

Abstract
Introduction
Abnormal brain amyloid beta (Aβ) is typically assessed in vivo using global concentrations from cerebrospinal fluid and positron emission tomography (PET). However, it is unknown whether the assessment of the topographical distribution of Aβ pathology can provide additional information to identify, among global Aβ positive individuals, those destined for dementia.
Methods
We studied 260 amnestic mild cognitive impairment (MCI) subjects who were Aβ‐PET positive with [18F]florbetapir. Using [18F]florbetapir, we assessed the percentage of voxels sowing Aβ abnormality as well as the standardized uptake value ratio (SUVR) values across brain regions. Regressions tested the predictive effect of Aβ on progression to dementia over 2 years.
Results
Neither global nor regional [18F]florbetapir SUVR concentrations predicted progression to dementia. In contrast, the spatial extent of Aβ pathology in regions comprising the default mode network was highly associated with the development of dementia over 2 years.
Discussion
These results highlight that the regional distribution of Aβ abnormality may provide important complementary information at an individual level regarding the likelihood of Aβ positive MCI to progress to dementia.
Keywords: Alzheimer's disease, amyloid beta, mild cognitive impairment, positron emission tomography
1. INTRODUCTION
Recently, the Imaging Dementia‐Evidence for Amyloid Scanning (IDEAS) study, a large‐scale multicenter cohort supported clinicians’ change in patient management or counseling in more than 60% of participants with mild cognitive impairment (MCI) or dementia as a result of amyloid beta positron emission tomography (Aβ‐PET) status information. 1 Smaller studies have also supported the use of Aβ‐PET for changes in clinical management, 2 including in specialized centers evaluating subjects with early‐onset dementia. 3 The use of Aβ‐PET also extends to clinical trial design: the enrolment of Aβ positive MCI individuals has been used for population enrichment of clinical trials due to their increased rate of progression to dementia, 4 to monitor target engagement, and to select individuals who are most likely to respond to specific interventions. 5 However, these studies invariably used global averaged values to represent Aβ pathology, neglecting information on the topographic extent of brain amyloidosis, which is the most important advantage of PET in relation to other Aβ biomarker techniques with non‐spatial resolution such as cerebrospinal fluid (CSF).
Previous studies support a link between the regional patterns of Aβ pathology and disease progression. For instance, Aβ deposition measured in vivo with PET imaging shows uptake quantitatively and topographically consistent with pathological descriptions of Aβ plaques in autopsy‐confirmed cases of Alzheimer's disease (AD). 6 , 7 , 8 Reports have demonstrated that elevated regional Aβ PET retention is associated with faster progression from MCI to AD dementia, 9 and Aβ‐PET can be used to distinguish between different forms of dementia. 10 In addition, several recent studies have demonstrated the value of topographic staging of Aβ pathology using PET. 11 , 12 Given the probable imminent use Aβ‐PET in clinical settings in the United States, 1 it is important to determine whether the topographical distribution of Aβ‐PET uptake can provide additional insights into probability of imminent clinical progression and therefore increase the utility of this biomarker in clinical practice.
Here, capitalizing on the spatial resolution of PET imaging, we tested the hypothesis that the extent of the Aβ abnormality across brain structures may provide complementary information to predict progression from Aβ positive MCI individuals to dementia.
RESEARCH IN CONTEXT
Systematic review: Given the possible use of amyloid beta positron emission tomography (Aβ‐PET) in clinical settings, it is important to determine whether the topographical distribution of Aβ‐PET uptake can provide additional insights into probability of imminent clinical progression and therefore increase the utility of this biomarker in clinical practice.
Interpretation: We tested the hypothesis that regional patterns of Aβ abnormality can provide additional information regarding probability of clinical progression from Aβ positive mild cognitive impairment (MCI) to Alzheimer's disease (AD) dementia. Our results support a framework in which the topographical information from Aβ‐PET imaging is useful: Aβ abnormality in the brain's default mode network is highly associated with clinical progression.
Future directions: Future clinical trials enrolling Aβ positive individuals could consider the topographical information conferred from an Aβ‐PET scan and balance groups accordingly. Future studies should validate this model using other available Aβ‐PET radioligands.
