

Abstract
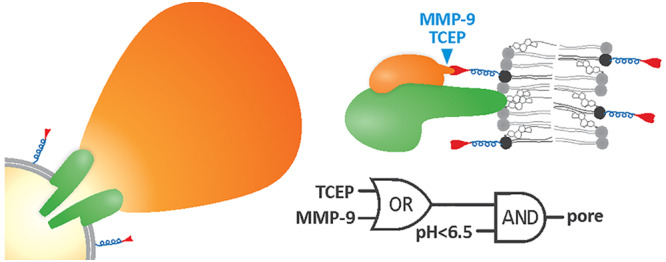
Lipid membranes are becoming increasingly popular in synthetic biology due to their biophysical properties and crucial role in communication between different compartments. Several alluring protein–membrane sensors have already been developed, whereas protein logic gates designs on membrane-embedded proteins are very limited. Here we demonstrate the construction of a two-level protein–membrane logic gate with an OR-AND logic. The system consists of an engineered pH-dependent pore-forming protein listeriolysin O and its DARPin-based inhibitor, conjugated to a lipid vesicle membrane. The gate responds to low pH and removal of the inhibitor from the membrane either by switching to a reducing environment, protease cleavage, or any other signal depending on the conjugation chemistry used for inhibitor attachment to the membrane. This unique protein logic gate vesicle system advances generic sensing and actuator platforms used in synthetic biology and could be utilized in drug delivery.
Keywords: pore-forming toxin, listeriolysin O, protein logic gates, lipid membrane, DARPin, pH
Synthetic biology is a rapidly developing field providing a wide variety of options for construction of biomolecular computers in vitro and in vivo with numerous applications in nanotechnology.1−3 Diverse systems have been developed employing mostly DNA,4−8 but also RNA,9 or enzymes10−15 to execute logic computing tasks using biomolecule-based Boolean logic gates. While a majority of the work has been done at the DNA or protein level, the engagement of lipid membranes and membrane proteins has been very limited in molecular-scale computational elements16−19 due to particular structural features of membrane proteins and the complex nature of the lipid bilayer membrane.20
Protein logic gates consist of an input component, which is sensitive to specific input signal, and an output component, which upon transduction of the incoming signal produces the perceivable effect.21 Engineering principles are very diverse as protein logic functions can be achieved with either single domain proteins, where the same domain has the input and the output capability, or by fusing two domains where one domain functions as a recognition domain (the input domain) and the other as an effector (the output domain).22 Proteins with a natural ability to form defined pores in membranes, so-called pore-forming proteins or pore-forming toxins (PFTs), offer an excellent option for logic gate design on membranes. These protein molecules are soluble as monomeric units, capable of binding to lipid membranes in a lipid-specific manner and consequently form oligomeric transmembrane pores, which are well-defined in terms of shape and size.23,24 Pore-formation is a complex process and is composed of succession of steps that can be manipulated in order to control the pore opening.25,26 Several applications of PFTs have been developed, e.g., for release of compounds encapsulated in lipid vesicles-based delivery systems,27 biosensing,28−32 and as integral parts of artificial cells.19,33,34 However, all those systems lack self-regulatory modules or produce only small (around 2 nm in diameter) membrane pores.
To bypass limitations of existing protein-vesicle systems, we present a unique multilevel system where we combined synthetic biology with the rational design and directed evolution based on a PFT listeriolysin O (LLO). This protein toxin is a major virulence factor of bacteria Listeria monocytogenes. It belongs to a protein family of cholesterol-dependent cytolysins that are able to form large pores on membranes of target cells, exceeding 20 nm in diameter.35 LLO is composed of four domains36 (Figure 1a), each playing a particular role in the pore-forming process. LLO predominately forms arc-shaped pores on the surface of target cells37−39 in a succession of steps, which involve binding to lipid bilayers with high cholesterol content by using domain 4 (D4; Figure 1a), oligomerization on the membrane surface, and final pore formation in which two β-hairpins are formed from two clusters of α-helices in domain 3 (D3) of each monomer (Figure 1b). We have recently reported an interesting mutant of LLO, Y406A (Figure 1a), which shows a unique pH-regulated pore-forming activity. While it binds to cholesterol-rich membranes in a wide pH range, it is able to form pores and is thus fully active only at low pH values.40
Figure 1.

Properties and permeabilizing activity of Y406A. (a) A ribbon model of LLO 3D structure (PDB ID: 4CDB) with each domain (D1–D4) labeled in different color. Position of residue 406 is denoted by an orange color and an arrow. (b) A model of pore formation by Y406A (monomer binding to the membrane, oligomerization, pore formation). Domains are colored as on panel a. (c) Calcein release from large unilamellar vesicles after 30 min is shown at different pH values for LLO (black) and Y406A (red). Data reproduced with permission from Kisovec et al.(40) (d) Relative rate of hemolysis by 2.3 nM CDCs at pH 5.7 in the presence of DARPins: 5 μM D22 (red bars), 5 μM D22M (blue bar), a variant that was used for immobilization to the lipid membrane (see below), and 5 μM D6 and D30 (green bars), DARPin clones that were selected with ribosome display as high affinity binders, but did not inhibit hemolytic activity of Y406A. Mean ± SD; n = 2–7.
We created a logic gate on lipid membrane by combining Y406A with an additional inhibitor of its activity, a designed ankyrin repeat protein (DARPin) variant 22, D22, which binds reversibly to Y406A. In the off state of the logic gate, Y406A is doubly inhibited by D22 covalently bound to the membrane, and pH > 7.4. The logical functioning of the protein gate was achieved with various cleavages of D22 from the membrane and pH activation of Y406A. Upon system activation, the inhibiting D22 dissociates from Y406A, which then, at a favorable pH, undergoes conformational changes and forms pores in membranes.
Results
DARPin D22 Binds Specifically to Y406A in Solution and in the Membrane-Bound State
Y406A is an interesting pH-dependent LLO mutant, which has a very narrow activity profile of pH dependence. It is active at low pH values, drastically loses activity in the pH range 6.0–7.4, and is not active at pH values >7.4 (Figure 1c). However, it is still capable of binding to the membrane at high pH values.40 Y406A is thus perfectly suited for controlled release in liposomal applications employing pH as an input signal. In order to provide another level of control over Y406A permeabilizing activity, we have developed a DARPin-based inhibitor. DARPins represent a useful tool for specific targeting of bigger molecules, such as proteins, due to their large interaction surface and high binding capacity. Specific DARPin inhibitor of permeabilizing activity of Y406A was gained with ribosome display.41,42 Forty clones among enriched variants after six rounds of ribosome display were isolated and tested with ELISA. Three clones, D6, D22, and D30 that exhibited highest affinity toward immobilized target protein were further selected and checked for permeabilizing activity. D22 was selected for further studies, because of its specific inhibition of hemolytic activity of Y406A, but not of the wild-type LLO (Figure 1d). To further prove the specificity of D22 for Y406A, inhibitory effect of D22 was tested toward another member of cholesterol-dependent cytolysin and a homologue of LLO, Perfringolysin O (PFO) from bacterium Clostridium perfringens, and its mutant Y381A, which is analogous to Y406A40 (Figure 1d). Indeed, D22 is selective only for Y406A by exhibiting no significant effect on hemolytic activity of these two homologous proteins (Figure 1d).
The size exclusion chromatography proved stable complex formation between Y406A and D22 (Figure 2a). Isothermal titration calorimetry suggested a stoichiometry ratio of Y406A-D22 complex to be 1 (0.96 ± 0.15) (Figure 2b), with a binding constant KD of 114 ± 43 nM (n > 4; average ± SD). LLO did not show any detectable binding at the same conditions (Figure 2b). Small-angle X-ray scattering (SAXS) measurements confirmed formation of Y406A-D22 complex (Figure 2c), indicating that D22 is most likely associated with the domain 2 (D2) of Y406A (Figure 2c inset).
Figure 2.

Binding of D22 to Y406A in solution. (a) Size exclusion chromatogram of LLO and Y406A in the absence or presence of D22. Triangles indicate positions of elution peaks for different proteins. Note that LLO travels aberrantly on the size exclusion column eluting with larger volumes of elution buffer than expected. (b) Binding of D22 to LLO (gray) or Y406A (black) in solution (22 mM MES, 150 mM NaCl and 5 mM 2-mercaptoethanol, pH 5.7), measured by isothermal titration calorimetry. Top panel represents raw data of injections of 54.9 μM D22 into a 5.9 μM solution of LLO or Y406A. Bottom panel shows normalized integrated enthalpies plotted against the molar ratio. Circles represent experimental points, and the solid line corresponds to the best fit obtained by one-site reaction model. (c) An overlay of experimental scattering data obtained by SAXS experiment of Y406A-D22 complex (red circles) with the calculated scattering curve from the representative DAMMIF model (χ2 = 1.125, black line). Inset, overlay of Y406A (blue ribbon) and D22 (red ribbon) refined by rigid body modeling (χ2 = 1.12), with the best SAXS bead model in surface representation and the average SAXS bead model in mesh representation.
