

Summary
Class-switched antibodies to double-stranded DNA (dsDNA) are prevalent and pathogenic in systemic lupus erythematosus (SLE), yet mechanisms of their development remain poorly understood. Humans and mice lacking secreted DNase DNASE1L3 develop rapid anti-dsDNA antibody responses and SLE-like disease. We report that anti-DNA responses in Dnase1l3−/− mice required CD40L-mediated T cell help but proceeded independently of germinal center formation via short-lived antibody-forming cells (AFCs) localized to extrafollicular regions. Type I interferon (IFN-I) signaling and IFN-I-producing plasmacytoid dendritic cells (pDCs) facilitated the differentiation of DNA-reactive AFCs in vivo and in vitro and were required for downstream manifestations of autoimmunity. Moreover, the endosomal DNA sensor TLR9 promoted anti-dsDNA responses and SLE-like disease in Dnase1l3−/− mice redundantly with another nucleic acid-sensing receptor, TLR7. These results establish extrafollicular B cell differentiation into short-lived AFCs as a key mechanism of anti-DNA autoreactivity, and reveal a major contribution of pDCs, endosomal TLRs and IFN-I to this pathway.
Graphical Abstract
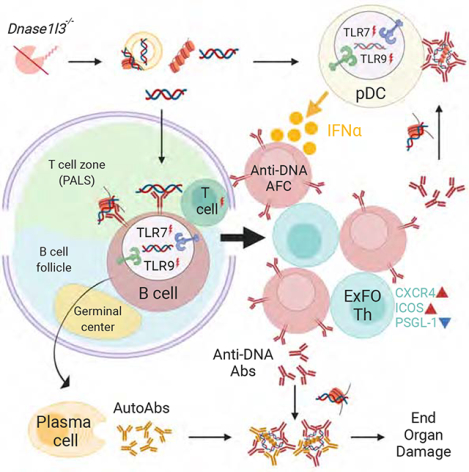
eTOC blurb
Autoantibodies to self-DNA are a defining feature of systemic lupus erythematosus (SLE), yet the mechanisms of their development remain poorly understood. Soni et al. show that anti-DNA autoreactivity is driven by extrafollicular B cell differentiation into short-lived plasmablasts, which is facilitated by plasmacytoid dendritic cells, type I interferon and endosomal Toll-like receptors 7 and 9.
Introduction
Systemic lupus erythematosus (SLE) is a systemic autoimmune disease defined by class-switched antibodies (Abs) to nuclear antigens. More than half of SLE patients develop high titers of IgG to double-stranded DNA (dsDNA), which correlate with disease activity, severity and lupus nephritis (Pisetsky, 2016; Yung and Chan, 2015). B cells that produce these Abs represent major targets of emerging therapies in SLE (Hale et al., 2018). Autoreactive B cell responses can be driven by the germinal center (GC) reaction (Vinuesa et al., 2009), which is supported by CD4+ follicular helper T cells (GC-Tfh) (Lesley et al., 2006; Seo et al., 2002; Vinuesa et al., 2016). Apart from the GC, autoreactive B cell differentiation can occur in the extrafollicular (ExFO) regions (Jenks et al., 2019; Jenks et al., 2018; William et al., 2002). The ExFO pathway in a lymphoproliferation-driven SLE model MRL.lpr is promoted by extrafollicularly localized CD4+ T-helper cells (ExFO-Th) expressing CXCR4, CD40L and ICOS (Odegard et al., 2008). An ExFO T cell population in patients with lupus nephritis has been recently defined (Caielli et al., 2019). However, the relative contribution of the ExFO pathway and its dedicated helper T cells to the initiation and propagation of anti-dsDNA responses remains to be explored.
High concentration of circulating type I interferons (IFN-I) and the expression of IFN-stimulated gene signature in SLE patients correlate with high anti-DNA Ab titers and disease severity (Bennett et al., 2003; Kirou et al., 2005; Weckerle et al., 2011). The overall pathogenic effect of IFN-I signaling in murine SLE has been demonstrated (Agrawal et al., 2009; Santiago-Raber et al., 2003), although in some cases it appears independent of autoAb production (Buechler et al., 2013). Furthermore, both the target cells of IFN-I signaling and the IFN-I-producing cell types in autoAb responses are poorly understood. Plasmacytoid dendritic cells (pDCs) are efficient producers of IFN-I that promote SLE pathogenesis (Rowland et al., 2014; Sisirak et al., 2014). However, other cell types such as stromal follicular dendritic cells (Das et al., 2017) or B cells themselves (Hamilton et al., 2017) have been proposed as IFN-I producers in SLE. Given the emerging therapeutic strategies to block IFN-I in SLE (Muskardin and Niewold, 2018), the precise cellular sources and mechanisms of IFN-I activity in anti-dsDNA responses warrant clarification.
Aberrant immune activation via innate pathways of nucleic acid (NA) sensing plays a fundamental role in SLE. The key extracellular NA sensing pathway involves MyD88-dependent endosomal Toll-like receptors TLR7 and TLR9 that recognize singlestranded RNA and unmethylated DNA, respectively. Both receptors are expressed in B cells and pDCs and facilitate autoAb production and IFN-I secretion by these respective cell types in response to endogenous NAs (Barrat et al., 2005; Leadbetter et al., 2002). TLR7 is necessary for autoreactivity in all tested models and its aberrant expression facilitates SLE in mice and humans (Celhar et al., 2012). The loss of TLR9 reduces anti-nucleosome Abs, yet exacerbates the disease in some SLE models, suggesting a net tolerogenic effect of this receptor (Christensen and Shlomchik, 2007; Sharma et al., 2015). These results remain to be reconciled with the activation of IFN-I production in human pDCs by extracellular DNA via TLR9 (Barrat and Su, 2019), and pose a fundamental question about the innate mechanisms driving anti-DNA responses in SLE.
DNASE1L3 is a secreted DNase that is a critical gatekeeper of tolerance to DNA. Homozygous null mutations in DNASE1L3 cause rare familial forms of SLE with anti-dsDNA Ab responses (Al-Mayouf et al., 2011; Ozcakar et al., 2013). Furthermore, a coding polymorphism that reduces DNASE1L3 activity is associated with SLE, scleroderma and arthritis (Westra et al., 2018; Zochling et al., 2014). Finally, Dnase1l3−/− mice develop rapid and specific anti-dsDNA responses, followed by generalized immune activation and SLE-like disease (Sisirak et al., 2016; Weisenburger et al., 2018). DNASE1L3 acts extracellularly to reduce the availability of antigenic cell-free DNA, e.g. by restricting DNA length (Serpas et al., 2019) and reducing its exposure on apoptotic cell microparticles (Sisirak et al., 2016). Thus, Dnase1l3−/− mice represent a clinically relevant model of anti-dsDNA responses driven by extracellular DNA, in which these responses can be studied independently of other autoAbs, lymphoproliferation or systemic inflammation.
Here we took advantage of the specificity and defined kinetics of this model to address the cellular and molecular basis of dsDNA-specific Ab responses. We identified a central role of CD40L-dependent ExFO differentiation in anti-DNA reactivity, and established pDC-derived IFN-I as an important signal for its maintenance. We also found that TLR9 promoted anti-DNA responses and the resulting pathology, and this effect was partially redundant with TLR7. Thus, extracellular self-DNA drives TLRdependent IFN-I production and continuous ExFO differentiation of antibody-forming cells (AFCs) that drive anti-DNA autoreactivity and pathology in SLE.
Results
Anti-DNA reactivity and autoimmunity in Dnase1l3-deficient mice require CD40L
To test whether rapid anti-nucleosome (anti-Nuc) and anti-dsDNA responses in Dnase1l3−/− mice (Sisirak et al., 2016) depend on T cell help via CD40 ligand (CD40L)-CD40 signaling (Lesley et al., 2006), we deleted Cd40lg in Dnase1l3−/− mice. While Dnase1l3−/− mice developed high titers of anti-dsDNA IgG, Dnase1l3−/−Cd40lg−/− mice did not develop any anti-dsDNA Abs detectable by ELISA or Crithidia luciliae immunofluorescence test (CLIFT) (Figures 1A, B). They also did not show reactivity to nuclear antigens in the anti-nuclear Ab (ANA) assay (Figures 1C, D). Accordingly, dsDNA- and Nuc-reactive AFCs in the BM and spleen were undetectable by ELISpot in Dnase1l3−/−Cd40lg−/− mice (Figures 1E, F). Whereas the numbers of mature follicular (FO) B cells were unaffected (Figure S1A), Dnase1l3−/−Cd40lg−/− mice harbored a reduced fraction of IgG+ AFC (Figures S1B, C) and lower total IgG titers compared to Dnase1l3−/− mice, although they were similar to those in wild-type (WT) mice (Figure S1D). However, unlike total IgG responses, anti-DNA AFCs and Abs were completely abrogated (Figures 1A, 1B, 1E and S1B).
Figure 1. CD40L deficiency in Dnase1l3−/− mice abolishes autoimmunity.
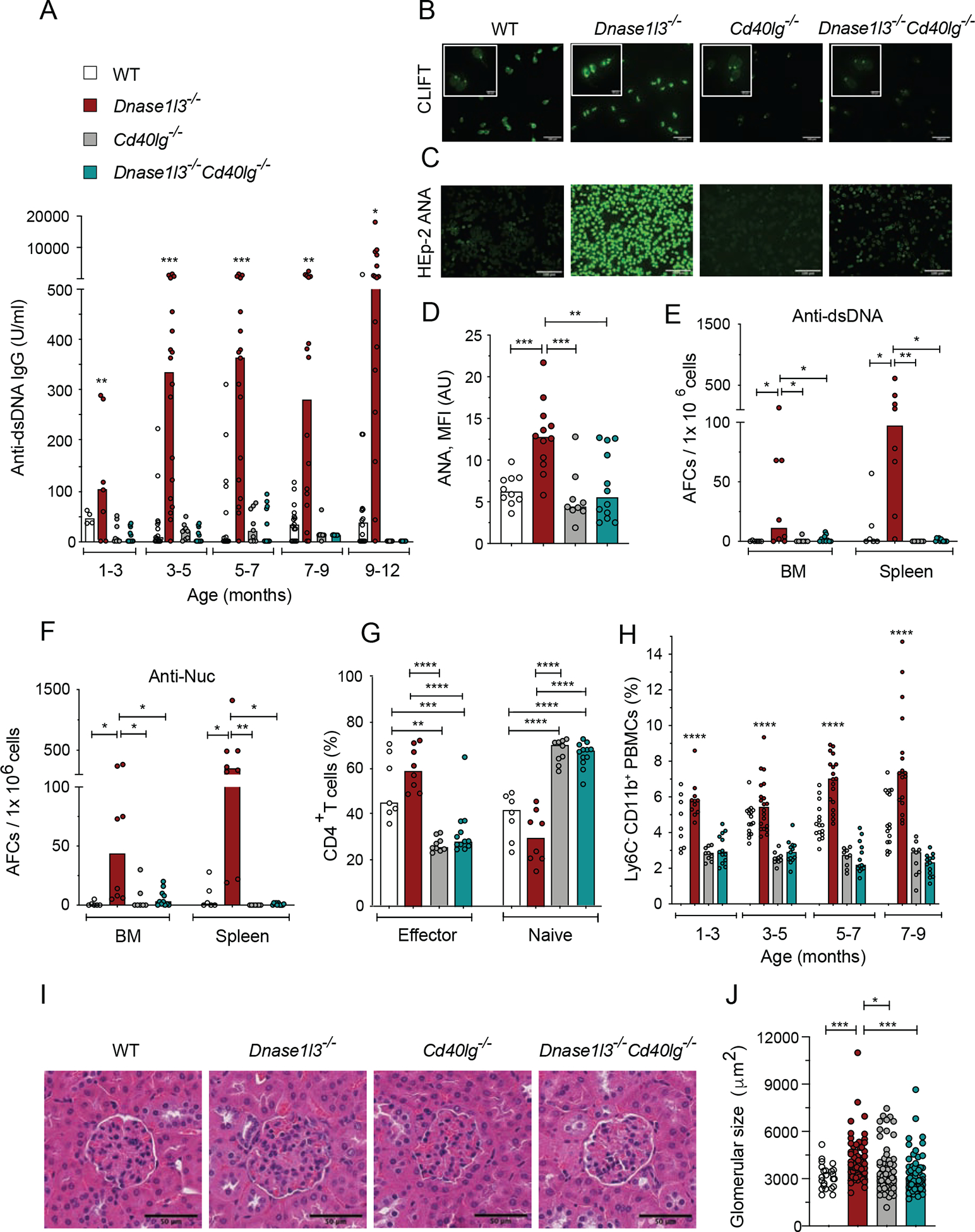
Wild-type (WT), Dnase1l3−/−, Cd40lg−/− and Dnase1l3−/−Cd40lg−/− mice were examined for Abs at the indicated ages or at the 12 month endpoint.
(A) Serum anti-dsDNA IgG titers measured by ELISA.
(B) CLIFT assay for anti-dsDNA IgG (representative of ≥4 mice per strain). Arrow indicates positive staining of the kinetoplast. Scale bar, 100 μm (insets, 20 μm).
(C, D) ANA assay, showing images representative of ≥8 mice per strain (C), and quantitation of fluorescence intensity (D). Scale bars, 100μm.
(E, F) Frequency of anti-dsDNA (E) and anti-nucleosome (F) AFCs in the bone marrow (BM) or spleen as determined by ELISpot.
(G) Fractions of effector and naive T cells among splenic CD4+ T cells at the endpoint.
(H) Fractions of CD11b+Ly6c− population among total PBMCs at indicated time points.
(I) Glomeruli from H&E-stained kidney sections (representative of ≥3 mice per group). Scale bars, 50 μm.
(J) Size of ≥ 20 glomeruli per kidney section from ≥3 mice per group.
In A and H, significant differences between Dnase1l3−/− vs Dnase1l3−/−Cd40lg−/−mice are shown. Symbols represent individual mice except in panel J, where they represent individual glomeruli. All bars indicate median.
* p ≤ 0.05, ** p ≤ 0.01, ***p ≤ 0.001 and **** p ≤ 0.0001.
See also Figure S1.
Consistent with disrupted CD40L signaling, the fractions of CD62L−CD44hi effector CD4+ T cells were significantly lower while those of naïve CD62+CD44−CD4+ T cells were higher in Dnase1l3−/−Cd40lg−/− compared to WT and Dnase1l3−/− mice (Figure 1G). In parallel, the frequency of CD4+CD25hiFoxp3+ regulatory T cells (Tregs) was significantly reduced in Dnase1l3−/−Cd40lg−/− mice (Figure S1E). As expected, GC B cells were completely absent in Dnase1l3−/−Cd40lg−/− mice (Figures S1F, G), and GC-Tfh cells were also reduced (Figure S1H). Notably, Dnase1l3−/−Cd40lg−/− mice lost manifestations of generalized immune activation such as progressive expansion of Ly6C− monocytes in the blood (Figure 1H) and splenomegaly (Figure S1I). Dnase1l3−/− mice on the C57BL/6 (B6) background do not develop overt glomerulonephritis but have enlarged glomeruli at 1 year of age (Sisirak et al., 2016). We observed a complete restoration to normal glomerular size in Dnase1l3−/−Cd40lg−/− mice (Figures 1I, J). Thus, anti-dsDNA responses and all disease manifestations in Dnase1l3−/− mice require CD40-CD40L signaling, suggesting a strict T cell dependence and revealing the primary role of autoAbs in this model of SLE-like disease.
Germinal center formation is dispensable for autoreactivity
Spontaneous formation of GC in the absence of infection or immunization requires TLR7, whose deletion abolishes GC formation and autoreactivity in several models of autoimmunity (Jackson et al., 2014; Soni et al., 2014). To test whether the T cell-dependent anti-dsDNA response requires GC formation, we generated Dnase1l3−/Tlr7−/− mice. The absence of TLR7 in both WT and Dnase1l3−/− mice significantly reduced the fraction of GC B cells and GC formation in the spleen (Figures 2A, B). Despite the loss of GCs, Dnase1l3−/−Tlr7−/− mice accumulated anti-dsDNA and anti-Nuc Abs (Figures 2C, D) and AFC (Figures 2E, F) comparably to GC-sufficient Dnase1l3−/− mice. Dnase1l3−/−Tlr7−/− mice also showed positive ANA with a characteristic perinuclear and homogeneous staining pattern (Figures 2G–I), and characteristic kinetoplast staining specific for dsDNA by CLIFT (Figure 2J). Accordingly, TLR7 deficiency did not rescue splenomegaly in old Dnase1l3−/− mice (Figure 2K).