2. MATERIAL AND METHODS
2.1. Database description and study participants
Data used in the preparation of this article were obtained from the Alzheimer's disease Neuroimaging Initiative (ADNI) database, phases ADNIGO and ADNI2 (adni.loni.usc.edu). ADNI was launched in 2003 as a public‐private partnership, led by Principal Investigator Michael W. Weiner, MD. The primary goal of ADNI has been to test whether serial magnetic resonance imaging (MRI), PET, CSF, and clinical assessment can be combined to measure the progression of MCI and early AD. We assessed 296 cognitively unimpaired (CU) individuals and 168 AD patients with baseline clinical evaluation, and MRI and [18F]florbetapir PET imaging. Cognitively unimpaired individuals had a Mini‐Mental State Examination (MMSE) score of 24 or higher and a clinical dementia rating (CDR) score of 0. AD dementia patients had a MMSE scores lower than or equal to 26, a CDR > 0.5, and met the National Institute of Neurological and Communicative Disorders and Stroke—Alzheimer's Disease and Related Disorders Association criteria for probable AD. 13 All patients with AD dementia had a positive [18F]florbetapir PET scan. 14 Also, we studied 365 single‐ or multi‐domain amnestic MCI individuals who underwent clinical evaluation, MRI, and [18F]florbetapir PET imaging at baseline as well as cognitive and clinical evaluations at 2‐year follow‐up. MCI participants had an MMSE score equal to or higher than 24, a CDR of 0.5, subjective and/or objective memory impairment, normal function in activities of daily living, and did not meet criteria for AD dementia. 13 All were free of other neuropsychiatric disorders. Detailed information about the ADNI inclusion/exclusion criteria may be found at www.adni-info.org.
2.2. Standard protocol approvals, registrations, and patient consents
The ADNI study was approved by the Institutional Review Boards of each participating institution. Informed written consent was obtained from all participants enrolled in this study.
2.3. PET/MRI methods
PET and MRI images were acquired in accordance with ADNI protocols (http://adni.loni.usc.edu/methods). The images were processed as previously described. 15 In summary, T1‐weighted MRI images were corrected for field distortions and processed using the CIVET image‐processing pipeline. T1‐weighted images were non‐uniformity corrected, 16 brain masked and segmented using the brain extraction based on nonlocal segmentation technique. 17 Then, images were co‐registered using a nine‐parameter affine transformation and non‐linearly spatially normalized to the MNI 152 reference template. PET images were blurred with a volumetric Gaussian kernel with a full‐width half‐maximum of 8 mm. Linear co‐registration and non‐linear spatial normalization to the MNI 152 template space were performed using the transformations derived from the PET/T1‐weighted image transformation and anatomical MRI registration for each subject. [18F]Florbetapir standardized uptake value ratio (SUVR) maps were generated using the cerebellar gray matter as reference regions. A global [18F]florbetapir SUVR value for each subject was estimated from the averaged frontal, parietal, temporal, and cingulate cortices based on the literature. 18 , 19 , 20 A published global [18F]florbetapir SUVR of 1.15 was used to determine Aβ positivity. 19 Using our PET pipeline, 30% of CU were Aβ positive using a [18F]florbetapir SUVR threshold of 1.15, which is consistent with ADNI publications. 15 The neuroanatomical regions in which we investigated regional Aβ deposition in this study were the posterior cingulate, precuneus, lateral temporal, inferior parietal, medial prefrontal, anterior cingulate, orbitofrontal, occipital, somatomotor, striatum, and thalamus, based on previous publications adapted from the Desikan‐Killiany‐Tourville (DKT) Atlas. 21 , 22
2.4. Statistical methods
Regressions were performed with R Statistical Software Package version 3.1.2 (http://www.r-project.org/). Voxel‐wise analyses were performed using MATLAB software version 9.2 (http://www.mathworks.com) with a novel computational framework, developed to perform complex statistical operations such as receiver operating characteristic (ROC) curves, in every brain voxel. 23 We compared biomarkers across groups using voxel‐wise t‐test and χ2 analyses. The parametric images were corrected for multiple testing using a false discovery rate (FDR) correction threshold of P < .05. To determine voxel‐wise thresholds for Aβ abnormality, we conducted voxel‐wise ROC analyses, contrasting [18F]florbetapir SUVR in every voxel between 296 CU elderly and 168 AD dementia individuals. For the voxel‐wise ROC, we first determined an optimal voxel‐wise threshold value for every brain voxel using the least distance from (0,1) point to the ROC curve contrasting CU and AD dementia cases, which provides the best trade‐off between sensitivity and specificity for a diagnosis of AD dementia. 24 Subsequently, the aforementioned voxel‐wise thresholds were applied to the population of Aβ+ individuals with MCI, which generated binarized brain maps of Aβ abnormality for each individual. Percentage of abnormal voxels (PAV) was determined for every individual's region as the ratio of abnormal voxels to the total number of voxels in a given region:
Then, logistic regression models were used to evaluate the ability of region's Aβ PAV values to predict likelihood of developing dementia during the following 2 years, with the probability of progression being , accounting for age, sex, 25 years of education, MMSE score, and apolipoprotein E (APOE) ε4 status. 26 We used standardized biomarker z‐score values to perform the logistic regression analysis. Logistic regression determined the risk of progression to AD dementia over a 2‐year time frame (Statistics in Medicine 2009).
