

Abstract
Free fatty acid receptor 3 (FFA3, previously GPR41) is activated by short-chain fatty acids, mediates health effects of the gut microbiota, and is a therapeutic target for metabolic and inflammatory diseases. The shortage of well-characterized tool compounds has however impeded progress. Herein, we report structure–activity relationship of an allosteric modulator series and characterization of physicochemical and pharmacokinetic properties of selected compounds, including previous and new tools. Two representatives, 57 (TUG-1907) and 63 (TUG-2015), showed improved solubility and preserved potency. Of these, 57, with EC50 = 145 nM and a solubility of 33 μM, showed high clearance in vivo but is a preferred tool in vitro. In contrast, 63, with EC50 = 162 nM and a solubility of 9 μM, showed lower clearance and seems better suited for in vivo studies. Using 57, we demonstrate for the first time that FFA3 activation leads to calcium mobilization in murine dorsal root ganglia.
Introduction
Short-chain fatty acids (SCFAs) are produced in large amounts by the lower gut microbiota and are known to affect human health in various and often beneficial ways.1 Free fatty acid receptors 2 and 3 (FFA2 and FFA3) are G protein-coupled receptors activated by SCFAs and mediate many of the physiological effects of SCFAs.2,3 The receptors were discovered and reported simultaneously in 2003 and are co-expressed in enteroendocrine cells, pancreatic β-cells, some immune cells, and certain cancers.4−7 Both FFA2 and FFA3 have been reported to be expressed in the adipose tissue, although most studies now suggest that only FFA2 is present.6−10 Of the two receptors, FFA2 has received more attention, showing promise as a target for the treatment of various metabolic and inflammatory conditions,6 with one compound reaching clinical trials for ulcerative colitis before being discontinued due to limited efficacy, despite the fact that the compound did inhibit neutrophil infiltration.11
Several studies have also suggested interesting therapeutic potential for FFA3. For example, Offermanns and co-workers demonstrated that deletion of FFA2 and FFA3 in combination, but not individually, improved insulin secretion and glucose tolerance in mice, indicating that dual antagonism of the receptors may counteract type 2 diabetes.12 Activation of both receptors is also reported to counteract cancer development.13,14 FFA3 is linked to hypoxia-induced apoptosis and may have potential as a target for ischemia/reperfusion-related injury.15 Marsland and co-workers found that FFA3 but not FFA2 mediates the protective effect of circulating SCFAs against allergic lung inflammation and is therefore of interest for treatment of allergic asthma.16 FFA3 has also been reported in both autonomic and somatic sensory ganglia.17 In sympathetic ganglia, propionate is found to promote sympathetic nervous system activation and to be involved in regulation of the body energy balance.18
Most of the studies involving FFA3 have relied on SCFAs as tools and/or knockout mice. These studies should be interpreted with caution because SCFAs are generally able to activate both FFA2 and FFA3 but with different profiles on human and rodent orthologues19 and because it has been found that knockout of one receptor may affect the expression of the other.10 More studies are therefore required to elucidate the therapeutic potential of FFA3 alone and in combination with FFA2. An important reason for the paucity in studies on FFA3 is the lack of well-characterized high-quality tool compounds for this receptor.20 Studies that use SCFAs often employ propionate (C3) as a dual agonist of FFA2 and FFA3, acetate as a FFA2-selective agonist, and butyrate as a FFA3-selective agonist; however, the selectivity for these compounds are modest at best.19 Recently, the FFA3-selective SCFA-analogue 1-methylcyclopropylcarboxylate (1-MCPC) has also been employed, but its potency remains very low.21
Currently, the only tools for FFA3 with potency in the single-digit micromolar range are from a series of tetrahydroquinolones originally disclosed by Arena Pharmaceuticals (represented by 1, Table 1) and that were subsequently shown to be allosteric modulators of the receptor.22,23 Although they have occasionally been used as tools,24−28 moderate potency and the lack of proper characterization have limited the use of these compounds. The compounds act as allosteric modulators and are not affected by mutation of arginine residues in the orthosteric site that are indispensable to the activity of propionate.22 Small structural changes of the tetrahydroquinolones have shown to affect the mode of action, ranging from pure allosteric agonists to modulators that either enhance the potency of propionate (positive allosteric modulators, PAMs), reduce the efficacy of propionate (negative allosteric modulators, NAMs), or acting as both agonists on their own but also enhance the potency of propionate (PAM agonists).
Table 1. Initial SAR Investigations of 1,4,7,8-Tetrahydroquinol-5-one-3-carboxamides.
![]()

Herein, we present the results from a thorough examination of the structure–activity relationships (SAR) within this compound series and further characterize bioavailability and pharmacokinetic properties of the most promising analogues. Moreover, we use a key compound to demonstrate the functional activity of FFA3 in cells of murine dorsal root ganglia.
Synthesis
The tetrahydroquinolone target compounds were typically synthesized from the appropriate 3-ketoamide, aldehyde, and 3-aminoenone using the Hantzsch dihydropyridine synthesis (Scheme 1). Heating at 80 °C in i-PrOH for up to 5 days generally gave the best outcome, with longer reaction times for more hindered substrates.29 Microwave heating or synthesis from 3-ketoamide, aldehyde, 1,3-dione, and ammonia provided the product in shorter time but at the expense of lower yield and purity. The 3-ketoamide substrates were most conveniently accessed by heating of the appropriate aniline in neat methyl acetoacetate. These intermediates were also synthesized by heating of the aniline with 2,2,6-trimethyl-4H-1,3-dioxin-4-one or in the presence of Lewis acid catalysts but generally with an inferior outcome. The preferred route to the typical tetrahydroquinolone is shown in Scheme 1.
Scheme 1. General Synthetic Route for Tetrahydroquinolone Ligands.

Results and Discussion
The ligands were initially screened in a human FFA3-dependent [35S]GTPγS binding assay as this assay reflects receptor-mediated activation of Gi/o proteins and is known to generally correlate well with ligand affinity.30 Analogues of particular interest were tested further in a cAMP inhibition assay as this is an important downstream effect of Gi-activation. The latter was applied as a standard assay because of higher reproducibility. It is furthermore performed in whole cells, is more downstream, includes G protein signal amplification, and therefore better reflects a more natural ligand–receptor response. Because the series binds to an allosteric site on FFA3,22 selected compounds were also tested together with a fixed submaximal concentration of the SCFA propionate to investigate potential allosteric effects on orthosteric agonist function.
Tetrahydroquinolones 1 and 2 were disclosed by Arena Pharmaceuticals in 2006.23 We resynthesized these compounds, confirming FFA3 agonist activity in the low micromolar range.22 2-Furyl derivative 1 showed agonist activity with approximately 2-fold higher potency than 3-furyl derivative 2 in the GTPγS assay (Table 1).
Replacing the 2-furyl with phenyl (3) led to significant deterioration of potency, whereas 2-thienyl (4), a group with a size and polarity that more closely resembles phenyl than furyl, largely preserved potency in the GTPγS assay.
The analogue with 2-bromophenyl (5) was previously characterized and found to be inactive alone but to act as a PAM of propionate, implying that 5 binds to FFA3 but is unable to activate the receptor directly.22 Introducing bromosubstituents in the 3- (6) or 4-position (7) of phenyl derivative 3 regained most of the activity relative to 2-furyl derivative 1 in the GTPγS assay, whereas the potency was essentially constant for 3–4 and 6–7 in the cAMP assay. Analogues with 3- (8) and 4-trifluoromethyl (9) and 4-methyl (10) substituents were full agonists with potencies similar to or lower than that of the unsubstituted 3, whilst extension to 4-ethyl (11) eroded potency.
Replacement of o-tolyl by phenyl (12) gave a >3-fold drop in potency, whereas m-tolyl (13) or p-tolyl (14) resulted in a more moderate drop, indicating the ortho-position as the most interesting. Introduction of methoxy (15) further eroded potency in the GTPγS binding assay but retained potency in the cAMP inhibition assay. The 2,5-dichloro-substituted AR420626 (16) also originates from Arena Pharmaceuticals and has been described as a tool compound in the literature and characterized by us.22,24,25 Like 1, this compound exhibited only moderate potency in the GTPγS assay22 but is an order of magnitude more potent in the cAMP assay. It thus represents one of the most potent compounds but is also known to have poor solubility.
We also wished to explore aliphatic R1 groups, and we were pleased to find that isobutyl (17) behaved as an FFA3 agonist with a potency similar to that of 1 in the [35S]GTPγS binding assay. Introducing 2,3-dimethyl (18), 2-iodo (19), 2-chloro (20), or 2,6-difluorophenyl (21) on the R2 phenyl while keeping R1 as isobutyl produced active compounds with potencies comparable to that of 17, with 20 representing an improvement in the cAMP but not in the GTPγS assay. In general, results from the cAMP assay corresponded satisfactorily with the GTPγS data, although some of the compounds deviated considerably. Notably, increased potency of 10-, 14-, and 50-fold was observed for 2, 3, and 15, respectively. In contrast, only 17 and 18 exhibited a lower potency of 5- and 2-fold, respectively. On the other hand, together with 1 μM propionate, 17 exhibited a potency that was equal with 1 (with or without propionate). As the cAMP assay is more downstream and showed a reproducibility that was at least as good as the GTPγS assay, this was chosen as the primary assay for the remaining compounds.
We next turned our attention to the other parts of the structure (Table 2). An extension of the ortho-methyl at the dihydropyridine to ethyl (22) led to an order of magnitude decrease of potency while a phenyl (23) produced a completely inactive compound. Oxidation of the dihydropyridine to pyridine (24) also produced an inactive compound, perhaps unsurprisingly because this compound represents substantial structural changes.
Table 2. Scaffold Exploration of Furyl and Isobutyl Analogues.

Mean of ≥3 independent experiments ± standard error. Efficacy (Emax) is relative to maximal response of propionate.
cAMP assay in the presence of 1 μM propionate.
No response.
GTPγS assay.
Using 17 as a starting point, opening of the cyclohexanone and formation of a phenone (25) or methyl ester (26) both produced compounds that were inactive (pEC50 < 4) alone but 26 acted as PAM with propionate, indicating that some larger changes in the structure also produced compounds with affinity for FFA3. Introduction of a nitrile (27) produced a compound that was active alone, albeit with low potency, but that was substantially potentiated by the presence of propionate. We also wished to explore the significance of the ketone; however, all attempts to reduce the cyclohexanone carbonyl or derivatize, for example, to form oximes, were unsuccessful, partly due to low electrophilicity due to stabilization by conjugation to the enamine of the dihydropyridine system and partly due to instability of products.
Replacement of the anilide part by a methyl ester (28) or a phenone (29) produced compounds that also were inactive alone but exhibited PAM properties. Introduction of a carboxylic acid in this position produced a compound that could not be characterized or tested due to insufficient solubility.
We next reverted to the R1 group to further explore aliphatic substituents (Table 3). Introduction of sec-butyl (30) resulted in a compound with very similar properties to isobutyl 17. The sterically more well-defined cyclohexyl (31) gave marginally increased potency which could be a result of a hydrophobic effect. Surprisingly, the slightly smaller cyclopentyl (32) increased potency 20-fold to 288 nM. Cyclopropyl (33) maintained good potency at 600 nM with an order of magnitude reduced lipophilicity. A series of n-alkyls from ethyl to pentyl (34–37) resulted in agonists with micromolar potency, with propyl (35) and butyl (36) being the most potent and the shorter ethyl (34) the least potent. Further extension with phenethyl (38), styryl (39), and pyrazolylethyl (40) continued this trend with pEC50 values <5, interestingly, with the less lipophilic 40 exhibiting the highest potency of the three. All compound were full agonists.
Table 3. SAR Exploration of Aliphatic and Heterocyclic Analogues.
![]()

Mean of ≥3 independent experiments ± standard error. Efficacy (Emax) is relative to maximal response of propionate.
cAMP assay in the presence of 1 μM propionate.
Calculated by ChemDraw Professional version 16.
Previously published.31
Identification of the cyclopentyl as the most potent aliphatic substituent motivated another venture into similarly sized aromatic substituents. N-Methyl-2-pyrazole (41) was a low-potency compound, in line with ortho-substituted phenyls such as 5. Furyls and thienyls with small substituents resulted in better potency, with 5-bromo-2-furyl (44) and 2-benzofuryl (45) as the most potent. The lower potency of 3-benzothienyl 46 is likely due to the positioning of the benzene ring.
In an attempt to increase aqueous solubility by decreasing lipophilicity, the 2-furyl of 16 was replaced by 2-thiazolyl (47), 5-thiazolyl (48), and 4-thiazolyl (49) (Table 3). This strategy failed, as the solubility dropped from 5 to below 2 μM for the thiazolyl analogues in a kinetic solubility assay, although the potency was only slightly decreased for 47 (Table 4). Also, even though clogP of 47–49 indicated an order of magnitude improvement of lipophilicity relative to 16 (log D7.4 = 3.19), measured log D7.4 revealed similar or increased lipophilicity. Replacement by 4-oxazolyl (50) improved solubility >10-fold but had a detrimental effect on potency. On the other hand, 2-oxazolyl (51) improved potency but reduced solubility to 1 μM.
Table 4. SAR Exploration of 2,5-Dichloro Analogues.
![]()

Mean of ≥3 independent experiments ± standard error. Efficacy (Emax) is relative to maximal response of propionate.
Calculated by ChemDraw Professional version 16.
Kinetic solubility at 25 °C in phosphate buffered saline pH 7.4 (PBS7.4).
No response.
With a focus on solving the solubility problem, we returned to the aliphatic substituents aiming to incorporate a positive charge. Unfortunately, both small polar and larger, lipophilic amine substituents (52–54) produced inactive compounds. Analogues with negatively charged carboxylate groups in the same position (not shown) were also explored but were completely inactive. Boc-protected intermediates (55–56) were tested and found to be inactive alone. 52–55 were also inactive in the presence of 3 μM propionate and were therefore also not PAMs. However, 56, representing the compound with the largest R1 substituent explored in this study, turned out to be a NAM, effectively inhibiting receptor signaling at 30 μM concentration (Figure 1). This is in line with previous compounds characterized as FFA3 NAMs, where R1 was 3- or 4-phenoxyphenyl and substantially larger than other R1-groups explored in the series. Thus, a larger R1 increases the chance of finding a NAM or antagonist.
Figure 1.

NAM 56 in the cAMP assay. (a) Concentration–response curves for propionate (C3) alone or together with 30 μM 52–56. (b) Concentration–response curves for C3 and 52–56 with 3 μM C3. (c) Concentration–response curves for C3 alone and with increasing concentrations of 56 (from 300 nM to 30 μM). Data represent the mean of three independent experiments and is normalized to forskolin.
We reasoned that the generally poor physicochemical properties of the 2,5-dichlorophenyl compounds are worsened with aromatic or larger, lipophilic R1-groups. With the failure of positively charged R1 groups to produce FFA3 agonists, we therefore proceeded with exploration of smaller neutral groups. The combination of n-propyl with 2,5-dichlorophenyl (57) indeed produced a compound with similar potency to 16 but 6-fold increased solubility (Table 5). Hoping to further increase solubility, we next investigated the requirements of chlorinated R2-groups in relation to potency. The 2-chloro (58) or 5-chloro (59) substituents alone were accompanied by 4–5-fold increased solubility compared to 57 but reduced potency down to a level similar to o-tolyl (35, Table 3). The solubility of 35 was 199 μM and its log D7.4 was 2.64 and thus favorable compared to all chlorinated compounds and comparable with 1 (190 μM solubility, log D7.4 2.05). A tendency toward higher potency for 58 indicated that the 2-chloro is more important than the 5-chloro, in line with observations for methyl substituents (cf. 1, 13, 14, Table 1). In an attempt to regain potency but keep the solubility properties, the 5-chloro of 57 was replaced by methyl (60) or methoxy (61). This indeed resulted in compounds with solubilities similar to the other monochlorinated compounds, but the potency was also similar or only marginally improved. Chemical stability was also tested, and all compounds (58–61) were completely recovered after 1 week at 37 °C in PBS. Finally, revisiting small aliphatic cycles, cyclopropyl (62) partly regained and cyclopentyl (63) fully regained the potency of the 2-furyl analogue 16, but with similar or only moderately increased solubility. Compounds 62 and 63 were also tested in the cAMP assay together with 1 μM propionate but did not reveal significant PAM effects. Overall, of the compounds tested as PAMs, none with pEC50 > 6 showed significantly enhanced potency in the presence of propionate.
Table 5. SAR of Neutral Aliphatic R1 and Chlorinated R2.

Mean of ≥3 independent experiments ± standard error. Efficacy (Emax) is relative to maximal response of propionate.
Calculated by ChemDraw Professional version 16.
No response.
To identify the most active enantiomer, racemic 1 was resolved by chiral HPLC and crystallized. The single crystal X-ray structures for one enantiomer (Figure 2) along with that of the racemate (Figure S1) were determined. The absolute configuration of the most active enantiomer (R)-1 was determined by anomalous dispersion effects with a Flack parameter of 0.04(12). However, the assignment must be viewed with some caution because of the high standard deviation in the Flack parameter. Crystal packing is influenced by the presence of a single or both enantiomers in the lattices of (R)-1 and (R,S)-1, respectively. Both structures show that the strongest intermolecular interaction between molecules is the H-bonding between the cyclic carbonyl (O1) and the amine NH group (N1) of the adjacent molecule (Figure S2). These H-bonds link the molecules in ribbons approximately parallel to the a- and c-axis in the structures of (R)-1 and (R,S)-1, respectively (Figures S5 and S2). The ribbons stack with the 2-furyl rings located on the same side of every molecule in (R)-1 (Figure S6). In (R,S)-1, they are alternating in the up and down positions in accordance with the H-bonded ribbons comprising alternating R and S enantiomers (Figure S3).
Figure 2.

Identification of the most potent enantiomer. (a) cAMP data for (R)-1 and (S)-1, data are mean of ≥3 independent experiments ± standard error. Structure and numbering scheme of one of the two molecules (molecule A) in the asymmetric unit of (R)-1.
The previously published compound 1 was not affected by mutation of either R185A or R258A in the orthosteric binding site, and the compounds were therefore believed to be allosteric modulators. In the search for the potential allosteric binding site, we mutated a third arginine residue in a neighboring site (R71A); however, this did not affect the potency of 1. After the publication of the FFA1 crystal structure in complex with an allosteric agonist located in the TM region close to the intracellular site,32 a homology model of hFFA3 was constructed using Modeller. The model revealed a similar potential binding site on FFA3 and initial docking studies of (R)-1 indicated three amino acids that might be involved in binding (Figure 3a). Mutation of E112A and R126A did not affect the potency of (R)-1 or propionate, whereas Q131A significantly reduced the potency of (R)-1 (>3-fold, p < 0.001) but did not affect the potency of propionate. Thus, additional docking of (R)-1 in this site were performed to find poses where Q131, but not R126, show a central role in binding. Seven out of 10 poses showed a hydrogen bond interaction between Q131 and either the amide carbonyl or the ketone without any constraints used and only one of the poses showed an additional hydrogen bonding to R126 (Figure 3b). The narrow binding cavity around the core scaffold is in agreement with the restrained SAR observed for this series and the furan moiety pointing out of the binding cavity might explain the flexibility in substituents at this site (Figure 3c,d). Although Q131A impacts the potency of (R)-1 with high significance, the magnitude of the effect is lower than what would be generally expected for the removal of a hydrogen bond interaction; thus, additional studies are required to confirm the proposed binding site.
