Samples were amplified 40 cycles using the Applied Biosystems 7500 Fast Real-Time PCR System. The ChIP-qPCR data was normalized using the “Percent Input Method”, which includes normalization for background and Input chromatin used for each ChIP. ChIP signals were divided by signals obtained from the Input sample (5% starting chromatin), which signifies the amount of chromatin used per ChIP. Values represent the percentage (%) of input DNA immunoprecipitated (IP DNA) and are the average of two to three independent experiments. Primers used for qPCR analysis are listed in Table S6.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-KAP1 | Abcam | Cat# Ab22553; RRID: AB_447151; lot #GR229743-12 |
| hFAB Rhodamine anti-Actin | Bio-Rad | Cat# 12004166 |
| Rabbit polyclonal anti-RPB3 (RNA Pol II) | MilliporeSigma | Cat# ABE999; lot #Q2550840 |
| Rat monoclonal anti-RNA Pol II CTD phospho Ser5 | Active Motif | Cat# 61085; lot #20017002; RRID: AB_2687451 |
| Rat monoclonal anti-RNA Pol II CTD phospho Ser2 | MillliporeSigma | Cat# 04-1571; lot # 2724427; RRID: AB_10627998 |
| Rabbit polyclonal anti-CDK9 (C-20) | Santa Cruz Biotechnology | Cat# Sc-484; lot # B2605; RRID: AB_2275986 |
| Rabbit monoclonal anti-CDK9 | Abcam | Cat# ab236045; lot # GR3235366-1 |
| Mouse monoclonal anti-RNA Pol II (4H8) | Active Motif | Cat# 39097; RRID: AB_2732926 |
| Rabbit polyclonal anti-SMAD2 | Thermo Fisher Scientific | Cat# PA5-29237; lot #UD2753805A RRID: AB_2546713 |
| Mouse monoclonal anti-FLAG M2 | MilliporeSigma | Cat# F3165; lot #SLBT6752 RRID: AB_259529 |
| Rabbit monoclonal anti-pSMAD2 | Cell Signaling Technology | Cat# 8828; lot #7; RRID: AB_2631089 |
| Rabbit polyclonal anti-GST | MilliporeSigma | Cat# G7781; lot #108M4775V; RRID: AB_259965 |
| Rabbit polyclonal anti-H3 | Abcam | Cat# ab1791; lot #940378; RRID: AB_302613 |
| Rabbit polyclonal anti-H4 | Abcam | Cat# ab7311; lot #GR325224-4; RRID: AB_305837 |
| Rabbit polyclonal anti-H2A | Active Motif | Cat# 39209; lot #09617014; RRID: AB_2793184 |
| Rabbit polyclonal anti-H2B | Active Motif | Cat# 39210; lot #34515005; RRID: AB_2793185 |
| Mouse monoclonal anti-CDK9 (D7) | Santa Cruz Biotechnology | Cat# sc-13130; lot #50917; RRID: AB_627245 |
| Mouse monoclonal anti-NELFE | Santa Cruz Biotechnology | Cat# sc-377052; lot #L0117 |
| Rabbit polyclonal anti-H4K16ac | Active Motif | Cat #39068 |
| Monoclonal anti-Strep-HRP | MilliporeSigma | Cat# 71591; lot #3150780; RRID: AB_11214448 |
| Anti-mouse-HRP | Cell Signaling Technology | Cat# 7076; lot #33; RRID: AB_330924 |
| Anti-rabbit-HRP | Cell Signaling Technology | Cat# 7074; lot #27; RRID: AB_2099233 |
| Anti-mouse-HRP | Rockland | Cat# 18-8817-33 RRID: AB_2610851 |
| Anti-rabbit-HRP | Rockland | Cat# 18-8816-33 RRID: AB_2610848 |
| Anti-mouse-Starbright Blue 700 | Bio-Rad | Cat# 12004159 |
| Anti-rabbit-Starbright Blue 700 | Bio-Rad | Cat# 12004162 |
| Anti-H2Av | Active Motif | Cat# 61686 |
| Bacterial and Virus Strains | ||
| E. coli BL21 DE3 | NEB | Cat# C2527I |
| E. coli DH5α | NEB | Cat# C2987I |
| E. coli XL-10 Gold | Agilent | Cat# 200522 (as part of the QuickChange II XL site-directed mutagenesis kit) |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM | HyClone | Cat# SH30022.FS |
| FBS | MilliporeSigma | Cat# 9268 |
| Penicillin/Streptomycin | MP Biomedicals | Cat# 091670049 |
| Tween-20 | Fisher Chemical | Cat# BP337-500 |
| Non-fat Dry Milk | LabScientific | Cat# M0841 |
| 0.45 μM Nitrocellulose Membrane | Bio-Rad | Cat# 1620115 |
| BsmB1 | NEB | Cat# R0580 |
| Quick Ligase | NEB | Cat# M2200 |
| Polybrene (Hexadimethrine Bromide) | MilliporeSigma | Cat# H9268 |
| 4-thio-uridine (4sU) | MilliporeSigma | Cat# T4509 |
| Biotin-HPDP | Thermo Fisher Scientific | Cat# 21341 |
| TRizol | Thermo Fisher Scientific | Cat# 15596026 |
| DNase | NEB | Cat# M0303 |
| Acid:Phenol:Chloroform | Thermo Fisher Scientific | Cat# AM9720 |
| Chloroform | Fisher Scientific | Cat# C606SK-1 |
| Dimethylformamide | Acros Organics | Cat# 279600010 |
| Dynabeads MyOne Streptavidin T1 | Thermo Fisher Scientific | Cat# 65601 |
| Polyvinylpyrrolidone | Fisher Scientific | Cat# BP431 |
| ERCC spike-ins | Thermo Fisher Scientific | Cat# 4456740 |
| Methanol-free formaldehyde | Thermo Fisher Scientific | Cat# 28908 |
| Protease Inhibitor Tablets | MilliporeSigma | Cat# 4693159001 |
| Protein G Dynabeads | Thermo Fisher Scientific | Cat# 10003D |
| Drosophila Spike-in Chromatin | Active Motif | Cat# 53083 |
| Proteinase K | MilliporeSigma | Cat# 3115879001 |
| SYBR Green | Thermo Fisher Scientific | Cat# 4385612 |
| Doxycycline Hydrochloride | Fisher Scientific | Cat# BP2653-1 |
| Flavopiridol | MilliporeSigma | Cat# F3055 |
| Clarity Western ECL Substrate | Bio-Rad | Cat# 1705061 |
| DMSO | Acros Organics | Cat# 61042-0010 |
| Micrococcal Nuclease | NEB | Cat# M0247S |
| PolyJet | SignaGen | Cat# SL100688 |
| StrepTactin Agarose | IBA Life Sciences | Cat# 2-1201-010 |
| 10X Strep Elution Buffer with Desthiobiotin | IBA Life Sciences | Cat# 2-1000-025 |
| Anti-FLAG M2 Affinity Gel | Millipore Sigma | Cat# A2220 |
| 3X FLAG Peptide | Millipore Sigma | Cat# F4799 |
| 27-gauge needle | BD Biosciences | Cat# 305109 |
| Trypan blue stain | Corning | Cat# 25-900-CI |
| Super Signal West Femto Luminol Enhancer | Thermo Fisher Scientific | Cat# 185022 |
| Opti-MEM | Thermo Fisher Scientific | Cat# 31985062 |
| RNAi Max | Thermo Fisher Scientific | Cat# 13778030 |
| Trypsin/EDTA | Thermo Fisher Scientific | Cat# R001100 |
| Trypsin Neutralizing Agent | Thermo Fisher Scientific | Cat# R002100 |
| TGFβ-1 | PeproTech | Cat# 100-21 Lot #0218209-3 B1918 |
| CutSmart Buffer | NEB | Cat# B7204S |
| Calf Intestinal alkaline phosphatase | NEB | Cat# M0290 |
| Paraformaldehyde | MilliporeSigma | Cat# P6148 |
| GST Agarose | MilliporeSigma | Cat# G4510 |
| L-Glutathione Reduced | MilliporeSigma | Cat# G4251 |
| Thrombin | MP Biomedicals | Cat# 0219492182 |
| Mononucleosomes, Recombinant Human Biotinylated | EpiCypher | Cat# 16-0006 |
| Histone H4 N-terminal peptide, biotinylated (1–23) | EpiCypher | Cat# 12-0029 |
| Histone H3 N-terminal peptide, biotinylated (1–20) | EpiCypher | Cat# 12-0001 |
| Histone H2A N-terminal peptide, biotinylated (1–17) | EpiCypher | Cat# 12-0012 |
| Histone H2B N-terminal peptide, biotinylated (1–24) | EpiCypher | Cat# 12-0077 |
| Histone H4K5ac peptide, biotinylated (1–23) | EpiCypher | Cat# 12-0030 |
| Histone H4K8ac peptide, biotinylated(1–23) | EpiCypher | Cat# 12-0031 |
| Histone H4K12ac peptide, biotinylated (1–23) | EpiCypher | Cat# 12-0032 |
| Histone H4K16ac peptide, biotinylated (1–23) | EpiCypher | Cat# 12-0033 |
| Histone H4Kac(4) peptide, biotinylated (1–23) | EpiCypher | Cat# 12-0103 |
| Histone H4R3me1 peptide, biotinylated (1–23) | EpiCypher | Cat# 12–0111 |
| Histone H4R3me2(s) peptide, biotinylated (1–23) | EpiCypher | Cat# 12-0059 |
| Histone H4R3me2(a) peptide, biotinylated (1–23) | EpiCypher | Cat# 12-0058 |
| Histone H4 N-terminal peptide, biotinylated (11–27) | EpiCypher | Cat# 12-0035 |
| Histone H4K20me1 peptide, biotinylated (11–27) | EpiCypher | Cat# 12–0036 |
| Histone H4K20me2 peptide, biotinylated (11–27) | EpiCypher | Cat# 12-0037 |
| Histone H4K20me3 peptide, biotinylated (11–27) | EpiCypher | Cat# 12-0038 |
| Histone H3 N-terminal peptide, biotinylated (1–30) | GenScript | Custom |
| Histone H3K23ac N-terminal peptide, biotinylated (1–30) | GenScript | Custom |
| Histone H3 N-terminal peptide, biotinylated (1–20) | EpiCypher | Cat# 12-0001 |
| Histone H3K9me3 peptide, biotinylated (1–20) | EpiCypher | Cat# 12-0012 |
| Histone H3K18ac Peptide, biotinylated (1–20) | EpiCypher | Cat# 12-0005 |
| Critical Commercial Assays | ||
| Plasmid MiniPrep Kit | Qiagen | Cat# 27104 |
| Zymo RNA Clean and Concentrator Kit | Zymo Research | Cat# R1013 |
| Qubit RNA HS Assay | Thermo Fisher Scientific | Cat# Q32852 |
| Tapestation RNA ScreenTape | Agilent | Cat# 5067-5576 |
| KAPA Stranded RNA-seq kit with RiboErase | KAPA Biosystems | Cat# KK8483 |
| Tapestation DNA ScreenTape | Agilent | 5067-5582 |
| Qubit dsDNA HS Assay | Thermo Fisher Scientific | Cat# Q32851 |
| KAPA Hyper Prep Kit | KAPA Biosystems | KK8502 |
| Zymo Quick-RNA MiniPrep Kit | Zymo Research | R1055 |
