

Abstract
Neonatal hypoxic ischemic (HI) brain injury causes lifelong neurologic disability. Therapeutic hypothermia (TH) is the only approved therapy that partially mitigates mortality and morbidity. Therapies specifically targeting HI-induced brain cell death are currently lacking. Intracellular calcium dysregulation, oxidative stress, and mitochondrial dysfunction through the formation of the mitochondrial permeability transition pore (mPTP) are drivers of HI cellular injury. GNX-4728, a small molecule direct inhibitor of the mPTP that increases mitochondrial calcium retention capacity, is highly effective in adult neurodegenerative disease models and could have potential as a therapy in neonatal HI. A dose of GNX-4728, equivalent to that used in animal models, 300 mg/kg, IP was highly toxic in p10 mice. We then tested the hypothesis that acute administration of 30 mg/kg, IP of GNX-4728 immediately after HI in a neonatal mouse model would provide neuroprotection. This non-lethal lower dose of GNX-4728 (30 mg/kg, IP) improved the respiratory control ratio of neonatal female HI brain tissue but not in males. Brain injury, assessed histologically with a novel metric approach at 1 and 30 days after HI, was not mitigated by GNX-4728. Our work demonstrates that a small molecule inhibitor of the mPTP has i) an age related toxicity, ii) a sex-related brain mitoprotective profile after HI but iii) this is not sufficient to attenuate forebrain HI neuropathology.
Keywords: neonatal hypoxia ischemia, neuronal cell death, mitochondrial respiratory control ratio, therapeutics, neuroprotection
INTRODUCTION:
Complications from hypoxic ischemic (HI) brain injury cause significant long-term neurological morbidities and contribute to approximately one quarter of neonatal deaths worldwide (Lawn et al., 2005, Lee et al., 2013, Obstet Gynecol, 2014). Whole body therapeutic hypothermia (TH) is the standard of care for treatment of neonatal encephalopathy (Roehr et al., 2011). TH has a significant impact on mortality; however, despite likely numerous modes of action, it is only partially neuroprotective (Shankaran et al., 2012, Azzopardi et al., 2014, Shankaran, 2014). Additionally, TH may even have more toxicity at lower gestational ages (Rao et al., 2017). Available therapies that selectively target HI-induced neuronal cell death are lacking (Northington et al., 2011). A hallmark of HI cellular injury is mitochondriopathy (Northington et al., 2007, Johnston et al., 2011), including the formation of the mitochondrial permeability transition pore (mPTP) (Rasola and Bernardi, 2007, Alavian et al., 2014). The mPTP opens under conditions of HI stress and calcium overload, resulting in permeabilization of the inner mitochondrial membrane (Martin et al., 2009, Martin, 2010a, Martin, 2010b). This loss of mitochondrial membrane potential creates osmotic changes within the mitochondrial matrix, disrupts ATP synthesis, and ultimately leads to cell death (Tsujimoto et al., 2006, Martin, 2012, Biasutto et al., 2016).
The mPTP is a possible pharmacological target for neuroprotection in neonatal HI given its critical role in cellular pathology. GNX-4728, a recently synthesized substituted cinnamic anilide, inhibits mPTP opening by increasing mitochondrial calcium retention capacity (Fancelli et al., 2014). When used prior to reperfusion in a rabbit model of acute myocardial infarction, GNX-4728 reduced infarct size by almost half (Fancelli et al., 2014). As a pre-symptomatic therapy in an adult mouse model of amyotrophic lateral sclerosis (ALS), GNX-4728 prolonged both lifespan and time to onset of motor symptoms (Martin et al., 2014). The drug also protected against mitochondrial and motor neuron degeneration, reduced spinal cord inflammation, and preserved neuromuscular junction innervation in the ALS susceptible mice (Martin et al., 2014). GNX-4728 has yet to be tested in acute settings of neurodegeneration in which mPTP formation is thought to be pathogenic. Forms of acute brain injury in which post-injury treatment is required, such as HI, present additional challenges to the implementation of mPTP inhibition.
We sought to examine therapeutic efficacy of GNX-4728 in a neonatal model of HI brain damage. We hypothesized that acute administration of GNX-4728 after HI would mitigate brain damage in neonatal mice. Proof of concept for neonatal efficacy of a drug already in testing for adult diseases would rapidly accelerate its progress toward clinical trials as an adjuvant therapy for neonatal HI brain injury.
METHODS AND MATERIALS:
Mice
C57BL6 mice from Charles River Laboratories (Wilmington, MA) were used with approval from the Institutional Animal Care and Use Committee at Johns Hopkins University School of Medicine. All animal studies were carried out with standards of care and housing in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, US Department of Health and Human Services 85–23, 2011.
Safety Trial for Drug Dosing
A safety trial was performed to determine appropriate drug dosing in postnatal day (p)10 mice. The brain of a p10 mouse brain has some similarities in developmental events to those of a term infant (Yager and Ashwal, 2009). Mice were administered intraperitoneal (IP) injections of GNX-4728 in 40% DMSO or vehicle (cyclodextrin-saline in 40% DMSO) at the same volume to deliver 300 mg/kg dose of GNX-4728 as used in adult mouse studies (Martin et al., 2014) (n = 7 GNX-4728, 5 vehicle). Lesser concentrations of DMSO were trialed but found to be unsuccessful in solubilizing GNX-4728. Doses of 100 mg/kg (n = 7 GNX-4728, 5 vehicle) and 30 mg/kg (n = 6 GNX-4728, 6 vehicle) were also tested. Survival at each dose was monitored for 30 days post-injection.
Neonatal HI Brain Injury Model
Following identification of a safe drug dose, we used the Rice-Vannucci model adapted for neonatal mice to induce HI in C57BL6 mice at p10 as previously published (Ditelberg et al., 1996, Graham et al., 2004). Mice were exposed to anesthesia using a mixture of isofluorane (3% for induction and 1% maintenance) and nitrous oxide to perform permanent unilateral right carotid artery ligation. After a one hour recovery, they were exposed to 45 min of hypoxia at FiO2 = 0.08. Sham mice were exposed to the same anesthesia as described above but not to surgery. Immediately after hypoxia, HI injured mice were randomized to receive IP GNX-4728 or vehicle. Mice were killed via exposure to 20% (v/v) mixture of isoflurane in propylene glycol via one drop exposure method (Markovic and Murasko, 1993) and used for the following experiments: brains were rapidly dissected to perform mitochondrial respiration experiments at p10; or following exsanguination with intracardiac cold 0.1 M phosphate buffered solution (PBS, pH 7.4, Amresco, Solon, OH), brains were perfused with 4% paraformaldehyde (4% PFA) in 0.1 M phosphate buffered saline for 15 min at 4 mL/min for brain fixation and histopathology with cresyl violet at p11 or p40. Mice used to validate the more quantitative scoring system for brain injury described below, were either assigned to sham (n = 4) or given HI at p10 as above and treated with either normothermia (n = 12) or therapeutic hypothermia (n = 6) as previously described (Burnsed et al., 2015, Chavez-Valdez et al., 2018). Animals were killed at p18 and brains perfused and prepared as above.
Mitochondrial Respiration
Mitochondrial respiration was measured in brains of sham, HI injured, and HI injured/GNX-4728 treated mice (sham, n = 14 [8 females, 6 males]; HI, n = 9 [5 females, 4 males]; HI treated with 30 mg/kg IP GNX-4728, n = 10 [5 females, 5 males]. To generate sufficient substrate for the experiment, 5–6 brain hemispheres were required to generate each data point. The ipsilateral hemisphere only was utilized in HI and HI/GNX-4728 measurements of mitochondrial respiration.