Figure 3.

Proposed allosteric binding site in a homology model of hFFA3 in complex with (R)-1. (a) Full receptor marked with the orthosteric binding site and potential allosteric binding site. (b) Overlay of 7 out of 10 docking poses. (c) Binding pocket surface. (d) Binding pose highlighting Q131.
With several compounds with improved properties in hand, we wished to investigate the suitability of the compounds as tools for in vivo studies in rodents. Because of the moderate potency and low solubility associated with this series, it has been presumed that they are poorly suited as in vivo tools. A basic requirement is preserved activity on the relevant species orthologues, which generally has been observed to be low within the free fatty acid receptor group.19 We confirmed that propionate maintained potency between human and rodent species. 1 and 16 both lost 3–10-fold potency from human to rodent species (1 pEC50 = 5.87 ± 0.07 on rFFA3, 5.42 ± 0.04 on mFFA3; 16 pEC50 = 6.34 ± 0.04 on rFFA3, 5.88 ± 0.05 on mFFA3), whereas 63 now tended toward the highest potency on the rodent orthologues (pEC50 = 6.39 ± 0.03 on rFFA3; pEC50 = 5.96 ± 0.04 on mFFA3).
We next investigated the chemical stability of the compounds and stability toward liver microsomes. Representative compounds were shaken in PBS at pH 7.4. Apart from some that precipitated, all compounds were quantitatively recovered after 1 week. On the other hand, the tested compounds exhibited varying stability towards mouse liver microsomes (MLM, Table 6). In the one end, 1 and 63 showed good stability, comparable with the propranolol reference compound (61%). In the other end, only 1−2% was recovered of 16, 47, 57, and 62. The remaining compounds were in the intermediate range. Apart from a generally higher stability of the o-tolyl compounds 1 and 35 than the remaining 2,5-dichlorophenyl compounds, it was difficult to see a clear relationship between structural or physicochemical properties and microsomal stability.
Table 6. In Vitro Stability and in Vivo Pharmacokinetic Properties of Selected Compounds in Mice.
| iv dosing (5 mg/kg) |
po
dosing (10 mg/kg) |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cAMP pEC50a | aq. sol. (μM)a | log D7.4a | MLM (%)b | t1/2 (min) | Vd (mL/kg) | CLtotal (mL/min/kg) | t1/2 (min) | tmax (min) | cmax (ng/mL) | AUCinf (ng.min/mL) | F (%) | |
| 1 | 6.28 | 190 | 2.05 | 52 | 62.2 | 759 | 8.45 | 89.7 | 15 | 7470 | 865 000 | 73 |
| 16 | 6.89 | 5 | 3.19 | 1 | 22.3 | 629 | 19.6 | 60.8 | 30 | 5740 | 423 000 | 88 |
| 35 | 5.92 | 199 | 2.64 | 23 | ||||||||
| 47 | 6.60 | 1 | 3.39 | 2 | ||||||||
| 51 | 6.72 | 1 | 2.59 | 12 | ||||||||
| 57 | 6.84 | 33 | 4.25 | 1 | 11.4 | 1150 | 70.3 | 63.5 | 30 | 561 | 33 800 | 36 |
| 62 | 6.60 | 5 | 3.73 | 1 | ||||||||
| 63 | 6.79 | 9 | >4.3 | 41 | 37.8 | 1110 | 20.3 | 51.8 | 15 | 3800 | 325 000 | 66 |
Based on the results so far, 63 appeared as a good compromise between the properties of 1 and 16, and 57 represented a good compromise between potency and solubility. We therefore selected these four compounds for pharmacokinetic studies in mice. Although the microsomal data for at least 16 and 57 indicated that these compounds would have a fast clearing, we decided to run the pharmacokinetic study using the same time frame for all compounds. Overall, the four compounds had surprisingly favorable PK properties and especially 1, 16, and 63 showed high bioavailability. Compound 1 exhibited the longest half-life, approximately an hour after iv dosing, and the lowest clearance, whereas 16 showed a half-life of 20 min. 63 was somewhat in between the two, and 57 had the shortest iv half-life. Half-life after oral dosing was satisfactory for all compounds. 1, 16, 57, and 63 were counterscreened on the related FFA receptors FFA1 and FFA2, the more distant FFA receptors FFA4 and GPR84, and the L-type calcium ion channels, a target for related dihydropyridine ligands. No significant activity was detected at any of the receptors at 10 μM concentration (see Supporting Information).
Because of its good compromise between solubility and potency (EC50 = 2 μM at mFFA3 vs 30 μM for propionate in the GTPγS assay), 57 was considered for further in vitro studies. To further ensure the suitability of 57 as a tool compound for in vitro studies, the chemical stability was also evaluated in dimethyl sulfoxide (DMSO). An NMR sample was stored at 4 °C, and recordings after 1 and 30 days showed complete stability. A sample stored at rt (not protected from light) was fully stable for 7 days, whereas minor decomposition was detected after 30 days (see Supporting Information).
Thus, 57 was selected for studies in cells from dorsal root ganglia (DRGs) isolated from wild-type and FFA3 KO mice. Addition of 57 (5 μM) gave a large elevation in intracellular calcium levels in wild type but not FFA3 KO cells, indicating a FFA3-specific effect of 57 on DRG cells (Figure 4). These results suggest, in line with previous studies,17,18 a key role for FFA3 in mediating effects for SCFAs from the gut microbiome within the peripheral nervous system. They furthermore for the first time demonstrate a pharmacological intervention at ganglionic FFA3, that 57 is able to evoke a response with high selectivity and that ganglionic FFA3 has potential as a therapeutic target for pain and metabolic disorders. Further studies of the role of FFA3 in dorsal root ganglions using 57 as a tool compound are in progress.
Figure 4.
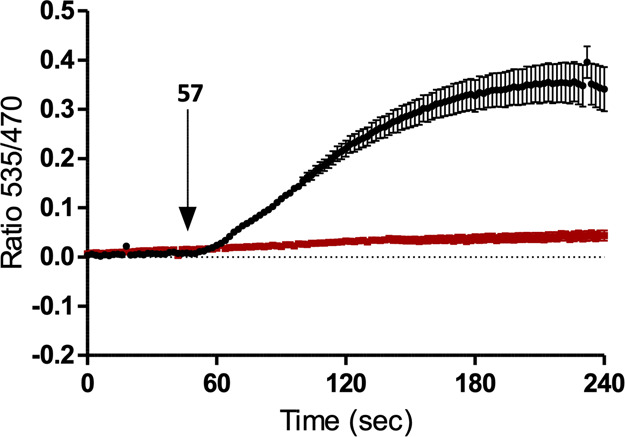
Compound 57 promotes FFA3-dependent elevation of intracellular Ca2+ in cells dissociated from mouse dorsal root ganglia. The arrow indicates addition of 57 (5 μM). Results are expressed as relative fluorescence; WT (black): N = 3 (number of mice), n = 34 (number of cells) and KO (red): N = 3, n = 28.
Conclusions
We have performed an extensive SAR investigation around a series of 1,4,7,8-tetrahydroquinol-5-one FFA3 allosteric modulators that has led to the identification of key structural parts required for inducing effects on FFA3 and parts where modifications are permitted. The studies revealed that the aromatic substituents at the 4-position (referred to as R1) can be replaced by small aliphatic substituents with fully preserved potency. Extension of the R1-group leads to loss of agonist activity but may produce antagonists or NAMs. The 2,5-dichlorophenyl at the amide (R2) has a beneficial effect on potency but a detrimental effect on solubility. The combination of N-2,5-dichlorophenyl with a smaller aliphatic substituent in the 4-position on the tetrahydroquinol-5-one resulted in compounds with preserved potency and improved solubility. The most potent enantiomer was found to have the (R)-configuration. We have for the first time evaluated the in vitro solubility, lipophilicity, and metabolic stability and in vivo pharmacokinetic properties of members of this compound series. Despite largely suboptimal result from in vitro studies, the compounds overall demonstrated surprisingly favorable in vivo pharmacokinetic properties. Compound 63 was found to have well preserved potency on rodent orthologues, good stability toward MLMs, and favorable PK properties for use as an in vivo tool compound. The previously characterized agonists 1 and 16 were also found to have surprisingly favorable PK profiles, although the former suffers from moderate potency and the latter from low solubility. Compound 57 was found to have sufficiently good aqueous solubility and good potency but showed the least favorable PK properties of the studied compounds; thus this is the preferred compound for in vitro but not in vivo studies. Studies of 57 in isolated DRGs demonstrated potent and FFA3-specific calcium mobilization in DRG cells. The study shows that compounds from this series, in the absence of highly potent FFA3 agonists, have merits as both in vitro and in vivo research tools.
Experimental section
All commercial starting materials and solvents were used without further purification, unless otherwise stated. Chemicals were obtained from Sigma-Aldrich, except 3-oxo-N-(o-tolyl)butanamide (Fluorochem), 2′,5′-dichloroacetoacetanilide (Alfa Aesar), oxazole-2-carboxaldehyde (Combi-Blocks), and 2-(dimethylamino)acetaldehyde (Combi-Blocks). THF was freshly distilled from sodium/benzophenone. DCM was freshly distilled and stored over 4 Å sieves. TLC was performed on TLC Silica gel 60 F254 plates and visualized at 254 nm or by staining with ninhydrin, phosphomolybdic acid, or KMnO4. Petroleum ether refers to alkanes with bp 60–80 °C. Purification by flash chromatography was carried out using silica gel 60 (0.040–0.063 mm, Merck) or in prepacked columns on a Reveleris X2. 1H and 13C NMR spectra were recorded at 400 (or 500) and 101 MHz at 300 K. Spectra were calibrated relative to residual solvent peaks: 1H NMR (CDCl3): 7.26 ppm; 13C NMR (CDCl3): 77.16 ppm; 1H NMR (DMSO-d6): 2.50 ppm; 13C NMR (DMSO-d6): 39.52 ppm; 1H NMR (CD3OD): 3.31 ppm; 13C NMR (CD3OD): 49.00 ppm; 1H NMR (acetone-d6): 2.05 ppm; 13C NMR (acetone-d6): 29.84 ppm. Purity was determined by HPLC and confirmed by inspection of NMR spectra (1H and 13C NMR). HPLC analysis was performed using a Dionex 120 C18 column (5 μm, 4.6 × 150 mm) or Gemini C18 column (5 μm, 4.6 × 150 mm); flow: 1 mL/min; 10% MeCN in water (0–1 min), 10–100% MeCN in water (1–10 min), and 100% MeCN (10–15 min), with both solvents containing 0.1% formic acid as modifier; UV detection at 254 nm. Purification of enantiomers was performed on a chiral LUX cellulose-1 column. High-resolution mass spectra (HRMS) were recorded on a Bruker micrOTOF-Q II (ESI). None of the test compounds showed patterns associated with PAINS or aggregators by screening in Zinc (http://zinc15.docking.org/patterns/home/). All test compounds were of ≥95% purity with the exception of 41 (82.1%) and 46 (94.3%).
X-ray crystal diffraction data were collected at 100(2) K on a Synergy, Dualflex, AtlasS2 diffractometer using CuKα radiation (λ = 1.54184 Å) and the CrysAlis PRO 1.171.38.43 suite. Using Olex2,33 the structures were solved with the ShelXT34 structure solution program using Intrinsic Phasing and refined with the ShelXL34 refinement package using Least Squares minimization. All nonhydrogen atoms were refined using anisotropic atomic displacement parameters, and hydrogen atoms were inserted at calculated positions using a riding model, except those belonging to the secondary amine and the amide groups. These hydrogen atoms were located in different electron density maps, and their positions were refined. The methyl group C22 in (R,S)-1 is disordered over both ortho positions on the aromatic ring, and the occupancy was refined to 76%:24% occupancy. Crystal data for (R,S)-1: C22H22N2O3 (M = 362.41 g/mol): monoclinic, space group P21/c (no. 14), a = 17.6367(2) Å, b = 7.53560(10) Å, c = 14.3717(2) Å, β = 111.5790(10)°, V = 1776.17(4) Å3, Z = 4, T = 100(2) K, μ(CuKα) = 0.732 mm–1, Dcalc = 1.355 g/cm3, 36 815 reflections were measured (10.788° ≤ 2Θ ≤ 133.202°), and 3132 are unique (Rint = 0.0364, Rsigma = 0.0139), which were used in all calculations. The final R1 was 0.0471 (I > 2σ(I)) and wR2 was 0.1318 (all data). Crystal data for (R)-1: C22H22N2O3 (M = 362.41 g/mol): triclinic, space group P1 (no. 1), a = 7.1817(3) Å, b = 7.4711(3) Å, c = 16.6298(4) Å, α = 87.768(3)°, β = 87.408(3)°, γ = 87.615(3)°, V = 889.98(6) Å3, Z = 2, T = 100(2) K, μ(CuKα) = 0.730 mm–1, Dcalc = 1.352 g/cm3, 16 586 reflections were measured (11.864° ≤ 2Θ ≤ 152.862°), and 6283 are unique (Rint = 0.0454, Rsigma = 0.0384), which were used in all calculations. The final R1 was 0.0371 (I > 2σ(I)) and wR2 was 0.0952 (all data). Flack parameter was 0.04(12). Further details of the X-ray diffraction data, including bond lengths and angles, can be found in the Supporting Information. CCDC 1905506 and 1905507 contain the supplementary crystallographic data for (R,S)-1 and (R)-1, respectively. These data can be obtained free of charge from the Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif.
General Tetrahydroquinoline (THQ) Procedure
A vial was charged with 3-aminocyclohex-2-en-1-one (1 equiv.), aldehyde (1–1.2 equiv.), 3-oxo-N-(o-tolyl)butanamide (1 equiv.), and isopropanol (IPA) (5 mL/mmol). The vial was capped and heated to 80 °C for 1–5 days. Afterward, the reaction mixture was cooled to room temperature, diluted with EtOAc, and concentrated on Celite before purification by flash chromatography.
General THQ-II Procedure
A MW vial was charged with aldehyde (1 equiv.), cyclohexane-1,3-dione (1 equiv.), 3-oxo-N-phenylbutanamide derivative (1 equiv.), and ammonium acetate (1 equiv.). The vial was heated to 150 °C until gas evolution had ceased and the mixture solidified. The residue was purified by flash column chromatography.
2-Methyl-5-oxo-4-phenyl-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (3)
Compound 3 was prepared from benzaldehyde (144 mg, 1.36 mmol), 3-aminocyclohex-2-en-1-one (167 mg, 1.50 mmol), and 3-oxo-N-(o-tolyl)butanamide (287 mg, 1.50 mmol) according to the general THQ procedure to give 257 mg (51%) of a yellow solid (tR = 10.43 min, purity 98.8% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1): 1H NMR (400 MHz, CDCl3) δ 7.77–7.68 (m, 1H), 7.52–7.43 (m, 2H), 7.31 (t, J = 7.0 Hz, 2H), 7.21 (t, J = 6.8 Hz, 1H), 7.13 (t, J = 7.4 Hz, 1H), 7.06–7.00 (m, 1H), 7.00–6.92 (m, 2H), 6.60–6.41 (m, 1H), 5.04 (s, 1H), 2.40 (s, 3H), 2.37–2.23 (m, 4H), 1.98–1.75 (m, 2H), 1.61 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 195.6, 166.4, 150.2, 145.1, 141.8, 136.2, 130.4, 129.2, 128.8, 128.3, 127.4, 126.5, 124.7, 122.9, 112.4, 107.8, 37.5, 37.1, 27.5, 21.0, 18.9, 17.1; ESI-HRMS calcd for C24H25N2O2 (M + H)+ 373.1912; found, 373.1911.
2-Methyl-5-oxo-4-(thiophen-2-yl)-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (4)
Compound 4 was prepared from thiophene-2-carbaldehyde (84 μL, 0.90 mmol), 3-aminocyclohex-2-en-1-one (100 mg, 0.90 mmol), and 3-oxo-N-(o-tolyl)butanamide (172 mg, 0.90 mmol) according to the general procedure THQ to give 132 mg (35%) of a red foam (tR = 10.30 min, purity 99.5% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 5:1): Rf = 0.19 (SiO2, EtOAc/petroleum ether, 5:1); 1H NMR (CDCl3) δ 7.75 (d, J = 7.9 Hz, 1H), 7.42 (s, 1H), 7.31 (s, 1H), 7.16 (dd, J = 5.1, 1.1 Hz, 1H), 7.15–7.09 (m, 1H), 7.08–7.01 (m, 2H), 6.97 (td, J = 7.5, 0.9 Hz, 1H), 6.91 (dd, J = 5.1, 3.5 Hz, 1H), 5.31 (s, 1H), 2.35 (s, 3H), 2.42–2.25 (m, 4H), 1.95–1.87 (m, 1H), 1.86–1.80 (m, 1H), 1.77 (s, 3H); 13C NMR (CDCl3) δ 195.5, 166.4, 151.0, 149.7, 142.5, 136.1, 130.5, 128.9, 127.2, 126.5, 125.3, 125.0, 124.7, 122.8, 111.5, 107.2, 36.9, 32.6, 27.1, 21.0, 18.6, 17.0. ESI-HRMS cald for C22H22N2NaO2S (M + Na)+ 401.1294; found, 401.1285.
4-(3-Bromophenyl)-2-methyl-5-oxo-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (6)
Compound 6 was prepared from 3-bromobenzaldehyde (278 mg, 1.50 mmol), 3-aminocyclohex-2-en-1-one (167 mg, 1.50 mmol), and 3-oxo-N-(o-tolyl)butanamide (287 mg, 1.50 mmol) according to the general THQ procedure to give 128 mg (19%) of a yellow solid (tR = 11.08 min, purity 96.6% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1): 1H NMR (400 MHz, CDCl3) δ 7.71 (d, J = 8.1 Hz, 1H), 7.56 (br s, 1H), 7.44 (d, J = 7.7 Hz, 1H), 7.35 (d, J = 8.0 Hz, 1H), 7.21–7.10 (m, 2H), 7.09–7.03 (m, 1H), 7.03–6.95 (m, 1H), 6.91 (s, 1H), 6.65–6.46 (m, 1H), 5.02 (s, 1H), 2.39 (s, 3H), 2.44–2.24 (m, 4H), 2.00–1.77 (m, 2H), 1.71 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 195.5, 166.2, 150.6, 147.5, 141.9, 136.0, 131.1, 130.7, 130.5, 128.9, 127.1, 126.6, 124.9, 123.4, 123.1, 111.7, 107.3, 37.4, 37.0, 27.5, 20.9, 19.0, 17.3; ESI-HRMS calcd for C24H24BrN2O2 (M + H)+ 451.1016; found, 451.1017.