| Zymo Quick DNA Clean & Concentrator Kit | Zymo Research | D5201 |
| Quick Ligase Kit | NEB | Cat# M2200L |
| M-MuLV Reverse Transcriptase and buffer | NEB | Cat# M02553L |
| Pierce BCA Protein Assay Kit | Thermo Fisher Scientific | Cat# 23225 |
| Quick Change II Site-Directed Mutagenesis Kit | Agilent | Cat# 3200522 |
| Deposited Data | ||
| Raw and analyzed NGS Data | This paper | GEO: GSE132705 |
| HEXIM and LARP7 ChIP-seq datasets | McNamara et al., 2016 | GEO: GSE72622 |
| Source data for western blots | This paper | Mendeley: http://dx.doi.org/10.17632/fc56g3m8ff.1 |
| KAP1-PHD-BD (apo) structure | Zeng et al., 2008 | PDB: 2RO1 |
| Chemical shift assignments for KAP1-PHD-BD | Zheng et al., 2008 | BMRB ID: 11036 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | CRL-11268 |
| HCT116 | ATCC | CCL-247 |
| MCF7 | ATCC | HTB-22 |
| HCT116 Ctrl | Laboratory of Iván D’Orso | Created in this study |
| HCT116 KAP1 KO1 | Laboratory of Iván D’Orso | Created in this study |
| HCT116 KAP1 KO2 | Laboratory of Iván D’Orso | Created in this study |
| 293 T-REx KAP1:S | Laboratory of Iván D’Orso | Created in this study |
| HCT116 shNT | Laboratory of Iván D’Orso | (McNamara, et al., 2016) |
| HCT116 shKAP1 | Laboratory of Iván D’Orso | (McNamara, et al., 2016) |
| HCEC 1CT | Laboratory of Jerry Shay | (Roig et al., 2010) |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| See Table S1 for all DNA oligonucleotide sequences used for RT-qPCR analysis and cloning | ||
| Illumina Adapters and Indexed Primers | NEB | Cat# E7335L |
| siKAP1_1 targeting KAP1’s CDS | Qiagen | Cat# SI03171903 |
| siKAP1_2 targeting KAP1’s 3’UTR | Qiagen | Cat# SI05121214 |
| siRNA targeting SMAD2: GAACAAACCAGGUCUCUUG | Dharmacon | Cat# D-003561-01 |
| siRNA targeting SMAD2: GCAGAACUAUCUCCUACUA | Dharmacon | Cat# D-003561-02 |
| siRNA targeting SMAD2: GAAGAGGAGUGCGCUUAUA | Dharmacon | Cat# D-003561-03 |
| siRNA targeting SMAD2: GGUGUUCGAUAGCAUAUUA | Dharmacon | Cat# D-003561-04 |
| shRNA non-target: CCGGCAACAAGATGAAGAGCACCAACTCGAGTTGGTGCTCTTCATCTTGTTGTTTTT | MilliporeSigma | Cat# SHC002 |
| shRNA targeting CDK9: CCGGGCACAGTTTGGTCCGTTAGAACTCGAGTTCTAACGGACCAAACTGTGCTTTTT | MilliporeSigma | Cat# TRCN0000000494 |
| shRNA targeting KAP1: CCGGCCTGGCTCTGTTCTCTGTCCTCTCGAGAGGACAGAGAACAGAGCCAGGTTTTT | MilliporeSigma | Cat# TRCN0000017998 |
| Recombinant DNA | ||
| Plasmid: LentiCRISPRv2 | Addgene | Cat# 52961 |
| Plasmid: pSPAX | Addgene | Cat# 12260 |
| Plasmid: pMD2.G | Addgene | Cat# 12259 |
| Plasmid: LentiCRISPRv2-KAP1-gRNA1 | This Paper | N.A. |
| Plasmid: LentiCRISPRv2-KAP1-gRNA2 | This Paper | N.A. |
| Plasmid: pLKO.1/Doxycycline Inducible | Addgene | Cat# 21915 |
| Plasmid: pLKO.1/Doxycycline Inducible-shNT | Addgene | Cat# 47541 |
| Plasmid: pLKO.1/Doxycycline Inducible-shCDK9 | This Paper | N.A. |
| Plasmid: pcDNA4/TO | Thermo Fisher Scientific | V102020 |
| Plasmid: pcDNA4/TO/Strep/FLAG-GFP | McNamara et al., 2016 | N.A. |
| Plasmid: pcDNA4/TO/Strep-KAP1 | McNamara et al., 2016 | N.A. |
| Plasmid: pcDNA4/TO/FLAG-SMAD2 | This Paper | N.A. |
| Plasmid: pGEX2T-GST | GE Life Sciences | Cat# 28954653 |
| Plasmid: pGEX2T/-GST-KAP1-PHD-BD | This Paper | N.A. |
| Plasmid: pGEX2T-GST-KAP1-PHD | This Paper | N.A. |
| Plasmid: pGEX2T-GST-KAP1-PHD-BD-E662/663A | This Paper | N.A. |
| Plasmid: pGEX2T-GST-KAP1-PHD-BD-L637A | This Paper | N.A. |
| Plasmid: pGEX2T-GST-TRIM24-PHD-BD | This Paper | N.A. |
| Plasmid: pGEX2T-GST-TRIM33-PHD-BD | This Paper | N.A. |
| Plasmid: pcDNA4/TO/FLAG-KAP1 | McNamara et al., 2016 | N.A. |
| Plasmid: pcDNA4/TO/FLAG-KAP1-ΔPHD-BD | This Paper | N.A. |
| Plasmid: pcDNA4/TO/FLAG-KAP1-ΔBD | This Paper | N.A. |
| Plasmid: pcDNA4/TO/FLAG-KAP1-ΔPHD | This Paper | N.A. |
| Plasmid: pcDNA4/TO/FLAG-KAP1-ΔCC | This Paper | N.A. |
| Plasmid: pcDNA4/TO/FLAG-KAP1-ΔRB | This Paper | N.A. |
| Plasmid: pcDNA4/TO/FLAG-KAP1-ΔRBCC | This Paper | N.A. |
| Software and Algorithms | ||
| STAR/2.6.1b | Dobin et al., 2013 | http://github.com/alexdobin/STAR |
| Subread/1.6.1 | N.A. | http://bioinf.wehi.edu.au/subread-package/ |
| RUVSeq/1.15 | Risso et al., 2014 | https://bioconductor.org/packages/release/bioc/html/RUVSeq.html |
| UCSC_userApps/v317 | N.A. | https://genome-source.gi.ucsc.edu/gitlist/kent.git/blob/master/src/userApps/README |
| Integrated Genome Viewer (IGV) | Robinson et al., 2011 | https://software.broadinstitute.org/software/igv/download |
| deepTools/2.5.0 | Ramirez et al., 2016 | https://deeptools.readthedocs.io/en/develop/content/installation.html |
| Java Treeview | Saldanha, 2004 | https://sourceforge.net/projects/jtreeview/files/ |
| Trimgalore v.0.4.1 | N.A. | https://github.com/FelixKrueger/TrimGalore |
| Fastqc/0.11.2 | N.A. | https://raw.githubusercontent.com/s-andrews/FastQC/master/INSTALL.txt |
| Bowtie2/2.1.0 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml |
| Picard/1.127 | https://github.com/broadinstitute/picard | |
| Macs/2.1.0-20151222 | Zhang et al., 2008 | https://github.com/taoliu/MACS/blob/master/INSTALL.md |
| SAMtools v1.6 | Li et al., 2009 | https://github.com/samtools/samtools/blob/develop/INSTALL |
| R/v3.4.1 | N.A. | http://www.r-project.org |
| bamR | N.A. | https://github.com/rchereji/bamR |
| GraphPad Prism v8.1.0 | N.A. | https://www.graphpad.com/scientific-software/prism/?gclid=Cj0KCQjwoInnBRDDARIsANBVyAST15UIY85-IY-6-_aDTwJl5hMTpw4Q4Nf16CZUjB1q_ld2oCjC97UaAjfCEALw_wcB |
| GROMACS 5 | Abraham, M.J., et al. 2015 | http://manual.gromacs.org/documentation/ |
| AMBER99SB-ILDN | Lindorff-Larsen, et al., 2010 | http://www.gromacs.org/Documentation/Terminology/Force_Fields/AMBER |
| UCSF Chimera | Pettersen et al., 2004 | https://www.cgl.ucsf.edu/chimera/ |
| Other | ||
| Detailed scripts and commands used for NGS data analysis in this study | N.A. | https://git.biohpc.swmed.edu/ivandorsolab/Curtis_Dissertation |
KAP1 and Pol II In vitro Binding Assay
STREP-tagged KAP1ΔRB and KAP1ΔRBCC were expressed as described below in the Nuclear Lysate Preparation and Affinity Purification (AP) section. STREP-tagged WT KAP1 was expressed using the 293 T-REx system. Briefly, the 293 T-REx cells (Thermo Fisher) were transfected with 1 μg of Ssp1-linearized pcDNA4/TO STREP-tagged KAP1 vector using Polyjet. Positive clones were selected with 10 μg/mL Blasticidin and 100 μg/mL Zeocin. To induce protein expression, the stable cell lines were treated with 1 μg/mL Doxycycline Hydrochloride (Fisher Scientific, BP2653–1) for 48 hr. Cells expressing WT and mutant proteins were then harvested and washed two times with 5X packed cell volume (PCV) of PBS. Cells were then resuspended in 5X PCV of modified passive lysis buffer (mPLB; 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5% NP-40, 5% Glycerol, 1 mM EDTA, 2 mM MgCl2, 1 mM PMSF, 1X protease inhibitor tablets (Roche), and 1X PhosStop tablets) and nutated for 30 min at 4°C. CaCl2 was then added to a final volume of 2 mM, along with 2,000 U of MNase, and samples were incubated for 15 min at room temperature to facilitate release of the chromatin fraction. Samples were centrifuged at 8,000 rpm for 10 min at 4°C. Protein lysates were then transferred to a new 1.5 mL tube. 75 μL of 50% StrepTactin agarose (IBA Life Sciences, 2–1201-010) preequilibrated with mPLB was added to samples. Samples were then nutated for 2 hr at 4°C. Samples were then washed 4X with 1 mL of mPLB + 750 mM NaCl and 4X with 1 mL of mPLB + 250 mM NaCl. Proteins were eluted from beads twice in 100 uL of Strep elution buffer with Desthiobiotin (IBA Life Sciences, 2-1000-025). RNA polymerase II (Pol II) was purified from hRPB9-FLAG nuclear extract as previously described (Kershnar et al., 1998). M2-agarose immobilized with FLAG-tagged Pol II (200 ng) or blocked with BSA was incubated with 3 μg of purified STREP-tagged KAP1 (WT or mutants) in 200 μL of BC100 (Kershnar et al., 1998) at 4°C for 12 hr. After three BC100 washes, protein-bound beads (20%) along with KAP1 input (5 ng) were analyzed on 4–15% Tris-HCl/SDS gradient gels and probed by western blotting with anti-Pol II (RPB1 or RPB3) and anti-STREP antibodies. Note that 0.1% Input and 20% elution used for western blot with STREP or RPB1 antibodies.