In the next step we wanted to test whether D22 interferes with the binding of Y406A to the lipid membrane. LLO and Y406A require high concentrations of lipid receptor cholesterol in membranes,40,43 and we used a mixture of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC):cholesterol 3:2 (mol:mol) in all model lipid vesicles systems presented in this paper, unless stated otherwise. Preincubated mixture of Y406A and D22 in solution showed larger surface plasmon resonance (SPR) response than Y406A alone, which indicates that Y406A-D22 complex can indeed bind to the membrane (Figure 3a). This is in agreement with proposed mode of binding of D22 to Y406A via D2 (Figure 2c), which leaves D4 free for interaction with the lipid membrane. D22 itself did not bind to the lipid membrane (Figure 3a). D22 also bound to the membrane-bound Y406A and the interaction was reversible, with slow dissociation of D22. As expected, D22 did not bind to the membrane-bound LLO (Figure 3b). This was also independently confirmed by vesicle sedimentation assays (Figures 3c and d). Thus, D22 specifically binds to soluble as well as membrane-bound Y406A.
Figure 3.

Interaction of D22 with Y406A in the lipid membrane environment. (a) SPR measurements showing binding of 100 nM Y406A, 5 μM D22 and preincubated Y406A-D22 complex (with same concentrations of individual proteins as used for single proteins injections) to large unilamellar vesicles. (b) SPR sensorgrams of 5 μM D22 binding to 100 nM membrane-inserted Y406A or LLO. Membrane denotes control experiment with D22 binding to vesicles only. (c) Vesicle sedimentation assays with multilamellar vesicles after preincubation of LLO and D22, or Y406A and D22 in solution (“preincubated”) or when LLO or Y406A were first preincubated with vesicles (“membrane bound”). p, pellet; s, supernatant; w, additional washing step, which was included when assaying membrane bound LLO or Y406A in order to check for the completeness of binding. Band at app. 60 kDa corresponds to LLO or Y406A, while band at ca. 18 kDa corresponds to D22. D22 is present at 5× molar excess; therefore, a large portion of it is always unbound in supernatant. (d) Quantification of the SDS-PAGE data from (c) by densitometry. Full binding of D22 to Y406A (100%) was considered when one-fifth of the applied D22 was bound to Y406A. Mean ± SD; two sample t test, ***P < 0.001 (n = 4–6). Amount of bound D22 is reported when preincubated with LLO (1) or Y406A (2) in solution or when LLO (3) or Y406A (4) were first bound to vesicles.
D22 Provides Additional Control of Y406A Permeabilizing Activity
We next verified ability of D22 to provide additional control of Y406A permeabilizing activity in different functional assays. For this purpose, we assayed permeability of giant unilamellar vesicles (GUVs) for fluorescently labeled dextran of 10 kDa size (FD10) and a Stokes radius of 23.6 Å at two pH values, 6.5 and 8.0. LLO was able to permeabilize GUVs at either pH and was not affected by D22 (Figure 4a). Y406A clearly showed activity only at low pH and in the absence of D22, while in the presence of D22, the permeabilizing activity of Y406A was inhibited (Figure 4b). We also assayed hemolytic activity of LLO or Y406A at two pH values, pH 5.7 and 7.4, and in the presence or absence of D22. Hemolysis results confirmed the results obtained in the GUVs system (Figures 4c and d). D22 itself was not able to induce any damage to the GUV lipid membrane at either pH (Figure 3e) and was not hemolytic up to 9.5 μM concentration (data not shown), which is in agreement with SPR results that showed a lack of membrane association for D22 (Figure 3). The experiments presented in Figure 4 convincingly show that activity of Y406A can be controlled by pH as expected,40 as well as with D22, which provides an additional level of control over Y406A activity. This system can thus conceptually be described as the NOR logic gate, when activity (pore formation) is observed only in the absence of the two signals, D22 and pH > 7.4 (Figure 4f).
Figure 4.

Modulation of Y406A permeabilization activity by D22 and pH. (a) Permeabilization of GUVs for FD10 at different conditions and induced by the 50 nM LLO. (b) Permeabilization induced by 50 nM Y406A. The graphs on the right in A and B show quantification of GUVs data from confocal microscopy images as represented on the left. Mean ± SD; n = 96–568. (c) Hemolysis induced by 18.2 nM LLO at different conditions. (d) Hemolysis induced by 18.2 nM Y406A. In C and D, mean ± SD is presented; n = 3. (e) 5 μM D22 by itself does not induce permeabilization of GUVs. Confocal images of GUVs on the left, and quantification is presented on the right. n is 256 and 458 for pH 6.5 and 8.0, respectively. (f) The truth table and schematic representation of a NOR logic gate for the Y406A-D22 system.
We further demonstrated the principle of selective inhibition of Y406A permeabilizing activity by assaying release of encapsulated fluorescent probe calcein from small and large unilamellar vesicles (SUVs and LUVs, respectively). In a SUVs system we essentially obtained similar results as in the GUVs system, with LLO showing similar activity regardless the condition at which it was assayed (Figure 5a), while Y406A only showed activity at pH 6.5 in the absence of D22 (Figure 5b). Y406A showed negligible release of calcein also in a LUVs system when assayed at high pH (8.0) (Figure 5c, traces A and D). Membrane-bound Y406A can be effectively activated by addition of small quantity of HCl (Figure 5c, blue trace D, pH drop to approximately 6.5) in agreement with previous work.40 D22 provided an effective control over Y406A permeabilizing activity also in this system. The pH activation of Y406A did not occur when D22 was added to solution containing vesicles and Y406A (Figure 5c, trace A), when Y406A was preincubated with D22 before addition to vesicles (Figure 5c, trace B) or when Y406A was added to vesicles and D22 in an acidic environment subsequently (Figure 5c, trace C). Interestingly, addition of D22 after activation of Y406A also inhibited further release of calcein, showing that D22 is efficient modulator that rapidly affects permeabilizing activity of Y406A (Figure 5c, trace E). In summary, permeabilizing experiments employing four independent model systems thus clearly exemplify a two-level control system of Y406A activity, modulated by the change in pH and the presence of D22.
Figure 5.
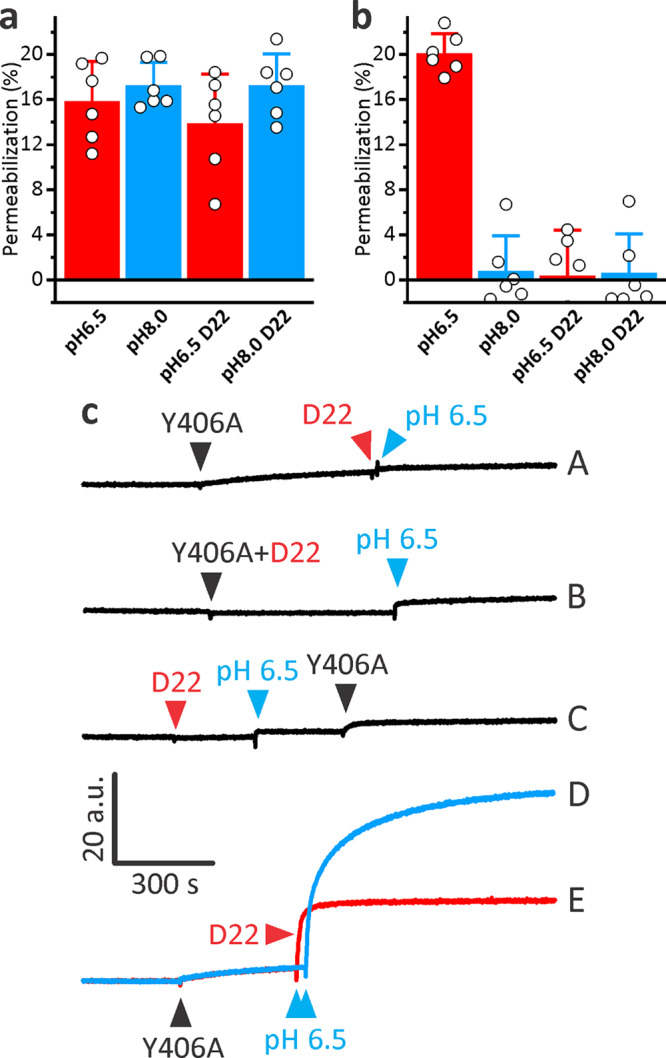
Modulation of Y406A activity in calcein release experiments. (a,b) Calcein release from SUVs composed of POPC:Chol, 3:2 (mol:mol) as a result of pore formation by 1 μM LLO (a) or Y406A (b), in presence or absence of 5 μM D22 and at pH values 6.5 and 8.0. (c) Calcein release from LUVs composed of POPC:Chol, 1:1 (mol:mol) monitored at different conditions. Vesicles were stirred in 10 mM HEPES, 150 mM NaCl, 1 mM EDTA, pH 8.0. Final concentration of 30 nM Y406A (black triangle), 1 μM D22 or 3 μL of 7% HCl (to reduce pH to approximately 6.5) were added, respectively, at times denoted by triangles. The scale bar is the same for all fluorescent traces.