Figure 2. TLR7-deficient Dnase1l3−/− mice do not develop germinal centers but retain anti-DNA reactivity.
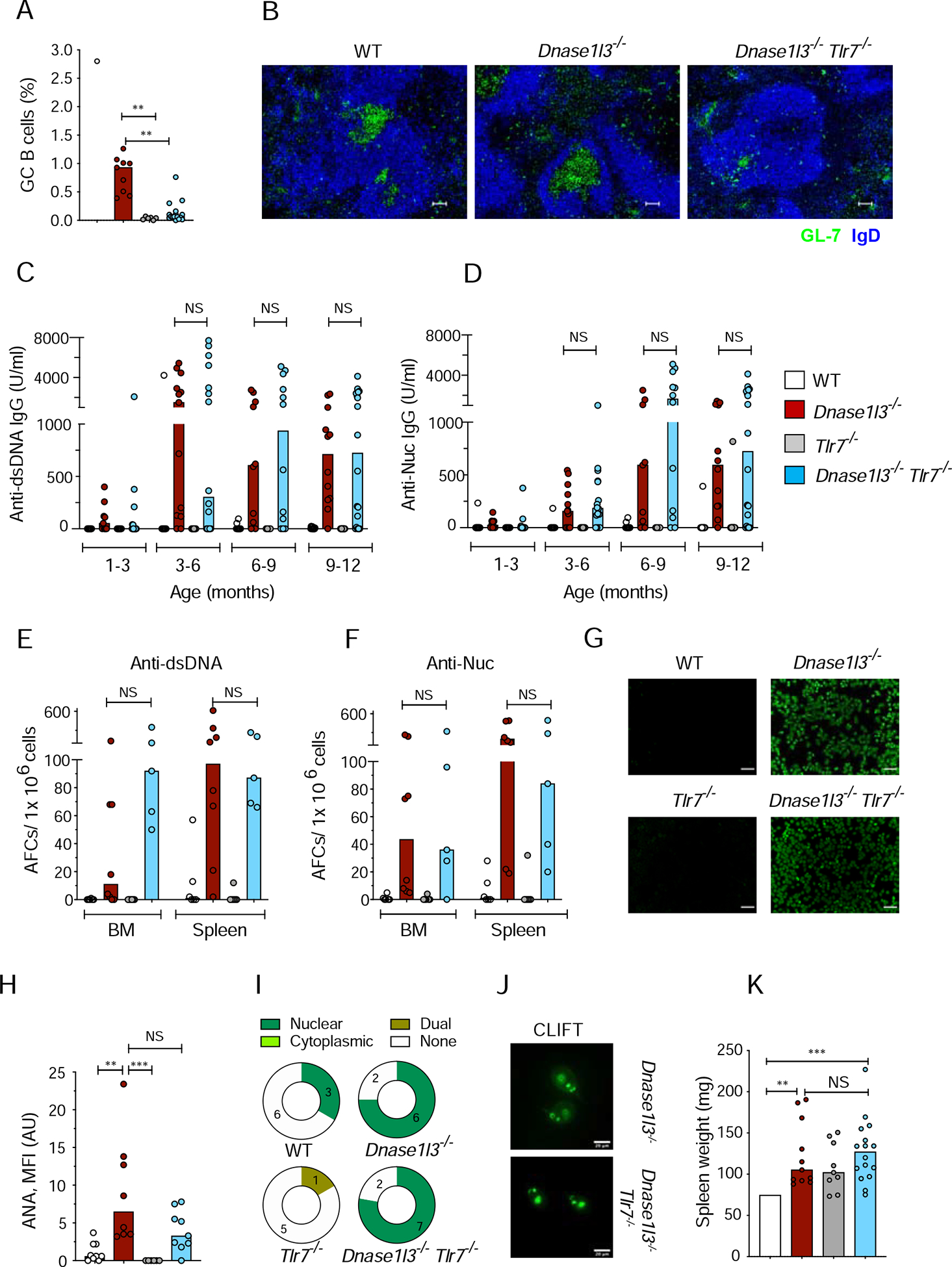
Wild-type (WT), Dnase1l3−/−, Tlr7−/− and Dnase1l3−/−Tlr7−/− mice were examined for Abs at the indicated ages or at the 12 month endpoint.
(A) Fraction of CD38− GL-7+ GC B cells among splenic B cells at the endpoint.
(B) Spleen sections stained for GC B cells (green) and follicular B cells (blue). Representative of ≥3 mice per group. Scale bars, 30 μm.
(C, D) Serum anti-dsDNA (C), and anti-Nucleosome (Nuc, D) IgG titers measured by ELISA.
(E, F) Frequency of anti-dsDNA (E), and anti-nucleosome (F) AFCs in the bone marrow (BM) or spleen as determined by ELISpot.
(G, H) ANA assay, showing images representative of ≥8 mice per strain (G), and quantitation of fluorescence intensity (H). Scale bars, 100μm.
(I) Distribution of ANA reactivity patterns in mice from each group.
(J) CLIFT assay for anti-dsDNA IgG (representative of ≥4 mice per strain). Scale bar, 20 μm.
(K) Spleen weights at the endpoint.
Symbols represent individual mice; bars indicate median.
NS= not significant, * p ≤ 0.05, ** p ≤ 0.01 and ***p ≤ 0.001
See also Figure S2.
We tested whether anti-dsDNA Abs in Dnase1l3−/−Tlr7−/− mice might be produced by GC B cells residing in the lymph nodes (LN). Unlike spleens, LNs of Dnase1l3−/−Tlr7−/− mice harbored a substantial number of GC B cells (Figure S2A), as well as total class-switched IgG+ B cells (Figure S2B). However, dsDNA and Nuc-reactive AFCs were virtually absent from the LN of both Dnase1l3−/− or Dnase1l3−/−Tlr7−/−mice (Figures S2C, D), suggesting that the bulk of autoreactive AFCs were generated in the spleen. Thus, the ablation of TLR7-dependent GC formation does not affect anti-dsDNA responses in Dnase1l3−/− mice, arguing against a major role of the GC pathway in this process.
Autoreactivity is driven by extrafollicular B cell differentiation
Given that DNA-specific AFCs were abundant in the spleens of Dnase1l3−/− mice, we stained spleen sections for markers of GCs (GL7), plasmablasts (PB) and plasma cells (CD138), and macrophages bordering the follicular areas (CD169). In WT spleens, small clusters of CD138+ cells were localized near the bridging channels between the follicular and marginal zones (Figures 3A). In contrast, Dnase1l3−/− mice showed an expansion of CD138+ cells outside of the follicular areas (Figures 3A and S3A). By flow cytometry, we noted a two-fold increase in the fraction of CXCR3+MHCIIhi AFC (Shi et al., 2015) amongst CD138+ B cells in Dnase1l3−/− splenocytes; these cells were almost undetectable in the absence of CD40L (Figures S3B and 3B). To target the proliferative PB rather than long-lived plasma cells, we treated Dnase1l3−/− mice with cyclophosphamide or PBS for 3 days. Four days later, mice were injected with thymidine analogue 5-ethynyl-2’-deoxyuridine (EdU) to label proliferating cells and examined 12 hours later. Cyclophosphamide treatment significantly reduced the fractions of total EdU+, CD138+ PB and GL7+CD38− GC B cells among total B cells (Figure S3C). Notably, cyclophosphamide caused a major reduction in splenic dsDNA-and Nuc-specific AFCs (Figure 3C), and a similarly significant reduction in anti-dsDNA and anti-Nuc Ab titers (Figure 3D) and ANA staining (Figure S3D). We also treated Dnase1l3−/− mice with a B cell-depleting Ab (anti-CD20), which rapidly reduced FO B cells but spared CD138+ PB and GC B cells (Figure S3E, F). Depletion of naïve B cells significantly reduced anti-dsDNA and anti-Nuc Abs only by 4 weeks post-treatment (Figure 3E), consistent with the depletion of B cells as a source of PB. Finally, ELISpot analysis of sorted B cell populations showed that dsDNA- and Nuc-specific AFCs were present only among CD138+ PB (Figure 3F), suggesting these short-lived cells as the primary source of autoreactivity.
Figure 3. DNA-reactive B cells in Dnase1l3−/− mice develop primarily through ExFO differentiation into plasmablasts.
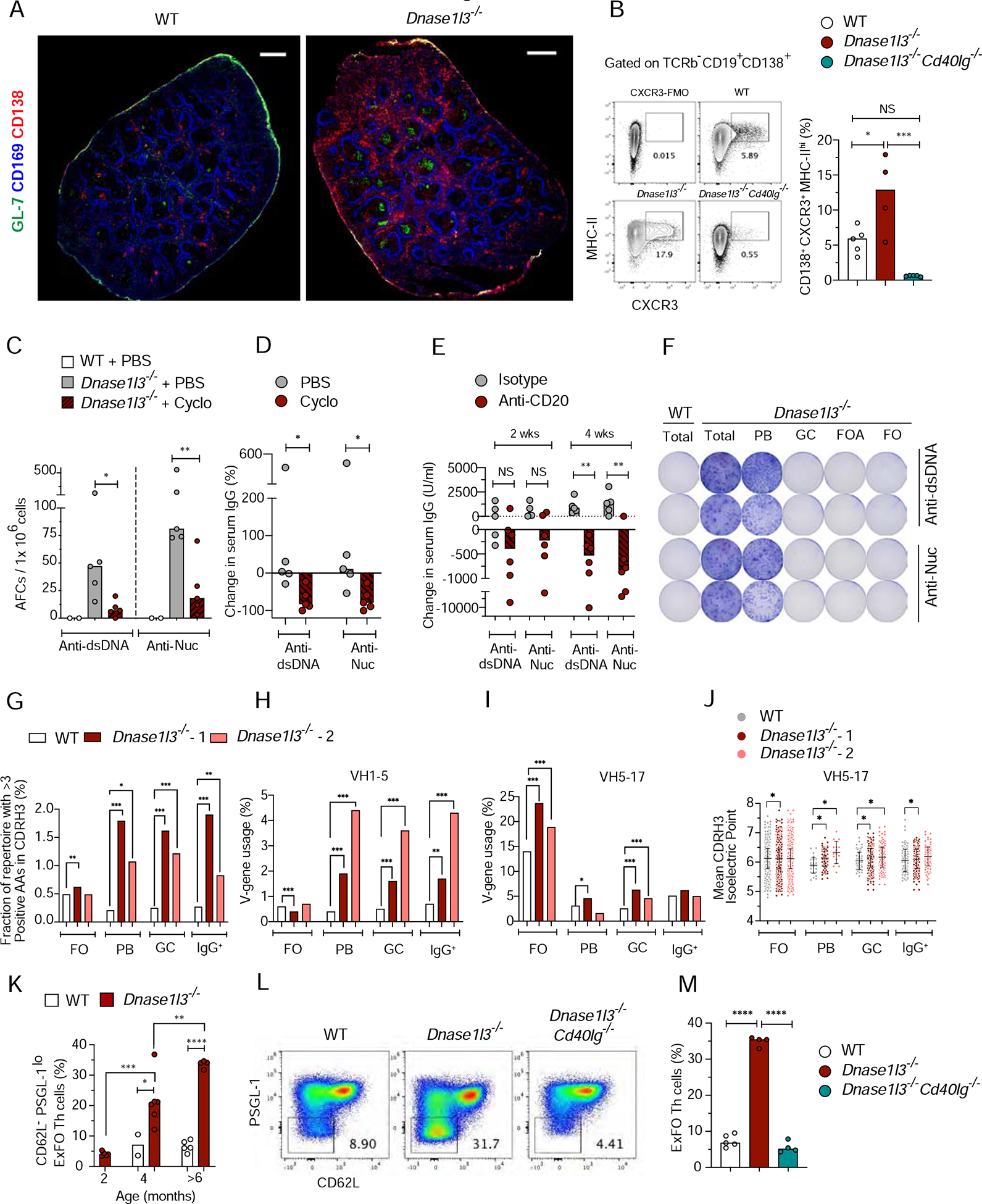
(A) Spleen sections representative of 3–5 WT or Dnase1l3−/− mice stained for CD138 (red), CD169 (blue) and GL-7 (green). Scale bar, 1000μm.
(B) Shown are representative flow plots and quantitation of the fraction of CXCR3+MHC-IIhi (ExFO) B cells, pre-gated on splenic TCRβ−B220+CD19+CD138+ cells from WT (open symbols), Dnase1l3−/− (red) and Dnase1l3−/−Cd40lg−/− (green) mice. CXCR3-FMO (fluorescence minus one) was used for gate allocation.
(C, D) Frequency of anti-dsDNA, and anti-nucleosome (Nuc) antibody-forming cells (AFCs), determined by ELISpot (C); and percent change in serum anti-dsDNA or antiNuc IgG titers determined by ELISA (D), in 6-mo-old WT mice (open) or Dnase1l3−/− mice treated with PBS (grey) or cyclophosphamide (cyclo, red).
(E) Change in serum titers of anti-dsDNA or anti-Nuc IgG in 6-mo-old Dnase1l3−/− mice after one dose (100μg/ mouse; i.p.) of treatment with IgG2a isotype control (grey) or anti-CD20 Ab (red) at 2 weeks and upon second dose of a similar treatment at 4 weeks.
(F) Image of ELISpot plate representative of 2 experiments with two mice per experiment. Shown are two serial dilutions of anti-dsDNA, and anti-nucleosome (Nuc) antibody-forming cells (AFCs) from total splenocytes of >6-mo-old WT and Dnase1l3−/− mice OR from flow sorted – Plasmablasts (PB); Germinal center B cells (GC); Follicular activated B cells (FOA) and Naïve Follicular B cells (FO) from >6 mo old Dnase1l3−/− mice. Sorted GC, FOA and FO cells were activated for 2h with 5 μg/ml anti-CD40 + 1 μg/ml LPS, and plated onto DNA or Nuc coated ELISpot plates.
(G) Percentage of clonotypes in each repertoire that contain >3 positively charged amino acids (AA) within the CDR-H3 region, in FO, PB, GC and IgG+ B cells from WT (open), Dnase1l3−/− −1 (red) and Dnase1l3−/− −2 (salmon).
(H, I) Percentage of clonotypes that use the V-gene IGHV1–5 (H), and V-gene IGH5–17 (I) in the indicated B cell subsets and mice.
(J) Mean isoelectric point of the CDR-H3 region among the clonotypes that use the V-gene IGH5–17. Each point represents one clonotype. Error bars represent the mean and S.D. of all clonotypes in each group.
(K) Flow analysis of the fraction of CD62L−PSGL-1loExFO Th cells among splenic B220TCRβ+CD4+ T cells of WT and Dnase1l3−/− mice at the indicated ages.
(L, M) Representative flow plots of pre-gated splenic B220−TCRβ+CD4+ cells with low ExFO Th cells shown within the gate (L); quantitation of the fraction of ExFO Th cells (M) from >6-mo-old WT, Dnase1l3−/− and Dnase1l3−/−Cd40lg−/− mice.
For panels A-E, K and M symbols represent individual mice and bars indicate median.
* p ≤ 0.05, ** p ≤ 0.01, ***p ≤ 0.001 and **** p ≤ 0.0001.
See also Figure S3.
To gain insight into the receptor repertoire of B cell fractions in Dnase1l3−/− mice, we sorted FO, GC, PB and total IgG+ B cells individually from two Dnase1l3−/− mice (Figures S3G, H) and one age- and sex-matched WT mouse, and sequenced in bulk immunoglobulin heavy chain variable regions (VH). Complementarity-determining regions (CDRs) of DNA-reactive hybridomas are enriched for positively charged amino acids arginine (Arg) and lysine (Lys), reflecting the binding to negatively charged DNA (Radic and Weigert, 1994). Indeed, the overall VH repertoire from PB, GC and IgG+ fractions from both Dnase1l3−/− mice had a 2–3-fold higher frequency of Arg and Lys in the CDRH3 region (Figure 3G). Among VH families, B cells from Dnase1l3−/− mice showed increased utilization of VH1–5 (Figure 3H), VH5–17 (Figure 3I) and VH1–81 (data not shown). The recurrent usage of VH5–17 was previously noted in anti-dsDNA hybridomas from a different Dnase1l3−/− strain (Weisenburger et al., 2018), warranting its further analysis. VH5–17 clonotypes contained more Arg and Lys residues in the CDRH3 region specifically in the PB of both Dnase1l3−/− mice (Figure S3I). Furthermore, in all fractions, the mean CDRH3 isoelectric point and mean CDRH3 hydrophobicity (Kyte-Doolittle index) were higher for the VH5–17 clonotypes in Dnase1l3−/− mice (Figures 3J and S3J). SHM in the VH regions were not notably higher in any of the B cell fractions from Dnase1l3−/− mice, and no dominant clones were observed (Figure S3K and data not shown). Similar to VH5–17, CDRH3 of VH1–5 clonotypes from PB and GC fractions of Dnase1l3−/− but not WT contained more positively charged amino acids (Figure S3L), supporting the involvement of VH5–17 and VH1–5 clones in DNA binding. Collectively, the observed selection for positive charges across all CDRH3 and within particular VH families suggests a polyclonal expansion of DNA-reactive B cells in Dnase1l3−/− mice. The expansion was observed in PB as well as in GC and did not involve extensive SHM typically associated with GC reaction, further supporting short-lived PBs as a source of anti-dsDNA reactivity in Dnase1l3−/− mice.