4-(4-Bromophenyl)-2-methyl-5-oxo-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (7)
Compound 7 was prepared from 4-bromobenzaldehyde (278 mg, 1.50 mmol), 3-aminocyclohex-2-en-1-one (167 mg, 1.50 mmol), and 3-oxo-N-(o-tolyl)butanamide (287 mg, 1.50 mmol) according to the general THQ procedure to give 226 mg (33%) of a yellow solid (tR = 11.14 min, purity 96.5% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1): 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 7.9 Hz, 1H), 7.45–7.38 (m, 2H), 7.38–7.30 (m, 2H), 7.12 (t, J = 7.5 Hz, 1H), 7.09–7.00 (m, 1H), 7.00–6.93 (m, 1H), 6.90 (br s, 1H), 6.52 (br s, 1H), 5.01 (s, 1H), 2.38 (s, 3H), 2.46–2.25 (m, 4H), 1.98–1.76 (m, 2H), 1.71 (s, 3H), 13C NMR (101 MHz, CDCl3) δ 195.5, 166.1, 150.4, 144.1, 141.7, 136.0, 132.2, 130.5, 130.0, 128.8, 126.6, 124.8, 122.9, 121.2, 111.9, 107.4, 37.1, 37.0, 27.5, 21.0, 18.9, 17.2; ESI-HRMS calcd for C24H24BrN2O2 (M + H)+ 451.1016; found, 451.1006.
2-Methyl-5-oxo-N-(o-tolyl)-4-(3-(trifluoromethyl)phenyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (8)
Compound 8 was prepared from 3-(trifluoromethyl)benzaldehyde (87 mg, 0.50 mmol), cyclohexane-1,3-dione (56 mg, 0.50 mmol), 3-oxo-N-(o-tolyl)butanamide (96 mg, 0.50 mmol), and ammonium acetate (39 mg, 0.51) according to the general THQ-II procedure to give 105 mg (48%) of a yellow solid (tR = 11.18 min, purity 97.5% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1): 1H NMR (400 MHz, CDCl3) δ 7.72–7.67 (m, 3H), 7.52–7.40 (m, 2H), 7.17–7.10 (m, 1H), 7.08–7.02 (m, 1H), 7.02–6.94 (m, 1H), 6.85 (br s, 1H), 6.42 (br s, 1H), 5.14 (s, 1H), 2.40 (s, 3H), 2.47–2.25 (m, 4H), 2.00–1.78 (m, 2H), 1.65 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 195.5, 166.1, 150.6, 146.1, 141.8, 135.9, 131.9 (q, J = 1.1 Hz), 131.4 (q, J = 32.1 Hz), 130.5, 129.6, 128.9, 126.7, 125.0, 124.7 (q, J = 3.8 Hz), 124.21 (q, J = 272.5 Hz), 124.19 (q, J = 3.7 Hz), 123.1, 111.7, 107.3, 37.6, 37.0, 27.5, 20.9, 18.9, 17.1; ESI-HRMS calcd for C25H24F3N2O2 (M + H)+ 441.1784; found, 441.1795.
2-Methyl-5-oxo-N-(o-tolyl)-4-(4-(trifluoromethyl)phenyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (9)
Compound 9 was prepared from 4-(trifluoromethyl)benzaldehyde (87 mg, 0.50 mmol), cyclohexane-1,3-dione (56 mg, 0.50 mmol), 3-oxo-N-(o-tolyl)butanamide (95 mg, 0.93 mmol), and ammonium acetate (39 mg, 0.51) according to the general THQ-II procedure to give 85 mg (39%) of a yellow solid (tR = 11.23 min, purity 96.8% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1): 1H NMR (400 MHz, CDCl3) δ 7.66 (d, J = 8.0 Hz, 1H), 7.62–7.53 (m, 4H), 7.13 (t, J = 7.6 Hz, 1H), 7.08–7.03 (m, 1H), 6.99 (t, J = 7.4 Hz, 1H), 6.86 (br s, 1H), 6.58 (br s, 1H), 5.12 (s, 1H), 2.37 (s, 3H), 2.45–2.24 (m, 4H), 1.97–1.77 (m, 2H), 1.67 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 195.6, 166.2, 150.9, 149.1 (q, J = 0.6 Hz), 141.4, 135.8, 130.5, 129.5 (q, J = 32.4 Hz), 129.1, 128.6, 126.6, 126.0 (q, J = 3.7 Hz), 125.1, 124.2 (q, J = 271.8 Hz), 123.2, 111.5, 107.6, 37.7, 37.0, 27.5, 20.9, 18.8, 17.1; ESI-HRMS calcd for C25H23F3N2NaO2 (M + Na)+ 463.1604; found, 463.1622.
2-Methyl-5-oxo-N-(o-tolyl)-4-(p-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (10)
Compound 10 was prepared from 4-methylbenzaldehyde (112 mg, 0.93 mmol), cyclohexane-1,3-dione (104 mg, 0.93 mmol), 3-oxo-N-(o-tolyl)butanamide (178 mg, 0.93 mmol), and ammonium acetate (72 mg, 0.93) according to the general THQ-II procedure to give 254 mg (71%) of a yellow solid (tR = 10.92 min, purity 96.3% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1): 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 8.0 Hz, 1H), 7.36 (d, J = 8.0 Hz, 2H), 7.16–7.09 (m, 3H), 7.05–6.99 (m, 2H), 6.99–6.93 (m, 1H), 6.57 (br s, 1H), 4.99 (s, 1H), 2.38 (s, 3H), 2.41–2.24 (m, 4H), 2.29 (s, 3H), 1.92–1.73 (m, 2H), 1.65 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 195.6, 166.5, 150.2, 142.2, 141.7, 137.0, 136.2, 130.4, 129.9, 128.8, 128.2, 126.5, 124.6, 122.9, 112.5, 107.8, 37.1, 37.1, 27.5, 21.2, 21.0, 18.8, 17.1; ESI-HRMS calcd for C25H27N2O2 (M + H)+ 387.2067; found, 387.2070.
4-(4-Ethylphenyl)-2-methyl-5-oxo-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (11)
Compound 11 was prepared from 4-ethylbenzaldehyde (134 mg, 1.00 mmol), cyclohexane-1,3-dione (112 mg, 1.00 mmol), 3-oxo-N-(o-tolyl)butanamide (191 mg, 1.00 mmol), and ammonium acetate (77 mg, 1.00 mmol) according to the general THQ-II procedure to give 77 mg (19%) of a yellow solid (tR = 11.44 min, purity 95.0% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1): 1H NMR (400 MHz, CDCl3) δ 7.75 (d, J = 7.9 Hz, 1H), 7.39 (d, J = 8.1 Hz, 2H), 7.18–7.09 (m, 3H), 7.04–6.99 (m, 2H), 6.98–6.92 (m, 1H), 6.57 (br s, 1H), 4.99 (s, 1H), 2.59 (q, J = 7.6 Hz, 2H), 2.37 (s, 3H), 2.41–2.24 (m, 4H), 1.92–1.75 (m, 2H), 1.58 (s, 3H), 1.19 (t, J = 7.6 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 195.6, 166.5, 150.2, 143.5, 142.5, 141.7, 136.3, 130.4, 128.7, 128.6, 128.3, 126.5, 124.5, 122.8, 112.5, 107.9, 37.1, 28.6, 27.5, 20.9, 18.8, 17.0, 15.8; ESI-HRMS calcd for C26H29N2O2 (M + H)+ 401.2224; found, 401.2241.
4-(Furan-2-yl)-2-methyl-5-oxo-N-(m-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (13)
2,2,6-Trimethyl-4H-1,3-dioxin-4-one (711 mg, 5.0 mmol) and m-toluidine (536 mg, 5.0 mmol) were dissolved in toluene (1 mL) and heated to 110 °C for 7 h. The reaction was cooled to room temperature, concentrated, and filtered through a small silica plug (EtOAc/petroleum ether, 1:4), and the crude product of 3-oxo-N-(m-tolyl)butanamide was used directly in the next step.
Compound 13 was prepared from furan-2-carbaldehyde (192 mg, 2.00 mmol), 3-aminocyclohex-2-en-1-one (222 mg, 2.00 mmol), and 3-oxo-N-(m-tolyl)butanamide (382 mg, 2.00 mmol) according to the general procedure THQ to give 173 mg (24%) of a brown solid (tR = 10.26 min, purity 98.3% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1): 1H NMR (400 MHz, CDCl3) δ 7.87 (br s, 1H), 7.36–7.30 (m, 2H), 7.19–7.14 (m, 2H), 6.91–6.85 (m, 1H), 6.36 (br s, 1H), 6.30 (dd, J = 3.2, 1.9 Hz, 1H), 6.14 (d, J = 3.2 Hz, 1H), 5.10 (s, 1H), 2.50–2.40 (m, 3H), 2.40–2.32 (m, 1H), 2.32 (s, 6H), 2.04–1.92 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 195.4, 166.1, 157.1, 151.4, 142.2, 142.1, 138.9, 138.4, 128.8, 125.0, 120.9, 117.3, 110.8, 108.8, 106.1, 105.8, 37.0, 31.2, 27.6, 21.6, 21.2, 19.2; ESI-HRMS calcd for C22H23N2O3 (M + H)+ 363.1703; found, 363.1694.
4-(Furan-2-yl)-2-methyl-5-oxo-N-(p-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (14)
2,2,6-Trimethyl-4H-1,3-dioxin-4-one (1422 mg, 10.0 mmol) and p-toluidine (1072 mg, 10.0 mmol) were dissolved in toluene (2 mL) and heated to 110 °C for 6 h. The reaction was cooled to room temperature and concentrated, and the residue was purified by flash chromatography (SiO2, EtOAc/petroleum ether, 1:4) to give 1456 mg (76%) of 3-oxo-N-(p-tolyl)butanamide as a light brown solid that was used directly in the next step.
Compound 14 was prepared from furan-2-carbaldehyde (192 mg, 2.00 mmol), 3-aminocyclohex-2-en-1-one (222 mg, 2.00 mmol), and 3-oxo-N-(p-tolyl)butanamide (382 mg, 2.00 mmol) according to the general procedure THQ to give 292 mg (40%) of a brown solid (tR = 10.23 min, purity 98.3% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1): 1H NMR (400 MHz, CDCl3) δ 7.86 (br s, 1H), 7.34–7.28 (m, 3H), 7.12–7.05 (m, 2H), 6.43 (br s, 1H), 6.29 (dd, J = 3.2, 1.9 Hz, 1H), 6.13 (d, J = 3.2 Hz, 1H), 5.10 (s, 1H), 2.50–2.40 (m, 3H), 2.40–2.32 (m, 1H), 2.30 (s, 3H), 2.29 (s, 3H), 2.03–1.88 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 195.4, 166.1, 157.2, 151.5, 142.0, 135.9, 133.7, 129.5, 120.3, 110.8, 108.7, 106.0, 105.8, 37.0, 31.2, 27.6, 21.2, 21.0, 19.1; ESI-HRMS calcd for C22H22N2NaO3 (M + Na)+ 385.1523; found, 385.1528.
4-(Furan-2-yl)-N-(2-methoxyphenyl)-2-methyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (15)
2,2,6-Trimethyl-4H-1,3-dioxin-4-one (711 mg, 5.0 mmol) and 2-methoxyaniline (616 mg, 5.0 mmol) were dissolved in dry toluene (1 mL) and heated to 110 °C for 6 h. The reaction was cooled to room temperature and filtrated. The solid was washed with ice cold toluene to give 268 mg (26%) of N-(2-methoxyphenyl)-3-oxobutanamide as a light brown solid that was used directly in the next step.
Compound 15 was prepared from furan-2-carbaldehyde (48 mg, 0.50 mmol), cyclohexane-1,3-dione (56 mg, 0.50 mmol), N-(2-methoxyphenyl)-3-oxobutanamide (104 mg, 0.50 mmol), and ammonium acetate (39 mg, 0.51) according to the general procedure THQ-II to give 32 mg (17%) of a yellow solid (tR = 10.27 min, purity 97.7% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1): 1H NMR (400 MHz, CDCl3) δ 8.33 (br s, 1H), 8.30 (dd, J = 8.0, 1.5 Hz, 1H), 7.33–7.28 (m, 1H), 6.99 (td, J = 7.8, 1.6 Hz, 1H), 6.91 (td, J = 7.8, 1.2 Hz, 1H), 6.82 (dd, J = 8.0, 1.1 Hz, 1H), 6.46 (br s, 1H), 6.29 (dd, J = 3.1, 1.9 Hz, 1H), 6.19 (d, J = 3.1 Hz, 1H), 5.13 (s, 1H), 3.78 (s, 3H), 2.50–2.38 (m, 3H), 2.37–2.26 (m, 1H), 2.33 (s, 3H), 2.06–1.88 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 195.4, 166.1, 156.7, 151.4, 148.4, 142.1, 142.0, 128.3, 123.5, 121.0, 120.1, 110.4, 110.1, 109.0, 106.2, 106.1, 55.7, 37.0, 31.2, 27.6, 21.2, 19.1; ESI-HRMS calcd for C22H23N2O4 (M + H)+ 379.1652; found, 379.1671.
4-Isobutyl-2-methyl-5-oxo-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (17)
Compound 17 was prepared from 3-aminocyclohex-2-en-1-one (111 mg, 1.00 mmol), 3-methylbutanal (110 μL, 1.02 mmol), and 3-oxo-N-(o-tolyl)butanamide (191 mg, 1.00 mmol) according to the general procedure THQ to give 96 mg (27%) of a pale yellow solid (tR = 10.74 min, purity 98.0% by HPLC) after purification by flash chromatography (SiO2, EtOAc): 1H NMR (400 MHz, DMSO-d6) δ 9.10 (s, 1H), 8.58 (s, 1H), 7.31–7.03 (m, 4H), 3.83 (t, J = 6.3 Hz, 1H), 2.46–2.14 (m, 7H), 2.06 (s, 3H), 1.98–1.55 (m, 3H), 1.33–0.96 (m, 2H), 0.83 (dd, J = 6.5, 1.9 Hz, 6H); 13C NMR (101 MHz, DMSO-d6) δ 194.1, 168.2, 152.7, 136.8, 134.2, 132.7, 130.1, 125.7, 125.6, 125.0, 111.5, 109.4, 47.0, 36.9, 29.6, 26.5, 23.6, 23.4, 23.0, 20.9, 18.1, 17.0; ESI-HRMS calcd for C22H28N2NaO2 (M + Na)+ 375.2043; found, 375.2052.
N-(2,3-Dimethylphenyl)-4-isobutyl-2-methyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (18)
A dry flask was charged with 2,3-dimethylaniline (0.12 mL, 1.0 mmol), 2,2,6-trimethyl-4H-1,3-dioxin-4-one (0.13 mL, 1.00 mmol), and toluene (0.2 mL) and heated to reflux under argon for 5 h. Afterward, the reaction was cooled to room temperature and concentrated in vacuo. The residue was purified by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1) to give 138 mg (67%) of N-(2,3-dimethylphenyl)-3-oxobutanamide as a beige solid: Rf = 0.20 (SiO2, EtOAc/petroleum ether, 1:1); 1H NMR (400 MHz, CDCl3) δ 9.01 (br s, 1H), 7.55 (d, J = 8.0 Hz, 1H), 7.08 (t, J = 7.8 Hz, 1H), 6.99 (d, J = 7.5 Hz, 1H), 3.60 (s, 2H), 2.31 (s, 3H), 2.29 (s, 3H), 2.18 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 205.7, 163.9, 137.5, 135.3, 128.9, 127.3, 125.9, 121.6, 49.5, 31.3, 20.7, 13.8; ESI-HRMS calcd for C12H15NNaO2 (M + Na+) 228.0995; found, 228.0991.
Compound 18 was prepared from 3-aminocyclohex-2-en-1-one (83 mg, 0.74 mmol), 3-methylbutanal (0.72 mL, 6.6 mmol), and N-(2,3-dimethylphenyl)-3-oxobutanamide (144 mg, 0.70 mmol) according to the general procedure THQ to give 22 mg (9%) of a white solid (tR = 11.08 min, purity 96.2% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1 → 1:0): 1H NMR (400 MHz, CDCl3) δ 7.42 (s, 1H), 7.39 (d, J = 7.9 Hz, 1H), 7.09 (t, J = 7.7 Hz, 1H), 7.00 (d, J = 7.4 Hz, 1H), 6.32 (br s, 1H), 3.92 (t, J = 6.6 Hz, 1H), 2.53–2.45 (m, 1H), 2.42–2.37 (m, 2H), 2.34–2.30 (m, 1H), 2.30 (s, 3H), 2.26 (s, 3H), 2.18 (s, 3H), 2.04–1.90 (m, 2H), 1.64–1.51 (m, 1H), 1.27–1.24 (m, 2H), 0.97 (d, J = 6.5 Hz, 3H), 0.88 (d, J = 6.6 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 196.3, 167.5, 152.0, 139.8, 137.6, 135.7, 130.3, 127.4, 125.9, 122.7, 111.8, 109.8, 77.2, 47.0, 37.2, 29.7, 27.7, 24.1, 23.7, 23.0, 21.2, 20.7, 18.7, 14.3; ESI-HRMS calcd for C23H30N2NaO2 (M + Na+) 389.2199; found, 389.2202.
N-(2-Iodophenyl)-4-isobutyl-2-methyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (19)
A dry flask was charged with 2-iodoaniline (258 mg, 1.18 mmol), 2,2,6-trimethyl-4H-1,3-dioxin-4-one (0.22 mL, 1.20 mmol), and toluene (0.25 mL) and heated to reflux under argon for 22 h. Afterward, the reaction was cooled to room temperature and concentrated in vacuo. The residue was purified by flash chromatography (SiO2, EtOAc/petroleum ether, 1:2) to give 113 mg (31%) of N-(2-iodophenyl)-3-oxobutanamide as a pale yellow solid: Rf = 0.19 (SiO2, EtOAc/petroleum ether, 1:2); 1H NMR (400 MHz, CDCl3) δ 9.16 (br s, 1H), 8.12 (dd, J = 8.2, 1.0 Hz, 1H), 7.80 (dd, J = 8.0, 1.4 Hz, 1H), 7.36–7.29 (m, 1H), 6.88–6.81 (m, 1H), 3.63 (s, 2H), 2.33 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 204.5, 163.8, 139.3, 138.5, 129.1, 126.5, 123.0, 90.2, 50.0, 31.4; ESI-HRMS calcd for C10H10INNaO2 (M + Na+) 325.9648; found, 325.9661.
Compound 19 was prepared from 3-aminocyclohex-2-en-1-one (40 mg, 0.36 mmol), 3-methylbutanal (39 μL, 0.36 mmol), and N-(2-iodophenyl)-3-oxobutanamide (106 mg, 0.35 mmol) according to the general procedure THQ to give 14 mg (9%) of a white solid (tR = 11.84 min, purity 98.4% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1): 1H NMR (400 MHz, CDCl3) δ 8.19 (dd, J = 8.2, 1.4 Hz, 1H), 7.82 (br s, 1H), 7.78 (dd, J = 7.9, 1.3 Hz, 1H), 7.36–7.30 (m, 1H), 6.82 (td, J = 7.6, 1.4 Hz, 1H), 6.12 (br s, 1H), 4.06 (dd, J = 7.9, 5.6 Hz, 1H), 2.53–2.42 (m, 3H), 2.36–2.26 (m, 1H), 2.30 (s, 3H), 2.11–1.93 (m, 2H), 1.60–1.50 (m, 1H), 1.35–1.23 (m, 2H), 0.99 (d, J = 6.5 Hz, 3H), 0.86 (d, J = 6.6 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 196.0, 167.0, 151.3, 140.6, 139.0, 138.9, 129.1, 125.8, 122.6, 112.2, 109.8, 90.8, 46.6, 37.2, 29.6, 27.8, 24.2, 23.8, 23.3, 21.2, 19.1; ESI-HRMS calcd for C21H25IN2NaO2 (M + Na+) 487.0853; found, 487.0875.