Brains were processed one hour after HI and treatment. Mice were killed and their forebrains rapidly removed and placed in ice-cold mitochondrial buffer (mannitol sucrose-bovine serum albumin-EGTA) (Robertson et al., 2004, Robertson et al., 2007). Mitochondria were isolated using digitonin to disrupt synaptosomal membranes (Starkov et al., 2004), and kept on ice throughout the experiment. Bio-Rad DC Protein Assay Kit (Hercules, CA) was used to quantify mitochondrial protein concentrations.
The respiratory functions of isolated mitochondria were measured polarimetrically with a Clark-type oxygen electrode (Hansatech Instruments/PP Systems, Amesbury, MA, USA). Mitochondrial assays were conducted at 37 °C at a pH of 7.0 in a KCl medium (125 mmol/L KCl, 2 mmol/L KH2PO4, 1 mmol/L MgCl2, and 20 mmol/L HEPES-KOH). The measurement chamber was supplemented with pyruvate (5 mmol/L), malate (0.2 mmol/L), and EGTA (1 μmol/L), in a total volume of 0.5 mL. Mitochondria were added to the chamber (0.5 mg/mL). State 2 is respiration rate in the presence of substrate, but prior to the addition of ADP. State 3 respiration was initiated by the addition of ADP (0.4 mmol/L). State 4oligomycin (4o) respiration was induced by the addition of the ATP synthetase inhibitor oligomycin (2.5 μg/mL). While the State 4o respiration measured in the presence of oligomycin is not equivalent to the classical State 4 rate obtained after a small bolus of ADP is almost completely converted to ATP, the use of oligomycin eliminates the contribution of ATP cycling via hydrolysis by contaminating ATPases and resynthesis by the mitochondrial ATP synthetase to State 4 respiration. The oligomycin-induced State 4o rate of respiration is therefore a more specific indicator of mitochondrial proton cycling limited by passive proton leakiness of the inner membrane. Mitochondrial respiratory energy coupling was evaluated by determining the respiratory control ratio (RCR) calculated as the rate of ADP-stimulated State 3 respiration to the State 4o rate in the presence of oligomycin. The mitochondrial respiration rates were calculated as nmol oxygen (O2)/min/mg of protein (Robertson et al., 2004).
HI Injury and Drug Treatment Protocol
HI injury was performed on neonatal pups as described above. Immediately following hypoxia, mice were randomized to be given 30 mg/kg body weight IP GNX-4728 or equivalent volume (40–60 μL) of cyclodextrin-saline/DMSO, after which they were then placed back in their cages with their mothers. They were killed for analysis of brain injury either at 24 h after intervention at p11 (GNX-4728, n = 29 [13 females, 16 males]; vehicle, n = 12 [5 females, 7 males]), or at p40 (GNX-4728, n = 21 [9 females, 12 males]; vehicle, n = 15 [7 females, 8 males]).
Histology and Brain Injury Scoring
Tissues were cryoprotected with graded immersion in 15% and 30% sucrose in PBS until they sank, frozen at −30 °C in isopentane and stored at −80 °C. Brains were sectioned at 50 μm on a freezing microtome and mounted on glass microscope slides for cresyl violet (CV) staining.
Three graders, unaware of the treatment group, scored ipsilateral cortex, hippocampus, and striatum 0–4 based on the amount of cell death at p11, or residual atrophy at p40 (Sheldon et al., 1998). For brain sections processed at p11; 0 = no detectable neuronal cell death; 1 = small, isolated, nonconfluent areas of neuronal cell death; 2 = moderate areas of neuronal cell death with some confluence or columnar damage; 3 = entirely confluent areas of neuronal cell death with still somewhat intact regional architecture; 4 = diffuse neuronal cell death without preservation of regional architecture (Sheldon et al., 1998) (Fig. 1). For brain sections processed at p40; 0 = no injury; 1 = isolated areas of cell loss; 2 = more confluent, clustered areas of atrophy, ventricular dilatation; 3 = obvious asymmetry, cystic atrophy with intact brain architecture; 4 = diffuse cystic atrophy without preservation of brain architecture. Scores from the three forebrain regions were summed to provide an overall qualitative brain injury score (range 0–12). A composite score representing both mortality and brain injury at p40 was created by assigning maximal injury scores (12) to mice that died between p10 and p40 and combining with the brain injury scores of mice that survived to p40.
Figure 1.
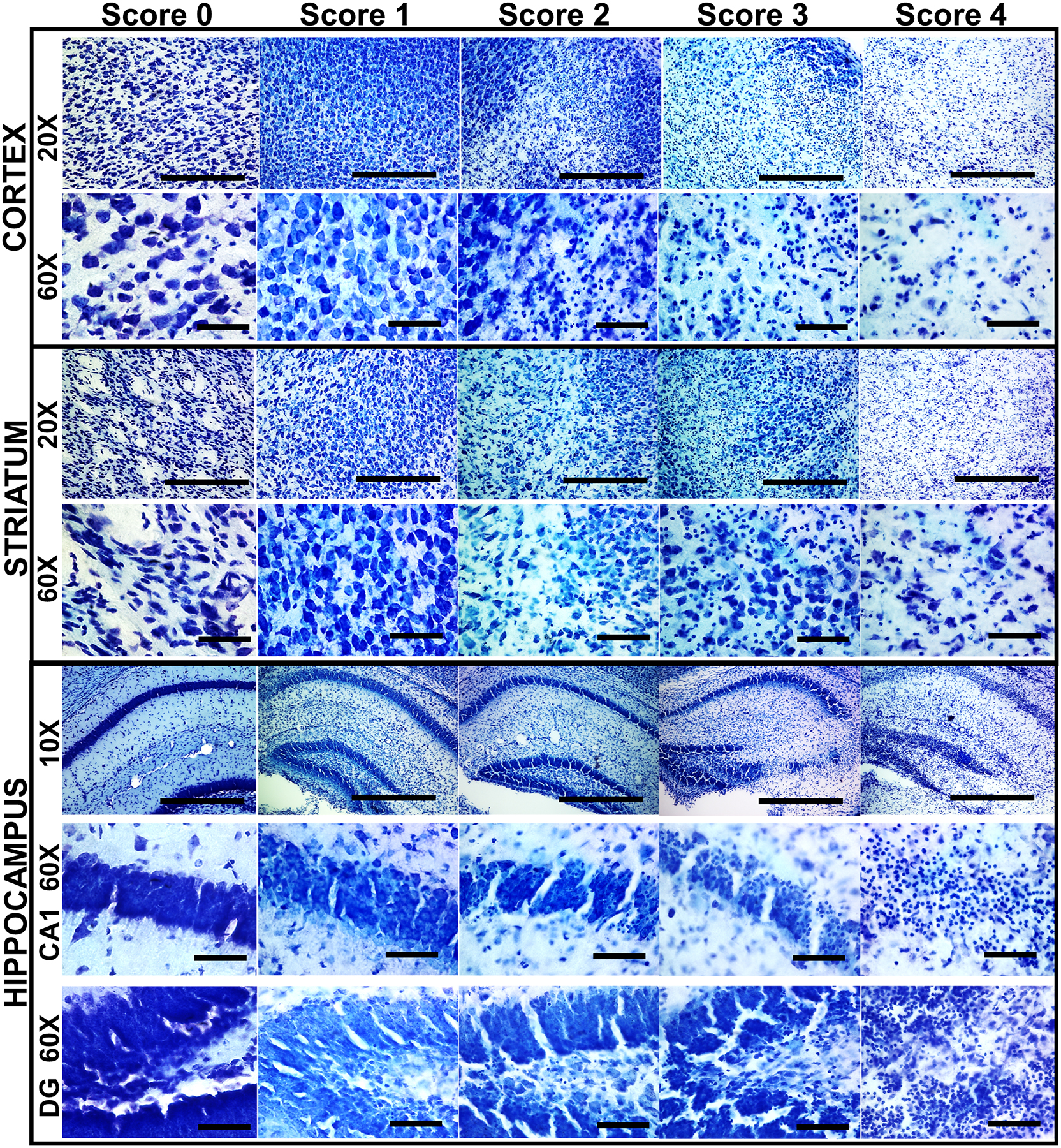
Qualitative assessment of regional brain injury after neonatal mouse HI. Representative sections displaying the features of each of the qualitative brain injury scores are shown for the cortex, striatum, and hippocampus at p11 after HI at p10. Both low (20x) magnification sections are shown to display overall cell loss, pallor, and massive cell loss and high (60x) magnification sections are shown to display individual cell appearance/pathology. Scale bars: 10x = 100 μm, 20x = 50 μm, 60x = 10 μm.