Given the T cell-dependent anti-dsDNA B cell response in Dnase1l3−/− mice, we analyzed ExFO-Th cells (CD62L− PSGL-1lo CXCR4hi ICOShi) (Figure S3M) and found that this population expanded with age in Dnase1l3−/− mice (Figure 3K) in a CD40L-dependent manner (Figure 3L, M). ExFO Th cells showed the highest expression of CXCR4, CD40L and ICOS compared to total T cells or CD62L− PSGL-1hi T cells (Figures S3N, O). In addition to their higher frequency (Figure S3P), ExFO Th cells from Dnase1l3−/− mice also had significantly higher expression of all three proteins compared to WT (Figures S3N, O). Thus, the expansion of DNA-reactive ExFO AFC in Dnase1l3−/− mice is associated with and supported by the expansion of ExFO Th cells. Overall, DNA-specific B cell response appears to proceed through short-lived autoAb-secreting cells with help from T cells in the extrafollicular regions.
IFN-I signaling facilitates extrafollicular anti-dsDNA responses
Given the importance of IFN-I signaling in SLE pathogenesis, we tested its role in ExFO-driven anti-DNA responses by generating Dnase1l3−/−Ifnar1−/− mice deficient in DNASE1L3 and the IFN-I receptor IFNAR1. The induction of IFN-I-inducible marker Sca-1 on T and B cells (Sisirak et al., 2016) was completely rescued in Dnase1l3−/Ifnar1−/− mice, confirming a functional blockade of IFN-I signaling (Figures S4A, B). Although Dnase1l3−/−Ifnar1−/−mice showed the initial emergence of anti-dsDNA and anti-Nuc IgG at 1–3 months, the titers were reduced in the majority of animals by 3–6 month and reaching significance at 9–12 month (Figure 4A, B). Accordingly, the numbers of anti-dsDNA and anti-Nuc AFCs at the endpoint were variable but reduced in Dnase1l3−/Ifnar1−/− mice, reaching significance for anti-dsDNA (Figures 4C, D). The reduction in anti-dsDNA reactivity was also observed by CLIFT (Figure S4C). Moreover, the frequency of total class-switched IgD− IgM− IgG2a.2b+ B cells, but not of GC B cells, was significantly reduced in Dnase1l3−/−Ifnar1−/− spleens (Figures S4D, E). Overall ANA reactivity was also significantly reduced in Dnase1l3−/−Ifnar1−/− mice, with more samples showing reactivity to cytoplasmic antigens (Figures 4E, F). Accordingly, all features of immune activation and pathology were abolished in Dnase1l3−/−Ifnar1−/− mice, including splenomegaly (Figure S4F), T cell activation (Figure S4G), expansion of Ly6C− monocytes (Figure S4H), the deposition of IgG and complement (C3) in the kidney glomeruli and the increase in glomerular size (Figure 4G–K). Thus, IFN-I signaling appears dispensable for the initial breach of tolerance to DNA, but facilitates the amplification of anti-DNA responses and their downstream pathogenic sequelae.
Figure 4. Autoreactivity and autoimmune manifestations in Dnase1l3−/− mice are facilitated by type I interferon signaling.
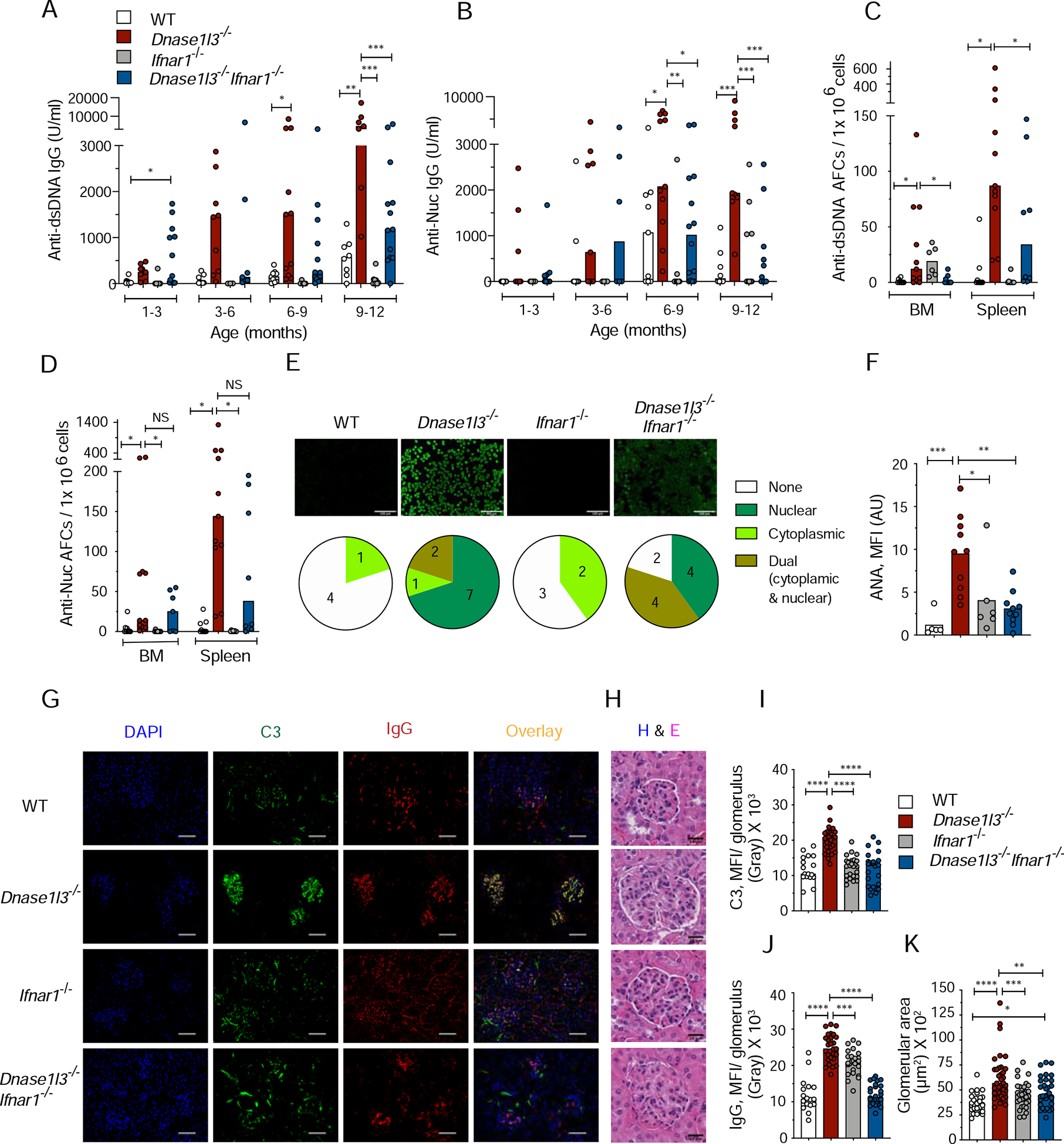
Wild-type (WT), Dnase1l3−/−, Ifnar1−/− and Dnase1l3−/− Ifnar1−/− mice were examined for Abs at the indicated ages or at the 12 month endpoint.
(A and B) Serum anti-dsDNA (A) and anti-Nucleosome (B) IgG titers determined by ELISA.
(C and D) ELISpot analysis of the number of anti-dsDNA (C), and anti-nucleosome (D) antibody forming cells (AFCs) in bone marrow (BM) or spleen cells.
(E) Images of HEp-2 cells stained with sera to detect ANAs (representative of n≥ 5 per strain). Scale bars, 100 μm. Pie charts with numbers indicate distribution of ANA reactivity patterns in mice from each group.
(F) Quantitation of ANA fluorescence intensity.
(G-K) Images of kidney sections representative of ≥ 3 mice per group stained for IgG, (red) and C3 (green) and DAPI (blue). Scale bars, 40 μm, (G). Images of glomeruli from H&E-stained kidney sections, representative of ≥3 mice per group. Scale bars 20 μm (H). Quantitation of mean fluorescence intensity of C3 (I), and IgG deposits (J), and size of ≥ 20 glomeruli per kidney section (K) from 3–4 mice per strain.
In panels A-D and F, symbols represent individual mice. Panels I-K each symbol represents an individual glomerulus. All bars indicate median.
NS= not significant, * p ≤ 0.05, ** p ≤ 0.01, ***p ≤ 0.001 and **** p ≤ 0.0001.
See also Figure S4.
Consistent with reduced anti-DNA responses, spleens of Dnase1l3−/−Ifnar1−/− mice showed an overall decrease of CD138+ PB in the extrafollicular regions (Figures 5A, B), with a significant reduction of CXCR3+ MHC-IIhi CD138+ PB (Figure 5C, D). Accordingly, the frequency of ExFO Th cells was significantly reduced in Dnase1l3−/−Ifnar1−/− mice (Figures 5E, F), and the elevated expression of ICOS and CD40L on these cells was completely rescued (Figures 5G,H). Dnase1l3−/− mouse spleens harbored multiple CD4+ T cells in close proximity with CD138+ PB clusters in the red pulp and bridging channels (Figure S5A) and a higher fraction of Ki67+ proliferating cells in the GC and T cell zones, with an elevated ratio of proliferating cells to CD4+ T cells (Figures 5I–K and S5B). Notably, this ratio was significantly reduced in Dnase1l3−/−Ifnar1−/− spleens (Figure 5K). Thus, IFN-I is necessary for the maintenance of ExFO B and T cell responses that drive autoreactivity in Dnase1l3−/− mice.
Figure 5. Type I IFN signaling promotes extrafollicular B cell proliferation and differentiation into antibody-forming cells.
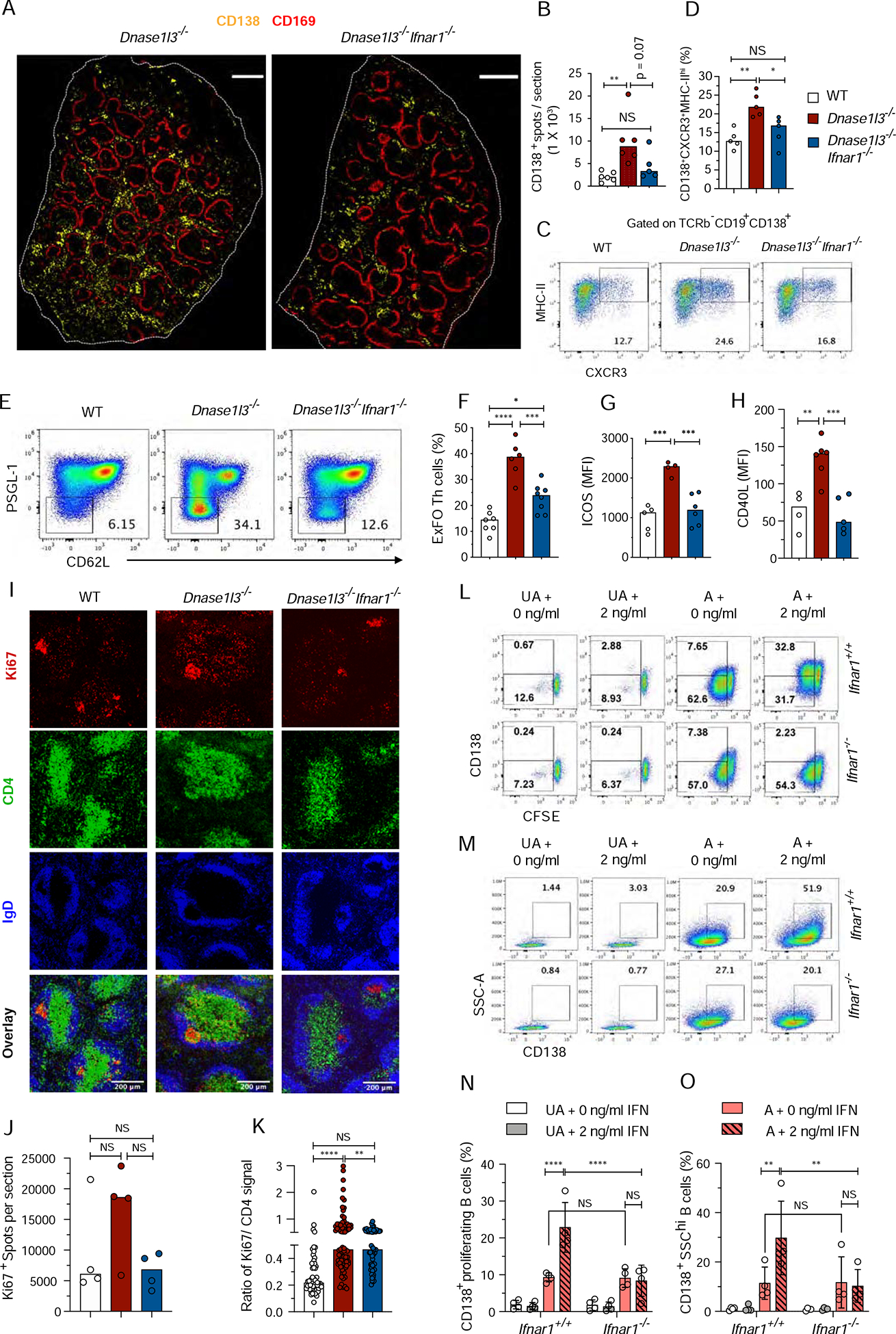
.
(A, B) Confocal images of half spleens representative of >4, 8–9-mo-old WT, Dnase1l3−/− and Dnase1l3−/−Ifnar1−/− mice, stained for CD169 (red) and CD138+ (yellow). Scale bar, 1000 μm (A); Total number of CD138+ spots per half spleen represented in A (B). Each symbol represents half a spleen section
(C, D) Staining profiles of gated splenic CD19+CD138+ cells (CXCR3+MHC-IIhi ExFO B cells highlighted) (C), and frequencies of ExFO B cells (D), in >10-mo-old WT, Dnase1l3−/− and Dnase1l3−/−Ifnar1−/− mice.
(E-H) Flow cytometric staining profiles of gated splenic CD4+ T cells (PSGL-1loCD62LloExFO Th cells highlighted) (E); Fraction of ExFO Th cells among splenic CD4+ T cells (F), mean fluorescence intensity of ICOS (G), and CD40L (H) expressed in ExFO Th cells of 6–8-mo-old WT, Dnase1l3−/− and Dnase1l3−/−Ifnar1−/− mice.
In panels C-H, wherever applicable, symbols represent individual mice and bars indicate median.
(I-K) Confocal images of spleen sections representative of three >8-mo-old WT, Dnase1l3−/− and Dnase1l3−/−Ifnar1−/− mice stained for Ki67+ proliferating cells (red), CD4+ T cells (green) and IgD+ B cells (blue). Scale bars, 200 μm (I); Quantitation of total number of Ki67+ spots per half spleen represented in Fig. S5B. Each symbol represents half a spleen section (J); Ratio of mean fluorescence intensity of Ki67 vs CD4 within a region of interest (ROI) marking CD4+ T cell zones excluding GC areas, from spleen sections represented in Fig.S5B. Each symbol represents a single ROI (K).