P values were interpreted after a Bonferroni correction for multiple comparisons at P < .05.
3. RESULTS
Voxel‐wise ROC analysis contrasting CU individuals with AD dementia patients determined the thresholds for a diagnosis of AD at every brain voxel. ROC area under the curve (AUC) values were the highest in the precuneus, posterior cingulate, lateral temporal, inferior parietal, and medial prefrontal cortices, and the lowest in the somatomotor cortex (Figure 1A). Also, this vowel‐wise analysis revealed a wide range of cortical [18F]florbetapir SUVR thresholds for a diagnosis of AD, with values ranging from 1.15 to 1.71 (Figure 1B). The highest neocortical SUVR threshold values were found in the precuneus, cingulate, and ventromedial prefrontal cortices, whereas the lowest were found in the medial temporal and occipital cortices (Figure 1B). Table 1 summarizes the key characteristics of CU elderly individuals and individuals with AD dementia patients.
FIGURE 1.
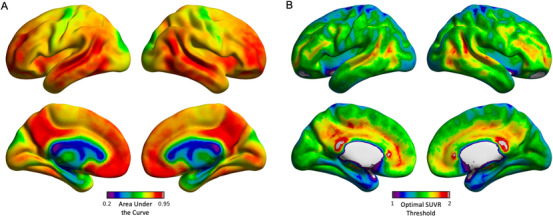
Area under the curve (AUC) and optimal amyloid beta (Aβ) thresholds separating cognitively unimpaired elderly and Alzheimer's disease (AD) patients based on voxel‐wise receiver operating characteristic curve (ROC) analysis. A, Voxel‐wise AUC the ROC values separating cognitively unimpaired controls and AD subjects based on Aβ [18F]florbetapir standardized uptake value ratio (SUVR). Values were the highest in voxels within the precuneus, posterior cingulate, lateral temporal, inferior parietal, and ventromedial prefrontal cortices. B, Voxel‐wise parametric maps showed [18F]florbetapir SUVR thresholds ranging from 1.15 to 1.71 across the cerebral cortex. The highest SUVR threshold values were found in the precuneus and cingulate cortices, whereas the lowest were located in the medial temporal and occipital cortices
TABLE 1.
Sample used to derive voxel‐wise cutoffs
| Characteristics | CU elderly | AD dementia | P value |
|---|---|---|---|
| No. | 296 | 168 | – |
| Age, years, mean (SD) | 71.24 (7.25) | 74.53 (7.6) | <.0001 |
| Female, no. (%) | 153 (51.6) | 83 (49.4) | .63 |
| APOE ε4, no. (%) | 86 (29) | 110 (65.5) | <.0001 |
|
Education, years, mean (SD) |
16.51 (2.65) |
16.04 (2.66) |
.07 |
|
MMSE score (SD) |
28.95 (1.32) |
22.77 (3.09) |
<.0001 |
| [18F]Florbetapir, mean SUVR (SD) | 1.13 (0.24) | 1.36 (0.18) | <.0001 |
P values indicate the values assessed with independent samples t‐tests for each variable except sex and APOE ε4, where contingency χ2 tests were performed.
Abbreviations: AD, Alzheimer's disease; APOE, apolipoprotein E; CU, cognitively unimpaired; MMSE, Mini‐Mental State Examination; SD, standard deviation; SUVR, standardized uptake value ratio
When examining all individuals with MCI regardless of amyloid‐PET status (n = 365), we observed that amyloid‐PET positivity has associated with a significantly increased risk of clinical progression to dementia over 2 years (odds ratio [OR]: 3.99; 95% confidence interval [CI]: 1.97 – 8.71; P = .0002). Of the 365 amnestic MCI individuals, 206 were classified as Aβ positive (56%; 206/365), of which 60 (29%; 60/206) progressed to dementia within 2 years. Table 2 summarizes the key characteristics of the 206 Aβ positive MCI individuals. There was no significant difference in global Aβ load between Aβ positive MCI nonprogressors ([18F]florbetapir SUVR = 1.413 [standard deviation (SD) 0.15]) and progressors ([18F]florbetapir SUVR = 1.379 [SD 0.24]) to dementia (student t‐test, P = .3246).
TABLE 2.
Demographics and key characteristics of the amyloid beta positive mild cognitive impairment individuals
| Characteristics | Progressors | Nonprogressors | P value |
|---|---|---|---|
| No. | 60 | 146 | — |
| Age, years, mean (SD) | 72.43 (6.9) | 72.87 (7.28) | .69 |
| Female, no. (%) | 89 (43.67) | 28 (46.67) | .76 |
| APOE ε4, no. (%) | 47 (78) | 72 (49.31) | .07 |
|
Education, years, mean (SD) |
15.85 (2.88) |
15.84 (2.72) |
.97 |
|
MMSE score (SD) |
27.05 (1.73) |
27.84(1.72) |
.004 |
| [18F]Florbetapir, mean SUVR (SD) | 1.38 (0.24) | 1.41 (0.15) | .32 |
P values indicate the values assessed with independent samples t‐tests for each variable except sex and APOE ε4, where contingency χ2 tests were performed.