Membrane-Based System Including Covalently Bound D22 Allows Employment of Various Signals for Activation of Y406A
We showed that D22 does not associate with lipid membrane by itself (Figure 3) and does not damage the lipid membrane (Figures 4 and 5), which is a convenient feature for development of logic gates, since it could be attached to the lipid bilayer without damaging or changing its properties. In order to bind D22 to lipid membranes, we prepared a variant of D22 by adding the matrix metalloproteinase 9 (MMP-9) cleavable peptide and an additional cysteine to the C-terminal end (D22M). This change in amino acid sequence did not affect inhibitory potential of D22M toward Y406A (Figure 1d). Such construct allowed attachment of D22 to the lipid membrane by conjugation via the introduced cysteine residue at the C-terminus and offered removal of D22 from the membrane surface by either proteolytic cleavage or addition of a reductant. Two alternative conjugations of D22M to lipid bilayers were thus prepared by using different functionalized lipids. In the case of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-2000] (DSPE-PEG2000Mal), D22 was attached to the lipid via stable irreversible thioether linkage and the subsequent release of D22 was achieved using MMP-9 cleavage (Figure 6a). In the case of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[PDP(polyethylene glycol)-2000] (DSPE-PEG2000PDP), D22M was attached to the lipid via reversible S–S bond and the subsequent release of D22 was achieved upon addition of a reducing agent such as Tris(2-carboxyethyl)phosphine hydrochloride (TCEP) (Figure 6c). Both lipids contain a PEG linker, which provides flexibility of membrane-anchored D22M needed for efficient inhibition of membrane-bound Y406A.
Figure 6.

Conjugation of D22M to vesicles and cleavage with different agents. Schematic diagrams of used systems and structures of employed lipids are shown. Y406A is presented with green color, D22 is shown in orange. Different parts of lipids used for conjugation are presented with different colors in the structural formulas and on the diagram. (a–c) A system employing DSPE-PEG2000Mal lipid, which attaches D22 to the lipid membrane and allows subsequent cleavage by MMP-9. (d–f) A system employing DSPE-PEG2000PDP lipid, which allows cleavage with reductant TCEP. The input of different proteins and reagents is shown above the sedimentation assay gels. The approximate positions of different proteins and lipids on the gels are indicated by arrows. i, 2 μg of D22M as an input; p and s denote pellet and supernatant after centrifugation of MLVs, respectively. (c) and (f) each represent one experiment where MLVs were first incubated with Y406A and then with MMP-9 or TCEP. 1, pellet after both incubations; 2, supernatant after centrifugation of MLVs after incubation with Y406A; 3, supernatant after centrifugation of MLVs with bound Y406A and incubation of MMP-9 or TCEP.
We optimized assays by (i) selecting the appropriate variant of D22 (N- terminal or C-terminal modification) and functionalized lipids (we tested lipids with or without flexible PEG linker) and (ii) determining the minimal proportion of functionalized lipids to show efficient inhibition of Y406A and cleavage of conjugated D22M from the MLVs by either MMP-9 or TCEP. We tested a range of Y406A (200 nM to 2 μM), conjugated lipids (from 2 to 10 mol %), TCEP (10 to 30 mM) and MMP-9 (0.002 μg, 0.02 μg and 0.1 μg) concentrations. We observed that the most optimal concentrations were the following, 500 nM Y406A, 2 mol % of conjugated lipids, 10 mM TCEP or 0.1 μg of MMP-9. When we used higher concentration of conjugated lipid, we needed more Y406A for efficient permeabilization, but also more TCEP or MMP-9, which we tried to avoid because of background effects of buffers in which these substances are stored and unwanted cleavage of proteins by MMP-9 (see below). After D22M conjugation, stability of D22M attachment to MLVs and cleavage from vesicles by different agents was checked by SDS-PAGE analysis (Figure 6). D22M was in both cases attached to the surface of vesicles and was successfully released from them after addition of either MMP-9 (Figure 6b) or TCEP (Figure 6e). We also checked whether membrane-conjugated D22M could be released from the membrane upon incubation of D22M-conjugated vesicles with Y406A. Indeed, the majority of the D22M was released from the vesicle membrane upon MMP-9 or TCEP cleavage with only minor amounts of D22M visible on the SDS-PAGE gels in the pellet fraction (Figures 6c and f), which might be due to the fact that not all of the D22M dissociated from Y406A despite the fact it was cleaved from the vesicles. On the other hand Y406A was not affected by such treatments and remained stably associated with the lipid membrane (Figures 6c and f), apart from minor degradation of Y406A in the pelleted fraction in the presence of MMP-9 (Figure 6c), which, however, did not have a great effect on Y406A’s ability to form pores (see below).
Membrane System with Logic Gate Based on Y406A and D22M
Conjugation of D22M to the lipid membrane of vesicles allowed development of a controllable system with different logic gates (Figure 7a). We followed calcein release from the vesicles to prove the gate opening (i.e., pore formation) of different systems. To develop an AND gate, D22M was attached by maleimide conjugation using DSPE-PEG2000Mal, and calcein was released from MLVs only upon both low pH and MMP-9 cleavage (Figure 7b). In the second system, an OR-AND gate could be conceptualized (Figure 7c). Employment of DSPE-PEG2000PDP was used to attach D22M to the vesicles, and the release of D22M from the vesicles was achieved by either the reducing agent or MMP-9, whereas system activation was achieved when one or both of those signals were present in addition to low pH in the surrounding buffer solution. This experiment was performed in microplate wells with ordered addition of different reagents and no possibility for washing away the components present in the reaction mixture. However, robust and significant differences between the closed (on) and the opened (off) states were found in both cases (Figure 7).
Figure 7.

A tunable vesicle system. (a) Y406A activity is controlled by pH and the reversible inhibitor D22. Vesicles are at pH 8.0; therefore, two input signals are needed for system activation: lowering pH with HCl and elimination of D22 from the system by release from vesicles induced by MMP-9 or TCEP. (b) A system based on DSPE-PEG2000Mal lipid. Bars represent fluorescence increase due to release of calcein from MLVs. A logic truth table and schematic representation of an AND gate is presented below the graph. (c) A system based on DSPE-PEG2000PDP lipid, which gives more flexibility for the removal of D22 from the vesicle membrane. A logic truth table and schematic representation of an OR-AND gate is presented beside the graph. n = 3–6, mean ± SD *P < 0.05; **P < 0.01.
Discussion
Existing protein logic gates are generally operated via proteolytic cleavage11,15 or allosteric regulation of conformationally stable proteins. Complex protein structures, enabling multidomain gating, are implemented in order to obtain higher efficiency of the system.10,11,13,21,44−47 However, a majority of the work on protein logic gates is done with enzymes in solution. A valuable contribution to synthetic biology represent PFTs,24 which are an excellent system since they are well characterized at the structural level, form stable pore complexes that are hard to dissociate once assembled, operate at the level of lipid membranes, and are amenable for rational design and mutagenesis to yield variants with changed useful properties. The incorporation of a stimuli-sensing PFT into lipid vesicles could thus enable a controlled opening of the membrane and consequently release of the encapsulated content at the desired site.
In this work we used a mutant of the PFT LLO, Y406A, to design protein logic gate at the surface of lipid membranes. Y406A is perfectly suited for such applications, since it forms transmembrane homo-oligomeric pores from soluble monomeric units in a controllable stepwise mechanism. We have shown before40 that single amino acid mutation in LLO D2, Y406A, drastically alters protein characteristics, most significantly its pH-dependent behavior, whereas the size of the formed pores remains generally the same. Here we employed a small protein molecule D22, which specifically and reversibly inhibits activity of Y406A by binding to the domain D2. The complex Y406A-D22 can still bind to lipid membranes (Figure 3); however, Y406A in such complex is not able to form pores. Our results suggest that D22 binds to the D2 of Y406A, which might prevent conformational changes in Y406A needed in the final stages of pore formation.48 An additional advantage of LLO-based systems is that LLO forms large pores with diameter of 25–40 nm in lipid membranes. This feature makes it extraordinary for synthetic biology and could be used for example in liposome-based delivery systems of larger proteins. In comparison to small pores of approximately 1.5–2 nm in diameter that are formed by some toxins, such as α-toxin from Staphylococcus aureus(49) and aerolysin-like proteins such as lysenin,50 Y406A can make much larger pores enabling release of larger compounds, such as 10 kDa dextran shown in Figure 4, or even larger 70 kDa dextran (∼12 nm in diameter).40 Y406A-D22 system could thus be used for liposome-based delivery of small proteins, such as nanobodies, or small enzymes.