(L-O) Flow cytometric analysis of proliferation and differentiation of B cells upon in-vitro activation with anti-IgM and anti-CD40 in the presence or absence of IFNα. Shown are representative flow plots from 4 independent experiments of purified CFSE labeled unactivated (UA) or activated (A) B cells from Ifnar+/+ or Ifnar1−/− mice cultured for 72h in the absence (0 ng/ml) or presence of 2 ng/ml of IFNα. The cells in the gates indicate CFSElo proliferating B cells, with (upper gate) or without (lower gate) CD138 expression (L); or CD138+SSChi B cells (M), under the indicated conditions. Fraction of CD138+ proliferating (N) and CD138+SSChi blasting (O) B cells. Each symbol represents an independent experiment. Error bars show mean ± SD. Two-way ANOVA followed by Tukey’s multiple-comparison test was used to compare different treatments within a genotype or 2-way ANOVA followed by Sidak’s multiple comparison test was used to compare different treatments between the two genotypes. NS= not significant, * p ≤ 0.05, ** p ≤ 0.01, ***p ≤ 0.001 and **** p ≤ 0.0001.
See also Figure S5.
Next, we investigated the effect of IFN-I on B cell differentiation into plasmablasts in vitro. Splenic B cells were activated with anti-IgM and anti-CD40 in the presence of IL-4 and increasing doses of IFN-α, which induced a dose-dependent expression of Sca-1 and CD69 on the resulting CD138+ PB (Figure S5C, D). A similar pattern of activation and responsiveness to IFN-α was observed between B cells from WT and Dnase1l3−/− mice, which were used interchangeably. Purified CFSE-labeled B cells from Ifnar1+/+ or Ifnar1−/− mice were left unactivated (UA) or activated (A) with anti-IgM, anti-CD40 and IL-4 with or without IFN-α for 3 days. The proliferation and differentiation of CFSE-labeled B cells into CD138+ PB were comparable between Ifnar1+/+ and Ifnar1−/− mice in the absence of IFN-α, but were increased by IFN-α in IFNAR-proficient B cells (Figure S5E and 5L–O). The enhancing effect of IFN-α was not observed when B cells were stimulated by lipopolysaccharide (LPS) in the presence of IL-4 (Figures S5F, G). Consistent with previous reports (Le Bon et al., 2001; Le Bon et al., 2006), these data suggest that IFN-I may directly facilitate antigen receptor-driven B cell differentiation into PB, supporting the observed role of IFN-I in the ExFO-driven differentiation of autoreactive B cells.
pDCs facilitate extrafollicular anti-dsDNA responses through IFN-I
Having established the role of IFN-I in anti-DNA reactivity in Dnase1l3−/− mice, we tested the role of IFN-I-producing pDCs by reducing the dosage of a pDC-specific transcription factor TCF4 (Cisse et al., 2008; Sisirak et al., 2014). Dnase1l3−/−Tcf4+/− mice on pure 129 background had reduced percentages of pDCs compared to Tcf4+/+ Dnase1l3−/− littermates (Figure S6A), which was also shown to be accompanied by profoundly impaired IFN-I production (Cisse et al., 2008). As expected, B cell subsets including naïve CD23hi CD21lo FO B cells and CD23lo CD21hi IgMhi marginal zone (MZ) B cells were comparable to the WT (Figures S6B, C). The induction of Sca-1 expression on B and T cells was rescued in Dnase1l3−/− Tcf4+/− mice (Figures S6D, E), suggesting a global decrease of IFN-I production. Similar to IFNAR1 deficiency, pDC impairment in Dnase1l3−/− mice caused a significant reduction of anti-dsDNA and anti-Nuc IgG titers starting at 5–6 months of age (Figures 6A, B). Accordingly, splenic anti-DNA AFCs were also significantly reduced in 1-year-old Dnase1l3−/−Tcf4+/− mice (Figure 6C), concomitant with reduced expansion of CD138+ PB in extrafollicular regions (Figures 6D, H) but normal GC B cell numbers (Figure S6F). Dnase1l3−/−Tcf4+/− mice also showed a tendency towards reduced ANA; although the effect was variable, ANA pattern showed a higher propensity for dual nuclear and cytoplasmic staining (Figures 6E, I, J). The reduction in anti-dsDNA titers was further confirmed by CLIFT, wherein only 2/7 sera from Dnase1l3−/−Tcf4+/− mice were positive compared to 5/7 samples from Dnase1l3−/− mice at a 1:25 dilution (Figure 6F). A similar reduction of anti-dsDNA titers and a shift towards cytoplasmic ANA staining was observed after the treatment of Dnase1l3−/− mice with anti-SiglecH mAb, which inhibits IFN-I production by pDCs (data not shown). All other manifestations of autoimmunity were abolished in Dnase1l3−/Tcf4+/− mice, including splenomegaly (Figure 6K), T cell activation (Figure S6G), glomerular deposition of C3 and IgG (Figure S6H) and increased size of kidney glomeruli (Figures 6G, L). Overall, pDC impairment by Tcf4 haplodeficiency phenocopied the loss of IFN-I signaling in Dnase1l3−/− mice, suggesting pDCs as the primary source of pathogenic IFN-I.
Figure 6. Functional impairment of pDCs in Dnase1l3−/− mice ameliorates autoreactivity and disease.
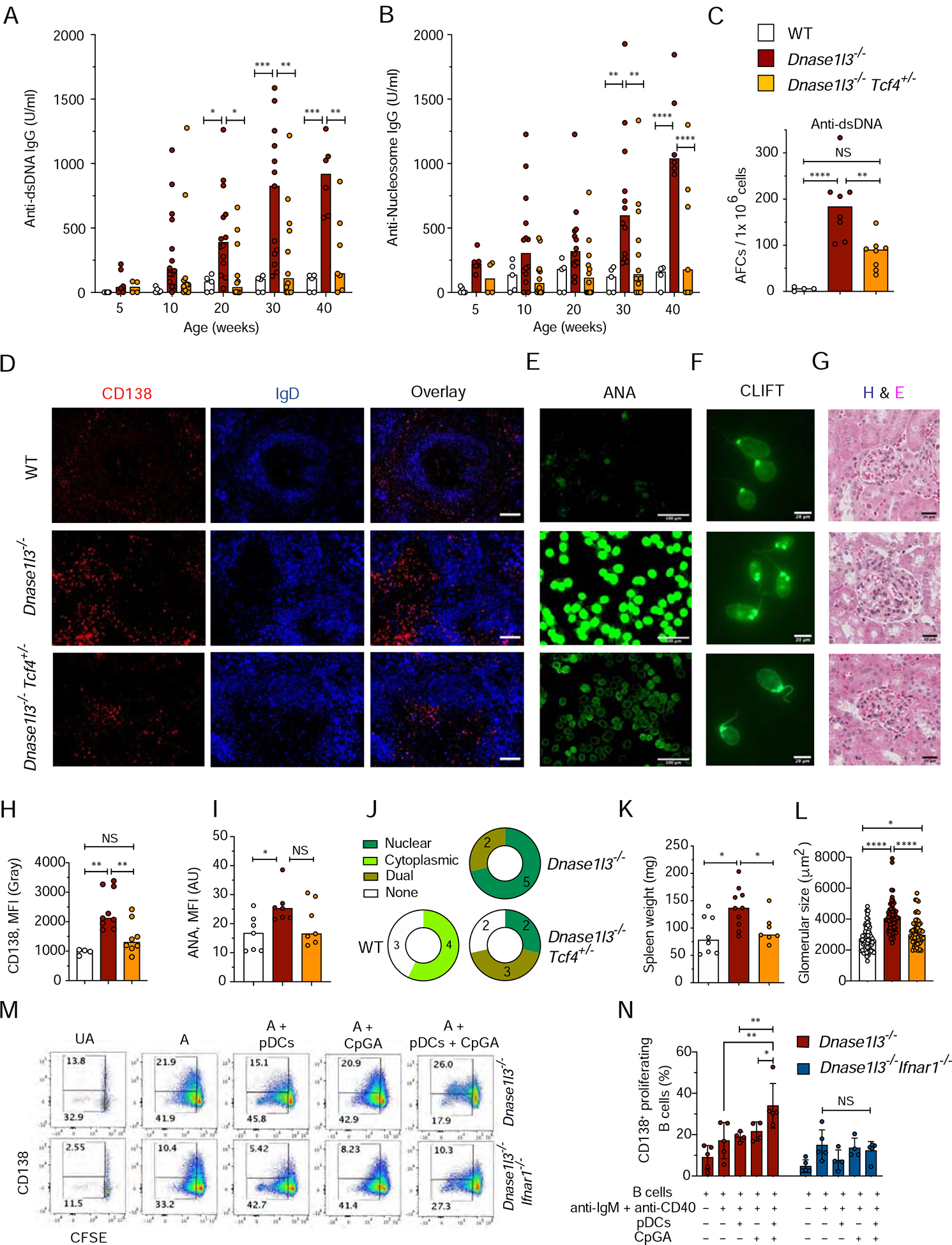
Wild-type (WT), Dnase1l3−/−, and Dnase1l3−/− mice with monoallelic deficiency of Tcf4 (Dnase1l3−/−Tcf4+/−) were examined for Abs at the indicated ages or at the 12 month endpoint.
(A-B) Serum anti-dsDNA IgG (A) and anti-Nucleosome (B) IgG titers by ELISA.
(C) Frequency of anti-dsDNA AFCs in the spleen by ELISpot.
(D) Spleen sections stained for CD138+ cells (red) and follicular B cells (blue). Representative of ≥3 mice per group. Scale bars 40 μm.
(E) ANA assay images, representative of 7 mice per group. Scale bar, 100 μm.
(F) CLIFT assay for anti-dsDNA IgG (representative of 5 mice per group). Scale bar: 20 μm.
(E) Glomeruli from H&E-stained kidney sections (representative of 3 mice per group). Scale bars, 20 μm.
(H) Intensity of CD138 fluorescence in ≥ 4 images of spleen sections from the indicated mice.
(I, J) Quantitation of ANA fluorescence intensity (I), and distribution of ANA reactivity patterns (J) in 7 mice per indicated group.
(K) Spleen weights of indicated mice at the endpoint.
(L) Quantitation of the size of ≥ 20 glomeruli per kidney section from 3 mice per group. Each symbol represents an individual glomerulus; bars indicate median.
(M-N) Purified CFSE labeled B cells from Dnase1l3−/− or Dnase1l3−/−Ifnar1−/− mice were left unactivated (UA); activated with anti-IgM + anti-CD40 (A); OR activated with anti-IgM + anti-CD40 in the presence of unstimulated pDCs (A+ pDCs); in the presence of 1μM CpGA (A+ CpGA) or in the presence of pDCs stimulated with 1μM CpGA (A+ pDCs+ CpGA) for 72h after which the B cells were analyzed for proliferation and differentiation using flow cytometry. Gating strategy (M) and quantitation (N) of CFSEloCD138+ proliferating B cells. Error bars show mean ± SD from four independent experiments (symbols). NS= not significant, * p ≤ 0.05, ** p ≤ 0.01, ***p ≤ 0.001 and **** p ≤ 0.0001.
See also Figure S6.
Immunofluorescent staining of Dnase1l3−/− spleens revealed the accumulation of B220+ SiglecH+ pDCs near the clusters of CD138+ B220lo− PB at the border of T cell zones, with frequent juxtaposition of the two cell types (Figure S6I). Although short of a formal proof, these data suggest that some pDCs are located in the extrafollicular region and may directly interact with proliferating plasmablasts. Finally, we activated primary pDCs to produce IFN-α with the TLR9 ligand CpG-A (Figure S6J, K) and tested their effect on B cell differentiation. CFSE-labeled B cells from Dnase1l3−/− or Dnase1l3−/Ifnar1−/− mice were stimulated for 3 days with anti-IgM, anti-CD40 and IL-4 in the absence or presence of CpG-A-stimulated pDCs. CpGA-stimulated pDCs significantly increased the proliferation and differentiation of activated B cells into CD138+ SSChi plasmablasts, and this effect was abolished by IFNAR1 deficiency (Figures 6M, N and S6L, M). We confirmed that IFNAR1 was expressed on activated B cells (Figure S6N), that IFN-α was detectable in the co-culture supernatant only when pDCs were activated with CpG (Figure S6O), and that CD69 was not upregulated on IFNAR1-deficient B cells (Figure S6P). Collectively, these data suggest that pDCs, via the production of IFN-I, directly facilitate anti-DNA responses driven by ExFO B cell differentiation.
Endosomal TLR signaling drives anti-DNA reactivity and SLE-like disease
The observed contribution of pDCs, along with the essential requirement for MyD88-dependent signaling (Sisirak et al., 2016), suggested the role of endosomal TLRs in anti-DNA reactivity. Having excluded the sole contribution of TLR7, we generated Dnase1l3−/−Tlr9−/− mice deficient in DNASE1L3 and the endosomal DNA sensor TLR9. Compared to Dnase1l3−/− mice, these mice showed only a modest reduction of anti-Nuc Abs but similar titers of anti-dsDNA Abs (Figure 7A, B) and similar numbers of anti-dsDNA and anti-Nuc AFCs (Figure 7C).Furthermore, CXCR3+ MHC-IIhi CD138+ ExFO PB and ExFO Th cells were not reduced in Dnase1l3−/−Tlr9−/− mice (Figures S7A, B), although a significant reduction of GC B cells was observed (Figure S7C). Total ANA titers in Dnase1l3−/−Tlr9−/− mice were not reduced (Figures 7D); however, the pattern of ANA reactivity shifted towards cytoplasmic or dual nuclear/cytoplasmic staining (Figure 7E). We also observed a significant decrease in immune activation including splenomegaly (Figure S7D) and expansion of Ly6c− CD11b+ myeloid cells (Figures S7E, F). Thus, TLR9 alone is largely dispensable for autoreactivity but contributes to downstream consequences of DNASE1L3 deficiency.
Figure 7. TLR9 and TLR7 are redundantly required for autoreactivity and autoimmunity.
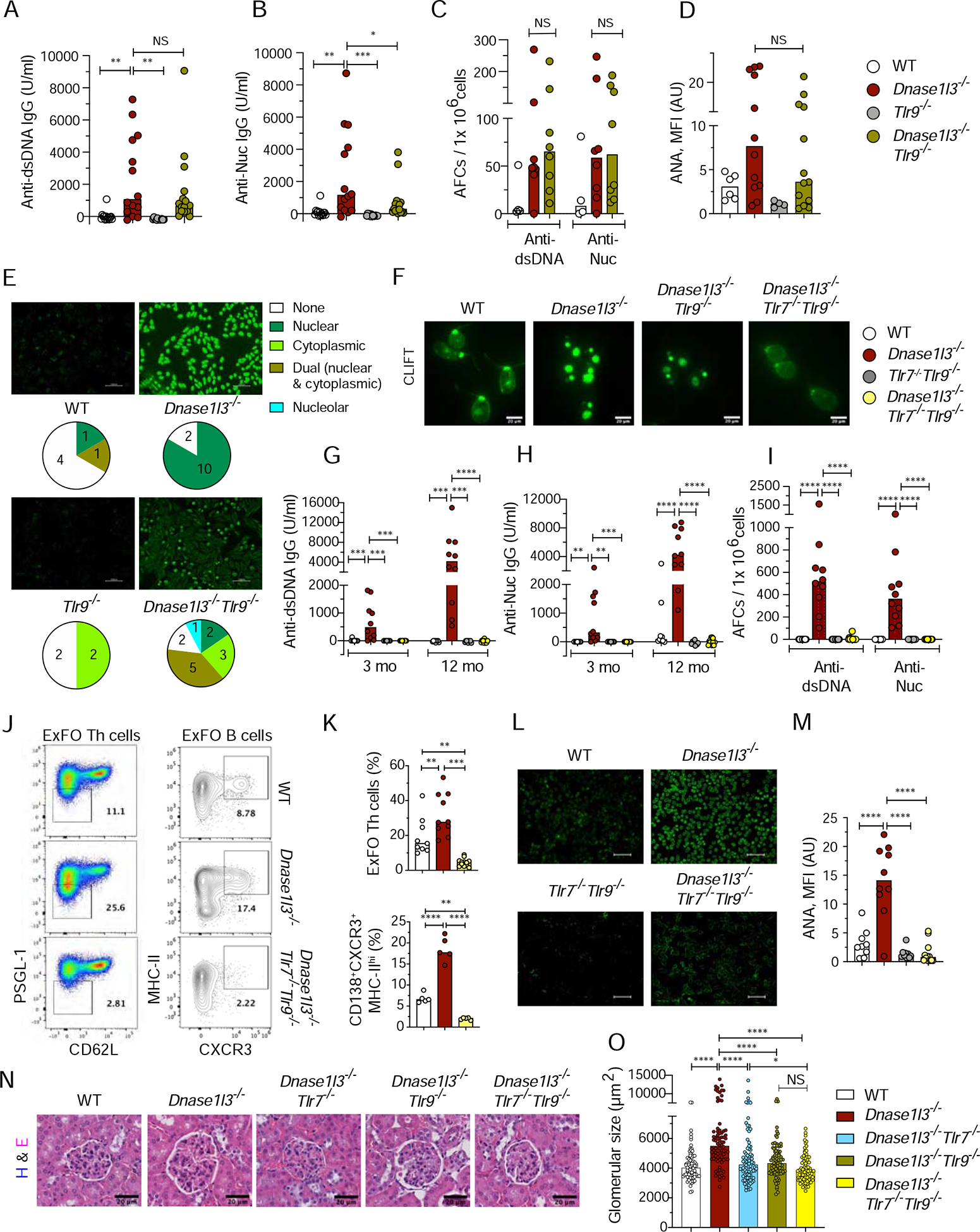
Wild-type (WT), Dnase1l3−/−, Tlr9−/−, Tlr7−/− mice or crosses thereof were examined for Abs at the indicated ages or at the 12 month endpoint.