Abbreviations: APOE, apolipoprotein E; MMSE, Mini‐Mental State Examination; SD, standard deviation
Applying the aforementioned voxel thresholds for AD to the Aβ positive MCI, we found that the regional topographic patterns of Aβ abnormality differentiate Aβ positive MCI progressors from nonprogressors at an individual level (Figure 2). Population probabilistic parametric maps showed that Aβ positive MCI nonprogressors presented a heterogeneous pattern of Aβ abnormality (Figure 3A). On the other hand, Aβ positive MCI progressors had the highest probability of Aβ abnormalities concentrated in regions confined to the brain's default mode network in the precuneus/posterior cingulate cortex, lateral temporal, and medial prefrontal cortices (Figure 3B). Voxel‐wise t‐test showed no significant differences in baseline [18F]florbetapir SUVR load between Aβ positive MCI progressors and nonprogressors (after FDR correction at P < .05; Figure 3C). Voxel‐wise χ2 comparison demonstrated that Aβ positive MCI individuals who progressed to dementia were significantly more likely to have Aβ abnormality in clusters in the precuneus/posterior cingulate cortex, lateral temporal, and medial prefrontal cortices (after FDR correction at P < .05; Figure 3C).
FIGURE 2.
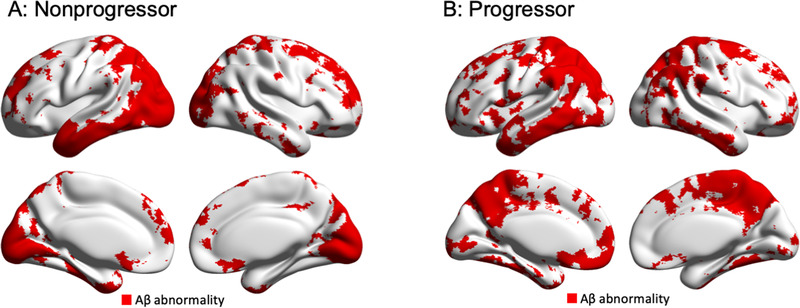
Regional patterns of amyloid beta (Aβ) abnormality differentiate Aβ positive mild cognitive impairment (MCI) progressors from nonprogressors to dementia at individual level. Voxel‐wise maps of Aβ positivity from two representative amnestic Aβ positive MCI individuals: (A) non‐progressor (global [18F]florbetapir standardized uptake value ratio [SUVR]: 1.42, 73‐year‐old, female, apolipoprotein E [APOE] ε4 positive, Mini‐Mental State Examination [MMSE] = 28) and (B) progressor (global [18F]florbetapir SUVR = 1.42, 79 year‐old, male, APOE ε4 positive, MMSE = 28). Whereas both individuals demonstrated similar global [18F]florbetapir SUVR values, the progressor showed more extensive Aβ abnormality in default mode network regions
FIGURE 3.
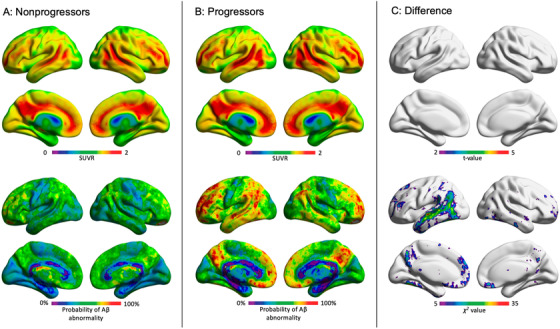
Amyloid beta (Aβ) positive mild cognitive impairment (MCI) progressors were more likely to have Aβ abnormality in the default mode network than nonprogressors. While all Aβ positive MCI individuals (progressors and non‐progressors) demonstrated similar voxel‐wise and global standardized uptake value ratios (SUVRs), they exhibited different patterns of voxel‐wise amyloid positivity. A, Bottom row: amyloid positive MCI individuals who remained stable over 2 years did not exhibit a consistent pattern of regional Aβ positivity. B, In contrast, individuals who progressed to dementia in a 2‐year time frame had higher probability of abnormality in the posterior cingulate/precuneus, lateral temporal, medial and lateral prefrontal cortices. C, Result of voxel‐wise statistical comparisons between Aβ positive MCI progressors versus non‐progressors. Top row: there was no significant difference in voxel‐wise [18F]florbetapir SUVR between progressors and non‐progressors after correcting for multiple comparisons with a false discovery rate of P < .05. Bottom row: Results of voxel‐wise χ2 comparing abnormal voxels (coded as: normal = 0 and abnormal = 1) between Aβ positive MCI progressors versus non‐progressors. Aβ positive MCI individuals who progressed to Alzheimer's disease were significantly more likely to have Aβ abnormality in large clusters in the posterior cingulate cortex/precuneus, medial prefrontal cortex, and lateral temporal cortices. Results are corrected for multiple comparisons with a false discovery rate of P < .05
Logistic regression showed that the Aβ PAV index in the posterior cingulate cortex/precuneus, lateral temporal, inferior parietal, and medial prefrontal cortices was highly predictive of progression to dementia among Aβ positive MCI individuals (Figure 4A,C). Table 3 summarizes the predictive value of Aβ PAV index in the context of other covariates (age, sex, MMSE, years of education and APOEε4). Aβ PAV index in other brain regions (anterior cingulate, orbitofrontal, occipital, somatomotor, striatum, and thalamus) did not associate with dementia over a 2‐year follow‐up. Also, Aβ SUVR values did not predict progression to dementia in these individuals in any tested brain region (Figure 4B).