In addition, p40 brains underwent a simple form of quantitative injury scoring to further assess the degree of residual brain in the ipsilateral hemisphere. Using an ocular filar micrometer, residual length (L) and height (H) of the ipsilateral and contralateral striatum and hippocampus and height of the hippocampal CA1 field were measured in matched sections from anterior hippocampus at the level of the dentate, just posterior to the striatum and from the striatum at the level of the anterior commissure. These sections were chosen because of the reliability with which can each be identified in the coronal sectioned mouse brain and because they are reliably injured in this model. In each region, the L and H measures were taken at 90° from one another at 40 × magnification (Fig. 4A). Overall areas of the hippocampus and striatum were estimated using the formula for a rhomboid [(L*H)/2] and circle ([π [(L + H)/4]2) and compared ipsilateral versus contralateral. One additional measure of pyramidal cell layer thickness in CA1 region was obtained in contralateral and ipsilateral hippocampus at 400 × magnification. To test this method of injury assessment, in a model with a treatment (therapeutic hypothermia) known to provide neuroprotection, % residual ipsilateral hippocampal area compared to contralateral hippocampal area was compared between sham, HI-normothermia treated mice and TH treated mice at P18 after HI at p10 (Burnsed et al., 2015). The results (Fig. 4A) showed the expected significant differences in treatment effect of normothermia and therapeutic hypothermia, thus providing validity of this simple measurement for use to further query our vehicle or GNX-4728 treated brains for treatment effect.
Figure 4:
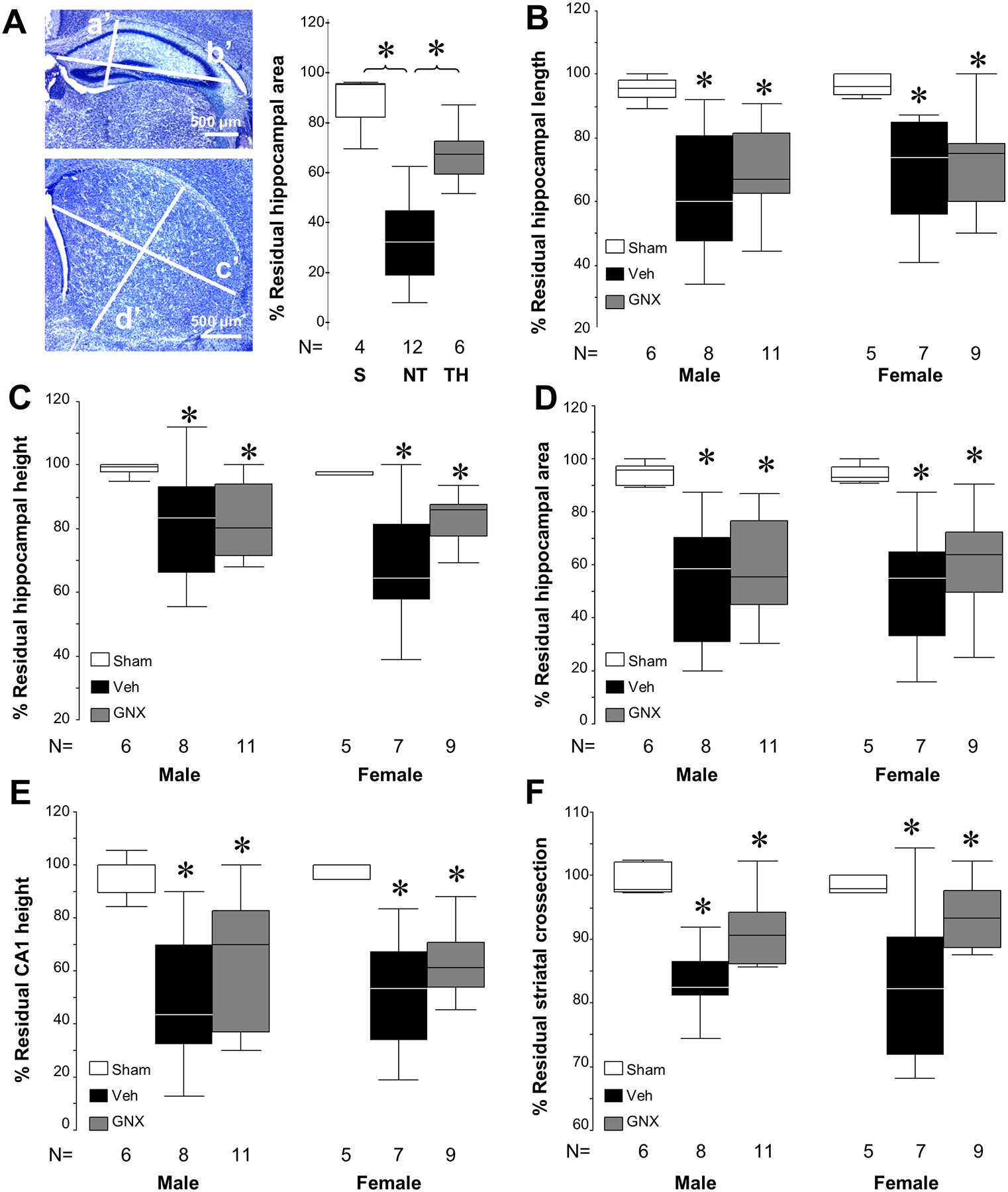
Regional measurements of brain injury in p40 survivors. [A] Schema of hippocampal (a’, height, b’, length) and striatal measurements (c’, length; d’, height) performed with calibrated ocular filar micrometer at p40 following HI and treatment with vehicle or GNX-4728. Using this analysis schema, significant injury after HI and neuroprotection with therapeutic hypothermia is demonstrated in % residual hippocampal area 8 days following HI at p10 (*P < .05 vs. sham) as expected. Scale bar = 500 μm. [B – F] Measurements with the filar ocular micrometer of the anterior hippocampus, CA1 pyramidal layer thickness and striatum are able to detect greater injury each area in both male and female injured mice exposed to HI than in shams (*P < .05). However, there are no detectable differences between vehicle and GNX-4728 treatment after HI in any of the individual regional measurements or composite measures of residual hippocampal or striatal area.