N-(2-Chlorophenyl)-4-isobutyl-2-methyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (20)
A dry flask was charged with 2-chloroaniline (0.50 mL, 4.75 mmol), 2,2,6-trimethyl-4H-1,3-dioxin-4-one (0.70 mL, 5.27 mmol), and toluene (1 mL) and heated to reflux under argon for 6 h. Afterward, the reaction was cooled to room temperature and concentrated in vacuo. The crude was dissolved in boiling acetone, and petroleum ether was added until precipitation started, and the solution was stored in the fridge overnight. The precipitate was isolated by filtration, washed with petroleum ether, and dried to give 342 mg (34%) of N-(2-chlorophenyl)-3-oxobutanamide as a beige solid, 90% pure by 1H NMR: Rf = 0.09 (SiO2, acetone/petroleum ether, 1:4); 1H NMR (400 MHz, CDCl3) δ 9.61 (br s, 1H), 8.33 (dd, J = 8.3, 1.3 Hz, 1H), 7.38 (dd, J = 8.0, 1.4 Hz, 1H), 7.28–7.22 (m, 1H), 7.05 (dt, J = 7.9, 1.5 Hz, 1H), 3.64 (s, 2H), 2.34 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 204.9, 163.7, 134.7, 129.3, 127.7, 125.1, 123.6, 122.1, 49.8, 31.4.
Compound 20 was prepared from 3-aminocyclohex-2-en-1-one (111 mg, 1.00 mmol), 3-methylbutanal (0.12 mL, 1.1 mmol), and N-(2-chlorophenyl)-3-oxobutanamide (214 mg, 1.01 mmol) according to the general procedure THQ to give 100 mg (27%) of a yellow foam (tR = 11.55 min, purity 98.7% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1 → 1:0): 1H NMR (400 MHz, CDCl3) δ 8.34 (dd, J = 8.3, 1.5 Hz, 1H), 7.99 (s, 1H), 7.36 (dd, J = 8.0, 1.5 Hz, 1H), 7.25 (dt, J = 8.2, 1.4 Hz, 1H), 7.04–6.98 (m, 1H), 6.36 (s, 1H), 3.96 (t, J = 6.8 Hz, 1H), 2.53–2.41 (m, 3H), 2.36–2.25 (m, 4H), 2.09–1.90 (m, 2H), 1.59–1.47 (m, 1H), 1.30–1.25 (m, 2H), 0.98 (d, J = 6.4 Hz, 3H), 0.85 (d, J = 6.6 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 196.0, 166.8, 151.5, 141.0, 135.2, 129.2, 127.7, 124.4, 123.5, 122.1, 112.1, 109.6, 46.7, 37.2, 29.4, 27.8, 24.1, 23.7, 22.9, 21.2, 19.1; ESI-HRMS calcd for C21H26ClN2O2 (M + H+) 373.1677; found, 373.1662.
N-(2,6-Difluorophenyl)-4-isobutyl-2-methyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (21)
A dry flask was charged with 2,6-difluoroaniline (0.18 mL, 1.67 mmol), 2,2,6-trimethyl-4H-1,3-dioxin-4-one (0.22 mL, 1.66 mmol), and toluene (0.3 mL) and heated to reflux under argon for 22 h. Afterwards the reaction was cooled to room temperature and concentrated in vacuo. The residue was purified by flash chromatography (SiO2, EtOAc/petroleum ether, 2:3) to give 182 mg (51%) of N-(2,6-difluorophenyl)-3-oxobutanamide as a pale yellow solid: Rf = 0.19 (SiO2, EtOAc/petroleum ether, 2:3); 1H NMR (400 MHz, CDCl3) δ 8.81 (br s, 1H), 7.18 (dq, J = 8.4, 6.2 Hz, 1H), 7.01–6.82 (m, 2H), 3.63 (s, 2H), 2.31 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 204.6, 164.5, 157.8 (dd, J = 250.9, 3.4 Hz), 127.9 (t, J = 9.1 Hz), 113.6 (t, J = 16.4 Hz), 111.8 (d, J = 22.1 Hz), 49.1, 30.9; ESI-HRMS calcd for C10H9F2NNaO2 (M + Na+) 236.0494; found, 236.0465.
Compound 21 was prepared from 3-aminocyclohex-2-en-1-one (58 mg, 0.52 mmol), 3-methylbutanal (56 μL, 0.52 mmol), and N-(2,6-difluorophenyl)-3-oxobutanamide (109 mg, 0.51 mmol) according to the general procedure THQ to give 8 mg (4%) of a white solid (tR = 10.50 min, purity 99.9% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 2:1): 1H NMR (400 MHz, CDCl3) δ 7.20 (br s, 1H), 7.19–7.12 (m, 1H), 6.98–6.91 (m, 2H), 6.06 (br s, 1H), 3.91 (t, J = 6.5 Hz, 1H), 2.53–2.40 (m, 3H), 2.35–2.23 (m, 1H), 2.28 (s, 3H), 2.10–1.92 (m, 2H), 1.67–1.54 (m, 1H), 1.31–1.21 (m, 2H), 0.95 (d, J = 6.5 Hz, 3H), 0.89 (d, J = 6.6 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 196.3, 167.1, 158.0 (dd, J = 250.2, 4.3 Hz), 151.5, 141.1, 127.2 (t, J = 9.4 Hz), 114.6 (t, J = 16.1 Hz), 112.4, 111.8 (d, J = 22.6 Hz), 108.6, 47.1, 37.2, 29.7, 27.8, 24.1, 23.5, 23.1, 21.2, 19.0; ESI-HRMS calcd for C21H24F2N2NaO2 (M + Na+) 397.1698; found, 397.1703.
2-Ethyl-4-(furan-2-yl)-5-oxo-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (22)
A dry 5 mL vial under argon atmosphere was charged with o-toluidine (0.21 mL, 1.98 mmol) and methyl 3-oxopentanoate (0.25 mL, 1.99 mmol) and heated to 100 °C for 17 h. The reaction was cooled to room temperature and purified by flash column chromatography (SiO2, EtOAc/petroleum ether, 1:2) to give 112 mg (28%) of 3-oxo-N-(o-tolyl)pentanamide as a yellow solid that was used directly in the next step.
Compound 22 was prepared from furan-2-carbaldehyde (55 μL, 0.66 mmol), 3-aminocyclohex-2-en-1-one (74 mg, 0.67 mmol), and 3-oxo-N-(o-tolyl)pentanamide (132 mg, 0.64 mmol) according to the general procedure THQ to give 56 mg (23%) of an orange foam (tR = 10.37 min, purity 95.7% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:2 → 1:0): 1H NMR (400 MHz, DMSO-d6) δ 9.04 (s, 1H), 8.80 (s, 1H), 7.43 (dd, J = 1.8, 0.9 Hz, 1H), 7.28–7.21 (m, 1H), 7.21–7.02 (m, 3H), 6.27 (dd, J = 3.1, 1.8 Hz, 1H), 5.93 (d, J = 3.1 Hz, 1H), 5.10 (s, 1H), 2.64–2.29 (m, 4H), 2.27–2.21 (m, 2H), 2.10 (s, 3H), 1.97–1.77 (m, 2H), 1.10 (t, J = 7.4 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 193.7, 166.9, 158.4, 153.1, 141.9, 141.0, 136.8, 132.5, 130.1, 125.7, 125.6, 125.0, 110.1, 106.8, 106.0, 104.5, 36.7, 31.9, 26.4, 23.7, 20.8, 17.8, 13.3; ESI-HRMS calcd for C23H24N2NaO3 (M + Na)+ 399.1679; found, 399.1685.
4-(Furan-2-yl)-5-oxo-2-phenyl-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (23)
A dry 5 mL vial under argon atmosphere was charged with o-toluidine (0.21 mL, 1.98 mmol) and ethyl 3-oxo-3-phenylpropanoate (0.35 mL, 2.02 mmol) and heated to 100 °C for 17 h. The reaction was cooled to room temperature and purified by flash column chromatography (SiO2, EtOAc/petroleum ether, 1:4) to give 291 mg (58%) of 3-oxo-3-phenyl-N-(o-tolyl)propanamide as a pale yellow solid that was used directly in the next step.
Compound 23 was prepared from furan-2-carbaldehyde (55 μL, 0.66 mmol), 3-aminocyclohex-2-en-1-one (71 mg, 0.64 mmol), and 3-oxo-3-phenyl-N-(o-tolyl)propanamide (160 mg, 0.63 mmol) according to the general procedure THQ to give 155 mg (58%) of a beige solid (tR = 10.83 min, purity 96.9% by HPLC) after washing the crude with cold EtOAc: 1H NMR (400 MHz, DMSO-d6) δ 9.06 (s, 1H), 8.33 (s, 1H), 7.47–7.41 (m, 6H), 7.23–7.15 (m, 1H), 7.08–7.00 (m, 2H), 6.99–6.92 (m, 1H), 6.30 (dd, J = 3.1, 1.8 Hz, 1H), 6.03 (d, J = 3.2 Hz, 1H), 5.14 (s, 1H), 2.62–2.45 (m, 2H), 2.33–2.23 (m, 2H), 2.00–1.76 (m, 2H), 1.66 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 194.0, 166.5, 158.2, 153.3, 141.0, 137.9, 136.3, 135.0, 131.0, 129.9, 129.1, 128.8, 128.3, 125.6, 124.6, 124.0, 110.2, 109.2, 106.1, 104.6, 36.8, 32.2, 26.4, 20.8, 16.9; ESI-HRMS calcd for C27H24N2NaO3 (M + Na)+ 447.1679; found, 447.1696.
4-(Furan-2-yl)-2-methyl-5-oxo-N-(o-tolyl)-5,6,7,8-tetrahydroquinoline-3-carboxamide (24)
4-(Furan-2-yl)-2-methyl-5-oxo-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (100 mg, 0.28 mmol) and MnO2 (400 mg, 4.60 mmol) were dissolved in CHCl3 (13 mL) and stirred at room temperature for 19 h. The reaction mixture was filtered through a pad of Celite and concentrated in vacuo to give 59 mg (60%) of 24 as a yellow solid (tR = 9.88 min, purity 97.0% by HPLC): 1H NMR (400 MHz, CDCl3) δ 7.59 (d, J = 7.9 Hz, 1H), 7.51 (d, J = 1.1 Hz, 1H), 7.24–7.07 (m, 3H), 6.98 (br s, 1H), 6.58 (dd, J = 3.4, 0.4 Hz, 1H), 6.47 (dd, J = 3.4, 1.8 Hz, 1H), 3.17 (t, J = 6.2 Hz, 2H), 2.73 (s, 3H), 2.71–2.65 (m, 2H), 2.25–2.15 (m, 2H), 2.04 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 196.7, 165.6, 164.7, 159.5, 148.0, 143.6, 136.0, 134.9, 132.3, 130.7, 130.0, 126.9, 126.2, 124.5, 123.7, 111.9, 111.2, 39.8, 33.5, 23.4, 21.5, 17.7; ESI-HRMS calcd for C22H21N2O3 (M + H)+ 361.1547; found, 361.1562.
5-Benzoyl-4-isobutyl-2,6-dimethyl-N-(o-tolyl)-1,4-dihydropyridine-3-carboxamide (25)
A microwave vial was charged with 1-phenylbutane-1,3-dione (163 mg, 1.01 mmol), IPA (2.5 mL), and NH4OAc (83 mg, 1.08 mmol) and stirred at room temperature under an argon atmosphere for two days. Afterward, 3-methylbutanal (0.12 mL, 1.11 mmol) and 3-oxo-N-(o-tolyl)butanamide (192 mg, 1.00 mmol) was added, and the microwave vial was capped and heated to 80 °C. After three days, the reaction was cooled to room temperature, diluted with EtOAc, and concentrated in vacuo on Celite. The residue was purified by flash column chromatography (SiO2, EtOAc/petroleum ether, 1:1) to give 128 mg (32%) of 25 as a bright yellow foam (tR = 12.54 min, 95.1% pure by HPLC): 1H NMR (400 MHz, CDCl3) δ 7.86–7.81 (m, 1H), 7.73–7.66 (m, 2H), 7.51–7.45 (m, 1H), 7.45–7.39 (m, 2H), 7.24–7.11 (m, 3H), 7.08–7.02 (m, 1H), 6.08 (br s, 1H), 3.77–3.69 (m, 1H), 2.37 (s, 3H), 2.15 (s, 3H), 2.02 (s, 3H), 1.67–1.55 (m, 1H), 1.44–1.35 (m, 1H), 1.15−1.08 (m, 1H), 0.78 (d, J = 6.6 Hz, 3H), 0.67 (d, J = 6.5 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 198.3, 166.8, 142.4, 141.9, 140.4, 136.3, 131.7, 130.6, 129.2, 128.7, 128.6, 126.8, 124.9, 123.4, 112.5, 106.7, 46.9, 35.5, 23.7, 23.6, 22.3, 19.2, 18.9, 18.1; ESI-HRMS calcd for C26H30N2NaO2 (M + Na)+ 425.2199; found, 425.2218.
Methyl 4-isobutyl-2,6-dimethyl-5-(o-tolylcarbamoyl)-1,4-dihydropyridine-3-carboxylate (26)
Compound 26 was prepared from methyl (E)-3-aminobut-2-enoate (112 mg, 0.97 mmol), 3-methylbutanal (0.15 mL, 1.39 mmol), and 3-oxo-N-(o-tolyl)butanamide (192 mg, 1.00 mmol) according to the general procedure THQ to give 207 mg (60%) of a pale yellow foam (tR = 12.03 min, 98.6% pure by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1): 1H NMR (400 MHz, acetone-d6) δ 8.21 (br s, 1H), 7.70–7.64 (m, 1H), 7.59 (br s, 1H), 7.22–7.12 (m, 2H), 7.06–7.00 (m, 1H), 3.89 (t, J = 6.7 Hz, 1H), 3.64 (s, 3H), 2.30 (s, 3H), 2.29 (s, 3H), 2.20 (s, 3H), 1.77–1.64 (m, 1H), 1.37–1.26 (m, 1H), 1.25–1.15 (m, 1H), 0.89 (dd, J = 6.5, 3.2 Hz, 6H); 13C NMR (101 MHz, acetone-d6) δ 168.8, 168.7, 147.5, 138.4, 138.3, 131.7, 131.1, 126.9, 125.4, 125.0, 110.1, 101.5, 50.7, 47.9, 34.1, 24.6, 23.7, 23.5, 19.1, 18.5, 17.7; ESI-HRMS calcd for C21H28N2NaO3 (M + Na)+ 379.1992; found, 379.1992.
5-Cyano-4-isobutyl-2,6-dimethyl-N-(o-tolyl)-1,4-dihydropyridine-3-carboxamide (27)
Compound 27 was prepared from (E)-3-aminobut-2-enenitrile (84 mg, 1.02 mmol), 3-methylbutanal (0.15 mL, 1.39 mmol), and 3-oxo-N-(o-tolyl)butanamide (192 mg, 1.01 mmol) according to the general procedure THQ to give 209 mg (64%) of a white solid (tR = 11.55 min, 97.6% pure by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1): 1H NMR (400 MHz, acetone-d6) δ 8.49 (br s, 1H), 7.79 (br s, 1H), 7.56–7.50 (m, 1H), 7.24–7.12 (m, 2H), 7.11–7.04 (m, 1H), 3.61–3.53 (m, 1H), 2.29 (s, 3H), 2.15 (s, 3H), 2.07 (s, 3H), 2.02–1.89 (m, 1H), 1.49–1.32 (m, 2H), 0.93 (dd, J = 15.5, 6.6 Hz, 6H); 13C NMR (101 MHz, acetone-d6) δ 168.3, 148.7, 137.9, 137.4, 132.7, 131.2, 126.9, 125.9, 125.8, 121.4, 108.7, 82.8, 48.3, 35.6, 24.7, 23.9, 22.8, 18.6, 18.1, 17.8; ESI-HRMS calcd for C20H26N3O (M + H)+ 324.2070; found, 324.2086.
Methyl 4-isobutyl-2-methyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxylate (28)
Compound 28 was prepared from 3-aminocyclohex-2-en-1-one (112 mg, 1.01 mmol), 3-methylbutanal (0.15 mL, 1.39 mmol), and methyl 3-oxobutanoate (0.15 mL, 1.39 mmol) according to the general procedure THQ to give 178 mg (64%) of a pale yellow solid (tR = 10.77 min, 96.0% pure by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1 → 1:0): 1H NMR (400 MHz, DMSO-d6) δ 9.02 (s, 1H), 3.83 (t, J = 6.7 Hz, 1H), 3.59 (s, 3H), 2.45–2.33 (m, 2H), 2.20 (s, 3H), 2.29–2.10 (m, 2H), 1.94–1.70 (m, 2H), 1.39–1.27 (m, 1H), 1.00–0.88 (m, 2H), 0.82 (d, J = 6.5 Hz, 3H), 0.78 (d, J = 6.6 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 194.9, 167.7, 151.9, 145.2, 111.2, 103.7, 50.5, 46.9, 36.8, 27.0, 26.2, 23.4, 23.2, 23.0, 20.9, 18.1; ESI-HRMS calcd for C16H23NNaO3 (M + Na)+ 300.1570; found, 300.1570.
3-Benzoyl-4-isobutyl-2-methyl-4,6,7,8-tetrahydroquinolin-5(1H)-one (29)
Compound 29 was prepared from 3-aminocyclohex-2-en-1-one (112 mg, 1.01 mmol), 3-methylbutanal (0.1 mL, 0.91 mmol), and 1-phenylbutane-1,3-dione (163 mg, 1.01 mmol) according to the general procedure THQ to give 100 mg (34%) of a yellow solid (tR = 11.38 min, purity 98.2% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1): 1H NMR (400 MHz, CDCl3) δ 7.70–7.64 (m, 2H), 7.50–7.43 (m, 1H), 7.42–7.35 (m, 2H), 6.88 (br s, 1H), 4.00 (dd, J = 7.7, 5.9 Hz, 1H), 2.52–2.39 (m, 3H), 2.39–2.28 (m, 1H), 2.08–1.89 (m, 2H), 1.97 (s, 3H), 1.52–1.37 (m, 1H), 1.22–1.12 (m, 1H), 1.02–0.91 (m, 1H), 0.73 (d, J = 6.5 Hz, 3H), 0.62 (d, J = 6.5 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 199.0, 196.2, 151.8, 139.4, 139.1, 132.0, 128.9, 128.6, 116.4, 112.5, 47.4, 37.4, 30.7, 27.7, 24.1, 23.4, 22.5, 21.4, 18.3; ESI-HRMS calcd for C21H26NO2 (M + H+) 324.1958; found, 324.1972.