We developed protein logic functioning for precise activation and release of lipid vesicles-encapsulated cargo. Our robust system consists of a vesicle with membrane-conjugated D22 that binds and reversibly inhibits Y406A. The system can be activated upon two different and very specific signals (low pH in conjunction with reductive environment or MMP-9), and therefore opens the vesicle in regulated manner and release the vesicle-encapsulated cargo. The nature of the system follows the “all-or-nothing” principle, where the response is fully expressed once activated. Two-level complex gates, especially with the possibility of different activating inputs that do not cancel each other out, function much better due to higher efficiency and simplicity compared to the sum of the individual gates. The modularity makes the logical functioning of this system suitable for diverse applications because of distinct operations being executed by separate components and the ease of expansion of input signals by incorporation of recognition sites for other proteolytic enzymes at the C-terminus of D22, or integration of other cleavable linkers, responsive to various reagents (reducing, oxidizing, nucleophilic/basic, electrophilic/acidic), metals, lysosomal enzymes, light, etc.51,52 Further development of logic gate system is also possible with utilization of other LLO variants. LLO undergoes extensive conformational rearrangements during pore formation, for example two clusters of helices in D3 rearrange and form two β-hairpins that are inserted in lipid membrane and form the β-barrel of the final pore (Figure 1b). Introduction of two cysteine residues in one of the helix clusters, A318C-L334C, prevents these rearrangements and pore formation upon disulfide formation.53 Such LLO variants could help to achieve additional logic operations using reductants as one of the input signals.
The presented system has a high potential to be used for therapeutic purposes, as it enables vesicle packing for directed administration of small molecules, biologics, or other protein or nonprotein cargos of high molecular weight, enabling very precise regulation and reliable delivery. The provided system has a potential in cancer treatment, where especially the differences in pH,54 redox potential,55 and presence of metalloproteinases56 between normal and cancerous tissue are significant57 and could provide input signals for the Y406A-D22 system.
Methods
Materials
The plasmid pRDV was kindly provided by dr. Plückthun’s lab.42,58 DARPin gene library was obtained from Eurofins Genomics, Germany. 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), cholesterol (Chol), 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide (polyethylene glycol)-2000] (DSPE-PEG2000Mal) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[3-(2-pyridyl)-dithiopropionyl (polyethylene glycol)-2000] (DSPE-PEG2000PDP) were from Avanti Polar Lipids (Alabaster, United States). Lissamine Rhodamine B 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (rhodamine DHPE) was purchased from Invitrogen (United States). Matrix Metalloproteinase 9 (MMP-9) was from Merck, Germany. All other chemicals were from Sigma, United States, unless stated otherwise.
D22 Design and Ribosome Display
The inhibitor that potently, specifically and selectively binds to Y406A, D22, was developed via directed evolution approach by using a randomized gene library of human ankyrin repeat consensus sequence.59 It contains 2 capping and 3 modular ankyrin repeats, each repeat forming a β-sheet followed by two α-helices. We applied diversity to the elements of secondary structure by randomizing 20 amino acid residues per each of 3 internal modular repeats: on the β-sheet and on the first α-helix. The resulting DARPin naïve library was cloned into pRDV vector and subjected to 6 rounds of ribosome display selection technique, following the protocol of Dreier and Plückthun,41 with LLO directly immobilized on plastic surface as a target (concentration decreasing from 200 nM at first round to 20 nM to sixth round of selection). Selection pressure was performed with the following washing steps: 5 short washes at the first round, gradually enhancing to 5 × short, 2 × 20 min, 40 min, 1 short wash at the sixth round.
Cloning, Expression, and Purification of LLO, Y406A, D22, and D22M
Genes for the wild-type LLO, Y406A, or D22 were multiplied by PCR, cleaved and inserted into a precleaved pProEXHTb expression vector. DARPin 22M (D22M) was created by substituting Cys50 and Cys167 to Ser (to avoid unspecific conjugation) and extending the protein at the C-terminal end with MMP-9 cleavage site sequence GPLGMLSQ,60 followed by the GGGSGGGS linker and the final residue Cys (for specific conjugation to the lipids). The gene for D22M was inserted into the precleaved pET28a expression vector. All constructs were verified by nucleotide sequencing. Expression of genes was performed in Escherichia coli BL21 (DE3) pLysS strain (Novagen, United States) in terrific broth (TB) supplemented with ampicillin. Cells were grown at 37 °C with shaking until optical density at 600 nm reached ∼1. The expression of desired genes was then induced with 0.5 mM IPTG (final concentration). Cells were grown for additional 5 h at 37 °C (D22) or 20 h at 20 °C, respectively (LLO and Y406A), centrifuged for 10 min at 2800g at 4 °C and frozen to −20 °C. Buffer containing 50 mM Na-phosphate, 250 mM NaCl, 5 mM 2-Mercaptoethanol and 2 mM phenylmethylsulfonyl fluoride (PMSF) at pH 6.5 was added to thawed lysate, which was then sonicated and centrifuged for 1 h at 35 000g at 4 °C. Supernatants were filtered through 0.45 and 0.22 μm filters. Filtrates were purified by immobilized metal affinity chromatography (IMAC), using a 9.6-ml Ni-NTA column (Qiagen, Germany), coupled to the Åkta FPLC system (Amersham Biosciences, United Kingdom). Column was equilibrated with buffer, containing 50 mM Tris-HCl, 500 mM NaCl at pH 7.4 following the application of lysate and washing of unbound proteins with buffer containing 10 mM and 60 mM of imidazole. The bound proteins were eluted by imidazole in the buffer (500 mM for D22 and D22M, and 300 mM for LLO wt and Y406A). His-tags were than cleaved off (0.6 mg Tobacco Etch Virus (TEV) protease to 1 mL of the sample) during overnight dialysis at 4 °C against 50 mM Tris-HCl, 500 mM NaCl at pH 7.4. Flow-through after additional IMAC, containing purified protein, was concentrated and buffer changed to 20 mM MES, 150 mM NaCl, pH 5.7 using Amicon Ultra 10 kDa MWCO (Merck, Germany). Content, size and purity of samples were observed by SDS-PAGE. Fractions containing desired proteins were pooled, concentration determined spectrophotometrically (Agilent 8453 UV–visible Spectroscopy System, United States), aliquoted and stored at −80 °C.
Enzyme-Linked Immunosorbent Assay (ELISA)
100 nM LLO was immobilized on Nunc Maxi-Sorp 96-well microtiter plate (Thermo Fisher, United States) overnight at 4 °C. Wells were washed after that three times with TBST buffer (10 mM Tris, 150 mM NaCl, 0.05% Tween-20 (Merck Millipore, United States), pH 7.4). After that, wells were blocked with buffer containing 0.5% BSA for 1 h and washed three times with TBST. Sample proteins (40 different clones of 5 μM DARPins with hexahistidine tags, isolated after six rounds of ribosome display) were added to wells and incubated for 1.5 h at room temperature with gentle shaking. After that, wells were washed 3 times with TBST and subsequently 1.6 nM mouse monoclonal anti-6His IgG antibodies (Santa Cruz Biotechnology, United States) were added and incubated in wells for 1.5 h at room temperature with gentle shaking. Wells were washed four times with TBST and 10 000 × diluted horseradish peroxidase-conjugated antimouse IgG antibodies (Sigma-Aldrich, United States) were added and incubated for 1 h at room temperature with gentle shaking. Wells were washed four times with TBST. 150 μL of the substrate system for ELISA detection TMB (3,3′,5,5′-tetramethylbenzidine)/H2O2 (Sigma-Aldrich, United States) was added per each well and incubated for 30 min in dark at room temperature. To stop the reaction, 75 μL of TMB substrate for ELISA (Stop solution, Sigma-Aldrich, United States) was added and absorbance was measured immediately with microplate reader Synergy MX (Biotek, United States) at 450 nm and at 600 nm for background subtraction.
Size Exclusion Chromatography (SEC)
Size exclusion chromatography was performed on UPLC Acquity Waters system (Waters Corporation, United States) with Acquity UPLC BEH200 1.7 μm column. The running buffer composition was 10 mM Tris-HCl, 150 mM NaCl, pH 7.4. Samples were injected at 0.4 mL/min flow rate at the concentration of single proteins being 26.6 μM; complexes were mixed at 1:1 molar ratio and preincubated for 30 min at room temperature before the injection. The protein elution profile was monitored with a UV-detector operated at 280 nm.
Isothermal Titration Calorimetry (ITC)
ITC experiments were conducted using a VP-ITC (MicroCal, United States). All protein samples for ITC measurements contained 5 mM 2-mercaptoethanol. 38 injections of 8 μL (first 2 injections 2 μL) of D22 were added by using a computer-controlled microsyringe at intervals of 300 s into the solution of LLO, Y406A, or buffer under constant stirring (307 rpm) at 25 °C. Reference power was set to 10 μcal/s and initial delay before injections was 180 s. The concentrations used for the experiments were 5.9 μM for LLO and Y406A, and 54.9 μM for D22 (the concentration in the syringe). Titrations were carried out in the buffer containing 22 mM MES, 150 mM NaCl and 5 mM 2-mercaptoethanol, at pH 5.7. Buffer was the same for all components to cancel out the possibility of the buffer background signal. Binding parameters were calculated by integration of individual titration peaks and presenting the resulting binding isotherm in a Wiseman plot. Isotherm was fitted to a model for one set of sites, which works for one site or n identical sites, binding constant (K) and enthalpy (ΔH). We calculated stoichiometry (n), ΔH and K for each data set using the one-site reaction model in the Origin 7 ITC software package (MicroCal, USA).