Statistics
Statistical analysis was performed using one way ANOVA stratified by sex and pair analysis by Tamhane’s T2 post hoc test to assume unequal variances for analysis of mitochondrial respiration, brain injury scoring and regional atrophy. Significance was assigned to a P < .05. Brain injury score, percent residual hippocampal and striatal measures, and mitochondrial respiration were represented as box-and-whisker plots, where the 25th and 75th percentiles limited the box and the solid line represented the median. Safety trial mortality was represented as Kaplan–Meier survival graphs and analyzed by χ2. No outliers were omitted from any analysis.
RESULTS:
Drug Dosing Trial
No p10 mice treated with 300 mg/kg of GNX-4728 survived, while 40% of mice treated with equivalent volumes (120–160 μl) of vehicle (DMSO/cyclodextrin-saline, 1:1.5) survived to p40. All deaths at the 300 mg/kg dose occurred within the first 24 h after treatment. Mice treated with 100 mg/kg of GNX-4728 or equivalent volume of vehicle had a similar survival rate, approximately 60%, at p40 and all deaths occurred within the first 96 h after treatment. In contrast, p10 mice treated with 30 mg/kg of GNX-4728 or equivalent volume of vehicle had a 100% survival, thus we chose this dose for the experiments with HI (Fig. 2A).
Figure 2:
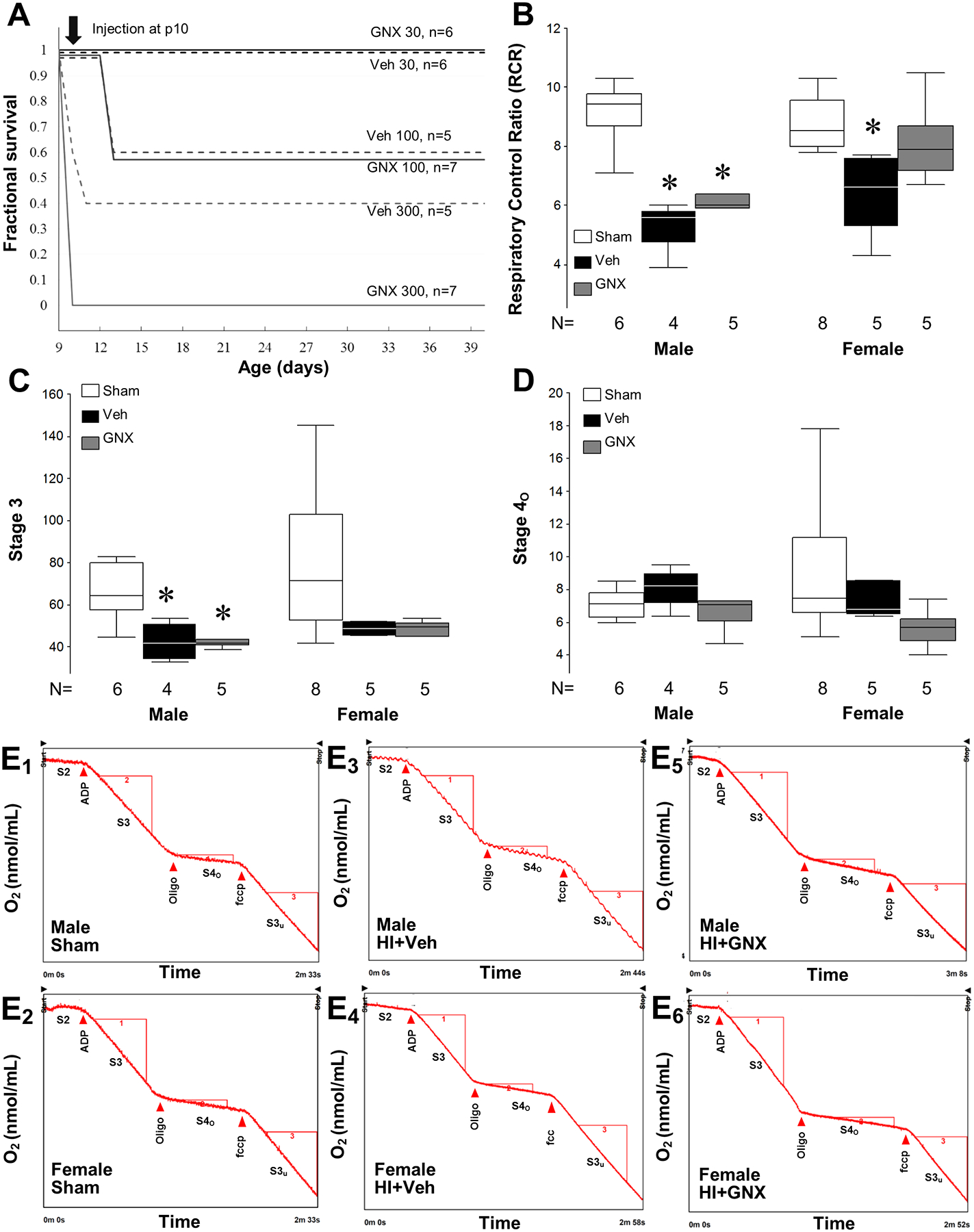
Effects of GNX-4728 treatment on mortality and mitochondrial respiration of neonatal mice. [A] Kaplan–Meier survival plot for age at death in p10 mice after varying doses of GNX-4728 or vehicle. Both 300 mg/kg and 100 mg/kg GNX-4728 and corresponding vehicle volumes in naive p10 mice cause significant mortality; 30 mg/kg GNX-4728 and corresponding vehicle volume do not cause mortality in uninjured p10 mice. [B] RCR for p10 HI injured female mice after treatment with vehicle or male mice treated with vehicle or GNX-4728 were lower than that of sham mice (*P < .05). RCR of GNX-4728 treated HI female mice was similar to shams. [C] State 3 respiration for injured neonatal male mice after treatment with vehicle or GNX-4728 was lower than that of sham mice (*P < .05), while there was no difference in State 3 respiration for injured neonatal female mice. [D] Values for State 4o respiration were similar across treatment groups and genders. [E1–E6] Representative mitochondrial respiration tracings for each group.
Mitochondrial Respiration
Twenty-four hours after HI and vehicle treatment, RCR (State 3/ State 4o) was 23 to 42% lower in injured hemispheres than sham hemispheres, indicating worse mitochondrial function (P < .01, Fig. 2B). Treatment with GNX-4728 prevented the decrease of mitochondrial RCR after HI in female mice as evidenced by the lack of significant difference in RCR value compared to sham (Fig. 2B). In contrast, GNX-4728 treatment did not restore brain mitochondrial RCR in HI male mice (Fig. 2B). State 3 respiration was decreased in both vehicle and GNX-4728 treated males after HI, (Fig. 2C) but State 4o respiration did not differ between groups in either males or females (Fig. 2D). Individual respiratory curves for each experimental condition are shown in E 1–6.