4-(sec-Butyl)-2-methyl-5-oxo-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (30)
Compound 30 was prepared from 3-aminocyclohex-2-en-1-one (112 mg, 1.01 mmol), 2-methylbutanal (108 μL, 1.01 mmol), and 3-oxo-N-(o-tolyl)butanamide (193 mg, 1.01 mmol) according to the general procedure THQ to give 18 mg (5%) of a white solid and mixture of diasteromers (tR = 10.6 min, 97.6% pure by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 4:1) followed by washing with petroleum ether: 1H NMR (400 MHz, acetone-d6) δ 8.40 (br s, 1H), 8.39 (br s, 1H), 7.75 (br s, 1H), 7.69 (br s, 1H), 7.66–7.58 (m, 2H), 7.20 (d, J = 7.5 Hz, 2H), 7.15 (t, J = 7.6 Hz, 2H), 7.04 (t, J = 7.5 Hz, 2H), 4.02 (d, J = 3.4 Hz, 1H), 3.99 (d, J = 3.2 Hz, 1H), 2.54–2.45 (m, 4H), 2.37–2.27 (m, 2H), 2.30 (s, 6H), 2.25–2.15 (m, 2H), 2.19 (d, J = 6.0 Hz, 6H), 2.02–1.87 (m, 4H), 1.57–1.37 (m, 4H), 1.19–1.01 (m, 2H), 0.85 (t, J = 7.3 Hz, 6H), 0.82 (d, J = 1.8 Hz, 3H), 0.80 (d, J = 1.8 Hz, 3H); ESI-HRMS calcd for C22H29N2O2 (M + H)+ 353.2224; found, 353.2239.
4-Cyclohexyl-2-methyl-5-oxo-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (31)
Compound 31 was prepared from 3-aminocyclohex-2-en-1-one (111 mg, 1.00 mmol), cyclohexanecarboxaldehyde (121 μL, 1.00 mmol), and 3-oxo-N-(o-tolyl)butanamide (191 mg, 1.00 mmol) according to the general procedure THQ to give 16 mg (4%) of a brownish solid (tR = 11.18 min, purity 95.7% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:3) followed by trituation with petroleum ether in EtOAc: 1H NMR (CDCl3) δ 7.77 (d, J = 7.9 Hz, 1H), 7.34 (s, 1H), 7.22–7.15 (m, 2H), 7.05 (td, J = 7.5, 0.9 Hz, 1H), 5.96 (s, 1H), 3.81 (d, J = 4.9 Hz, 1H), 2.56–2.49 (m, 1H), 2.46–2.42 (m, 2H), 2.36–2.31 (m, 1H), 2.30 (s, 3H), 2.28 (s, 3H), 2.07–1.95 (m, 2H), 1.74–1.53 (m, 6H), 1.13–0.94 (m, 5H); 13C NMR (CDCl3) δ 196.4, 167.9, 152.1, 139.9, 136.2, 130.6, 129.6, 126.8, 125.0, 123.4, 109.8, 108.0, 46.1, 37.5, 37.3, 29.5, 29.1, 27.8, 26.7, 26.6, 21.2, 18.7, 18.2; ESI-HRMS calcd for C24H31N2O2 (M + H)+ 379.2380; found, 379.2393.
4-Cyclopentyl-2-methyl-5-oxo-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (32)
Compound 32 was prepared from cyclopentanecarbaldehyde (110 μL, 1.03 mmol), 3-aminocyclohex-2-en-1-one (111 mg, 1.00 mmol), and 3-oxo-N-(o-tolyl)butanamide (192 mg, 1.00 mmol) according to the general procedure THQ to give 16 mg (4%) of a white solid (tR = 10.78 min, purity 98.3% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1 → 0:1): 1H NMR (500 MHz, DMSO-d6) δ 9.06 (s, 1H), 8.59 (s, 1H), 7.24 (d, J = 6.8 Hz, 1H), 7.20 (d, J = 7.4 Hz, 1H), 7.17–7.13 (m, 1H), 7.08 (td, J = 7.4, 1.1 Hz, 1H), 3.89 (d, J = 6.0 Hz, 1H), 2.47–2.33 (m, 2H), 2.29–2.12 (m, 2H), 2.20 (s, 3H), 2.09 (s, 3H), 1.95–1.70 (m, 2H), 1.59–1.40 (m, 4H), 1.40–1.28 (m, 2H), 1.27–1.15 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 194.6, 168.8, 153.2, 137.0, 135.2, 132.9, 130.2, 125.8 (two CH), 125.1, 110.0, 108.2, 48.0, 37.1, 34.2, 28.0, 27.9, 26.6, 24.3, 24.2, 20.9, 18.2, 17.1; ESI-HRMS calcd for C23H29N2O2 (M + H)+ 365.2224; found, 365.2234.
4-Cyclopropyl-2-methyl-5-oxo-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (33)
Compound 33 was prepared from 3-aminocyclohex-2-en-1-one (116 mg, 1.05 mmol), cyclopropanecarboxaldehyde (78 μL, 1.05 mmol), and 3-oxo-N-(o-tolyl)butanamide (200 mg, 1.05 mmol) according to the general procedure THQ to give 44 mg (13%) of a white solid (tR = 9.74 min, purity 98.3% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:5): 1H NMR (CDCl3) δ 7.73 (d, J = 7.9 Hz, 1H), 7.48 (s, 1H), 7.23–7.16 (m, 2H), 7.10–7.04 (m, 1H), 5.92 (s, 1H), 3.79 (d, J = 6.7 Hz, 1H), 2.55–2.47 (m, 1H), 2.45–2.40 (m, 2H), 2.38–2.32 (m, 1H), 2.30 (s, 3H), 2.29 (s, 3H), 2.08–1.99 (m, 2H), 1.09–0.96 (m, 1H), 0.47–0.31 (m, 3H), 0.30–0.18 (m, 1H); 13C NMR (CDCl3) δ 196.3, 167.2, 151.1, 140.8, 136.2, 130.7, 129.9, 126.8, 125.1, 123.7, 111.3, 107.1, 37.2, 32.8, 27.7, 21.4, 19.0, 18.3, 2.9, 2.8; ESI-HRMS calcd for C21H24N2NaO2 (M + Na)+ 359.1730; found, 359.1717.
4-Ethyl-2-methyl-5-oxo-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (34)
Compound 34 was prepared from 3-aminocyclohex-2-en-1-one (111 mg, 1.00 mmol), propionaldehyde (0.08 mL, 1.10 mmol), and 3-oxo-N-(o-tolyl)butanamide (193 mg, 1.01 mmol) according to the general procedure THQ to give 46 mg (14%) of a pale yellow foam (tR = 9.65 min, 96.6% pure by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1): 1H NMR (400 MHz, CDCl3) δ 7.73–7.68 (m, 1H), 7.38 (br s, 1H), 7.21–7.15 (m, 2H), 7.09–7.03 (m, 1H), 6.24 (br s, 1H), 3.92 (t, J = 5.1 Hz, 1H), 2.51–2.29 (m, 4H), 2.27 (s, 3H), 2.26 (s, 2H), 2.03–1.91 (m, 2H), 1.62–1.50 (m, 1H), 1.50–1.38 (m, 1H), 0.82 (t, J = 7.5 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 196.1, 167.2, 152.3, 140.1, 136.1, 130.6, 130.1, 126.8, 125.2, 123.8, 110.3, 108.4, 37.2, 33.0, 29.0, 27.6, 21.4, 18.7, 18.1, 9.4; ESI-HRMS calcd for C20H25N2O2 (M + H)+ 325.1911; found, 325.1925.
2-Methyl-5-oxo-4-propyl-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (35)
Compound 35 was prepared from 3-aminocyclohex-2-en-1-one (113 mg, 1.01 mmol), butyraldehyde (91 μL, 1.01 mmol), and 3-oxo-N-(o-tolyl)butanamide (193 mg, 1.01 mmol) according to the general procedure THQ to give 92 mg (27%) of a pale yellow solid (tR = 10.25 min, 98.4% pure by HPLC) after purification by flash chromatography (SiO2, EtOAc) followed by washing with petroleum ether: 1H NMR (400 MHz, acetone-d6) δ 8.36 (br s, 1H), 7.71 (br s, 1H), 7.66–7.58 (m, 1H), 7.23–7.11 (m, 2H), 7.08–7.02 (m, 1H), 3.96 (t, J = 4.5 Hz, 1H), 2.49–2.44 (m, 2H), 2.30 (s, 3H), 2.34–2.16 (m, 4H), 2.19 (s, 3H), 2.01–1.83 (m, 2H), 1.54–1.41 (m, 1H), 1.41–1.23 (m, 3H), 0.87–0.80 (m, 3H); 13C NMR (101 MHz, acetone-d6) δ 195.1, 168.4, 153.3, 153.2, 138.2, 138.1, 132.4, 131.1, 126.8, 125.59, 125.55, 125.4, 110.9, 110.9, 110.84, 110.81, 110.6, 110.5, 39.8, 38.0, 32.8, 27.7, 27.6, 22.2, 18.9, 18.5, 17.82, 17.75, 14.8 (contains rotamers); ESI-HRMS calcd for C21H27N2O2 (M + H)+ 339.2067; found, 339.2077.
2-Methyl-5-oxo-4-butyl-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (36)
Compound 36 was prepared from 3-aminocyclohex-2-en-1-one (111 mg, 1.00 mmol), pentanal (0.15 mL, 1.41 mmol), and 3-oxo-N-(o-tolyl)butanamide (191 mg, 1.00 mmol) according to the general procedure THQ to give 145 mg (41%) of a yellow solid (tR = 10.75 min, 96.5% pure by HPLC) after purification by flash chromatography (SiO2, EtOAc) followed by washing with petroleum ether: 1H NMR (400 MHz, CDCl3) δ 7.72–7.66 (m, 1H), 7.39 (s, 1H), 7.21–7.15 (m, 2H), 7.09–7.03 (m, 1H), 6.44 (s, 1H), 3.91 (t, J = 5.4 Hz, 1H), 2.46 (dt, J = 16.7, 4.6 Hz, 1H), 2.41–2.28 (m, 3H), 2.27 (s, 3H), 2.25 (s, 3H), 2.04–1.90 (m, 2H), 1.56–1.45 (m, 1H), 1.45–1.31 (m, 1H), 1.31–1.17 (m, 4H), 0.84 (t, J = 6.8 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 196.1, 167.2, 152.2, 140.1, 136.1, 130.6, 130.1, 126.7, 125.3, 123.9, 110.9, 108.8, 37.2, 36.5, 32.0, 27.5, 27.3, 23.1, 21.3, 18.7, 18.2, 14.3; ESI-HRMS calcd for C22H29N2O2 (M + H)+ 353.2224; found, 353.2206.
2-Methyl-5-oxo-4-pentyl-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (37)
Compound 37 was prepared from 3-aminocyclohex-2-en-1-one (111 mg, 1.00 mmol), hexanal (0.12 mL, 0.99 mmol), and 3-oxo-N-(o-tolyl)butanamide (193 mg, 1.01 mmol) according to the general procedure THQ to give 105 mg (29%) of a yellow solid (tR = 11.36 min, 96.5% pure by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 2:3) followed by washing with petroleum ether: 1H NMR (400 MHz, acetone-d6) δ 8.40 (br s, 1H), 7.74 (br s, 1H), 7.62–7.56 (m, 1H), 7.22–7.18 (m, 1H), 7.18–7.12 (m, 1H), 7.08–7.02 (m, 1H), 4.00–3.93 (m, 1H), 2.49–2.43 (m, 2H), 2.30 (s, 3H), 2.35–2.20 (m, 2H), 2.18 (s, 3H), 2.03–1.83 (m, 2H), 1.40–1.17 (m, 8H), 0.84 (t, J = 6.9 Hz, 3H); 13C NMR (101 MHz, acetone-d6) δ 195.2, 168.5, 153.3, 153.2, 138.2, 138.1, 132.5, 132.3, 131.1, 126.8, 125.62, 125.60, 125.5, 110.9, 110.6, 38.0, 37.2, 33.1, 32.9, 27.7, 27.6, 25.4, 23.4, 22.2, 18.5, 17.82, 17.75, 14.4 (contains rotamers); ESI-HRMS calcd for C23H31N2O2 (M + H)+ 367.2380; found, 367.2363.
2-Methyl-5-oxo-4-phenethyl-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (38)
Compound 38 was prepared from 3-aminocyclohex-2-en-1-one (111 mg, 1.00 mmol), 3-phenylpropanal (0.15 mL, 1.14 mmol), and 3-oxo-N-(o-tolyl)butanamide (191 mg, 1.00 mmol) according to the general procedure THQ to give 123 mg (31%) of a pale yellow solid (tR = 11.08 min, 97.1% pure by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:2 → 1:1); 1H NMR (400 MHz, CDCl3) δ 7.70–7.63 (m, 1H), 7.33 (br s, 1H), 7.24–7.03 (m, 9H), 6.41 (br s, 1H), 4.04 (t, J = 5.2 Hz, 1H), 2.65–2.56 (m, 2H), 2.48–2.28 (m, 4H), 2.26 (s, 3H), 2.22 (s, 3H), 2.02–1.83 (m, 3H), 1.81–1.68 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 196.1, 167.1, 152.4, 142.6, 139.9, 136.0, 130.6, 130.3, 128.4, 128.3, 126.7, 125.7, 125.4, 124.0, 110.6, 108.7, 38.0, 37.2, 32.0, 31.5, 27.6, 21.3, 18.7, 18.1; ESI-HRMS calcd for C26H29N2O2 (M + H)+ 401.2224; found, 401.2206.
4-(2-(1H-Pyrazol-1-yl)ethyl)-N-(2,5-dichlorophenyl)-2-methyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (40)
Compound 40 was prepared from 3-(1H-pyrazol-1-yl)propanal (62 mg, 0.5 mmol), 3-aminocyclohex-2-en-1-one (56 mg, 0.5 mmol), and N-(2,5-dichlorophenyl)-3-butanamide (123 mg, 0.5 mmol) according to the general procedure THQ to give 8 mg (4%) of a pale gray solid (tR = 10.75 min, purity 98.2% by HPLC) after purification by flash chromatography (Reveleris X2, 12 g, MeOH in DCM, 0% to 10%): 1H NMR (400 MHz, CDCl3) δ 8.36 (d, J = 4.9 Hz, 1H), 8.17 (s, 1H), 7.45–7.40 (m, 1H), 7.29 (d, J = 8.6 Hz, 1H), 7.21 (s, 1H), 702 (dd, J = 8.6, 2.5 Hz, 1H), 6.19 (t, J = 2.1 Hz, 1H), 4.19–4.03 (m, 3H), 2.51–2.39 (m, 3H), 2.37–2.13 (m, 5H), 2.07–1.81 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 196.0, 166.3, 152.6, 141.5, 138.9, 135.8, 133.3, 129.7, 129.5, 124.6, 122.4, 122.0, 110.0, 107.5, 105.3, 48.7, 37.0, 36.5, 29.5, 27.4, 21.1, 18.8; ESI-HRMS calcd for C22H22Cl2N4NaO2 (M + Na)+ 467.1012; found, 467.1005.
2-Methyl-4-(1-methyl-1H-pyrrol-2-yl)-5-oxo-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (41)
Compound 41 was prepared from 1-methyl-1H-pyrrole-2-carbaldehyde (55 μL, 0.51 mmol), 3-aminocyclohex-2-en-1-one (57 mg, 0.51 mmol), and 3-oxo-N-(o-tolyl)butanamide (90 mg, 0.51 mmol) according to the general procedure THQ to give 8 mg (4%) of a yellow foam (tR = 10.17 min, purity 82.1% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 2:1): 1H NMR (400 MHz, DMSO-d6) δ 8.84 (s, 1H), 8.72 (s, 1H), 7.20–7.01 (m, 4H), 6.42–6.37 (m, 1H), 5.80–5.77 (m, 1H), 5.70 (dd, J = 3.5, 1.9 Hz, 1H), 4.89 (s, 1H), 3.61 (s, 3H), 2.48–2.43 (m, 2H), 2.21–2.15 (m, 2H), 2.08 (s, 3H), 1.93–1.70 (m, 2H), 1.89 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 194.1, 167.4, 151.4, 138.7, 136.6, 133.3, 132.5, 130.1, 125.7, 125.4, 125.0, 120.4, 110.4, 109.0, 106.9, 106.1, 36.8, 33.5, 29.7, 26.4, 20.8, 17.6, 17.0; ESI-HRMS calcd for C23H25N3NaO2 (M + Na)+ 398.1839; found, 398.1856.
2-Methyl-4-(5-methylfuran-2-yl)-5-oxo-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (42)
Compound 42 was prepared from 5-methylfuran-2-carbaldehyde (50 μL, 0.50 mmol), 3-aminocyclohex-2-en-1-one (57 mg, 0.51 mmol), and 3-oxo-N-(o-tolyl)butanamide (89 mg, 0.50 mmol) according to the general procedure THQ to give 69 mg (37%) of a pale yellow foam (tR = 10.28 min, purity 99.9% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 2:1): 1H NMR (400 MHz, DMSO-d6) δ 9.03 (s, 1H), 8.81 (s, 1H), 7.27–7.23 (m, 1H), 7.21–7.11 (m, 2H), 7.10–7.04 (m, 1H), 5.88–5.83 (m, 1H), 5.73 (d, J = 3.0 Hz, 1H), 5.02 (s, 1H), 2.54–2.43 (m, 2H), 2.29–2.20 (m, 2H), 2.16 (d, J = 0.7 Hz, 3H), 2.15 (s, 3H), 2.09 (s, 3H), 1.98–1.81 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 193.8, 167.1, 156.8, 152.9, 149.3, 136.9, 135.9, 132.7, 130.1, 125.7, 125.6, 125.0, 107.8, 106.1, 105.9, 105.0, 36.8, 31.5, 26.4, 20.9, 17.9, 17.2, 13.4; ESI-HRMS calcd for C23H24N2NaO3 (M + Na)+ 399.1679; found, 399.1696.
2-Methyl-4-(4-methylthiophen-2-yl)-5-oxo-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (43)
Compound 43 was prepared from 4-methylthiophene-2-carbaldehyde (65 μL, 0.53 mmol), 3-aminocyclohex-2-en-1-one (56 mg, 0.50 mmol), and 3-oxo-N-(o-tolyl)butanamide (89 mg, 0.50 mmol) according to the general procedure THQ to give 66 mg (34%) of a pale orange foam (tR = 10.89 min, purity 98.6% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 2:1): 1H NMR (400 MHz, DMSO-d6) δ 8.93 (s, 1H), 8.90 (s, 1H), 7.32–7.25 (m, 1H), 7.14 (dt, J = 9.1, 4.6 Hz, 2H), 7.05 (td, J = 7.4, 1.3 Hz, 1H), 6.80–6.76 (m, 1H), 6.60 (s, 1H), 5.19 (s, 1H), 2.52–2.44 (m, 2H), 2.28–2.21 (m, 2H), 2.15 (s, 3H), 2.10 (d, J = 0.9 Hz, 3H), 2.02 (s, 3H), 1.96–1.75 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 193.9, 166.9, 152.0, 151.1, 136.7, 136.2, 136.1, 132.3, 130.1, 125.7, 125.4, 125.3, 125.0, 118.8, 109.4, 108.9, 36.7, 32.7, 26.3, 20.8, 17.7, 17.3, 15.5; ESI-HRMS calcd for C23H24N2NaO2S (M + Na)+ 415.1451; found, 415.1457.