(A-B) Serum anti-dsDNA IgG (A) and anti-Nuc (B) IgG titers by ELISA.
(C) Frequency of anti-dsDNA and anti-Nuc AFCs in the spleen by ELISpot.
(D, E) ANA assay, showing quantitation of fluorescence intensity (D), representative images and the distribution of reactivity patterns in mice from each group (E).
(F) CLIFT assay for anti-dsDNA IgG (representative of ≥4 mice per strain). Scale bar: 20 μm.
(G-H) Serum anti-dsDNA IgG (A) and anti-Nucleosome (B) IgG titers by ELISA.
(I) Frequency of anti-dsDNA and anti-nucleosome AFCs in the spleen as determined by ELISpot.
(J) Staining profiles of gated splenic CD4+ T cells (PSGL-1loCD62Llo ExFO Th cells highlighted) and CD19+ CD138+ cells (CXCR3+ MHC-IIhi ExFO B cells highlighted).
(K) Frequencies of populationss highlighted in J at the endpoint.
(L-M) ANA assay, showing images representative of ≥8 mice per strain (L), and quantitation of fluorescence intensity (M). Scale bars, 100μm.
(N) Glomeruli from H&E-stained kidney sections (representative of 4 mice per group). Scale bars, 20 μm.
(O) Size of ≥ 20 glomeruli per kidney section from 4 mice per group.
Symbols represent individual mice except in panel O, where they represent individual glomeruli. All bars indicate median.
NS= not significant, * p ≤ 0.05, ** p ≤ 0.01, ***p ≤ 0.001 and **** p ≤ 0.0001.
See also Figure S7.
To test whether the contribution of TLR9 might be partially compensated by TLR7, we generated Dnase1l3−/− mice deficient in both TLR7 and TLR9 (Dnase1l3−/−Tlr7−Tlr9−/−). Unlike Dnase1l3−/−Tlr9 mice, Dnase1l3−/−Tlr7/− Tlr9−/− mice completely lost the reactivity to pure dsDNA in the CLIFT assay (Figure 7F), all anti-dsDNA and anti-Nuc reactivity by ELISA (Figures 7G, H), and anti-dsDNA and anti-Nuc AFCs (Figure 7I). This was accompanied by a near-complete loss of ExFo Th cells and of CD138+ PB cells expressing CXCR3 (Figures 7J, K), in addition to the expected loss of GC B cells (Figure S7G). Similarly, ANA reactivity was completely abolished (Figures 7L, M), as was splenomegaly (Figure S7H) and the expansion of CD11b+Ly6c− myeloid cells (data not shown). Finally, increased glomerular size (Figure 7N, O) and the glomerular deposition of IgG and C3 (Figure S7I) were fully rescued in Dnase1l3−/−Tlr7−/−Tlr9−/− mice at the endpoint. Notably, these kidney manifestations were partially reduced in Dnase1l3−/−Tlr9−/−and particularly in Dnase1l3−/−Tlr7−/− mice (Figure 7N, O and Figure S7I), suggesting independent contributions by both TLR9 and TLR7. Altogether, these results reveal essential but partially redundant contributions of TLR9 and TLR7 to autoimmunity driven by extracellular DNA.
Discussion
Dnase1l3−/− mice model the human genetic DNASE1L3 deficiency that results in SLE, and may be relevant for severe sporadic SLE that is frequently associated with reduced extracellular DNase activity (Bruschi et al., 2019). In addition to its clinical relevance, the Dnase1l3−/− model manifests prominent anti-dsDNA responses in the absence of additional pathogenetic features such as lymphoproliferation, systemic inflammation or defects of antigen receptor signaling. Importantly, immunopathology in Dnase1l3−/− mice comprises three distinct phases: i) initial breach of tolerance to DNA (1–3 months); ii) amplification of anti-DNA Ab responses without reactivity to other self-antigens (3–9 months); iii) spreading of autoreactivity, immune activation and immune complex deposition (9–12 months). This separation of anti-DNA Ab responses from their consequences or other confounding factors make Dnase1l3−/− mice an attractive reductionist model of DNA-specific reactivity in SLE.
We found that the ExFO pathway represented the predominant route of autoreactive B cell differentiation in Dnase1l3−/− mice, generating short-lived AFCs that produced anti-nucleosome and anti-dsDNA Abs. ExFO B cell responses are generally associated with infections, rapidly generating Abs to protect against pathogens (Cunningham et al., 2007; Finke et al., 2001). Although the ExFO-generated plasmablasts have been implicated in a lymphoproliferation-driven model of autoreactivity (William et al., 2002), long-lived plasma cells and the GC pathway leading to them are thought to predominate in other models (Degn et al., 2017; Hoyer et al., 2004). Our results do not rule out the role of GC, which may be prominent at late stages of the disease accompanied by broad autoreactivity (Sisirak et al., 2016) and contribute to the generation of pathogenic Abs that synergize with anti-dsDNA Abs to induce renal disease. Furthermore, anti-dsDNA response in Dnase1l3−/− mice can be greatly enhanced by additional genetic lesions that cause the expansion of GC (Weisenburger et al., 2018). Collectively, our analysis suggests the ExFO pathway as the primary route of differentiation of DNA-reactive B cells, which continuously generates anti-DNA and can be amplified by additional factors that elicit GC-derived plasma cells.
We found that autoreactivity and inflammation in Dnase1l3−/− mice required CD40L signaling and were associated with the expansion of CXCR4hiICOShi ExFO-Th cells (Odegard et al., 2008), which was also CD40L-dependent. This is consistent with the role of T cells in general and ExFO-Th cells in particular in other models of ExFO-driven autoreactive responses (Deng et al., 2017; Sweet et al., 2011). Notably, a population of T cells supporting ExFO B cell expansion in human SLE patients has been described recently (Caielli et al., 2019). Together with this and other descriptions of the ExFO B cell differentiation in human SLE (Jenks et al., 2018; Tipton et al., 2015), our data emphasize the predominant role of the ExFO pathway in autoimmunity, including in polyclonal B cell responses driven by DNA. This is consistent with the dynamic nature of anti-dsDNA responses and may also explain the low efficiency of B cell-focused therapies, as these are often inefficient against plasmablasts (Hale et al., 2018). _Indeed, a short 3-day treatment with an anti-proliferative drug cyclophosphamide effectively reduced anti-dsDNA titers, while a month-long treatment with B cell depleting anti-CD20 Ab was required for a similar reduction._Therefore, targeting actively proliferating short-lived plasmablasts in lupus patients with high anti-dsDNA titers represents an important therapeutic goal (Soni and Reizis, 2018).
Given the critical role of IFN-I signaling in SLE pathogenesis and specifically in GC-driven B cell autoreactivity (Domeier et al., 2018), we tested the potential role of this pathway in ExFO-driven anti-dsDNA responses. Our analysis in Dnase1l3−/− mice revealed that IFN-I was dispensable for the initial emergence of anti-dsDNA Abs, but facilitated their subsequent maintenance and amplification. Notably, IFN-I was required for the optimal expansion of ExFO CD138+ B cells and ICOShiExFO-Th cells. These data agree with enhanced generation of short-lived plasmablasts after overexpression of IFN-I in lupus-prone mice (Mathian et al., 2011), and establish the role of endogenous IFN-I in the ExFO pathway. In vitro, IFN-I enhanced B cell differentiation into CD138+ plasmablasts that was driven by BCR and CD40 signaling but not by LPS, which presumably mimics B cell responses driven by bacterial pathogens. Thus, continuous activation of self-reactive B cells by antigenic self-DNA, alongside costimulatory signals from T cells, makes B cells responsive to IFN-I-mediated differentiation into AFCs, and therefore may be particularly important for autoAb responses in SLE.
Although the responsiveness of human pDCs to self-DNA (Barrat et al., 2005; Caielli et al., 2016) and the important role of murine pDCs in several SLE models (Rowland et al., 2014; Sisirak et al., 2014) are well established, the mechanism of pDC activity in SLE remains unclear. Furthermore, pDCs appear dispensable in certain models of SLE-like autoreactivity such as Wiskott-Aldrich syndrome deficiency (Sawai et al., 2018). We found that the functional impairment of pDCs phenocopied IFN-I blockade, including the impairment of ExFO-driven anti-DNA responses prior to the abrogation of all downstream pathology. The production of IFN-I by pDCs in vivo has been difficult to detect in this or any other SLE model, likely because of its low and/or transient nature. Nevertheless, in addition to genetic evidence, we were able to show that activated pDCs facilitated CD40-driven plasmablast differentiation in vitro, and this effect was IFN-I-dependent. Together with similar observations for human pDCs in vitro (Jego et al., 2003), these results underscore the emerging role of pDCs as essential IFN-I producers in SLE. They further support antibody-mediated depletion or functional impairment of pDCs as a viable and potentially specific therapeutic strategy in SLE (Barrat and Su, 2019).
Autoreactivity in Dnase1l3−/− mice is independent of STING-dependent cytosolic DNA sensing and is completely dependent on MyD88 (Sisirak et al., 2016). We found that the loss of the sole known endosomal DNA receptor, TLR9, had no major effect on anti-dsDNA response as noticed in another Dnase1l3-deficient mouse strain (Weisenburger et al., 2018), and caused a minor albeit significant reduction of immune activation. However, the loss of TLR9 in the absence of TLR7 completely abrogated autoreactivity and pathology, suggesting that TLR9 promoted anti-DNA response yet its pathogenic function was partially compensated by TLR7. The mechanism of such compensation may include a functional competition between the two receptors, whereby TLR9 deletion creates a hyperactive TLR7 (Fukui et al., 2009). This scenario is consistent with the observed shift in autoantibody reactivity by ANA staining pattern as also observed in other SLE models (Christensen et al., 2006; Nickerson et al., 2010). Another possible mechanism includes the ability of TLR7 to recognize both RNA and DNA degradation products such as guanosine and deoxyguanosine, respectively (Shibata et al., 2016). Indeed, the self-antigen in DNASE1L3 deficiency and presumably other SLE forms comprises chromatin in apoptotic microparticles (Nielsen et al., 2011; Pisetsky et al., 2011; Sisirak et al., 2016), which are likely to contain DNA and RNA and their degradation products. Irrespective of the mechanism, the synergistic yet partially redundant role of TLR9 and TLR7 in anti-DNA autoreactivity is consistent with i) the redundancy between the two receptors in an Ab-independent kidney inflammation driven by monocytes (Kuriakose et al., 2019); ii) the requirement for endosomal TLR sensing but not for TLR9 alone in DNA-driven autoimmunity in Dnase2-deficient mice (Pawaria et al., 2015). Our results help reconcile the neutral or even pathogenic effects of deleting TLR9 alone in several SLE models (Celhar et al., 2012; Christensen and Shlomchik, 2007; Sharma et al., 2015) with the emerging pathogenic role of TLR9 in human SLE (Barrat et al., 2005; Caielli et al., 2016). Collectively, our analysis of autoreactivity driven by extracellular DNA supports a prominent role for extrafollicular B cell differentiation into AFCs that requires endosomal TLR signaling and is facilitated by pDCs and IFN-I.
STAR Methods
LEAD CONTACT
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Boris Reizis (boris.reizis@nyulangone.org).
MATERIALS AVAILABILITY
All animal strains are available from repositories described above. No new animal strains or reagents were generated.
EXPERIMENTAL MODEL DETAILS
Mouse strains
All experiments were performed in accordance with the animal protocol approved by the Institutional Animal Care and use committee of Columbia University and New York University. All the mouse strains were on a C57BL/6 background and C57BL/6J (Stock 000664) mice were used as WT controls unless otherwise mentioned. As described previously (Sisirak et al., 2016), mice with a targeted germline disruption of Dnase1l3 with Dnase1L3LacZ (Dnase1l3−/−) were purchased from Taconic Animal repository (Model TF2732) and backcrossed onto C57BL/6 or 129SvEvTac for >10 generations and the colonies were maintained in house. Breeding pairs of wild type (WT), Cd40lg−/− [002770 - B6.129S2-Cd40lgtm1Imx/J], Tlr7−/− [008380 – B6.129S1Tlr7tm1Flv/J], Tlr9−/− [034329-JAX C57BL/6J-Tlr9M7Btlr/Mmjax] and Ifnar1−/− [032045-JAX B6.129S2-Ifnar1tm1Agt/Mmjax] mice were purchased from Jackson Laboratories (Bar Harbor, ME) and bred and maintained in house. Dnase1l3−/− mice on a pure B6 background were crossed with Cd40lg−/−, Tlr7−/−, Tlr9−/−or Ifnar1−/− to generate Dnase1l3−/−Cd40lg−/−, Dnase1l3−/−Tlr7−/−, Dnase1l3−/−Tlr9−/and Dnase1l3−/−Ifnar1−/− mice respectively. Dnase1l3−/−Tlr7−/− and Dnase1l3−/Tlr9−/−mice were bred to generate Dnase1l3−/−Tlr7−/−Tlr9−/− triple deficient strain. All the double and triple deficient mice were bred and maintained in house. Dnase1l3−/− mice on a pure 129Sv background were crossed with Tcf4+/− animals (Zhuang et al., 1996) on pure 129SvEvTac (129Sv) background to generate Dnase1l3−/−Tcf4+/− mice and bred and maintained in house. 129SvEvTac mice (Taconic farms) were used as WT controls for this cohort.
METHOD DETAILS
Serology: Ig and autoantibody titers
Relative titers of anti-dsDNA, anti-Nucleosome in sera were measured by ELISA. Briefly, Nunc-Immuno maxisorp 96 well flat bottom plates were coated with 50μl/ well of 0.01% poly-L lysine (Sigma Aldrich) for one hour at RT, washed with PBS and coated with 50μl/ well of 10μg/ml calf-thymus DNA (Calbiochem) or 1μg/ml calf-thymus nucleosomes (Arotec diagnostics) in PBS, overnight at 4°C. Excess antigen was washed off using 2–3 washes with PBS. Antigen-coated plates were blocked with 250μl/ well of PBS with 4% non-fat dry milk (NFDM) for 3h at RT. Blocked plates were washed once with PBS and coated with diluted serum in PBS and incubated overnight at 4°C. Unbound serum antibodies were washed off with 3 washes of PBS + 1% NFDM. The bound IgG was detected using a 1:1000 dilution of goat anti-mouse IgG-AP-conjugate (Jackson Immunoresearch) diluted in PBS + 1% NFDM. Unbound secondary antibody was washed with PBS+ 1% NFDM and developed using diethanolamine substrate buffer (Thermo Fisher Scientific, 34064) and PNPP phosphatase substrate tablets (Sigma Aldrich, SIGMAS0942). For relative quantitation of antigen-specific IgG titers, serum from an anti-DNA or anti-Nuc positive animal was used as a standard using serial double dilutions. O.D. at the lowest serum dilution was arbitrarily assigned a value of 100 U/ml. Standard curve was plotted as O.D. at 405nm vs antigen concentration (U/ml). Relative IgG titers of samples were calculated using the straight-line equation and multiplied with the dilution factor. In all ELISA assays, in each plate, controls and test samples were run together and analyzed using the same standard curve.
Total serum IgG titers were determined by coating the ELISA plates with 5 μg/ml mouse IgG (Southern Biotech) and probing the bound serum antibody with anti-mouse IgG-AP (Jackson Immunoresearch).