Doxycycline Inducible shRNA Knockdown
shCDK9 (Sigma, TRCN0000000494) oligos were annealed by mixing 11.25 μL of each oligo (Top and Bottom) with 2.5 μL of 10X annealing buffer (1M NaCl, 100mM Tris-HCl, pH 7.5) in a tube and heated at 95°C for 5 min followed by cooling down to room temperature. The annealed oligo mixture was diluted in 0.5X annealing buffer at 1:400 (1 μL in 399ul 0.5X buffer) and cloned into pLKO-Tet-On vector (Addgene, 21915) using the 5’-AgeI and 3’-EcoRI restriction sites. The pLKO-Tet-On vector with CDK9 shRNA and one encoding scrambled shRNA (Addgene, 47541) was transfected along with gag/pol (pSPAX) and VSV-G (pDM2) into HEK 293T cells for expression of competent lentiviruses. To concentrate the lentiviruses, cell supernatants were collected two days post-transfection and subjected to ultracentrifugation at 25000 RPM for 90 min at 4°C. The viral pellet was resuspended in 1X resuspension buffer (100 mM NaCl + 10 mM HEPES pH 7.4) and incubated overnight at 4°C. Viral transduction was done by spinoculation using 1×105 HCT116 cells, 10 μg/mL polybrene, and unsupplemented DMEM to a final volume of 300 μL at room temperature for 2 hr at 400 g. Infected cells were selected with 1 μg/mL puromycin for 3 days post-infection. Knockdowns were generated by inducing the shRNA using 4×105 puromycin-selected cells, 1 μg/mL of doxycycline hydrochloride and incubated for 4 days. Knockdown efficiency was determined through standard western blot and RT-qPCR. For western blots, 1:1000 dilution of anti-CDK9 (D-7) (Santa Cruz Biotechnology, #sc-484) and 1:10,000 dilution of anti-Rhodamine Actin (Bio-Rad, #12004166) was used.
FP Treatment
1×106 HCT116 cells were seeded into 6-well plates and incubated overnight at 37°C to achieve ~70% confluency. A time course was conducted (0, 1, 2, 4, 8, 16 hr) using 1 μM Flavopiridol (Millipore Sigma, F3055) and DMSO (Acros Organics, 61042–0010) as vehicle control to determine the CDK9 inhibition efficiency by blotting for Pol II Ser2 CTD phosphorylation and RPB3 (1:5000 dilutions of both primary anti-RNA Pol II CTD phospho Ser2 (Millipore, #04–1571) and anti-RPB3 were used). RNA was extracted from control, 2 and 4 hr treatments and RT-qPCR was performed.
MNase-seq
HCT116 Ctrl and KO1 cells were crosslinked with 0.5% methanol-free formaldehyde for 10 min and reaction was quenched for 5 min with 150 mM glycine before harvesting. Cells were washed twice with PBS buffer and nuclei were gathered by lysing the cells in Farnham’s lysis buffer (5 mM PIPES pH 8.0, 85 mM KCl and 0.5% NP-40 freshly supplemented with 1 mM PMSF and EDTA free-Protease inhibitor cocktail). Nuclei were washed once with cold MNase buffer (20 mM Tris-HCl pH 7.5,15 mM NaCl, 60 mM KCl, 2 mM CaCl2) and resuspended in MNase buffer with concentration of 10×106 cells/mL before digestion. Micrococcal nuclease (NEB, M0247S) digestion was performed (240 U/106 cells in 500 μL of MNase buffer) at 37°C to achieve 80% mono-nucleosomes populations and reaction was stopped with 10 mM EDTA, 20 mM EGTA, and 0.4% SDS solution. Samples were centrifuged for 5 min at 14,000 rpm, supernatants were collected and reverse crosslinked in de-crosslinking buffer (20 mM Tris-HCl pH 6.8, 500 mM NaCl, 2mM EDTA, and 0.5 mg/mL Proteinase K) overnight at 65°C. Following phenol-chloroform extraction and ethanol precipitation, DNA samples were run on 1.5% agarose gel and fragments of ~150 bp size were excised and purified. Libraries were quantified, prepared, and sequenced as described for ChIP-seq except the MNase libraries were not size selected. Libraries were deep sequenced with 75-bp bases paired-end on a HiSeq 2000 (Illumina) platform with >200 million reads per sample.
Nuclear Lysate Preparation and Affinity Purification (AP)
HCT116 cells were grown to ~60% confluency in 2×10 cm plates per AP the day before transfection. 7 μg of plasmid DNA encoding tagged proteins of interest were transfected into cells by first complexing the DNA with 21 μL of PolyJet reagent (SignaGen, SL100688) in 1 mL of serum-free DMEM for 10 min at room temperature. The DNA:PolyJet mix was then added dropwise to cells in 5 mL of complete tissue culture media. Cells were incubated for 5 hr at 37°C with 5% CO2 and then media was replaced with 10 mL of fresh DMEM (10% FBS, 1% Penicillin/Streptomycin). Cells were then incubated 48 hr at 37°C with 5% CO2 and harvested by scraping and pelleting at 3000 rpm for 5 min at 4°C. Cell pellets were washed twice with 1 mL of cold PBS. Cell pellets were quickly resuspended in 5X packed cell volume (PCV) of cold Buffer A (10 mM HEPES pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.2 mM PMSF, 1X protease inhibitor tablet) and centrifuged at 3000 rpm for 2 min at 4°C. Cells were then resuspended in 3X PCV of Buffer A and allowed to swell on ice for 10 min. Cells were further lysed by passing through a 27-gauge needle (BD Biosciences, 305109) 20 times. ~90% plasma membrane lysis was confirmed by trypan blue staining (Corning, 25–900-CI). Nuclei were pelleted by centrifugation at 4000 rpm for 15 min at 4°C. The 100,000 g soluble fraction (S-100) was removed and nuclei were resuspended in 1 packed nuclear volume (PNV) of low salt buffer C (10 mM HEPES pH 7.9, 1.5 mM MgCl2, 25% glycerol, 200 mM NaCl, 0.2 mM PMSF, 1X protease inhibitor tablet). The NaCl concentration was gradual brought to 420 mM by slowly adding an equal volume of high salt buffer C (same as low salt buffer C but with 650 mM NaCl). Nuclei were extracted for 45 min with slow rotation at 4°C. To facilitate further solubilization of chromatin, 5 mM CaCl2 was added and samples were treated with 200 U of MNase per uL of cell lysate for 10 min at 37°C. Extracted nuclei were pelleted by centrifuging for 30 min at 21,000 g at 4°C. The nuclear extract was transferred to new tubes and used for AP.
For STREP AP, 40 μL of 50% slurry StrepTactin agarose equilibrated with buffer C containing 420 mM NaCl was added to each sample in Protein LoBind tubes. Samples were bound to beads for 2 hr at 4°C with rotation. Beads were washed 4X with 500 μL of wash buffer (50 mM Tris-HCl pH 7.5, 250 mM NaCl, 0.2% NP-40, 1.5 mM MgCl2, 5% glycerol). Before the final wash, samples were transferred to fresh protein LoBind tubes. Samples were eluted in 50 μL of 1 X Strep Elution buffer with Desthiobiotin by continuous vortexing for 15 min at 4°C.
For FLAG AP, 40 μL of anti-FLAG M2 Affinity Gel (Millipore Sigma, A2220) was added to each sample, which were bound and washed as described for the STREP AP. Proteins bound to beads were eluted in 50 μL of FLAG elution buffer (Tris-HCl pH 8.0, 150 mM NaCl, 1 mM, EDTA, 0.1% NP-40, 200 μg/mL 3X FLAG peptide (Millipore Sigma, F4799)). For western blots, membranes were probed with 1:10,000 anti-Strep-HRP (Millipore Sigma, 71591), 1:20,000 anti-FLAG (MiiliporeSigma, F3165), 1:5000 anti-KAP1 (Abcam, ab22553), 1:1000 anti-CDK9 (Abcam, ab236045), and 1:1000 anti-SMAD2 (Thermo Fisher Scientific, PA5–29237). Anti-KAP1, anti-CDK9, and anti-SMAD2 blots were exposed using Super Signal West Femto Luminol Enhancer (Thermo Fisher Scientific, 185022). Note that 1% input was loaded for all blots, 10% of the elution was loaded for the tagged protein blot, and 50% of the elution was loaded for the co-purified factor blot.
Endogenous immunoprecipitation (IP)
For endogenous IP of KAP1 and SMAD2, 4×15 cm plates of HCT116 cells were grown to confluency. Nuclear extracts were obtained as described in Nuclear Lysate Preparation and Affinity Purification (AP). 50 μL of Protein G beads per sample were equilibrated by washing 3 times in 500 μL of 1X PBS + 0.05% Tween. Beads were then resuspended in 250 μL of 1X PBS + 0.05% tween per sample and aliquoted to Protein LoBind tubes. 10 μg of either α-IgG, α-KAP1, or anti-SMAD2 was then added and bound to Protein G beads and nutated for 1 hr at 4°C. Beads were washed 1 time with 500 μL of 1X PB + 0.05% Tween and then 2 times with buffer C containing 420 mM NaCl. Antibody bound beads were then block for 1 hr with 500 μL of buffer C (420 mM NaCl) + 5% BSA. Nuclear extract was then pre-cleared for 1 hr with 50 μL of Protein G beads and added to the antibody bound, blocked Protein G beads. Samples nutated for 2 hr at 4°C. Beads were then washed 4 times with 1 mL of wash buffer (50 mM Tris-HCl pH 7.5, 250 mM NaCl, 0.2% NP-40, 1.5 mM MgCl2, 5% glycerol). Sample was eluted from beads by boiling in 40 μL of 2X Laemmli buffer for 10 min. For western blots, membranes were probed with 1:1000 anti-SMAD2, 1:5000 anti-KAP1, and 1:5000 anti-Pol II (RPB1, 4H8). TruBlot anti-Mouse and anti-Rabbit HRP conjugated secondary antibodies were used (1:1,000 dilution; Rockland #18-8816-33 and #18-8817-33). The Pol II blot was exposed using Super Signal West Femto Luminol Enhancer and the others were exposed with Clarity Western ECL. Note that 1% input was loaded for all blots, 10% of the elution was loaded for the protein that was purified, and 50% of the elution was loaded for the co-purified factor blot.
siRNA knockdown
HCT116 cells were seeded into 6-well plates the night before transfection to achieve ~50% confluency. 50 pmol of 4-pooled siRNAs targeting SMAD2 and a single siRNA targeting KAP1 were diluted in 100 μL Opti-MEM media (Thermo Fisher Scientific, 31985062). 10 μL of RNAiMax Transfection Reagent (Thermo Fisher Scientific, 13778030) was diluted in 100 μL of Opti-MEM media. The RNAiMax solution was added to the siRNA solution and incubated at room temperature for 5 min and then added dropwise to cells for a final siRNA concentration of 25 nM. Cells treated with siSMAD2 were incubated for 24 hr, while cells treated with siKAP1 were incubated for 72 hr. Knockdown efficiency was determined by western blot and RT-qPCR analysis. For western blot, whole cell lysates were electrophoresed on an 8% SDS-PAGE and blots were probed with 1:1000 dilution of anti-SMAD2, 1:5000 dilution of anti-KAP1, and 1:10,000 of anti-Rhodamine actin. Anti-SMAD2 blot was exposed using Super Signal West Femto Luminol Enhancer.