Effect of GNX-4728 on Brain Injury, Survival and Composite Injury and Survival
The qualitative injury scale (Ditelberg et al., 1996) used for assessing injury and neuroprotection following HI and treatment with vehicle or GNX-4728 is demonstrated in Fig. 1 with individual regions shown at low and high magnification and along the ordinate and the representative examples of the worsening injury scores along the abscissa. High magnification is necessary to demonstrate rare injured and dying neurons comprising score 1 while the more severe scores of injury, with large numbers of pyknotic nuclei, apoptotic bodies and overall cell loss, are easily seen at lower magnification. Using this scale, neonatal HI mice treated with 30 mg/kg GNX-4728 had similar overall brain injury and striatal and hippocampal injury compared to HI mice treated with vehicle when assessed 1 day after HI (Fig. 3A and B). In addition, there was no difference in qualitative hippocampal, striatal, or overall brain injury between injured mice treated with GNX-4728 or vehicle that survived for 30 days after HI (Fig. 3C and D). GNX-4728 offered no neuroprotection to either sex.
Figure 3:
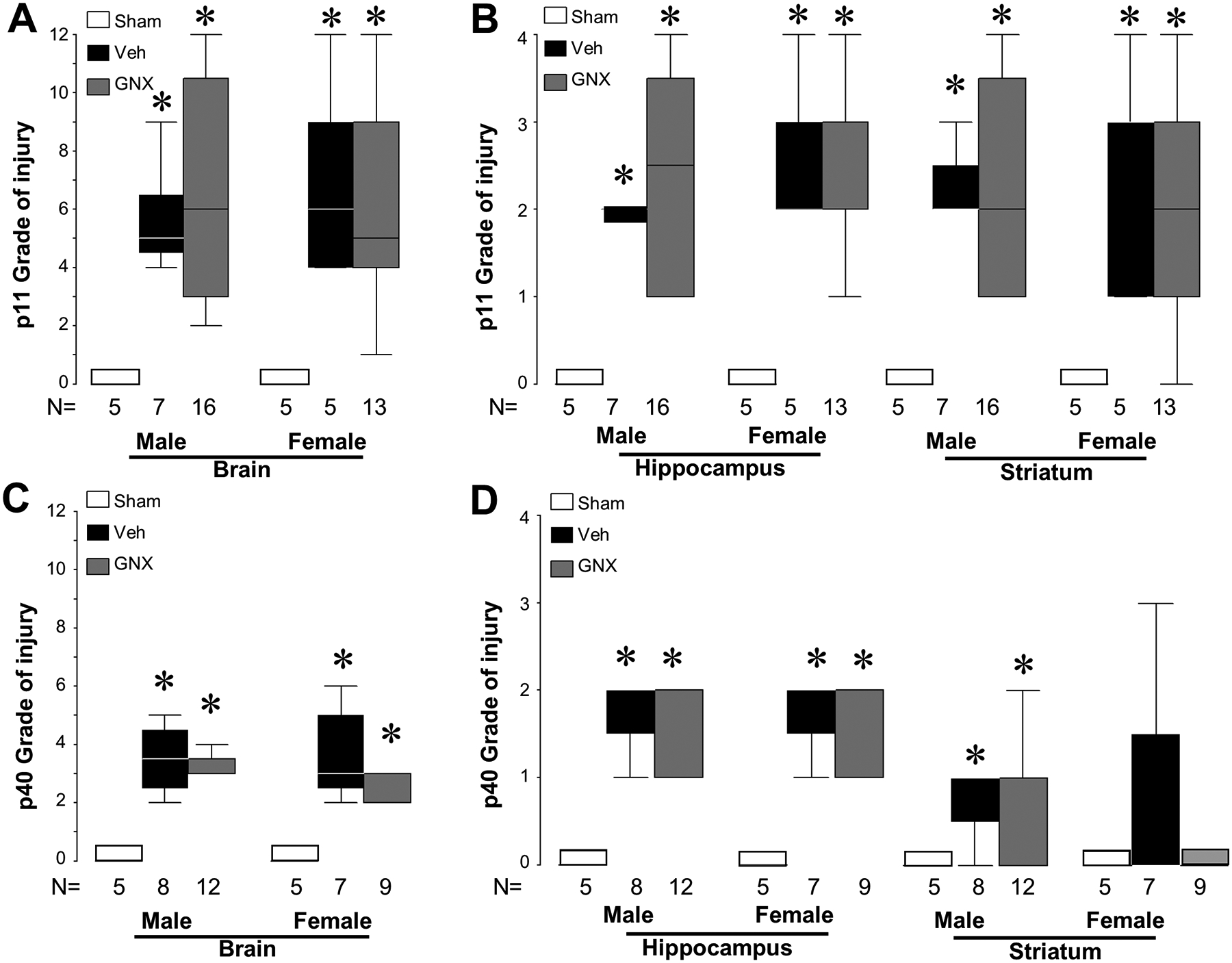
Lack of neuroprotection is seen in HI injured neonatal mice after GNX-4728 treatment. HI caused overall and regional brain injury in both male and female vehicle and GNX-4728 treated mice compared to sham when analyzed at 24 h (p11) [A, B] and at 1 month (p40) following injury [C, D] HI (*P < .05, vs. sham). GNX-4728 did not provide neuroprotection compared to vehicle in either males or females at either time point of analysis.
Because of the potential insensitivity of the scale-based scoring of neuropathological outcome for detecting evidence of neuroprotection, we used measurements with filar optical micrometry to evaluate ipsilateral hippocampal and striatal injury on specific coronal brain sections. Ipsilateral/contralateral ratios were used for comparison between treatment groups for each measure. The sensitivity and validation of this neuropathological assessment approach was established by data showing that this more simple technique is able to show that therapeutic hypothermia attenuates the hippocampal atrophy 8 days after HI (Fig. 4A), as previously reported using MRI determined measures of regional brain volume (Burnsed et al., 2015). We used this simple tool to further identify potential effects of GNX-4728 on brain injury. No differences between GNX-4728 or vehicle treated HI survivors were observed at p40 in hippocampal width, height, hippocampal area determined from the width and height, or CA1 pyramidal cell layer width or in striatal cross sectional area (Fig. 4B–F). Injured mice regardless of treatment group showed significant hippocampal, CA1 and striatal atrophy compared to sham mice at p40. No sex differences were evident (Fig. 4B–F).
Survival was measured in all mice using an intention to treat design. Mice given vehicle or GNX-4728 following HI on p10 and randomized to the p40 analysis time point were followed for survival (Fig. 5A). By p20, all deaths that were to occur in the groups had occurred and resulted in a mortality of 11.8% for the injured vehicle treated group and 27.6% for the injured GNX-4728 treated group. These mortality rates were not different (P = .209). Assigning an injury score of 12 to mice in each group that died and combining the qualitative grade of brain injury at p40 with the mortality data as a composite death or injury outcome (Fig. 5B) failed to identify differences in the vehicle or GNX-4728 treated HI mice.
Figure 5. [A].