ANA analysis
For detection of ANA and assessment of ANA staining patterns, HEp-2 human tissue culture substrate slides (Antibodies Incorporated, Davis, CA) were coated with sera at a 1:50 dilution in PBS for 1h at RT in a hydrated chamber and probed with rat anti-mouse κ- FITC conjugate (Southern Biotechnologies Associates, Birmingham, AL) at a 1:200 dilution. Immunofluorescence images were captured using a Keyence BZ-X710 fluorescence microscope at 20X magnification. The color intensity of images was enhanced slightly using Adobe Photoshop CC (Adobe Systems, San Jose, CA) or ImageJ. This was necessary for better visualization and was carried out consistently between all images while maintaining the integrity of the data. Immunofluorescence intensity was quantitated using ImageJ 1.51s image processing and analysis software (NIH, USA).
Crithidia luciliae Immunofluorescence test – CLIFT
Crithidia luciliae is a protozoan routinely used in the clinic as a substrate to specifically detect antibodies to pure dsDNA (without proteins). A positive test shows staining of the kinetoplast of the protozoan, while staining for basal body or nucleus or both without it, is considered a negative test. The substrate was purchased from Euroimmun (Lubeck, Germany). Serum samples from 12-month-old mice were tested at 1:25 dilution. Anti-dsDNA binding was detected using rat anti-mouse Igκ-FITC conjugate (Southern Biotechnologies Associates, Birmingham, AL). Fluorescence images were acquired using a Zeiss Ax10 fluorescence microscope at 63X magnification and processed using ImageJ software.
ELISpot
ELISpot plates (Millipore, Ref# MSIPS4W10; Multiscreen HTS) were coated with 0.01% poly-L lysine for 1h at RT, followed by coating with 100 μg/ ml calf thymus DNA (Sigma Aldrich) or 10 μg/ ml calf thymus nucleosomes (Arotec diagnostics) overnight at 4°C. Blocked with PBS + 5% FCS for 2–3h at RT. Freshly isolated splenocytes or BM cells were resuspended in freshly prepared warm 15% RPMI 1640 media + antibiotics + 1mM L-glutamine at 20 × 106 cells/ ml. 1×106 total cells were plated on the top wells and serially double diluted (1:2). ELISpot plates were then incubated for 18h at 37°C with 5% CO2. Washed with PBS + 1% FCS to remove the cells and probed with a 1:500 dilution of goat anti-mouse IgG AP (Jackson Immunoresearch) for 3–4h at 4°C. After washing, plates were developed using the Vector Blue AP Substrate Kit III (Vector Labs). Spots were captured and counted using ImmunoSpot S6 Analyzer (Cellular Technology Limited, Shaker Heights, OH). For analysis of total IgG-secreting AFCs, ELISPot plates were coated with goat anti-mouse IgG (SouthernBiotech) at 5μg/ml and blocked with PBS + 5% BSA. Plates were washed with PBS + 1% BSA and probed with a 1:500 dilution of goat anti-mouse IgG AP (Jackson Immunoresearch).
Flow cytometry
Cell suspensions of peripheral blood, mesenteric and inguinal lymph nodes or splenocytes were subjected to red blood cell lysis, washed and resuspended in staining buffer (1% FCS + 2% BSA + 1mM EDTA in PBS). Cells were stained with indicated cell surface markers. For intracellular staining, cells were surface stained and subsequently fixed and permeabilized using FOXP3 intracellular staining kit (eBioscience) and stained with anti-FOXP3-FITC conjugate. EdU positive cells were stained with Click-iT EdU Alexa Fluor 647 Flow cytometry assay kit (Thermo Fisher Scientific # C10424), as per instructions. Samples were acquired using Attune NxT (Thermo Fisher) flow cytometer and analyzed using FLowJo software version 9 or 10 (Tree Star).
Kidney histopathology
For imaging of kidneys, we use at least 4–5 mice per genotype that are closest to the average values of anti-DNA titers and spleen weight for that genotype. One half from each kidney was fixed with 10% neutral formalin for 24h at RT, and stored in 75% ethanol and subsequently embedded in paraffin. 5 μm thick sections were stained with Hematoxylin and Eosin and captured using the PerkinElmer Vectra multispectral imaging system at 40X magnification and glomerular size was analyzed using Slidepath software (Leica Biosystems digital image hub).
Immunofluorescence analysis of kidney and spleen sections
For imaging of the spleens and kidneys we use at least 4–5 mice per genotype that are closest to the average values of anti-DNA titers and spleen weight for that genotype. One half from both kidneys were frozen in OCT (Tissue Tek). 5–10 μm thick kidney sections were fixed using chilled acetone for 10 min. Sections were stained with DAPI, goat anti-mouse IgG-PE conjugate (eBioscience) and goat anti-mouse C3-FITC conjugate (Immunology consultant’s Laboratory Inc., Oregon). Images were captured using Keyence BZ-X710 fluorescence microscope at 40X magnification. Immunofluorescence intensity was quantitated using ImageJ 1.51s image processing and analysis software (NIH, USA). The color intensity of images was enhanced slightly using Adobe Photoshop CC (Adobe Systems, San Jose, CA). This was necessary for better visualization and was carried out consistently on whole images of control and test sections.
Spleen were either frozen in OCT and then sectioned and fixed using chilled acetone or were fixed in 4% PFA overnight followed by 8–10 hours of dehydration in 30% sucrose, after which they were frozen in OCT for cryosectioning. 5–10 μm thick spleen sections were stained with indicated anti mouse Ab-conjugates: GL-7-FITC, IgDAPC, CD138-PE, CD169-APC, Ki67-PE, CD4-FITC/ APC, SiglecH-APC and B220-FITC. Images were captured using Keyence BZ-X710 fluorescence microscope at 4X or 20X magnification or using the Zeiss 880 confocal microscope, maintained by NYU Langone Health’s Microscopy Laboratory. Scanning of whole sections was achieved using ZEN imaging software– tile and stitch functions. All images were acquired with similar functions. Image analysis was done using Bitplane’s Imaris version 9.2.0. Quantification of in situ mean fluorescence intensity sum of CD138 staining was determined using isosurfacing function with subtraction of background staining of spleen capsule. Quantitative analysis of total number of CD138+ or Ki67+ spots per half spleen represented, were calculated by rendering CD138+ or Ki67+ cells with spots using Imaris image analysis software.
Cyclophosphamide treatment
Ten 6-mo-old Dnase1l3−/− mice were bled on day 0 to get pretreatment serum titers of anti-dsDNA and anti-Nucleosome antibodies. 5 mice each were subsequently injected intraperitonially with equal volume of either PBS or 500μg cyclophosphamide monohydrate (CYTOPAC, Sigma Aldrich) reconstituted in PBS per mouse for three consecutive days. Mice were rested for 4 days. Subsequently mice were treated with 1 mg EdU (Carbosynth) intravenously for 12 h after which they were euthanized.
Anti-CD20 treatment
Serum was collected from five-month-old Dnase1l3−/− female mice (pretreatment). Subsequently they were injected (i.p) with 100μg of anti-mouse CD20 (clone 5D2), a kind gift from Genentech, or an IgG2a isotype control antibody (C1.18.4, Bio × cell, cat # BE0085), ~ every 15 days for a month. Serum was collected after two weeks of each treatment. Mice were euthanized at six-months of age and spleen was analyzed for AFCs and cellular populations.
Sorting of B cell subsets
Plasmablasts (PB): CD19+ B220+/− SiglecH− TCRb− CD138hi; Germinal center B cells (GC): CD19+ B220+ SiglecH− TCRb− CD138− CD38− GL-7hi ; Naïve Follicular B cells (FO): CD19+ B220+ SiglecH− TCRb− CD138− CD38+ GL-7− CD44− Sca1− ; Follicular activated B cells (FOA): CD19+ B220+ SiglecH− TCRb− CD138− CD38+ GL-7− CD44hi Sca1hi and class-switched B cells (IgG+): CD19+ B220+ SiglecH− TCRb− IgD− IgM− IgG2a/2b+ were sorted from total splenocytes of > 6 mo old WT or Dnase1l3−/− mice using the BD FACSAria III Sorter. Sorted cells were used for ELISPot analysis or BCR repertoire analysis.
BCR sequencing and analysis
VH Sequencing.
Sorted B cell subsets FO, PB, GC and IgG+ were lysed in TRIzol® Reagent (Invitrogen) and total RNA was extracted and purified using the RNeasy Mini Kit (Qiagen). cDNA was synthesized from 500 ng total RNA using the SuperScript™ IV First-Strand Synthesis System (Invitrogen). Variable heavy (VH) transcript was amplified with a multiplex primer set (primer details in table 1) using FastStart™ High Fidelity PCR System (Roche) under the following conditions: 2 min at 95C; 4 cycles of 92C for 30 s, 50C for 30 s, 72C for 1 min; 4 cycles of 92C for 30 s, 55C for 30 s, 72C for 1 min; 22 cycles of 92C for 30 s, 63C for 30 s, 72C for 1 min; 72C for 7 min; hold at 4C, and sequenced by 2×300 paired-end Illumina MiSeq.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| V450- anti-mouse CD19 (1D3) | BD Biosciences | Cat # 560375; RRID:AB_1645269 |
| PE/Cy7 anti-mouse CD4 (RM4–5) | eBioscience | Cat # 25-0042-82; RRID:AB_469578 |
| APC-anti-mouse CD4 (GK1.5) | eBioscience | Cat # 17-0041-81; RRID:AB_469319 |
| BV711-anti-mouse CD4 (RM4–5) | BioLegend | Cat # 100549; RRID:AB_11219396 |
| PE anti-mouse CD11b (M1/70) | eBioscience | Cat # 12-0112-81; RRID:AB_465546 |
| FITC anti-mouse CD11c (N418) | eBioscience | Cat # 11-0114-82; RRID:AB_464940 |
| AF700 anti-mouse CD44 (IM7) | BD Pharmingen | Cat # 560567; RRID:AB_1727480 |
| APC anti-mouse CD44 (IM7) | eBioscience | Cat # 17-0441-83; RRID:AB_469391 |
| PeCy7 anti-mouse CD69 (H1.2F3) | eBioscience | Cat # 25-0691-81; RRID:AB_469636 |
| PE anti mouse PD-1 (29F.IA12) | BioLegend | Cat # 135205; RRID:AB_1877232 |
| BV605 anti-mouse ICOS (C398.4A) | BioLegend | Cat # 313537; RRID:AB_2687078 |
| PE anti-mouse CD162 (PSGL-1) (2PH1) | BD Pharmingen | Cat # 555306; RRID:AB_395719 |
| BV711 anti-mouse CD40L (MR1) | BD Bioscience | Cat # 740685; RRID:AB_2740371 |
| PE anti-mouse CD138 (281–2) | BD Pharmingen | Cat # 561070; RRID:AB_2033998 |
| PEy7 anti-mouse CD138 (281–2) | BioLegend | Cat # 142514; RRID:AB_2562198 |
| FITC anti-mouse CD62L (MEL-14) | eBioscience | Cat # 11-0621-82; RRID:AB_465109 |
| PerCP/Cy5.5 anti-mouse CD25 (PC61) | BioLegend | Cat # 102029; RRID:AB_893291 |
| Pacific Blue anti-mouse B220 (RA3–6B2) | BioLegend | Cat # 103227; RRID:AB_492876 |
| BV605 anti-mouse B220 (RA3–6B2) | BioLegend | Cat # 103243; RRID:AB_11203907 |
| Alexa Fluor 488 anti-mouse FOXP3 (FJK-16S) | eBioscience | Cat # 53-5773-82; RRID:AB_763537 |
| FITC Rat and mouse T and B cell activation antigen (GL-7) | BD Pharmingen | Cat # 562080; RRID:AB_10894953 |
| APC anti-mouse IgD (11–26c.2a) | BioLegend | Cat # 405714; RRID:AB_10643423 |
| FITC anti-mouse IgM (II/41) | eBioscience | Cat # 11-5790-81; RRID:AB_465244 |
| PerCP/Cy5.5 anti-mouse Sca-I (D7) | eBioscience | Cat # 45-5981-82; RRID:AB_914372 |
| PE/Cy anti-mouse Sca-I (D7) | eBioscience | Cat # 25-5981-81 ; RRID:AB_469668 |
| AF700 anti-mouse MHC-II (M5/114.15.2) | eBioscience | Cat # 56-5321-82; RRID:AB_494009 |
| PE/Cy7 anti-mouse Ly6G (1A8) | BioLegend | Cat # 127617; RRID:AB_1877262 |
| APC anti-mouse Ly6C (HK1.4) | eBioscience | Cat # 17-5932-82; RRID:AB_1724153 |
| PE anti-mouse Ki67 (16A8) | BioLegend | Cat # 652403; RRID:AB_2561524 |
| PE anti-mouse IFNAR (AR1–5A3) | BioLegend | Cat # 127312; RRID:AB_2248800 |
| PerCP/Cy5.5 anti-mouse CXCR4 (2B11) | eBioscience | Cat # 46-9991-80; RRID:AB_10670192 |
| BV711 anti-mouse Siglec-H (440c) | BD Bioscience | Cat # 747671; RRID:AB_2744232 |
| FITC goat anti-mouse C3 | Immunology Consultants Laboratory Inc | Cat # GC3–90F-Z |
| FITC rat anti-mouse kappa (187.1) | SouthernBiotech | Cat # 1170–02; RRID:AB_2794663 |
| PE goat anti-mouse IgG F(ab)’ Polyclonal | eBioscience | Cat # 12-4010-82; RRID:AB_11063706 |
| Biotin anti-mouse IgG polyclonal | eBioscience | Cat # 13-4013-85; RRID:AB_466650 |
| Biotin anti-mouse IgM (II/41) | eBioscience | Cat # 13-5790-81; RRID:AB_466674 |
| Biotin anti-mouse IgD (11–26c (11–26) | eBioscience | Cat # 13-5993-81; RRID:AB_466859 |
| Biotin anti-mouse CD19 (1D3) | eBioscience | Cat # 13-0193-82; RRID:AB_657656 |
| Biotin anti-mouse CD93 (AA4.1) | eBioscience | Cat # 13-5892-81; RRID:AB_466766 |
| Biotin anti-mouse CD5 (53–7.3) | eBioscience | Cat # 13-0051-81; RRID:AB_466338 |
| Biotin anti-mouse Ly6G | BioLegend | Cat # 127603; RRID:AB_118610 |
| Biotin anti-mouse Ter-119 (TER-119) | eBioscience | Cat # 13-5921-81; RRID:AB_466796 |
| Biotin anti-mouse CD41 (MWReg30) | BioLegend | Cat # 133930; RRID:AB_2572133 |
| Biotin anti-mouse NK1.1 (PK136) | eBioscience | Cat # 13-5941-81; RRID:AB_466803 |
| Biotin anti-mouse TCRß (H57–597) | eBioscience | Cat # 13-5961-81; RRID:AB_466818 |
| Biotin anti-mouse CD3e (145–2C11) | BioLegend | Cat # 100303; RRID:AB_350220 |
| Biotin anti-mouse CD11b (M1/70) | eBioscience | Cat # 13-0112-81; RRID:AB_466358 |