FIGURE 4.
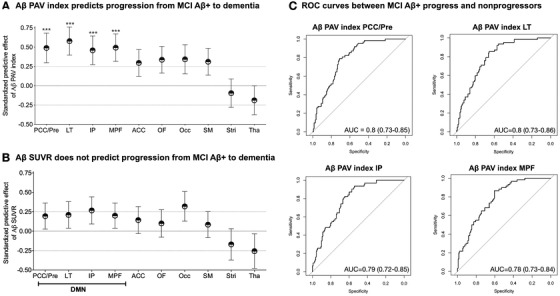
Amyloid beta (Aβ) percentage of abnormal voxels, rather than standardized uptake value ratio (SUVR) load, in brain's default mode network regions predicts the progression of Aβ positive mild cognitive impairment (MCI) individuals to dementia. The dots represent predictive effects (β estimates) of standardized baseline biomarkers (z‐scores) to progression to dementia using logistic regressions. The bars represent standard errors of β estimates. A, In Aβ positive MCI individuals, Aβ percentage of abnormal voxels (PAV) index in the posterior cingulate and precuneus (PCC/Pre), lateral temporal (LT), inferior parietal (IP), and medial prefrontal (MPF) predicted progression to dementia over 2 years. B, In Aβ positive MCI individuals, Aβ SUVR did not predict progression to dementia. C, Aβ PAV index accurately separated Aβ positive MCI (MCI Aβ+) progressors from non‐progressors. The models were adjusted for age, sex, years of formal education, apolipoprotein E (APOE) ε4 status, general cognitive performance at baseline (Mini‐Mental State Examination [MMSE] score), and Bonferroni corrected at P < .05. ACC, anterior cingulate; Occ, occipital; OF, orbitofrontal; SM, somatomotor; Stri, striatum; Tha, thalamus. *** P < .0001
TABLE 3.
Summary statistics of variables predicting likelihood of conversion to dementia from amyloid beta positive mild cognitive impairment individuals over 2 years
| Beta estimate | P‐value | OR | 95% CI | |
|---|---|---|---|---|
| PCC/precuneus Aβ PAV index | 0.49 | .005 | 1.63 | 1.17‐2.32 |
| Lat temporal Aβ PAV index | 0.57 | .0007 | 1.77 | 1.28‐2.48 |
| Inf parietal Aβ PAV index | 0.45 | .008 | 1.57 | 1.13‐2.2 |
| mPFC Aβ PAV index | 0.49 | .004 | 1.62 | 1.17‐2.28 |
| Sex (male) | 0.001 | .98 | 1.01 | 0.53‐1.91 |
| APOE ε4 status | 1.37 | .0002 | 3.94 | 1.95‐8.49 |
| Education (years) | 0.12 | .06 | 1.13 | 0.99‐1.28 |
| Age (years) | 0.001 | .97 | 0.99 | 0.95‐1.05 |
| MMSE score | ‐0.40 | <.0001 | 0.67 | 0.56‐7.89 |
Abbreviations: Aβ, amyloid beta; APOE, apolipoprotein E; CI, confidence interval; MMSE, Mini‐Mental State Examination; OR, odds ratio; PAV, percentage of abnormal voxels; PCC, posterior cingulate cortex; PFC, prefrontal cortex; SD, standard deviation
4. DISCUSSION
We showed here that the evaluation of the regional extent of Aβ may provide important information regarding the clinical fate of Aβ positive MCI cases, which has been overlooked by traditional measures of overall Aβ concentrations. Specifically, our results revealed that although Aβ positive MCI progressors and nonprogressors had similar global SUVR loads, progressors have a consistent pattern of Aβ abnormalities concentrated in default mode network hubs, whereas nonprogressors do not.