HCEC 1CT cells (Roig et al., 2010) were transfected with siRNA’s through a reverse transfection protocol. 100 pmol siKAP1 and siNT and 5 uL RNAiMax were added to the bottom of a 6-well primaria plate and incubated with 1 mL Opti-MEM for 20 min at room temperature. Cells were trypsinized with Trypsin/EDTA (Thermo Fisher Scientific, R001100) and neutralized with trypsin neutralizing agent (Thermo Fisher Scientific, R002100). Cells were centrifuged at 1,000 g for 5 min, were resuspended in basal X media, and 200,000 cells were plated on each well. On the third day-post transfection, cells were stimulated for 2 and 8 hr with 10 ng/mL TGFβ−1 (PeproTech, 100–21) in 2 mL basal X media with water as vehicle. Following stimulation, cells were harvested using trypsin/trypsin neutralizing solution, and collected for both western blot and RT-qPCR analysis as described above. For detection of SMAD2 and PSMAD2, blots were probed with 1:1000 of anti-SMAD2 or anti-P-SMAD2 (Cell Signaling Technology, 8828).
MCF7 cells were transfected with siRNA’s through a reverse transfection protocol. 150 pmol siNT/siKAP1 (Qiagen, final concentration of ~18 nM) and 30 uL RNAiMAX (Thermo Fisher) were added to the bottom of a 10 cm plate and incubated with 5 mL OPTI-MEM (Thermo Fisher) for 20 min at room temperature. MCF7 Cells were trypsinized and resuspended in complete media, and 3.5×106 cells were added to each OPTIMEM mix in each plate (final volume 10 mL). Cells were collected three days post-transfection for western blotting, RT-qPCR and ChIP-qPCR (after crosslinking).
For siSMAD2 followed by ChIP-qPCR, 4.5×106 HCT116 cells were seeded into 10 cm plates and incubated overnight. The next day, 2.5 mL of media was removed from cells (final volume 7.5 mL). 150 pmol of the siSMAD2 pool and siNT were diluted in 500 uL Opti-MEM. 30 uL RNAiMax Transfection Reagent was also diluted in 500 uL Opti-MEM. The RNAiMax solution was added to the siRNA solution and incubated at room temperature for 5 min and then added dropwise to cells for a final siRNA concentration of ~18 nM. After 5 hr, 2.5 mL of new complete media was added to cells to a final volume of 10 mL. 48 hr post-transfection, cells were harvested for ChIP-qPCR.
KAP1 Deletion Construct Cloning
SMAD2 and WT KAP1 and deletion constructs were cloned into the pcDNA/4TO-FLAG vector (Thermo Fisher Scientific, V102020) (Jager et al., 2011) using the 5’-HindIII and 3’-XhoI restriction sites. Deletion construct inserts were PCR amplified by designing custom-made primers (Millipore Sigma, Table S6) targeting specific regions (as indicated in Figure 6A) of WT KAP1 that had been previously cloned into pcDNA4/TO (McNamara et al., 2016). Primers targeting SMAD2 were designed and used to amplify SMAD2 cDNA cloned into pENTR221 vector that was purchased from the UTSW McDermott Center for Human Genetics and obtained from The Ultimate ORF Lite human cDNA collection (Thermo Fisher Scientific). Amplified inserts and pcDNA4/TO-FLAG vector were then digested for 3 hr with HindIII and XhoI (NEB, R310L and R0146L, respectively) using CutSmart Buffer (NEB, B7204S). After 3 hr, 1,000 units of calf intestinal alkaline phosphatase (NEB, M0290) was added to pcDNA4/TO-FLAG and incubated overnight. Both the insert and pcDNA4/TO-FLAG vector were purified from the reactions using the Zymo DNA Clean and Concentrator Kit (Zymo Research, D4003). DNA concentrations were measured using the DeNovix DS-II FX+ Spectrophotometer. Inserts were then ligated into the pcDNA4/TO-FLAG vector at a 5:1 insert:vector ratio using the NEB Quick Ligase Kit (NEB, M2200L). Ligated DNA was then transformed into E. coli DH5α cells (NEB, 2987I) and grown overnight on LB + Ampicillin agar selection plates. Individual colonies were selected and grown in LB + Ampicillin liquid media cultures. Plasmids were purified using the Plasmid MiniPrep Kit. Purified plasmids were verified to contain the correct inserts by both HindIII/XhoI restriction analysis and Sanger Sequencing at the UTSW McDermott Sanger Sequencing Core. Expression of deletion constructs in shKAP1 HCT116 cells was confirmed by western blot. Blots were probed with 1:5000 anti-KAP1, 1:20,000 anti-FLAG, or 1:10,000 Rhodamine anti-Actin.
KAP1 PHD-BD (residues 614–835), KAP1 PHD (residues 624–673), TIF1α PHD-BD (residues 824–1011), and TIF1γ PHD-BD (residues 882–1087) were cloned into the pGEX2T N-terminal GST tagged vector (GE Life Sciences, 28954653) using the same method described above except EcoRI and BamHI restriction sites were used.
KAP1 shRNA Knockdown
pLKO.1 non-target (Sigma, SHC002) and KAP1 (Sigma, TRCN0000017998) vectors encoding directed shRNA’s were transfected along with gag/pol (pSPAX) and VSV-G (pDM2) into HEK 293T cells for expression of competent lentiviruses. Cell supernatants were collected two days post-transfection. Viral transduction was done by spinoculation using 2×105 cells, 8 μg/mL polybrene, and unsupplemented DMEM to a final volume of 1 mL at room temperature for 2 hr at 400 g. Transduced cells were selected with 1 μg/mL puromycin for 3 days post-infection. Cells were monitored for KD efficiency through standard western blot and RT-qPCR. For western blot, membranes were probed with 1:5000 diluted anti-KAP1 and 1:10,000 Rhodamine anti-Actin.
Reconstitution Assays
5.0×105 shNT and shKAP1 HCT116 cells were seeded into 6-well plates with 2 mL of complete cell culture media and grown for ~24 hr to ~70% confluency. The next day media was replaced with 1 mL of DMEM + 10% FBS. 1 μg of plasmid DNA encoding epitope-tagged proteins of interest were transfected into HCT116 cells by first diluting the plasmid in 100 μL of serum-free DMEM and diluting 3 μL of the PolyJet reagent in a separate tube of 100 μL serum-free DMEM. The PolyJet dilution was added to the plasmid dilution for a final volume of 200 μL and incubated for 10 min at room temperature. The Plasmid:PolyJet mix was then added dropwise to cells. Cells were incubated for 5 hr at 37°C with 5% CO2 and then media was replaced with 2 mL of fresh DMEM (10% FBS, 1% Pen/Strep). Cells were then incubated 48 hr at 37°C with 5% CO2. 1 mL of media was removed and then cells were harvested by scraping and pelleting at 1000 g for 5 min at 4°C. RT-qPCR was then used to analyze KAP1 target gene expression.
Recombinant Protein Purification for In Vitro Binding Assays
pGEX2T plasmids encoding GST, KAP1 PHD-BD, KAP1 PHD, KAP1 PHD-BD E662/663A, KAP1 PHD-BD L637A, TFI1α PHD-BD, and TIF1γ PHD-BD were transformed into BL21 DE3 E. coli (NEB, 2527I). Cultures were grown overnight in LB and then diluted 1:50 in LB. Cultures were grown to ~0.6 O.D. and then induced with 0.1 mM IPTG (Research Products International, 367-93-1) for 3 hr at 37°C. Cultures were centrifuged at 4,500 rpm at 4°C. Pellets were then washed 2X with cold 1X PBS and stored at −80°C. Bacteria pellets were thawed and resuspended in 5X pellet cell volume (PCV) of Buffer A (20 mM Tris-HCl pH 8.0, 150 mM NaCl, 1 mM EDTA, 10% glycerol, 0.1 mM ZnCl2, 1X Roche Protease Inhibitor Tablet). Lysozyme (Fisher Scientific, BP535) was then added to a final concentration of 1 mg/mL and samples incubated at 4°C for 15 min. Samples were then sonicated for 7 cycles with a tip sonicator (15 sec on, 15 sec off with the output setting at 5 and 50% duty cycle). Lysates then centrifuged for 15 min at 14,000 rpm and 4°C. Lysates filtered using a syringe and 0.25 μm syringe filter. For purification, 200 μL of a 50% GST agarose slurry (Millipore Sigma, G4510) was applied to a Bio-Rad Poly-Prep Chromatography Column (Bio-Rad, 731–1550) and equilibrated with 20 column volumes of Buffer A. Lysates were then applied to the column 2X to bind GST tagged proteins. Columns were washed with 20 column volumes of Buffer A. Proteins were eluted in four fractions in Buffer A + 10 mM L-Glutathione Reduced (Millipore Sigma, G4251) pH 9. A portion of each fraction was used to check for purity by Coomassie staining. Fractions were combined, concentrated, and buffer exchanged with Buffer A pH 8.0 using Amicon Ultra Spin Filters with a 10 kDa cutoff (Millipore Sigma, UFC501024). Protein concentration was determined using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, 23225). Proteins were aliquoted and stored at −80°C.
Nucleosome Binding Assays
2 μg of purified GST-tagged protein was incubated with 2 μg of recombinant biotinylated mono-nucleosomes (Epicypher, 16–0006) in protein LoBind tubes overnight at 4°C with rotation in 250 μL of nucleosome binding buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 2 mM DTT, 20% glycerol, 0.1 mM ZnCl2). Samples were then incubated with 40 μL of equilibrated 50% slurry StrepTactin agarose for 1 hr at 4°C with rotation. Beads were then washed 4X with 500 μL of nucleosome binding buffer and samples were then eluted by boiling in 50 μL of 2X Laemmli Buffer for 10 min. 5% of Input and 20% of elution used for western blotting. Western blots were probed with 1:5000 diluted anti-GST (Millipore Sigma, G7781).