4-(5-Bromofuran-2-yl)-2-methyl-5-oxo-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (44)
Compound 44 was prepared from 5-bromofuran-2-carbaldehyde (88 mg, 0.50 mmol), 3-aminocyclohex-2-en-1-one (56 mg, 0.50 mmol), and 3-oxo-N-(o-tolyl)butanamide (89 mg, 0.50 mmol) according to the general procedure THQ to give 84 mg (38%) of a pale yellow solid (tR = 10.59 min, purity 99.9% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 2:1): 1H NMR (400 MHz, DMSO-d6) δ 9.18 (s, 1H), 8.88 (s, 1H), 7.27–7.04 (m, 4H), 6.35 (d, J = 3.2 Hz, 1H), 5.94 (dd, J = 3.3, 0.7 Hz, 1H), 5.09 (s, 1H), 2.55–2.42 (m, 2H), 2.30–2.19 (m, 2H), 2.14 (s, 3H), 2.09 (s, 3H), 1.98–1.81 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 193.8, 166.9, 160.8, 153.4, 136.7, 135.7, 133.1, 130.1, 126.0, 125.7, 125.3, 118.3, 112.0, 107.4, 107.3, 105.0, 36.7, 32.1, 26.4, 20.8, 18.0, 17.2; ESI-HRMS calcd for C22H21BrN2NaO3 (M + Na)+ 463.0628; found, 463.0617.
4-(Benzofuran-2-yl)-2-methyl-5-oxo-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (45)
Compound 45 was prepared from benzofuran-2-carbaldehyde (65 μL, 0.54 mmol), 3-aminocyclohex-2-en-1-one (57 mg, 0.51 mmol), and 3-oxo-N-(o-tolyl)butanamide (89 mg, 0.50 mmol) according to the general procedure THQ to give 67 mg (32%) of a pale yellow foam (tR = 10.89 min, purity 96.1% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 2:1): 1H NMR (400 MHz, DMSO-d6) δ 9.22 (s, 1H), 8.93 (s, 1H), 7.52–7.38 (m, 2H), 7.27–7.04 (m, 6H), 6.38 (s, 1H), 5.26 (s, 1H), 2.57–2.48 (m, 2H), 2.32–2.25 (m, 2H), 2.12 (s, 3H), 2.11 (s, 3H), 1.98–1.88 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 193.9, 167.0, 161.6, 154.0, 153.6, 136.7, 135.9, 133.0, 130.1, 128.5, 125.9, 125.7, 125.2, 123.1, 122.4, 120.4, 110.7, 107.4, 105.1, 101.2, 36.7, 32.4, 26.5, 20.9, 17.9, 17.2; ESI-HRMS calcd for C26H24N2NaO3 (M + Na)+ 435.1679; found, 435.1686.
4-(Benzo[b]thiophen-3-yl)-2-methyl-5-oxo-N-(o-tolyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (46)
Compound 46 was prepared from benzo[b]thiophene-3-carbaldehyde (83 mg, 0.51 mmol), 3-aminocyclohex-2-en-1-one (56 mg, 0.50 mmol) and 3-oxo-N-(o-tolyl)butanamide (89 mg, 0.50 mmol) according to the general procedure THQ to give 60 mg (28%) of a pale yellow foam (tR = 11.10 min, purity 94.3% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 2:1): 1H NMR (400 MHz, DMSO-d6) δ 8.96 (s, 1H), 8.91 (s, 1H), 8.09–8.03 (m, 1H), 7.91–7.85 (m, 1H), 7.35–7.27 (m, 2H), 7.24 (s, 1H), 7.11–6.98 (m, 5H), 5.39 (s, 1H), 2.59–2.46 (m, 2H), 2.27–2.15 (m, 2H), 2.11 (s, 3H), 1.97–1.79 (m, 2H), 1.77 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 193.9, 167.3, 152.7, 142.4, 139.9, 137.8, 136.5, 133.9, 132.6, 130.0, 125.60, 125.58, 125.1, 123.7, 123.6, 123.5, 123.0, 122.5, 110.5, 108.2, 36.8, 32.1, 26.5, 20.8, 17.5, 17.0; ESI-HRMS calcd for C26H24N2NaO2S (M + Na)+ 451.1451; found, 451.1429.
N-(2,5-Dichlorophenyl)-2-methyl-5-oxo-4-(thiazol-2-yl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (47)
Compound 47 was prepared from thiazole-2-carbaldehyde (50 μL, 0.57 mmol), 3-aminocyclohex-2-en-1-one (56 mg, 0.50 mmol), and N-(2,5-dichlorophenyl)-3-oxobutanamide (123 mg, 0.50 mmol) according to the general procedure THQ to give 95 mg (44%) of a pale yellow solid (tR = 11.68 min, purity 98.2% by HPLC) after purification by filtration followed by washing with IPA: 1H NMR (500 MHz, DMSO-d6) δ 10.76 (s, 1H), 9.52 (s, 1H), 8.11 (d, J = 2.6 Hz, 1H), 7.76 (d, J = 3.3 Hz, 1H), 7.56 (d, J = 3.3 Hz, 1H), 7.54 (d, J = 8.6 Hz, 1H), 7.21 (dd, J = 8.6, 2.6 Hz, 1H), 5.23 (s, 1H), 2.61 (dd, J = 7.5, 4.7 Hz, 2H), 2.34 (dd, J = 8.0, 5.0 Hz, 2H), 2.13 (s, 3H), 2.08–1.88 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 194.2, 177.4, 166.2, 154.0, 143.2, 142.4, 137.2, 131.5, 130.7, 124.6, 123.4, 123.0, 119.9, 107.4, 105.7, 36.6, 34.1, 26.2, 20.8, 17.9; ESI-HRMS calcd for C20H18Cl2N3O2S (M + H)+ 434.0491; found, 434.0510.
N-(2,5-Dichlorophenyl)-2-methyl-5-oxo-4-(thiazol-5-yl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (48)
Compound 48 was prepared from thiazole-5-carbaldehyde (50 μL, 0.58 mmol), 3-aminocyclohex-2-en-1-one (56 mg, 0.50 mmol), and N-(2,5-dichlorophenyl)-3-oxobutanamide (123 mg, 0.50 mmol) according to the general procedure THQ to give 138 mg (63%) of a white solid (tR = 10.51 min, purity 96.2% by HPLC) after purification by filtration followed by washing with IPA: 1H NMR (500 MHz, DMSO-d6) δ 9.21 (s, 1H), 9.19 (s, 1H), 8.82 (s, 1H), 7.76 (d, J = 2.6 Hz, 1H), 7.57 (s, 1H), 7.50 (d, J = 8.6 Hz, 1H), 7.24 (dd, J = 8.6, 2.6 Hz, 1H), 5.30 (s, 1H), 2.49–2.46 (m, 2H), 2.30–2.24 (m, 2H), 2.23 (s, 3H), 1.96–1.74 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 194.0, 166.5, 152.9, 152.3, 144.7, 140.1, 139.4, 136.5, 131.4, 130.7, 125.6, 125.4, 125.0, 109.0, 106.8, 36.6, 30.0, 26.2, 20.8, 17.7; ESI-HRMS calcd for C20H18Cl2N3O2S (M + H)+ 434.0491; found, 434.0508.
N-(2,5-Dichlorophenyl)-2-methyl-5-oxo-4-(thiazol-4-yl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (49)
Compound 49 was prepared from thiazole-4-carbaldehyde (58 mg, 0.51 mmol), 3-aminocyclohex-2-en-1-one (56 mg, 0.50 mmol), and N-(2,5-dichlorophenyl)-3-oxobutanamide (124 mg, 0.50 mmol) according to the general procedure THQ to give 130 mg (60%) of a white solid (tR = 11.31 min, purity 98.3% by HPLC) after purification by filtration followed by washing with IPA: 1H NMR (500 MHz, DMSO-d6) δ 10.92 (s, 1H), 9.32 (s, 1H), 9.14 (d, J = 2.1 Hz, 1H), 8.16 (d, J = 2.5 Hz, 1H), 7.54 (d, J = 8.6 Hz, 1H), 7.19 (dd, J = 8.6, 2.4 Hz, 1H), 7.11 (d, J = 1.3 Hz, 1H), 5.04 (s, 1H), 2.65–2.53 (m, 2H), 2.37–2.25 (m, 2H), 2.10 (s, 3H), 2.07–1.95 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 194.3, 166.7, 159.6, 154.8, 153.5, 143.8, 137.4, 131.5, 130.7, 124.2, 123.0, 122.7, 114.1, 107.3, 106.0, 36.7, 32.3, 26.3, 20.8, 18.0; ESI-HRMS calcd for C20H18Cl2N3O2S (M + H)+ 434.0491; found, 434.0513.
N-(2,5-Dichlorophenyl)-2-methyl-4-(oxazol-4-yl)-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (50)
Compound 50 was prepared from oxazole-4-carbaldehyde (49 mg, 0.50 mmol), 3-aminocyclohex-2-en-1-one (56 mg, 0.50 mmol), and N-(2,5-dichlorophenyl)-3-oxobutanamide (123 mg, 0.50 mmol) according to the general procedure THQ to give 77 mg (37%) of a brown solid (tR = 10.77 min, purity 97.8% by HPLC) after purification by filtration followed by washing with IPA: 1H NMR (500 MHz, DMSO-d6) δ 10.11 (s, 1H), 9.24 (s, 1H), 8.34 (d, J = 1.0 Hz, 1H), 8.10 (d, J = 2.6 Hz, 1H), 7.69–7.62 (m, 1H), 7.53 (d, J = 8.6 Hz, 1H), 7.20 (dd, J = 8.6, 2.6 Hz, 1H), 4.83 (s, 1H), 2.58–2.44 (m, 2H), 2.33–2.20 (m, 2H), 2.12 (s, 3H), 2.04–1.88 (m, 2H); 13C NMR (101 MHz, DMSO-d6) δ 194.1, 166.5, 153.4, 152.2, 143.1, 142.8, 137.1, 135.0, 131.5, 130.7, 124.6, 123.4, 123.3, 106.3, 105.3, 36.6, 28.4, 26.3, 20.7, 17.9; ESI-HRMS calcd for C20H18Cl2N3O3 (M + H)+ 418.0720; found, 418.0733.
N-(2,5-Dichlorophenyl)-2-methyl-4-(oxazol-2-yl)-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (51)
Compound 51 was prepared from oxazole-2-carbaldehyde (49 mg, 0.50 mmol), 3-aminocyclohex-2-en-1-one (56 mg, 0.50 mmol), and N-(2,5-dichlorophenyl)-3-butanamide (123 mg, 0.50 mmol) according to the general procedure THQ to give 12 mg (6%) of a pale yellow solid (tR = 10.43 min, purity 99.0% by HPLC) after purification by flash chromatography (Reveleris X2, 12 g, EtOAc in petroleum ether, 0 to 100%): 1H NMR (400 MHz, CDCl3) δ 9.69 (s, 1H), 9.23 (s, 1H), 7.91 (d, J = 0.7 Hz, 1H), 7.88 (d, J = 2.5 Hz, 1H), 7.52 (d, J = 8.6 Hz, 1H), 7.24 (dd, J = 8.6, 2.6 Hz, 1H), 7.08 (d, J = 0.7 Hz, 1H), 5.19 (s, 1H), 2.58–2.47 (m, 2H), 2.34–2.24 (m, 2H), 2.15 (s, 3H), 2.03–1.81 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 194.1, 166.4, 165.7, 153.3, 141.1, 139.0, 136.8, 131.4, 130.7, 126.7, 125.2, 124.6, 104.9, 103.9, 36.5, 31.8, 26.3, 20.9, 17.7; ESI-HRMS calcd for C20H17Cl2N3NaO3 (M + Na)+ 440.0539; found, 440.0539.
4-(Aminomethyl)-N-(2,5-dichlorophenyl)-2-methyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide Hydrochloride (52)
Compound 52 was prepared from tert-butyl ((3-((2,5-dichlorophenyl)carbamoyl)-2-methyl-5-oxo-1,4,5,6,7,8-hexahydroquinolin-4-yl)methyl)carbamate (8 mg, 17 μmol) which was dissolved in 1 mL of 4 M HCl in 1,4-dioxane at 0 °C. The mixture was stirred overnight at room temperature and concentrated to give 7 mg (quant.) of a yellow solid (tR = 7.00 min, purity 96.4% by HPLC): 1H NMR (400 MHz, DMSO-d6) δ 9.75 (s, 1H), 9.15 (s, 1H), 7.86 (d, J = 2.5 Hz, 1H), 7.78 (s, 2H), 7.55 (d, J = 8.7 Hz, 1H), 7.30 (dd, J = 8.6, 2.6 Hz, 1H), 4.00 (t, J = 4.2 Hz, 1H), 2.80–2.69 (m, 1H), 2.64–2.54 (m, 1H), 2.46–2.21 (m, 4H), 2.18 (s, 3H), 2.06–1.97 (m, 1H), 1.94–1.83 (m, 1H); 13C NMR (101 MHz, DMSO-d6) δ 195.1, 167.3, 155.2, 139.6, 136.3, 131.3, 130.7, 126.08, 126.07, 125.8, 104.3, 104.2, 42.9, 36.6, 30.9, 26.4, 20.4, 17.8; ESI-HRMS calcd for C18H20Cl2N3O2 (M + H)+ 380.0927; found, 380.0925.
N-(2,5-Dichlorophenyl)-2-methyl-5-oxo-4-(piperidin-4-ylmethyl)-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide Hydrochloride (53)
Compound 53 was prepared from tert-butyl 4-((3-((2,5-dichlorophenyl)carbamoyl)-2-methyl-5-oxo-1,4,5,6,7,8-hexahydroquinolin-4-yl)methyl)piperidine-1-carboxylate (30 mg, 55 μmol) which was dissolved in 2 mL of 4 M HCl in 1,4-dioxane at 0 °C. The mixture was stirred overnight at room temperature and concentrated to give 26 mg (quant.) of a yellow solid (tR = 7.27 min, purity 95.1% by HPLC): 1H NMR (400 MHz, DMSO-d6) δ 9.23 (s, 1H), 9.01 (s, 1H), 8.82 (d, J = 9.6 Hz, 1H), 8.62–8.48 (m, 1H), 7.80 (d, J = 2.5 Hz, 1H), 7.54 (d, J = 8.6 Hz, 1H), 7.26 (dd, J = 8.6, 2.6 Hz, 1H), 3.85–3.77 (m, 2H), 3.24–3.10 (m, 2H), 2.75–2.59 (m, 2H), 2.53–2.36 (m, 2H), 2.35–2.18 (m, 2H), 2.15 (s, 3H), 2.07–1.68 (m, 4H), 1.52–1.38 (m, 2H), 1.32–1.06 (m, 4H); 13C NMR (101 MHz, DMSO-d6) δ 194.6, 167.5, 152.9, 138.4, 136.7, 131.3, 130.7, 125.51, 125.50, 125.0, 109.3, 108.8, 43.6, 43.1, 43.0, 36.8, 29.2, 28.8, 28.4, 28.1, 26.4, 20.8, 17.5; ESI-HRMS calcd for C23H28Cl2N3O2 (M + Na)+ 448.1553; found, 448.1568.
N-(2,5-Dichlorophenyl)-4-((2-(diethylamino)ethoxy)methyl)-2-methyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (54)
2-(Diethylamino)ethan-1-ol (500 mg, 4.27 mmol) was added dropwise to a suspension of NaH (266 mg, 50%) in 5 mL of dry THF. The mixture was heated at 50 °C for 10 min, before dropwise addition of 2-bromo-1,1-diethoxyethane (0.77 mL, 5.12 mmol). The mixture was afterward heated to 100 °C overnight. The reaction mixture was cooled down, quenched with water, and extracted with EtOAc (×3). The organic phases were combined, washed with brine, dried over Na2SO4, and concentrated. The crude 2-(2,2-diethoxyethoxy)-N,N-diethylethan-1-amine was used directly in the next step.
N,N-Diethyl-2-(2-oxoethoxy)ethan-1-aminium 2,2,2-trifluoroacetate was prepared from 2-(2,2-diethoxyethoxy)-N,N-diethylethan-1-amine (730 mg, 3.13 mmol) and TFA (5 mL), and the mixture was stirred at rt overnight. Afterward, the reaction mixture was concentrated and dried, and the crude was used in the next step without any further purification: 1H NMR (400 MHz, CDCl3) δ 9.61 (s, 1H), 9.18 (s, 1H), 4.31 (s, 2H), 3.93–3.86 (m 2H), 3.45–3.25 (m, 6H), 1.37 (t, J = 7.3 Hz, 6H); ESI-HRMS calcd for C8H18NO2 (M + H)+ 160.1332; found, 160.1325.
Compound 54 was prepared from N,N-diethyl-2-(2-oxoethoxy)ethan-1-aminium 2,2,2-trifluoroacetate (138 mg, 0.51 mmol), 3-aminocyclohex-2-en-1-one (56 mg, 0.51 mmol), and N-(2,5-dichlorophenyl)-3-butanamide (125 mg, 0.51 mmol) according to the general procedure THQ to give 15 mg (6%) of a pale yellow solid (tR = 7.38 min, purity 99.4% by HPLC) after purification by flash chromatography (SiO2, MeOH in EtOAc, 0% to 8%): 1H NMR (400 MHz, CDCl3) δ 9.21 (s, 1H), 8.34 (d, J = 2.5 Hz, 1H), 7.28 (d, J = 8.6 Hz, 1H), 6.99 (dd, J = 8.6, 2.5 Hz, 1H), 6.21 (s, 1H), 4.18–4.11 (m, 1H), 3.67–3.52 (m, 2H), 3.31–3.22 (m, 2H), 2.69–2.62 (m, 2H), 2.59–2.40 (m, 8H), 2.37–2.25 (m, 5H), 2.10–2.91 (m, 3H), 0.97 (t, J = 7.2 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 196.6, 167.3, 160.5, 155.1, 143.1, 136.3, 133.6, 130.0, 124.4, 121.7, 121.3, 107.2, 106.4, 74.9, 65.6, 56.7, 56.3, 51.5, 48.6, 47.9, 47.8, 36.9, 32.6, 27.3, 21.3, 18.9, 8.9; ESI-HRMS calcd for C24H32Cl2N3O2 (M + Na)+ 480.1815; found, 480.1828.
tert-Butyl((3-((2,5-dichlorophenyl)carbamoyl)-2-methyl-5-oxo-1,4,5,6,7,8-hexahydroquinolin-4-yl)methyl)carbamate (55)
Compound 55 was prepared from tert-butyl (2-oxoethyl)carbamate (80 mg, 0.50 mmol), 3-aminocyclohex-2-en-1-one (56 mg, 0.50 mmol), and N-(2,5-dichlorophenyl)-3-oxobutanamide (125 mg, 0.51 mmol) according to the general procedure THQ to give 41 mg (17%) of a white solid (tR = 11.76 min, purity 97.2% by HPLC) after purification by flash chromatography (SiO2, EtOAc): 1H NMR (400 MHz, CD3OD) δ 8.13 (d, J = 1.4 Hz, 1H), 7.45 (d, J = 8.6 Hz, 1H), 7.19 (dd, J = 8.6, 2.4 Hz, 1H), 4.07–3.94 (m, 1H), 3.16–2.93 (m, 2H), 2.60–2.25 (m, 4H), 2.16 (s, 3H), 2.12–1.94 (m, 2H), 1.41 (s, 9H); 13C NMR (101 MHz, CD3OD) δ 198.9, 170.5, 159.0, 157.4, 140.0, 137.4, 133.9, 131.5, 126.8, 125.9, 125.6, 108.9, 108.2, 79.9, 45.5, 37.8, 35.1, 28.8, 27.9, 22.2, 17.7; ESI-HRMS calcd for C23H28Cl2N3O4 (M + H)+ 480.1451; found, 480.1445.
tert-Butyl 4-((3-((2,5-dichlorophenyl)carbamoyl)-2-methyl-5-oxo-1,4,5,6,7,8-hexahydroquinolin-4-yl)methyl)piperidine-1-carboxylate (56)
Compound 56 was prepared from tert-butyl 4-(2-oxoethyl)piperidine-1-carboxylate (115 mg, 0.50 mmol), 3-aminocyclohex-2-en-1-one (113 mg, 1.02 mmol), and N-(2,5-dichlorophenyl)-3-oxobutanamide (125 mg, 0.51 mmol) according to the general procedure THQ to give 142 mg (52%) of a white solid (tR = 13.21 min, purity 99.1% by HPLC) after purification by flash chromatography (SiO2, EtOAc): 1H NMR (400 MHz, acetone-d6) δ 8.50 (d, J = 2.5 Hz, 0.3H), 8.49 (d, J = 2.5 Hz, 0.7H), 8.39 (br s, 1H), 8.15 (br s, 1H), 7.49 (d, J = 8.6 Hz, 1H), 7.15 (dd, J = 8.6, 2.6 Hz, 1H), 4.05–3.88 (m, 3H), 2.65–2.43 (m, 4H), 2.30 (s, 3H), 2.41–2.18 (m, 2H), 2.03–1.87 (m, 3H), 1.60 (d, J = 13.0 Hz, 1H), 1.40 (s, 9H), 1.46–1.17 (m, 3H), 1.06–0.90 (m, 2H); 13C NMR (101 MHz, acetone-d6) δ 195.4, 167.7, 155.1, 152.8, 152.7, 142.4, 137.7, 133.5, 131.1, 124.9, 124.8, 122.7, 122.6, 111.7, 109.2, 109.1, 79.0, 45.1, 44.7 (br s), 37.8, 33.8, 33.3, 32.8, 28.6, 27.64, 27.56, 22.0, 18.5, 18.4 (contains rotamers); ESI-HRMS calcd for C28H35Cl2N3NaO4 (M + Na)+ 570.1897; found, 570.1880.