Small-Angle X-ray Scattering (SAXS) Data Collection, Ab Initio Shape Determination and Molecular Modeling
Y406A and D22 were mixed in a 1:1 molar ratio and stored at −70 °C. Buffer composition was 20 mM MES, 150 mM NaCl, 1 mM DTT, pH 5.7. After thawing, the sample was centrifuged and serial dilution from 164 μM to 5.5 μM was prepared. SAXS measurements were performed at the high brilliance synchrotron beamline P12 at the European Molecular Biology Laboratory (EMBL, DESY, Hamburg). The X-ray wavelength was λ = 1.24 Å. Detector (Pilatus 2M) to sample distance was 3 m, resulting in a q range of 0.003–0.5 Å–1. Measurements were performed at 23 °C, samples storage temperature before the measurement was 10 °C. Exposure time was 45 ms and 20 frames were recorded in one second with constant flow through the quartz capillary. The flow ensures that a certain sample volume is illuminated only for a certain time and is displaced by the fresh sample volume. On the basis of the comparison of successive frames, no detectable radiation damage was observed. Frames were normalized to the transmitted beam intensity, azimuthally integrated and averaged, resulting in scattering curves l(q) versusq. The background from the quartz capillary and sample buffer was subtracted. These averaged and subtracted difference curves were next normalized by their concentrations. Low quality data and very low and very high q values were discarded. Valid q range was between 0.006 and 0.48 Å–1. Two approaches to data analysis were tested. First, concentrations from 37.5 μM to 5.5 μM were used to extrapolate data and in parallel concentrations of 73 μM (high-s) and 9.7 μM (low-s) were merged for modeling. We saw no major differences between extrapolated data and merged data and we report data from the merged data set. All data manipulations were performed with PRIMUS, part of ATSAS software suite.61 Low-resolution shape envelopes were determined using the ab initio bead modeling program DAMMIF62 as part of the ATSAS online software suite. The results of 20 independent DAMMIF runs were clustered and analyzed online using DAMAVER63 to identify the most representative models. Rigid body molecular modeling was conducted using in silico mutated crystal structures of Y406A and D22. LLO (PDB ID: 4CDB(36)) residue Tyr406 was mutated to Ala using Pymol.64 First 19 residues of LLO were absent in the crystal structure and were modeled using Modeler software65 to neutralize the difference between the measured and modeled Y406A. D22 was modeled with SWISS-MODEL66 and model PDB ID: 2P2C(67) was used as the template. Rigid body modeling was conducted in ATSAS online service using the program SASREF.68 Merged data set and two subunits were inserted and modeled without any symmetry. Models were aligned in Pymol with SASpy plugin (supalm).69
Lipid Vesicles Preparation
POPC and cholesterol were dissolved in chloroform and mixed together in a molar ratio 3:2 or 1:1. The solvent was removed under reduced pressure by rotary evaporator (Büchi, Switzerland) and the resulting lipid film on glass flask wall was left to dry for additional 4 h in a vacuum (SpeedVac, Thermo Scientific, United States). The film was then resuspended in the buffer (20 mM MES, 150 mM NaCl, pH 5.7, or 20 mM Tris, 150 mM NaCl, pH 7.4) by alternatingly vortexing and heating (65 °C, water bath) the sample for 10–15 min. To get multilamellar vesicles (MLVs), this sample was additionally freeze–thawed in liquid N2 for 6 times. To obtain SUVs, MLVs were pulse-sonicated on ice using a Cole Parmer CPX500 500 W sonicator for 15 min (10 s on/10 s off duty cycle) at 38% amplitude. MLVs were extruded through 100 nm pore size polycarbonate membranes by the mini extruder (Avanti Polar Lipids, United States) to yield large unilamellar vesicles (LUVs) of 100 nm in diameter. Giant unilamellar vesicles (GUVs) were prepared by electroformation method, as described previously by Ruan et al.(37) Shortly, lipid mixture POPC:Chol 3:2 (mol:mol) with addition of 1 mol % rhodamine-DHPE was placed on ITO-slides and dried under the nitrogen stream. Electroformation was carried out between two conductive ITO-slides (Vesicle Prep Pro, Nanion Technologies, Germany) in sucrose solution (300 mM sucrose, 1 mM MES, pH 6.5, or 300 mM sucrose, 1 mM Tris, pH 8.0) for 4 h with 3 V amplitude and 5 Hz frequency. Afterward, GUVs were sedimented with glucose solution (300 mM glucose, 1 mM MES, pH 6.5, or 300 mM glucose, 1 mM Tris, pH 8.0) and addition of buffer (10 mM MES, 150 mM NaCl, pH 6.5, or 20 mM Tris, 150 mM NaCl, pH 8.0). The osmolarity of all solutions was adjusted with Osmomat 3000 (Gonotec GmbH, Germany).
Surface Plasmon Resonance (SPR)
SPR measurements were performed on a Biacore X100 (GE Healthcare, Biacore AB, Sweden) at room temperature on an L1 sensor chip. Large unilamellar vesicles (LUVs) were loaded on equilibrated sensor chip (20 mM MES, 150 mM NaCl, pH 5.7). Reference flow cell contained LUVs composed only of POPC (representing negative control as LLO requires cholesterol for efficient membrane binding), whereas active flow cell contained LUVs with POPC and cholesterol at 3:2 (mol:mol) ratio. The LUV-coated chip surface was prepared as described.70,71 All protein samples for SPR measurements contained 10 mM DTT. For studies regarding binding of proteins or protein complexes to membranes, 100 nM Y406A, 5 μM D22, or premixed samples of 5 μM D22 and 100 nM Y406A (incubated for 45 min at room temperature) were injected over the LUV-coated surfaces for 3 min at a flow rate 5 μL/min and dissociated for 20 min. For studies of interactions of D22 with membrane-bound proteins, 100 nM LLO or Y406A was injected over LUV-coated surfaces for 3 min at flow rate 5 μL/min, followed by dissociation for 5 min. Following one blank injection, 5 μM D22 was injected over LLO bound to vesicles for 3 min at a flow rate 5 μL/min and left to dissociate for 20 min. Blank injections were subtracted from sample sensorgrams to eliminate the influence of buffer and DTT. Data was processed with Biaevaluation v3.2 (GE Healthcare, United Kingdom) software. Three individual experiments were conducted for each case.
Vesicle Sedimentation Assay
Sedimentation assays were carried out in the buffered system (20 mM MES, 150 mM NaCl) at pH 5.7. For studying binding of D22 complexed with binding partner to membranes, protein pairs (LLO + D22 or Y406A + D22) with D22 in 5 × molar excess were first incubated for 15 min at room temperature and then multilamellar vesicles (MLVs) in 2000 × molar excess of lipids were added. For studying interaction of D22 with membrane-inserted LLO or Y406A, LLO or Y406A was first incubated with MLVs for 15 min, followed by addition of D22. Those mixtures were left to incubate for an additional 15 min at room temperature and then centrifuged at 16 100g for 15 min. Supernatants were transferred to fresh microtubes and centrifuged again, whereas pellets were washed with 50 μL of the buffer (resuspended and centrifuged again) and finally resuspended in 15 μL of the buffer. Samples were analyzed in the presence of 10 mM DTT with SDS-PAGE, showing MLV-bound portion in pellet and unbound molecules in supernatant. Proteins were visualized with SimplyBlue SafeStain (Thermo Fisher Scientific, United States) and the amount of bound D22 was determined by densitometry by using ImageJ.72
Hemolytic Assay
Bovine erythrocytes, stored at 4 °C in Alsevier preservative, were washed four times with resuspension in the erythrocyte buffer (20 mM MES, 140 mM NaCl, pH 5.7) and centrifugation at 800g for 5 min at room temperature. Erythrocytes were diluted with erythrocyte buffer to yield absorbance of 1.0 at 630 nm, which was determined by microplate reader Synergy MX (Biotek, United States). 50 μL of LLO or Y406A (final concentration 2.3 nM) was mixed with 50 μL of D22 (final concentration 5 μM) and incubated for 45 min at room temperature. Afterward, 100 μL of erythrocyte suspension was added to each well. The final volume in all wells was 200 μL. Absorbance at 630 nm was measured every 20 s for 20 min at 25 °C. Relative rate of hemolytic activity was determined as the ratio of maximal hemolytic rate in the presence and absence of D22, respectively.
Permeabilization Experiments
GUVs were mixed with the buffer (10 mM MES, 150 mM NaCl, pH 6.5, or 20 mM Tris, 150 mM NaCl, pH 8.0), fluorescent dextran of 10 kDa in size (Sigma; FD10, final concentration of 1 mg/mL) and proteins in the following combinations: 50 nM Y406A alone, 5 μM D22M alone or premixed 50 nM Y406A + 5 μM D22M. Mixtures were incubated for 30 min at room temperature before imaging. Images were recorded on DMI6000 CS inverted microscope with TCS SP5 laser scanning system (both Leica Microsystems, Germany) with a 63 × oil-immersion objective (numerical aperture = 1.4). The rhodamine-containing GUV membrane was excited at 550 nm, and emission was detected from 570 to 600 nm. FD10 were excited at 488 nm, and emission was detected from 497 to 527 nm. Percent of permeabilization was calculated from green channel fluorescent intensities in ImageJ software, namely fluorescent intensities inside the vesicles were divided by background intensities outside the vesicles. For each condition 50 to 500 GUVs were analyzed.