Histone Peptide Binding Assays
2 μg of GST-tagged protein was incubated with 1 μg of histone peptide overnight in protein LoBind tubes at 4°C with rotation in 250 μL of peptide binding buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.1% NP-40, 0.1 mM ZnCl2). After binding, samples were incubated with 40 μL of equilibrated 50% slurry StrepTactin agarose for 1 hr at 4°C with rotation. Beads were then washed 4X with 1 mL of peptide wash buffer (50 mM Tris-HCl pH 7.5, 300 mM NaCl, 0.1% NP-40, 0.1 mM ZnCl2). Samples were eluted by boiling in 60 μL of 2X Laemmli Buffer for 10 min and 5% of Input and 20% of elution were then used for western blotting. Blots were performed as described for the Nucleosome Binding Assays.
NMR Titrations
The KAP1 PHD-BD construct was expressed in E. coli BL21 DE3 RIL cells grown in 15NH4Cl supplemented minimal media with 100 μM ZnCl2. Bacteria were harvested by centrifugation after induction with IPTG and lysed by sonication. The 15N-labeled GST-fusion protein was purified on glutathione Sepharose 4B beads, and ~1 mg/mL of GST tagged protein was cleaved with 125 U of Thrombin protease (MP Biomedicals, 0219492182) in 10 mL at 4°C overnight. The protein was concentrated into 20 mM Tris pH 6.8, in the presence of 150 mM NaCl and 5 mM DTT. NMR experiments were performed at 298 K on a Varian 600 MHz spectrometer at the University of Colorado School of Medicine NMR core facility. 1H,15N HSQC spectra of 0.2 mM uniformly 15N-labelled KAP1 BD-PHD were collected while histone H4 peptide (aa 1–23) was gradually added. Histograms were generated by calculating the normalized chemical shift change between apo and peptide-bound (1:5 ratio) protein states per residue and using the equation:
where δ is the chemical shift in p.p.m.
Molecular Dynamics (MD) Simulations
All molecular dynamics simulations steps reported in this work were performed using GROMACS 5 with the AMBER99SB-ILDN (Lindorff-Larsen et al., 2010) force field and simulation step size of 1 fs (where relevant). An extended, 23 amino acid long H4 peptide was generated in UCSF Chimera (Pettersen et al., 2004) and placed in a 302 nm3 dodecahedron simulation box containing one of the models from the NMR ensemble of KAP1 (PDB: 2RO1). Since there were 20 such models in the PDB file, we started with 20 separate simulation boxes for generating the binding model. Each of these 20 boxes were solvated with TIP3P water model and the charge on the system was neutralized by addition of five chloride ions. The system was then energy minimized using steepest descent and conjugate gradient methods followed by 50 ps of position restraints on the solute (KAP1 and H4) while the temperature of the box was raised to 200 K (by coupling to an external bath). Next, the position restraints were removed, and the system was simulated under NVT [constant number of particles in the system (symbol: N), system’s volume (symbol: V) and system’s absolute temperature in K (symbol:T)] conditions for 100 ps. Finally, the box was heated to the final simulation temperature of 298.15 K and equilibrated under NPT [constant number of particles in the system (symbol: N), system’s pressure (symbol: P) and system’s absolute temperature in K (symbol T)] conditions for a further 5 ns. During all of these steps, the ZN atoms were restrained at their original position to maintain the Zn fingers as much as possible. The equilibration was monitored using the pairwise root mean square distance (RMSD) of the solute configuration during the simulation with the corresponding model structure in 2RO1.pdb. At the end of equilibration, the 20 trajectories were ranked on the following criteria – (i) absence of unusual contacts between the solute and solvent molecules that could lead to simulation crashes, (ii) stable PHD and BD domain structures, and (ii) non-redundant contact modes between H4 and PHD – and chose the top four systems as the starting KAP1:H4 complex structure for further production runs. Here, the four trajectories were restrained in parallel with the 168 amide chemical shifts measured for the KAP1:H4 complex as described in section above. The instantaneous chemical shifts during the restrained simulations were calculated as ensemble average of the configurations of the KAP1:H4 complex in the four replicas using the PLUMED 2.1 patch for GROMACS. Another set of unrestrained molecular dynamics simulation was also run for a total of 250 ns for comparing the effect of the chemical shift restraints on the system.
Site-directed Mutagenesis
KAP1 PHD domain point mutants were created using the Quick Change II Site-Directed Mutagenesis Kit (Agilent, 200522). Briefly, mutagenesis primers were designed using the Agilent QuickChange Primer Design online tool (http://www.agilent.com/store/primerDesignProgram.jsp). Manufacturer instructions were followed to amplify KAP1 point mutant plasmids. Amplified plasmids were then transformed into E. coli XL-10 Gold (included with mutagenesis kit). Point mutation sequences were confirmed using Sanger Sequencing performed at the UTSW McDermott Sanger Sequencing Core. Expression of KAP1 point mutants was confirmed through western blot analysis of whole cell lysate of HCT116 shKAP1 cells transiently expressing each point mutant (see Reconstitution Assays). Blots were probed with 1:5000 diluted anti-KAP1, 1:10,000, Rhodamine anti-Actin, or 1:20,000 anti-FLAG.
QUANTIFICATION AND STATISTICAL ANALYSIS
All qPCR, cell growth and colony formation data was processed and visualized using GraphPad Prism v8.1.0. For all cell growth assays, P-values were determined by 2-way ANOVA. For the rest of the assays (RT-qPCR, ChIP-qPCR) P-values were determined by unpaired Student’s t-test. P-values were indicated in the following manner: *P<0.05, *P<0.01, ***P<0.001, n.s. denotes non-significant. We considered P<0.05 to be statistically significant.
4sU-seq and RNA-seq Analysis
Raw data files for Ctrl and KO1 samples were checked for quality control with fastqc/0.11.2. Low quality reads and adapter contaminations were removed using Trimgalore v0.4.1. Reads were mapped to the human reference genome, hg38, using STAR/2.6.1b (Dobin et al., 2013). Spike-ins were mapped to a fasta file of ERCC sequences provided by the manufacturer. featureCounts (as part of the subread/1.6.1 package) was used to calculate counts across the entire gene body of protein coding genes, as well as, ERCC spike-ins (Liao et al., 2014). RUVSeq/1.15 was used for differential gene expression analysis, PCA analysis, and to create MA plots (Risso et al., 2014). A gene was considered a KAP1 target gene if it was differentially expressed in the same direction (FDR<0.05) in both the 4sU- and RNA-seq datasets, as well as, if it contained KAP1 in its promoter region as determined by our group’s previously generated KAP1 ChIP-seq dataset in HCT116 cells (McNamara et al., 2016). Signal tracks were generated using RPM normalized bedGraph outputs from STAR and bedGraphtTobigWig (as part of UCSC_userApps/v317) then visualized with the Integrated Genome Viewer (Robinson et al., 2011). Δ4sU-seq heatmap matrices were generated using the bigwigCompare function in deepTools/2.5.0 (Ramirez et al., 2016) sorting to total Pol II heatmap matrices (see ChIP-seq Analysis). Matrices were loaded into Java Treeview for visualization (Saldanha, 2004). The KO2 sample was analyzed like the KO1 sample except instead of normalizing to ERCC spike-ins, the RUVr function of RUVSeq/1.15 was used.
ChIP-seq Analysis
Raw data files were checked for quality control with fastqc/0.11.2. Low quality reads and adapter contaminations were removed using Trimgalore v0.4.1. Reads were mapped to the human reference genome, hg38, using bowtie2/2.1.0. (Langmead and Salzberg, 2012). Reads were also mapped to the dm6 Drosophila genome to extract reads originating from the spike-ins chromatin. Duplicates were marked and removed using picard/1.127. Input subtracted peak and bedgraph files for both human aligned and Drosophila aligned reads were generated with macs/2.1.0–20151222 (FDR<0.05) (Zhang et al., 2008). For normalization, a normalization factor was generated by dividing the Drosophila read counts from samples with the highest read counts to the sample containing the least read counts (Egan et al., 2016; Orlando et al., 2014). This normalization factor was then used to normalize the signal in the sample with the higher Drosophila read counts to the sample with the lowest. BigWig tracks were generated from the Drosophila normalized bedGraph files using bedGraphToBigWig, as part of UCSC_userApps/v317, and visualized with the Integrated Genome Viewer (Robinson et al., 2011). Genes selected for downstream analysis had to be differentially expressed in the KO1 cells (see 4sU-/RNA-seq Analysis) and contain a total Pol II peak in their promoter region (−100/+500 bp relative to the TSS). If multiple isoforms of a gene contained total Pol II in their promoter region then the isoform with the highest peak signal was selected. ΔTotal Pol II bigwig files were created from Ctrl and KO1 bigWig files using the bigwigCompare function in deepTools/2.5.0. k-means clustered Δtotal Pol II heatmap matrices were then generated using deepTools/2.5.0 and centered on the TSS. Multiple flanking region widths (−/+ 0.5 kb, −/+ 1 kb, and −/+ 2 kb were used to determine the optimal window for analysis. While all three windows tested gave similar outputs (data not shown), 2 kb flanking regions were chosen for the final analysis because we observed Pol II occupancy further downstream the typical −0.1/+0.5 kb window used for PI analysis. All remaining heatmap matrices were then arranged into the same clusters with each cluster sorted by decreasing total Pol II using deepTools/2.5.0. All heatmaps were visualized using Java Treeview. Log2(S2P/total) bigwig signal tracks were created using the bigwigCompare function in deepTools/2.5.0. All metagene plots were generated using the scale-regions function in deepTools/2.5.0. Pausing indices were calculated by first measuring the promoter bound Pol II (−100/+500 bp relative to the TSS) and gene body (+501 bp to the end of gene) signals using the scale-regions function in deepTools/2.5.0. The resulting matrix file was then loaded into R/3.4.1 and the pausing index was taken to be the log2 ratio of promoter bound to gene body bound Pol II. ECDF plots were created in R/3.4.1 using the ecdf function. KEGG pathway analysis of genes from each cluster was performed using Metascape (http://metascape.org) (Tripathi et al., 2015).