We found that the extent of Aβ pathology, rather than its load, across vulnerable brain regions relates to the likelihood of Aβ positive MCI cases developing dementia over 2 years. The negative results of several disease‐modifying therapies that did not consider Aβ positivity as a population enrichment strategy were used to suggest that inclusion of Aβ biomarkers for selection purposes is critical for including appropriate subjects. 27 , 28 Indeed, Aβ positivity increases the probability that MCI subjects will progress to dementia and therefore increases the statistical power of these trials, allowing potential pharmacological effects to be successfully tested using a smaller number of individuals. 29 To overcome the fact most of Aβ positive MCI individuals do not progress to dementia over a typical clinical trial period, 30 studies have used different techniques to propose more liberal or conservative global Aβ PET thresholds, resulting in a constant trade‐off between sensitivity and specificity. 29 , 31 However, although statistical techniques of threshold optimization have the potential to increase the probability of patients’ progression to dementia, if two individuals have the same averaged Aβ burden across cortical regions, they will invariably receive the same Aβ classification. Our findings suggest that the assessment of the extent of Aβ abnormality across brain regions may significantly increase the statistical power of future disease‐modifying trials by helping to select among Aβ positive MCI cases those with the highest likelihood of developing dementia over 2 years.
Global Aβ measures using either CSF or PET are considered optimal and equivalent estimates to determine individuals at risk of dementia. 18 , 32 , 33 , 34 It has been suggested that the regional assessment of Aβ using PET provides little benefit for selecting individuals that are the most likely to progress to dementia. 35 In contrast with the aforementioned literature, our results show a significant predictive effect of the regional extent of Aβ abnormality, which can only be achieved using techniques with spatial resolution, such as PET. In fact, it is important to mention that our results suggest that the aforementioned prediction cannot be obtained by the currently favored approach using global or regional Aβ PET SUVR values, which do not take into account the spatial extent of Aβ pathology in the regions used for the quantification. For example, an Aβ positive MCI individual presenting Aβ pathology across the whole extension of the vulnerable brain regions presented here has a higher probability of developing dementia over 2 years than an individual with Aβ pathology concentrated in a subset of these regions. Differentiation of two such subjects can be achieved assessing Aβ pathology regional extent. On the other hand, a global averaged SUVR composite may not differentiate these two individuals if the individual showing Aβ pathology concentrated in a subset of vulnerable regions presents a high Aβ load in these regions. In other words, the assessment of the regional extent of Aβ abnormality may prove to offer complementary information to averaged SUVR load values regarding the potential for progression of Aβ positive MCI individuals to dementia over 2 years.
The question of whether the topographical distribution of Aβ deposits across anatomical structures or the regional selective vulnerability drives AD dementia progression has been extensively debated. 36 The concept of selective neuronal vulnerability suggests that certain neurons are intrinsically more vulnerable to the underlying Aβ deposition, which would determine AD progression. 36 Alternatively, the pathogenic spread hypothesis suggests that the progressive accumulation of Aβ aggregates is the determinant factor of the disease symptoms. In this theory, the pathogenesis of AD occurs from the movement of Aβ deposits across neuronal populations and anatomical regions. 36 At first glance, the results presented in this study suggests that topographical distribution of Aβ abnormality across anatomical structures drives the progression of AD. However, our results also suggest that dementia occurs when this Aβ abnormality converges on vulnerable neuronal populations in the brain's default mode network, supporting the conceptual framework suggesting that the presence of Aβ in this brain network serving as a hub of neuronal activity is a key element of AD progression. 37 , 38 , 39 Altogether, one may argue that our results support that the combination of Aβ abnormality and regional vulnerability determines whether Aβ deposits associate with dementia symptoms in AD. 36
The main methodological strength of this study is the fact that the results were obtained in a voxel‐wise manner and are therefore free of anatomical assumptions. The main limitation of this study was the lack of autopsy confirmation of the presence of brain Aβ deposition in our subjects. 40 The lack of a replication cohort for the results obtained in the Aβ positive MCI limits the interpretation of the predictive models presented here. Additionally, because Aβ deposition occurs on a continuum, all forms of dichotomization are invariably subject to conceptual and analytical idiosyncrasies and are furthermore likely to vary depending on the analytical method, including the Aβ‐PET imaging agent used. ADNI participants represent a group of self‐selected individuals showing enough motivation to participate in a study focusing on dementia. Thus, because these individuals might not represent the general population, it would be desirable to replicate our results in a separate population‐based cohort. It is important to emphasize that we aimed to assess here the influence of the regional patterns of Aβ pathology in the progression from MCI to dementia, rather than determine the method with the best sensitivity and specificity for clinical progression to dementia, for which biomarkers of tau and neurodegeneration would need to be included.