N-(2,5-Dichlorophenyl)-2-methyl-5-oxo-4-propyl-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (57)
Compound 57 was prepared from butyraldehyde (65 μL, 0.72 mmol), 3-aminocyclohex-2-en-1-one (70 mg, 0.63 mmol), and N-(2,5-dichlorophenyl)-3-oxobutanamide (155 mg, 0.63 mmol) according to the general procedure THQ to give 85 mg (34%) of a pale yellow solid (tR = 12.34 min, purity 96.1% by HPLC) after purification by flash chromatography (SiO2, EtOAc/petroleum ether, 1:1): 1H NMR (500 MHz, DMSO-d6) δ 9.14 (s, 1H), 8.72 (s, 1H), 7.80 (d, J = 2.5 Hz, 1H), 7.53 (d, J = 8.6 Hz, 1H), 7.25 (dd, J = 8.6, 2.5 Hz, 1H), 3.85 (t, J = 4.7 Hz, 1H), 2.44–2.33 (m, 2H), 2.30–2.15 (m, 2H), 2.12 (s, 3H), 1.94–1.77 (m, 2H), 1.39–1.12 (m, 4H), 0.79 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, DMSO-d6) δ 194.5, 167.5, 152.8, 138.8, 136.7, 131.4, 130.6, 125.51, 125.46, 125.1, 108.9, 108.1, 38.6, 36.9, 31.1, 26.3, 21.0, 17.6, 17.5, 14.3; ESI-HRMS calcd for C20H23Cl2N2O2 (M + H)+ 393.1131; found, 393.1112.
N-(2-Chlorophenyl)-2-methyl-5-oxo-4-propyl-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (58)
Compound 58 was prepared from butyraldehyde (36 mg, 0.50 mmol), 3-aminocyclohex-2-en-1-one (55 mg, 0.50 mmol), and N-(2-chlorophenyl)-3-oxobutanamide (105 mg, 0.50 mmol) according to the general procedure THQ to give 38 mg (21%) of a pale yellow solid (tR = 11.07 min, purity 97.3% by HPLC) after purification by flash chromatography (Reveleris X2, 24 g, EtOAc/petroleum ether, 3:2): 1H NMR (400 MHz, CDCl3) δ 8.34 (dd, J = 1.5, 8.3 Hz, 1H), 8.03 (s, 1H), 7.37 (dd, J = 1.5, 8.0 Hz, 1H), 7.29–7.23 (m, 1H), 7.05–6.99 (m, 1H), 6.42 (s, 1H), 3.96 (t, J = 5.5 Hz, 1H), 2.53–2.40 (m, 3H), 2.37–2.27 (m, 4H), 2.08–1.93 (m, 2H), 1.64–1.20 (m, 4H), 0.86 (t, J = 7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 195.9, 166.7, 151.7, 140.9, 135.1, 129.1, 127.5, 124.3, 123.4, 122.0, 111.2, 108.6, 39.0, 37.1, 31.5, 27.5, 21.2, 18.9, 18.3, 14.3; ESI-HRMS calcd for C20H24ClN2O2 (M + Na)+ 359.1521; found, 359.1537.
N-(3-Chlorophenyl)-2-methyl-5-oxo-4-propyl-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (59)
N-(3-Chlorophenyl)-3-oxobutanamide was prepared from 2-chloroaniline (1.10 g, 8.61 mmol) and 2 mL of methyl acetoacetate. The mixture was stirred at 110 °C overnight, cooled down, and dry loaded on silica. Purification by flash chromatography (Reveleris X2, 12 g, EtOAc in petroleum ether, 0 to 100%) gave 536 mg (29%) of an off-white solid: 1H NMR (400 MHz, CDCl3) δ 9.28 (s, 1H), 7.68 (t, J = 2.0 Hz, 1H), 7.40–7.34 (m, 1H), 7.28–7.20 (m, 1H), 7.12–7.06 (m, 1H), 3.59 (s, 2H), 2.32 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 205.3, 163.5, 138.6, 134.7, 130.0, 124.6, 120.3, 118.1, 49.4, 31.3; ESI-HRMS calcd for C10H10ClNNaO2 (M + Na)+ 234.0292; found, 234.0302.
Compound 59 was prepared from butyraldehyde (36 mg, 0.50 mmol), 3-aminocyclohex-2-en-1-one (55 mg, 0.50 mmol), and N-(3-chlorophenyl)-3-oxobutanamide (105 mg, 0.50 mmol) according to the general procedure THQ to give 25 mg (14%) of a pale yellow solid (tR = 11.27 min, purity 97.5% by HPLC) after purification by flash chromatography (Reveleris X2, 24 g, MeOH in DCM, 0 to 10%): 1H NMR (400 MHz, CDCl3) δ 7.88 (s, 1H), 7.74 (t, J = 2 Hz, 1H), 7.35–7.30 (m, 1H), 7.22 (t, J = 7.1 Hz, 1H), 7.08–7.03 (m, 1H), 6.18 (s, 1H), 3.88 (t, J = 5.9 Hz, 1H), 2.52–2.39 (m, 3H), 2.35–2.26 (m, 1H), 2.25 (s, 3H), 2.07–1.94 (m, 2H), 1.49–1.19 (m, 4H), 0.83 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 196.2, 167.3, 151.9, 140.0, 139.5, 134.6, 129.8, 124.0, 120.4, 118.2, 111.0, 108.9, 39.1, 37.1, 31.7, 27.6, 21.2, 18.6, 18.2, 14.3; ESI-HRMS calcd for C20H23ClN2NaO2 (M + Na)+ 381.1340; found, 381.1343.
N-(2-Chloro-5-methylphenyl)-2-methyl-5-oxo-4-propyl-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (60)
N-(2-Chloro-5-methylphenyl)-3-oxobutanamide was prepared from 2-chloro-5-methylaniline (1.0 g, 7.06 mmol), 2 mL of methyl acetoacetate, and a catalytic amount of potassium t-butoxide. The mixture was heated in a MW reactor at 120 °C for 2 h. After completion, the reaction mixture was cooled to rt, dry loaded on silica, and purified by flash chromatography (SiO2, EtOAc/petroleum ether, 1:4) to yield 640 mg (40%) of a yellow solid: 1H NMR (400 MHz, CDCl3) δ 9.49 (s, 1H), 8.15 (d, J = 1.4 Hz, 1H), 7.24 (d, J = 8.2 Hz, 1H), 6.88–6.84 (m, 1H), 3.63 (s, 2H), 2.34 (s, 3H), 2.32 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 204.6, 163.6, 137.7, 134.1, 128.7, 125.8, 122.6, 120.5, 49.9, 31.2, 21.3; ESI-HRMS calcd for C11H12ClNNaO2 (M + Na)+ 248.0449; found, 248.0457.
Compound 60 was prepared from butyraldehyde (34 mg, 0.47 mmol), 3-aminocyclohex-2-en-1-one (52 mg, 0.47 mmol), and N-(2-chloro-5-methylphenyl)-3-oxobutanamide (105 mg, 0.47 mmol) according to the general procedure THQ to give 39 mg (22%) of a pale yellow solid (tR = 11.63 min, purity 98.7% by HPLC) after purification by flash chromatography (SiO2, EtOAc in petroleum ether, 50% to 70%): 1H NMR (400 MHz, CDCl3) δ 8.18 (d, J = 1.6 Hz, 1H), 7.99 (s, 1H), 7.23 (d, J = 8.2 Hz, 1H), 6.86–6.80 (m, 1H), 6.56 (s, 1H), 3.96 (t, J = 5.5 Hz, 1H), 2.54–2.39 (m, 3H), 2.37–2.26 (m, 7H), 2.07–1.93 (m, 2H), 1.55–1.34 (m, 2H), 1.33–1.21 (m, 2H), 0.91–0.81 (m, 3H); 13C NMR (101 MHz, CDCl3) δ 196.0, 166.8, 151.8, 140.8, 137.7, 134.6, 128.6, 125.2, 122.5, 120.4, 111.1, 108.7, 39.0, 37.1, 31.5, 27.5, 21.3, 21.2, 18.9, 18.3, 14.3; ESI-HRMS calcd for C21H25ClN2NaO2 (M + Na)+ 395.1497; found, 395.1513.
N-(2-Chloro-5-methoxyphenyl)-2-methyl-5-oxo-4-propyl-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (61)
N-(2-Chloro-5-methoxyphenyl)-3-oxobutanamide was prepared from 2-chloro-5-methoxylaniline (1.0 g, 7.06 mmol), 2 mL of methyl acetoacetate, and a catalytic amount of potassium t-butoxide. The mixture was heated in a MW reactor at 120 °C for 2 h. After completion, the reaction mixture was cooled to rt, dry loaded on silica, and purified by flash chromatography (SiO2, EtOAc/petroleum ether, 1:4) to yield 617 mg (30%) of a yellow solid: 1H NMR (400 MHz, CDCl3) δ 9.59 (s, 1H), 8.04 (d, J = 3.0 Hz, 1H), 7.25 (d, J = 8.9 Hz, 1H), 6.61 (dd, J = 8.9, 3.0 Hz, 1H), 3.79 (s, 3H), 3.64 (s, 2H), 2.34 (s, 3H); 13C NMR (101 MHz, CDCl3) δ 204.6, 163.6, 158.8, 135.2, 129.3, 114.6, 111.2, 107.1, 55.6, 49.8, 31.2; ESI-HRMS calcd for C11H12ClNNaO3 (M + Na)+ 264.0398; found, 264.0389.
Compound 61 was prepared from butyraldehyde (36 mg, 0.50 mmol), 3-aminocyclohex-2-en-1-one (55 mg, 0.50 mmol), and N-(2-chloro-5-methoxyphenyl)-3-oxobutanamide (120 mg, 0.50 mmol) according to the general procedure THQ to give 48 mg (25%) of a pale yellow solid (tR = 11.42 min, purity 97.6% by HPLC) after purification by flash chromatography (SiO2, EtOAc in petroleum ether, 50% to 70%): 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 3.0 Hz, 1H), 8.03 (s, 1H), 7.24 (d, J = 8.9 Hz, 1H), 6.66 (s, 1H), 6.58 (dd, J = 8.9, 3.0 Hz, 1H), 3.96 (t, J = 5.5 Hz, 1H), 3.81 (s, 3H), 2.53–2.40 (m, 3H), 2.37–2.26 (m, 4H), 2.07–1.89 (m, 2H), 1.55–1.45 (m, 1H), 1.44–1.34 (m, 1H), 1.33–1.20 (m, 2H), 0.85 (t, J = 7.2 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 196.0, 166.8, 158.9, 151.9, 140.9, 135.7, 129.2, 114.5, 111.1, 110.7, 108.8, 106.9, 55.7, 39.0, 37.1, 31.5, 27.5, 21.2, 18.8, 18.3, 14.3; ESI-HRMS calcd for C21H25ClN2NaO3 (M + Na)+ 411.1446; found, 411.1467.
4-Cyclopropyl-N-(2,5-dichlorophenyl)-2-methyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (62)
Compound 62 was prepared from cyclopropanecarbaldehyde (36 mg, 0.51 mmol), 3-aminocyclohex-2-en-1-one (56 mg, 0.51 mmol), and N-(2,5-dichlorophenyl)-3-oxobutanamide (125 mg, 0.51 mmol) according to the general procedure THQ to give 125 mg (63%) of a pale yellow solid (tR = 11.85 min, purity 99.4% by HPLC) after purification by flash chromatography (Reveleris X2, 24 g, EtOAc in petroleum ether, 0 to 100%): 1H NMR (400 MHz, CDCl3) δ 8.46 (d, J = 2.5 Hz, 1H), 8.15 (s, 1H), 7.29 (d, J = 8.6 Hz, 1H), 7.00 (dd, J = 8.6, 2.5 Hz, 1H), 5.93 (s, 1H), 3.82 (d, J = 6.7 Hz, 1H), 2.57–2.42 (m, 3H), 2.39–2.29 (m, 4H), 2.11–2.01 (m, 2H), 1.10–1.00 (m, 1H), 0.41–0.35 (m, 3H), 0.28–0.18 (m, 1H); 13C NMR (101 MHz, CDCl3) δ 196.0, 166.8, 150.4, 142.2, 136.1, 133.4, 129.7, 124.1, 121.7, 121.2, 111.7, 106.3, 37.1, 32.3, 27.6, 21.2, 19.2, 18.0, 2.8, 2.7; ESI-HRMS calcd for C20H20Cl2N2NaO2 (M + Na)+ 413.0794; found, 413.0793.
4-Cyclopentyl-N-(2,5-dichlorophenyl)-2-methyl-5-oxo-1,4,5,6,7,8-hexahydroquinoline-3-carboxamide (63)
Compound 63 was prepared from cyclopentanecarbaldehyde (50 mg, 0.51 mmol), 3-aminocyclohex-2-en-1-one (56 mg, 0.51 mmol), and N-(2,5-dichlorophenyl)-3-oxobutanamide (125 mg, 0.51 mmol) according to the general procedure THQ to give 10 mg (5%) of a pale yellow solid (tR = 12.83 min, purity 95.6% by HPLC) after purification by flash chromatography (Reveleris X2, 24 g, EtOAc in petroleum ether, 0 to 100%): 1H NMR (400 MHz, DMSO-d6) δ 9.07 (s, 1H), 8.81 (s, 1H), 7.84 (d, J = 2.6 Hz, 1H), 7.53 (d, J = 8.6 Hz, 1H), 7.24 (dd, J = 8.6, 2.6 Hz, 1H), 3.90 (d, J = 6.1 Hz, 1H), 2.47–2.35 (m, 2H), 2.34–2.12 (m, 5H), 1.97–1.87 (m, 1H), 1.86–1.71 (m, 2H), 1.55–1.08 (m, 8H); 13C NMR (101 MHz, DMSO-d6) δ 194.7, 168.1, 152.8, 139.0, 136.7, 131.4, 130.6, 125.2, 124.9, 124.5, 108.6, 107.6, 48.0, 37.0, 33.7, 27.9, 27.8, 26.5, 24.1, 24.0, 20.8; ESI-HRMS calcd for C22H24Cl2N2NaO2 (M + Na)+ 441.1107; found, 441.1090.
Inhibition of cAMP Assay
HEK293 Flp-In T-Rex cells stably transfected with FFA3-eYFP receptors were incubated overnight with doxycycline to induce expression, then harvested, and cryopreserved. On the day of each experiment, cells were thawed and resuspended in stimulation buffer. Assays were carried out using a 384-well plate format with 3000 cells/well using the CisBio cAMP GS dynamic kit as per manufacturer’s protocol. Compounds were tested for the ability to inhibit 0.3 μM forskolin-induced cAMP production after co-incubation for 1 h. Reactions were measured using a PHERAstar FS plate reader (BMGLabtech, Aylesbury, UK).
Membrane Preparation
Membranes were isolated from Flp-In T-REx HEK293 cells treated with 100 ng/mL doxycycline to induce receptor expression. Cells were washed with ice-cold phosphate-buffered saline and removed from dishes by scraping before being centrifuged for 5 min at 4 °C, 3000 rpm. Pellets were resuspended in TE buffer (75 mM Tris–HCl, 5 mM EDTA; pH 7.5) with the addition of a protease inhibitor mixture (Roche Applied Science, West Sussex, UK). This was then homogenized with a 5 mL hand-held homogenizer and subsequently centrifuged at 1500 rpm for 5 min at 4 °C, from which the supernatant was collected and further centrifuged at 50 000 rpm for 30 min at 4 °C. The resulting pellet was resuspended in TE buffer, and the protein content was measured using a BCA protein assay kit (Pierce, Fisher Scientific, Loughborough, UK).
[35S]GTPγS Incorporation Assay
[35S]GTPγS binding assays were performed in reactions with 5 μg of cell membrane protein preincubated for 15 min at 25 °C in assay buffer (50 mM Tris–HCl, pH 7.4, 10 mM MgCl2, 100 mM NaCl, 1 mM EDTA, 1 μM GDP, and 0.1% fatty acid-free bovine serum albumin) containing the indicated concentrations of ligands. Reactions were initiated with the addition of [35S]GTPγS at 50 nCi per tube; reactions were terminated after 1 h of incubation at 25 °C by rapid filtration through GF/C glass filters using a 24-well Brandel cell harvester (Alpha Biotech, Glasgow, UK). Unbound radioligand was removed from filters by washing three times with ice-cold wash buffer (50 mM Tris–HCl, pH 7.4, and 10 mM MgCl2), and [35S]GTPγS binding was determined by liquid scintillation spectrometry.
Physicochemical Properties
Solubility and log D7.4 measurements were performed in accordance to the previously published procedures.35
Microsomal Stability
The study was performed by Bienta (www.bienta.net). Mouse hepatic microsomes were isolated from pooled and perfused livers of male BALB/c mice (n = 50). Isolation was performed according to the standard protocol.36 The batches of microsomes were tested for quality control using Imipramine and Propranolol as reference compounds.