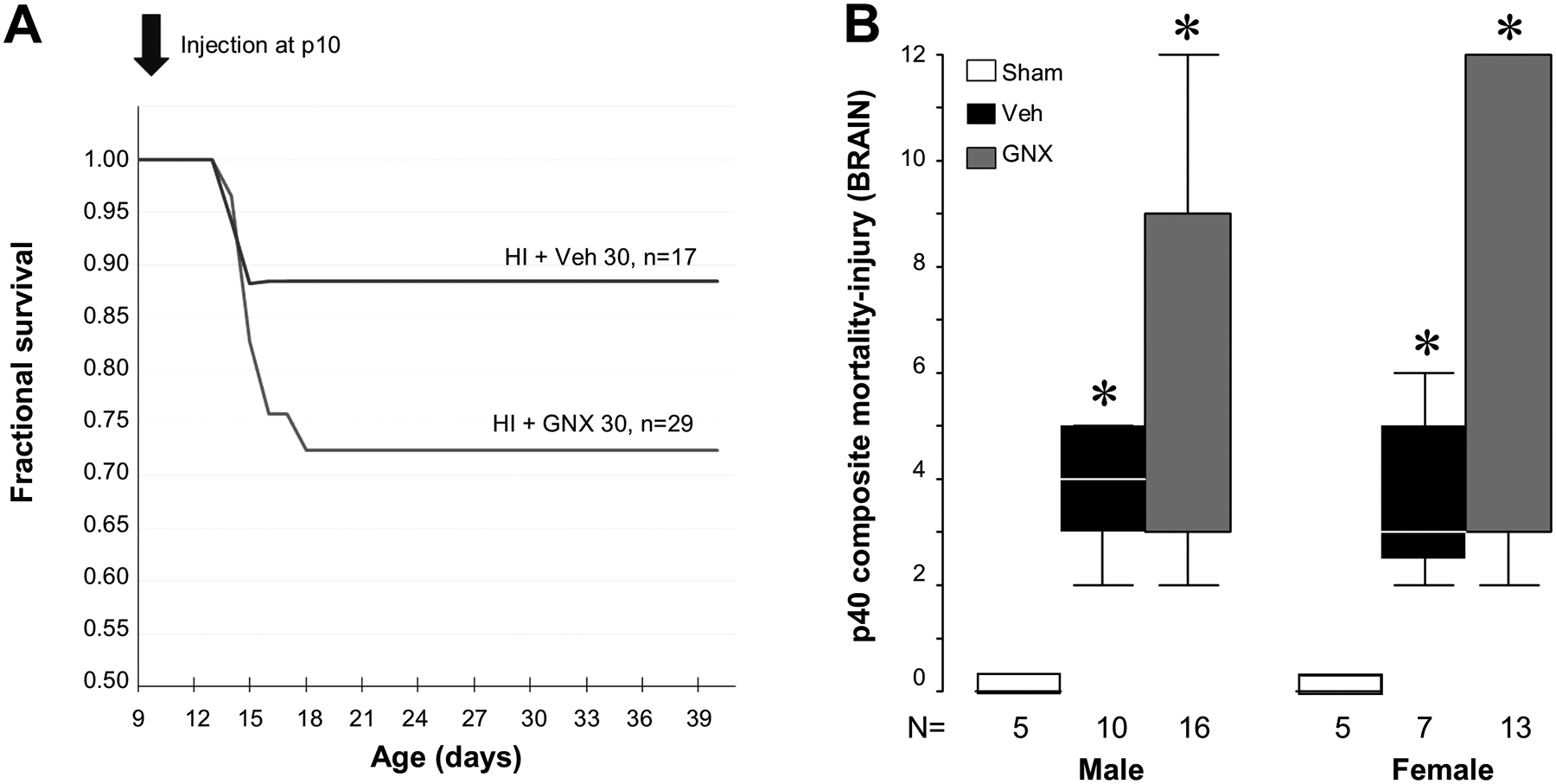
Kaplan–Meier survival plot for age at death in p10 mice after HI and treatment with vehicle or 30 mg/kg (IP) of GNX-4728 showed no differences between groups. [B] Assigning a maximum injury score of 12 to mice that died prior to analysis at p40 and combining this with the p40 injury score to create a combined composite score revealed significantly more brain injury in both male and female GNX-4728 and vehicle treated HI mice than in shams (*P < .05 vs. shams. There was no difference in composite scores between vehicle or GNX-4728 treated groups.
DISCUSSION
We tested GNX-4728, a small molecule mPTP inhibitor, as a potential neuroprotectant in a mouse model of neonatal HI. Our results support the following conclusions: i) GNX-4728, a small molecule inhibitor of the mPTP, and its vehicle, are highly toxic to neonatal mice at doses that are therapeutic in adult mice; ii) at a dose an order of magnitude lower than the therapeutic dose in adults, GNX-4728, provides sex-dependent brain mitochondrial protection (in females) after neonatal HI; iii) however, in neither males nor females, after neonatal HI, is acute brain damage or more chronic brain damage mitigated by the tolerated dose of GNX-4728 administered immediately after the insult.
Both GNX-4728 and vehicle (DMSO/cyclodextrin-saline, 1:1.5), when given at doses therapeutic in adult models, 300 mg/kg, and even 60% lower, 100 mg/kg, had significant toxicity in neonatal mice. In adults, 300 mg/kg is tolerated on a multidose regimen as chronic therapy. (Martin et al., 2014). GNX-4728 has low solubility, so the vehicle is a critical consideration and at the highest equivalent doses, the vehicle was toxic in our neonatal mice as well. Toxicity in our mouse neonatal model highlights the need for testing of all drugs found to be helpful in adult neuroprotection (and their vehicles) in different appropriate preclinical neonatal animal models and immature human neural cell culture models as a first stage of adapting a therapy for potential therapeutic use in neonates.
The most significant effects on mitochondrial respiration measured in our assay were a decrease in RCR in 3 of the 4 groups exposed to HI. In our assay, the lower dose of 30 mg/kg GNX-4728, which showed no apparent toxicity in our safety trial in uninjured mice, restored brain mitochondrial RCR of HI female mice to sham levels when assayed one hour after HI and treatment. This effect was not seen in males with both GNX-4728 and vehicle-treated HI males having RCRs significantly lower than sham. The effect to depress brain mitochondrial RCR following HI in males is clearly due to a decrease in State 3 respiration. Neither HI nor GNX-4728 treatment have an effect on State 4o respiration in either males or females.
Despite the protective effect of GNX-4728 at 30 mg/kg as acute, single-dose treatment on brain mitochondrial RCR, GNX-4728 had no neuropathological benefit to injured mice assessed at 24 h or at 1 month after neonatal HI. When no neuroprotection was found using a qualitative review of the tissues with scores assigned as in Fig. 1 and initially described by Ditelberg, (Ditelberg et al., 1996), consideration was given to the possibility that this tool developed for analysis of acute brain injury might be insensitive to the appearance of injury at p40 when acute injury has resolved. We then did a brief measurement of residual tissue area remaining in ipsilateral anterior hippocampus, the CA1 pyramidal cell layer and mid-striatum compared to the contralateral region as described and no effect of GNX-4728 could be identified with this measure either. Before undertaking this analysis of GNX-4728 or vehicle treated mice, we used the filar ocular measurements on tissue from HI mice treated with either normothermia or hypothermia and found that the expected injury and therapeutic effects of HI and hypothermia were detectable as expected. These data are consistent with results from MRI measurement of therapeutic hypothermia neuroprotection seen previously (Burnsed et al., 2015) When the GNX-4728 results were further stratified by sex, no differences were detectable. Finally, any trend toward possible neuroprotection by GNX-4728 in a single region, i.e. the striatum Fig. 4F, is negated when mortality of HI + vehicle or HI + GNX-4728 mice is combined with the qualitative injury scores at p40 (Fig. 5) for a composite assessment of death and injury.