| Biotin anti-mouse CD24 (M1/69) | eBioscience | Cat # 13-0242-81; RRID:AB_466396 |
| Biotin anti-mouse F4/80 (BM8) | eBioscience | Cat # 13-4801-81; RRID:AB_466656 |
| Biotin anti-mouse CXCR5 (2G8) | BD Biosciences | Cat # 551960; RRID:AB_394301 |
| Biotin anti-mouse IgG2a/2b (R2–40) | BD Biosciences | Cat # 553398; RRID:AB_394836 |
| Biotin anti-mouse CD43 (eBioR2/60) | eBioscience | Cat # 13-0431-82; RRID:AB_466439 |
| Alkaline phosphatase conjugated goat anti-mouse IgG | Jackson Immunoresearch | Cat # 115-055-071; RRID:AB_2338535 |
| Mouse IgG-UNLB (Unconjugated) normal mouse serum | SouthernBiotech | Cat # 0107–01 ; RRID:AB_2732898 |
| Goat Anti-Mouse IgG-UNLB | SouthernBiotech | Cat # 1010–01; RRID:AB_2794121 |
| TruStain FcX anti-mouse CD16/32 (93) | BioLegend | Cat # 101319; RRID:AB_1574973 |
| BV421 anti-mouse CD183 (CXCR3–173) | BioLegend | Cat # 126529; RRID:AB_2563100 |
| InVivoMAb mouse IgG2a isotype control (C1.18.4) | Bio × Cell | Cat # BE0085; RRID:AB_1107771 |
| Purified mouse-anti-CD20 (5D2) | Genentech, Inc. | NA |
| F(ab)’2 fragment goat anti-mouse IgM, μ chain specific | Jackson ImmunoResearch | Cat # 115-006-075 |
| LEAF purified anti-mouse CD40 | BioLegend | Cat # 102810 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Nucleosome antigen, calf thymus | Arotec Diagnostics | Cat # ATN02 |
| Deoxyribonucleic acid, calf thymus | Calbiochem | Cat # 2618 |
| Poly-L-lysine solution | Sigma Aldrich | Cat # P8920–100ML |
| Non-Fat Dry Milk | LabScientific | Cat # M-0841 |
| Pierce Diethanolamine Substrate Buffer (5X) | Thermo Scientific | Cat # 34064 |
| Phosphatase substrate | Sigma Aldrich | Cat # S0942–200TAB |
| ANA substrate slides | Antibodies Incorporated | Cat # 15–123 |
| Crithidia luciliae sensitive (anti-dsDNA) indirect immunofluorescence test | Euroimmun | Cat # FA 1572-1010-1 |
| ELISPot plates | Millipore | Cat # MSIPS4W10 |
| Fetal Bovine Serum | Sigma Aldrich | Cat # F0926–500 |
| VECTOR Blue alkaline phosphatase (Blue AP) substrate kit | Vector Laboratories | Cat # SK-5300 |
| EDTA | Fisher | Cat # BP24821 |
| Neutral Buffered Formalin (10%) | Thermo Scientific | Cat # 22-050-104 (5701) |
| Ethanol, Absolute | Fisher Scientific | Cat # BP2818–500 |
| Tissue Plus O.C.T. Compound | Thermo Fisher Scientific | Cat # 23-730-571 |
| Acetone | Fisher Scientific | Cat # A18–1 |
| DAPI (4’,6 Diamidino 2 Phenylindole, Dilactate) | Thermo Fisher Scientific | Cat # D3571 |
| Paraformaldehyde 4% in PBS | Thermo Fisher Scientific (Alfa Aesar) | Cat # J19943-K2 |
| Cyclophosphamide monohydrate (CYTOPAC) | Sigma Aldrich | Cat # C7397–1G |
| 2’-deoxy-ethyryluridine (EdU) | Carbosynth | Cat # NE08701 |
| Recombinant mouse IFNa1 (carrier free) | BioLegend | Cat # 751804 |
| ODN2216 (CpGA) | Invivogen | Cat # Tlrl-2216–1 |
| Recombinant mouse IL-4 protein | R&D systems | Cat # 404-ML-010 |
| RPMI medium 1640 with L-glutamine | Gibco | Cat # 11875–119 |
| L-glutamine 200mM | Thermo Fisher Scientific | Cat # 25030–164 |
| Beta-mercaptoethanol | Sigma Aldrich | Cat # 444203–250ML |
| Penicillin-Streptomycin 10,000U/ml | Gibco | Cat # 15140122 |
| RBC lysis buffer | BioLegend | Cat # 420301 |
| Live/Dead fixable aqua dead cell stain | Thermo Fisher Scientific | Cat # L34965 |
| Bovine serum albumin | Sigma Aldrich | Cat # A9647–100G |
| Sucrose | Sigma Aldrich | Cat # S0389–1 KG |
| LPS-EK Ultrapure | Invivogen | Cat # Tlrl-peklps |
| Trizol LS reagent | Ambion | Cat # 10296028 |
| Streptavidin PerCP/Cy5.5 | eBioscience | Cat # 45-4317-82 |
| Streptavidin PE/Cy7 | eBioscience | Cat # 25-4317-82 |
| Prolong Diamond antifade | Thermo Fisher Scientific | Cat # P36961 |
| Critical Commercial Assays | ||
| Click-iT™ EdU Alexa Fluor™ 647 Flow Cytometry Assay Kit | Thermo Scientific | Cat # C10419 |
| RNeasy Mini Kit | Qiagen | Cat # 74104 |
| SuperScript IV First strand synthesis system | Thermo Fisher Scientific | Cat # 18091050 |
| FastStart high fidelity PCR system | Roche Applied Science | N/A |
| Lumikine mIFNa bioluminescence ELISA kit | Invivogen | Cat # lumi-mifna |
| Streptavidin microbeads | Miltenyi Biotec | Cat # 130-048-101 |
| FOXP3/transcription factor staining buffer set | eBioscience | Cat # 00–5523 |
| Deposited Data | ||
| Experimental Models: Organisms/Strains | ||
| Mouse: WT: C57BL/6J | The Jackson Laboratory | Stock # 000664; RRID:IMSR_JAX:000664 |
| Mouse: WT: 129SvEvTac | Taconic Farms | Model # 129SVE-F or 129SVE-M; RRID:IMSR_TAC:129sve |
| Mouse: Dnase1l3−/− [B6NCrl.Cg-Dnase1l3tm1c(KOMP)Wtsi/TwinMmucd] | Taconic animal repository | Model TF2732; RRID:MMRRC_065348-UCD |
| Mouse: Cd40lg−/− [B6.129S2-Cd40lgtm1Imx/J] | The Jackson Laboratory | Stock # 002770; RRID:IMSR JAX:002770 |
| Mouse: Tlr7−/− [B6.129S1-Tlr7tm1Flv/J] | The Jackson Laboratory | Stock # 008380; RRID:IMSR JAX:008380 |
| Mouse: Ifnar1−/− [B6.129S2-Ifnar1tm1Agt/Mmjax] | The Jackson Laboratory | Stock # 032045-JAX; RRID:MMRRC 032045-JAX |
| Mouse: Tcf4+/− [B6;129- Tcf4tm1Zhu/J] | The Jackson Laboratory | Stock No: 013598 RRID:MGI :3040597 |
| Mouse: C57BL/6J-Tlr9M7Btir/Mmjax | The Jackson Laboratory | MMRRC Stock No. 34329-JAX/CpG11 ; RRID:MMRRC 034329-JAX |
| Oligonucleotides | ||
| VH Forward Primers Primer (5’ -> 3’) | ||
| mVH-Fwd1 GAKGTRMAGCTTCAGGAGTC |
Sigma Aldrich | N/A |
| mVH-Fwd2 GAGGTBCAGCTBCAGCAGTC |
Sigma Aldrich | N/A |
| mVH-Fwd3 CAGGT GCAGCT GAAGSASTC |
Sigma Aldrich | N/A |
| mVH-Fwd4 GAGGTCCARCTGCAACARTC |
Sigma Aldrich | N/A |
| mVH-Fwd5 CAGGTYCAGCTBCAGCARTC |
Sigma Aldrich | N/A |
| mVH-Fwd6 CAGGTYCARCTGCAGCAGTC |
Sigma Aldrich | N/A |
| mVH-Fwd7 CAGGT CCACGT GAAGCAGT C |
Sigma Aldrich | N/A |
| mVH-Fwd8 GAGGT GAASST GGTGGAATC |
Sigma Aldrich | N/A |
| mVH-Fwd9 GAVGTGAWGYTG GTGGAGTC |
Sigma Aldrich | N/A |
| mVH-Fwd10 GAGGT GCAGSKGGT GGAGT C |
Sigma Aldrich | N/A |
| mVH-Fwd11 GAKGTGCAMCTGGTGGAGTC |
Sigma Aldrich | N/A |
| mVH-Fwd12 GAG GT GAAG CTGATGGARTC |
Sigma Aldrich | N/A |
| mVH-Fwd13 GAGGT GCARCTT GTT GAGT C |
Sigma Aldrich | N/A |
| mVH-Fwd14 GARGTRAAGCTTCTCGAGTC |
Sigma Aldrich | N/A |
| mVH-Fwd15 GAAGT GAARSTT GAGGAGT C |
Sigma Aldrich | N/A |
| mVH-Fwd16 CAGGTTACTCTRAAAGWGTSTG |
Sigma Aldrich | N/A |
| mVH-Fwd17 CAGGTCCAACTVCA GCARCC | Sigma Aldrich | N/A |
| mVH-Fwd18 GAT GT GAACTTGGAAGT GT C |
Sigma Aldrich | N/A |
| mVH-Fwd19 GAG GT GAAG GT CAT CGAGTC |
Sigma Aldrich | N/A |
| VH Reverse Primers Primer (5’ -> 3’) | ||
| mVH-Rev-IgG CCARKGGATAGACHGATGGGG |
Sigma Aldrich | N/A |
| mVH-Rev-IgA TGGTGGGATTTCTCGCAGAC |
Sigma Aldrich | N/A |
| mVH-Rev-IgM GCAGGAGACGAGGGGGAAGA |
Sigma Aldrich | N/A |
| Software and Algorithms | ||
| ImageJ | https://imaqei.nih.qov/ij/ | RRID:SCR_001935 |
| CTL Immunospot analyzer | http://www.immunosDot.com/ImmunoSpot-analyzers-software | RRID:SCR_011082 |
| Adobe Photoshop CC2018 | https://www.adobe.com/products/photoshop.html | RRID: SCR_014199 |
| Adobe Illustrator CC2018 | https://www.adobe.com/products/illustrator.html | RRID: SCR_010279 |
| FLowjo v10 | Treestar Inc | RRID: SCR_008520 |
| GraphPad Prism 7 | Graphpad Inc | RRID: SCR_002798 |
| Imaris | http://www.bitplane.com/imaris/imaris | RRID: SCR 007370 |
| Keyence BZ Analyzer software | https://www.keyence.co.iD/Droducts/microscoDe/fluorescence-microscoDe/bz-8100/models/bz-h1a/ | RRID:SCR_017205 |
| Slidepath Gateway digital pathology | Leica Microsystems | RRID:SCR_005597 |
Bioinformatic Analysis.
Raw 2×300 reads were trimmed depending on sequence quality using Trimmomatic (Bolger et al., 2014) and annotated using MiXCR (Bolotin et al., 2015). Sequences with ≥ 2 reads were grouped into clonotypes by clustering those with ≥ 95% CDR-H3 amino acid identity (Edgar, 2010). CDR-H3 characteristics were analyzed using custom Python scripts.
Statistics.
p-values for the analysis of positively charged amino acids within the CDR-H3 and V-gene usage were determined using Fisher’s Exact test. p-values for the analysis of the mean CDR-H3 isoelectric point and hydrophobicity were determined using Student’s t-test or Mann-Whitney U test.
In vitro B cell proliferation/ differentiation assay
Naïve B cells were MACS-purified by negative selection from splenocytes of WT, Dnase1l3−/−, Ifnar1−/− or Dnase1l3−/−Ifnar1−/− mice using CD43 microbeads. Purified B cells were labelled with 3 μM Cell trace CFSE (Invitrogen), in PBS+ 2% BSA for 10 mins at RT. CFSE labelled cells were washed twice with PBS and subsequently suspended in RPMI + 10% FBS + 2mM L-glutamine + 10μM β-Mercaptoethanol and plated in round bottom 96 well plates at a concentration of 0.5×106 cells/ well in 200μl media. Cells were either left unactivated or activated with: (1) 5 μg/ml anti-IgM (Jackson Immunoresearch) + anti-CD40 (LEAF-purified, BioLegend) and 5 ng/ml recombinant mouse IL-4 (R&D systems) OR (2) 1μg/ml LPS (LPS-EK Ultrapure, InvivoGen) + 5 ng/ml recombinant mouse IL-4. Unactivated (UA) and activated (A) B cells were supplemented or not with recombinant mouse IFNα1 (BioLegend) at indicated concentrations. B cells were analyzed for activation, proliferation and differentiation after 3 days of culture.
pDC purification
RBC lysed splenocytes or bone marrow cells of Dnase1l3−/− mice were resuspended in RPMI + 10% FCS and were allowed to adhere to sterile tissue culture treated 100 mm petriplates for 1h at 37°C after which adherent cells were discarded. Floating cells were resuspended at 10 ×106 cells in 90μl of wash buffer and labelled with the following cocktail of biotinylated antibodies: Anti-IgG, anti-IgM, anti-IgD, anti-CD19, anti-CD93, anti-CD5, anti Ly6G, anti-Ter119, anti-CD41, anti NK1.1, anti-TCRb, anti CD3, anti CD11b, anti-CD24 and anti-F4/80 each at 1: 200 dilution for 20 mins at 4°C. Labeled cells were washed and stained with Streptavidin microbeads for 20 mins at 4°C. Unlabeled cells were collected by negative selection. Up-to 45–50% pure pDCs (B220lo SiglecH+ CD11clo) could be routinely identified by flow cytometry.
pDC, B cell co-culture assay
pDCs were purified from Dnase1l3−/− BM cells using MACS negative selection (described above) and B cells were purified from Dnase1l3−/ or Dnase1l3−/−Ifnar1−/− mice by negative selection using CD43 microbeads and were labeled with cell trace CFSE. 0.5 × 106 purified B cells per well were left unactivated (UA) or activated (A) with 5 μg/ml anti-IgM (Jackson Immunoresearch) + anti-CD40 (BioLegend) and 5 ng/ml recombinant mouse IL-4 (R&D systems). Activated B cells were cultured in the presence of ~2×104 purified unactivated pDCs (A + pDCs) OR treated with 1μg/ml CpGA (ODN 2216, InvioGen) (A + CpG) OR co-cultured with ~2×104 purified pDCs activated with 1μg/ml CpGA (A + pDCs + CpGA). Cells were co-cultured for three days after which B cells were analyzed for activation, proliferation and differentiation by flow cytometry and supernatant was analyzed for IFNα production by ELISA.
IFNα ELISA
Purified BM or splenic pDCs at various concentrations were left unstimulated or stimulated with 1μg/ml CpGA (ODN 2216, InvioGen) for 18h and supernatant was collected. Supernatants were collected from pDC + B cells cocultures as described above. Secreted IFNα in culture supernatants was measured using mouse IFN-alpha bioluminescence ELISA kit (InvivoGen) following manufacturer’s instructions.
QUANTIFICATION AND STATISTICAL ANALYSIS
Comparisons between multiple groups were performed by one-way ANOVA followed by multiple comparisons analysis, Tukey’s test, unless otherwise mentioned. Comparisons between two groups were performed by non-parametric student’s t-test with Mann-Whitney analysis. P values less than or equal to 0.05 were considered significant and significance was assigned according to the following breakdown: *p < 0.05, ** p < 0.01, *** p< 0.001 and **** p< 0.0001. Graph Pad Prism software vs. 7 or 8 was used for all statistical analysis.
DATA AND CODE AVAILABILITY
VH repertoire sequencing data have been deposited in the NCBI SRA database (accession code pending)
Supplementary Material
Highlights.
Anti-DNA antibody response is driven by T-dependent extrafollicular plasmablasts
IFN-I signaling propagates anti-DNA responses and SLE-like disease
IFN-I produced by pDCs promotes plasmablast proliferation and differentiation
TLR9 drives anti-DNA responses and autoimmunity redundantly with TLR7
Acknowledgements
Supported by the NIH grants AI072571 (B.R.), AR071703 (B.R., G.C.I.), AR070591 (B.R.), CA232666 (O.A.P.), AI100853 (O.A.P., L.S.), AR069515 (L.S.), HL145997 (J.N.P), CA009161 (J.N.P), CA110624 (G.C.I.) and AR071703 (G.C.I.). Also supported by the Lupus Research Alliance (B.R.), the Colton Center for Autoimmunity (B.R.), Idex Junior Chair program from the Bordeaux University (V.S.) and Cancer Research Institute CLIP grant (V.S).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing financial interest.