Microsomal incubations were carried out in 96-well plates in 5 aliquots of 40 μL each (one for each time point). Liver microsomal incubation medium comprised PBS (100 mM, pH 7.4), MgCl2 (3.3 mM), NADPH (3 mM), glucose-6-phosphate (5.3 mM), and glucose-6-phosphate dehydrogenase (0.67 units/mL) with 0.42 mg of liver microsomal protein per ml. In the control reactions, the NADPH-cofactor system was substituted with PBS. Test compounds (2 μM, final solvent concentration 1.6%) were incubated with microsomes at 37 °C, shaking at 100 rpm. Each reaction was performed in duplicates. Five time points over 40 min were analyzed. The reactions were stopped by adding 12 volumes of 90% acetonitrile–water to incubation aliquots, followed by protein sedimentation by centrifuging at 5500 rpm for 3 min. Each reaction was performed in duplicates. Supernatants were analyzed using HPLC-MS/MS on an API 3000 PE instrument.
In Vivo Pharmacokinetics
The study was performed by Bienta (www.bienta.net). Study design, animal selection, handling, and treatment were all in accordance with the Enamine PK study protocols and Institutional Animal Care and Use Guidelines. Animal treatment and plasma samples preparation were conducted by the Animal Laboratory personnel at Enamine/Bienta. Male BALB/c mice (8–10 weeks old, body weight: 1 17.2 g to 29.5 g and average body weight across all groups 22.4 ± 2.95 g; 16 18.0 g to 28.6 g and average body weight across all groups 22.8 ± 2.34 g; 57 20.0 g to 37.4 g and average body weight across all groups 27.0 ± 4.71 g; 63 17.6 g to 28.1 g and average body weight across all groups 22.0 ± 2.61 g) were used in this study. The animals were randomly assigned to the treatment groups before the pharmacokinetic study; all animals were fasted for 4 h before dosing. Nine time points (5, 15, 30, 60, 120, 240, 360, 480, and 1440 min) were set for this pharmacokinetic study. Each of the time point treatment group included four animals. There was also a control group of two animals. Compound 1, 16, 57, and 63 in a vehicle (DMSO–Cremophor EL–water w/5% mannitol, 1:1:8) was dosed at 10 mg/kg po or 5 mg/kg iv. Dosing volumes of compounds or vehicle was 5 mL/kg. Mice were injected iv with 2,2,2-tribromoethanol at the dose of 150 mg/kg prior to drawing the blood. Blood collection was performed from the orbital sinus in microtainers containing K2EDTA. Animals were sacrificed by cervical dislocation after the blood sample collection. All samples were immediately processed, flash-frozen, and stored at −70 °C until subsequent analysis. Before analysis, plasma samples (50 μL) were added acetonitrile-methanol (1:1, v/v, 200 μL) containing an internal standard (IS-10784, C20H26N8, Mw 378.47) 200 ng/mL for 1, 63 400 ng/mL for 16, or 16 200 ng/mL for 57 and 63, respectively). After mixing by pipetting and centrifuging for 4 min at 6000 rpm, 0.5–1 μL of each supernatant was analyzed by HPLC-MS/MS on an API 3000 PE instrument.
Dorsal Root Ganglion Assay
Colonic innervating DRGs were isolated from the T9-L2 region of the spinal cord of C57/BL6 mice and from FFA3 knock-out animals and immediately placed in cold Hanks’ balanced salt solution (HBSS; Sigma-Aldrich). Isolated DRGs were initially digested with HBSS containing l-cysteine (0.3 mg/mL) and papain (2.0 mg/mL) for 20 min at 37 °C. The solution was removed and replaced with HBSS contain collagenase (4.0 mg/mL) and dispase (4.0 mg/mL) (20 min at 37 °C) for further digestion. The collagenase solution was then replaced with DMEM to stop the reaction. The DRGs were finally dissociated by mechanical trituration using a pipette. Dissociated cells were plated on matrigel-coated coverslips and placed in an incubator (37 °C and 5% CO2). Following a 2 h incubation, cells were flooded with 90% DMEM (Sigma) supplemented with 10% fetal calf serum and 1% PenStrep and further incubated overnight at 37 °C and 5% CO2.
To measure intracellular calcium and its potential regulation, dissociated cells on the coverslips were loaded with Fura 8-AM (3 μM) (Stratech Scientific Limited) for 20 min at 37 °C in the dark. Coverslips were then placed in a recording chamber and mounted onto an inverted fluorescent microscope (Nikon TE2000-E; Nikon Instruments, Melville, NY) equipped with a (NA = 1.3) oil-immersion Super Fluor objective lens (×40), an Optoscan monochromator (Cairn Research, Faversham, Kent, UK), and a digital Cool Snap-HQ CCD camera (Roper Scientific/Photometrics, Tucson, AZ). Illumination of the preparation was achieved by a Meta Fluor imaging software (Molecular Devices, San Jose CA, version 7.8.8).
Cluster of cells were randomly selected for real time imaging and continuously perfused with HEPES buffer (composition: HEPES 10 mM, NaCl 135 mM, glucose 10 mM, KCl 5 mM, CaCl2 2 mM and MgCl2 1 mM, pH 7.4) for 20 min at room temperature. All test ligands were diluted in HEPES buffer and perfused through the chamber for 3 min, followed by a final application of the Ca2+ ionophore ionomycin (5 μM), as a positive control.
Acknowledgments
We thank Ole Buch Madsen, Rikke Linnet Jørgensen, and Lone Overgaard Storm for assistance with synthesis and Loukas Ieremias for assistance with counterscreening.
Glossary
Abbreviations
- C3
propionate
- DRG
dorsal root ganglion
- FFA2
free fatty acid receptor 2 (GPR43)
- FFA3
free fatty acid receptor 3 (GPR41)
- IPA
isopropanol
- 1-MCPC
1-methylcyclopropylcarboxylate
- MLM
mouse liver microsomes
- SCFA
short-chain fatty acids
- THQ
tetrahydroquinoline
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.9b02036.
Accession Codes
Crystallographic data (CIF) for (R)-1 and (R,S)-1 have been deposited at the Cambridge Crystallographic Data Centre, CCDC1873629
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was supported by the Lundbeck Foundation (grant R181-2014-3247, R307-2018-2950), the Danish Council for Strategic Research/Innovation Fund Denmark (grant 11-116196, 0603-00452B), and the Biotechnology and Biosciences Research Council (grant numbers BB/L027887/1 and BB/LO278X/1).
The authors declare no competing financial interest.
Supplementary Material
References
- Topping D. L.; Clifton P. M. Short-chain fatty acids and human colonic function: Roles of resistant starch and nonstarch polysaccharides. Physiol. Rev. 2001, 81, 1031–1064. 10.1152/physrev.2001.81.3.1031. [DOI] [PubMed] [Google Scholar]
- Kuwahara A. Contributions of colonic short-chain fatty acid receptors in energy homeostasis. Front. Endocrinol. 2014, 5, 144. 10.3389/fendo.2014.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulven T. Short-chain free fatty acid receptors FFA2/GPR43 and FFA3/GPR41 as new potential therapeutic targets. Front. Endocrinol. 2012, 3, 111. 10.3389/fendo.2012.00111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Poul E.; Loison C.; Struyf S.; Springael J.-Y.; Lannoy V.; Decobecq M.-E.; Brezillon S.; Dupriez V.; Vassart G.; Van Damme J.; Parmentier M.; Detheux M. Functional characterization of human receptors for short chain fatty acids and their role in polymorphonuclear cell activation. J. Biol. Chem. 2003, 278, 25481–25489. 10.1074/jbc.m301403200. [DOI] [PubMed] [Google Scholar]
- Brown A. J.; Goldsworthy S. M.; Barnes A. A.; Eilert M. M.; Tcheang L.; Daniels D.; Muir A. I.; Wigglesworth M. J.; Kinghorn I.; Fraser N. J.; Pike N. B.; Strum J. C.; Steplewski K. M.; Murdock P. R.; Holder J. C.; Marshall F. H.; Szekeres P. G.; Wilson S.; Ignar D. M.; Foord S. M.; Wise A.; Dowell S. J. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–11319. 10.1074/jbc.m211609200. [DOI] [PubMed] [Google Scholar]
- Milligan G.; Shimpukade B.; Ulven T.; Hudson B. D. Complex pharmacology of free fatty acid receptors. Chem. Rev. 2017, 117, 67–110. 10.1021/acs.chemrev.6b00056. [DOI] [PubMed] [Google Scholar]
- Tang C.; Offermanns S. FFA2 and FFA3 in metabolic regulation. Handb. Exp. Pharmacol. 2017, 236, 205–220. 10.1007/164_2016_50. [DOI] [PubMed] [Google Scholar]
- Steensels S.; Cools L.; Avau B.; Vancleef L.; Farré R.; Verbeke K.; Depoortere I. Supplementation of oligofructose, but not sucralose, decreases high-fat diet induced body weight gain in mice independent of gustducin-mediated gut hormone release. Mol. Nutr. Food Res. 2017, 61, 1600716. 10.1002/mnfr.201600716. [DOI] [PubMed] [Google Scholar]
- Hudson B. D.; Due-Hansen M. E.; Christiansen E.; Hansen A. M.; Mackenzie A. E.; Murdoch H.; Pandey S. K.; Ward R. J.; Marquez R.; Tikhonova I. G.; Ulven T.; Milligan G. Defining the molecular basis for the first potent and selective orthosteric agonists of the FFA2 free fatty acid receptor. J. Biol. Chem. 2013, 288, 17296–17312. 10.1074/jbc.m113.455337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaibi M. S.; Stocker C. J.; O’Dowd J.; Davies A.; Bellahcene M.; Cawthorne M. A.; Brown A. J. H.; Smith D. M.; Arch J. R. S. Roles of GPR41 and GPR43 in leptin secretory responses of murine adipocytes to short chain fatty acids. FEBS Lett. 2010, 584, 2381–2386. 10.1016/j.febslet.2010.04.027. [DOI] [PubMed] [Google Scholar]
- Vermeire S.; Kojecký V.; Knoflícek V.; Reinisch W.; Van Kaem T.; Namour F.; Beetens J.; Vanhoutte F. GLPG0974, an FFA2 antagonist, in ulcerative colitis: efficacy and safety in a multicenter proof-of-concept study. J. Crohn’s Colitis 2015, 9, S39. 10.1093/ecco-jcc/jju027.058. [DOI] [Google Scholar]
- Tang C.; Ahmed K.; Gille A.; Lu S.; Gröne H.-J.; Tunaru S.; Offermanns S. Loss of FFA2 and FFA3 increases insulin secretion and improves glucose tolerance in type 2 diabetes. Nat. Med. 2015, 21, 173–177. 10.1038/nm.3779. [DOI] [PubMed] [Google Scholar]
- Thirunavukkarasan M.; Wang C.; Rao A.; Hind T.; Teo Y. R.; Siddiquee A. A.-M.; Goghari M. A. I.; Kumar A. P.; Herr D. R. Short-chain fatty acid receptors inhibit invasive phenotypes in breast cancer cells. PLoS One 2017, 12, e0186334 10.1371/journal.pone.0186334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkins M. M.; Meier K. E. Free fatty acid receptors and cancer: From nutrition to pharmacology. Handb. Exp. Pharmacol. 2017, 236, 233–251. 10.1007/164_2016_48. [DOI] [PubMed] [Google Scholar]
- Kimura M.; Mizukami Y.; Miura T.; Fujimoto K.; Kobayashi S.; Matsuzaki M. Orphan G protein-coupled receptor, GPR41, induces apoptosis via a p53/bax pathway during ischemic hypoxia and reoxygenation. J. Biol. Chem. 2001, 276, 26453–26460. 10.1074/jbc.m101289200. [DOI] [PubMed] [Google Scholar]
- Trompette A.; Gollwitzer E. S.; Yadava K.; Sichelstiel A. K.; Sprenger N.; Ngom-Bru C.; Blanchard C.; Junt T.; Nicod L. P.; Harris N. L.; Marsland B. J. Gut microbiota metabolism of dietary fiber influences allergic airway disease and hematopoiesis. Nat. Med. 2014, 20, 159–166. 10.1038/nm.3444. [DOI] [PubMed] [Google Scholar]
- Nøhr M. K.; Egerod K. L.; Christiansen S. H.; Gille A.; Offermanns S.; Schwartz T. W.; Møller M. Expression of the short chain fatty acid receptor GPR41/FFAR3 in autonomic and somatic sensory ganglia. Neuroscience 2015, 290, 126–37. 10.1016/j.neuroscience.2015.01.040. [DOI] [PubMed] [Google Scholar]
- Kimura I.; Inoue D.; Maeda T.; Hara T.; Ichimura A.; Miyauchi S.; Kobayashi M.; Hirasawa A.; Tsujimoto G. Short-chain fatty acids and ketones directly regulate sympathetic nervous system via G protein-coupled receptor 41 (GPR41). Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 8030–8035. 10.1073/pnas.1016088108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson B. D.; Tikhonova I. G.; Pandey S. K.; Ulven T.; Milligan G. Extracellular ionic locks determine variation in constitutive activity and ligand potency between species orthologs of the free fatty acid receptors FFA2 and FFA3. J. Biol. Chem. 2012, 287, 41195–41209. 10.1074/jbc.m112.396259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan G.; Bolognini D.; Sergeev E. Ligands at the Free Fatty Acid Receptors 2/3 (GPR43/GPR41). Handb. Exp. Pharmacol. 2017, 236, 17–32. 10.1007/164_2016_49. [DOI] [PubMed] [Google Scholar]
- Schmidt J.; Smith N. J.; Christiansen E.; Tikhonova I. G.; Grundmann M.; Hudson B. D.; Ward R. J.; Drewke C.; Milligan G.; Kostenis E.; Ulven T. Selective orthosteric free fatty acid receptor 2 (FFA2) agonists. Identification of the structural and chemical requirements for selective activations of FFA2 versus FFA3. J. Biol. Chem. 2011, 286, 10628–10640. 10.1074/jbc.m110.210872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson B. D.; Christiansen E.; Murdoch H.; Jenkins L.; Hansen A. H.; Madsen O.; Ulven T.; Milligan G. Complex pharmacology of novel allosteric free fatty acid 3 receptor ligands. Mol. Pharmacol. 2014, 86, 200–210. 10.1124/mol.114.093294. [DOI] [PubMed] [Google Scholar]
- Leonard J. N.; Chu Z. L.; Bruce M. A.; Boatman P. D.. GPR41 and Modulators Thereof for the Treatment of Insulin-Related Disorders. WO2006052566A2, 2006.
- Nøhr M. K.; Pedersen M. H.; Gille A.; Egerod K. L.; Engelstoft M. S.; Husted A. S.; Sichlau R. M.; Grunddal K. V.; Seier Poulsen S.; Han S.; Jones R. M.; Offermanns S.; Schwartz T. W. GPR41/FFAR3 and GPR43/FFAR2 as cosensors for short-chain fatty acids in enteroendocrine cells vs FFAR3 in enteric neurons and FFAR2 in enteric leukocytes. Endocrinology 2013, 154, 3552–3564. 10.1210/en.2013-1142. [DOI] [PubMed] [Google Scholar]
- Engelstoft M. S.; Park W.-m.; Sakata I.; Kristensen L. V.; Husted A. S.; Osborne-Lawrence S.; Piper P. K.; Walker A. K.; Pedersen M. H.; Nøhr M. K.; Pan J.; Sinz C. J.; Carrington P. E.; Akiyama T. E.; Jones R. M.; Tang C.; Ahmed K.; Offermanns S.; Egerod K. L.; Zigman J. M.; Schwartz T. W. Seven transmembrane G protein-coupled receptor repertoire of gastric ghrelin cells. Mol. Metab. 2013, 2, 376–392. 10.1016/j.molmet.2013.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Said H.; Akiba Y.; Narimatsu K.; Maruta K.; Kuri A.; Iwamoto K.-i.; Kuwahara A.; Kaunitz J. D. FFA3 activation stimulates duodenal bicarbonate secretion and prevents NSAID-induced enteropathy via the GLP-2 pathway in rats. Dig. Dis. Sci. 2017, 62, 1944–1952. 10.1007/s10620-017-4600-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaji I.; Akiba Y.; Konno K.; Watanabe M.; Kimura S.; Iwanaga T.; Kuri A.; Iwamoto K.-i.; Kuwahara A.; Kaunitz J. D. Neural FFA3 activation inversely regulates anion secretion evoked by nicotinic ACh receptor activation in rat proximal colon. J. Physiol. 2016, 594, 3339–3352. 10.1113/jp271441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaji I.; Akiba Y.; Furuyama T.; Adelson D. W.; Iwamoto K.; Watanabe M.; Kuwahara A.; Kaunitz J. D. Free fatty acid receptor 3 activation suppresses neurogenic motility in rat proximal colon. Neurogastroenterol. Motil. 2018, 30, e13157 10.1111/nmo.13157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll W. A.; Agrios K. A.; Altenbach R. J.; Buckner S. A.; Chen Y.; Coghlan M. J.; Daza A. V.; Drizin I.; Gopalakrishnan M.; Henry R. F.; Kort M. E.; Kym P. R.; Milicic I.; Smith J. C.; Tang R.; Turner S. C.; Whiteaker K. L.; Zhang H.; Sullivan J. P. Synthesis and structure–activity relationships of a novel series of tricyclic dihydropyridine-based KATP openers that potently inhibit bladder contractions in vitro. J. Med. Chem. 2004, 47, 3180–3192. 10.1021/jm030357o. [DOI] [PubMed] [Google Scholar]
- Harrison C.; Traynor J. R. The [35S]GTPgammaS binding assay: approaches and applications in pharmacology. Life Sci. 2003, 74, 489–508. 10.1016/j.lfs.2003.07.005. [DOI] [PubMed] [Google Scholar]
- Domljanovic I.; Rexen Ulven E.; Ulven T.; Thomsen R. P.; Okholm A. H.; Kjems J.; Voss A.; Taskova M.; Astakhova K. Dihydropyridine fluorophores allow for specific detection of human antibodies in serum. ACS Omega 2018, 3, 7580–7586. 10.1021/acsomega.8b00424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J.; Byrne N.; Wang J.; Bricogne G.; Brown F. K.; Chobanian H. R.; Colletti S. L.; Di Salvo J.; Thomas-Fowlkes B.; Guo Y.; Hall D. L.; Hadix J.; Hastings N. B.; Hermes J. D.; Ho T.; Howard A. D.; Josien H.; Kornienko M.; Lumb K. J.; Miller M. W.; Patel S. B.; Pio B.; Plummer C. W.; Sherborne B. S.; Sheth P.; Souza S.; Tummala S.; Vonrhein C.; Webb M.; Allen S. J.; Johnston J. M.; Weinglass A. B.; Sharma S.; Soisson S. M. Structural basis for the cooperative allosteric activation of the free fatty acid receptor GPR40. Nat. Struct. Mol. Biol. 2017, 24, 570–577. 10.1038/nsmb.3417. [DOI] [PubMed] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/s0021889808042726. [DOI] [Google Scholar]
- Sheldrick G. M. SHELXT- Integrated space-group and crystal-structure determination. Acta Crystallogr., Sect. A: Found. Adv. 2015, 71, 3–8. 10.1107/s2053273314026370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rexen Ulven E.; Trauelsen M.; Brvar M.; Lückmann M.; Bielefeldt L. Ø.; Jensen L. K. I.; Schwartz T. W.; Frimurer T. M. Structure-Activity Investigations and Optimisations of Non-metabolite Agonists for the Succinate Receptor 1. Sci. Rep. 2018, 8, 10010. 10.1038/s41598-018-28263-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill J. R. In vitro drug metabolism using liver microsomes. Curr. Protoc. Pharmacol. 2003, 23, 7.8.1–7.8.11. 10.1002/cpph.9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.