TCGA Analysis
Clinical information, gene expression and copy number analysis (RNA-seq) data for the indicated genes were obtained from The Cancer Genome Atlas (TCGA) for CRC using the FIREBROWSE portal (http://firebrowse.org). The cohort consists of tissues from 623 CRC patients with primary tumors who underwent tumor resection. For Tumor (T) vs No Tumor (NT) comparison, a sub-cohort of 50 CRC cases (a combination of colon adenocarcinoma and rectal adenocarcinoma patient data) of paired samples from both types of tissues were used to calculate the individual fold changes. Then, log2 fold change values were used for calculating the average of fold change expression for KAP1. Similarly, paired data for Bladder Urothelial Carcinoma (BLCA, n=19), Breast Invasive Carcinoma (BRCA, n=112), Cholangiocarcinoma (CHOL, n=9), Head and Neck Squamous Cell Carcinoma (HNSC, n=43), Pan-Kidney Cancer Cohort (KIPAN, n=129), Liver Hepatocellular Carcinoma (LIHC, n=50), Lung Adenocarcinoma (LUAD, n=58), Lung Squamous Cell Carcinoma (LUSC, n=51), Prostate Adenocarcinoma (PRAD, n=52), Stomach and Esophageal Carcinoma (STES, n=43), Thyroid Carcinoma (THCA, n=59) and Uterine Corpus Endometrial Carcinoma (UCEC, n=23) obtained from the TCGA was analyzed. For survival analysis, the longest time the patients were followed up or known to be alive or free of tumor was considered for the overall survival analysis (OS, n=619) or disease-free survival analysis (DFS, n=539), respectively. Gene expression levels were separated in quartiles and 1st and 4th quartile was considered as group of patients with high-expression and low-expression (OS n=154, DFS=135), respectively. Gene upregulation was correlated with the overall survival using Kaplan-Meier tables and compared with the log-rank test using GraphPad prism. Hazard ratios were calculated using Cox regression. For correlation between KAP1 and cluster 1 genes a Pearson correlation analysis was performed using the expression data of 623 CRC patients.
MNase-seq Analysis
Raw data files were checked for quality control; low quality reads and adapter contaminations were removed using Trimgalore v0.4.1. High quality reads (MAPQ >20) were aligned to the human genome assembly hg38 using Bowtie2 v2.3.3 and further processed using SAMtools v1.6 (Li et al., 2009). R v3.4.1 scripts adapted from bamR package (https://github.com/rchereji/bamR) were used for analysis of MNase-seq data. To precisely map nucleosome positions, mono-nucleosomal DNA fragments with a size of 120–180 bp were used for the analysis. Normalized nucleosome dyad distributions were computed by rescaling the average occupancy to 1, for each chromosome. A custom R script was used to calculate average dyads density around TSS of Pol II-transcribed genes and for plotting of nucleosome profiles in HCT116 Ctrl and KO1 cells. R script from bamR package (Ocampo et al., 2016) was used to estimate the average nucleosome spacing from aligned nucleosome dyads. The algorithm used to detect location of the nucleosome free region (NFR) and of the two nucleosomes upstream the TSS of each gene is described (Ocampo et al., 2016) and provided in the GitHub repository (https://github.com/rchereji/Detect_NDRs_and_flanking_nucleosomes). The nucleosome coordinates were determined in two replicates separately and only coordinates that differ in less than 50 bp difference between replicates were selected for further analysis. Shifts in the NFR width were calculated for each gene by subtracting the values of control from KAP1 KO samples. Heatmaps were generated using custom R script with nucleosome dyads aligned to center of the NFR.
Motif Analysis
Motif analysis was performed on promoter regions (−100/+100 relative to the TSS) of genes from each cluster using the CentriMo tool and using the HOCOMOCO motif database (Kulakovskiy et al., 2018) as a reference as part of the MEME suite (Bailey et al., 2009; Bailey and Machanick, 2012).
RT-qPCR Analysis
All RT-qPCR data was analyzed using the ΔΔCt method. Graphs were created using GraphPad Prism v8.1.0. Statistical significance was determined using an unpaired t-test in GraphPad Prism unless otherwise noted.
ChIP-qPCR Analysis
Graphs and statistical analysis was performed as described for RT-qPCR Analysis.
Western Blot Quantification
Western blots were quantified by determining the band intensity using Image Lab 6.0. If comparing protein levels in cell lysates, band intensity was first normalized to β-actin, to account for differences in loading, and then to the control condition. For quantification of in vitro nucleosome and peptide binding assays, band intensity was normalized to unmodified H4 or WT KAP1 where appropriate.
Supplementary Material
Table S1. Genes Differentially Expressed After KAP1 Depletion. Related to Figure 1
Table S2. Genes with KAP1 Bound Promoters. Related to Figure 1
Table S3. KEGG Pathway Analysis of KAP1 Regulated Genes in MCF7 Breast Cancer Cells. Related to Figure 2
Table S4. Tissue Samples Used for TCGA Analysis. Related to Figure 3
Table S5. TCGA Analysis. Related to Figure 3
Highlights.
KAP1 sustains transcriptional programs commonly dysregulated in cancer patients
KAP1’s chromatin reader domain directly binds unmodified and hypo-acetylated H4 tails
KAP1 binds chromatin to scaffold a pathway-specific transcription complex
KAP1-H4 binding couples the establishment of promoter-bound Pol II with pause release
ACKNOWLEDGEMENTS
We thank members of the D’Orso laboratory for critical reading of the manuscript, L. Kraus, N. Conrad and D. Corey for discussions, Y. Zhang for help with NMR data analysis, and the UTSW McDermott Center. Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases (NIAID) of the NIH under award number R01AI114362, Welch Foundation grant number I-1782, and CPRIT grant number RP170572 (to I.D.), C.B. was supported by The Graduate Research Fellowship Program from the National Science Foundation (No. 2016220513). U.H. was partially funded through the Green Fellows program and the SURF program at the University of Texas at Dallas and UTSW, respectively. A.N.B. was supported by the Sir Henry Wellcome Postdoctoral Fellowship from the Wellcome Trust, UK under award reference 103963/Z/14/Z and the Anne McLaren Fellowship at University of Nottingham. T.G.K. is supported by grants from the NIH. C.-M.C.’s research is supported by NIH grant 1RO1CA251698-01, CPRIT grants RP180349 and RP190077, and Welch Foundation grant I-1805.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests
SUPPLEMENTAL INFORMATION
Supplemental Information contains 6 figures and can be found online.
References
- Agricola E, Randall RA, Gaarenstroom T, Dupont S, and Hill CS (2011). Recruitment of TIF1gamma to chromatin via its PHD finger-bromodomain activates its ubiquitin ligase and transcriptional repressor activities. Mol Cell 43, 85–96. [DOI] [PubMed] [Google Scholar]
- Ahn SH, Kim M, and Buratowski S (2004). Phosphorylation of serine 2 within the RNA polymerase II C-terminal domain couples transcription and 3’ end processing. Mol Cell 13, 67–76. [DOI] [PubMed] [Google Scholar]
- Allis CD, and Jenuwein T (2016). The molecular hallmarks of epigenetic control. Nat Rev Genet 17, 487–500. [DOI] [PubMed] [Google Scholar]
- Bacon CW, and D’Orso I (2018). CDK9: a signaling hub for transcriptional control. Transcription, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, and Noble WS (2009). MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37, W202–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, and Machanick P (2012). Inferring direct DNA binding from ChIP-seq. Nucleic Acids Res 40, e128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barboric M, Nissen RM, Kanazawa S, Jabrane-Ferrat N, and Peterlin BM (2001). NF-kappaB binds P-TEFb to stimulate transcriptional elongation by RNA polymerase II. Mol Cell 8, 327–337. [DOI] [PubMed] [Google Scholar]
- Berg KCG, Eide PW, Eilertsen IA, Johannessen B, Bruun J, Danielsen SA, Bjornslett M, Meza-Zepeda LA, Eknaes M, Lind GE, et al. (2017). Multi-omics of 34 colorectal cancer cell lines - a resource for biomedical studies. Mol Cancer 16, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunch H, Zheng X, Burkholder A, Dillon ST, Motola S, Birrane G, Ebmeier CC, Levine S, Fargo D, Hu G, et al. (2014). TRIM28 regulates RNA polymerase II promoter-proximal pausing and pause release. Nat Struct Mol Biol 21, 876–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buratowski S (2012). Gene expression: transcription initiation unwrapped. Nature 483, 286–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calon A, Espinet E, Palomo-Ponce S, Tauriello DV, Iglesias M, Cespedes MV, Sevillano M, Nadal C, Jung P, Zhang XH, et al. (2012). Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell 22, 571–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N (2012). Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capili AD, Schultz DC, Rauscher IF, and Borden KL (2001). Solution structure of the PHD domain from the KAP-1 corepressor: structural determinants for PHD, RING and LIM zinc-binding domains. EMBO J 20, 165–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman RD, Heidemann M, Albert TK, Mailhammer R, Flatley A, Meisterernst M, Kremmer E, and Eick D (2007). Transcribing RNA polymerase II is phosphorylated at CTD residue serine-7. Science 318, 1780–1782. [DOI] [PubMed] [Google Scholar]
- Chen F, Gao X, and Shilatifard A (2015). Stably paused genes revealed through inhibition of transcription initiation by the TFIIH inhibitor triptolide. Genes Dev 29, 39–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen FX, Smith ER, and Shilatifard A (2018). Born to run: control of transcription elongation by RNA polymerase II. Nat Rev Mol Cell Biol. [DOI] [PubMed] [Google Scholar]
- Chen L, Munoz-Antonia T, and Cress WD (2014). Trim28 contributes to EMT via regulation of E-cadherin and N-cadherin in lung cancer cell lines. PLoS One 9, e101040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng B, Ren X, and Kerppola TK (2014). KAP1 represses differentiation-inducible genes in embryonic stem cells through cooperative binding with PRC1 and derepresses pluripotency-associated genes. Mol Cell Biol 34, 2075–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho EJ, Kobor MS, Kim M, Greenblatt J, and Buratowski S (2001). Opposing effects of Ctk1 kinase and Fcp1 phosphatase at Ser 2 of the RNA polymerase II C-terminal domain. Genes Dev 15, 3319–3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Core LJ, Waterfall JJ, Gilchrist DA, Fargo DC, Kwak H, Adelman K, and Lis JT (2012). Defining the status of RNA polymerase at promoters. Cell Rep 2, 1025–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Y, Yang S, Fu X, Feng J, Xu S, and Ying G (2014). High levels of KAP1 expression are associated with aggressive clinical features in ovarian cancer. Int J Mol Sci 16, 363–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Orso I, and Frankel AD (2010). RNA-mediated displacement of an inhibitory snRNP complex activates transcription elongation. Nat Struct Mol Biol 17, 815–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalgaard K, Landgraf K, Heyne S, Lempradl A, Longinotto J, Gossens K, Ruf M, Orthofer M, Strogantsev R, Selvaraj M, et al. (2016). Trim28 Haploinsufficiency Triggers Bistable Epigenetic Obesity. Cell 164, 353–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David CJ, and Massague J (2018). Contextual determinants of TGFbeta action in development, immunity and cancer. Nat Rev Mol Cell Biol 19, 419–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JM, Gao J, Wang J, Yang M, and Potts PR (2010). MAGE-RING protein complexes comprise a family of E3 ubiquitin ligases. Mol Cell 39, 963–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan B, Yuan CC, Craske ML, Labhart P, Guler GD, Arnott D, Maile TM, Busby J, Henry C, Kelly TK, et al. (2016). An Alternative Approach to ChIP-Seq Normalization Enables Detection of Genome-Wide Changes in Histone H3 Lysine 27 Trimethylation upon EZH2 Inhibition. PLoS One 11, e0166438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eick D, and Geyer M (2013). The RNA polymerase II carboxy-terminal domain (CTD) code. Chem Rev 113, 8456–8490. [DOI] [PubMed] [Google Scholar]