References
- Agrawal H, Jacob N, Carreras E, Bajana S, Putterman C, Turner S, Neas B, Mathian A, Koss MN, Stohl W, et al. (2009). Deficiency of type I IFN receptor in lupus-prone New Zealand mixed 2328 mice decreases dendritic cell numbers and activation and protects from disease. J Immunol 183, 6021–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Mayouf SM, Sunker A, Abdwani R, Abrawi SA, Almurshedi F, Alhashmi N, Al Sonbul A, Sewairi W, Qari A, Abdallah E, et al. (2011). Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat Genet 43, 1186–1188. [DOI] [PubMed] [Google Scholar]
- Barrat FJ, Meeker T, Gregorio J, Chan JH, Uematsu S, Akira S, Chang B, Duramad O, and Coffman RL (2005). Nucleic acids of mammalian origin can act as endogenous ligands for Toll-like receptors and may promote systemic lupus erythematosus. J Exp Med 202, 1131–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrat FJ, and Su L (2019). A pathogenic role of plasmacytoid dendritic cells in autoimmunity and chronic viral infection. J Exp Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett L, Palucka AK, Arce E, Cantrell V, Borvak J, Banchereau J, and Pascual V (2003). Interferon and granulopoiesis signatures in systemic lupus erythematosus blood. J Exp Med 197, 711–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, and Chudakov DM (2015). MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods 12, 380–381. [DOI] [PubMed] [Google Scholar]
- Bruschi M, Bonanni A, Petretto A, Vaglio A, Pratesi F, Santucci L, Migliorini P, Bertelli R, Galetti M, Belletti S, et al. (2019). Neutrophil Extracellular Traps (NETs) profiles in patients with incident SLE and lupus nephritis. J Rheumatol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buechler MB, Teal TH, Elkon KB, and Hamerman JA (2013). Cutting edge: Type I IFN drives emergency myelopoiesis and peripheral myeloid expansion during chronic TLR7 signaling. J Immunol 190, 886–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, Baisch J, Phelps K, Clayton S, Gong M, et al. (2016). Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caielli S, Veiga DT, Balasubramanian P, Athale S, Domic B, Murat E, Banchereau R, Xu Z, Chandra M, Chung CH, et al. (2019). A CD4(+) T cell population expanded in lupus blood provides B cell help through interleukin-10 and succinate. Nat Med 25, 75–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celhar T, Magalhaes R, and Fairhurst AM (2012). TLR7 and TLR9 in SLE: when sensing self goes wrong. Immunol Res 53, 58–77. [DOI] [PubMed] [Google Scholar]
- Christensen SR, and Shlomchik MJ (2007). Regulation of lupus-related autoantibody production and clinical disease by Toll-like receptors. Semin Immunol 19, 11–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, and Shlomchik MJ (2006). Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity 25, 417–428. [DOI] [PubMed] [Google Scholar]
- Cisse B, Caton ML, Lehner M, Maeda T, Scheu S, Locksley R, Holmberg D, Zweier C, den Hollander NS, Kant SG, et al. (2008). Transcription factor E2–2 is an essential and specific regulator of plasmacytoid dendritic cell development. Cell 135, 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham AF, Gaspal F, Serre K, Mohr E, Henderson IR, Scott-Tucker A, Kenny SM, Khan M, Toellner KM, Lane PJ, and MacLennan IC (2007). Salmonella induces a switched antibody response without germinal centers that impedes the extracellular spread of infection. J Immunol 178, 6200–6207. [DOI] [PubMed] [Google Scholar]
- Das A, Heesters BA, Bialas A, O’Flynn J, Rifkin IR, Ochando J, Mittereder N, Carlesso G, Herbst R, and Carroll MC (2017). Follicular Dendritic Cell Activation by TLR Ligands Promotes Autoreactive B Cell Responses. Immunity 46, 106–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degn SE, van der Poel CE, Firl DJ, Ayoglu B, Al Qureshah FA, Bajic G, Mesin L, Reynaud CA, Weill JC, Utz PJ, et al. (2017). Clonal Evolution of Autoreactive Germinal Centers. Cell 170, 913–926 e919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng R, Hurtz C, Song Q, Yue C, Xiao G, Yu H, Wu X, Muschen M, Forman S, Martin PJ, and Zeng D (2017). Extrafollicular CD4(+) T-B interactions are sufficient for inducing autoimmune-like chronic graft-versus-host disease. Nat Commun 8, 978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domeier PP, Chodisetti SB, Schell SL, Kawasawa YI, Fasnacht MJ, Soni C, and Rahman ZSM (2018). B-Cell-Intrinsic Type 1 Interferon Signaling Is Crucial for Loss of Tolerance and the Development of Autoreactive B Cells. Cell Rep 24, 406–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. [DOI] [PubMed] [Google Scholar]
- Finke D, Baribaud F, Diggelmann H, and Acha-Orbea H (2001). Extrafollicular plasmablast B cells play a key role in carrying retroviral infection to peripheral organs. J Immunol 166, 6266–6275. [DOI] [PubMed] [Google Scholar]
- Fukui R, Saitoh S, Matsumoto F, Kozuka-Hata H, Oyama M, Tabeta K, Beutler B, and Miyake K (2009). Unc93B1 biases Toll-like receptor responses to nucleic acid in dendritic cells toward DNA- but against RNA-sensing. J Exp Med 206, 1339–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale M, Rawlings DJ, and Jackson SW (2018). The long and the short of it: insights into the cellular source of autoantibodies as revealed by B cell depletion therapy. Curr Opin Immunol 55, 81–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton JA, Wu Q, Yang P, Luo B, Liu S, Hong H, Li J, Walter MR, Fish EN, Hsu HC, and Mountz JD (2017). Cutting Edge: Endogenous IFN-beta Regulates Survival and Development of Transitional B Cells. J Immunol 199, 2618–2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer BF, Moser K, Hauser AE, Peddinghaus A, Voigt C, Eilat D, Radbruch A, Hiepe F, and Manz RA (2004). Short-lived plasmablasts and long-lived plasma cells contribute to chronic humoral autoimmunity in NZB/W mice. J Exp Med 199, 1577–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson SW, Scharping NE, Kolhatkar NS, Khim S, Schwartz MA, Li QZ, Hudkins KL, Alpers CE, Liggitt D, and Rawlings DJ (2014). Opposing impact of B cell-intrinsic TLR7 and TLR9 signals on autoantibody repertoire and systemic inflammation. J Immunol 192, 4525–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jego G, Palucka AK, Blanck JP, Chalouni C, Pascual V, and Banchereau J (2003). Plasmacytoid dendritic cells induce plasma cell differentiation through type I interferon and interleukin 6. Immunity 19, 225–234. [DOI] [PubMed] [Google Scholar]
- Jenks SA, Cashman KS, Woodruff MC, Lee FE, and Sanz I (2019). Extrafollicular responses in humans and SLE. Immunol Rev 288, 136–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenks SA, Cashman KS, Zumaquero E, Marigorta UM, Patel AV, Wang X, Tomar D, Woodruff MC, Simon Z, Bugrovsky R, et al. (2018). Distinct Effector B Cells Induced by Unregulated Toll-like Receptor 7 Contribute to Pathogenic Responses in Systemic Lupus Erythematosus. Immunity 49, 725–739 e726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirou KA, Lee C, George S, Louca K, Peterson MG, and Crow MK (2005). Activation of the interferon-alpha pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum 52, 1491–1503. [DOI] [PubMed] [Google Scholar]
- Kuriakose J, Redecke V, Guy C, Zhou J, Wu R, Ippagunta SK, Tillman H, Walker PD, Vogel P, and Hacker H (2019). Patrolling monocytes promote the pathogenesis of early lupus-like glomerulonephritis. J Clin Invest 129, 2251–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Bon A, Schiavoni G, D’Agostino G, Gresser I, Belardelli F, and Tough DF (2001). Type i interferons potently enhance humoral immunity and can promote isotype switching by stimulating dendritic cells in vivo. Immunity 14, 461–470. [DOI] [PubMed] [Google Scholar]
- Le Bon A, Thompson C, Kamphuis E, Durand V, Rossmann C, Kalinke U, and Tough DF (2006). Cutting edge: enhancement of antibody responses through direct stimulation of B and T cells by type I IFN. J Immunol 176, 2074–2078. [DOI] [PubMed] [Google Scholar]
- Leadbetter EA, Rifkin IR, Hohlbaum AM, Beaudette BC, Shlomchik MJ, and Marshak-Rothstein A (2002). Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature 416, 603–607. [DOI] [PubMed] [Google Scholar]
- Lesley R, Kelly LM, Xu Y, and Cyster JG (2006). Naive CD4 T cells constitutively express CD40L and augment autoreactive B cell survival. Proc Natl Acad Sci U S A 103, 10717–10722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathian A, Gallegos M, Pascual V, Banchereau J, and Koutouzov S (2011). Interferon-alpha induces unabated production of short-lived plasma cells in preautoimmune lupus-prone (NZBxNZW)F1 mice but not in BALB/c mice. Eur J Immunol 41, 863–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muskardin TLW, and Niewold TB (2018). Type I interferon in rheumatic diseases. Nat Rev Rheumatol 14, 214–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson KM, Christensen SR, Shupe J, Kashgarian M, Kim D, Elkon K, and Shlomchik MJ (2010). TLR9 regulates TLR7- and MyD88-dependent autoantibody production and disease in a murine model of lupus. J Immunol 184, 1840–1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen CT, Ostergaard O, Johnsen C, Jacobsen S, and Heegaard NH (2011). Distinct features of circulating microparticles and their relationship to clinical manifestations in systemic lupus erythematosus. Arthritis Rheum 63, 3067–3077. [DOI] [PubMed] [Google Scholar]
- Odegard JM, Marks BR, DiPlacido LD, Poholek AC, Kono DH, Dong C, Flavell RA, and Craft J (2008). ICOS-dependent extrafollicular helper T cells elicit IgG production via IL-21 in systemic autoimmunity. J Exp Med 205, 2873–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozcakar ZB, Foster J 2nd, Diaz-Horta O, Kasapcopur O, Fan YS, Yalcinkaya F, and Tekin M (2013). DNASE1L3 mutations in hypocomplementemic urticarial vasculitis syndrome. Arthritis Rheum 65, 2183–2189. [DOI] [PubMed] [Google Scholar]
- Pawaria S, Moody KL, Busto P, Nundel K, Baum R, Sharma S, Gravallese EM, Fitzgerald KA, and Marshak-Rothstein A (2015). An unexpected role for RNA-sensing toll-like receptors in a murine model of DNA accrual. Clin Exp Rheumatol 33, S70–73. [PMC free article] [PubMed] [Google Scholar]
- Pisetsky DS (2016). Anti-DNA antibodies--quintessential biomarkers of SLE. Nat Rev Rheumatol 12, 102–110. [DOI] [PubMed] [Google Scholar]
- Pisetsky DS, Gauley J, and Ullal AJ (2011). Microparticles as a source of extracellular DNA. Immunol Res 49, 227–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radic MZ, and Weigert M (1994). Genetic and structural evidence for antigen selection of anti-DNA antibodies. Annu Rev Immunol 12, 487–520. [DOI] [PubMed] [Google Scholar]
- Rowland SL, Riggs JM, Gilfillan S, Bugatti M, Vermi W, Kolbeck R, Unanue ER, Sanjuan MA, and Colonna M (2014). Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J Exp Med 211, 1977–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santiago-Raber ML, Baccala R, Haraldsson KM, Choubey D, Stewart TA, Kono DH, and Theofilopoulos AN (2003). Type-I interferon receptor deficiency reduces lupus-like disease in NZB mice. J Exp Med 197, 777–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawai CM, Serpas L, Neto AG, Jang G, Rashidfarrokhi A, Kolbeck R, Sanjuan MA, Reizis B, and Sisirak V (2018). Plasmacytoid Dendritic Cells Are Largely Dispensable for the Pathogenesis of Experimental Inflammatory Bowel Disease. Front Immunol 9, 2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo SJ, Fields ML, Buckler JL, Reed AJ, Mandik-Nayak L, Nish SA, Noelle RJ, Turka LA, Finkelman FD, Caton AJ, and Erikson J (2002). The impact of T helper and T regulatory cells on the regulation of anti-doublestranded DNA B cells. Immunity 16, 535–546. [DOI] [PubMed] [Google Scholar]
- Serpas L, Chan RWY, Jiang P, Ni M, Sun K, Rashidfarrokhi A, Soni C, Sisirak V, Lee WS, Cheng SH, et al. (2019). Dnase1l3 deletion causes aberrations in length and end-motif frequencies in plasma DNA. Proc Natl Acad Sci U S A 116, 641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma S, Fitzgerald KA, Cancro MP, and Marshak-Rothstein A (2015). Nucleic Acid-Sensing Receptors: Rheostats of Autoimmunity and Autoinflammation. J Immunol 195, 3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi W, Liao Y, Willis SN, Taubenheim N, Inouye M, Tarlinton DM, Smyth GK, Hodgkin PD, Nutt SL, and Corcoran LM (2015). Transcriptional profiling of mouse B cell terminal differentiation defines a signature for antibody-secreting plasma cells. Nat Immunol 16, 663–673. [DOI] [PubMed] [Google Scholar]
- Shibata T, Ohto U, Nomura S, Kibata K, Motoi Y, Zhang Y, Murakami Y, Fukui R, Ishimoto T, Sano S, et al. (2016). Guanosine and its modified derivatives are endogenous ligands for TLR7. Int Immunol 28, 211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisirak V, Ganguly D, Lewis KL, Couillault C, Tanaka L, Bolland S, D’Agati V, Elkon KB, and Reizis B (2014). Genetic evidence for the role of plasmacytoid dendritic cells in systemic lupus erythematosus. J Exp Med 211, 1969–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sisirak V, Sally B, D’Agati V, Martinez-Ortiz W, Ozcakar ZB, David J, Rashidfarrokhi A, Yeste A, Panea C, Chida AS, et al. (2016). Digestion of Chromatin in Apoptotic Cell Microparticles Prevents Autoimmunity. Cell 166, 88–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soni C, and Reizis B (2018). DNA as a self-antigen: nature and regulation. Curr Opin Immunol 55, 31–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soni C, Wong EB, Domeier PP, Khan TN, Satoh T, Akira S, and Rahman ZS (2014). B cell-intrinsic TLR7 signaling is essential for the development of spontaneous germinal centers. J Immunol 193, 4400–4414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweet RA, Ols ML, Cullen JL, Milam AV, Yagita H, and Shlomchik MJ (2011). Facultative role for T cells in extrafollicular Toll-like receptor-dependent autoreactive B-cell responses in vivo. Proc Natl Acad Sci U S A 108, 7932–7937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tipton CM, Fucile CF, Darce J, Chida A, Ichikawa T, Gregoretti I, Schieferl S, Hom J, Jenks S, Feldman RJ, et al. (2015). Diversity, cellular origin and autoreactivity of antibody-secreting cell population expansions in acute systemic lupus erythematosus. Nat Immunol 16, 755–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vinuesa CG, Linterman MA, Yu D, and MacLennan IC (2016). Follicular Helper T Cells. Annu Rev Immunol 34, 335–368. [DOI] [PubMed] [Google Scholar]
- Vinuesa CG, Sanz I, and Cook MC (2009). Dysregulation of germinal centres in autoimmune disease. Nat Rev Immunol 9, 845–857. [DOI] [PubMed] [Google Scholar]
- Weckerle CE, Franek BS, Kelly JA, Kumabe M, Mikolaitis RA, Green SL, Utset TO, Jolly M, James JA, Harley JB, and Niewold TB (2011). Network analysis of associations between serum interferon-alpha activity, autoantibodies, and clinical features in systemic lupus erythematosus. Arthritis Rheum 63, 1044–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenburger T, von Neubeck B, Schneider A, Ebert N, Schreyer D, Acs A, and Winkler TH (2018). Epistatic Interactions Between Mutations of Deoxyribonuclease 1-Like 3 and the Inhibitory Fc Gamma Receptor IIB Result in Very Early and Massive Autoantibodies Against Double-Stranded DNA. Front Immunol 9, 1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westra HJ, Martinez-Bonet M, Onengut-Gumuscu S, Lee A, Luo Y, Teslovich N, Worthington J, Martin J, Huizinga T, Klareskog L, et al. (2018). Finemapping and functional studies highlight potential causal variants for rheumatoid arthritis and type 1 diabetes. Nat Genet 50, 1366–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- William J, Euler C, Christensen S, and Shlomchik MJ (2002). Evolution of autoantibody responses via somatic hypermutation outside of germinal centers. Science 297, 2066–2070. [DOI] [PubMed] [Google Scholar]
- Yung S, and Chan TM (2015). Mechanisms of Kidney Injury in Lupus Nephritis - the Role of Anti-dsDNA Antibodies. Front Immunol 6, 475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang Y, Cheng P, and Weintraub H (1996). B-lymphocyte development is regulated by the combined dosage of three basic helix-loop-helix genes, E2A, E2–2, and HEB. Mol Cell Biol 16, 2898–2905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zochling J, Newell F, Charlesworth JC, Leo P, Stankovich J, Cortes A, Zhou Y, Stevens W, Sahhar J, Roddy J, et al. (2014). An Immunochip-based interrogation of scleroderma susceptibility variants identifies a novel association at DNASE1L3. Arthritis Res Ther 16, 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
VH repertoire sequencing data have been deposited in the NCBI SRA database (accession code pending)