The refractoriness of HI neurodegeneration in neonatal mouse to systemic GNX-4728 is important considering that this drug protects the adult heart and spinal cord in pre-injury treatment models (Fancelli et al., 2014, Martin et al., 2014) albeit at higher doses. A single dose of GNX-4728 protected the heart from myocardial infarct in rabbit (Fancelli et al., 2014). Chronic treatment of adult mice with GNX-4728 had astonishing disease-modifying therapeutic mitigation of ALS-like fatal disease in a trangenic human superoxide dismutase-1 mouse model and also in neurons, mitochondria, and neuromuscular junctions (Martin et al., 2014). A key caveat of this comparison of neuroprotection in different mouse models is that treatment in neonatal mice was a single acute bolus administered after the insult while in adult mice GNX-4728 treatment was chronic and initiated preinsult/presymptomatically.
Our findings are consistent with the concept that post-injury therapies targeted at improving mitochondrial outcomes may be too late. Mitochondrial failure occurs during the period of hypoxia and inhibition of mPTP opening, likely, must occur then to be protective (Bernardi et al., 1994, Bernardi et al., 1998, Di Lisa and Bernardi, 2009, Azzolin et al., 2010, Rasola et al., 2010). Thus, effective therapeutic actions of mPTP drugs might be predicated on administration prior to an excessive Ca2+ loading event. In pharmacologic studies with a related compound, GNX-4975, Ki increases dramatically from 1.8 nM when administered prior to Ca2+ loading to 140 nM after Ca2+ loading (Richardson and Halestrap, 2016). Binding on the mPTP with GNX-4975 when administered prior to an excessive Ca2+ load then prevents Ca2+ binding that triggers mitochondrial permeability transition pore opening (Richardson and Halestrap, 2016). Smaller increases in Ca2+, and oxygen free radicals appear to increase binding sites for GNX-4975. These data likely partially explain the efficacy of GNX-4728 in treatment of ALS susceptible mice and other pre-treatment/preconditioning paradigms and the lack of efficacy in our experiments.
The toxicity of “safe” adult doses of GNX-4728 in naïve uninjured mice is perhaps the most important finding in the present study. High mortality from administration of adult levels of a mitochondria-active drug in a neonate could be due to the differences in mitochondrial biology and pathology based on age of the animal. There is a developmental difference in mitochondrial calcium uptake capacity in isolated mitochondria from normal (uninjured) brain. In physiologic conditions (pH = 7.0, with ATP), mitochondria isolated from adult rats have a higher Ca2+ uptake capacity than mitochondria from immature rats (Robertson et al., 2004). Furthermore acidosis (pH = 6.5) significantly reduces maximal Ca2+ uptake at both ages (Robertson et al., 2004), suggesting that post-injury acidosis could be particularly harmful in the developing brain. This pH vulnerability of immature mitochondria finding replicated data from immature and mature cortical brain sliced in which both alkalotic and acidic environments inhibited mitochondrial respiration in immature cortical brain slices, but not in adult brain slices (Holtzman et al., 1987). Further developmental differences pertinent to our findings include evidence that genetic deletion of the mPTP activator, cyclophilin-D (CypD), is neuroprotective in adult mice after HI, but worsens brain injury after HI in neonatal mice (Wang et al., 2009). Adult CypD knockout mice were protected from HI injury compared to wildtypes, indicating that CypD-dependent permeability transition is a critical step in injury development in adults. However, neonatal CypD knockout mice exposed to HI had more brain volume loss than wildtype, suggesting that regulation of the mPTP in neonatal brain is distinctly different from that in adult brain.
Limitations for our study were identified. The multifactorial mechanisms contributing to mitochondrial dysfunction, such as oxidative stress, dysregulation of calcium homeostasis, and oxidative phosphorylation failure, have likely caused irreparable damage in the brain by the time GNX-4728 was administered in our study (Chang et al., 1992, Rizzuto et al., 1992, Kuroda et al., 1996, Fiskum et al., 1999, Starkov et al., 2004, Blomgren and Hagberg, 2006). Repeated, chronic dosing of the drug might provide some benefit compared to the one-time dose that we used and it is possible that GNX-4728 could have a role as therapy prior to neonatal HI injury. Given the very concerning toxicity data unveiled in the current study, use of GNX-4728 in its current formulation is untenable even in other animal studies of neonatal HI. Other drugs for neonatal brain injury that require DMSO/cyclodextrin as a diluent/vehicle because of solubility issues, should also have a toxicity study done as a part of their investigation as well. The mortality from the adult doses of vehicle may reflect either toxicity of the DMSO (Hanslick et al., 2009), or that the amount of volume administered was excessive for a neonatal model. GNX-4728 is highly insoluble and no other diluents were found that could successfully solubilize the drug.
In summary, our study highlights the need for careful testing of any drug shown to have promise in adult models in relevant neonatal models in which mortality can be carefully followed, in addition to determining its efficacy separately in neonatal models. It is unclear whether inhibition of mPTP after HI will be neuroprotective in neonatal models. New approaches to inhibiting pathologic mitochondrial permeability transition are needed in the neonate.
Acknowledgements
The authors thank Congenia Srl-Genextra Group (Milan, Italy) for providing drug.
Financial support and sponsorship
FJN is supported by HD086058, HD070996 and HD074593, RCV by NIH NS096115, Johns Hopkins University Clinician Scientist Award and Sutland Pakula Endowment for Neonatal Research, CR by NS092747, and LJM by NS079348.
ABBREVIATIONS:
- ALS
amyotrophic lateral sclerosis
- CV
cresyl violet
- CypD
cyclophilin-D
- HI
hypoxic ischemic
- IP
intraperitoneal
- mPTP
mitochondrial permeability transition pore
- RCR
respiratory control ratio
- TH
therapeutic hypothermia
Footnotes
Conflicts of interest
None.