- Erickson B, Sheridan RM, Cortazar M, and Bentley DL (2018). Dynamic turnover of paused Pol II complexes at human promoters. Genes Dev 32, 1215–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong KW, Zhao JC, Song B, Zheng B, and Yu J (2018). TRIM28 protects TRIM24 from SPOP-mediated degradation and promotes prostate cancer progression. Nat Commun 9, 5007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonti G, Marcaida M, Bryan L, Traeger S, Kalantzi A, Helleboid P-Y, Demurtas D, Tully M, Grudinin S, Trono D, et al. (2019). KAP1 is an antiparallel dimer with a natively functional asymmetry. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JR, Fredericks WJ, Jensen DE, Speicher DW, Huang XP, Neilson EG, and Rauscher FJ 3rd (1996). KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev 10, 2067–2078. [DOI] [PubMed] [Google Scholar]
- Gaertner B, Johnston J, Chen K, Wallaschek N, Paulson A, Garruss AS, Gaudenz K, De Kumar B, Krumlauf R, and Zeitlinger J (2012). Poised RNA polymerase II changes over developmental time and prepares genes for future expression. Cell Rep 2, 1670–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gawel DR, Lee EJ, Li X, Lilja S, Matussek A, Schafer S, Olsen RS, Stenmarker M, Zhang H, and Benson M (2019). An algorithm-based meta-analysis of genome- and proteome-wide data identifies a combination of potential plasma biomarkers for colorectal cancer. Sci Rep 9, 15575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist DA, Dos Santos G, Fargo DC, Xie B, Gao Y, Li L, and Adelman K (2010). Pausing of RNA polymerase II disrupts DNA-specified nucleosome organization to enable precise gene regulation. Cell 143, 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist DA, Nechaev S, Lee C, Ghosh SK, Collins JB, Li L, Gilmour DS, and Adelman K (2008). NELF-mediated stalling of Pol II can enhance gene expression by blocking promoter-proximal nucleosome assembly. Genes Dev 22, 1921–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes NP, Bjerke G, Llorente B, Szostek SA, Emerson BM, and Espinosa JM (2006). Gene-specific requirement for P-TEFb activity and RNA polymerase II phosphorylation within the p53 transcriptional program. Genes Dev 20, 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gressel S, Schwalb B, Decker TM, Qin W, Leonhardt H, Eick D, and Cramer P (2017). CDK9-dependent RNA polymerase II pausing controls transcription initiation. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grunberg S, and Hahn S (2013). Structural insights into transcription initiation by RNA polymerase II. Trends Biochem Sci 38, 603–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudipaty SA, McNamara RP, Morton EL, and D’Orso I (2015). PPM1G Binds 7SK RNA and Hexim1 To Block P-TEFb Assembly into the 7SK snRNP and Sustain Transcription Elongation. Mol Cell Biol 35, 3810–3828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hector S, Chen H, Kijanka G, Murray F, and Prehn JH (2012). A reverse-ELISA for the detection of TRIM28/KAP1 serum autoantibodies in colorectal cancer patients. Acta Oncol 51, 394–396. [DOI] [PubMed] [Google Scholar]
- Henriques T, Gilchrist DA, Nechaev S, Bern M, Muse GW, Burkholder A, Fargo DC, and Adelman K (2013). Stable pausing by RNA polymerase II provides an opportunity to target and integrate regulatory signals. Mol Cell 52, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herquel B, Ouararhni K, Khetchoumian K, Ignat M, Teletin M, Mark M, Bechade G, Van Dorsselaer A, Sanglier-Cianferani S, Hamiche A, et al. (2011). Transcription cofactors TRIM24, TRIM28, and TRIM33 associate to form regulatory complexes that suppress murine hepatocellular carcinoma. Proc Natl Acad Sci U S A 108, 8212–8217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CS (2016). Transcriptional Control by the SMADs. Cold Spring Harb Perspect Biol 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho J, Kong JW, Choong LY, Loh MC, Toy W, Chong PK, Wong CH, Wong CY, Shah N, and Lim YP (2009). Novel breast cancer metastasis-associated proteins. J Proteome Res 8, 583–594. [DOI] [PubMed] [Google Scholar]
- Imhof A, Schuierer M, Werner O, Moser M, Roth C, Bauer R, and Buettner R (1999). Transcriptional regulation of the AP-2alpha promoter by BTEB-1 and AP-2rep, a novel wt-1/egr-related zinc finger repressor. Mol Cell Biol 19, 194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov AV, Peng H, Yurchenko V, Yap KL, Negorev DG, Schultz DC, Psulkowski E, Fredericks WJ, White DE, Maul GG, et al. (2007). PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol Cell 28, 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager S, Cimermancic P, Gulbahce N, Johnson JR, McGovern KE, Clarke SC, Shales M, Mercenne G, Pache L, Li K, et al. (2011). Global landscape of HIV-human protein complexes. Nature 481, 365–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung M, Philpott M, Muller S, Schulze J, Badock V, Eberspacher U, Moosmayer D, Bader B, Schmees N, Fernandez-Montalvan A, et al. (2014). Affinity map of bromodomain protein 4 (BRD4) interactions with the histone H4 tail and the small molecule inhibitor JQ1. J Biol Chem 289, 9304–9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kershnar E, Wu SY, and Chiang CM (1998). Immunoaffinity purification and functional characterization of human transcription factor IIH and RNA polymerase II from clonal cell lines that conditionally express epitope-tagged subunits of the multiprotein complexes. J Biol Chem 273, 34444–34453. [DOI] [PubMed] [Google Scholar]
- Kulakovskiy IV, Vorontsov IE, Yevshin IS, Sharipov RN, Fedorova AD, Rumynskiy EI, Medvedeva YA, Magana-Mora A, Bajic VB, Papatsenko DA, et al. (2018). HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res 46, D252–D259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Ilin S, Wang W, Duncan EM, Wysocka J, Allis CD, and Patel DJ (2006). Molecular basis for site-specific read-out of histone H3K4me3 by the BPTF PHD finger of NURF. Nature 442, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Xi Y, Li W, McCarthy RL, Stratton SA, Zou W, Li W, Dent SY, Jain AK, and Barton MC (2017). TRIM28 interacts with EZH2 and SWI/SNF to activate genes that promote mammosphere formation. Oncogene 36, 2991–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, and Shaw DE (2010). Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins 78, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lis JT, Mason P, Peng J, Price DH, and Werner J (2000). P-TEFb kinase recruitment and function at heat shock loci. Genes Dev 14, 792–803. [PMC free article] [PubMed] [Google Scholar]
- Liu L, Xu Y, He M, Zhang M, Cui F, Lu L, Yao M, Tian W, Benda C, Zhuang Q, et al. (2014). Transcriptional pause release is a rate-limiting step for somatic cell reprogramming. Cell Stem Cell 15, 574–588. [DOI] [PubMed] [Google Scholar]
- Liu L, Zhao E, Li C, Huang L, Xiao L, Cheng L, Huang X, Song Y, and Xu D (2013a). TRIM28, a new molecular marker predicting metastasis and survival in early-stage non-small cell lung cancer. Cancer Epidemiol 37, 71–78. [DOI] [PubMed] [Google Scholar]
- Liu W, Ma Q, Wong K, Li W, Ohgi K, Zhang J, Aggarwal A, and Rosenfeld MG (2013b). Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell 155, 1581–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maniatis T, and Reed R (2002). An extensive network of coupling among gene expression machines. Nature 416, 499–506. [DOI] [PubMed] [Google Scholar]
- Marshall NF, Peng J, Xie Z, and Price DH (1996). Control of RNA polymerase II elongation potential by a novel carboxyl-terminal domain kinase. J Biol Chem 271, 27176–27183. [DOI] [PubMed] [Google Scholar]
- Massague J (2008). TGFbeta in Cancer. Cell 134, 215–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J (2012). TGFbeta signalling in context. Nat Rev Mol Cell Biol 13, 616–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNamara RP, Reeder JE, McMillan EA, Bacon CW, McCann JL, and D’Orso I (2016). KAP1 Recruitment of the 7SK snRNP Complex to Promoters Enables Transcription Elongation by RNA Polymerase II. Mol Cell 61, 39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miles DC, de Vries NA, Gisler S, Lieftink C, Akhtar W, Gogola E, Pawlitzky I, Hulsman D, Tanger E, Koppens M, et al. (2017). TRIM28 is an Epigenetic Barrier to Induced Pluripotent Stem Cell Reprogramming. Stem Cells 35, 147–157. [DOI] [PubMed] [Google Scholar]
- Miller TE, Liau BB, Wallace LC, Morton AR, Xie Q, Dixit D, Factor DC, Kim LJY, Morrow JJ, Wu Q, et al. (2017). Transcription elongation factors represent in vivo cancer dependencies in glioblastoma. Nature 547, 355–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muse GW, Gilchrist DA, Nechaev S, Shah R, Parker JS, Grissom SF, Zeitlinger J, and Adelman K (2007). RNA polymerase is poised for activation across the genome. Nat Genet 39, 1507–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musselman CA, Lalonde ME, Cote J, and Kutateladze TG (2012). Perceiving the epigenetic landscape through histone readers. Nat Struct Mol Biol 19, 1218–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nechaev S, Fargo DC, dos Santos G, Liu L, Gao Y, and Adelman K (2010). Global analysis of short RNAs reveals widespread promoter-proximal stalling and arrest of Pol II in Drosophila. Science 327, 335–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen VT, Kiss T, Michels AA, and Bensaude O (2001). 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 414, 322–325. [DOI] [PubMed] [Google Scholar]
- Nielsen AL, Ortiz JA, You J, Oulad-Abdelghani M, Khechumian R, Gansmuller A, Chambon P, and Losson R (1999). Interaction with members of the heterochromatin protein 1 (HP1) family and histone deacetylation are differentially involved in transcriptional silencing by members of the TIF1 family. EMBO J 18, 6385–6395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ocampo J, Chereji RV, Eriksson PR, and Clark DJ (2016). The ISW1 and CHD1 ATP-dependent chromatin remodelers compete to set nucleosome spacing in vivo. Nucleic Acids Res 44, 4625–4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlando DA, Chen MW, Brown VE, Solanki S, Choi YJ, Olson ER, Fritz CC, Bradner JE, and Guenther MG (2014). Quantitative ChIP-Seq normalization reveals global modulation of the epigenome. Cell Rep 9, 1163–1170. [DOI] [PubMed] [Google Scholar]