Calcein Release from SUVs and LUVs
POPC:Chol 3:2 (mol:mol) lipids were used to prepare SUVs and POPC:Chol 1:1 (mol:mol) lipids for LUVs, both in 10 mM HEPES, 150 mM NaCl, 1 mM EDTA, 80 mM calcein, pH 8.0. Excess calcein was removed from vesicles suspension by gravity gel filtration on the Sephadex G-50 matrix (GE Healthcare, United Kingdom). Concentration of POPC and cholesterol was enzymatically determined with Phospholipids C kit and Free Cholesterol E kit (Wako Diagnostics, United States), respectively. Size and uniformity of SUVs and LUVs were checked with Dynamic Light Scattering (Zetasizer Nano, Malvern Panalytical, United Kingdom). We followed calcein release from SUVs in 96-well nontransparent microtiter plates (Costar, United States) with microplate reader Synergy MX (Biotek, United States). Vesicles with calcein were complemented with various combinations of 1 μM LLO or Y406A, and 5 μM D22 in buffer containing 10 mM HEPES, 150 mM NaCl, 1 mM EDTA at pH 6.5 or 8.0. From obtained fluorescence signals, we subtracted the fluorescence of the background, containing only vesicles with calcein and D22. Total release of calcein from SUVs was obtained by adding 2 mM detergent Triton X-100. To follow calcein release from LUVs, we used fluorimeter LS 55 Fluorescence Spectrometer (PerkinElmer, United States), measuring in a cuvette with final volume of 1 mL. Final concentration of lipids was 50 μM. Baseline fluorescence was followed for 300 s before the addition of proteins Y406A (30 nM final concentration) or D22 (1 μM final concentration). For the Y406A protein activation, pH was lowered to pH 6.5 by addition of 3 μL 7% HCl. For the Y406A protein inhibition, D22 was used either by immediate addition (10 s) after activation of Y406A or by preincubation of D22 and Y406A before or after addition to LUVs before or after lowering the pH. Fluorescence was then followed for at least 400 s before the addition of Triton X-100 (final concentration of 2 mM) to achieve full release of calcein.
Conjugation and Cleavage of D22M from MLVs
POPC:Chol:DSPE-PEG2000Mal 60:38:2 (molar ratio) and POPC:Chol:DSPE-PEG2000PDP 60:38:2 (molar ratio) mixtures were used to prepare MLVs in 20 mM HEPES, 300 mM NaCl, pH 8.0. D22M was conjugated on MLVs via maleimide conjugation (for MMP-9 cleavage) or PDP (for cleavage with the reducing agent) at 25 °C and shaking at 500 rpm under argon atmosphere overnight. Excess D22M was removed by centrifugation (800g, 8 min, 20 °C) and addition of buffer. First, the cleavage success of D22M from MLVs was analyzed by addition of 0.1 μg MMP-9 or 10 mM TCEP and incubation at 25 °C for 30 min. Afterward, the sample was centrifuged, and supernatant and pellet were analyzed by SDS-PAGE. Second, the binding of Y406A to D22M conjugated-MLVs and additional cleavage was analyzed. For this experiment, 1 μg of Y406A was added to MLVs with previously conjugated D22M and incubated for 30 min at 25 °C. Then the sample was centrifuged, the supernatant was saved for SDS-PAGE analysis, the pellet was resuspended in the buffer, and 0.1 μg MMP-9 or 10 mM TCEP were added and incubated for 30 min at 25 °C. The sample was centrifuged, and the supernatant and pellet were analyzed by SDS-PAGE.
Calcein Release from MLVs with Conjugated D22M
Calcein release was followed by using 96 well microtiter plates (Costar, United States). Here, DSPE-PEG2000Mal 60:38:2 MLVs conjugated with D22M were used for the cleavage experiments of D22M with MMP-9 and DSPE-PEG2000PDP 60:38:2 MLVs were used for the cleavage experiments of D22M with reducing agent TCEP. Final volume of reactions was 50 μL. First, the background fluorescence of 0.5 mM MLVs and buffer 20 mM HEPES, 300 mM NaCl, pH 8.0 (in the case of experiments with MMP-9 buffer also contained 10 mM CaCl2, 100 μM ZnSO4) was measured. Then Y406A was added at a final 500 nM concentration to the MLVs, and incubated for 30 min at 25 °C and 600 rpm. Afterward, 0.1 μg MMP-9 or 10 mM TCEP was added (incubated at 25 °C and 600 rpm for 30 min). Finally, 0.5 μL 3.5% HCl was added (incubated at 25 °C and 600 rpm for 30 min). Triton X-100 was added to achieve full release of the calcein. Fluorescence signal was measured by the Synergy MX microplate reader (Biotek, United States).
Statistical Analysis
For all experiments, raw data points are presented as circles on graphs, together with mean values ± standard deviation (SD). Unpaired two-tailed t tests assuming equal variances were performed using Origin 8.1 to present the differences between samples. Equalities of variances were assessed by two-sample F-test. A significant difference was determined by P-value <0.05. All the experiments were replicated at least three times and represented biological replicates.
Acknowledgments
The work presented in this paper was supported by The Slovenian Research Agency Grants P1-0391 and J4-8225. We thank Andreas Plückthun for the pRDV vector. We thank Alexey Kikhney from EMBL c/o DESY Synchrotron, Germany, to assist us with the SAXS experiments, Proposal No. SAXS-429.
Author Contributions
§ N.O. and S.A. equally contributed to the work presented in this paper. N.O., S.A., M.P., and G.A. designed the study and experiments, N.O. and S.A. performed all experiments except SAXS, M.K. and M.P. performed SAXS experiments, N.O. and G.A. wrote the manuscript. All authors commented and approved the manuscript.
The authors declare no competing financial interest.
References
- Evans A. C.; Thadani N. N.; Suh J. (2016) Biocomputing nanoplatforms as therapeutics and diagnostics. J. Controlled Release 240, 387–393. 10.1016/j.jconrel.2016.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil A. S.; Collins J. J. (2010) Synthetic biology: Applications come of age. Nat. Rev. Genet. 11, 367–379. 10.1038/nrg2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyamoto T.; Razavi S.; Derose R.; Inoue T. (2013) Synthesizing biomolecule-based boolean logic gates. ACS Synth. Biol. 2, 72–82. 10.1021/sb3001112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaber R.; Lebar T.; Majerle A.; Šter B.; Dobnikar A.; Benčina M.; Jerala R. (2014) Designable DNA-binding domains enable construction of logic circuits in mammalian cells. Nat. Chem. Biol. 10, 203–208. 10.1038/nchembio.1433. [DOI] [PubMed] [Google Scholar]
- Lebar T.; Bezeljak U.; Golob A.; Jerala M.; Kadunc L.; Pirš B.; Stražar M.; Vučko D.; Zupančič U.; Benčina M.; Forstnerič V.; Gaber R.; Lonzarić J.; Majerle A.; Oblak A.; Smole A.; Jerala R. (2014) A bistable genetic switch based on designable DNA-binding domains. Nat. Commun. 5, 5007. 10.1038/ncomms6007. [DOI] [PubMed] [Google Scholar]
- Lebar T., and Jerala R. (2018) Designed transcriptional regulation in mammalian cells based on TALE- and CRISPR/dCas9, in Synthetic Biology Methods and Protocols, Methods in Molecular Biology, (Braman J. C., Ed.), pp 191–203, Humana Press, New York, NY. [DOI] [PubMed] [Google Scholar]
- Qian L.; Winfree E. (2011) Scaling up digital circuit computation with DNA strand displacement cascades. Science 332, 1196–1201. 10.1126/science.1200520. [DOI] [PubMed] [Google Scholar]
- Yasuga H.; Kawano R.; Takinoue M.; Tsuji Y.; Osaki T.; Kamiya K.; Miki N.; Takeuchi S. (2016) Logic gate operation by DNA translocation through biological nanopores. PLoS One 11, e0149667 10.1371/journal.pone.0149667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faulhammer D.; Cukras A. R.; Lipton R. J.; Landweber L. F. (2000) Molecular computation: RNA solutions to chess problems. Proc. Natl. Acad. Sci. U. S. A. 97, 1385–1389. 10.1073/pnas.97.4.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J. H.; Ostermeier M. (2015) Rational design of a fusion protein to exhibit disulfide-mediated logic gate behavior. ACS Synth. Biol. 4, 400–406. 10.1021/sb500254g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink T.; Lonzarić J.; Praznik A.; Plaper T.; Merljak E.; Leben K.; Jerala N.; Lebar T.; Strmšek Ž.; Lapenta F.; Benčina M.; Jerala R. (2019) Design of fast proteolysis-based signaling and logic circuits in mammalian cells. Nat. Chem. Biol. 15, 115–122. 10.1038/s41589-018-0181-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moseley F.; Halámek J.; Kramer F.; Poghossian A.; Schöning M. J.; Katz E. (2014) An enzyme-based reversible CNOT logic gate realized in a flow system. Analyst 139, 1839–1842. 10.1039/c4an00133h. [DOI] [PubMed] [Google Scholar]