To conclude, our results highlight that the analysis of the regional extent of Aβ pathology might provide important information at an individual level to assess the probability of Aβ positive MCI individuals destined to develop dementia over 2 years.
CONFLICTS OF INTEREST
All authors report no conflicts of interest.
ACKNOWLEDGMENTS
This work was supported by the Canadian Institutes of Health Research (CIHR; MOP‐11‐51‐31), the Alan Tiffin Foundation, the Alzheimer's Association (NIRG‐12‐ 92090, NIRP‐12‐259245), Prevent‐AD scholarship (TAP), Fonds de Recherche du Québec—Santé (FRQS; Chercheur Boursier, P.R.‐N.); S.G. and P.R‐N. are members of the CIHR‐CCNA Canadian Consortium of Neurodegeneration in Aging. Data collection and sharing for this project was funded by the ADNI (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer's Association; Alzheimer's Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research also provides funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the AD Cooperative Study at the University of California, San Diego. ADNI data were disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Pascoal TA, Therriault J, Mathotaarachchi S, et al. Topographical distribution of Aβ predicts progression to dementia in Aβ positive mild cognitive impairment. Alzheimer's Dement. 2020;12:e12037 10.1002/dad2.12037
Tharick A. Pascoal and Joseph Therriault contributed equally to the study.
REFERENCES
- 1. Rabinovici GD, Gatsonis C, Apgar C, et al. Association of amyloid positron emission tomography with subsequent change in clinical management among Medicare beneficiaries with mild cognitive impairment or dementia. JAMA. 2019;321:1286‐1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boccardi M, Altomare D, Ferrari C, et al. Assessment of the incremental diagnostic value of florbetapir f 18 imaging in patients with cognitive impairment: the incremental diagnostic value of amyloid pet with [18f]‐florbetapir (INDIA‐FBP) study. JAMA Neurol. 2016;73:1417‐1424. [DOI] [PubMed] [Google Scholar]
- 3. Zwan MD, Bouwman FH, Konijnenberg E, et al. Diagnostic impact of [(18)F]flutemetamol PET in early‐onset dementia. Alzheimers Res Ther. 2017;9:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jack CR Jr., Wiste HJ, Vemuri P, et al. Brain beta‐amyloid measures and magnetic resonance imaging atrophy both predict time‐to‐progression from mild cognitive impairment to Alzheimer's disease. Brain. 2010;133:3336‐3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sevigny J, Chiao P, Bussiere T, et al. The antibody aducanumab reduces Abeta plaques in Alzheimer's disease. Nature. 2016;537:50‐56. [DOI] [PubMed] [Google Scholar]
- 6. Bacskai BJ, Frosch MP, Freeman SH, et al. Molecular imaging with Pittsburgh Compound B confirmed at autopsy: a case report. Arch Neurol. 2007;64:431‐434. [DOI] [PubMed] [Google Scholar]
- 7. Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer's disease with Pittsburgh Compound‐B. Ann Neurol. 2004;55:306‐319. [DOI] [PubMed] [Google Scholar]
- 8. Ikonomovic MD, Klunk WE, Abrahamson EE, et al. Post‐mortem correlates of in vivo PiB‐PET amyloid imaging in a typical case of Alzheimer's disease. Brain. 2008;131:1630‐1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Okello A, Koivunen J, Edison P, et al. Conversion of amyloid positive and negative MCI to AD over 3 years: an 11C‐PIB PET study. Neurology. 2009;73:754‐760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Laforce R Jr., Rabinovici GD. Amyloid imaging in the differential diagnosis of dementia: review and potential clinical applications. Alzheimers Res Ther. 2011;3:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sakr FA, Grothe MJ, Cavedo E, et al. Applicability of in vivo staging of regional amyloid burden in a cognitively normal cohort with subjective memory complaints: the INSIGHT‐preAD study. Alzheimers Res Ther. 2019;11:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mattsson N, Palmqvist S, Stomrud E, Vogel J, Hansson O. Staging beta‐amyloid pathology with amyloid positron emission tomography. JAMA Neurol. 2019;76(11):1319–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology. 1984;34:939‐944. [DOI] [PubMed] [Google Scholar]
- 14. Jack CR Jr., Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pascoal TA, Mathotaarachchi S, Mohades S, et al. Amyloid‐beta and hyperphosphorylated tau synergy drives metabolic decline in preclinical Alzheimer's disease. Mol Psychiatry. 2017;22(2):306‐311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zijdenbos AP, Forghani R, Evans AC. Automatic “pipeline” analysis of 3‐D MRI data for clinical trials: application to multiple sclerosis. IEEE Trans Med Imaging. 2002;21:1280‐1291. [DOI] [PubMed] [Google Scholar]
- 17. Eskildsen SF, Coupe P, Fonov V, et al. BEaST: brain extraction based on nonlocal segmentation technique. Neuroimage. 2012;59:2362‐2373. [DOI] [PubMed] [Google Scholar]