- Pena PV, Davrazou F, Shi X, Walter KL, Verkhusha VV, Gozani O, Zhao R, and Kutateladze TG (2006). Molecular mechanism of histone H3K4me3 recognition by plant homeodomain of ING2. Nature 442, 100–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H, Begg GE, Schultz DC, Friedman JR, Jensen DE, Speicher DW, and Rauscher FJ 3rd (2000). Reconstitution of the KRAB-KAP-1 repressor complex: a model system for defining the molecular anatomy of RING-B box-coiled-coil domain-mediated protein-protein interactions. J Mol Biol 295, 1139–1162. [DOI] [PubMed] [Google Scholar]
- Peng H, Feldman I, and Rauscher FJ 3rd (2002). Hetero-oligomerization among the TIF family of RBCC/TRIM domain-containing nuclear cofactors: a potential mechanism for regulating the switch between coactivation and corepression. J Mol Biol 320, 629–644. [DOI] [PubMed] [Google Scholar]
- Peng J, Marshall NF, and Price DH (1998). Identification of a cyclin subunit required for the function of Drosophila P-TEFb. J Biol Chem 273, 13855–13860. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Rabani M, Levin JZ, Fan L, Adiconis X, Raychowdhury R, Garber M, Gnirke A, Nusbaum C, Hacohen N, Friedman N, et al. (2011). Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat Biotechnol 29, 436–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahl PB, Lin CY, Seila AC, Flynn RA, McCuine S, Burge CB, Sharp PA, and Young RA (2010). c-Myc regulates transcriptional pause release. Cell 141, 432–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen EB, and Lis JT (1993). In vivo transcriptional pausing and cap formation on three Drosophila heat shock genes. Proc Natl Acad Sci U S A 90, 7923–7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risso D, Ngai J, Speed TP, and Dudoit S (2014). Normalization of RNA-seq data using factor analysis of control genes or samples. Nat Biotechnol 32, 896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roig AI, Eskiocak U, Hight SK, Kim SB, Delgado O, Souza RF, Spechler SJ, Wright WE, and Shay JW (2010). Immortalized epithelial cells derived from human colon biopsies express stem cell markers and differentiate in vitro. Gastroenterology 138, 1012–1021 e1011–1015. [DOI] [PubMed] [Google Scholar]
- Ross S, Cheung E, Petrakis TG, Howell M, Kraus WL, and Hill CS (2006). Smads orchestrate specific histone modifications and chromatin remodeling to activate transcription. EMBO J 25, 4490–4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg AJ, Li H, Milne TA, Dewell S, McGinty RK, Yuen M, Ueberheide B, Dou Y, Muir TW, Patel DJ, et al. (2011). Recognition of a mononucleosomal histone modification pattern by BPTF via multivalent interactions. Cell 145, 692–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan RF, Schultz DC, Ayyanathan K, Singh PB, Friedman JR, Fredericks WJ, and Rauscher FJ 3rd (1999). KAP-1 corepressor protein interacts and colocalizes with heterochromatic and euchromatic HP1 proteins: a potential role for Kruppel-associated box-zinc finger proteins in heterochromatin-mediated gene silencing. Mol Cell Biol 19, 4366–4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha AJ (2004). Java Treeview--extensible visualization of microarray data. Bioinformatics 20, 3246–3248. [DOI] [PubMed] [Google Scholar]
- Saxton RA, and Sabatini DM (2017). mTOR Signaling in Growth, Metabolism, and Disease. Cell 168, 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz DC, Friedman JR, and Rauscher FJ 3rd (2001). Targeting histone deacetylase complexes via KRAB-zinc finger proteins: the PHD and bromodomains of KAP-1 form a cooperative unit that recruits a novel isoform of the Mi-2alpha subunit of NuRD. Genes Dev 15, 428–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki Y, Kurisaki A, Watanabe-Susaki K, Nakajima Y, Nakanishi M, Arai Y, Shiota K, Sugino H, and Asashima M (2010). TIF1beta regulates the pluripotency of embryonic stem cells in a phosphorylation-dependent manner. Proc Natl Acad Sci U S A 107, 10926–10931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao W, and Zeitlinger J (2017). Paused RNA polymerase II inhibits new transcriptional initiation. Nat Genet 49, 1045–1051. [DOI] [PubMed] [Google Scholar]
- Shiromizu T, Kume H, Ishida M, Adachi J, Kano M, Matsubara H, and Tomonaga T (2017). Quantitation of putative colorectal cancer biomarker candidates in serum extracellular vesicles by targeted proteomics. Sci Rep 7, 12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shogren-Knaak M, Ishii H, Sun JM, Pazin MJ, Davie JR, and Peterson CL (2006). Histone H4-K16 acetylation controls chromatin structure and protein interactions. Science 311, 844–847. [DOI] [PubMed] [Google Scholar]
- Smith E, Lin C, and Shilatifard A (2011). The super elongation complex (SEC) and MLL in development and disease. Genes Dev 25, 661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sripathy SP, Stevens J, and Schultz DC (2006). The KAP1 corepressor functions to coordinate the assembly of de novo HP1-demarcated microenvironments of heterochromatin required for KRAB zinc finger protein-mediated transcriptional repression. Mol Cell Biol 26, 8623–8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh H, Ficarro SB, Kang UB, Chun Y, Marto JA, and Buratowski S (2016). Direct Analysis of Phosphorylation Sites on the Rpb1 C-Terminal Domain of RNA Polymerase II. Mol Cell 61, 297–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Parmely TJ, Sato S, Tomomori-Sato C, Banks CA, Kong SE, Szutorisz H, Swanson SK, Martin-Brown S, Washburn MP, et al. (2011). Human mediator subunit MED26 functions as a docking site for transcription elongation factors. Cell 146, 92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka S, Pfleger C, Lai JF, Roan F, Sun SC, and Ziegler SF (2018). KAP1 Regulates Regulatory T Cell Function and Proliferation in Both Foxp3-Dependent and -Independent Manners. Cell Rep 23, 796–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taverna SD, Li H, Ruthenburg AJ, Allis CD, and Patel DJ (2007). How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat Struct Mol Biol 14, 1025–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MC, and Chiang CM (2006). The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol 41, 105–178. [DOI] [PubMed] [Google Scholar]
- Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LC, et al. (2015). Meta- and Orthogonal Integration of Influenza “OMICs” Data Defines a Role for UBR4 in Virus Budding. Cell Host Microbe 18, 723–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai WW, Wang Z, Yiu TT, Akdemir KC, Xia W, Winter S, Tsai CY, Shi X, Schwarzer D, Plunkett W, et al. (2010). TRIM24 links a non-canonical histone signature to breast cancer. Nature 468, 927–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Takagi T, Yamaguchi Y, Ferdous A, Imai T, Hirose S, Sugimoto S, Yano K, Hartzog GA, Winston F, et al. (1998a). DSIF, a novel transcription elongation factor that regulates RNA polymerase II processivity, is composed of human Spt4 and Spt5 homologs. Genes Dev 12, 343–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Takagi T, Yamaguchi Y, Watanabe D, and Handa H (1998b). Evidence that P-TEFb alleviates the negative effect of DSIF on RNA polymerase II-dependent transcription in vitro. EMBO J 17, 7395–7403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Jiang J, Li Q, Ma H, Xu Z, and Gao Y (2016). KAP1 is overexpressed in hepatocellular carcinoma and its clinical significance. Int J Clin Oncol 21, 927–933. [DOI] [PubMed] [Google Scholar]
- Wang YY, Li L, Zhao ZS, and Wang HJ (2013). Clinical utility of measuring expression levels of KAP1, TIMP1 and STC2 in peripheral blood of patients with gastric cancer. World J Surg Oncol 11, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams LH, Fromm G, Gokey NG, Henriques T, Muse GW, Burkholder A, Fargo DC, Hu G, and Adelman K (2015). Pausing of RNA polymerase II regulates mammalian developmental potential through control of signaling networks. Mol Cell 58, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright WE, and Shay JW (2006). Inexpensive low-oxygen incubators. Nat Protoc 1, 2088–2090. [DOI] [PubMed] [Google Scholar]
- Wu CH, Yamaguchi Y, Benjamin LR, Horvat-Gordon M, Washinsky J, Enerly E, Larsson J, Lambertsson A, Handa H, and Gilmour D (2003). NELF and DSIF cause promoter proximal pausing on the hsp70 promoter in Drosophila. Genes Dev 17, 1402–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu GJ, Pen J, Huang Y, An S, Liu Y, Yang Y, Hao Q, Guo XX, and Xu TR (2018). KAP1 inhibits the Raf-MEK-ERK pathway to promote tumorigenesis in A549 lung cancer cells. Mol Carcinog 57, 1396–1407. [DOI] [PubMed] [Google Scholar]
- Xi Q, Wang Z, Zaromytidou AI, Zhang XH, Chow-Tsang LF, Liu JX, Kim H, Barlas A, Manova-Todorova K, Kaartinen V, et al. (2011). A poised chromatin platform for TGF-beta access to master regulators. Cell 147, 1511–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Lamouille S, and Derynck R (2009). TGF-beta-induced epithelial to mesenchymal transition. Cell Res 19, 156–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada T, Yamaguchi Y, Inukai N, Okamoto S, Mura T, and Handa H (2006). P-TEFb-mediated phosphorylation of hSpt5 C-terminal repeats is critical for processive transcription elongation. Mol Cell 21, 227–237. [DOI] [PubMed] [Google Scholar]
- Yamaguchi Y, Takagi T, Wada T, Yano K, Furuya A, Sugimoto S, Hasegawa J, and Handa H (1999). NELF, a multisubunit complex containing RD, cooperates with DSIF to repress RNA polymerase II elongation. Cell 97, 41–51. [DOI] [PubMed] [Google Scholar]
- Yang Z, Zhu Q, Luo K, and Zhou Q (2001). The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 414, 317–322. [DOI] [PubMed] [Google Scholar]
- Zeitlinger J, Stark A, Kellis M, Hong JW, Nechaev S, Adelman K, Levine M, and Young RA (2007). RNA polymerase stalling at developmental control genes in the Drosophila melanogaster embryo. Nat Genet 39, 1512–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Yap KL, Ivanov AV, Wang X, Mujtaba S, Plotnikova O, Rauscher FJ 3rd, and Zhou MM (2008). Structural insights into human KAP1 PHD finger-bromodomain and its role in gene silencing. Nat Struct Mol Biol 15, 626–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Genes Differentially Expressed After KAP1 Depletion. Related to Figure 1
Table S2. Genes with KAP1 Bound Promoters. Related to Figure 1
Table S3. KEGG Pathway Analysis of KAP1 Regulated Genes in MCF7 Breast Cancer Cells. Related to Figure 2
Table S4. Tissue Samples Used for TCGA Analysis. Related to Figure 3
Table S5. TCGA Analysis. Related to Figure 3
Data Availability Statement
Original western blot data has been deposited at Mendeley Data (http://dx.doi.org/10.17632/fc56g3m8ff.1). Raw and analyzed NGS datasets have been deposited at NCBI GEO under accession number GSE132705. Detailed scripts and commands used for all NGS analysis can be found on the D’Orso Lab public GitHub repository (https://git.biohpc.swmed.edu/ivandorsolab/Curtis_Dissertation).