- Stein V.; Alexandrov K. (2014) Protease-based synthetic sensing and signal amplification. Proc. Natl. Acad. Sci. U. S. A. 111, 15934–15939. 10.1073/pnas.1405220111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Arugula M. A.; Halámek J.; Pita M.; Katz E. (2009) Enzyme-based NAND and NOR logic gates with modular design. J. Phys. Chem. B 113, 16065–16070. 10.1021/jp9079052. [DOI] [PubMed] [Google Scholar]
- Gao X. J.; Chong L. S.; Kim M. S.; Elowitz M. B. (2018) Programmable protein circuits in living cells. Science 361, 1252–1258. 10.1126/science.aat5062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilburger C. E.; Jacobs M. L.; Lewis K. R.; Peruzzi J. A.; Kamat N. P. (2019) Controlling Secretion in Artificial Cells with a Membrane and Gate. ACS Synth. Biol. 8, 1224–1230. 10.1021/acssynbio.8b00435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamala K. P.; Martin-Alarcon D. A.; Guthrie-Honea K. R.; Boyden E. S. (2017) Engineering genetic circuit interactions within and between synthetic minimal cells. Nat. Chem. 9, 431–439. 10.1038/nchem.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Einfalt T.; Goers R.; Dinu I. A.; Najer A.; Spulber M.; Onaca-Fischer O.; Palivan C. G. (2015) Stimuli-Triggered Activity of Nanoreactors by Biomimetic Engineering Polymer Membranes. Nano Lett. 15, 7596–7603. 10.1021/acs.nanolett.5b03386. [DOI] [PubMed] [Google Scholar]
- Einfalt T.; Witzigmann D.; Edlinger C.; Sieber S.; Goers R.; Najer A.; Spulber M.; Onaca-Fischer O.; Huwyler J.; Palivan C. G. (2018) Biomimetic artificial organelles with in vitro and in vivo activity triggered by reduction in microenvironment. Nat. Commun. 9, 1127. 10.1038/s41467-018-03560-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G.; Voelker D. R.; Feigenson G. W. (2008) Membrane lipids: where they are and how they behave. Nat. Rev. Mol. Cell Biol. 9, 112–124. 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golynskiy M. V.; Koay M. S.; Vinkenborg J. L.; Merkx M. (2011) Engineering protein switches: sensors, regulators, and spare parts for biology and biotechnology. ChemBioChem 12, 353–361. 10.1002/cbic.201000642. [DOI] [PubMed] [Google Scholar]
- Arosio D.; Ricci F.; Marchetti L.; Gualdani R.; Albertazzi L.; Beltram F. (2010) Simultaneous intracellular chloride and pH measurements using a GFP-based sensor. Nat. Methods 7, 516–518. 10.1038/nmeth.1471. [DOI] [PubMed] [Google Scholar]
- Dal Peraro M.; van der Goot F. G. (2016) Pore-forming toxins: ancient, but never really out of fashion. Nat. Rev. Microbiol. 14, 77–92. 10.1038/nrmicro.2015.3. [DOI] [PubMed] [Google Scholar]
- Anderluh G.; Lakey J. H. (2008) Disparate proteins use similar architectures to damage membranes. Trends Biochem. Sci. 33, 482–490. 10.1016/j.tibs.2008.07.004. [DOI] [PubMed] [Google Scholar]
- Laventie B.; Potrich C.; Atmanène C.; Saleh M.; Joubert O.; Viero G.; Bachmeyer C.; Antonini V.; Mancini I.; Cianferani-Sanglier S.; Keller D.; Colin D. A.; Bourcier T.; Anderluh G.; van Dorsselaer A.; Dalla Serra M.; Prévost G. (2013) p-Sulfonato-calix[n]arenes inhibit staphylococcal bicomponent leukotoxins by supramolecular interactions. Biochem. J. 450, 559–571. 10.1042/BJ20121628. [DOI] [PubMed] [Google Scholar]
- Booth M. J.; Schild V. R.; Graham A. D.; Olof S. N.; Bayley H. (2016) Light-activated communication in synthetic tissues. Sci. Adv. 2, e1600056 10.1126/sciadv.1600056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Provoda C. J.; Stier E. M.; Lee K. D. (2003) Tumor cell killing enabled by listeriolysin O-liposome-mediated delivery of the protein toxin gelonin. J. Biol. Chem. 278, 35102–35108. 10.1074/jbc.M305411200. [DOI] [PubMed] [Google Scholar]
- Robertson J. W. F.; Rodrigues C. G.; Stanford V. M.; Rubinson K. A.; Krasilnikov O. V.; Kasianowicz J. J. (2007) Single-molecule mass spectrometry in solution using a solitary nanopore. Proc. Natl. Acad. Sci. U. S. A. 104, 8207–8211. 10.1073/pnas.0611085104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke J.; Wu H.; Jayasinghe L.; Patel A.; Reid S.; Bayley H. (2009) Continuous base identification for single-molecule nanopore DNA sequencing. Nat. Nanotechnol. 4, 265–270. 10.1038/nnano.2009.12. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Montana V.; Grubišić V.; Stout R. F.; Parpura V.; Gu L. Q. (2015) Nanopore sensing of botulinum toxin type B by discriminating an enzymatically cleaved peptide from a synaptic protein synaptobrevin 2 derivative. ACS Appl. Mater. Interfaces 7, 184–192. 10.1021/am5056596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Zheng D.; Tan Q.; Wang M.; Gu L. (2011) Nanopore-based detection of circulating microRNAs in lung cancer patients. Nat. Nanotechnol. 6, 668–674. 10.1038/nnano.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braha O.; Bayley H.; Gu L. Q.; Zhou L.; Lu X.; Cheley S. (2000) Simultaneous stochastic sensing of divalent metal ions. Nat. Biotechnol. 18, 1005–1007. 10.1038/79275. [DOI] [PubMed] [Google Scholar]
- Villar G.; Graham A. D.; Bayley H. (2013) A tissue-like printed material. Science 340, 48–52. 10.1126/science.1229495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elani Y.; Law R. V.; Ces O. (2014) Vesicle-based artificial cells as chemical microreactors with spatially segregated reaction pathways. Nat. Commun. 5, 5305. 10.1038/ncomms6305. [DOI] [PubMed] [Google Scholar]
- Hamon M. A.; Ribet D.; Stavru F.; Cossart P. (2012) Listeriolysin O: the Swiss army knife of Listeria. Trends Microbiol. 20, 360–368. 10.1016/j.tim.2012.04.006. [DOI] [PubMed] [Google Scholar]
- Köster S.; van Pee K.; Hudel M.; Leustik M.; Rhinow D.; Kühlbrandt W.; Chakraborty T.; Yildiz O. (2014) Crystal structure of listeriolysin O reveals molecular details of oligomerization and pore formation. Nat. Commun. 5, 3690. 10.1038/ncomms4690. [DOI] [PubMed] [Google Scholar]
- Ruan Y.; Rezelj S.; Bedina Zavec A.; Anderluh G.; Scheuring S. (2016) Listeriolysin O membrane damaging activity involves arc formation and lineaction - implication for Listeria monocytogenes escape from phagocytic vacuole. PLoS Pathog. 12, e1005597 10.1371/journal.ppat.1005597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podobnik M.; Marchioretto M.; Zanetti M.; Bavdek A.; Kisovec M.; Cajnko M. M.; Lunelli L.; Serra M. D.; Anderluh G. (2015) Plasticity of lysteriolysin O pores and its regulation by pH and unique histidine. Sci. Rep. 5, 9623. 10.1038/srep09623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulvihill E.; Van Pee K.; Mari S. A.; Müller D. J.; Yildiz Ö. (2015) Directly observing the lipid-dependent self-assembly and pore-forming mechanism of the cytolytic toxin listeriolysin O. Nano Lett. 15, 6965–6973. 10.1021/acs.nanolett.5b02963. [DOI] [PubMed] [Google Scholar]
- Kisovec M.; Rezelj S.; Knap P.; Cajnko M.; Caserman S.; Flašker A.; Žnidaršič N.; Repič M.; Mavri J.; Ruan Y.; Scheuring S.; Podobnik M.; Anderluh G. (2017) Engineering a pH responsive pore forming protein. Sci. Rep. 7, 42231. 10.1038/srep42231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier B., and Plückthun A. (2011) Ribosome Display: A Technology for Selecting and Evolving Proteins from Large Libraries, in PCR Protocols. Methods in Molecular Biology (Methods and Protocols), (Park D. J., Ed.), pp 283–306, Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- Binz H. K.; Amstutz P.; Kohl A.; Stumpp M. T.; Briand C.; Forrer P.; Grütter M. G.; Plückthun A. (2004) High-affinity binders selected from designed ankyrin repeat protein libraries. Nat. Biotechnol. 22, 575–82. 10.1038/nbt962. [DOI] [PubMed] [Google Scholar]