References
- Alavian KN, et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A, 111 (2014), pp. 10580–10585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzolin L, et al. The mitochondrial permeability transition from yeast to mammals. FEBS Lett, 584 (2010), pp. 2504–2509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azzopardi D, et al. Effects of hypothermia for perinatal asphyxia on childhood outcomes. N Engl J Med, 371 (2014), pp. 140–149 [DOI] [PubMed] [Google Scholar]
- Bernardi P, et al. Recent progress on regulation of the mitochondrial permeability transition pore; a cyclosporin-sensitive pore in the inner mitochondrial membrane. J Bioenerg Biomembr, 26 (1994), pp. 509–517 [DOI] [PubMed] [Google Scholar]
- Bernardi P, et al. The mitochondrial permeability transition. Biofactors, 8 (1998), pp. 273–281 [DOI] [PubMed] [Google Scholar]
- Biasutto L, et al. The mitochondrial permeability transition pore in AD 2016: an update. Biochim Biophys Acta, 1863 (2016), pp. 2515–2530 [DOI] [PubMed] [Google Scholar]
- Blomgren K, Hagberg H Free radicals, mitochondria, and hypoxia-ischemia in the developing brain. Free Radic Biol Med, 40 (2006), pp. 388–397 [DOI] [PubMed] [Google Scholar]
- Burnsed JC, et al. Hypoxia-ischemia and therapeutic hypothermia in the neonatal mouse brain – a longitudinal study. PLoS One, 10 (2015), Article e0118889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang LH, et al. Effect of dichloroacetate on recovery of brain lactate, phosphorus energy metabolites, and glutamate during reperfusion after complete cerebral ischemia in rats. J Cereb Blood Flow Metab, 12 (1992), pp. 1030–1038 [DOI] [PubMed] [Google Scholar]
- Chavez-Valdez R, Emerson P, Goffigan-Holmes J, Kirkwood A, Martin LJ, Northington FJ. Delayed injury of hippocampal interneurons after neonatal hypoxia-ischemia and therapeutic hypothermia in a murine model. Hippocampus, 28 (8) (2018), pp. 617–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lisa F, Bernardi P A CaPful of mechanisms regulating the mitochondrial permeability transition. J Mol Cell Cardiol, 46 (2009), pp. 775–780 [DOI] [PubMed] [Google Scholar]
- Ditelberg JS, et al. Brain injury after perinatal hypoxia-ischemia is exacerbated in copper/zinc superoxide dismutase transgenic mice. Pediatr Res, 39 (1996), pp. 204–208 [DOI] [PubMed] [Google Scholar]
- Executive summary: Neonatal encephalopathy and neurologic outcome, second edition. Report of the American College of Obstetricians and Gynecologists’ task force on neonatal encephalopathy, Obstet Gynecol, 123 (2014), pp. 896–901 [DOI] [PubMed] [Google Scholar]
- Fancelli D, et al. Cinnamic anilides as new mitochondrial permeability transition pore inhibitors endowed with ischemia-reperfusion injury protective effect in vivo. J Med Chem, 57 (2014), pp. 5333–5347 [DOI] [PubMed] [Google Scholar]
- Fiskum G, et al. Mitochondria in neurodegeneration: acute ischemia and chronic neurodegenerative diseases. J Cereb Blood Flow Metab, 19 (1999), pp. 351–369 [DOI] [PubMed] [Google Scholar]
- Graham EM, et al. Neonatal mice lacking functional Fas death receptors are resistant to hypoxic-ischemic brain injury. Neurobiol Dis, 17 (2004), pp. 89–98 [DOI] [PubMed] [Google Scholar]
- Hanslick JL, et al. Dimethyl sulfoxide (DMSO) produces widespread apoptosis in the developing central nervous system. Neurobiol Dis, 34 (2009), pp. 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtzman D, et al. Brain cellular and mitochondrial respiration in media of altered pH. Metab Brain Dis, 2 (1987), pp. 127–137 [DOI] [PubMed] [Google Scholar]
- Johnston MV, et al. Treatment advances in neonatal neuroprotection and neurointensive care. Lancet Neurol, 10 (2011), pp. 372–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuroda S, et al. Secondary bioenergetic failure after transient focal ischaemia is due to mitochondrial injury. Acta Physiol Scand, 156 (1996), pp. 149–150 [DOI] [PubMed] [Google Scholar]
- Lawn JE, et al. 4 million neonatal deaths: when? Where? Why?. Lancet, 365 (2005), pp. 891–900 [DOI] [PubMed] [Google Scholar]
- Lee AC, et al. Intrapartum-related neonatal encephalopathy incidence and impairment at regional and global levels for 2010 with trends from 1990. Pediatr Res, 74 (Suppl. 1) (2013), pp. 50–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markovic SN, Murasko DM Anesthesia inhibits interferon-induced natural killer cell cytotoxicity via induction of CD8 + suppressor cells. Cell Immunol, 151 (1993), pp. 474–480 [DOI] [PubMed] [Google Scholar]
- Martin LJ. Mitochondrial and cell death mechanisms in neurodegenerative diseases. Pharmaceuticals (Basel), 3 (2010), pp. 839–915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ. The mitochondrial permeability transition pore: a molecular target for amyotrophic lateral sclerosis therapy. Biochim Biophys Acta, 1802 (2010), pp. 186–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ. Biology of mitochondria in neurodegenerative diseases. Prog Mol Biol Transl Sci, 107 (2012), pp. 355–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, et al. The mitochondrial permeability transition pore in motor neurons: involvement in the pathobiology of ALS mice. Exp Neurol, 218 (2009), pp. 333–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin LJ, et al. GNX-4728, a novel small molecule drug inhibitor of mitochondrial permeability transition, is therapeutic in a mouse model of amyotrophic lateral sclerosis. Front Cell Neurosci, 8 (2014), p. 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northington FJ, et al. Failure to complete apoptosis following neonatal hypoxia-ischemia manifests as “continuum” phenotype of cell death and occurs with multiple manifestations of mitochondrial dysfunction in rodent forebrain. Neuroscience, 149 (2007), pp. 822–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Northington FJ, Chavez-Valdez R, Martin LJ. Neuronal cell death in neonatal hypoxia-ischemia. Ann Neurol, 69 (5) (2011), pp. 743–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao R, et al. Safety and short-term outcomes of therapeutic hypothermia in preterm neonates 34–35 weeks gestational age with hypoxic-ischemic encephalopathy. J Pediatr, 183 (2017), pp. 37–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasola A, Bernardi P The mitochondrial permeability transition pore and its involvement in cell death and in disease pathogenesis. Apoptosis, 12 (2007), pp. 815–833 [DOI] [PubMed] [Google Scholar]
- Rasola A, et al. Signal transduction to the permeability transition pore. FEBS Lett, 584 (2010), pp. 1989–1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson AP, Halestrap AP. Quantification of active mitochondrial permeability transition pores using GNX-4975 inhibitor titrations provides insights into molecular identity. Biochem J, 473 (2016), pp. 1129–1140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, et al. Rapid changes of mitochondrial Ca2 + revealed by specifically targeted recombinant aequorin. Nature, 358 (1992), pp. 325–327 [DOI] [PubMed] [Google Scholar]
- Robertson CL, et al. Mitochondrial response to calcium in the developing brain. Brain Res Dev Brain Res, 151 (2004), pp. 141–148 [DOI] [PubMed] [Google Scholar]
- Robertson CL, et al. Mitochondrial dysfunction early after traumatic brain injury in immature rats. J Neurochem, 101 (2007), pp. 1248–1257 [DOI] [PubMed] [Google Scholar]
- Roehr CC, et al. The 2010 guidelines on neonatal resuscitation (AHA, ERC, ILCOR): similarities and differences – what progress has been made since 2005? Klin Padiatr, 223 (2011), pp. 299–307 [DOI] [PubMed] [Google Scholar]
- Shankaran S. Outcomes of hypoxic-ischemic encephalopathy in neonates treated with hypothermia. Clin Perinatol, 41 (2014), pp. 149–159 [DOI] [PubMed] [Google Scholar]
- Shankaran S, et al. Childhood outcomes after hypothermia for neonatal encephalopathy. N Engl J Med, 366 (2012), pp. 2085–2092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon RA, et al. Strain-related brain injury in neonatal mice subjected to hypoxia–ischemia. Brain Res, 810 (1998), pp. 114–122 [DOI] [PubMed] [Google Scholar]
- Starkov AA, et al. Mitochondrial alpha-ketoglutarate dehydrogenase complex generates reactive oxygen species. J Neurosci, 24 (2004), pp. 7779–7788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujimoto Y, et al. Mitochondrial membrane permeability transition and cell death. Biochim Biophys Acta, 1757 (2006), pp. 1297–1300 [DOI] [PubMed] [Google Scholar]
- Wang X, Carlsson Y, Basso E, Zhu C, Rousset CI, Rasola A, Johansson BR, Blomgren K, Mallard C, Bernardi P, Forte MA, Hagberg H Developmental shift of cyclophilin D contribution to hypoxic-ischemic brain injury. J. Neurosci, 29 (8) (2009), pp. 2588–2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yager JY, Ashwal S Animal models of perinatal hypoxic-ischemic brain damage. Pediatr Neurol, 40 (2009), pp. 156–167. [DOI] [PubMed] [Google Scholar]