- 18. Landau SM, Mintun MA, Joshi AD, et al. Amyloid deposition, hypometabolism, and longitudinal cognitive decline. Ann Neurol. 2012;72:578‐586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pascoal TA, Mathotaarachchi S, Shin M, et al. Amyloid and tau signatures of brain metabolic decline in preclinical Alzheimer's disease. Eur J Nucl Med Mol Imaging. 2018;45:1021‐1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jack CR Jr., Wiste HJ, Weigand SD, et al. Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimers Dement. 2017;13:205‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jack CR Jr., Lowe VJ, Senjem ML, et al. 11C PiB and structural MRI provide complementary information in imaging of Alzheimer's disease and amnestic mild cognitive impairment. Brain. 2008;131:665‐680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Klein A, Tourville J. 101 labeled brain images and a consistent human cortical labeling protocol. Front Neurosci. 2012;6:171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mathotaarachchi S, Wang S, Shin M, et al. VoxelStats: A MATLAB Package for Multi‐Modal Voxel‐Wise Brain Image Analysis. Front Neuroinform. 2016;10:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fawcett T. An introduction to ROC analysis. Pattern Recognit Lett. 2006;27:861‐874. [Google Scholar]
- 25. Buckley RF, Mormino EC, Amariglio RE, et al. Sex, amyloid, and APOE epsilon4 and risk of cognitive decline in preclinical Alzheimer's disease: findings from three well‐characterized cohorts. Alzheimers Dement. 2018;14:1193‐1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mattsson N, Groot C, Jansen WJ, et al. Prevalence of the apolipoprotein E epsilon4 allele in amyloid beta positive subjects across the spectrum of Alzheimer's disease. Alzheimers Dement. 2018;14:913‐924. [DOI] [PubMed] [Google Scholar]
- 27. Vellas B, Carrillo MC, Sampaio C, et al. Designing drug trials for Alzheimer's disease: what we have learned from the release of the phase III antibody trials: a report from the EU/US/CTAD Task Force. Alzheimers Dement. 2013;9:438‐444. [DOI] [PubMed] [Google Scholar]
- 28. Karran E, Hardy J. A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann Neurol. 2014;76:185‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mattsson N, Insel PS, Landau S, et al. Diagnostic accuracy of CSF Ab42 and florbetapir PET for Alzheimer's disease. Ann Clin Transl Neurol. 2014;1:534‐543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schreiber S, Landau SM, Fero A, Schreiber F, Jagust WJ, Alzheimer's Disease Neuroimaging I . Comparison of visual and quantitative florbetapir f 18 positron emission tomography analysis in predicting mild cognitive impairment outcomes. JAMA Neurol. 2015;72(10):1183‐1190. [DOI] [PubMed] [Google Scholar]
- 31. Villeneuve S, Rabinovici GD, Cohn‐Sheehy BI, et al. Existing Pittsburgh Compound‐B positron emission tomography thresholds are too high: statistical and pathological evaluation. Brain. 2015;138(Pt 7):2020‐2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hansson O, Zetterberg H, Buchhave P, Londos E, Blennow K, Minthon L. Association between CSF biomarkers and incipient Alzheimer's disease in patients with mild cognitive impairment: a follow‐up study. The Lancet Neurology. 2006;5:228‐234. [DOI] [PubMed] [Google Scholar]
- 33. Jagust WJ. Amyloid imaging: coming to a PET scanner near you. Ann Neurol. 2010;68:277‐278. [DOI] [PubMed] [Google Scholar]
- 34. Hansson O, Seibyl J, Stomrud E, et al. CSF biomarkers of Alzheimer's disease concord with amyloid‐beta PET and predict clinical progression: A study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement. 2018;14:1470‐1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Altmann A, Ng B, Landau SM, Jagust WJ, Greicius MD, Alzheimer's Disease Neuroimaging I . Regional brain hypometabolism is unrelated to regional amyloid plaque burden. Brain. 2015;138:3734‐3746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Walsh DM, Selkoe DJ. A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat Rev Neurosci. 2016;17:251‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sperling RA, Laviolette PS, O'Keefe K, et al. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 2009;63:178‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Buckner RL, Snyder AZ, Shannon BJ, et al. Molecular, structural, and functional characterization of Alzheimer's disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 2005;25:7709‐7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Palmqvist S, Scholl M, Strandberg O, et al. Earliest accumulation of beta‐amyloid occurs within the default‐mode network and concurrently affects brain connectivity. Nat Commun. 2017;8:1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol. 2009;66:200‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]