- Bavdek A.; Gekara N. O.; Priselac D.; Gutiérrez Aguirre I.; Darji A.; Chakraborty T.; Macek P.; Lakey J. H.; Weiss S.; Anderluh G. (2007) Sterol and pH interdependence in the binding, oligomerization, and pore formation of Listeriolysin O. Biochemistry 46, 4425–4437. 10.1021/bi602497g. [DOI] [PubMed] [Google Scholar]
- Sallee N. A.; Yeh B. J.; Lim W. A. (2007) Engineering modular protein interaction switches by sequence overlap. J. Am. Chem. Soc. 129, 4606–4611. 10.1021/ja0672728. [DOI] [PubMed] [Google Scholar]
- Karginov A. V.; Ding F.; Kota P.; Dokholyan N. V.; Hahn K. M. (2010) Engineered allosteric activation of kinases in living cells. Nat. Biotechnol. 28, 743–747. 10.1038/nbt.1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards W. R.; Busse K.; Allemann R. K.; Jones D. D. (2008) Linking the functions of unrelated proteins using a novel directed evolution domain insertion method. Nucleic Acids Res. 36, e78 10.1093/nar/gkn363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha J. H.; Butler J. S.; Mitrea D. M.; Loh S. N. (2006) Modular enzyme design: Regulation by mutually exclusive protein folding. J. Mol. Biol. 357, 1058–1062. 10.1016/j.jmb.2006.01.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tweten R. K.; Hotze E. M.; Wade K. R. (2015) The unique molecular choreography of giant pore formation by the cholesterol-dependent cytolysins of Gram-positive bacteria. Annu. Rev. Microbiol. 69, 323–340. 10.1146/annurev-micro-091014-104233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L.; Hobaugh M. R.; Shustak C.; Cheley S.; Bayley H.; Gouaux J. E. (1996) Structure of staphylococcal alpha-hemolysin, a heptameric transmembrane pore. Science 274, 1859–1866. 10.1126/science.274.5294.1859. [DOI] [PubMed] [Google Scholar]
- Podobnik M.; Savory P.; Rojko N.; Kisovec M.; Wood N.; Hambley R.; Pugh J.; Wallace E. J.; McNeill L.; Bruce M.; Liko I.; Allison T. M.; Mehmood S.; Yilmaz N.; Kobayashi T.; Gilbert R. J. C.; Robinson C. V.; Jayasinghe L.; Anderluh G. (2016) Crystal structure of an invertebrate cytolysin pore reveals unique properties and mechanism of assembly. Nat. Commun. 7, 11598. 10.1038/ncomms11598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchikama K.; An Z. (2018) Antibody-drug conjugates: recent advances in conjugation and linker chemistries. Protein Cell 9, 33–46. 10.1007/s13238-016-0323-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leriche G.; Chisholm L.; Wagner A. (2012) Cleavable linkers in chemical biology. Bioorg. Med. Chem. 20, 571–582. 10.1016/j.bmc.2011.07.048. [DOI] [PubMed] [Google Scholar]
- Cajnko M. M.; Marušić M.; Kisovec M.; Rojko N.; Benčina M.; Caserman S.; Anderluh G. (2015) Listeriolysin O affects the permeability of Caco-2 monolayer in a pore-dependent and Ca2+-independent manner. PLoS One 10, e0130471 10.1371/journal.pone.0130471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerweck L. E.; Seetharaman K. (1996) Cellular pH gradient in tumor versus normal tissue: Potential exploitation for the treatment of cancer. Cancer Res. 56, 1194–1198. [PubMed] [Google Scholar]
- Cerutti P. A. (1985) Prooxidant states and tumor promotion. Science 227, 375–381. 10.1126/science.2981433. [DOI] [PubMed] [Google Scholar]
- Bergers G.; Brekken R.; McMahon G.; Vu T. H.; Itoh T.; Tamaki K.; Tanzawa K.; Thorpe P.; Itohara S.; Werb Z.; Hanahan D. (2000) Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat. Cell Biol. 2, 737–744. 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimi M.; Ghasemi A.; Sahandi Zangabad P.; Rahighi R.; Moosavi Basri S. M.; Mirshekari H.; Amiri M.; Shafaei Pishabad Z.; Aslani A.; Bozorgomid M.; Ghosh D.; Beyzavi A.; Vaseghi A.; Aref A. R.; Haghani L.; Bahrami S.; Hamblin M. R. (2016) Smart micro/nanoparticles in stimulus-responsive drug/gene delivery systems. Chem. Soc. Rev. 45, 1457–1501. 10.1039/C5CS00798D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zahnd C.; Amstutz P.; Plückthun A. (2007) Ribosome display: selecting and evolving proteins in vitro that specifically bind to a target. Nat. Methods 4, 269–279. 10.1038/nmeth1003. [DOI] [PubMed] [Google Scholar]
- Zahnd C.; Wyler E.; Schwenk J. M.; Steiner D.; Lawrence M. C.; McKern N. M.; Pecorari F.; Ward C. W.; Joos T. O.; Plückthun A. (2007) A designed ankyrin repeat protein evolved to picomolar affinity to Her2. J. Mol. Biol. 369, 1015–28. 10.1016/j.jmb.2007.03.028. [DOI] [PubMed] [Google Scholar]
- Liu S.; Netzel-Arnett S.; Birkedal-Hansen H.; Leppla S. H. (2000) Tumor cell-selective cytotoxicity of matrix metalloproteinase-activated anthrax toxin. Cancer Res. 60, 6061–6067. [PubMed] [Google Scholar]
- Franke D.; Petoukhov M. V.; Konarev P. V.; Panjkovich A.; Tuukkanen A.; Mertens H. D. T.; Kikhney A. G.; Hajizadeh N. R.; Franklin J. M.; Jeffries C. M.; Svergun D. I. (2017) ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 50, 1212–1225. 10.1107/S1600576717007786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke D.; Svergun D. I. (2009) DAMMIF, a program for rapid ab-initio shape determination in small-angle scattering. J. Appl. Crystallogr. 42, 342–346. 10.1107/S0021889809000338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkov V. V.; Svergun D. I. (2003) Uniqueness of ab initio shape determination in small-angle scattering. J. Appl. Crystallogr. 36, 860–864. 10.1107/S0021889803000268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The PyMOL Molecular Graphics System, Version 2.0, Schrödinger, LLC.
- Webb B., and Sali A. (2017) Protein Structure Modeling with MODELLER, in Functional Genomics: Methods and Protocols, Methods in Molecular Biology, (Kaufmann M., Klinger C., and Savelsbergh A., Eds.), pp 39–54, Humana Press, New York, NY. [DOI] [PubMed] [Google Scholar]
- Waterhouse A.; Bertoni M.; Bienert S.; Studer G.; Tauriello G.; Gumienny R.; Heer F. T.; De Beer T. A. P.; Rempfer C.; Bordoli L.; Lepore R.; Schwede T. (2018) SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303. 10.1093/nar/gky427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweizer A.; Roschitzki-Voser H.; Amstutz P.; Briand C.; Gulotti-Georgieva M.; Prenosil E.; Binz H. K.; Capitani G.; Baici A.; Plückthun A.; Grütter M. G. (2007) Inhibition of caspase-2 by a designed ankyrin repeat protein: specificity, structure, and inhibition mechanism. Structure 15, 625–36. 10.1016/j.str.2007.03.014. [DOI] [PubMed] [Google Scholar]
- Petoukhov M. V.; Svergun D. I. (2005) Global rigid body modeling of macromolecular complexes against small-angle scattering data. Biophys. J. 89, 1237–1250. 10.1529/biophysj.105.064154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panjkovich A.; Svergun D. I. (2016) SASpy: A PyMOL plugin for manipulation and refinement of hybrid models against small angle X-ray scattering data. Bioinformatics 32, 2062–2064. 10.1093/bioinformatics/btw071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodnik V., and Anderluh G. (2013) Surface Plasmon Resonance for Measuring Interactions of Proteins with Lipid Membranes, in Lipid-Protein Interactions: Methods and Protocols, Methods in Molecular Biology, (Kleinschmidt J. H., Ed.), pp 23–36, Humana Press, Totowa, NJ. [DOI] [PubMed] [Google Scholar]
- Anderluh G.; Beseničar M.; Kladnik A.; Lakey J. H.; Maček P. (2005) Properties of nonfused liposomes immobilized on an L1 Biacore chip and their permeabilization by a eukaryotic pore-forming toxin. Anal. Biochem. 344, 43–52. 10.1016/j.ab.2005.06.013. [DOI] [PubMed] [Google Scholar]
- Schneider C. A.; Rasband W. S.; Eliceiri K. W. (2012) NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]